

Abstract
Chimeric antigen receptor (CAR) redirected T cells are potent therapeutic options against hematological malignancies. The current dominant manufacturing approach for CAR T cells depends on retroviral transduction. With the advent of gene editing, insertion of a CD19-CAR into the T cell receptor (TCR) alpha constant (TRAC) locus using adeno-associated viruses for gene transfer was demonstrated, and these CD19-CAR T cells showed improved functionality over their retrovirally transduced counterparts. However, clinical-grade production of viruses is complex and associated with extensive costs. Here, we optimized a virus-free genome-editing method for efficient CAR insertion into the TRAC locus of primary human T cells via nuclease-assisted homology-directed repair (HDR) using CRISPR-Cas and double-stranded template DNA (dsDNA). We evaluated DNA-sensor inhibition and HDR enhancement as two pharmacological interventions to improve cell viability and relative CAR knockin rates, respectively. While the toxicity of transfected dsDNA was not fully prevented, the combination of both interventions significantly increased CAR knockin rates and CAR T cell yield. Resulting TRAC-replaced CD19-CAR T cells showed antigen-specific cytotoxicity and cytokine production in vitro and slowed leukemia progression in a xenograft mouse model. Amplicon sequencing did not reveal significant indel formation at potential off-target sites with or without exposure to DNA-repair-modulating small molecules. With TRAC-integrated CAR+ T cell frequencies exceeding 50%, this study opens new perspectives to exploit pharmacological interventions to improve non-viral gene editing in T cells.
Keywords: gene editing, CRISPR-Cas9, chimeric antigen receptor, non-viral cell manufacturing, CAR T cells, TRAC, knockin, adoptive T cell therapy, HDR
Graphical abstract
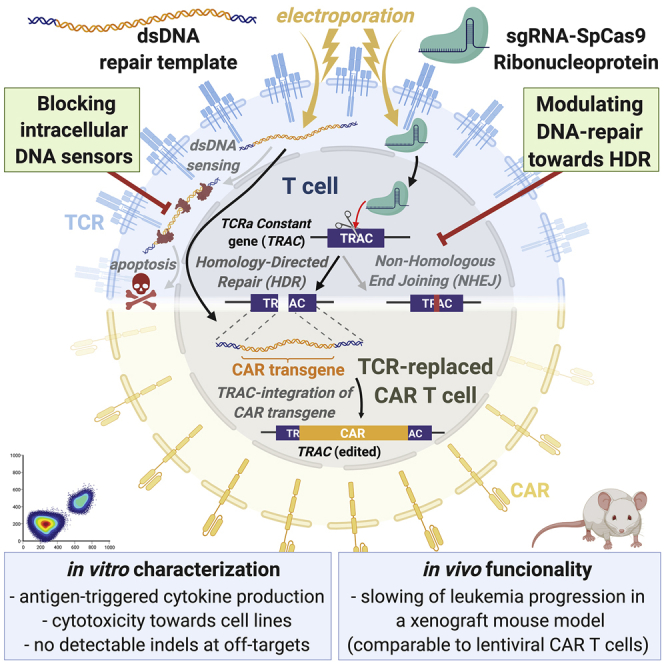
Virus-free generation of TRAC-replaced CAR T cells by electroporation of CRISPR-Cas ribonucleoproteins and linear double-stranded DNA is limited by DNA dose-dependent toxicity. Blocking cellular DNA sensing and modulating DNA repair increases CAR integration rate into the TRAC gene of primary human T cells without affecting their functionality or inducing off-target indel formation.
Introduction
Chimeric antigen receptor (CAR) redirected T cells are a potent and approved treatment option for certain hematological malignancies, such as relapsed acute B cell leukemia, aggressive B cell lymphoma,1,2 and treatment-refractory multiple myeloma.3,4 Furthermore, CAR T cells are under development for solid tumors.5,6 Different types of CAR T cells are currently being investigated preclinically beyond oncology for regeneration of liver and cardiac fibrosis,7,8 for amelioration of autoimmune diseases,9,10 for mitigation of allogeneic immune responses in transplantation medicine,11,12 and as anti-viral adoptive cell therapies.13,14 Hence, CAR T cells represent an exciting immunotherapeutic platform for both personalized and off-the-shelf T cell therapies.
Lentiviral (LV)- and retroviral vector (RV)-mediated gene transfer are the current gold standards for clinical manufacturing of CAR T cells.15 High CAR expression levels after random viral integration are achieved by the transcriptional activities of viral or human promoters present in the vectors. Excessive and persistent CAR expression, however, may contribute to tonic signaling and potentiate T cell susceptibility to activation-induced cell death16,17 and exhaustion.18 Moreover, the technological demands and high costs associated with the production and testing of clinical grade LV and RV still remain as limiting logistical and financial factors for manufacturing of new CAR T cell therapies.19
Preclinical evidence suggests that targeted integration of CAR transgenes into the T cell receptor (TCR) alpha constant chain (TRAC) gene locus allows for optimally regulated expression and superior functionality through reduced T cell exhaustion in vitro and in vivo.18,20 In addition, resulting TRAC-replaced and thus TCR/CD3-deficient CAR T cells avoid the risk of hazardous allogeneic graft-versus-host disease and offers a potential route toward off-the-shelf products.21 Furthermore, the precise insertion of CAR transgenes into specific transcriptionally active sites using genome-editing techniques can reduce the risk of insertional mutagenesis and can also avoid CAR expression in contaminating leukemia cells.22
TRAC integration of CAR expression modules has been achieved through nuclease-assisted homology-directed repair (HDR).18,20 In previous studies, Streptococcus pyogenes (Sp)Cas9-single guide (sg)RNA ribonucleoprotein (RNP) complexes were transfected into anti-CD3/CD28-stimulated T cells followed by transduction with recombinant adeno-associated virus serotype 6 (rAAV6) for HDR donor template (HDRT) delivery.18,20 Other nucleases have also been used in this context to induce DNA double-strand breaks to initiate HDR.23,24 While RNP-based gene editing has been commonly used to modify T cells25 and retrovirally transduced CAR T cells,26,27 fewer research publications have reported the use of T cells with CARs integrated at the TRAC locus.18,20,23,24,28, 29, 30 Most of the aforementioned studies have used methods requiring high titers of rAAV6 for HDRT delivery, thus necessitating virological expertise and labor-intensive production, even at preclinical scale. Therefore, we conclude that there is a need for methods that circumvent the practical hurdles of viral vectors for manufacturing of gene-edited CAR T cells.
Non-viral reprogramming is an attractive alternative to the conventional approaches that circumvents the need of rAAV6 for effective donor DNA delivery.18,31 Roth et al.32 achieved efficient site-specific transgene insertions by co-electroporation of CRISPR-Cas RNPs and single- or double-stranded DNA (dsDNA) HDR templates. Small fluorescent tags (∼700 bp) were efficiently integrated into different genetic loci in primary human T cells.32 However, this approach is less efficient when delivering therapeutically relevant transgenes for adoptive T cell transfer with lengths exceeding 1,500 bp.32 Integration rates of TCRs reactive against epitopes present in tumor-associated antigens range from 5% (∼2.1 kb insert)33 to 15% (∼1.5 kb insert).32,34 Hence, harnessing and optimizing the efficacy of CAR knockin into specific genomic loci are highly relevant for CAR T cell product development and ultimately their batch release for clinical use based on identity parameters.
In addition, even under optimized conditions, significant T cell toxicity of up to 70%–80% occurs.35 The toxicity of non-viral reprogramming in T cells is likely dependent on the physical strain produced by co-electroporation of dsDNA and RNP aggregates into cells. This issue has been partially addressed by the use of anionic polymers, such as poly-L-glutamic acid (PGA), which was recently shown to improve knockin rates and reduce toxicity by physically dispersing large RNP aggregates into smaller complexes.36 In addition, Nguyen and colleagues suggested a modification of HDR-donor DNA using truncated Cas9 target sequences (tCTSs) flanking both homology arms (HAs). The tCTSs create binding sites for the co-delivered RNPs. The bound RNPs shuttle the DNA into the cell nucleus via the nuclear localization sequences (NLS) of the SpCas9 nuclease, leading to improved knockin rates; however, it also increases toxicity when higher amounts of tCTS-modified donors are used.36 Another potential driver of toxicity of non-viral reprogramming of T cells are the endogenous immune responses triggered by cytosolic dsDNA mediated by innate DNA-sensor protein pathways,37 such as cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS), which is activated in a DNA-length-dependent manner,38 promoting apoptosis via its downstream effector stimulator of interferon genes (STING).39 To the best of our knowledge, pharmacological interference with DNA sensing has not been used for modulation of virus-free gene editing in human T cells.
In this study, we provide a guide to fast and efficient virus-free integration of CARs into the TRAC locus of primary human T cells using an optimized method adapted from prior works.32,36 We hypothesized that cell viability is primarily compromised by toxicity of dsDNA donor templates. We therefore assessed whether temporary inhibition of cytosolic DNA sensors increases the viability and CAR insertion rates after DNA transfection. In addition, we evaluated whether HDR-enhancing substances can amplify CAR integration rates at low concentrations of dsDNA donors using different nucleases in primary human T cells. We show that transient dsDNA-sensor inhibition synergizes with two commercially available HDR enhancers for effective CAR T cell generation. In vitro assays provide proof of principle for the functionality of CD19- and B cell maturation antigen (BCMA)-CAR T cells generated under optimized conditions for TRAC integration. Finally, CD19-CAR T cells generated by optimized knockin of the CD19-CAR into TRAC showed a trend toward superior in vivo performance in a xenograft pre-B lymphoblastic leukemia mouse model compared with LV-transduced counterparts. These results underscore the advance and relevance of non-viral gene-editing approaches for CAR T cell engineering.
Results
sgRNA selection to insert a CD19-specific CAR via CRISPR-Cas9-assisted HDR
In order to perform optimization experiments with a clinically relevant transgene, we rationally designed a second-generation, CD19-specific CAR with an intermediate length immunoglobulin G1 (IgG1) hinge for detection purposes, a CD28 costimulatory domain, followed by the CD3 zeta effector chain (Figure 1A; Table S1). Since careful selection of guide RNAs (gRNAs) is required to avoid off targets and to ensure a high level of safety for future clinical translation,40 we chose the single-guide RNA (sgRNA)-targeting TRAC exon 1 with the fewest in silico predicted off targets for our experiments.24 Aiming to further decrease the risk for off-target editing, we then compared the original sgRNA with a modified sequence carrying a mismatch G base at its second position (Table S2). Using genome-wide, unbiased identification of double strand breaks enabled by sequencing (GUIDE-seq), empirical nomination of off-target activity indicated five potential off-target sites for the original sgRNA and identified only one off-target event for the modified sgRNA (Figure S1). To insert our 2,015-bp-sized CAR transgene in frame with TRAC exon 1, we created a donor template for HDR (Figure 1A; DNA and amino acid sequences disclosed in Table S1). The insert was flanked with symmetric HAs (400 bp each) and consisted of a P2A-self-cleavage site, the CD19-CAR cDNA, and a bovine growth hormone (bGH)-derived polyadenylation sequence (pA). Two types of HDRT were prepared as previously described: one with regular HAs (reg. HA) and one with tCTS-modified HAs (tCTS-HA) (Table S1).36
Figure 1.

