

Abstract
T cells engineered to express HIV-specific chimeric antigen receptors (CARs) represent a promising strategy to clear HIV-infected cells, but to date have not achieved clinical benefits. A likely hurdle is the limited T cell activation and persistence when HIV antigenemia is low, particularly during antiretroviral therapy (ART). To overcome this issue, we propose to use a cytomegalovirus (CMV) vaccine to stimulate CMV-specific T cells that express CARs directed against the HIV-1 envelope protein gp120. In this study, we use a GMP-compliant platform to engineer CMV-specific T cells to express a second-generation CAR derived from the N6 broadly neutralizing antibody, one of the broadest anti-gp120 neutralizing antibodies. These CMV-HIV CAR T cells exhibit dual effector functions upon in vitro stimulation through their endogenous CMV-specific T cell receptors or the introduced CARs. Using a humanized HIV mouse model, we show that CMV vaccination during ART accelerates CMV-HIV CAR T cell expansion in the peripheral blood and that higher numbers of CMV-HIV CAR T cells were associated with a better control of HIV viral load and fewer HIV antigen p24+ cells in the bone marrow upon ART interruption. Collectively, these data support the clinical development of CMV-HIV CAR T cells in combination with a CMV vaccine in HIV-infected individuals.
Keywords: chimeric antigen receptor T cell (CAR T), cytomegalovirus (CMV) vaccine, broadly neutralizing antibody (bNAb), N6, HIV/AIDS, immunotherapy
Graphical abstract
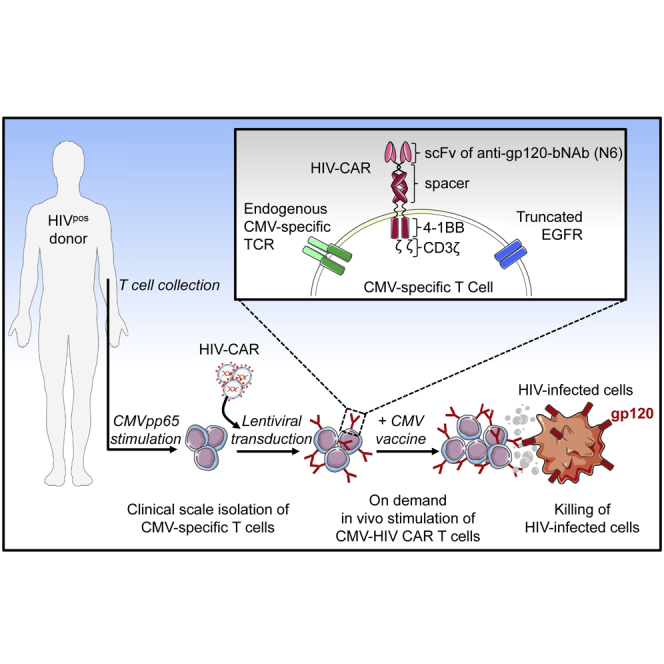
X. Wang and colleagues have manufactured at clinical-scale a novel CMV-specific T cells modified to express CARs derived from the HIV broadly neutralizing antibody N6. These bispecific CMV-HIV CAR T cells expand upon stimulation with a CMV vaccine and control HIV in a mouse model of HIV.
Introduction
Combination antiretroviral therapy (cART) achieves undetectable plasma viremia,1 but fails to cure HIV infection owing to the persistence of a latent virus reservoir containing replication-competent HIV-1.2 Alternative cellular immune strategies of adoptive immunotherapy have been proposed using either expansion of endogenous HIV-specific T cells or autologous T cells or natural killer redirected to HIV-infected cells.3, 4, 5 The first-generation of HIV CAR T cells was developed almost 25 years ago by engineering the extracellular domain of the CD4 receptor on the surface of T cells.6,7 These CD4-based CAR T cells were tested in three clinical trials in HIV-seropositive individuals,4,8, 9, 10 but this strategy was aborted owing to negligible clinical efficacy. Several factors may explain the lack of or minimal antiviral efficacy, such as limited CAR activity in the absence of an intracellular co-stimulatory signaling domain, low expansion, and minimal persistence of CAR T cells owing to insufficient exposure to HIV antigens because CAR recipients remained on ART, or the susceptibility of CD4 CAR-expressing T cells to HIV infection. Of note, a low level of these first-generation CD4 CAR T cells was detected a decade later in these recipients, suggesting that long-term persistence is possible.11 Since then, CARs have been optimized by adding the co-stimulatory domains CD28 or 4-1BB, and in the setting of B cell malignancies, have shown improved efficacy and persistence with promising therapeutic results.12, 13, 14, 15 In addition, a series of broadly neutralizing antibodies (bNAbs) directed to the HIV-1 envelope glycoprotein gp120 were identified in HIV-infected non-progressors and used to develop bNAbs-derived CAR T cells that efficiently kill gp120-expressing cells in vitro.16,17 To limit the emergence of resistance to HIV, these bNAb-based CAR T cells should be effective against nearly all strains of HIV. Notably, Huang et al.18 isolated a bNAb named N6 that potently neutralizes 98% of HIV-1 isolates including 16 of 20 that evolved to circumvent common mechanisms of resistance. As a current best-in-class for bNAbs targeting the CD4 binding site, we hypothesize that N6-derived CAR T cells represent a promising strategy to redirect T cells to HIV-infected cells, enforcing specificity and minimizing the likelihood of immune escape. However, the lack of HIV antigens in individuals on ART, or until HIV rebounds upon analytical treatment interruption, remains a major challenge for CAR T cell activation and persistence in vivo. It may require high CAR T cell number to achieve therapeutic benefit, as currently tested in a CD4-based CAR T cell clinical trial (NCT03617198). Another approach to enhance CAR T cell persistence involves the engagement of viral antigens as stimulatory factors for CAR T cells.19 This strategy has been evaluated for other diseases using Epstein-Barr virus-specific cytotoxic T lymphocytes (CTL) engineered to express CARs targeting GD2+ neuroblastoma cells20 or CD30+ tumors of neural crest origin,21 or using influenza A matrix protein 1-specific T cells modified to express CD19 CARs targeting B cell malignancies.22 These virus and CAR-bispecific T cells demonstrated superior survival and antitumor activity compared with non-virus-specific CAR T cells, possibly owing to a more potent co-stimulation of virus-specific T cells after engagement of their native T cell receptors. Our group has also shown that cytomegalovirus (CMV)-specific T cells expressing CD19 CARs exhibited enhanced antitumor activity and persistence in CD19+ tumor-bearing NSG mice upon in vivo stimulation with a CMVpp65 vaccine.23 Similarly, here we test the concept that bispecific CMV-HIV CAR T cells, stimulated with CMV vaccine to enhance CAR T cell activity, can expand and retain anti-HIV function.
In this study, we used peripheral blood mononuclear cells (PBMCs) collected from HIV-seronegative (HIVneg) or seropositive (HIVpos) donors on ART to manufacture at clinical scale CMV-specific T cells engineered to express a second-generation CAR containing the single-chain variable fragment (scFv) of N6, with the 4-1BB co-stimulatory and CD3ζ intracellular signaling domains. CMV-HIV CAR T cell products derived from HIVpos donors were mostly CD8+, making the gene-modified T cells naturally resistant to HIV infection. CMV-HIV CAR T cell products demonstrated in vitro potent and specific effector functions upon stimulation with gp120 or CMVpp65 antigens. We also evaluated the safety of the cell product by assessing the potential off-target of the N6-scFv-Fc on healthy human tissues. Finally, using a humanized mouse model for HIV infection, in vivo stimulation during ART with CMVpp65 peptide-pulsed autologous PBMCs was shown to accelerate CMV-HIV CAR T cell expansion in the peripheral blood. An inverse relationship was found between CMV-HIV CAR T cell number and both the plasma viral load and the percentage of cells expressing HIV-1 p24 antigen in the bone marrow. Altogether, these proof-of-concept experiments suggest that N6-based CAR immunotherapy using CMV-specific T cells in combination with a CMV vaccine warrants clinical evaluation in people living with HIV (PLWH).
