

Abstract
LncRNAs have been shown to be involved in the biological and pathological processes of acute myeloid leukemia (AML). Hox antisense intergenic RNA myeloid 1 (HOTAIRM1) was reported to be highly expressed in AML. However, the detailed role and molecular mechanism of HOTAIRM1 in AML pathogenesis remain undefined. In the present study, HOTAIRM1 and miR-148b expressions in AML patients and healthy controls were detected by qRT-PCR. Cell proliferation and apoptosis were evaluated by CCK-8 and flow cytometry assays, respectively. The regulatory interaction between HOTAIRM1 and miR-148b was explored by bioinformatics analysis using starBase v3.0 software and The Cancer Genome Atlas (TCGA) AML dataset. We found that the miR-148/miR-152 family members including miR-148a, miR-148b, and miR-152 were predicted to be potential targets of HOTAIRM1. HOTAIRM1 expression was negatively correlated with miR-148b expression but had no correlation with miR-148a/miR-152 expressions in AML samples from the TCGA dataset. HOTAIRM1 expression was higher while miR-148b expression was lower in AML patients than in healthy controls. A negative correlation between HOTAIRM1 and miR-148b in AML patients was observed. HOTAIRM1 silencing and miR-148b overexpression both suppressed cell proliferation and induced apoptosis in AML cells. miR-148b was identified as a target of HOTAIRM1 in AML cells. Moreover, HOTAIRM1 knockdown inhibited proliferation and induced apoptosis in AML cells by negatively regulating miR-148b. In summary, HOTAIRM1 was involved in the progression of AML through targeting miR-148b, shedding light on the biological function and molecular mechanism of HOTAIRM1 in AML.
LncRNAs have been shown to be involved in the biological and pathological processes of acute myeloid leukemia (AML).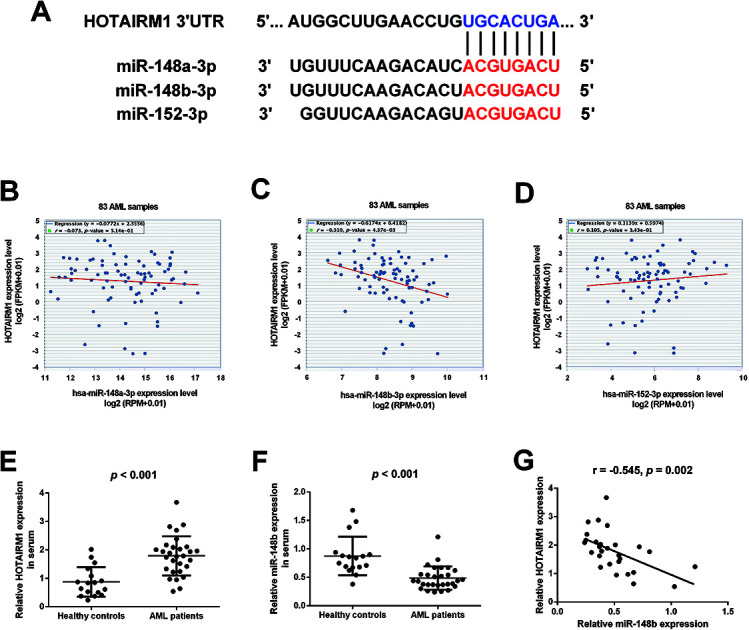
1. Introduction
Acute myeloid leukemia (AML), the most prevalent type of adult leukemia, is an aggressive heterogeneous hematological malignancy associated with several cytogenetic, molecular, and epigenetic aberrations.1,2 AML is characterized by the unlimited proliferation and blocked apoptosis of aberrantly differentiated myeloid cells in the bone marrow (BM) and peripheral blood, which ultimately leads to hematopoietic insufficiency and poor prognosis.3 According to the 2017 report by the American Cancer Society, there were an estimated 21 380 new AML cases and 10 590 deaths due to AML in the USA.4 Even with the great progress in current therapeutic approaches including chemotherapy and hematopoietic stem cell transplants, the clinical outcome of AML remains unsatisfactory due to the occurrence of chemoresistance and high rates of relapse following the standard therapy.5 Therefore, elucidating the molecular mechanisms underlying AML leukemogenesis is critical for the development of novel therapeutic approaches for AML to improve the survival of AML patients.
