

Abstract
Complement opsonization is among the biggest challenges facing nanomedicine. Nearly instantly after injection into blood, nanoparticles are opsonized by the complement protein C3, leading to clearance by phagocytes, fouling of targeting moieties, and release of anaphylatoxins. While surface polymers such as polyethylene glycol (PEG) partially decrease complement opsonization, most nanoparticles still suffer from extensive complement opsonization, especially when linked to targeting moieties. To ameliorate the deleterious effects of complement, we have conjugated two of mammals’ natural regulators of complement activation (RCAs), Factors H & I, to the surface of nanoparticles. In vitro, Factor H or I conjugation to PEG-coated nanoparticles decrease their C3 opsonization, and markedly reduce nanoparticle uptake by phagocytes. In an in vivo mouse model of sepsis-induced lung injury, Factor I-conjugation abrogates nanoparticle uptake by intravascular phagocytes in the lungs, allowing the blood concentration of the nanoparticle to remain elevated much longer. For nanoparticles targeted to the lung’s endothelium by conjugation to anti-ICAM antibodies, Factor I-conjugation shifts the cell type distribution away from phagocytes and towards endothelial cells. Finally, Factor I-conjugation abrogates the severe anaphylactoid responses common to many nanoparticles, preventing systemic capillary leak and preserving blood flow to visceral organs and the brain. Thus, conjugation of RCAs, like Factor I, to nanoparticles is likely to help in nanomedicine’s long battle against complement, improving several key parameters critical for clinical success.
Keywords: nanoparticles, nanomedicine, complement, opsonization, C3, reticulo-endothelial system, anaphylaxis, CARPA
Graphical Abstract
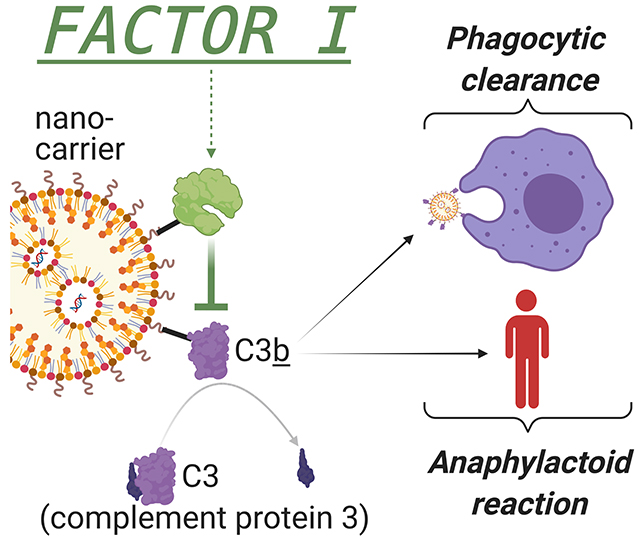
When nano-scale drug carriers enter the blood, they are rapidly bound by complement proteins, such as C3, which mark the nanocarriers for clearance by phagocytes and produce an anaphylaxis-like reaction. Here we conjugate nanocarriers to the complement-cleaving enzyme Factor I, and show it abrogates several major problems facing nanomedicine.
1. Introduction
Nanomedicine has long sought an arsenal of nanoparticles that, upon intravascular injection, display a prolonged circulation time, the ability to target specific cells and organs, and minimal side effects from the nanoparticle itself. Unfortunately, each of these goals has been continually impeded by one of the oldest parts of the immune system, the complement protein cascade[1–3]. Complement proteins comprise ~40 proteins in the blood that evolved over 500 million years to rapidly opsonize (bind to the surface of) microbes in order to mobilize the immune system to clear the pathogen[4]. Given that engineered nanoparticles share with microbes a similar size scale and many similarities in surface chemistry, it is not surprising that complement similarly attacks therapeutic nanoparticles, causing increased phagocytosis by the reticulo-endothelial system (RES), decreased circulation time, fouling of targeting antibodies, and anaphylactoid side effects. Thus, engineering new materials to evade complement has been among nanomedicine’s longest and most-sought-after goals[5].