Results
N6-CAR T cells exhibit potent effector functions in vitro
We first developed novel N6-based CAR T cell products by transducing primary T cells isolated from HIVneg donors with a lentiviral vector encoding a CAR containing the scFv of the bNAb N6 (Figure 1A). The IgG4 spacer between the scFv ectodomain and the transmembrane domain was mutated to prevent Fc receptor binding and improve T cell persistence and efficacy.24 The 4-1BB co-stimulatory domain was selected as it has been shown to increase CAR T cell persistence25 and to promote a central memory phenotype26 in various animal studies,27,28 including a humanized mouse model of HIV.29 A truncated human epidermal growth factor receptor (EGFRt) was added to the CAR construct to serve as an element for immunomagnetic purification, cell tracking by flow cytometry and immunohistochemistry, and potential in vivo cell ablation with the anti-EGFR antibody cetuximab.30 After approximately 15 days of in vitro expansion, the final T cell products contained on average 38.16% ± 8.87% EGFR+ CAR T cells, of which 69.63% ± 16.63% were CD4+ and 31.57% ± 17.76%, were CD8+ T cells (mean ± standard deviation [SD]; n = 4) (Figure 1B shows representative FACS plots). We examined whether N6-CAR T cells elicited cytotoxic function by performing a 96h killing assay against 8E5-gp120 cells. These 8E5-gp120 cells were obtained by engineering 8E5 cells to express eGFP-ffLuc and then sorting for co-expression of eGFP and surface gp120 (Figure S1). N6-CAR T cell products and 8E5-gp120 cells were cocultured at various effector-to-target (E:T) ratios. Flow cytometric analysis of the remaining target cells demonstrated that N6-CAR T cells, normalized to mock T cells, efficiently killed 8E5-gp120 cells (Figure 1C). In a separate experiment, N6-CAR T cells were co-cultured at various E:T ratios with purified gp120-negative or gp120-positive 8E5 cells. Efficient and gp120-specific killing was observed against gp120-positive 8E5 cells, but not against the gp120-negative 8E5 cells (Figure S2). Finally, only gated EGFR+ CAR T cells from a mixed T cell population, but not the gated CAR-negative T cell fraction, exhibited proliferative capacity after stimulation with 8E5-gp120 cells (Figure 1D). Flow cytometric analysis revealed that these stimulated N6-CAR T cells maintained sustained memory (CD62L = 66.07%; CD127 = 51.42%; and CD27 = 87.02%, average of three donors), and low exhaustion features (programmed cell death-1 [PD-1] = 10.84%, lymphocyte-activation gene-3 [LAG-3] = 0.26%, and T cell immunoglobulin and mucin domain-3 [Tim-3] = 4.71%, average of three donors) (Figure S3).
Figure 1.

Functional characterization of N6-CAR T cells
(A) Schematic diagram of the N6-CAR construct. The construct contains the GMCSFRss under the control of the EF1α promoter, the scFv of the anti-gp120 bNAb N6 linked to the CD4 transmembrane (tm) and 4-1BB costimulatory domains through an IgG4 (EQ) spacer, followed by CD3ζ and a T2A linked EGFRt. (B) Primary T cells were activated with CD3/CD28 microbeads and transduced with a lentiviral vector encoding N6-CAR. After approximately 15-day in vitro expansion, CAR expression and T cell subsets were analyzed by flow cytometry using antibodies against EGFR, CD3, CD4, and CD8. Representative FACS plots of four HIVneg donor-derived CAR T cell products are presented. (C) N6-CAR T cells or mock T cells derived from HIVneg donors were cocultured with eGFP-expressing 8E5-gp120 cells at various total T cells:target (E:T) ratios for 96 h followed by immunostaining for CD3 and eGFP. Cytotoxicity is calculated as follows: 100% – (% of remaining tumor cells in CAR T cell group/% of remaining tumor cells in mock T or negative target); n = 3 donors. (D) N6-CAR T cell products were labeled with CellTrace Violet dye (CTV) and stimulated at an E:T ratio of 1:1 with 8,000 rads irradiated 8E5-gp120 for 8 days before CTV analysis (blue line). The 8,000 rads irradiated lymphoblastoid cells that express the CD3 agonist OKT3 (LCL-OKT3) were used as a positive control (red line) and media as a negative control (black line). CTV dilutions from EGFR+ gated cells (bottom) and EGFR−-gated cells (top) are depicted. Representative data of five HIVneg donor-derived CAR T cell products are presented.
N6 scFv-Fc does not cross-react with normal human tissues
To assess the potential off-target effects of N6-CARs, immunostaining with soluble N6 scFv-Fc was performed on normal human tissues. As expected, a concentration-dependent immunostaining was observed using either the N6 scFv-Fc or a positive control (the anti-gp120 bNAb VRC01 obtained from the NIH HIV Reagent Program) on 8E5-gp120 cells, but not on gp120-negative leukemic KG-1a cells (Figure S4). Then, N6 scFv-Fc was used for pan-immunostaining on 37 frozen human tissues from three unrelated normal donors (Charles River Labs, CRL study no:20182940). The immunopathological analysis did not reveal membrane signals on these tissues. However, cytoplasmic staining was observed in epithelial cells in the esophagus (mucosa), kidney (renal pelvis), pituitary (adenohypophysis), salivary gland (ducts), skin (sweat glands), thymus (epithelial-reticular), and ureter (mucosa) and in the colloid in the thyroid. The binding to cytoplasmic sites is considered of little to no toxicologic significance owing to the limited ability of antibody-based therapeutics to access the cytoplasmic compartment in vivo.31,32 Overall, the immunohistochemistry staining analysis supports the low risk of clinically relevant off-target tissue cross-reactivity of N6-CAR.
CMV-HIV CAR T cells can be manufactured at clinical scale from HIVneg and HIVpos donors
CMV-specific T cells were isolated using a GMP-compliant CliniMACS Prodigy automated closed system as previously described33,34 and transduced with lentiviral vector encoding N6-CAR (Figure 2A). Briefly, PBMCs were collected from CMVpos HIVneg or HIVpos donors on ART (Table S1) and processed in the CliniMACS Prodigy Cytokine Capture System (CCS) by stimulation with a PepTivator overlapping CMVpp65 peptide pool, followed by labeling with Catchmatrix reagent and anti-IFN-γ microbeads. IFN-γ+ cells were then isolated via magnetic selection in the CliniMACS Prodigy system (Figure 2B shows representative FACS plots). CMV-specific T cells (IFN-γ+CD3+) from HIVneg donors were enriched from 4.33% ± 3.46% to 74.71% ± 9.17% of the total viable T cells (mean ± SD; n = 6) (Figure 2C). Similarly, IFN-γ+ T cells from HIVpos donors were enriched from 1.91% ± 0.94% to 67.28% ± 17.04% of the total viable T cells (mean ± SD, n = 7) (Figure 2C). Unlike CMV-specific T cells isolated from HIVneg donors that had similar proportions of CD4+ and CD8+ T cells (42.34% ± 20.30% and 46.18% ± 18.86%, respectively), CMV-specific T cells from HIVpos donors had a higher content in CD8+ cells (78.04% ± 9.21%) as compared with CD4+ cells (25.59% ± 12.19%; mean ± SD) (Figure 2C). This observation is consistent with previous reports showing that PLWH have a higher proportion of CD8+ CMV-specific T cells as compared with HIVneg individuals.35,36 Of note, the overall composition in memory T cell subsets was similar between CMV-specific T cells isolated from HIVneg and HIVpos donors (Figure S5). Recovered IFN-γ+ T cells (approximately 1 × 106) were then transduced with the N6-CAR lentiviral vector at MOI 3 to generate CMV-HIV CAR T cells and expanded in the presence of IL-2 (50 U/mL) and IL-15 (1 ng/mL) for approximately 15 days. To assess the effect of endogenous reactivation of HIV, we first expanded the CAR T cells in the absence of antiretroviral drugs (ARV). At the end of the culture, the total cell number was 209.5 × 106 ± 97.62 × 106 for HIVneg donors and 13.49 × 106 ± 12.17 × 106 for HIVpos donors (mean ± SD) (Figure 2D). ARV (43 nM darunavir and 279 nM enfuvirtide) were supplemented to the medium during the CAR T cell expansion of three HIVpos donors to inhibit HIV replication. This cocktail was shown to prevent HIV replication in vitro (Figure S6A), without affecting lentiviral transduction efficiency (Figure S6B) and cell expansion (Figure S6C). Interestingly, the three best cell expansions from HIVpos donors (64.1×106, 237.2×106, and 269.53×106) occurred in the presence of ARV (Figure 2D, red dotted lines). Thus, with ARV, HIVpos donor cells can expand as well as HIVneg donor cells. To further explore the difference between products made from HIVneg and HIVpos donors, transduction efficiency was assessed by measuring EGFR expression with flow cytometry in the final cell products. Similar EGFR expression levels were observed in cell products derived from HIVneg (24.68% ± 18.34%) and HIVpos donors (21.45% ± 12.65%; mean ± SD) (Figure 2E). As expected, CMV-HIV CAR T cells from HIVpos donors consisted of a higher percentage of CD8+ cells (86.46% ± 16.52%) as compared to CD4+ cells (10.49% ± 6.62%), whereas the proportion of CD8+ and CD4+ cells within CMV-HIV CAR T cells manufactured from HIVneg donors was 61.35% ± 40.63% and 43.07% ± 42.56%, respectively (mean ± SD) (Figure 2E). Finally, we assessed the final number of CAR T cells manufactured per campaign (Figure 2F). The average CAR T cell number in HIVpos-derived cell product expanded in presence of ARV was 34.19×106.
Figure 2.

Clinical scale manufacturing of CMV-HIV CAR T cells derived from HIVneg and HIVpos donors
(A) Manufacturing workflow to generate CMV-HIV CAR T cells as described in the Methods. (B) Representative FACS plots of CMV-specific T cells isolated from an HIVpos donor before and after IFN-γ+ cell enrichment using the CliniMACS Prodigy platform. (C) Flow cytometric analysis of the percentage of enriched IFN-γ+ CMV-specific T cells and their relative CD4 and CD8 expression. n = 6–7 donors per group. (D) Growth curves of total cell number in final products derived from HIVneg (n = 6) and HIVpos (n = 7) donors over approximately 15-day expansion. The expansion curves in presence of ARVs (darunavir and enfuvirtide) are presented with red dotted lines. (E) Flow cytometric analysis of the percentage of CMV-HIV CAR T cell in the final cell products and their relative CD4 and CD8 expression. n = 6–7 donors per group. (F) Total number of CMV-HIV CAR T cells in each final cell product.