Low dosages of transfected dsDNA donor templates reduce toxicity during non-viral CAR T reprogramming
Virus-free insertion of a 2-kb-sized CD19-CAR transgene into the human TRAC locus. (A) Design of dsDNA donor templates is shown. The transgene is composed of a P2A self-cleavage site, the CAR encoding sequence followed by a STOP codon, and a bGH-derived polyadenylation site (pA). Two template formats are used: the transgene flanked by regular homology arms (reg. HA, black) or homology arms with additional truncated Cas9 target sequences (tCTS-HA, red). (B) Experimental setup to evaluate co-electroporation of RNPs and escalating doses of dsDNA donor templates with modified (tCTS-HA) and reg. HA is shown. (C and D) Representative flow cytometry plots showing editing outcomes 2 days after co-electroporation of RNP and escalating amounts of dsDNA donor templates of both HA formats. (E) Summary of flow cytometric analysis 2 days after electroporation (n = 4 healthy donors in two independent experiments). Black and red indicate the use of dsDNA HDRTs of reg. HA or tCTS-HA format, respectively. Thick lines indicate mean values. Error bars indicate standard deviation. Thin lines connect single data points from one healthy donor over the different dsDNA amounts. Statistical analysis was performed using ordinary one-way ANOVA with subsequent Dunn’s correction (for multiple testing) comparing values for each HDRT amount with 0.5 μg HDRT as reference. Asterisks in this and all further figures represent different p values calculated in the respective statistical tests (not significant [ns]: p > 0.5; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
Optimization of electroporation conditions and pre-electroporation culture of T cells
First, we re-evaluated multiple parameters of the original protocols32,36 to generate an optimized version with decreased cost by replacing or reducing expensive reagents. In line with previous reports, 1 × 106 activated primary human T cells per 20 μL electroporation solution were optimal for cell survival and knockin rates (Figure S2A). We found that the amount of Cas9 could be reduced from 8 to 4 μg/electroporation (E′) without decreasing knockin or knockout efficiencies as determined by flow cytometry (Figure S2B). Furthermore, we confirmed the positive effects of PGA on cell survival and relative CAR insertion rates for both dsDNA donor formats (reg. HA and tCTS-HA; Figure S2C). Aiming to replace expensive reagents, we compared T cell activator beads32,36 with plate-bound anti-CD3/CD28 antibodies and the commercial electroporation buffer P3 with the self-made electroporation solution 1M. 1M buffer has a known formulation and can be qualified for GMP use (Figure S2D).41 T cell stimulation on plate-bound antibodies significantly increased CAR T cell yields both on day 2 and day 7 post-electroporation. Electroporation in the self-made buffer 1M had a minor but significant negative impact on CAR insertion rates detected on day 2 and day 7 post-electroporation and also significantly reduced CAR T cell expansion between day 2 and day 7 post-electroporation. Nonetheless, all further electroporation experiments were carried out using plate-bound antibody stimulation and the self-made (thus inexpensive) buffer 1M despite its moderately reduced performance compared with buffer P3. Importantly, transgene expression was detectable by flow cytometry 2 days after electroporation only when the dsDNA HDR templates were co-electroporated with RNP and not in the absence of RNP (Figure S2E). This indicates that transient expression from non-integrated HDR templates did not take place due to their lack of exogenous promotor sequences.
Cell toxicity is dependent on the amount of transfected dsDNA donor templates
Using our cost-optimized protocol (T cell stimulation in anti-CD3/CD28-coated wells; cell count: 1 × 106/E′; amount of RNP: 25 μM/E′; volume PGA: 0.5 μL/E′; buffer 1M), we investigated the effects of dsDNA HDR template dosage and format as well as different pharmacological interventions on gene insertion rates and cell survival 2 days after electroporation (Figure 1A). We detected significant dose-dependent toxicity with reg. HA HDRT 2 days after electroporation (Figures 1B–1E). As expected from the original report, this dsDNA dose-dependent toxicity was more pronounced with tCTS-HA donor templates likely owing to enhanced DNA delivery.36 Mean toxicity at the highest HDRT amount tested (reg. HA: 4 μg; tCTS: 2.5 μg) was 87% for reg. HA and 90% for tCTS-HA compared with mock-electroporated controls (Figures 1D and 1E). After delivery of the lowest HDRT dose tested (0.5 μg), an average of 20% (for reg. HA) and 24% (for tCTS-HA) of living cells showed CAR surface expression (Figure 1E). Using higher HDRT doses did not significantly increase overall CAR knockin rates, but within the resulting population of CD3-negative T cells, we observed a significant increase in relative CAR knockin rates up to 35% (for reg. HA) and 42% (for tCTS-HA) at the respective highest HDRT amount tested. However, CD3 knockout rates decreased when using large HDRT amounts, indicating inhibitory DNA-RNP interaction with both HDRT formats. Application of tCTS HDRTs showed a trend toward increased CAR T cell counts at 0.5 μg/Eʹ. In subsequent dose-titration experiments, further reduction of HDRT amount/E′ resulted in decreased CAR knockin rates and CAR T cell counts without further improvements of cell survival (Figure S2F). Taken together, dsDNA HDRT amounts exceeding 0.5 μg decreased CAR T cell yield through increased toxicity and reduced knockout efficiency.
DNA sensing can impact cell viability after virus-free gene editing
Transfected dsDNA activates innate immune pathways within T cells, thereby contributing to a loss in viability after electroporation of large amounts of dsDNA HDRT.38,39 Aiming to improve T cell viability, we investigated whether temporary inhibition of intracellular dsDNA sensors or associated downstream pathways could increase the CAR T cell yield after non-viral gene editing. Initially, four different DNA-sensor inhibitors (ODN A151, RU.521 BX785, and H151) were individually tested for their propensity to increase cell viability and CAR integration rates in moderate and highly toxic conditions (1 μg HA-HDRT versus 2 μg tCTS-HDRT; Figures S3A–S3D). Two compounds were selected for a combined approach based on different beneficial effects: on the one hand, treating T cells for 6 h prior to electroporation with ODN A151, a modified DNA oligonucleotide resembling the telomere repeat quadruplex 5′-TTAGGG-3′, significantly increased cell survival over untreated T cells in highly toxic conditions (2 μg tCTS-HDRT; Figure S3C). However, this effect was not statistically significant in the condition with low-moderate toxicity (1 μg HA-HDRT; Figure S3D). On the other hand, pretreatment of T cells with RU.521, a small-molecule antagonist of cGas, tended to increase the relative CAR insertion rate in both conditions without significantly affecting cell survival compared with drug-free controls.
Treating T cells with two DNA-sensor inhibitors prior to electroporation improves CAR T cell generation at low dsDNA dose
Consequently, we evaluated whether a combined pretreatment with ODN A151 and RU.521 could increase TCR-to-CAR replacement rates while retaining high T cell recovery with escalating amounts of reg. HA and tCTS-modified HDRT (Figures 2A–2C; Figures S3E and S3F). We observed a small but consistent increase in the relative CAR expression rate as well as an increase in the absolute CAR T cell recovery at the lowest HDRT concentration (Figure 2C). Cytokine analysis of cell culture supernatants collected 24 h after electroporation revealed dsDNA dose-dependent release of interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α), two cytokines that have been associated with intracellular sensing of dsDNA (Figure 2D).37 In contrast, IL-1β was barely above the lower limit of detection and without a clear trend, indicating minimal AIM2-mediated inflammasome activation in T cells (Figure 2A). In samples treated with DNA-sensor inhibitors, we observed moderate suppression of IL-6 release by combined DNA-sensor inhibition but no effects on TNF-α and IL-1β. Surprisingly, we observed a DNA-independent effect of DNA-sensor inhibition on Th2-associated cytokines IL-10 and IL-13 (Figure 2D), suggesting differential sensitivity of various T helper pathways. Similar effects were observed with tCTS-HA HDRT, indicating that occupation of dsDNA ends by RNP is unlikely to affect innate immune recognition of the dsDNA donors (Figure S3G). As cell cycle is a key determinant of DNA repair outcome,42 we explored the effects of this drug combination on cell cycling prior to electroporation as previously described (Figures S4A–S4C).43 Flow cytometric evaluation revealed an increased proportion of T cells within S phase after 6 h of combined DNA-sensor inhibition, which may explain the increase in relative TCR-to-CAR replacement (Figure 2C). As DNA toxicity was not effectively prevented through transient DNA-sensor inhibition, we concluded that low dsDNA amounts (0.5–1.0 μg HDRT) were superior for effective CAR T cell generation.
Figure 2.

DNA-sensor inhibition increases relative CAR insertion rate with minimal improvement of survival at optimized dsDNA donor dosage
(A) Illustration of common DNA-sensing pathways (in immune cells) that induce downstream cytokine production and the presumed mode of action of different DNA sensor inhibitors used in subsequent experiments. (B) Experimental setup for the combined addition of TLR-9 inhibitor ODN A151 and the cGAS-inhibitor RU.521 with escalating amounts of dsDNA donor templates is shown (as in Figure 1B). DNA sensor inhibitors were supplemented together into the medium 6 h prior to co-electroporation of RNP and reg. HA dsDNA donor templates. (C) Summary of flow cytometric analysis 2 days after electroporation. Data were obtained in parallel to controls presented in Figures 1D–1G (n = 4 healthy donors in two independent experiments). Editing outcomes of T cells that received combined DNA-sensor inhibition prior to electroporation are shown in orange. Black indicates the control values from Figure 1E. Thick lines indicate mean values; error bars indicate standard deviation. Light dots represent individual values. Light lines connect these for each donor. Descriptive statistical analysis was performed using paired, two-tailed Student’s t tests comparing values for DNA-sensor inhibition with values for no intervention. (D) Summary of supernatant analysis 24 h post-electroporation for cytokines associated with DNA sensing: IL-1β (lower limit of detection [LOD]: 0.85 pg/mL), IL-6 (lower LOD: 0.13 pg/mL), and TNF-α (lower LOD: 0.05 pg/mL) as well as the Th2-associated cytokines IL-10 (lower LOD: 0.01 pg/mL) and IL-13 (lower LOD: 0.27 pg/mL). Descriptive statistical analysis was performed as for (C).
HDR enhancer improves CAR insertion rates with different nucleases and templates
Pharmacologic interventions in DNA repair processes (typically aiming to inhibit non-homologous end joining [NHEJ] pathways in order to favor HDR) represent an alternative strategy to obtain higher HDR rates, as previously demonstrated for gene-editing procedures in other cell types.44, 45, 46, 47, 48, 49, 50, 51, 52 Therefore, we evaluated whether we could increase TCR-to-CAR replacement rates by exposing the T cells to a commercially available HDR-modulating agent (Alt-R HDR enhancer version 1 [v.1]) after electroporation (Figure 3). To identify the optimal HDR enhancer dose regarding efficacy and toxicity, we performed a titration experiment (Figures 3B and 3C). Two days after electroporation, we found a significant dose-dependent increase in the relative CAR insertion rate up to a mean of 38% (range: 28%–44%) at the highest concentration tested (30 μM). Due to the toxicity observed in some donors at this dose, we chose the concentration of 15 μM for subsequent experiments. The increase of CAR insertion rates mediated by HDR enhancer v.1 was reproduced with another TRAC-HDR template32 and with different Cas nucleases and guide RNAs (Figures 3D and 3E). Importantly, suboptimal gene editing with an engineered Cas12a molecule from Acidaminococcus species (Alt-R A.s. Cas12a (Cpf1) Ultra [AsCas12a Ultra])53 as well as the HiFi SpCas954 could be increased by HDR enhancer v.1. Thus, Alt-R HDR enhancer (v.1) promoted high rates of HDR-mediated CAR integration into the TRAC gene in primary human T cells.
Figure 3.

HDR-enhancing compound improves CAR integration rates in primary human T cells
(A) Experimental setup. (B) Representative flow cytometry plots showing the effects of HDR enhancer (v.1), supplemented in different concentrations, on editing outcomes 2 days after electroporation are shown. (C) Data summary from experiments presented in (A) and (B), 2 and 7 days after electroporation (n = 9 biol. replicates in three independent experiments) are shown. Purple indicates mean ± SD. Light gray indicates individual data points. Statistical analysis was performed using repeated-measures one-way ANOVA with Geisser-Greenhouse correction followed by Dunnett’s multiple comparison test. (D) Scheme illustrating two dsDNA donor templates of regular HA format for integration of a CD19-CAR at two distinct sites of the TRAC locus is shown. CAR integration is assisted by three different Cas nucleases pre-complexed with different guide RNAs, summing up to six distinct RNPs. (E) Representative flow cytometry and summary plots from experiments outlined in (D) show the effect of HDR enhancer (v.1) on CAR integration rates 7 days after electroporation of six different HDRT/RNP combinations. For RNP/HDRT combinations 1 and 2, statistical analysis was performed using a paired, two-tailed Student’s t test comparing values for no intervention with values for HDR enhancer (v.1).