The focal point of the complement system is the protein C3, whose highly reactive thioester forms covalent bonds to nucleophiles on non-self surfaces such as microbes and nanoparticles (Figure 1a)[6]. This reaction leaves the surface covalently bound to a large protein fragment, C3b, which initiates rapid interaction with other complement proteins to form an enzymatic complex (C3bBb) which catalyzes other C3 molecules to bond to nearby surface nucleophiles. This positive feedback loop, called the amplification loop, produces rapid spreading of C3-adducts across the non-self surface, coating it with C3b and its further cleavage fragments such as iC3b . On engineered nanoparticles, C3 opsonization causes 3 major problems[2]: 1) C3b/iC3b avidly bind to complement receptors on phagocytes, resulting in decreased nanoparticle circulation time and increased deposition in RES organs. Indeed, for the vast majority of engineered nanoparticles, a supermajority of the injected dose ends up in the RES, instead of the target tissue[7]. 2) C3b/iC3b can theoretically foul targeting moieties such as antibodies (or their fragments), inhibiting targeting of the nanoparticle to the organs and cell types of interest. 3) The C3-surface activation produces a cascade of reactions that release anaphylatoxins (C3a, C5a), producing an anaphylactoid syndrome called complement-activation-related pseudoallergy (CARPA), which includes systemic capillary leak, hypotension, and even death[8]. CARPA is dose-limiting for many nanoparticle applications, and may be prohibitive for some patient populations, such as those in the intensive care unit (ICU). ICU patients are usually very sensitive to any cause of hypotension, so the sudden capillary leak and decreased blood flow caused by CARPA could be catastrophic (e.g., stroke patients would likely suffer expanded infarct volume). Thus, C3 opsonization of nanoparticles represents one of the biggest challenges for nanomedicine to realize its full potential.
Figure 1.
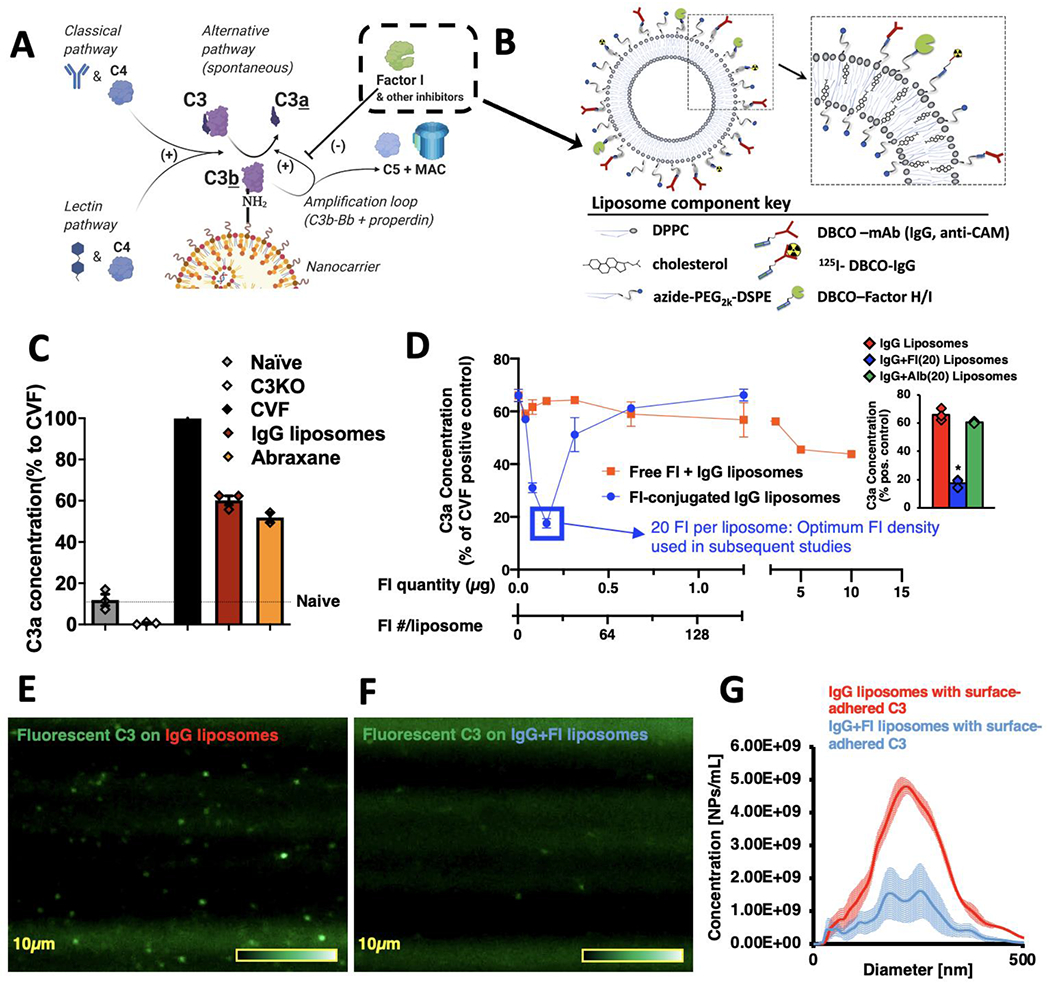
Conjugation of Factors I to nanoparticles inhibits C3-opsonization in vitro. a) Diagram of the reactions leading to C3 adducts forming on the surface of nanoparticles. The reactive thioester of C3 attacks surface nucleophiles, such as amines, resulting in a covalent C3b-surface adduct, and releasing the small protein C3a. This slow C3-surface reaction is catalytically accelerated by enzymes of the classical, lectin, and alternative pathways. C3b-surface adducts form a complex (C3bBb) that catalyzes further C3b to deposit on adjacent nucleophiles (the “amplification loop”), which allows C3b to rapidly spread across a surface. On mammalian cell surfaces, C3b is rapidly broken down