Aanalysis of T cell memory subsets in the final T cell products showed that HIVneg donor-derived cell products had 19.03% ± 24.52% CD27+CD45RA+ stem cell memory T cells (Tscm), 18.92% ± 27.40% CD27+CD45RA− central memory, 18.78% ± 22.41% CD27−CD45RA+ effector memory RA(TEMRA), and 43.26% ± 37.37% CD27−CD45RA− effector memory T cells (Tem) (mean ± SD) (Figure 3A). In comparison, HIVpos donors-derived cell products contained fewer Tscm (1.71% ± 2.39%) than those from HIVneg donors. Similar observations were made when looking at the cell memory subset composition within the EGFR+ CAR T cells (Figure 3B). Finally, only low expression levels of the exhaustion markers LAG-3, PD-1, and Tim-3 were observed in the final T cell products (Figure 3C) or in EGFR + CAR T cells derived from either HIVpos or HIVneg donors (Figure 3D).
Figure 3.

Phenotypic characterization of CMV-HIV CAR T cell products derived from HIVneg and HIVpos donors
T cell memory markers: CD27+CD45RA+ Tscm, CD27+CD45RA− central memory (Tcm), CD27−CD45RA+ effector memory RA (TEMRA), and CD27−CD45RA− Tem were analyzed by flow cytometry after INF-r enrichment (A) and in the final CAR T cell products (B). Exhaustion markers (LAG-3, PD-1 and Tim-3) were analyzed by flow cytometry in the final cell products (C) or within the EGFR+ CAR T cell fractions (D). Lines indicate means ± SD; n = 2–6 donors per group.
CMV-HIV CAR T cells exhibit HIV and CMV antigen-specific effector functions
We first showed that CMV-HIV CAR T cells from HIVneg donors were specifically cytotoxic against gp120-expressing cells by performing a 96-h long-term killing assay using 8E5-gp120 cells as target cells (Figure 4A). We then evaluated if the CMV-HIV CAR T cells were reactive to CMV antigen stimulation via signaling of their endogenous CMV-specific T cell receptors (TCR). A proliferation assay by CTV dye dilution showed that CMV-HIV CAR T cells proliferate only when co-cultured with CMVpp65 peptide-pulsed autologous PBMCs (CMVpp65-PBMCs) as antigen-presenting cells (APCs), or with LCL-OKT3 cells that engage all the TCRs, but not when exposed to KG-1a cells or media (Figure 4B). Accordingly, higher IFN-γ expression was measured in CMV-HIV CAR T cells after overnight stimulation with either LCL-OKT3, CMVpp65 PBMCs, or 8E5-gp120 expressing cells, as compared with stimulation with KG-1a cells or media (Figure 4C). As expected, CMV-specific T cells only expressed IFN-γ after stimulation with LCL-OKT3 cells and CMVpp65-PBMCs, but not with 8E5-gp120 cells, KG-1a cells, or media (Figure 4C). The relatively low IFN-γ expression in CMV-HIV CAR T cell products after overnight stimulation with 8E5-gp120 suggests that the CAR T cells are slowly killing their target cells.
Figure 4.

Effector functions of CMV-HIV CAR T cells derived from HIVneg donors
(A) Specific cytotoxicity against gp120-expressing target cells was determined by culturing CMV-HIV CAR T cell products with eGFP+ 8E5-gp120 or eGFP+ KG-1a cells at different E:T ratios 2:1, 1:1, 1:2 or 1:5) for 96 h followed by immunostaining for CD3 and eGFP. Percentages of remaining eGFP+ tumor cells were measured by flow cytometry and cytotoxicity was calculated. N = 5. (B) CMV-HIV CAR T cell products were labeled with CTV and cultured for 8 days with CMVpp65 peptide-pulsed and 3,500 rads irradiated PBMCs (CMVpp65-PBMC), 8,000 rads irradiated LCL-OKT3 or KG-1a cells or media. CMV-HIV CAR T cell proliferation was determined by CTV dilution. Representative data of four donors are shown. (C) CMV-HIV CAR T cell or CMV-specific T-cell products derived from the same donor were stimulated overnight with CMVpp65 peptide-pulsed autologous PBMC (CMVpp65-PBMC), LCL-OKT3, 8E5-gp120, KG-1a cells or media. Cocultures were stained for surface CD8 followed by intracellular IFN-γ. Representative data of three different donors are shown.
Similarly, we assessed whether CMV-HIV CAR T cells derived from HIVpos donors maintained their effector functions. CMV-HIV CAR T cell products were predominantly CD8+ and were reactive to CMVpp65 antigen stimulation, as shown by their high IFN-γ expression after overnight stimulation with CMVpp65 PBMCs (Figure 5A). In addition, we observed dose-dependent cytotoxicity (Figure 5B) against 8E5-gp120 cells after both short-term (24 h, left) and long-term (96 h, right) co-cultures. We observed better cytotoxicity with 96-h co-culture than 24 h, supporting the optimal killing kinetics with 96 h in the context of HIV CAR and gp120 target. After 24 h and 96 h co-culture, T cell products and CAR T cells had similar low exhaustion phenotype (Figure 5C), was similar to its final product indicating our CMV-HIV CAR T cells remain functional and potent after target engagement (Figures 3C and 3D). Cytotoxicity of the final cell product against HIV infected cells was further assessed by co-culturing for 7 days CMV-HIV CAR T cells or CMV-CD19 CAR T cells derived from the same donor, with HIVNL4-3-infected eGFP+ Jurkat cells at various E:T ratios (Figure 5D). Compared with CMV-CD19 CAR T cells, CMV-HIV CAR T cells were cytotoxic against HIV-infected cells. In the same experiment, HIV-1 p24 levels were measured by ELISA in the cell supernatants and showed a decrease in p24 release in the presence of CMV-HIV CAR T cells, as compared to CMV-CD19 CAR T cells (Figure 5E). Finally, higher levels of p24 were detected in the supernatant of HIVpos donor-derived CMV-CD19 CAR T cells as compared with CMV-HIV CAR T cells derived from the same donor (Figure 5F). Since the only source of HIV in these cultures is from the HIVpos donor, who was aviremic at the time of blood collection, this result suggests that the therapeutic product can eliminate detectable HIV after endogenous reactivation. We next tested whether CMV-HIV CAR T cells have the potential to control HIV viremia and expand in vivo in response to CMVpp65 vaccine in a humanized mouse model of HIV.
Figure 5.

Effector functions of CMV-HIV CAR T cells derived from HIVpos donors
(A) Representative FACS plots of CMV-specific T cells enriched from an HIVpos donor and transduced with a lentiviral vector expressing N6-CAR (n = 7). The transduction efficiency was assessed on day 7 by measuring EGFR expression in T cells. The CMV-HIV CAR T cell products were then stimulated overnight with CMVpp65 peptide-pulsed autologous PBMC and analyzed by flow cytometry for the expression of IFN-γ, CD3, CD4 and CD8. Representative data of three different donors (n = 3) are shown. (B, C) Specific cytotoxicity was determined by co-culturing CMV-HIV CAR T cells (n = 3) with eGFP+ 8E5-gp120 or eGFP+ KG-1a cells at different E:T ratios (2:1, 1:1, or 1:5) for 24 h (n = 2) and 96 h (n = 3) followed by immunostaining for CD3, EGFR as well as LAG-3, PD-1 and Tim-3 exhaustion markers. Percentages of remaining eGFP+ tumor cells were measured by flow cytometry and cytotoxicity was calculated as described in Methods. The graph presents the cytotoxicity of CAR T cells from two or three donors against 8E5-gp120 target cells at different E:T ratios. (D) CMV-HIV CAR T cells or CMV-CD19 CAR T cells were manufactured from the same HIVpos donor and cultured with HIVNL4-3-infected eGFP+ Jurkat cells at different E:T ratios (1:1, 1:2, and 1:4) for 7 days. The cytotoxicity of the CAR T cell products against HIVNL4-3-infected eGFP+ Jurkat cells was calculated and normalized to an untreated control well. The levels of HIV p24 in the cell supernatants on day 7 were measured by ELISA and normalized to the p24 levels in the control condition at an E:T ratio of 1:1 (E). (F) CMV-HIV CAR T cells and CMV-CD19 CAR T cells were manufactured from an HIVpos donor on ART. Levels of p24 were measured in the culture supernatant by ELISA after 20-day expansion and normalized to the p24 level in supernatant of CMV-CD19 CAR T cells. Data from one HIVpos donor are shown in (D), (E), and (F).