Non-coding RNAs (ncRNAs), a group of new regulatory molecule with no or limited protein-coding potential, are attracting increasing attention due to their important roles in AML pathogenesis.6 Long ncRNAs (lncRNAs) is a class of evolutionarily conserved ncRNAs longer than 200 nucleotides.7 Recently, substantive studies regarding lncRNAs have reported that lncRNAs are involved in a variety of physiological and pathological processes, such as cellular proliferation, differentiation, apoptosis, and metastasis.8 Several recent pieces of evidence have demonstrated that lncRNAs are frequently deregulated, which contribute to the pathogenesis and development of various human malignancies including AML and may serve as novel therapeutic targets.9 Hox antisense intergenic RNA myeloid 1 (HOTAIRM1), located in the 5′ end of homeobox A (HOXA) genomic cluster which is highly conserved in evolution, is transcribed antisense to the HOXA1/2 intergenic region.10 As a myeloid-specific lncRNA, HOTAIRM1 regulates gene expression in the HOXA cluster during myelopoiesis and is associated with myeloid formation and differentiation.11 It was demonstrated that HOTAIRM1 was highly expressed in AML and associated with poor prognosis, shorter overall survival and leukemia-free survival.11 However, the detailed role and molecular mechanism of HOTAIRM1 in AML pathogenesis remain undefined.
As one of the best-studied ncRNAs, microRNAs (miRNAs, 18–25 nucleotides in length) have been widely recognized as important regulators of hematopoiesis and their dysregulation is correlated with the pathogenesis of hematologic malignancies such as AML.12 miR-148a, miR-148b, and miR-152 are three highly homologous members of the miR-148/152 family with a common stem-loop structure and the same seed region, which are located on human chromosome 7, 12, and 17, respectively.13 Previous reports showed that the expression of the miR-148/152 family was lower in patients with AML and associated with AML clinicopathological parameters.14 Recently, a novel regulatory mechanism about the competitive endogenous RNA (ceRNA) hypothesis has been proposed that the interaction between lncRNAs and mRNAs widely exists in various biological processes and lncRNAs function as a ceRNA to suppress the expression and biological functions of miRNAs.15,16 Our study aimed to investigate whether HOTAIRM1 and the miR-148/152 family were involved in the ceRNA regulatory network in AML as well as the underlying mechanism.
2. Materials and methods
2.1. Patients and blood samples
Peripheral blood samples were collected from 29 AML patients and 17 age-matched healthy individuals at Huaihe Hospital of Henan University. Our study was performed in accordance with the Guidelines of Huaihe Hospital of Henan University and was approved by the Ethic Committee at Huaihe Hospital of Henan University. All participants signed the written informed consent before the study.
2.2. Cell culture and transfection
Human AML cell lines HL60, THP-1, and NB4 were purchased from the American Type Culture Collection (Manassas, VA, USA) and grown in RPMI-1640 medium (HyClone, Logan, Utah, USA) containing 10% heat-inactivated fetal bovine serum (FBS; Gibco, Grand Island, NY, USA), and 1% penicillin/streptomycin (Sigma-Aldrich, St Louis. MO, USA) at 37 °C in a 5% CO2 humidified atmosphere. siRNA specifically targeting HOTAIRM1 (si-HOTAIRM1), miR-148b agomir (miR-148b), miR-148b antagomir (anti-miR-148b), and their matched controls (si-con, miR-con, and anti-miR-con) were synthesized from GenePharma Co., Ltd. (Shanghai, China). For transfection, cells were seeded at 3 × 105 cells per well into 6-well plates or 5 × 103 cells per well into 96-well plates. After 24 h, cells were transfected with si-HOTAIRM1 (25 nM), si-con (25 nM), miR-148b (50 nM), miR-con (50 nM), anti-miR-148b (50 nM), or anti-miR-con (50 nM) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Cells were harvested 48 h after transfection.