by nearby regulators of complement activity (RCAs), such as Factor I, breaking the amplification loop and preventing formation of the most potent anaphylatoxin, C5a. b) Schematic of the nanoparticles on which we tested the effects of Factors H & I conjugation. All surface proteins were first conjugated to DBCO via its NHS ester, and then were conjugated onto the surface of liposomes by cycloaddition with azides on the end of the liposomes’ phospholipid DSPE-PEG2k-azide. c) Clinically used nanoparticles induce complement activation in vitro. Into mouse plasma, we mixed either Abraxane (nab-paclitaxel) or liposomes conjugated to random IgG molecules (to represent nanoparticles conjugated to targeting moieties). Reactions were EDTA-quenched after 10 minutes, and C3-nanoparticle adduct formation was measured indirectly by C3a ELISA. Data is presented as the % of C3a compared to the positive control, cobra venom factor (CVF), which rapidly cleaves all C3 in solution. Compared to naive serum (no nanoparticles), both nanoparticles induced large amounts of C3a. d) Factor-I-conjugation to nanoparticles efficiently reduces C3a production. Onto IgG-liposomes, we conjugated varying amounts of Factor I (blue trace), incubated in serum using the protocol of (c), and measured C3a production. In separate experiments, we incubated IgG-liposomes with free Factor I (not conjugated to the nanoparticles; red trace). Conjugating 20 Factor I molecules onto the liposomes reduced C3a production 3.7-fold (blue box), while free Factor I molecules at a dose equivalent to 1280 Factor I per liposome led to a 27% reduction in C3a. (n=3, per condition). Inset: Comparison of C3a production by IgG liposomes with 20 Factor I per liposome (blue) vs. C3a production by IgG liposomes without Factor I or vs. C3a production by IgG liposomes with 20 albumin per liposome. Factor I induced a 73.4% reduction in C3a vs. IgG alone and a 71.0% reduction in C3a vs. irrelevant protein. e) Direct measurement of C3-nanoparticle surface adducts. Purified C3 was conjugated to Alexa-488, added to serum, and then nanoparticles (IgG-liposomes) were added to allow opsonization. This mixture was then flowed through a Malvern Nanosight to image and size individual nanoparticles that had become bound to fluorescent C3. f) The same process as (e), but for Factor I-conjugated IgG-liposomes, showing far fewer fluorescent-C3 labeled nanoparticles. g) Histogram of the data from (e) & (f), showing Factor I conjugation leads to far fewer fluorescent-C3-opsonized nanoparticles. Statistics: For (d), inset: n=3; * = p=2.23x10−6 for comparison of IgG+FI(20) liposomes vs. IgG liposomes, p=4.43x10−6 for comparison of IgG+FI(20) liposomes vs. IgG+Albumin(20) liposomes.