CMV-HIV CAR T cells exhibit anti-HIV activity in a humanized mouse model of HIV
HIVneg donors were used to generate high numbers of CMV-HIV CAR T cells. The HIV-infected NSG humanized-PBMC (NSG hu-PBMC) mouse model, summarized in Figure 6A, was established by transplant with autologous PBMCs in 3- to 5-week-old NSG mice (day 0). On day 7, mice were challenged with HIV-1 BaL via intraperitoneal (IP) injection, and on day 12 initiated on a 3-week-long oral ART regimen (emtricitabine, tenofovir, raltegravir), which decreased the plasma viral load from low to undetectable levels. On day 21, during ART, two cohorts were treated with a single infusion of low dose CMV-HIV CAR T cells (0.1 × 106 EGFR+ T cells), and either with or without CMVpp65 immunization on day 28. A group of mice was treated with high dose CMV-HIV CAR T cells (1 × 106 EGFR+ T cells) followed by CMVpp65 vaccine on day 28. Two control cohorts included mice treated with CMV-negative T cells (1 × 106 cells) from the same HIVneg donor, either with or without CMVpp65 vaccine on day 28. CMV-HIV CAR T cells were well tolerated in all the mice, and no differences in body weight and temperature were observed between the groups (Figure S7). At day 28, when mice were on ART before vaccination, high dose of CMV-HIV CAR T cells was found to significantly control HIV plasma viremia compared to control T cell- and to low dose CAR T cell-treated mice (Figure 6B). EGFR+ CAR T cells were then measured in the peripheral blood (Figure 6C), and for each mouse, the slope of the linear regression lines for EGFR+ CAR T cell number/μL between day 33 (i.e., the final day of ART) and day 42 (i.e., 9 days after ART interruption) transformed to a log10 scale was calculated (Figure S8). The mean slope, which represents the rate of expansion of CAR T cells after interruption of ART, was significantly higher for the two vaccinated CAR T cell-treated groups, compared to the unvaccinated low dose CAR T cell-treated cohort. This suggests that the vaccine induced CAR T cell expansion even after ART interruption. At day 42, after ART interruption and viral rebound, mice that received both a high dose of CMV-HIV CAR T cells and the CMVpp65 vaccine were the only cohort with controlled plasma viremia compared with vaccinated or unvaccinated low dose CAR T cell-treated mice (Figure 6D). This suggests the importance of CAR T cell dose such that the CMVpp65 vaccine-driven expansion of low dose CAR T cells was not sufficient to reach a therapeutic effect when viremia was high. CAR T cells were also detected in the bone marrow of mice at sacrifice (Figure 6E), and, notably, an inverse relationship (p = 0.045) was observed in the frequency of T cells with active HIV-1 infection (i.e., p24+ T cells) versus the frequency of EGFR+ CAR T cells in the bone marrow, suggesting a CAR-mediated reduction of HIV-infected cells (Figure 6F).
Figure 6.

CMVpp65-driven expansion of CMV-HIV CAR T cells and dose-dependent control of HIV viremia in hu-PBMC-NSG mice on ART
(A) NSG humanized PBMCs (hu-PBMC) mouse model of HIV on ART and experimental design. HIV-infected mice on oral ART were treated with a low dose CMV-HIV CAR T cells (0.1 × 106 EGFR+ T cells), with or without CMVpp65 vaccine, or a high dose of CMV-HIV CAR T cells (1 × 106 EGFR+ T cells) with CMVpp65 vaccine on day 28. Mice treated with CMV-negative T cells (1 × 106 cells) from the same HIVneg donor, with or without CMVpp65 vaccine, were used as controls. (B) HIV viral load in the peripheral blood on day 28. Additive models with the baseline HIV viral load (day 21), log10CD3, log10CD4 and treatment group were considered, and the best model was chosen based on Akaike information criteria. This model included treatment groups only so analysis of variance followed by the Tukey method for all possible one-sided comparisons (family-wise error rate = 0.05) was used to assess if there were treatment differences among the control (T cell-treated mice), low-dose and high-dose CMV-HIV CAR T cell-treated cohorts; ∗∗∗p < 0.001; ∗∗p = 0.002; n = 8–17/group. (C) Flow cytometric analysis of EGFR+ CAR T cell expansion in the peripheral blood between day 33 and day 42. n = 8/group. Note that one female mouse in the low-dose CAR T + vaccine group did not have a day 42 measure and was not included in this analysis. (D) HIV viral load in the peripheral blood on day 42, after vaccine stimulation and ART interruption. The best model was an analysis of covariance including log10CD3, log10CD4 and treatment group followed by the Tukey method for all possible one-sided comparisons. ∗∗p < 0.01, ∗p = 0.03; n = 6–8/group. (E) Flow cytometric analysis of the frequency of EGFR+ CAR T cells in the bone marrow at the time of sacrifice. The data was transformed using a logit transformation. ANOVA followed by the Tukey method for all possible one-sided comparisons (family-wise error rate = 0.05) was used to assess if there were treatment differences among the CAR T cell-treated cohorts; ∗∗p < 0.01; ∗p = 0.02; n = 7–8/group. (F) Percentage of EGFR+ CAR T cells in the bone marrow plotted against the percentage of p24+ T cells in the bone marrow. Staining for surface antibodies (CD45, CD3, and EGFR) were performed as in panel (C), while intracellular p24 HIV-1 antigen was stained with KC57-FITC antibody after fixation and permeabilization. The simple least squares model with only percent of EGFR+ CAR T cells was best, both percent of EGFR+ CAR T cells, and percent of p24+ T cells were transformed using a logit. Box and whisker plots were used to present the data in (B), (D), and (E). The black box represents the quartiles and black line represent the quartiles and median and the plus sign represents the mean, and values outside the whiskers are considered outliers.
Finally, in a separate experiment, we assessed whether CMV-HIV CAR T cells derived from an HIVpos donor could migrate to the bone marrow, as memory T cells from the bone marrow are long lasting and persist long after the dissipation of circulating antigen-specific memory T cells.37 EGFR+ CAR T cells (50 × 103) were infused into hu-PBMC-NSG mice 14 days after engraftment of HIV-challenged PBMCs (day 0) and in the absence of ART. On day 55, EGFR+ CAR T cells were detected in the peripheral blood and in the bone marrow. As anticipated, these CAR T cells were mostly CD8+ (Figure 7A). Importantly, they still expressed the memory cell markers CD62L and CD27 (Figures 7B and 7C). Thus, these results demonstrate CMV-HIV CAR T cells derived from an HIVpos individual can establish persistent T cell memory in the marrow of HIV-infected mice. No significant difference in HIV viral load was observed in the peripheral blood (data not shown), possibly owing to the limited number of infused CAR T cell and the high level of active HIV infection at the time of CAR T cell infusion.
Figure 7.

Distribution and phenotype of HIVpos donor-derived CMV-HIV CAR T cells in a humanized PBMC-NSG mouse model
Flow cytometric analyses of EGFR + CAR T cells 6 weeks post-CAR T cell infusion. (A) Frequency of CD4+ and CD8+ T cells, and (B) CD62L+ and (C) CD27+ within the EGFR+ CAR T cell fraction. Lines represent means ± SD; n = 5.
Altogether, these data demonstrate that CMVpp65 vaccine stimulates CMV-HIV CAR T cell expansion during ART, and that these cells can traffic to the bone marrow, and locally control viremia, in a CAR T cell dose-dependent manner. These data demonstrate the feasibility of our therapeutic approach and support the development of a clinical trial using N6-based CAR immunotherapy combined with a CMV vaccine to efficiently eliminate gp120-expressing cells in PLWH with the potential for long-term treatment interruption.