2.3. Quantitative real-time PCR (qRT-PCR)
Total RNA from peripheral blood samples and cultured cells were extracted using TRIzol Reagent (Invitrogen) and reversely transcribed to cDNAs using PrimeScript RT Reagent Kit (Takara, Dalian, China). Then, HOTAIRM1 and miR-148b expressions were detected by SYBR® Green Real-Time PCR Master Mixes (Thermo Fisher Scientific, Waltham, MA, USA) and TaqMan microRNA assay (Ambion, Austin, TX, USA) on the StepOnePlus qPCR system (Thermo Fisher Scientific). GAPDH and U6 small nuclear RNA were chosen as the endogenous control for HOTAIRM1 and miR-148b, respectively. The 2−ΔΔCt method was applied to calculate the relative gene expression. Amplification condition consisted of a 5 min hot start at 95 °C and then 40 cycles of 95 °C for 5 s, 60 °C for 30 s, and 72 °C for 10 s. The primers used were synthesized by Invitrogen and the primer sequences are as below: HOTAIRM1, forward 5′-CCCACCGTTCAATGAAAG-3′, and reverse 5′-GTTTCAAACACCCACATTTC-3′; GAPDH, forward 5′-TGCACCACCAACTGCTTAG-3′ and reverse 5′-GACGCAGGGATGATGTTC-3′; miR-148b, forward 5′-TGCTCAGTGCATCACAGAA-3′, and reverse 5′-GTGCAGGGTCCGAGGT-3′; U6, forward 5′-GCTTCGGCAGCACATATACTAAAAT-3′, and reverse 5′-CGCTTCAGAATTTGCGTGTCAT-3′.
2.4. Cell counting kit-8 (CCK-8) assay
HL60, THP-1, and NB4 cells were seeded at 5 × 103 cells per well into 96-well plates in triplicates and transfected with si-HOTAIRM1, si-con, miR-148b, miR-con, si-HOTAIRM1 + anti-miR-148b, si-HOTAIRM1 + anti-miR-con, or si-con + anti-miR-con. Following culturing for indicated times, 10 μl of CCK-8 working solution (Beyotime, Shanghai, China) was added into each well and cells were incubated for another 2 h at 37 °C. Then, the optical density of each well was measured at 450 nm on a microplate reader (Bio-Rad, Hercules, CA, USA).
2.5. Apoptosis analysis by flow cytometry
At 48 h post-transfection, HL60 and THP-1 cells were harvested and washed twice with ice-cold PBS. Then, the cells were resuspended in 100 μl 1 × binding buffer and double-stained with 5 μl annexin V and 10 μl propidium iodide (PI) for 15 min in the dark from the Annexin-VFITC Apoptosis Detection Kit (KeyGen, Nanjing, China). The apoptotic cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
2.6. Luciferase reporter assay
The wild-type fragments of HOTAIRM1 containing the predicted binding sites of miR-148b and its corresponding mutant were synthesized by PCR and inserted into the pmirGLO dual luciferase miRNA target expression vector (Promega, Madison, WI, USA). The recombinant reporter plasmids were named as WT-HOTAIRM1 and MUT-HOTAIRM1. For luciferase reporter assay, HL60 cells were seeded into 24-well plates with 3 × 104 cells per well and transiently cotransfected with the constructed luciferase reporter plasmids and miR-148b or miR-con using Lipofectamine 2000 reagent (Invitrogen). HL60 cells were harvested at 48 h post-transfection and the firefly and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay kit (Promega).
2.7. Statistical analysis
All experimental data are shown as mean ± standard deviation (SD). Statistical analyses were conducted using SPSS Statistics (version 19.0; SPSS, Inc., Chicago, IL, USA). Student's t test and one-way analysis of variance were used to evaluate significant differences. P < 0.05 was considered as statistically significant.
3. Results
3.1. The expression of HOTAIRM1 and miR-148b in AML
Bioinformatics analysis by starBase v3.0 software predicted that HOTAIRM1 could target the miR-148/miR-152 family members including miR-148a, miR-148b, and miR-152, and the predicted complementary binding sites of the miR-148/miR-152 family members to HOTAIRM1 were shown in Fig. 1A. The correlation between HOTAIRM1 and the miR-148/miR-152 family member expressions in a cohort of AML patients from The Cancer Genome Atlas (TCGA) dataset were further explored. Analysis of TCGA AML dataset revealed that HOTAIRM1 expression was negatively correlated with the expression of miR-148b but not miR-148a/miR-152 in AML samples (Fig. 1B–D), suggesting that miR-148b may be a potential target of HOTAIRM1 in AML. Subsequently, we analyzed the expressions of HOTAIRM1 and miR-148b in the peripheral blood samples from 29 AML patients and 17 age-matched healthy individuals by qRT-PCR. The results showed that HOTAIRM1 expression was higher (Fig. 1E) while miR-148b expression was lower (Fig. 1F) in AML patients than healthy controls. Interestingly, Pearson's correlation analysis revealed a negative correlation between HOTAIRM1 and miR-148b expression in AML patients (Fig. 1G).