Several attempts have been made to create nanomaterials that avoid C3 opsonization[5]. The most frequent approach is to orthogonally append to the nanoparticle surface a “brush” coating of (usually linear) polymers. The most common of these in clinical use is polyethylene glycol (PEG), though dozens of others have shown similar or somewhat better effects. By creating a hydration shell around the particle, these polymers increase the time it takes for C3 to penetrate to the nanoparticle surface and react with surface nucleophiles. PEG and similar polymer brushes certainly do increase the time to C3 opsonization [9], increase circulation time, and decrease CARPA, but these effects are far from optimal. Even clinically-approved PEG-or dextran-coated nanoparticles suffer from eventual (within minutes) complement opsonization and CARPA[8], as elegant studies have shown that such nanoparticles rapidly develop a shell (“corona”) of physisorbed plasma proteins, and C3 then covalently bonds to these corona proteins, possibly with the corona proteins intercalated in between polymer chains[10,11]. These limitations of PEG and other polymer brushes are dramatically more pronounced when targeting moieties (e.g., antibodies or their fragments) are conjugated onto the surface of nanocarriers, as the targeting moieties must extend beyond the polymer brush to engage their receptors, and this allows for C3 adduct formation. Thus, while polymer brushes are very useful and clever, they have not come close to fully winning the nano-war against complement.
Here we introduce a new approach to fight off C3 from the nanoparticle surface: surface conjugation of a class of proteins known as regulators of complement activation (RCA). Several RCAs circulate in blood and are expressed on the surface of mammalian cells, where they inhibit C3 and its upstream and downstream complement proteins. Perhaps the central RCA is Factor I, an 88 kDa serine protease circulating in human blood at just 35 μg/mL (compared to C3 at 1.2 mg/mL), which cleaves and inactivates C3b when Factor I is brought in close proximity to a surface[6,12,13]. Mammalian cells avoid complement attacking themselves by recruiting Factor I, and its soluble cofactor, Factor H, to their cell surface (along with expressing similar cofactors on their surface). To harness the power of RCAs for nanomedicine, prior studies have elegantly shown that adding high doses of RCAs to in vitro serum can reduce complement activation upon nanoparticle addition[14,15]. However, clinically, co-injection of long-acting complement inhibitors with nanoparticles comes with the significant danger of systemic complement inhibition, which is a powerful immunosuppressant that is dangerous in many patient populations. Instead, nanoengineers can borrow an idea from pathogens: multiple unrelated bacterial species recruit Factor H to their surface to avoid complement activation[16]. Indeed, a prior study non-specifically physisorbed multiple proteins, including Factor H, to silicon nanoparticles, but unfortunately this physisorption did not significantly reduce nanoparticle activation of C3 in whole serum, and failed to reduce phagocyte uptake, suggesting that Factor H is very sensitive to the method of adsorption onto nanoparticles [17]. Therefore, drawing inspiration from the precise and flexible manner in which bacteria recruit RCAs, here we covalently conjugated both Factors H and I separately onto nanoparticles using flexible PEG spacers. We showed that, as with microbes, precisely appending RCAs to nanomaterials’ surfaces is highly protective against complement activation by nanoparticles.