Discussion
The CTLs response is a key component of host immunity against HIV infection.38 It is believed that viral control in HIV-infected individuals, which in “elite controllers” can suppress viral loads for years without ART, is mediated largely by HIV-specific CD8+ T cell responses that exert continuous suppression of the HIV reservoir.39, 40, 41 A study suggests that this prolonged suppressive immune pressure alters the characteristics of the latent virus within the reservoir, gradually selecting for defective latent virus that results in a population of infected cells with an inability to reactivate.42,43 Thus, a strategy for a functional HIV cure maintains that suppression of latent virus infection requires T cell immunity that is both effective and prolonged. How best to duplicate this effective and sustained control mechanism observed in elite controllers?44 One approach, tested in HIV-infected individuals on ART (NCT03485963 and NCT04561258) uses ex vivo expanded HIV-specific T cells that are able to recognize multiple HIV epitopes.5 However, this approach may still be limited by the lack of continuous viral antigens which is required to stimulate and maintain the T cell number necessary for a prolonged effect, while avoiding immune escape by HIV. The engineering of T cells with a CD4-based CAR has been developed to overcome the potential for HIV resistance. The early studies of these first-generation CAR T cells produced no convincing anti-HIV effect but were safe.4,8, 9, 10 The in vitro and in vivo potency was significantly improved with the development of second-generation CD4-based CAR T cells14,26,27 that are being currently tested in the clinic (NCT03617198). An alternative approach to design HIV-specific CAR T cells16,17 relies on scFv of gp120-specific bNAb.45 A study even combined both gp120-and CD4 CAR T cells to induce in vivo expansion in non-human primates after gp120 antigen pulsing.46 A bispecific anti-HIV CAR T cell that responds to endogenous signals could solve the problem of in vivo maintenance of T cell levels over the long period that is likely required for a functional cure.44 Our CMV-HIV bispecific CAR T products are shown in this study to be responsive to both CMV and gp120 antigenic stimulations, in vitro and in vivo, and could meet this requirement. We first showed that CMV-HIV CAR T cells from HIVneg donors maintain the capacity to proliferate in vitro in response to a CMV vaccine through TCR stimulation47 and to specifically target gp120-expressing cells. To assess if CMV-HIV CAR T cells can control HIV reactivation in vivo, we then optimized a humanized mouse model of HIV on ART and administered CMV-HIV CAR T cells from HIVneg donors followed by CMVpp65 vaccination. As expected, we found that CMV-HIV CAR T cells expanded faster upon administration of the CMVpp65 vaccine compared with unvaccinated mice.23 Notably, we found that mice that received both a high dose of CAR T cells and the CMVpp65 vaccine showed control of plasma viral load compared with untreated HIV-infected mice, suggesting that CMV vaccine-stimulated CAR T cells are not losing their effector function in vivo owing to exhaustion. The vaccine-driven expansion of a low dose of CMV-HIV CAR T cells was, however, not enough to exert an anti-HIV activity in the peripheral blood. We also observed a CAR T cell dose-dependent control of the frequency of p24+ T cells in the bone marrow, suggesting that CMV-HIV CAR T cells have potential to traffic to this site and to eliminate HIV reservoirs. Unfortunately, this mouse model is limited by the occurrence of graft-versus-host disease and does not allow long-term CAR T cell persistence studies.48 Interestingly, in a non-human primate model, animals treated with simian immunodeficiency virus-specific CAR T cells expressing the follicular homing receptor CXCR5 maintained lower viral loads and follicular viral RNA levels than untreated control animals. These findings hold promise of CAR T cell therapy for the durable remission of HIV.49
Unlike other immunotherapies that require additional gene engineering to protect CD4+ CAR T cells from HIV infection,16,50, 51, 52 most of our HIVpos donor-derived gene-modified T cells are CD8+ T cells and thus naturally resistant to HIV infection. The PBMCs used from HIV-positive donors are capable of supporting HIV infection but, within the combination of T cells, it is not known if the CAR T cells themselves become infected. However, if ARVs are used during manufacturing this aborts any HIV infection (data not shown). In this regard, we found that the cell expansion from HIVpos donors was lower compared with HIVneg donors if performed in the absence of ARV. For this reason, we have optimized the ex vivo culture conditions by adding ARV during the manufacturing, leading to better cell expansion and showing the feasibility of clinical scale production of the CMV-HIV CAR T cells under good manufacturing practice (GMP) guidelines. We found that HIVpos donor-derived CMV-HIV CAR T cells maintain in vitro cytotoxicity against various gp120-expressing cells (8E5-gp120, HIVNL4-3-infected Jurkat cells and autologous T cells) and the ability to proliferate and secrete IFN-ɣ upon stimulation with CMVpp65-PBMCs, suggesting bifunctionality specific for both HIV and CMV. These cell products also express low levels of exhaustion markers. Finally, we found that HIVpos-derived CMV-HIV CAR T cells were able to migrate to the bone marrow of HIV-infected NSG humanized-PBMC mice and still maintained expression of early memory cell markers.51
Regarding the induction of HIV resistance during CAR T cell therapy, we have chosen the gp120-specific bNAb N6 because it has been shown to neutralize in vitro HIV strains that are resistant to other antibodies of the same class.18 However, N6 has been used in a simian model and shown to induce resistant SHIV indicating a cautionary note.53 Additional information regarding the risk of resistance induction will come from the study of N6LS (i.e., N6 modified to extend its half-life in serum) in HIV-1 infected adults (NCT04871113). It remains to be determined whether the N6-based CAR T cell strategy will be safe in humans while evading immune escape mechanisms of HIV. In case of resistance in a N6-CAR T cell trial, the CAR construct could be modified to include other approaches to dual-targeted CAR T cells to decrease the potential for virus immune escape.54, 55, 56, 57, 58
In summary, we have designed and manufactured at clinical scale a novel bispecific CMV-HIV CAR T cell that confers anti-HIV specificity to T cells that have immunologic and antiviral responses both in vitro and in vivo. This CMV vaccine-driven approach to enhance expansion and maintenance of these CAR T cells deserves clinical evaluation. The CMV Triplex vaccine, which is an investigational vaccine made using a modified vaccinia Ankara platform containing CMV antigens, has been shown to be immunogenic, safe and effective at expanding CMV-specific cells when used in healthy individuals59 or marrow transplant patients.60 This vaccine is currently being evaluated in PLWH.61 As the level of in vivo expansion upon TCR and CAR stimulation is unknown, we plan to start with a low cell dose (25 × 106 autologous CMV-CAR T cells per participant) to assess the safety and feasibility of our approach. Our studies here demonstrated that we were able to generate sufficient CMV-HIV CAR T cells within a 14-day ex vivo expansion period without further stimulation. We have the capacity to generate more CAR T cells for higher dose levels by using simultaneously two Prodigy platforms. The shortened ex vivo expansion of CMV-HIV CAR T cells could prevent differentiation and promote effector functions after infusion. We hope this approach will help to achieve sustained immunologic control of HIV-1 infection in the absence of ART.
Materials and methods
DNA constructs
The N6-CAR construct was modified from the previously described CD19-specific scFvFc:ζ chimeric immunoreceptor.62 The HIV:41BB:ζ/EGFRt-epHIV7 lentiviral vector contains the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor-α chain signal sequence (GMCSFRss), which enhances CAR surface expression, the CAR sequence consisting of the VH and VL gene segments of the N6 bNAb, the IgG4 hinge with two site mutations (L235E; N297Q) within the CH2 region, the CD4 transmembrane and 4-1BB co-stimulatory domains, the cytoplasmic domain of the CD3ζ chain,24 the ribosomal skip T2A sequence, and the EGFRt sequence as previously described to allow for CAR T cell enrichment, tracking, and potential cell ablation through ADCC.63 The full CAR sequence is available upon request. The lentiviral vector encoding eGFP and ffLuc was created by removing the STOP codon in the eGFP open reading frame from pFUGW (Addgene plasmid #14883) and inserting a P2A-ffLuc-STOP cassette in frame with eGFP.
Clinical scale production of CMV-HIV CAR T cells
Fresh blood products were obtained from CMVpos HIVneg donors (StemCell Technologies, Vancouver, Canada) and HIVpos donors on ART (Zen-Bio Inc, Research Triangle Park, NC) (Table S1). All procedures were performed in accordance with the Declaration of Helsinki Protocols (KC15TISI0494). CMV-specific T cells were isolated on the CliniMACS Prodigy and CCS (Miltenyi Biotec, Bergisch Gladbach, the Netherlands) according to the manufacturer’s instruction. Briefly, PBMCs were isolated and purified by density gradient centrifugation over Ficoll-Paque (Pharmacia Biotech, Uppsala, Sweden). After PBMCs (109) were added to the application bag connected to the tubing set, the CliniMACS Prodigy device automatically performed successive processes, including sample washing, antigen stimulation with PepTivator CMVpp65, Catchmatrix labeling, anti-IFN-γ microbead labeling, magnetic enrichment, and elution. CMV-specific cells and non-CMV-specific cells were eluted in separate bags after magnetic enrichment. After overnight rest in RPMI medium containing 10% human AB serum (Gemini Bio Products, Sacramento, CA), IL-2 (50 U/mL) and IL-15 (1 ng/mL), recovered IFN-γ+ cells (approximately 1 × 106) were transduced at MOI 3 with the research (for HIVneg donors and HIVpos donors #551 and #552) or GMP-grade (for HIVpos donors #553, #572, #573, IEQR#2, IEQR#3) lentiviral vector HIV:41BB:ζ/EGFRt-epHIV7. Fresh culture medium and cytokines were added every other day for approximately 15 days. ARVs (43 nM darunavir and 279 nM enfuvirtide) were added twice per week during the expansion of HIVpos #573, IEQR#2 and IEQR#3 derived CMV-HIV CAR T cells. Cultures were maintained at 37°C under 5% (v/v) CO2.
Cell lines
The 8E5 cells contain a single defective proviral genome of HIV and therefore are not infectious but express most of the HIV viral proteins including gp120. 8E5 (CRL-8993) cells were purchased from ATCC (Manassas, VA) and maintained in RPMI 1640 (Irvine Scientific, Santa Ana, CA) medium supplemented with 10% heat-inactivated FCS (Hyclone Laboratories, Inc, Logan, UT). To generate 8E5 cell lines expressing enhanced green fluorescent protein (eGFP) and firefly luciferase (ffLuc), 8E5 cells were transduced with a lentiviral vector encoding eGFP-ffLuc, and GFP+gp120+ cells were sorted and expanded. Acute myeloid leukemia cell line KG-1a (CCL-246.1) cells were purchased from ATCC and maintained in 10% FCS IMDM medium and used as negative target cells. LCL-OKT3 cells were generated as previously described and served as positive T cell stimulator.23 Cells were grown in complete medium supplemented with 0.4 mg/mL hygromycin. eGFP+ Jurkat cells were infected with the HIVNL4-3 and maintained in culture for 2 weeks. HEK293-eGFP-ffLuc-gp160 cells used as positive control cell lines for the tissue cross-reactivity study were obtained by stably transfecting HEK-293T cell lines with C97ZA012 gp160 construct to express the cell surface gp160 (eventually cleaved into gp120 and gp41 HIV-1 envelope proteins). Banks of all cell lines were authenticated for the desired antigen/marker expression by flow cytometry before cryopreservation, and thawed cells were cultured for less than 3 months before use in assays.