HDR-enhancer and DNA sensor inhibition synergize for efficient TRAC-CAR T cell generation
We subsequently tested whether the combination of DNA-sensor inhibition (ODN A151 and RU.521) and HDR enhancer (v.1) could further improve CAR T cell generation (Figure 4A). We observed a synergistic effect on the relative TCR-to-CAR replacement rate as well as on absolute CAR T cell recovery (Figure 4B). The increase in relative CAR knockin rates was independent of the donor template HA format, but the drug combination only significantly improved the CAR T cell yield when reg. HA HDRTs were used. For tCTS-HA HDRTs, we observed a similar but non-significant trend, presumably due to higher inter-donor variability in these conditions. The synergy between DNA-sensor inhibitors and HDR enhancer (v.1) was also observed after electroporation of larger HDRT amounts, although absolute CAR T cell yields were lower (Figure S5A). We also compared magnetic-activated cell sorting (MACS)-enriched T cells with peripheral blood mononuclear cells (PBMCs) as the starting material for this pharmacologically assisted transgene transfer (Figure S5B). The use of PBMCs was inferior in all conditions with or without pharmacological interventions.
Figure 4.

Synergy of DNA-sensor inhibition and HDR enhancer improves efficiency and yield of TRAC-replaced CAR T cell generation
(A) Experimental setup adding the combination of DNA-sensor inhibition and HDR enhancement as an additional dimension to the setup originally presented in Figure 1, Figure 2E and 2C. DNA-sensor inhibition was performed with the compounds ODN A151 and RU.521 6 h prior to electroporation. After electroporation, cells were cultured in HDR enhancer v.1 supplemented medium (15 μM) for 16 h. (B) Summary of relative CAR integration rates and CAR T cell counts on day 4 and day 9 with 0.5 μg or 1.0 μg of dsDNA donor templates of both formats is shown (reg. HA, black; tCTS-HA, red). Data were obtained in parallel to controls presented in Figure 1E from four biological replicates. Furthermore, data from one additional experiment with two biological replicates only analyzed on day 9 were also included. Bars respresent mean ± SD. Statistical analysis was performed using paired, two-tailed Student's t test comparing values for no intervention (“none”) with values for a combined pharmacological interventino (“both”).
Further optimization of HDR modulation in T cells
To identify even more effective compounds that increase CAR-insertion rates in T cells, we screened 15 small molecules for their ability to enhance HDR in Jurkat cells, a T cell leukemia cell line that mimics many aspects of primary human T cell biology (Figures S6A and S6B). Jurkat cells were co-electroporated with RNP and single-stranded oligonucleotides (ssODNs) to introduce an exogeneous DNA motif (EcoRI site) into the HPRT1 gene via HDR (Tables S2 and S3). Amplicon sequencing was performed to distinguish between short insertions and deletions (indels) and perfect repair (HDR). Nine compounds increased HDR at least 2-fold on average, with nedisertib and Alt-R HDR enhancer v.2 being the most efficacious. Despite previous success in other human cell lines,44 the compounds SCR7, wortmannin, and L755507 showed no effect on HDR in Jurkat cells, indicating that different cell lines may differ in their response to HDR-modulating agents. Similar to its effects in Jurkat cells, HDR-enhancer v.2 was superior to Alt-R HDR enhancer (v.1) at improving TCR-to-CAR replacement rates in primary human T cells (Figures S6C and S6D). When combined with dual DNA-sensor inhibition, CAR insertion rates were further increased up to 68% (Figure S6E). As gene editing with RNPs is likely to happen immediately after electroporation,55, 56, 57 we evaluated the effects of timing and cell-culture conditions on pharmacological HDR enhancement. HDR enhancers showed reduced effects when the supplemented medium was not pre-warmed to 37°C and thus exhibit a temperature-dependent activity (Figure S6F). Of note, extended time between electroporation and transfer into HDR-enhancer-containing medium decreased the desired effects (Figure S6G). Exposing T cells to HDR enhancers longer than the first 3 h after electroporation did not improve CAR integration rates (Figure S6H). To test whether these two HDR-enhancing drugs have similar effect on cell cycle as the DNA-sensor inhibitors, cell cycle analysis was performed but did not show any relevant effects (Figures S6I and S6K). Thus, HDR enhancer v.2 or other small molecules like nedisertib showed promising results to further optimize virus-free gene-editing outcomes with large transgenes, such as CARs.
Functional evaluation of TRAC-replaced CAR T cells in vitro
Next, we assessed whether these pharmacological interventions during CAR T cell production have any influence on their functional characteristics in vitro (Figure 5). Target-specific cytotoxicity was tested in a 4-h VITAL assay58 by co-culturing CD19-CAR T cells with fluorescently labeled CD19+ target cells (Nalm6 cell line) and fluorescently labeled CD19− control cells (Jurkat cell line; Figure 5A). CD19-CAR T cells that were treated with DNA-sensor inhibitors and HDR enhancer (v.1) showed slightly increased target-specific cytotoxicity against Nalm6 tumor cells compared with CAR T cells that received none or only one of the two described interventions. This, however, is presumably a bias that results from the higher relative CAR+ rates in these conditions, as this parameter alone can increase the likelihood of cellular engagement between effector and target cells. Apart from the slightly different VITAL assay performance, CAR T cells generated with or without pharmacological assistance did not differ in their capacity to produce cytokines upon target cell challenge (Figures 5D–5G), their CD4/CD8 ratio (Figure 5H), or their T cell memory phenotype (Figure 5I). We also generated TRAC-replaced CAR T cells directed against BCMA59 using the combined transient DNA-sensor inhibition together with HDR enhancer (v.1) and compared them with similarly produced CD19-CAR T cells in vitro (Figure S7). We observed antigen-specific, dose-dependent lysis of the respective target cells in 4-h VITAL assays (Figures S7A and S7B) and target-specific cytokine production in CD4+ and CD8+ T cell subsets for both CARs (Figures S7C–S7F), highlighting intact effector function. To sum up, the described pharmacological interventions for enhanced non-viral CAR T cell production are applicable to different CARs and have no relevant impact on CAR T cell function in vitro.
Figure 5.

Functional in vitro characterization of TRAC-integrated CD19-CAR T cells after drug-assisted gene transfer
CAR T cell generated with no drug assistance (none), with HDR enhancer (v.1) (“HDR-E.”), DNA-sensor inhibition (“HDR-Sens. Inh.”), or the combination of both approaches (both) were assessed for possible differences in cytotoxicity, cytokine production, and phenotype. (A) Experimental setup of a flow cytometric VITAL assay is shown. (B) Representative flow cytometry dot plots of viable target (T) and control (C) cells after 4 h co-culture with effecter (E) CD19-CAR T cells for the highest E:T:C ratio tested (8:1:1) are shown. Nalm-6 cells (CD19+, GFP+) served as target cells. Jurkat cells (CD19−, CellTrace Far Red labeled) served as control cells. (C) Summary of VITAL assay results as shown in (B) (n = 3 techn. replicates) is shown. (D) Experimental setup for detection of intracellular cytokines after T cell stimulation is shown. (E) Summary of intracellular cytokine staining of bulk (CAR) T cells after co-culture with CD19+ Nalm-6 cells is shown. Boolean gating was used to identify cells that produce one, two, or three of the following cytokines: interferon (IFN)-γ, IL-2, and TNFα (n = 3 donors). (F and G) Summary of intracellular cytokine staining for CD4+ (F) and CD8+ (G) (CAR) T cells alone or after co-culture with target (Nalm6) or control (Jurkat) cells is shown. (H and I) CD4/CD8 ratio (H) and summary of CAR T cell phenotype (I) on day 9 after electroporation (n = 2 donors in techn. duplicates). TEMRA: CD45RA+, CCR7−; Tnaive-like: CD45RA+ CCR7+; TCM: CD45RA, CCR7+; TEM: CD45RA−, CCR7−.
TRAC-replaced CAR T cells are less prone to exhaustion than lentiviral controls
In line with previous reports,18 TRAC-integrated CD19-CAR T cells displayed a lower basal CAR expression in comparison to lentivirally transduced CAR T cells (Figures S9A and S9B). To assess whether lower CAR expression level also correlated with reduced T cell exhaustion, we performed a serial tumor rechallenge assay as previously described18,20 and analyzed the expression of exhaustion markers on the CAR T cell surface (Figure S9C). After four stimulations, we observed higher expression of TIM3, LAG3, and PD1 on CD4+ CAR+ T cells. Similarly, expression of TIM3 and LAG3 were also elevated in CD8+ CAR+ T cells; however, no significant difference was found for PD1 expression.
TRAC-replaced CD19-CAR T cells slow tumor progression in vivo
The knockin of the CAR into TRAC locus results in the regulation of the CAR expression by the orthotopic TCR promoter. This could potentially influence the regulation of the CAR expression and therefore the potency of the CAR T cells in vivo. Hence, we performed a proof-of-concept experiment to evaluate the potency of our completely virus-free generated TRAC-replaced CD19-CAR T cells in vivo in Nod.Rag.gamma (NRG) mice xenografted with Nalm-6/GFP/fLuc pre-B acute lymphoblastic leukemia (Figure 6A). As controls, we used non-treated mice challenged with leukemia and leukemia-challenged mice treated with LV-transduced, TCR-deficient CD19-CAR T cells (LV CD19-CAR-T). Four days after leukemia challenge, mice were treated with CAR T cells and monitored for a total of 5 weeks. During this observation period, the leukemia development and distribution was assessed longitudinally and non-invasively by weekly bioluminescence imaging (BLI) analyses. The mice in all cohorts showed comparable leukemia engraftment 1 week after challenge (Figure 6B). Non-treated controls showed a steady leukemia growth and systemic bio-distribution, coinciding with a decrease in body weight, until the terminal analyses at week 5 after challenge (Figures 6B and S9). Compared with untreated controls, mice treated with LV CD19-CAR-T showed significant reduction of the leukemia growth until week 4 after challenge, but the differences were no longer significant at week 5 (Figures 6B and 6C). On the other hand, mice treated with TRAC CD19-CAR T cells maintained a significantly lower leukemia growth at week 5 (p < 0.05). In summary, these results indicate a trend toward superior in vivo functionality of virus-free TRAC-replaced CAR T cells over LV-transduced CAR T cells.
Figure 6.

Virus-free generated TRAC-replaced CAR T cells slow leukemia progression in vivo
Comparison of the therapeutic efficacies between TRAC-replaced and lentivirally transduced CD19-CAR T cells in a pre-B acute lymphoblastic leukemia xenograft mouse model. (A) Experimental setup for generation of TCR-negative CAR T cells by virus-free TRAC replacement with CD19-CAR or lentiviral transduction (LV CAR T cells) followed by TRAC-KO is shown. Prior to formulation, remaining TCR/CD3-positive T cells were depleted by MACS cell separation technology. (B) Experimental setup for Nalm-6 xenograft model. Eight-week-old Nod.Rag.Gamma (NRG) mice were challenged i.v. with 5 × 105 Nalm-6/GFP/fLuc cells. Mice not receiving leukemia (“no tumor”) were used as controls for analyses. Four days after leukemia challenge, mice were randomized among three cohorts: (1) injected i.v. with phosphate-buffered saline (PBS) (corresponding to the “tumor only” group), (2) injected i.v. with 5 × 105 CD19-CAR T cells generated via lentiviral transfer and consecutive TRAC KO (“LV-CD19CAR”), and (3) injected i.v. with 5 × 105 CD19-specific TCR-deficient CAR T cells generated by TRAC replacement (“TRAC CD19-CAR”). The residual CD3+ T cells were depleted from both types of CAR T cell types prior to infusion. Mice were monitored for disease severity every 2 to 3 days, and body weights were measured weekly. Leukemia engraftment, bio-distribution, and progression were assessed by weekly bioluminescence imaging (BLI) analyses. The experimental endpoint was 5 weeks after leukemia challenge. (C) BLI of all mice over a 5-week observation period is shown. BLI pictures were generated sequentially from weeks 1 to 5. Frontal pictures of mice are shown, with bioluminescence signal radiance (photons/s/cm2/sr) depicted by the color barcode on the right side. (D) Quantification of leukemia spread by BLI is shown. Radiance (photons/s) depicts each pixel summed over the regions of interest (ROIs) area containing the whole frontal side of the body. Statistical analysis comparing each cohort with the tumor only cohort at each time point was performed after log transformation using a repeated-measures two-way ANOVA with Geisser-Greenhouse correction followed by Dunnett’s multiple comparison test.