2. Results
2.1. Conjugation to Factors H & I dramatically decreases C3-nanoparticle adduct formation
Within the enormous space of nanomaterials, we chose to focus on the two classes of nanoparticles which together represent a plurality of clinically-approved nanomedicine therapeutics: lipid-based nanoparticles (liposomes and lipid nanoparticles/LNPs) and protein-nanoparticles. We began by measuring the extent to which C3-surface adducts form on these nanomaterials’ surfaces. Into in vitro mouse serum, we added either the one clinically approved protein nanoparticle, Abraxane (130 nanometer albumin particles loaded with paclitaxel) or a liposome conjugated to a targeting moiety (here, just random IgG). We quenched the reaction with EDTA (complement enzymes are Ca2+-dependent) after 10 minutes. To quantify C3-surface adduct formation, we employed the classic technique of measuring, by ELISA, the release of the small protein fragment C3a into the bulk solution. Note that C3a production is precisely stoichiometric with C3-surface adduct formation: when one C3 molecule covalently bonds to a surface nucleophile, it releases one C3a, and leaves one C3b covalently bound to the surface, usually via an amide or ester linkage. As shown in Figure 1c, both Abraxane and IgG-liposomes increase C3-surface adduct formation, by 5.1-fold (p<0.0001) and 4.4-fold (p<0.0001), respectively. Thus, these nanoparticles elicit C3a levels that are not far below that of the positive control, cobra venom factor (CVF), which cleaves all the available soluble C3 to release C3a, and thus serves as a ceiling for the maximal amount of C3 that can react. Notably, PEG-coated liposomes without surface-conjugated IgG induced only a 2.0-fold increase in C3a relative to naive serum, underscoring the role of surface-conjugated targeting moieties in provoking complement activation (Supplementary Figure 1). Thus, for testing strategies to inhibit complement opsonization of nanoparticles, Abraxane can serve as a facile test material that is FDA-approved, and IgG-liposomes can represent the numerous targeted nanocarriers that have been brought to clinical studies.
Next, to test our core hypothesis about the utility of conjugating RCAs to nanoparticles, we constructed liposomes that possessed on their surface not just a targeting moiety, but also Factors H or I (Figure 1b). We used a standard liposome formulation composed of the lipids DPPC and cholesterol (similar to clinical products like Doxil and Arykace), and also low molar fractions of lipids linked to PEG-2000 (linear PEG of molecular weight 2 kDa), end-capped by azide for bioorthogonal conjugation chemistry. Importantly, these long PEG chains are currently the field’s standard for preventing nanoparticle opsonization, allowing us to determine if Factors H & I can improve beyond the current state of the art. To the surface azides, we employed strain-promoted-alkyne-azide cycloaddition (SPAAC; a copper-free “click chemistry”) to conjugate on select proteins which had previously been conjugated to the alkyne-containing linker dibenzocyclooctyne (DBCO) [18]. In this manner, we conjugated IgG molecules (random or anti-ICAM IgG; sometimes 125I-labeled for radiotracing) and varying numbers of Factor I molecules. Supplementary Figure 2 provides materials characterization of all the key nanoparticles including naive unconjugated liposomes (136 +/− 8.9 nm), IgG/mAb conjugated liposomes (144 +/− 9.5 nm) and those containing complement proteins Factor H (154 +/− 13 nm) or I (143 +/− 8.9 nm) in addition to IgG including size (z-average, by dynamic light scattering), polydispersity index, and the efficiency of conjugation to IgG and Factor I. Additionally, Supplementary Figure 2 shows example traces from the gel exclusion chromatography used to purify the protein-conjugated liposomes away from unconjugated protein and quantify extent of conjugation of bound moieties.