Cytotoxicity assay
CAR T cell products (2.5 × 105) and eGFP-positive target cells (8E5-gp120, 8E5, LCL-OKT3, or KG-1a) were cocultured at various E:T ratios of total T cells:target (2:1, 1:1, 1:2 or 1:5) for 4 days. Cocultures with LCL-OKT3 and 8E5 or KG-1a were used as positive and negative target controls. The cells were stained with anti-CD3 antibody. The percentages of viable eGFP+ CD3– tumor cells were measured using multicolor flow cytometry. The percent of cytotoxicity was calculated as following: 100% – (% of remaining tumor cells in CAR T cell group/% of remaining tumor cells in mock T or negative target groups). eGFP+ HIVNL4-3-infected Jurkat cells (2.5 × 105) were cocultured with CMV-HIV CAR T cells at various E:T ratios (1:1, 1:2, 1:4) for 7 days. Cells then were stained with Viability Dye eFluor 450 (Miltenyi Biotec) for flow cytometric analysis of viable eGFP+ cells.
Proliferation assay
CAR T cell products (2.5 × 105) were labeled with 0.5 μM CellTrace Violet dye and cocultured with 8,000 cGy-irradiated stimulator cells LCL-OKT3, 8E5-gp120 and KG-1a, or autologous CMVpp65-peptide pulsed PBMCs which had been 3,500 cGy-irradiated at 1:1 ratio for 8 days. Cocultures with LCL-OKT3, KG-1a cells, and media were used as positive and negative controls. The proliferation of CD3+ and EGFR+ populations was determined using multicolor flow cytometry.
Intracellular IFN-γ staining
CAR T cell products (105) were activated overnight with LCL-OKT3, 8E5-gp120, or KG-1a cells (105) in 96-well tissue culture plates, or with CMVpp65 peptide-pulsed autologous PBMC cells (105) in the presence of Brefeldin A (BD Biosciences, Franklin Lakes, NJ). The cell mixture was then stained with anti-CD8 antibody, anti-EGFR antibody cetuximab, and streptavidin to analyze surface expression of CD8 and CAR, respectively. Cells were then fixed and permeabilized using the BD Cytofix/Cytoperm kit (BD Biosciences). After fixation, the T cells were stained with an anti-IFN-γ antibody. Cells were then analyzed using multicolor flow cytometry on MACSQuant (Miltenyi Biotec).
Mice
Studies were performed with male and female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (JAX stock #005557) aged 3–5 weeks at the initiation of studies. Mice were group housed in individually ventilated cages (OptiCages, Animal Care Systems) on corn-cob bedding (‘Bed-o’-Cobs 1/8 in., The Andersons, Maumee, OH) with a square nestlet and PVC tube provided for enrichment. Mice were allowed free access to rodent chow (LabDiet 5350) and autoclaved acidified reverse osmosis purified water (pH 2.4–2.8) in bottles. After inoculation with HIV, mice were housed under animal biosafety level-2 conditions, group housed in static disposable cages (Innocage, Innovive, San Diego, CA). The room temperature was held at a range of 68°F –79 °F and the room humidity range was 30%–70%. Mice were designated as specific-pathogen-free for mouse rotavirus, Sendai virus, pneumonia virus of mice, mouse hepatitis virus, minute virus of mice, mice parvovirus, Theiler murine encephalomyelitis virus, mouse reovirus type 3, mouse norovirus, lymphocytic choriomeningitis virus, mouse thymic virus, mouse adenovirus types 1 and 2, mouse CMV, polyomavirus, K virus, ectromelia virus, Hantavirus, Prospect Hill virus, Filobacterium rodentium, Encephalitozoon cuniculi, and Mycoplasma pulmonis, Helicobacter spp., Clostridium piliforme, and free of any endo- and ectoparasites. Mice were maintained in accordance with the Guide for the Care and Use of Laboratory Animals in a facility accredited by the American Association for the Accreditation of Laboratory Animal Care. All experiments were performed according to the guidelines of the Institutional Animal Committee of the Beckman Research Institute of the City of Hope, IACUC 16095.
Engraftment of hu-PBMC-NSG mice and HIV challenge
PBMCs were collected from an HIVneg or HIVpos donor to manufacture CMV-HIV CAR T cells. The CMV-negative fraction of autologous PBMCs was cryopreserved as control T cells. CMV-negative PBMCs (1 × 106) were mixed with CMV-negative resting PBMCs (9 × 106) before transplantation on day 0 in each mouse. Cells (1 × 107 per mouse) were resuspended in sterile saline and injected IP into NSG mice. Before treatment, mice were randomized to assure similar engraftment and gender proportions across groups. On day 7, mice were challenged with HIV-1 BaL via IP injection. Longitudinal blood collections were performed using retro-orbital bleeding on anesthetized mice and peripheral blood cell populations and plasma viral loads were analyzed periodically using flow cytometry and quantitative real-time PCR. Mice that did not engraft huCD45+ cells (defined as >30 cells/μL huCD45+ cells in peripheral blood) were excluded for analysis. Mice showing severe signs of graft-versus-host disease were immediately humanely euthanized.
Oral ART therapy
Infected mice with detectable viral infection (defined as >103 cp/mL of HIV in blood) were treated orally for 3 weeks with ART composed of drugs that block new infections, without inhibiting viral production in infected cells. The ART regimen consisting of Truvada (tenofovir disoproxil fumarate [300 mg/tablet], emtricitabine [200 mg/tablet; Gilead Sciences, Foster City, CA]) and Isentress (raltegravir 400 mg/tablet (Merck, Kenilworth, NJ), scaled down to the equivalent mouse dosage using the appropriate conversion factor, was administered in a drinking water formulation (sweetened water gel, Medidrop Sucralose, ClearH20). For 400 mL Medidrop, ½ Truvada tablet, and ½ Isentress tablet were crushed to powder and mixed by shaking bottle to a homogenous solution; medicated water was changed weekly. Doses of ART drugs were calculated based on previous studies using the same delivery system.64
CMV-HIV CAR T cells and in vivo CMVpp65 stimulation
Mice received CMV-HIV CAR T cells (0.05–1 × 106 EGFR+ T cells), CMV-negative T cells (1 × 106); or autologous PBMCs as control T cells by retro-orbital injection under general isoflurane anesthesia. Autologous PBMCs were pulsed with CMVpp65 peptide mix (#PM-PP65, JPT Peptide Technologies, Berlin, Germany) as APCs as CMVpp65 vaccine. When indicated, mice received CMVpp65 peptide-pulsed and irradiated (3,500 rads) autologous CMV-negative PBMCs (5 × 106) by retro-orbital injection under general isoflurane anesthesia.
Statistical analysis
Analyses were performed using Prism (GraphPad Software Inc.) or R version 4.0.265 and are described in the individual figure legends. A significance level of 0.05 was used for all analyses.
Acknowledgments
We thank staff members of the Flow Cytometry Core, the Animal Facility Core, and the Pathology Core in the Beckman Research Institute at the City of Hope Comprehensive Cancer Center for excellent technical assistance. Figures were modified with text, markings, and annotation after adaptation from image bank of Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 Unported License. The following reagent was obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: Human Immunodeficiency Virus 1 (HIV-1) VRC01 Monoclonal Antibody Light Chain Expression Vector, ARP-12036, contributed by John Mascola. This work was supported by a grant from the California Institute for Regenerative Medicine (CLIN1-11223, PI: X.W).
Author contributions
Designing research studies: M.G., L.H., R.U., P.Y., A.A.C., J.C.B., and X.W.; conducting experiments: M.G., L.L., L.H., R.U., V.V., T.H., S.L., W.C., and A.M.; data acquisition: M.G., L.L., L.H., R.U., V.V., T.H., S.L., W.C., and A.M.; analysis and interpretation: M.G., L.H., R.U., T.S., P.Y., K.V.M., A.A.C., J.C.B., S.M.B., V.L.V., J.A.Z, and X.W.; writing manuscript: M.G., V.L.V., J.A.Z, and X.W.; supervision: P.Y., J.B., J.A.Z, and X.W.; Concept: X.W., J.C.B; J.A.Z. and S.J.F.
Declaration of interests
Competing interests: A patent associated with this study covering the work has been held and submitted by City of Hope (US2016/024560) with X.W. and S.J.F. as inventors who could potentially receive licensing royalties in the future. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.04.007.