Fig. 1. The expressions of HOTAIRM1 and miR-148b in AML. (A) The predicted complementary binding sites of the miR-148/miR-152 family members to HOTAIRM1 by bioinformatics analysis using starBase v3.0 software. (B–D) The correlation between HOTAIRM1 and the miR-148/miR-152 family member expressions in a cohort of AML patients from TCGA dataset. qRT-PCR analysis of HOTAIRM1 (E) and miR-148b (F) in the peripheral blood samples from 29 AML patients and 17 age-matched healthy individuals. (G) Pearson's correlation analysis of the correlation between HOTAIRM1 and miR-148b expression in AML patients.

3.2. The effects of HOTAIRM1 on AML cell proliferation and apoptosis
To determine the biological functions of HOTAIRM1 in AML cells, HL60, THP-1, and NB4 cells were transfected with si-HOTAIRM1 or si-con. The transfection efficiency was determined by qRT-PCR and the results showed that HOTAIRM1 expression was remarkably repressed by transfection with si-HOTAIRM1 in HL60 (Fig. 2A), THP-1 (Fig. 2B), and NB4 cells (Fig. 2C). The subsequent CCK-8 assay demonstrated that downregulation of HOTAIRM1 suppressed the proliferation of HL60 (Fig. 2D), THP-1 (Fig. 2E), and NB4 cells (Fig. 2F) compared with control groups. Flow cytometry analysis revealed that the proportion of apoptotic HL60 and THP-1 cells was effectively promoted after HOTAIRM1 silencing (Fig. 2G). Therefore, we concluded that HOTAIRM1 silencing suppressed cell proliferation and induced apoptosis in AML cells.
Fig. 2. HOTAIRM1 silencing suppressed cell proliferation and induced apoptosis in AML cells. (A–C) qRT-PCR was applied to examine HOTAIRM1 expression in HL60, THP-1, and NB4 cells after transfection with si-HOTAIRM1 or si-con for 48 h. (D–F) Cell proliferation was evaluated by CCK-8 assay after HL60, THP-1, and NB4 cells were treated with si-HOTAIRM1 or si-con for 24, 48, and 72 h. (G) Apoptosis of si-HOTAIRM1 or si-con-transfected HL60 and THP-1 cells was analyzed by flow cytometry analysis. *P < 0.05 vs. si-con.

3.3. The effects of miR-148b on AML cell proliferation and apoptosis
We also determined the biological functions of miR-148b in AML cells, HL60, THP-1, and NB4 cells were transfected with miR-148b or miR-con. The transfection efficiency was determined by qRT-PCR and the results manifested that miR-148b expression was upregulated following introduction with miR-148b in HL60 (Fig. 3A), THP-1 (Fig. 3B), and NB4 cells (Fig. 3C). The subsequent CCK-8 assay demonstrated that augmented expression of miR-148b impeded the proliferation of HL60 (Fig. 3D), THP-1 (Fig. 3E), and NB4 cells (Fig. 3F) compared with control groups. Meanwhile, flow cytometry analysis further revealed that the apoptosis rates of HL60 and THP-1 cells were increased after miR-148b overexpression (Fig. 3G). These findings suggested that miR-148b overexpression suppressed cell proliferation and induced apoptosis in AML cells.
Fig. 3. miR-148b overexpression suppressed cell proliferation and induced apoptosis in AML cells. (A–C) miR-148b expression was detected by qRT-PCR in HL60, THP-1, and NB4 cells transfected with miR-148b or miR-con for 48 h. (D–F) Cell proliferation was assessed by CCK-8 assay in HL60, THP-1, and NB4 cells transfected with miR-148b or miR-con for 24, 48, and 72 h. (G) Apoptosis of miR-148b- or miR-con-transfected HL60 and THP-1 cells was analyzed by flow cytometry analysis. *P < 0.05 vs. si-con.