Gene editing of TRAC in primary T cells is highly specific, with no measurable impact from small-molecule, DNA-repair modulators
Off-target gene editing is a major concern in the clinical translation of gene-edited cell therapies. Therefore, we examined 58 potential off-target editing sites identified by GUIDE-seq (Figure S1) and COSMID60 via next-generation sequencing (NGS) in primary human T cells to investigate the impact of our A > G mismatch and the exposure to HDR enhancer v.1 or v.2. Since indel-editing signal from NGS is subject to variable levels of experimental noise, we used a statistical approach to classify an off target as significant above a defined limit of detection (editing >0.5%; see materials and methods). Using this approach, only the on-target site was classified as edited for two different donor cell lines for both the original and A > G mismatch gRNA, suggesting that there are no off-target editing events or they are below the limit of detection (0.5%) for our method (Figures 7A and 7B). In addition, when HDR enhancer v.1 or v.2 was introduced to the cells, there was no significant increase in editing at the nominated off-target loci (Figure 7C). Since small-molecule HDR enhancers have been shown to modulate DNA-repair pathways, it is possible that introduction of these compounds can affect the mutation spectrum at an on-target locus. We investigated this by comparing the spectra of mutations produced under variable conditions with those predicted by the FORECasT in silico repair profile tool using symmetric Kullback-Leibler (KL) divergence to calculate similarity, as has been done previously.61,62 FORECasT was able to predict the most prevalent mutation correctly under all conditions (−8-bp deletion), but there was no significant difference (p < 0.05) between the KL divergence of samples with or without HDR enhancer treatments (Figures 7D and 7E).
Figure 7.

Characterization of off-target editing and effects of DNA-repair modulators
Editing was quantified using CRISPAltRations at all nominated on- or off-target loci for two healthy donors with paired treatment or controls after being edited with the TRAC guide (A) without a mismatch in the gRNA or (B) with a mismatch in the gRNA. Sites were classified as edited (orange circle) or not edited (blue circle) using a thresholded Fisher’s exact test (p < 0.05) with classification limitations. Green areas indicate areas capable of classification, whereas red areas indicate areas outside classification limits (indels in treatment <0.5%; indels in control >0.4%; <5,000 reads). Using the gRNA with no mismatch, the effects of HDR enhancing small molecules on (C) off-target editing and (D) on-target indel profiles, measured as KL divergence from FORECasT predictions, were investigated (one-way ANOVA with post-hoc Tukey’s correction; p < 0.05). (E) The contributions of the top five indels for the no enhancer sample are shown for each donor and treatment (cut site shown as red line).
Discussion
Here, we showed that the manufacturing of TCR-replaced CAR T cells using a virus-free technology could be significantly improved by pharmacological means. We showed that two different pharmacological interventions synergistically enhanced both the relative knockin rate and the absolute CAR T cell yield. Under our optimized gene-editing conditions, we increased the efficacy of a >2-kb CAR transgene insertion by 4- to 11-fold compared with previously published virus-free approaches with similar CARs sizes. Finally, we confirmed that TRAC-replaced CAR T cells performed effector functions in vitro and in vivo. These results support the perception that the complex and costly rAAV6-mediated CAR T cell manufacturing can be fully switched toward a non-viral approach. Furthermore, the enhancement of the gene editing can be readily extrapolated to improve orthotopic TCR replacement,33 to repurpose endogenous transcriptional pathways,28 or for applications of pooled knockin screens, such as pooled knockin sequencing (PoKi-seq).34
We have independently replicated findings by Nguyen et al.,36 reporting that the anionic nanoparticle PGA increases efficiency and decreases toxicity during electroporation of RNP and dsDNA HDRT. Nonetheless, in our hands, the enhancement effects were lower than originally reported. Similarly, tCTS-modified dsDNA HDRTs displayed an advantage over the reg. HA configuration regarding knockin efficiency. However, the overall CAR T cell yield was only improved when the tCTS-modified HDRT was transfected in small doses (0.5 μg). These differences may result from changes resulting in the incremental developments in our protocols, such as the reduction of the co-transfected RNP amount, as well as the plate-bound stimulation.25 Notably, the latter potentially reduces costs and may circumvent cell loss caused by bead depletion prior to electroporation.
Of note, increased amounts of transfected dsDNA donor templates resulted in substantially reduced cell survival and coincided with increased secretion of IL-6 and TNF-α and overall decreased CAR T cell yields. Combined DNA-sensor inhibition (ODN A151 and RU.521) partially reduced IL-6 release, but it did not affect TNF-α secretion. In line with our initial hypotheses, these findings indicate that the toxicity of the virus-free approach is caused mainly by electroporation of dsDNA donor templates and is at least partially mediated via classical DNA-sensing pathways. Independent of transfected DNA amount, our combined DNA-sensor inhibition reduced the release of Th2-associated cytokines, such as IL-13 and IL-10, although the underlying mechanism remains to be elucidated. DNA-sensor inhibition provides a route toward further improvements of this method, potentially through inhibitors with higher potency or interference with additional (non-classical) DNA-sensing pathways.63
Lowering the amount of transfected dsDNA donor may reduce toxicity by decreasing the physical strain imposed on the cell as well as by provoking a weaker innate immune response. While this is advantageous in terms of absolute CAR T cell yield, it also results in decreased relative HDR rates. Thus, in order to increase CAR integration rates with low amounts of dsDNA donors, modulation of DNA repair toward HDR is desirable. Many HDR-enhancing drugs have been previously identified.44, 45, 46, 47, 48, 49, 50, 51, 52 As only a few of these compounds have been validated for use in T cells, we assessed 15 HDR-enhancing small molecules in Jurkat cells. Nine compounds increased relative HDR more than 2-fold on average. The two commercially available HDR enhancers that we tested in primary human T cells increased relative knockin rates in a dose-dependent manner. Importantly, they did not increase off-target editing or significantly alter non-HDR repair outcomes for the investigated TRAC-targeting sgRNAs (Figure 7). The observed effects mirrored the preliminary data obtained with the cell line, indicating that Jurkat cells may be an accessible model to study DNA-repair modulation in T cells. Intriguingly, suboptimal editing with a Cas12a derivative could be improved in primary T cells, which may be of interest for groups targeting AT-rich regions in the genome.64 Suboptimal editing with HiFi SpCas9 was worsened by the intended mismatch in our TRAC sgRNA (Figure S6D). However, even with the original sgRNA, HiFi SpCas9 yielded less than 2-fold lower CAR-knockin (KI) rates (Figures S6C and S6D), which may be explained by the low amount of RNP used in our study, because others can achieve highly efficient editing with this nuclease and rAAV6 in T cells.24,54
We discovered that pre-electroporation DNA-sensor inhibition treatment increased relative KI rates of the CAR constructs. As HDR is cell cycle dependent,42,65 the improved KI rates seen here may in part be attributed to a small increase in the proportion of T cells within S phase at the time of electroporation. Alternatively, this may be directly related to the use of the cGAS-inhibitor, RU.521. Recent studies reported that nuclear cGAS is a general inhibitor of DNA repair and prevents homology-directed DNA repair through direct condensation of genomic DNA.66,67 In particular, this may explain the synergy between cGAS-directed DNA sensor inhibition and HDR enhancers observed in our experiments. Before implementation in clinical manufacturing, HDR enhancers and DNA-sensor inhibitors must be available with GMP-compatible documentation and carefully evaluated regarding their effects on genomic DNA integrity. This will require future studies investigating the effects of these modulators at diverse and numerous genomic loci while interrogating additional complex genomic rearrangement events (i.e., translocations, chromothripsis, homology-independent integration events, etc.).
Since the reagents used in our protocol are commercially available, the techniques are easily transferable to other groups for preclinical research and development aiming at a high efficiency of a non-viral CAR KI (exceeding 50%). These methods circumvent the need for the laborious, time-consuming, and costly manufacturing of viral vectors. By removing the practical hurdle of virus production, we hope our described technique may boost the development of TRAC-replaced CAR T cells. The proof-of-concept animal study did not show any adverse effects and even a trend toward improved therapeutic efficacy in comparison to LV-driven CAR, although further in vivo preclinical studies will be required for evaluation of safety and establish stronger evidence for enhanced in vivo performance. This method can be upscaled to suit GMP-compliant CAR T cell production and translational uses in an allogeneic setting. Our straight-forward technology platform could pave the way for “off-the-shelf” CAR T cell products. Lower biosafety and manufacturing requirements and potentially competitive lower costs would enable a decentralized manufacturing for the point-of-care clinical use. In doing so, we believe this platform may accelerate innovation for adoptive T-cell-based therapies for the treatment of several malignancies, autoimmune diseases, and chronic infections.
Materials and methods
GUIDE-seq
GUIDE-Seq68 for unbiased identification of CRISPR-Cas off targets was performed as previously described.54,69 Briefly, SpCas9-expressing HEK293 cells were co-transfected with dsDNA oligonucleotides (GUIDE-seq tag; for sequence, see Tsai et al.68) and either the original24 or target-sequence-modified TRAC sgRNA (this study). Subsequently, genomic DNA was isolated after 72 h using QuickExtract (Lucigen, USA) and fragmented using the LOTUS DNA library kit (Integrated DNA Technologies [IDT], Coralville, USA). Libraries were generated according to the original protocol,68 followed by Illumina-based NGS. Read alignment and data analysis was performed based on GUIDE-seq software.68
PBMC isolation and T cell enrichment
The study was performed in accordance with the Declaration of Helsinki. Peripheral blood was obtained from healthy human adults after informed consent (Charité ethics committee approval EA4/091/19). PBMCs were isolated using density-gradient centrifugation. Fresh heparinized whole blood was diluted 1:1 with sterile phosphate-buffered saline (PBS) (Gibco) and layered onto PANcoll separation medium (PAN Biotech, Germany) in 50-mL tubes (Falcon). Centrifugation was performed, with the brake function turned off, at 800g for 20 min. Subsequently, the mononuclear cell layer was harvested and diluted in sterile PBS. After an initial centrifugation at 450g for 10 min, supernatant was discarded. The pellet was resuspended in sterile PBS and centrifuged at 300g for 10 min. Afterward, the supernatant was discarded. PBMC pellets were resuspended in 10–20 mL PBS and counted using a Neubauer hemocytometer. Unless otherwise stated, PBMCs were positively enriched for CD3+ T cells using magnetic column enrichment with human CD3 microbeads according to the manufacturer’s recommendations (LS columns, Miltenyi Biotec, Germany).
Cell culture
PBMCs or enriched T cells were cultured in RPMI 1640 (PAN Biotech), 10% heat-inactivated fetal calf serum (FCS) (Biochrom), and recombinant IL-2 (200 IU/mL), IL-7 (10 ng/mL), and IL-15 (5 ng/mL). T cell stimulation was performed for 48 h on anti-CD3/28-coated tissue culture plates unless stated otherwise. Coating of vacuum gas plasma-treated polystyrene 24-Well-Tissue-Culture plates (Corning) was performed overnight with 500 μL/well of sterile ddH2O supplemented with 1 μg/mL anti-CD3 monoclonal antibody (mAb) (clone OKT3; Invitrogen) and 1 μg/mL anti-CD28 mAb (clone CD28.2; BioLegend). Plates were washed twice in PBS and once in RPMI without letting the wells dry out. T cells were seeded at a density of 1–1.5 × 106 per well in a 24-well-plate. For some experiments, stimulation was performed with Dynabeads Human T-Activator CD3/CD28 beads (Invitrogen) according to the manufacturer’s protocol. Prior to nucleofection, beads were removed by incubation on strong magnet stands. The tumor cell lines Nalm-6 and MM.1S were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ) and cryopreserved for later use. The MM.1S cell line was genetically manipulated to overexpress GFP and Firefly Luciferase (by A.R. and U.E.H.). All cell lines were freshly thawed and passaged 2–6 times prior to use in assays. Cell lines were cultured in RPMI1640 supplemented with 10% heat-inactivated FCS, 100 IU/mL penicillin, and 100 μg/mL streptomycin. Tumor cell lines were split every 2 to 3 days. All cell culture was performed at 37°C and 5% CO2.