Supplemental information
References
- 1.Günthard H.F., Saag M.S., Benson C.A., del Rio C., Eron J.J., Gallant J.E., Hoy J.F., Mugavero M.J., Sax P.E., Thompson M.A., et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2016 recommendations of the international antiviral society-USA panel. JAMA. 2016;316:191–210. doi: 10.1001/jama.2016.8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadowski I., Hashemi F.B. Strategies to eradicate HIV from infected patients: elimination of latent provirus reservoirs. Cell Mol. Life Sci. 2019;76:3583–3600. doi: 10.1007/s00018-019-03156-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hege K.M., Cooke K.S., Finer M.H., Zsebo K.M., Roberts M.R. Systemic T cell-independent tumor immunity after transplantation of universal receptor-modified bone marrow into SCID mice. J. Exp. Med. 1996;184:2261–2269. doi: 10.1084/jem.184.6.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner T.A. Quarter century of anti-HIV CAR T cells. Curr. HIV/AIDS Rep. 2018;15:147–154. doi: 10.1007/s11904-018-0388-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel S., Hanajiri R., Grant M., Saunders D., Van Pelt S., Keller M., Hanley P.J., Simon G., Nixon D.F., Hardy D., et al. HIV-specific T cells can Be generated against non-escaped T cell epitopes with a GMP-compliant manufacturing platform. Molecular therapy. Methods Clin. Dev. 2020;16:11–20. doi: 10.1016/j.omtm.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romeo C., Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell. 1991;64:1037–1046. doi: 10.1016/0092-8674(91)90327-u. [DOI] [PubMed] [Google Scholar]
- 7.Roberts M.R., Qin L., Zhang D., Smith D.H., Tran A., Duil T.J., Groopman J.E., Capon D.J., Byrn R.A., Finer M.H. Targeting of human immunodeficiency virus-infected cells by CD8+T lymphocytes armed with universal T-cells receptors. Blood. 1994;84:2878–2889. [PubMed] [Google Scholar]
- 8.Mitsuyasu R.T., Anton P.A., Deeks S.G., Scadden D.T., Connick E., Downs M.T., Bakker A., Roberts M.R., June C.H., Jalali S., et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96:785–793. [PubMed] [Google Scholar]
- 9.Deeks S.G., Wagner B., Anton P.A., Mitsuyasu R.T., Scadden D.T., Huang C., Macken C., Richman D.D., Christopherson C., June C.H., et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther.: J. Am. Soc. Gene Ther. 2002;5:788–797. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 10.Walker R.E., Bechtel C.M., Natarajan V., Baseler M., Hege K.M., Metcalf J.A., Stevens R., Hazen A., Blaese R.M., Chen C.C., et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96:467–474. [PubMed] [Google Scholar]
- 11.Scholler J., Brady T.L., Binder-Scholl G., Hwang W.T., Plesa G., Hege K.M., Vogel A.N., Kalos M., Riley J.L., Deeks S.G., et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012;4:132ra153. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Stegen S.J., Hamieh M., Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhlmann A.S., Peterson C.W., Kiem H.P. Chimeric antigen receptor T-cell approaches to HIV cure. Curr. Opin. HIV AIDS. 2018;13:446. doi: 10.1097/COH.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ying Z., He T., Wang X., Zheng W., Lin N., Tu M., Xie Y., Ping L., Zhang C., Liu W., et al. Parallel comparison of 4-1BB or CD28 Co-stimulated CD19-targeted CAR-T cells for B cell non-hodgkin's lymphoma. Mol. Ther. Oncolytics. 2019;15:60–68. doi: 10.1016/j.omto.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong Q., Zhu Y.M., Zheng L.L., Shen H.J., Ou R.M., Liu Z., She Y.L., Chen R., Li C., Huang J., et al. Chimeric antigen receptor-T cells with 4-1BB Co-stimulatory domain present a superior treatment outcome than those with CD28 domain based on bioinformatics. Acta Haematol. 2018;140:131–140. doi: 10.1159/000492146. [DOI] [PubMed] [Google Scholar]
- 16.Hale M., Mesojednik T., Romano Ibarra G.S., Sahni J., Bernard A., Sommer K., Scharenberg A.M., Rawlings D.J., Wagner T.A. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol. Ther.: J. Am. Soc. Gene Ther. 2017;25:570–579. doi: 10.1016/j.ymthe.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mu W., Carrillo M.A., Kitchen S.G. Engineering CAR T cells to target the HIV reservoir. Front. Cell. Infect. Microbiol. 2020;10:410. doi: 10.3389/fcimb.2020.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J., Kang B.H., Ishida E., Zhou T., Griesman T., Sheng Z., Wu F., Doria-Rose N.A., Zhang B., McKee K., et al. Identification of a CD4-binding-site antibody to HIV that evolved near-Pan neutralization breadth. Immunity. 2016;45:1108–1121. doi: 10.1016/j.immuni.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lapteva N., Gilbert M., Diaconu I., Rollins L.A., Al-Sabbagh M., Naik S., Krance R.A., Tripic T., Hiregange M., Raghavan D., et al. T-cell receptor stimulation enhances the expansion and function of CD19 chimeric antigen receptor-expressing T cells. Clin. Cancer Res. 2019;25:7340–7350. doi: 10.1158/1078-0432.ccr-18-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pule M.A., Savoldo B., Myers G.D., Rossig C., Russell H.V., Dotti G., Huls M.H., Liu E., Gee A.P., Mei Z., et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savoldo B., Rooney C.M., Di Stasi A., Abken H., Hombach A., Foster A.E., Zhang L., Heslop H.E., Brenner M.K., Dotti G. Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood. 2007;110:2620–2630. doi: 10.1182/blood-2006-11-059139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooper L.J., Al-Kadhimi Z., Serrano L.M., Pfeiffer T., Olivares S., Castro A., Chang W.C., Gonzalez S., Smith D., Forman S.J., Jensen M.C. Enhanced antilymphoma efficacy of CD19-redirected influenza MP1-specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood. 2005;105:1622–1631. doi: 10.1182/blood-2004-03-1208. [DOI] [PubMed] [Google Scholar]
- 23.Wang X., Wong C.W., Urak R., Mardiros A., Budde L.E., Chang W.C., Thomas S.H., Brown C.E., La Rosa C., Diamond D.J., et al. CMVpp65 vaccine enhances the antitumor efficacy of adoptively transferred CD19-redirected CMV-specific T cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015;21:2993–3002. doi: 10.1158/1078-0432.ccr-14-2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonnalagadda M., Mardiros A., Urak R., Wang X., Hoffman L.J., Bernanke A., Chang W.C., Bretzlaff W., Starr R., Priceman S., et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol. Ther.: J. Am. Soc. Gene Ther. 2015;23:757–768. doi: 10.1038/mt.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philipson B.I., O'Connor R.S., May M.J., June C.H., Albelda S.M., Milone M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci. Signal. 2020;13:eaay8248. doi: 10.1126/scisignal.aay8248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frigault M.J., Lee J., Basil M.C., Carpenito C., Motohashi S., Scholler J., Kawalekar O.U., Guedan S., McGettigan S.E., Posey A.D., Jr., et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015;3:356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brentjens R.J., Riviere I., Park J.H., Davila M.L., Wang X., Stefanski J., Taylor C., Yeh R., Bartido S., Borquez-Ojeda O., et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porter D.L., Hwang W.T., Frey N.V., Lacey S.F., Shaw P.A., Loren A.W., Bagg A., Marcucci K.T., Shen A., Gonzalez V., et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leibman R.S., Richardson M.W., Ellebrecht C.T., Maldini C.R., Glover J.A., Secreto A.J., Kulikovskaya I., Lacey S.F., Akkina S.R., Yi Y., et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. 2017;13:e1006613. doi: 10.1371/journal.ppat.1006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X., Chang W.C., Wong C.W., Colcher D., Sherman M., Ostberg J.R., Forman S.J., Riddell S.R., Jensen M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall W.C., Price-Schiavi S.A., Wicks J., Rojko J.L. 2010. Tissue Cross-Reactivity Studies for Monoclonal Antibodies: Predictive Value and Use for Selection of Relevant Animal Species for Toxicity Testing. [DOI] [Google Scholar]
- 32.Leach M.W., Halpern W.G., Johnson C.W., Rojko J.L., MacLachlan T.K., Chan C.M., Galbreath E.J., Ndifor A.M., Blanset D.L., Polack E., Cavagnaro J.A. Use of tissue cross-reactivity studies in the development of antibody-based biopharmaceuticals: history, experience, methodology, and future directions. Toxicol. Pathol. 2010;38:1138–1166. doi: 10.1177/0192623310382559. [DOI] [PubMed] [Google Scholar]
- 33.Kumaresan P., Figliola M., Moyes J.S., Huls M.H., Tewari P., Shpall E.J., Champlin R., Cooper L.J. Automated cell enrichment of cytomegalovirus-specific T cells for clinical applications using the cytokine-capture system. J. Vis. Exp.: JoVE. 2015;104:e52808. doi: 10.3791/52808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X., Urak R., Walter M., Guan M., Han T., Vyas V., Chien S.H., Gittins B., Clark M.C., Mokhtari S., et al. Large-scale manufacturing and characterization of CMV-CD19CAR T cells. J. Immunother. Cancer. 2022;10:e003461. doi: 10.1136/jitc-2021-003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naeger D.M., Martin J.N., Sinclair E., Hunt P.W., Bangsberg D.R., Hecht F., Hsue P., McCune J.M., Deeks S.G. Cytomegalovirus-specific T cells persist at very high levels during long-term antiretroviral treatment of HIV disease. PLoS One. 2010;5:e8886. doi: 10.1371/journal.pone.0008886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stone S.F., Price P., Khan N., Moss P.A., French M.A. HIV patients on antiretroviral therapy have high frequencies of CD8 T cells specific for Immediate Early protein-1 of cytomegalovirus. AIDS. 2005;19:555–562. doi: 10.1097/01.aids.0000163931.68907.7e. [DOI] [PubMed] [Google Scholar]