3.4. miR-148b is a target of HOTAIRM1 in AML cells
To confirm whether HOTAIRM1 could act as a molecular sponge of miR-148b, luciferase reporter plasmids containing the WT or MUT fragment of HOTAIRM1 were constructed (Fig. 4A) and cotransfected with miR-148b or miR-con into HL60 cells. As shown in Fig. 4B, promotion of miR-148b significantly decreased the luciferase activity of WT-HOTAIRM1 relative to miR-con group, but did not change the luciferase activity of MUT-HOTAIRM1. qRT-PCR analysis further implicated that miR-148b expression was increased in si-HOTAIRM1-transfected HL60 and THP-1 cells versus si-con-introduced group (Fig. 4C). These results proved that miR-148b is a target of HOTAIRM1 in AML cells.
Fig. 4. The interaction between HOTAIRM1 and miR-148b in AML cells. (A) The WT or MUT fragments of HOTAIRM1 carrying the predicted miR-148b binding sites. (B) HL60 cells were cotransfected with WT-HOTAIRM1 or MUT-HOTAIRM1 and miR-148b or miR-con for 48 h, and luciferase activity was then measured by luciferase reporter assay. (C) qRT-PCR analysis was performed to detect miR-148b expression in HL60 and THP-1 cells transfected with si-HOTAIRM1 or si-con for 48 h. *P < 0.05.

3.5. HOTAIRM1 knockdown inhibited proliferation and induced apoptosis in AML cells by negatively regulating miR-148b
qRT-PCR results hinted that anti-miR-148b evidently reduced miR-148b expression in HL60 and THP-1 cells (Fig. 5A). We also observed that anti-miR-148b elevated HOTAIRM1 expression in HL60 and THP-1 cells (Fig. 5B). To determine whether HOTAIRM1 exerted its role in AML via miR-148b, rescue experiments were performed in HL60 and THP-1 cells after transfection with si-HOTAIRM1 + anti-miR-con, si-con + anti-miR-con, or si-HOTAIRM1 + anti-miR-148b. CCK-8 assay uncovered that HL60 and THP-1 cells transfected with si-HOTAIRM1 and anti-miR-con showed a remarkable inhibition of cell proliferation compared with control group, which was greatly reversed following reintroduction with anti-miR-148b (Fig. 5C and D). Furthermore, flow cytometry analysis demonstrated that HOTAIRM1 knockdown conspicuously induced apoptosis of HL60 and THP-1 cells with respect to control group, while this effect was attenuated by cotreatment with HOTAIRM1 knockdown and miR-148b inhibition (Fig. 5E and F). These data suggested that HOTAIRM1 knockdown inhibited proliferation and induced apoptosis in AML cells by upregulating miR-148b.
Fig. 5. Effects of HOTAIRM1 or along with miR-148b on AML cell proliferation and apoptosis. (A) qRT-PCR analysis of miR-148b expression in HL60 and THP-1 cells transfected with anti-miR-con or anti-miR-148b for 48 h. (B) qRT-PCR analysis of HOTAIRM1 expression in HL60 and THP-1 cells transfected with anti-miR-con or anti-miR-148b for 48 h. (C and D) CCK-8 assay was performed to detect cell viability in HL60 and THP-1 cells transfected with si-HOTAIRM1 + anti-miR-con, si-con + anti-miR-con, or si-HOTAIRM1 + anti-miR-148b for 48 h. (E and F) Flow cytometry analysis was performed to evaluate apoptosis of HL60 and THP-1 cells after transfection with si-HOTAIRM1 + anti-miR-con, si-con + anti-miR-con, or si-HOTAIRM1 + anti-miR-148b for 48 h. *P < 0.05.

4. Discussion
More recently, the incidence and mortality rates of AML have risen dramatically with age in China.17 Therefore, much more research is required to identify new available therapeutic targets and develop effective therapeutic strategies for AML. In recent decades, accumulating previous studies suggest that lncRNAs could modulate gene expression through several molecular mechanisms such as epigenetic control, transcriptional and post-transcriptional regulation.18 Moreover, lncRNAs have been shown to be involved in the biological and pathological processes of AML, acting as oncogenes or tumor suppressors. Our study is the first direct investigation of the role and molecular mechanism of HOTAIRM1 in AML. Herein, we demonstrated that HOTAIRM1 was negatively correlated with miR-148b expression in AML tissues and miR-148b was a target of HOTAIRM1 in AML. Moreover, we identified that HOTAIRM1 knockdown inhibited proliferation and induced apoptosis in AML cells by acting as a ceRNA of miR-148b.