Generation of dsDNA donor template for homology-directed insertion of a chimeric antigen receptor
Design of homology arms was performed as recently described.32 Cloning of HDR donor templates with over 2,000-bp insert size was performed with multiple fragment In-Fusion cloning according to the manufacturer’s protocol (Clontech, Takara). In brief, synthesis of 400-bp dsDNA sequences homologous to the targeted locus was commissioned (gBlocks, IDT) with a total overlap of 16 bp with either the insert or the pUC19 vector backbone. Similarly, inserts consisting of a P2A self-cleaving peptide, CAR/reporter transgene, and a bovine-growth-hormone-derived polyadenylation sequence were designed and synthesis was commissioned (gBlocks, IDT). As a model insert for a therapeutically relevant transgene, a CD19-specific CAR was designed based on the original FMC63 scFv. We chose an intermediate-length IgG1 hinge (for staining purposes) and a CD28 transmembrane and costimulatory domain linked to a cytosolic CD3 zeta domain. The CAR sequence was rationally designed and subsequently codon optimized using two separate algorithms: first using the COOL-algorithm;70 in a second step, IDT’s codon optimization tool was used to eliminate any complexities and allow DNA synthesis. In-Fusion cloning strategies were planned with SnapGene (from Insightful Science; snapgene.com). For other experiments, CAR transgenes were PCR amplified (Kapa Hotstart HiFi Polymerase Readymix, Roche) from lenti- and retroviral expression plasmids with primers that allowed In-Fusion reaction with the existing donor template backbones (TRAC). The BCMA.4-1BB-zeta CAR was previously described,59 and the plasmid was provided by A.R. and U.E.H. (MDC, Berlin). In-Fusion reactions were performed in 5-μL reactions at the recommended volume ratios. One microliter of In-Fusion reaction mixtures were transformed into Stellar Competent E. coli in 10-μL reactions and plated on ampicillin containing (LB) broth agar plates. After performing colony PCR for size validation with universal primers adjacent to the pUC19 insertion site (M13-for: 5′-GTAAAACGACGGCCAG-3′; M13-rev: 5′-CAGGAAACAGCTATGAC-3′), 3–5 mL ampicillin-containing bacterial cultures of preferred clones were incubated at 37°C overnight. Plasmids were purified using ZymoPURE Plasmid Mini Prep Kit (Zymo Research). Sequence validation of HDR-donor-template-containing plasmids was performed by Sanger Sequencing (LGC Genomics, Berlin). HA-flanked transgenes were amplified from the plasmids by PCR using the KAPA HiFi HotStart 2x Readymix (Roche) with reaction volumes >500 μL. For PCR amplification of TRAC CD19-CAR HDR template no. 1, either regular primers (Table S1, line 20 and 21) or overhang primers adding tCTSs (Table S1, line 22 and 23) to the PCR product were used. PCR products were purified and concentrated using paramagnetic beads (AMPure XP, Beckman Coulter Genomics). This purification process included two washing steps in 70% ethanol. Resulting HDRT concentrations were adjusted to 2 μg/μL in nuclease-free water. For quantification, HDRTs were diluted 1:20 and measured on Nanodrop 1000 (Thermo Fisher Scientific). For optimal quantification accuracy, we recommend using Qubit fluorometer or similar.
Electroporation buffer
One hundred milliliters of electroporation buffer 1M were made according to the original publication41 by dissolving 37.3 mg KCl, 142.8 mg MgCl2, and 910 mg mannitol in 40-mL sterile ddH2O (Ampuwa) prior to adding 60 mL of 0.2 M phosphate buffer solution (Na2HPO4/NaH2PO4; pH 7.2). The buffer was sterile filtered using 0.2-μm syringe filters, aliquoted, and stored at −20°C. Prior to use, it was thawed, shaken to dissolve intermittently formed precipitates, and placed on ice.
Formulation of ribonucleoproteins and mix with dsDNA templates
Per electroporation of 106 primary human T cells, 0.5 μL of an aqueous solution of 15- to 50-kDa poly(L-glutamic acid) (PGA) (Sigma-Aldrich, 100 μg/μL) was mixed with 0.48 μL of synthetic modified sgRNA (2′-O-methyl at three first and last bases and 3′ phosphorothioate bonds between first three and last two bases; Synthego, 3.2 μg/μL = 100 μM in TE buffer; in some experiments, modified sgRNA was provided by IDT) by pipetting thoroughly. We added 0.4 μL recombinant Streptococcus pyogenes Cas9 protein (Alt-R S.p. Cas9 Nuclease V3; IDT; 10 μg/μL = 61 μM) and again mixed by thorough pipetting. The molar ratio of Cas9 and sgRNA was thus 1:2. The mixture was incubated for 15 min at room temperature (RT) to allow for RNP formation and placed on ice afterward. For DNA-escalation studies, 0.25–2.00 μL of HDRT (stock concentration: 2 μg/μL) per 106 cells was added just prior to electroporation. For all other experiments, 0.5 μL HDRT per 106 cells was used. Details on sgRNA target sequence are presented in Table S2.
Electroporation
Anti-CD3- and anti-CD28-stimulated primary human T cells were resuspended, pooled, and washed twice in sterile PBS at 100g for 10 min, RT. Afterward, they were resuspended in 20 μL/106 cells ice-cold electroporation buffer (1M or P3 [Lonza] as indicated). The exposure time to the electroporation buffers was kept as short as possible. For electroporation of 1 × 106 cells, 20 μL of resuspended cells were transferred to 1.88 μL of RNP/HDRT (except during DNA-escalation studies where different volumes were used) and mixed thoroughly. Afterward, the T cell/RNP/HDRT mixture was transferred onto a 16-well electroporation strip (20 μL = 106 cells per well, Lonza) or an electroporation cartridge (100 μL = 5 × 106 cells, Lonza). Cells were carefully transferred onto the electroporation strips using 200-μL tips to avoid trapping air in the solution. Electroporation strips and cartridges were tapped onto the bench several times to ensure correct placement of fluids within the electroporation vessel. Electroporation was performed on a 4D-Nucleofector Device (Lonza) using the program EH-115. Directly after electroporation, pre-warmed T cell medium was added onto the cells (90 μL per well and 450 μL per cartridge). Afterward, resuspended cells were transferred to 96-well round-bottom plates (50 μL/well) containing 150 μL pre-warmed T cell medium per well (with or without HDR-enhancing supplements) at a density of 0.5 × 106 cells per well. For large-scale CAR T cell generation by electroporation in cartridges, 950 μL of pre-warmed T cell medium was used for initial resuspension. Ten minutes after electroporation, T cells were transferred into 24-well flat-bottom plates (500 μL/well) containing 1.5 mL of T cell medium (with or without HDR-enhancing supplements) at a density of 2.5 × 106 cells per well.
HDR-enhancing small-molecule screen in Jurkat cells
A series of small-molecule compounds was screened for the ability to increase CRISPR HDR rates in Jurkat cells. Small molecules were purchased from commercial sources, dry powders solubilized in DMSO as directed, or solutions handled as instructed. Jurkat cells (Clone E6-1, ATCC TIB-152) were cultured in RPMI media (ATCC) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% Pen Strep (Gibco), maintained at a density between 105 and 106 cells/mL. Cas9 gRNA targeting HPRT1 was prepared by mixing equimolar amounts of Alt-R CRISPR-RNA (crRNA) and Alt-R trans-activating CRISPR RNA (tracrRNA) (IDT, Coralville, IA, USA) in Tris-EDTA (TE), heating to 95°C, and slow cooling at room temperature. A single-stranded DNA oligo consisting of 40 nt of homology flanking the Cas9 cleavage site and a 6-base EcoRI (5′-GAATTC-3′) restriction site inserting at the Cas9 cleavage site was designed (Table S3); the non-targeting (guide-containing) sequence strand was ordered as an Alt-R HDR Donor Oligo (IDT). The RNP was formed by complexation of 56-pmol IDT Cas9 protein with 67.2-pmol gRNA complex in a total volume of 7.5 μL with PBS to adjust to the final volume. All reagents were delivered to Jurkat cells using the Lonza Nucleofector System. Cells were counted and pelleted by centrifugation (300g, 10 min at room temperature) and washed with 10 mL 1× PBS. The cells were again pelleted and resuspended in Lonza Nucleofection Solution SE at a density of 1 × 108 cells/mL. For each electroporation, 5 μL of RNP was added to 20 μL of Jurkat cells in Nucleofection Solution SE (5 × 105 cells/nucleofection). Donor DNA and IDT Alt-R Cas9 Electroporation Enhancer were added to achieve a final concentration of 1 and 3 μM, respectively, in a total reaction volume of 28 μL. The solution was mixed by pipetting, and 25 μL was transferred to an electroporation cuvette plate. The cells were electroporated according to the manufacturer’s protocol using the Lonza 96-well Shuttle and nucleofection protocol 96-CL-120. After electroporation, the cells were resuspended in 75 μL pre-warmed RMPI media in the electroporation cuvette. Triplicate aliquots of 25 μL of resuspended cells were further cultured in 175 μL pre-warmed RPMI media containing either PBS, DMSO, or a small molecule dissolved in DMSO. Twenty-four hours after electroporation, the media containing DMSO or small molecule was aspirated away and replaced with fresh media. The cells were allowed to grow for 72 h in total, after which genomic DNA was isolated with QuickExtract (Lucigen, Middleton, WI, USA). The experiment was repeated two more times for three total biological replicates. Perfect HDR of all experiments was quantified by NGS. The genomic region was amplified first with target-specific primers and then a second round of PCR to incorporate P5 and P7 Illumina adapters to the ends of the amplicons as previously described. Libraries were purified using Agencourt AMPure XP system (Beckman Coulter Genomics, Brea, CA, USA) and quantified with qPCR before loading onto the Illumina MiSeq platform (Illumina, San Diego, CA, USA). Paired end, 150-bp reads were sequenced using v.2 chemistry and data analysis done via IDT’s in-house data analysis pipeline (CRISPAltRations).
Lentivirus production and transduction
Propagation of lentiviruses for CD19-CAR overexpression were prepared as previously described using transient transfection of 293T cells.71 Briefly, the CD19 construct was cloned into the SIN epHIV7 lentiviral vector plasmid. Supernatant from transfected 293T cells was filtered and concentrated by ultra-centrifugation. Titers were determined by transduction of H9 cells with a dilution series of concentrated lentivirus from supernatants. For T cell transduction, primary human T cells were enriched and plated on anti-CD3/CD28-coated tissue culture plates in T cell medium. After 1 day, transduction was performed by centrifugation at 800g for 30 min at 32°C with lentiviral supernatant (MOI of 1) supplemented with 1 mg/mL protamine sulfate (APP Pharmaceuticals, Barceloneta, Puerto Rico). One day later (after 2 days of plate-bound CD3/CD28 stimulation), transduced T cells were taken off the stimulation plate.
CAR T cell expansion
First medium change or first splitting of cells was performed 18 h after electroporation unless stated otherwise. Cells were expanded in T cell medium either on 96-well round-bottom plates (electroporation volume of 20 μL) or on 24-well plates (electroporation volume of 100 μL). For further expansion, cells were subsequently transferred to G-Rex10 devices (Wilson Wolf). On day 14 after blood collection, cell products were frozen in FCS supplemented with 10% DMSO at a density of 107 cells/mL.