- 37.Okhrimenko A., Grun J.R., Westendorf K., Fang Z., Reinke S., von Roth P., Wassilew G., Kuhl A.A., Kudernatsch R., Demski S., et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc. Natl. Acad. Sci. U S A. 2014;111:9229–9234. doi: 10.1073/pnas.1318731111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker B., McMichael A. The T-cell response to HIV. Cold Spring Harbor Perspect. Med. 2012;2:a007054. doi: 10.1101/cshperspect.a007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okulicz J.F., Lambotte O. Epidemiology and clinical characteristics of elite controllers. Curr. Opin. HIV AIDS. 2011;6:163–168. doi: 10.1097/COH.0b013e328344f35e. [DOI] [PubMed] [Google Scholar]
- 40.Loffredo J.T., Sidney J., Bean A.T., Beal D.R., Bardet W., Wahl A., Hawkins O.E., Piaskowski S., Wilson N.A., Hildebrand W.H., et al. Two MHC class I molecules associated with elite control of immunodeficiency virus replication, Mamu-B∗08 and HLA-B∗2705, bind peptides with sequence similarity. J. Immunol. 2009;182:7763–7775. doi: 10.4049/jimmunol.0900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saez-Cirion A., Lacabaratz C., Lambotte O., Versmisse P., Urrutia A., Boufassa F., Barre-Sinoussi F., Delfraissy J.F., Sinet M., Pancino G., Venet A. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc. Natl. Acad. Sci. U S A. 2007;104:6776–6781. doi: 10.1073/pnas.0611244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chomont N., El-Far M., Ancuta P., Trautmann L., Procopio F.A., Yassine-Diab B., Boucher G., Boulassel M.R., Ghattas G., Brenchley J.M., et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009;15:893–900. doi: 10.1038/nm.1972. nm.1972 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang C., Lian X., Gao C., Sun X., Einkauf K.B., Chevalier J.M., Chen S.M.Y., Hua S., Rhee B., Chang K., et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature. 2020;585:261–267. doi: 10.1038/s41586-020-2651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chomont N. HIV enters deep sleep in people who naturally control the virus. Nature. 2020;585:190–191. doi: 10.1038/d41586-020-02438-7. [DOI] [PubMed] [Google Scholar]
- 45.Hua C.K., Ackerman M.E. Engineering broadly neutralizing antibodies for HIV prevention and therapy. Adv. Drug Deliv. Rev. 2016;103:157–173. doi: 10.1016/j.addr.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rust B.J., Kean L.S., Colonna L., Brandenstein K.E., Poole N.H., Obenza W., Enstrom M.R., Maldini C.R., Ellis G.I., Fennessey C.M., et al. Robust expansion of HIV CAR T cells following antigen boosting in ART-suppressed nonhuman primates. Blood. 2020;136:1722–1734. doi: 10.1182/blood.2020006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hebeis B.J., Klenovsek K., Rohwer P., Ritter U., Schneider A., Mach M., Winkler T.H. Activation of virus-specific memory B cells in the absence of T cell help. J. Exp. Med. 2004;199:593–602. doi: 10.1084/jem.20030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holguin L., Echavarria L., Burnett J.C. Novel humanized peripheral blood mononuclear cell mouse model with delayed onset of graft-versus-host disease for preclinical HIV research. J. Virol. 2022;96:e0139421. doi: 10.1128/jvi.01394-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pampusch M.S., Abdelaal H.M., Cartwright E.K., Molden J.S., Davey B.C., Sauve J.D., Sevcik E.N., Rendahl A.K., Rakasz E.G., Connick E., et al. CAR/CXCR5-T cell immunotherapy is safe and potentially efficacious in promoting sustained remission of SIV infection. PLoS Pathog. 2022;18:e1009831. doi: 10.1371/journal.ppat.1009831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhen A., Carrillo M.A., Mu W., Rezek V., Martin H., Hamid P., Chen I.S.Y., Yang O.O., Zack J.A., Kitchen S.G. Robust CAR-T memory formation and function via hematopoietic stem cell delivery. PLoS Pathog. 2021;17:e1009404. doi: 10.1371/journal.ppat.1009404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maldini C.R., Gayout K., Leibman R.S., Dopkin D.L., Mills J.P., Shan X., Glover J.A., Riley J.L. HIV-resistant and HIV-specific CAR-modified CD4(+) T cells mitigate HIV disease progression and confer CD4(+) T cell help in vivo. Mol. Ther.: J. Am. Soc. Gene Ther. 2020;28:1585–1599. doi: 10.1016/j.ymthe.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maldini C.R., Claiborne D.T., Okawa K., Chen T., Dopkin D.L., Shan X., Power K.A., Trifonova R.T., Krupp K., Phelps M., et al. Dual CD4-based CAR T cells with distinct costimulatory domains mitigate HIV pathogenesis in vivo. Nat. Med. 2020;26:1776–1787. doi: 10.1038/s41591-020-1039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Julg B., Pegu A., Abbink P., Liu J., Brinkman A., Molloy K., Mojta S., Chandrashekar A., Callow K., Wang K., et al. Virological control by the CD4-binding site antibody N6 in simian-human immunodeficiency virus-infected rhesus monkeys. J. Virol. 2017;91:e00498-17. doi: 10.1128/JVI.00498-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anthony-Gonda K., Bardhi A., Ray A., Flerin N., Li M., Chen W., Ochsenbauer C., Kappes J.C., Krueger W., Worden A., et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci. Transl. Med. 2019;11:eaav5685. doi: 10.1126/scitranslmed.aav5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Larrea C.F., Staehr M., Lopez A.V., Ng K.Y., Chen Y., Godfrey W.D., Purdon T.J., Ponomarev V., Wendel H.G., Brentjens R.J., Smith E.L. Defining an optimal dual-targeted CAR T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape-driven relapse in multiple myeloma. Blood Cancer Discov. 2020;1:146–154. doi: 10.1158/2643-3230.bcd-20-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qin H., Ramakrishna S., Nguyen S., Fountaine T.J., Ponduri A., Stetler-Stevenson M., Yuan C.M., Haso W., Shern J.F., Shah N.N., Fry T.J. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol. Ther. Oncolytics. 2018;11:127–137. doi: 10.1016/j.omto.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang L., Zhang J., Li M., Xu N., Qi W., Tan J., Lou X., Yu Z., Sun J., Wang Z., et al. Characterization of novel dual tandem CD19/BCMA chimeric antigen receptor T cells to potentially treat multiple myeloma. Biomark Res. 2020;8:14. doi: 10.1186/s40364-020-00192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zah E., Nam E., Bhuvan V., Tran U., Ji B.Y., Gosliner S.B., Wang X., Brown C.E., Chen Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020;11:2283. doi: 10.1038/s41467-020-16160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.La Rosa C., Longmate J., Martinez J., Zhou Q., Kaltcheva T.I., Tsai W., Drake J., Carroll M., Wussow F., Chiuppesi F., et al. MVA vaccine encoding CMV antigens safely induces durable expansion of CMV-specific T-cells in healthy adults. Blood. 2017;129:114–125. doi: 10.1182/blood-2016-07-729756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aldoss I., La Rosa C., Baden L.R., Longmate J., Ariza-Heredia E.J., Rida W.N., Lingaraju C.R., Zhou Q., Martinez J., Kaltcheva T., et al. Poxvirus vectored cytomegalovirus vaccine to prevent cytomegalovirus viremia in transplant recipients: a phase 2, randomized clinical trial. Ann. Intern. Med. 2020;172:306–316. doi: 10.7326/M19-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diamond, C.S.G.W.a.D. (A5355) Phase II, Double-Blind, Randomized, Placebo-Controlled Trial to Evaluate the Safety and Immunogenicity of a Modified Vaccinia Ankara (MVA)-based Anti-cytomegalovirus (CMV) Vaccine (Triplex®), in Adults with Both Human Immunodeficiency Virus (HIV)-1 and CMV Who Are on Potent Combination ART with Conserved Immune Function. NIAID.
- 62.Kowolik C.M., Topp M.S., Gonzalez S., Pfeiffer T., Olivares S., Gonzalez N., Smith D.D., Forman S.J., Jensen M.C., Cooper L.J. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 63.Wang X., Li H., Matte-Martone C., Cui W., Li N., Tan H.S., Roopenian D., Shlomchik W.D. Mechanisms of antigen presentation to T cells in murine graft-versus-host disease: cross-presentation and the appearance of cross-presentation. Blood. 2011;118:6426–6437. doi: 10.1182/blood-2011-06-358747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Satheesan S., Li H., Burnett J.C., Takahashi M., Li S., Wu S.X., Synold T.W., Rossi J.J., Zhou J. HIV replication and latency in a humanized NSG mouse model during suppressive oral combinational antiretroviral therapy. J. Virol. 2018;92:e02118-17. doi: 10.1128/JVI.02118-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.R Core Team, R.F.f.S.C. 2020. R: A Language and Environment for Statistical Computing.https://www.gbif.org/tool/81287/r-a-language-and-environment-for-statistical-computing [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.