Increasing experimental data have demonstrated that HOTAIRM1 plays crucial roles in several types of tumor. HOTAIRM1 was abnormally upregulated in glioblastoma multiforme (GBM) tissues and cells, and its silencing caused tumor suppressive effects in GBM in vitro and in vivo by epigenetic regulation of HOXA1 gene.19 In contrast, HOTAIRM1 was downregulated in gastric cancer (GC) and suppressed the progression of GC in vitro and in vivo by regulating miR-17-5p/phosphatase and tensin homolog (PTEN) axis.20 HOTAIRM1, which was downregulated in hepatocellular carcinoma (HCC), might suppress the proliferative ability and promote the apoptosis of HCC cells by inhibiting the Wnt pathway.21 These results suggested the context-dependent role of HOTAIRM1 in different types of tumors. In the present study, we demonstrated that HOTAIRM1 was upregulated in AML patients, in line with the previous report.11 Functional analysis revealed that depletion of HOTAIRM1 suppressed proliferation and induced apoptosis in AML cells, suggesting that HOTAIRM1 played an oncogenic role in AML.
It has been proposed that lncRNAs function as endogenous miRNA sponges to competitively target miRNAs and suppress the function of miRNAs.16 To further elucidate the molecular mechanism underlying the oncogenic role of HOTAIRM1 in AML, we aimed to predict the potential miRNAs that can be targeted by HOTAIRM1. Our bioinformatics analysis by starBase v3.0 software predicted that the miR-148/miR-152 family members including miR-148a, miR-148b, and miR-152, which were reported to be lowly expressed in AML,14 were the predicted potential targets of HOTAIRM1. Moreover, HOTAIRM1 expression was negatively correlated with miR-148b expression but had no correlation with miR-148a/miR-152 expressions in AML samples from TCGA dataset, suggesting that miR-148b may be a potential target of HOTAIRM1 in AML. miR-148b, located on chromosome 12q13, is well-documented to be highly expressed in some tumors including ovarian cancer22 and lung cancer,23 but frequently downregulated in other malignancies, such as pancreatic cancer,24 GC,25 and AML.14,26 However, the detailed role of miR-148b in AML remains unclear. In our study, we suggested that miR-148b expression was downregulated in AML patients and overexpression of miR-148b inhibited proliferation and induced apoptosis in AML cells, suggesting the tumor suppressive role of miR-148b in AML. Furthermore, Pearson's correlation analysis suggested a negative correlation between miR-148b and HOTAIRM1 in AML patients. Luciferase reporter assay and qRT-PCR analysis indicated that HOTAIRM1 suppressed miR-148b expression by acting as a ceRNA in AML cells. Rescue experiments implied that inhibition of miR-148b reversed the effects of HOTAIRM1 knockdown on proliferation and apoptosis in AML cells. Collectively, these results suggested that HOTAIRM1 was involved in the progression of AML through targeting miR-148b.
5. Conclusion
In summary, we provided the first evidence that HOTAIRM1 functioned as a molecular sponge of miR-148b in AML. Our findings indicated that HOTAIRM1 knockdown inhibited proliferation and induced apoptosis in AML cells by negatively regulating miR-148b, shedding light on the biological function and molecular mechanism of HOTAIRM1 in AML. Therefore, HOTAIRM1 may be a promising therapeutic target for AML.
Conflicts of interest
Authors declare that there are no conflicts of interest.