Drug treatments before and after nucleofection
To test the individual effects of selected DNA-sensor inhibitors, inhibitors were added to the T cells cultured in 24-well stimulation plates at the following concentrations 6–8 h prior to electroporation: ODN A151 (Invivogen): 5 μM; RU.521 (Invivogen): 9.64 μM; H151 (Invivogen): 3.58 μM; and BX795 (Invivogen): 5 μM. The medium was mixed by pipetting, and the resuspended T cells were incubated at 37°C and 5% CO2 until electroporation.
For combined DNA-sensor inhibition, 6 h prior to electroporation, 1 mL of the T cell medium per well (50% of the medium) was removed from the 24-well stimulation plates and 10 μL of the ODN A151 and 1 μL RU.521 was added to the remaining medium in the stimulation plate at the following concentrations: ODN A151: 5 μM and Ru.521: 4.82 μM. The medium was mixed by pipetting, and the resuspended T cells were incubated at 37°C and 5% CO2 until electroporation.
To test the effects of HDR-enhancing substances (Alt-R HDR enhancer v.1 and HDR enhancer v.2, both available at IDT), electroporated and resuspended T cells were transferred onto cell-culture plates containing pre-warmed T cell medium with HDR-enhancing supplements in a volume ratio of 1:3 (50 μL cell suspension was added to 150 μL supplemented T cell medium). Concentrations tested ranged from 3.25 μM to 30 μM for HDR-Enhancer v.1 and from 0.25 μM to 2 μM for HDR-Enhancer v.2. All concentrations stated in this publication refer to the supplemented T cell medium before seeding of T cells.
Multiplex cytokine analysis of cell-culture supernatants
To characterize the cytokine response to transfected DNA, 100 μL of supernatant was harvested 24 h after electroporation. Supernatants were transferred to a sterile 96-well plate, sealed, and stored at −80°C until analysis. After thawing of supernatants, measurements were performed using the human Proinflammatory Panel 1 kit (for cytokines such as IL-1β, IL-6, IL-10, IL-13, and TNFα) from Meso Scale Diagnostics according to the manufacturer’s instructions. Analysis was performed at the Immunological Study Lab of CheckImmune on a Mesoscale Discovery platform. For each sample, the respective value of the electrochemiluminescence (relative fluorescent unit [RFU] signal) of the analyte concentration was calculated on the basis of a calibration curve and the blank control values were subtracted (fresh T cell medium). Measurements of the Meso Scale Diagnostics assay were performed and evaluated in accordance with the International Conference on Harmonisation Good Clinical Practice Guideline under “Validation of analytical procedures.”
Flow cytometry
Unless stated otherwise, flow cytometric assessments were carried out on a Cytoflex LX device (Beckman Coulter Genomics) using the 96-well plate format. Measurements of cell concentrations were performed in 96-well, flat-bottom, cell-culture plates; other measurements were performed in 96-well, round-bottom, cell-culture plates. All staining panels are specified in Table S4. Representative gating strategies for flow cytometry panels are depicted in Figure S10. Cell concentrations were assessed by acquiring 20 μL of resuspended cells diluted 1/10 in PBS without any prior washing steps.
For detection of CAR expression after KI, 40 μL of cell suspension was transferred onto the 96-U-bottom-well plate and assessed following a series of successive washing and staining steps. For phenotyping, approximately 100,000 T cells were aliquoted per well. Each washing step included adding 240 μL PBS, centrifuging the plates at 400g for 5 min at RT, discarding the supernatants, and resuspending the pellets in the remaining volume by vortexing briefly. For any individual staining procedure, a mastermix of the antibodies and dyes diluted in PBS was prepared. Twenty microliters of the mastermix was added per well. The plates were vortexed briefly, incubated for 15 min at 4°C, and briefly vortexed again. Because anti-Fc (anti-IgG1, Fcγ part) antibodies used to detect CAR protein can potentially bind to other detection antibodies, a first extracellular staining step with anti-Fc antibody only and a live-dead discriminating dye and subsequent washing step were performed prior to any other staining steps. The phenotype assessment presented in Figures S6A and S6B was performed on an LSR-II Fortessa flow cytometer (BD Biosciences). The staining for this experiment was performed in 5 mL fluorescence-activated cell sorting (FACS) tubes (Corning). Apart from this difference, it was carried out analogously to the other staining procedures.
Cell cycle analysis
Stimulation of CD3-enriched T cells as well as DNA-sensor inhibition treatment with RU.521 and ODN A151 were performed as described above. For the staining, 200,000 cells were harvested, washed, and stained with Live/Dead Fixable Blue (Invitrogen) for discrimination of dead cells. Then, cells were stained as described previously.43 Briefly, the cells were fixed and permeabilized using the Intracellular Fixation & Permeabilization Buffer Set (Thermo Fisher Scientific, 88-8824). A mastermix containing 7AAD and KI67 Alexa Fluor 647 (BioLegend) was added to the cells for 30 min at 4°C in the dark. After washing, cells were resuspended in PBS and run on the Cytoflex LX cytometer (Beckman Coulter Genomics). The 7AAD dye stains DNA and indicates individual cell genomic DNA content to allow for differentiation of cells in resting (G0) and G1 phase versus cells in S and G2 phase. Ki-67 is an activation and proliferation marker that indicates cells outside resting phase (G0).
VITAL assay to assess cytotoxicity
Effector T cells (TRAC-CD19-CAR T cells, TRAC-BCMA-CAR T cells, and unedited [wild-type] T cells; each from the same three donors) were co-cultured with target cells expressing the surface protein recognized by the CAR and control cells negative for this marker. For the CD19 VITAL assay, Nalm6 cells (CD19+ BCMA−) engineered to express green fluorescent protein (GFP) served as target cells and CD19-knockout Nalm6 cells engineered to express red fluorescent protein (RFP) served as control cells. For the BCMA VITAL assay, MM.1S (CD19− BCMA+) engineered to express GFP served as target cells, while the same control cells were used. A 1:1 suspension of target and control cells was added to 25,000 (CAR) T cells in 96-well, round-bottom, cell-culture plates at three different effector:target:control cell ratios (1:2:2, 1:1:1, and 1:0.2:0.2). The plates were centrifuged at 100g, 3 min, RT and then incubated at 37°C, 5% CO2 for 4 h. Afterward, the plates were centrifuged at 400g, 10 min, RT and the supernatant was discarded. Cell pellets were resuspended in PBS and the plates centrifuged again at 400g, 10 min, RT. The supernatant was discarded, and the cells were stained with LIVE/DEAD Fixable Blue Dead Cell Stain (Invitrogen, L23105) diluted 1:60 in 20 μL PBS per well for 10 min at 4°C. Afterward, 240 μL PBS/well was added and the plates were centrifuged at 400g, 15 min, RT. The supernatant was discarded, and the cells were resuspended in 50 μL PBS and analyzed by flow cytometry. Effector-cell-mediated cytotoxicity was calculated from shifts in the target:control cell ratio relative to control conditions without effector cells. The experiment was performed on day 12 after blood collection (day 10 after electroporation).
Intracellular cytokine analysis
CAR T cells cryopreserved on day 14 after peripheral blood collection (day 12 after electroporation) were thawed by washing twice in pre-warmed RPMI1640 medium supplemented with 20% FCS and 10 ng/mL DNAse I (Roche, 11,284,932,001). Thawed CAR T cells were rested in cytokine-free complete medium (RPMI1640 medium containing 10% FCS and 1% penicillin-streptomycin) overnight at 37°C, 5% CO2. Nalm6 and Jurkat cells were labeled with 2.5 μM carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) (Thermo Fisher Scientific, V12883) and rested in complete medium overnight. One hundred thousand CAR T cells were co-cultured with carboxyfluorescein succinimidyl ester (CFSE)-labeled Nalm6 or Jurkat cells (ratio 1:1) in 96-well, round-bottom plates for 6 h. CAR T cells stimulated with 10 ng/mL phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, P8139-1MG) and 2.5 μg/mL ionomycin (Sigma Aldrich, I3909) were included as positive control for polyclonal stimulation independent of TCR expression, and CAR T cells cultured alone were included as negative control. Brefeldin A (Sigma Aldrich, B5936) was added at a concentration of 10 μg/mL after 1 h of co-culture and stimulation. After 6 h of stimulation, cells were harvested and stained with fixable blue dead cell stain dye. Subsequently, cells were fixed and permeabilized using the Intracellular Fixation & Permeabilization Buffer Set (Thermo Fisher Scientific, 88-8824). A first intracellular staining with anti-Fc AF647 was performed prior to washing twice with 1× permeabilization buffer. A second intracellular staining was performed with all other antibodies as stated in Table S4.
Nalm-6 tumor cell rechallenge assay and exhaustion panel
CD19-CAR T cells (produced by lentiviral gene transfer or TRAC integration as described above) were seeded into a flat-bottom, 96-well plate at a density of 25,000 CAR+ cells/well using X-vivo 15 medium supplemented with 10% FCS and 50 IU/mL IL-2. Every 12 h, gamma-irradiated (30 Gy) GFP+ Nalm-6 cells were added to the CAR T cells in a 1:1 cellular ratio. Twenty-four hours after the fourth round of co-culture, CAR T cells were analyzed by flow cytometry for expression of the inhibitory receptors PD-1, LAG-3, and Tim-3 (Figure S10; Table S4).
In vivo testing of CAR T cells in a xenograft leukemia model
The in vivo CAR T cell potency analyses were performed in accordance with the German Animal Welfare Act and the EU-directive 2010/63 and approved by the Lower Saxony Office for Consumer Protection and Food Safety–LAVES (permit numbers 16/2222, 16/2374, and 21/3791). Breeding pairs of nonobese diabetic (NOD).Cg-Rag1tm1MomIL-2Rγctm1Wjl mice (NRG) (stock number 007799) were obtained from the Jackson Laboratory (JAX) (Bar Harbor, USA) and bred under pathogen-free conditions. For the sake of accessing the variability of responses among mouse sexes, we used both female and male mice. Eight-week-old NRG mice were infused with 5 × 105 Nalm-6/GFP/fLuc cells via tail vein injection. Four days later, 5 × 105 CD19-specific, TCR-deficient CAR+ T cells generated by TRAC replacement (using TRAC CD19-CAR HDR template no. 1 [Table S1], Alt-R SpCas9 V3, TRAC (gG) v1 sgRNA [Table S2], or by LV gene transfer) and virus-transduced counterparts with TRAC knockouts (KOs) were infused intravenously (i.v.). Prior to T cell infusion, residual CD3+ T cells were depleted using MACS technology (CD3+ Microbeads and LD columns, Miltenyi Biotec) according to the manufacturer’s instructions. An IVIS SpectrumCT apparatus (PerkinElmer, Waltham, MA, USA) was used for weekly dynamic BLI analyses. Before imaging, mice were anesthetized using isoflurane. Mice were imaged 5 min after luciferin administration (2.5 mg D-luciferin potassium salt intraperitoneally [SYNCHEM, Elk Grove Village, IL, USA] reconstituted in 100 μL PBS). Data were analyzed using the Living Image Software (PerkinElmer, Waltham, MA, USA). Terminal analyses were performed 5 weeks after leukemia challenge, and mice were euthanized afterward according to the animal experiment protocol.
Quantification of off targets in primary human T cells by targeted sequencing using rhAmpSeq CRISPR
Off-target editing at sites nominated by both GUIDE-seq and in silico nomination69 was measured in primary human T cells using NGS. For these experiments, gDNA was isolated from healthy donor T cells in edited and non-edited matched controls (two biological replicates per condition) using the Quick-DNA Miniprep Kit (Zymo Research). Sequencing libraries were then prepared using a previously described rhAmpSeq amplification-based method (IDT, Coralville, USA).72,73 Briefly, locus-specific amplification was performed for 14 cycles followed by a 1× SPRI bead clean-up (Beckman-Coulter). An indexing round of PCR was performed for 24 cycles to incorporate sample-unique P5 and P7 indexes followed by a 1× SPRI bead clean-up and library quantification by Qubit (Thermo Fisher Scientific). PCR amplicons were sequenced on an Illumina MiSeq instrument (v.2 chemistry, 150-bp paired end reads; Illumina). Data were analyzed and editing quantified using CRISPAltRations,61 utilizing the default window size for Cas9 (8 bp).
Data analysis, statistics, and presentation
Flow cytometry data were analyzed with FlowJo software v.10 (BD Biosciences). Data from different assays were sorted in Excel (Microsoft). Graphs were created using Prism 9 (GraphPad). Off-target editing data were binarily classified as edited using a thresholded Fisher’s exact test (p < 0.05) with limitations for classification (%indels in treatment >0.5%; %indels in control <0.4%; >5,000 reads per site). Significance of HDR-enhancer effects was calculated using one-way repeated measures ANOVA with a post hoc Tukey’s correction for multiple comparisons (p < 0.05). Conditions with failed electroporation (indicated by 4D-Nucleofector Device) were recorded during experiment and later excluded from analysis. Exploratory statistics were performed with Prism 9 (GraphPad). Schemes and graphs in the presented figures were created using www.biorender.com.
Data availability statement
All underlying data of this manuscript can be obtained from the corresponding author upon request. The plasmid used for the generation of dsDNA HDR template for the insertion of the CD19-specific CAR into the TRAC gene can be obtained from Addgene (ID: 183473). NGS data from GUIDE-seq experiment and HDR-enhancer compound screen in Jurkat cells were deposited in the Sequencing Read Archive (SRA) repository of the National Center for Biotechnological Information (NCBI) under the bioproject accession code PRJNA701496.
Acknowledgments
We would like to thank Dr. Alexander Marson and Dr. Theodore L. Roth for protocols on their original work on non-viral T cell reprogramming. We show gratitude to Lonza GmbH for temporarily providing the 4D-Nucleofector 96-well unit for the DNA-escalation experiment presented in Figures 1, 2, 4, and S5; in particular, we would like to thank Dr. Melanie Homberg and Dr. Nina Novak for technical assistance. We thank Jan Csupor for helping with multiplex-cytokine analysis of cell culture supernatants. We thank Silke Schwiebert (Charité) for technical assistance with lentivirus preparation and titration and Andreas Schneider (MHH) for his excellent technical expertise in the mouse experiment. Norohiro Watanabe (Baylor College of Medicine, USA) provided insights for the cell cycle analysis protocols. We thank Dr. Ciaran M. Lee and Dr. Kathleen Anders for the critical revision of the manuscript. Finally, we would like to express our gratitude to Dr. Nicola R. Brindle for scientific language editing our manuscript. This study was generously supported in part by the German Federal Ministry of Education and Research (BIH Center for Regenerative Therapies, 10178 Berlin—J.K., W.D., L. Amini, M.S., T.Z., S.M., H.-D.V., M.S.-H., P.R., and D.L.W.), a kick-box grant for young scientists and a research grant by the Einstein Center for Regenerative Therapies (J.K., S.M., and D.L.W.), and a Berlin Institute of Health (BIH) Translation-Mission-Fund and a Crossfield project fund of the BIH Research Focus Regenerative Medicine (H.-D.V., M.S.-H., P.R., and D.L.W.). This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 825392 (ReSHAPE-h2020.eu). A.K. is a participant in the BIH-Charité Advanced Clinician Scientist pilot program funded by the Charité – Universitätsmedizin Berlin and the Berlin Institute of Health. The mouse study was financed by a grant of the German Center for Infections Research (DZIF-TTU07.912; R.S.) and by a research grant of the German Cancer Aid (Stiftung Deutsche Krebshilfe; no. 70114234; R.S.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
J.K. designed this study, planned and performed experiments, analyzed results, interpreted the data, and wrote the manuscript. W.D. performed experiments, analyzed results, interpreted data, and edited the manuscript. A.Pruene. and T.B. performed the in vivo xenograft experiment, BLI measurements, and analyzed the results. B.T. and R.T. provided reagents, performed GUIDE-seq experiments and HDR-enhancing small-molecule screen in Jurkat cells, analyzed results, interpreted data, and edited the manuscript. M.L.S. and G.L.K. performed and interpreted analyses on off-target editing and indel profile characterization and edited the manuscript. L. Amini, M.S., T.Z., S.M., L.O., A.W., and L. Akyüz performed experiments and analyzed results. A.R. and U.E.H. provided reagents (BCMA-CAR and MM1S cell line), interpreted data, and edited the manuscript. A.P. provided reagents and interpreted data. A.K. provided reagents (CD19-CAR lentivirus and Nalm-6 cell line), interpreted data, and edited the manuscript. A.M.J. planned GUIDE-seq, HDR-enhancer screen in Jurkat cells and off-target analysis, interpreted data, and edited the manuscript. H.-D.V. provided reagents, interpreted data, and edited the manuscript. M.S.-H. planned experiments, interpreted data, and edited the manuscript. R.S. designed and coordinated the mouse experiments, interpreted data, and edited the manuscript. P.R. supervised the study, interpreted data, and edited the manuscript. D.L.W. designed and led the study, planned and performed experiments, analyzed results, interpreted data, and wrote the manuscript. All authors discussed, commented on, and approved the manuscript in its final form.
Declaration of interests
As part of a collaboration agreement between Charité Universitätsmedizin Berlin and Integrated DNA Technologies (IDT), IDT provided certain reagents (HDR enhancer v.2 and TRAC sgRNA used in some experiments) and performed GUIDE-seq analysis, HDR-enhancing small-molecule screen in Jurkat cells, and targeted sequencing of potential off-target sites. R.T., B.T., M.L.S., G.L.K., and A.M.J. are employees of IDT, which offers reagents for sale similar to some of the compounds described in the manuscript. Products and tools supplied by IDT are for research use only and not intended for diagnostic or therapeutic purposes. Purchaser and/or user are solely responsible for all decisions regarding the use of these products and any associated regulatory or legal obligations. Lonza GmbH provided 96-well 4D-Nucleofector unit and some nucleofection reagents. A.W. and L. Akyüz are part-time employees of CheckImmune GmbH. A.R. and U.E.H. filed a patent application WO 2017211900A1 “Chimeric antigen receptor and CAR T cells that bind BCMA” related to the work with the BCMA-CAR disclosed in this paper. A.R. and U.E.H. have received research funding from Fate Therapeutics for work unrelated to the data generated in the manuscript.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.03.018.
Supplemental information
References
- 1.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F., et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 3.Raje N., Berdeja J., Lin Y., Siegel D., Jagannath S., Madduri D., Liedtke M., Rosenblatt J., Maus M.V., Turka A., et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019;380:1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brudno J.N., Maric I., Hartman S.D., Rose J.J., Wang M., Lam N., Stetler-Stevenson M., Salem D., Yuan C., Pavletic S., et al. T cells genetically modified to express an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J. Clin. Oncol. 2018;36:2267–2280. doi: 10.1200/JCO.2018.77.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A., et al. HER2-Specific chimeric antigen receptor–modified virus-specific T cells for progressive glioblastoma. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Straathof K., Flutter B., Wallace R., Jain N., Loka T., Depani S., Wright G., Thomas S., Cheung G.W.-K., Gileadi T., et al. Antitumor activity without on-target off-tumor toxicity of GD2–chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020;12:eabd6169. doi: 10.1126/scitranslmed.abd6169. [DOI] [PubMed] [Google Scholar]
- 7.Amor C., Feucht J., Leibold J., Ho Y.-J., Zhu C., Alonso-Curbelo D., Mansilla-Soto J., Boyer J.A., Li X., Giavridis T., et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583:127–132. doi: 10.1038/s41586-020-2403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aghajanian H., Kimura T., Rurik J.G., Hancock A.S., Leibowitz M.S., Li L., Scholler J., Monslow J., Lo A., Han W., et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573:430–433. doi: 10.1038/s41586-019-1546-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellebrecht C.T., Bhoj V.G., Nace A., Choi E.J., Mao X., Cho M.J., Di Zenzo G., Lanzavecchia A., Seykora J.T., Cotsarelis G., et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353:179–184. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kansal R., Richardson N., Neeli I., Khawaja S., Chamberlain D., Ghani M., Ghani Q., Balazs L., Beranova-Giorgianni S., Giorgianni F., et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 2019;11:eaav1648. doi: 10.1126/scitranslmed.aav1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacDonald K.G., Hoeppli R.E., Huang Q., Gillies J., Luciani D.S., Orban P.C., Broady R., Levings M.K. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Invest. 2016;126:1413–1424. doi: 10.1172/JCI82771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fritsche E., Volk H.-D., Reinke P., Abou-El-Enein M. Toward an optimized process for clinical manufacturing of CAR-treg cell therapy. Trends Biotechnol. 2020;38:1099–1112. doi: 10.1016/j.tibtech.2019.12.009. S016777991930304X. [DOI] [PubMed] [Google Scholar]
- 13.Olbrich H., Theobald S.J., Slabik C., Gerasch L., Schneider A., Mach M., Shum T., Mamonkin M., Stripecke R. Adult and cord blood-derived high-affinity gB-CAR-T cells effectively react against human cytomegalovirus infections. Hum. Gene Ther. 2020;31:423–439. doi: 10.1089/hum.2019.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slabik C., Kalbarczyk M., Danisch S., Zeidler R., Klawonn F., Volk V., Krönke N., Feuerhake F., Ferreira de Figueiredo C., Blasczyk R., et al. CAR-T cells targeting epstein-barr virus gp350 validated in a humanized mouse model of EBV infection and lymphoproliferative disease. Mol. Ther. Oncolytics. 2020;18:504–524. doi: 10.1016/j.omto.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abou-El-Enein M., Elsallab M., Feldman S.A., Fesnak A.D., Heslop H.E., Marks P., Till B.G., Bauer G., Savoldo B. Scalable manufacturing of CAR T cells for cancer immunotherapy. Blood Cancer Discov. 2021;2:408–422. doi: 10.1158/2643-3230.BCD-21-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frigault M.J., Lee J., Basil M.C., Carpenito C., Motohashi S., Scholler J., Kawalekar O.U., Guedan S., McGettigan S.E., Posey A.D., et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015;3:356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomes-Silva D., Mukherjee M., Srinivasan M., Krenciute G., Dakhova O., Zheng Y., Cabral J.M.S., Rooney C.M., Orange J.S., Brenner M.K., et al. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector dependent. Cell Rep. 2017;21:17–26. doi: 10.1016/j.celrep.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eyquem J., Mansilla-Soto J., Giavridis T., van der Stegen S.J.C., Hamieh M., Cunanan K.M., Odak A., Gönen M., Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Comisel R.-M., Kara B., Fiesser F.H., Farid S.S. Lentiviral vector bioprocess economics for cell and gene therapy commercialization. Biochem. Eng. J. 2020;167:107868. [Google Scholar]
- 20.MacLeod D.T., Antony J., Martin A.J., Moser R.J., Hekele A., Wetzel K.J., Brown A.E., Triggiano M.A., Hux J.A., Pham C.D., et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol. Ther. 2017;25:949–961. doi: 10.1016/j.ymthe.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Depil S., Duchateau P., Grupp S.A., Mufti G., Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 2020;19:185–199. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 22.Ruella M., Xu J., Barrett D.M., Fraietta J.A., Reich T.J., Ambrose D.E., Klichinsky M., Shestova O., Patel P.R., Kulikovskaya I., et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018;24:1499–1503. doi: 10.1038/s41591-018-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai X., Park J.J., Du Y., Kim H.R., Wang G., Errami Y., Chen S. One-step generation of modular CAR-T cells with AAV-Cpf1. Nat. Methods. 2019;16:247–254. doi: 10.1038/s41592-019-0329-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiebking V., Lee C.M., Mostrel N., Lahiri P., Bak R., Bao G., Roncarolo M.G., Bertaina A., Porteus M.H. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: optimizing αβ T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica, Haematol. 2020;2019:233882. doi: 10.3324/haematol.2019.233882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gundry M.C., Brunetti L., Lin A., Mayle A.E., Kitano A., Wagner D., Hsu J.I., Hoegenauer K.A., Rooney C.M., Goodell M.A., et al. Highly efficient genome editing of murine and human hematopoietic progenitor cells by CRISPR/Cas9. Cell Rep. 2016;17:1453–1461. doi: 10.1016/j.celrep.2016.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomes-Silva D., Srinivasan M., Sharma S., Lee C.M., Wagner D.L., Davis T.H., Rouce R.H., Bao G., Brenner M.K., Mamonkin M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–296. doi: 10.1182/blood-2017-01-761320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kagoya Y., Guo T., Yeung B., Saso K., Anczurowski M., Wang C.-H., Murata K., Sugata K., Saijo H., Matsunaga Y., et al. Genetic ablation of HLA class I, class II, and the T-cell receptor enables allogeneic T cells to Be used for adoptive T-cell therapy. Cancer Immunol. Res. 2020;8:926–936. doi: 10.1158/2326-6066.CIR-18-0508. [DOI] [PubMed] [Google Scholar]