Supplementary Material
References
- Dohner H. Weisdorf D. J. Bloomfield C. D. N. Engl. J. Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- Kern W. Haferlach C. Haferlach T. Schnittger S. Cancer. 2008;112:4–16. doi: 10.1002/cncr.23128. [DOI] [PubMed] [Google Scholar]
- Grove C. S. Vassiliou G. S. Dis. Models Mech. 2014;7:941–951. doi: 10.1242/dmm.015974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzi V. Lakkala J. Devarajan R. Savolainen E. R. Koistinen P. Heljasvaara R. Pihlajaniemi T. Leukemia Research Reports. 2018;9:9–13. doi: 10.1016/j.lrr.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estey E. Dohner H. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- Zebisch A. Hatzl S. Pichler M. Wolfler A. Sill H. Int. J. Mol. Sci. 2016;17:2080. doi: 10.3390/ijms17122080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer T. R. Dinger M. E. Mattick J. S. Nat. Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- Shi X. Sun M. Liu H. Yao Y. Song Y. Cancer Lett. 2013;339:159–166. doi: 10.1016/j.canlet.2013.06.013. [DOI] [PubMed] [Google Scholar]
- Huarte M. Nat. Med. 2015;21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- Wei S. Zhao M. Wang X. Li Y. Wang K. J. Hematol. Oncol. 2016;9:44. doi: 10.1186/s13045-016-0274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Beya M. Brunet S. Nomdedeu J. Pratcorona M. Cordeiro A. Gallardo D. Escoda L. Tormo M. Heras I. Ribera J. M. Duarte R. de Llano M. P. Bargay J. Sampol A. Nomdedeu M. Risueno R. M. Hoyos M. Sierra J. Monzo M. Navarro A. Esteve J. Oncotarget. 2015;6:31613–31627. doi: 10.18632/oncotarget.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Chen H. Bai J. He A. Cell Biol. Int. 2017;41:936–945. doi: 10.1002/cbin.10770. [DOI] [PubMed] [Google Scholar]
- Calin G. A. Sevignani C. Dumitru C. D. Hyslop T. Noch E. Yendamuri S. Shimizu M. Rattan S. Bullrich F. Negrini M. Croce C. M. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. X. Zhang R. Li Y. Med. Sci. Monit. 2017;23:4768–4778. doi: 10.12659/MSM.902689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalali S. Bhartiya D. Lalwani M. K. Sivasubbu S. Scaria V. PLoS One. 2013;8:e53823. doi: 10.1371/journal.pone.0053823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R. Agarwal V. Stefano J. Bartel D. P. Stoffel M. Mol. Cell. 2014;54:766–776. doi: 10.1016/j.molcel.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. Zheng R. Zeng H. Zhang S. Chin. J. Cancer. 2015;34:502–507. doi: 10.1186/s40880-015-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista P. J. Chang H. Y. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q. Dong C. Cui J. Wang Y. Hong X. J. Exp. Clin. Cancer Res. 2018;37:265. doi: 10.1186/s13046-018-0941-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R. Zhao G. Yang Y. Jiang Z. Cai J. Zhang Z. Hu H. J. Cell. Biochem. 2019;120:4952–4965. doi: 10.1002/jcb.27770. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Mi L. Xuan Y. Gao C. Wang Y. H. Ming H. X. Liu J. Eur. Rev. Med. Pharmacol. Sci. 2018;22:4861–4868. doi: 10.26355/eurrev_201808_15622. [DOI] [PubMed] [Google Scholar]
- Chang H. Zhou X. Wang Z. N. Song Y. X. Zhao F. Gao P. Chiang Y. Xu H. M. Mol. Med. Rep. 2012;5:1277–1280. doi: 10.3892/mmr.2012.794. [DOI] [PubMed] [Google Scholar]
- Nymark P. Guled M. Borze I. Faisal A. Lahti L. Salmenkivi K. Kettunen E. Anttila S. Knuutila S. Genes, Chromosomes Cancer. 2011;50:585–597. doi: 10.1002/gcc.20880. [DOI] [PubMed] [Google Scholar]
- Bloomston M. Frankel W. L. Petrocca F. Volinia S. Alder H. Hagan J. P. Liu C. G. Bhatt D. Taccioli C. Croce C. M. JAMA. 2007;297:1901–1908. doi: 10.1001/jama.297.17.1901. [DOI] [PubMed] [Google Scholar]
- Song Y. X. Yue Z. Y. Wang Z. N. Xu Y. Y. Luo Y. Xu H. M. Zhang X. Jiang L. Xing C. Z. Zhang Y. Mol. Cancer. 2011;10:1. doi: 10.1186/1476-4598-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejcik Z. Belickova M. Hrustincova A. Votavova H. Jonasova A. Cermak J. Dyr J. E. Merkerova M. D. Cancer Biomarkers. 2018;22:101–110. doi: 10.3233/CBM-171029. [DOI] [PubMed] [Google Scholar]