- 28.Sachdeva M., Busser B.W., Temburni S., Jahangiri B., Gautron A.-S., Maréchal A., Juillerat A., Williams A., Depil S., Duchateau P., et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat. Commun. 2019;10:5100. doi: 10.1038/s41467-019-13088-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feucht J., Sun J., Eyquem J., Ho Y.-J., Zhao Z., Leibold J., Dobrin A., Cabriolu A., Hamieh M., Sadelain M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019;25:82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jing R., Jiao P., Chen J., Meng X., Wu X., Duan Y., Shang K., Qian L., Huang Y., Liu J., et al. Cas9-Cleavage sequences in size-reduced plasmids enhance nonviral genome targeting of CARs in primary human T cells. Small Methods. 2021;5:2100071. doi: 10.1002/smtd.202100071. [DOI] [PubMed] [Google Scholar]
- 31.Bak R.O., Porteus M.H. CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Rep. 2017;20:750–756. doi: 10.1016/j.celrep.2017.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roth T.L., Puig-Saus C., Yu R., Shifrut E., Carnevale J., Li P.J., Hiatt J., Saco J., Krystofinski P., Li H., et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559:405–409. doi: 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schober K., Müller T.R., Gökmen F., Grassmann S., Effenberger M., Poltorak M., Stemberger C., Schumann K., Roth T.L., Marson A., et al. Orthotopic replacement of T-cell receptor α- and β-chains with preservation of near-physiological T-cell function. Nat. Biomed. Eng. 2019;3:974–984. doi: 10.1038/s41551-019-0409-0. [DOI] [PubMed] [Google Scholar]
- 34.Roth T.L., Li P.J., Blaeschke F., Nies J.F., Apathy R., Mowery C., Yu R., Nguyen M.L.T., Lee Y., Truong A., et al. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell. 2020;181:728–744.e21. doi: 10.1016/j.cell.2020.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Odé Z., Condori J., Peterson N., Zhou S., Krenciute G. CRISPR-mediated non-viral site-specific gene integration and expression in T cells: protocol and application for T-cell therapy. Cancers. 2020;12:1704. doi: 10.3390/cancers12061704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen D.N., Roth T.L., Li P.J., Chen P.A., Apathy R., Mamedov M.R., Vo L.T., Tobin V.R., Goodman D., Shifrut E., et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020;38:44–49. doi: 10.1038/s41587-019-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paludan S.R., Bowie A.G. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luecke S., Holleufer A., Christensen M.H., Jønsson K.L., Boni G.A., Sørensen L.K., Johannsen M., Jakobsen M.R., Hartmann R., Paludan S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017;18:1707–1715. doi: 10.15252/embr.201744017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zierhut C., Yamaguchi N., Paredes M., Luo J.-D., Carroll T., Funabiki H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell. 2019;178:302–315.e23. doi: 10.1016/j.cell.2019.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee C.M., Cradick T.J., Fine E.J., Bao G. Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing. Mol. Ther. 2016;24:475–487. doi: 10.1038/mt.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chicaybam L., Sodre A.L., Curzio B.A., Bonamino M.H. An efficient low cost method for gene transfer to T lymphocytes. PLoS One. 2013;8:e60298. doi: 10.1371/journal.pone.0060298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hustedt N., Durocher D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016;19:1–9. doi: 10.1038/ncb3452. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe N., Bajgain P., Sukumaran S., Ansari S., Heslop H.E., Rooney C.M., Brenner M.K., Leen A.M., Vera J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology. 2016;5:e1253656. doi: 10.1080/2162402X.2016.1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- 45.Wienert B., Nguyen D.N., Guenther A., Feng S.J., Locke M.N., Wyman S.K., Shin J., Kazane K.R., Gregory G.L., Carter M.A.M., et al. Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Nat. Commun. 2020;11:2109. doi: 10.1038/s41467-020-15845-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maruyama T., Dougan S.K., Truttmann M.C., Bilate A.M., Ingram J.R., Ploegh H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delacôte F., Han M., Stamato T.D., Jasin M., Lopez B.S. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res. 2002;30:3454–3463. doi: 10.1093/nar/gkf452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu Y.-W., Dai X.-Y., Wang W.-T., Yang Z.-X., Zhao J.-J., Zhang J.-P., Wen W., Zhang F., Oberg K.C., Zhang L., et al. Dynamics and competition of CRISPR-Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res. 2021;49:969–985. doi: 10.1093/nar/gkaa1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bertino E.M., Otterson G.A. Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin. Investig. Drugs. 2011;20:1151–1158. doi: 10.1517/13543784.2011.594437. [DOI] [PubMed] [Google Scholar]
- 50.Shin H.R., See J.-E., Kweon J., Kim H.S., Sung G.-J., Park S., Jang A.-H., Jang G., Choi K., Kim I., et al. Small-molecule inhibitors of histone deacetylase improve CRISPR-based adenine base editing. Nucleic Acids Res. 2021;49:2390–2399. doi: 10.1093/nar/gkab052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu C., Liu Y., Ma T., Liu K., Xu S., Zhang Y., Liu H., La Russa M., Xie M., Ding S., et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell. 2015;16:142–147. doi: 10.1016/j.stem.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leahy J.J.J., Golding B.T., Griffin R.J., Hardcastle I.R., Richardson C., Rigoreau L., Smith G.C.M. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg. Med. Chem. Lett. 2004;14:6083–6087. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
- 53.Zhang L., Zuris J.A., Viswanathan R., Edelstein J.N., Turk R., Thommandru B., Rube H.T., Glenn S.E., Collingwood M.A., Bode N.M., et al. AsCas12a ultra nuclease facilitates the rapid generation of therapeutic cell medicines. Nat. Commun. 2021;12:3908. doi: 10.1038/s41467-021-24017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J., et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim S., Kim D., Cho S.W., Kim J., Kim J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Overbeek M., Capurso D., Carter M.M., Thompson M.S., Frias E., Russ C., Reece-Hoyes J.S., Nye C., Gradia S., Vidal B., et al. DNA repair profiling reveals nonrandom outcomes at cas9-mediated breaks. Mol. Cell. 2016;63:633–646. doi: 10.1016/j.molcel.2016.06.037. [DOI] [PubMed] [Google Scholar]
- 57.Brinkman E.K., Chen T., de Haas M., Holland H.A., Akhtar W., van Steensel B. Kinetics and fidelity of the repair of cas9-induced double-strand DNA breaks. Mol. Cell. 2018;70:801–813.e6. doi: 10.1016/j.molcel.2018.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hermans I.F., Silk J.D., Yang J., Palmowski M.J., Gileadi U., McCarthy C., Salio M., Ronchese F., Cerundolo V. The VITAL assay: a versatile fluorometric technique for assessing CTL- and NKT-mediated cytotoxicity against multiple targets in vitro and in vivo. J. Immunol. Methods. 2004;285:25–40. doi: 10.1016/j.jim.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 59.Bluhm J., Kieback E., Marino S.F., Oden F., Westermann J., Chmielewski M., Abken H., Uckert W., Höpken U.E., Rehm A. CAR T cells with enhanced sensitivity to B cell maturation antigen for the targeting of B cell non-hodgkin’s lymphoma and multiple myeloma. Mol. Ther. J. Am. Soc. Gene Ther. 2018;26:1906–1920. doi: 10.1016/j.ymthe.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: a web-based tool for identifying and validating CRISPR/cas off-target sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurgan G., Turk R., Li H., Roberts N., Rettig G.R., Jacobi A.M., Tso L., Sturgeon M., Mertens M., Noten R., et al. CRISPAltRations: a validated cloud-based approach for interrogation of double-strand break repair mediated by CRISPR genome editing. Mol. Ther. Methods Clin. Dev. 2021;21:478–491. doi: 10.1016/j.omtm.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P., Páleníková P., Khodak A., Kiselev V., Kosicki M., et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2018 doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burleigh K., Maltbaek J.H., Cambier S., Green R., Gale M., James R.C., Stetson D.B. Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol. 2020;5:eaba4219. doi: 10.1126/sciimmunol.aba4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A., et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin J.J., Schröder M.S., Caiado F., Wyman S.K., Bray N.L., Bordi M., Dewitt M.A., Vu J.T., Kim W.-T., Hockemeyer D., et al. Controlled cycling and Quiescence enables efficient HDR in engraftment-enriched adult hematopoietic stem and progenitor cells. Cell Rep. 2020;32:108093. doi: 10.1016/j.celrep.2020.108093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang H., Xue X., Panda S., Kawale A., Hooy R.M., Liang F., Sohn J., Sung P., Gekara N.O. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J. 2019;38:e102718. doi: 10.15252/embj.2019102718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu H., Zhang H., Wu X., Ma D., Wu J., Wang L., Jiang Y., Fei Y., Zhu C., Tan R., et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. 2018;563:131–136. doi: 10.1038/s41586-018-0629-6. [DOI] [PubMed] [Google Scholar]
- 68.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shapiro J., Iancu O., Jacobi A.M., McNeill M.S., Turk R., Rettig G.R., Amit I., Tovin-Recht A., Yakhini Z., Behlke M.A., et al. Increasing CRISPR efficiency and measuring its specificity in HSPCs using a clinically relevant system. Mol. Ther. Methods Clin. Dev. 2020;17:1097–1107. doi: 10.1016/j.omtm.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chin J.X., Chung B.K.-S., Lee D.-Y. Codon Optimization OnLine (COOL): a web-based multi-objective optimization platform for synthetic gene design. Bioinformatics. 2014;30:2210–2212. doi: 10.1093/bioinformatics/btu192. [DOI] [PubMed] [Google Scholar]
- 71.Ausubel L.J., Hall C., Sharma A., Shakeley R., Lopez P., Quezada V., Couture S., Laderman K., McMahon R., Huang P., et al. Production of CGMP-grade lentiviral vectors. Bioprocess Int. 2012;10:32–43. [PMC free article] [PubMed] [Google Scholar]
- 72.Dobosy J.R., Rose S.D., Beltz K.R., Rupp S.M., Powers K.M., Behlke M.A., Walder J.A. RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011;11:80. doi: 10.1186/1472-6750-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacobi A.M., Rettig G.R., Turk R., Collingwood M.A., Zeiner S.A., Quadros R.M., Harms D.W., Bonthuis P.J., Gregg C., Ohtsuka M., et al. Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods San Diego Calif. 2017;121–122:16–28. doi: 10.1016/j.ymeth.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All underlying data of this manuscript can be obtained from the corresponding author upon request. The plasmid used for the generation of dsDNA HDR template for the insertion of the CD19-specific CAR into the TRAC gene can be obtained from Addgene (ID: 183473). NGS data from GUIDE-seq experiment and HDR-enhancer compound screen in Jurkat cells were deposited in the Sequencing Read Archive (SRA) repository of the National Center for Biotechnological Information (NCBI) under the bioproject accession code PRJNA701496.