After mixing these nanoparticles with serum in vitro, we then measured C3-adduct formation using the above C3a ELISA. Figure 1d (blue trace) shows that the conjugation of Factor I onto the nanoparticle surface efficiently decreases the production of C3-nanoparticle adducts, with the optimal Factor I surface concentration (20 Factor I molecules liposome) reducing C3a levels by 3.7-fold (p<0.0001). By contrast, adding free Factor I (not conjugated to the nanoparticle surface) to the serum had minimal effect, with 1,280 free Factor I molecules required to reduce the C3a production by just 27% (Figure 1d, red trace). We created similar particles with Factor H instead of Factor I, and achieved similar suppression of C3a production (Supplementary Fig 3). However, while Factor-I-conjugated nanoparticles were very stable in terms of size and PDI for up to 1 week, Factor H nanoparticles have a tendency to flocculate, with ~50% of batches flocculating (aggregating to a macroscopic size and settling out) at 24 hours at 4°C (Supplementary Fig 4). Therefore, for subsequent experiments, we used 20 molecules of Factor I per liposome. As further controls, we replaced Factor I with albumin, and found no reduction C3a levels (Fig 1D, inset, Supplementary Figure 5). Thus, these experiments show that Factor I conjugation onto nanoparticles efficiently prevents complement opsonization.
To further confirm this result, we developed a new assay of C3-nanoparticle adduct formation. The classical C3a ELISA technique is, of course, an indirect measurement of C3-nanoparticle adduct formation, as it is detecting release of a soluble product, not the adducts themselves. Therefore, we sought to directly quantify C3-nanoparticle adduct formation. To do this, we conjugated a small-molecule fluorophore to the surface of purified C3 (Supplementary Figure 6), which we added to mouse serum, and then mixed in nanoparticles for 20 minutes, followed by EDTA-quenching. We then injected diluted nanoparticles-in-serum suspensions, under continuous flow, into a nanoparticle tracking analysis (NTA) device (Malvern NanoSight) to detect C3-fluorescence on individual IgG-liposomes (Figure 1f–g) and count and size the nanoparticles with detectable C3-fluorescence (Figure 1h). As expected, nanoparticles with detectable fluorescent C3 are slightly larger than the nanoparticles before incubation with serum (Supplementary Figure 7). Having established this first-ever direct visualization of C3-adducts on nanoparticles, we measured the C3-fluorescence signals on IgG-liposomes that also had Factor I conjugated to their surface. Factor I clearly reduced the number of nanoparticles that had detectable C3-fluorescence (Figure 1e–g), to a degree closely resembling the Factor I effect in C3a measurements of complement activation (Supplementary Figure 8). Thus, this novel NTA-based measurement shows directly that conjugation of RCA proteins like Factor I to the nanoparticle surface prevents C3-nucleophile surface adduct formation.
In summary, these results confirm that, in vitro, C3-surface adducts form rapidly and extensively on clinically used nanoparticles, but conjugation of RCA proteins (here Factors H & I) markedly reduces the C3-surface reaction. We showed this both via the classical indirect assay of a C3a ELISA, and a newly introduced NTA-based direct measurement of C3-surface adducts.
2.2. Factor I surface-conjugation prevents RES-organ uptake of nanoparticles in mouse models of disease, dramatically improving the pharmacokinetics and biodistribution
We next sought to investigate how Factor H & I conjugation onto nanoparticles could affect in vivo pharmacokinetics and biodistribution. To investigate this in an animal model of disease, we sought a mouse model in which complement has already been shown to drive changes in biodistribution. Previously it has been shown that in mouse models of sepsis (dysregulated immune response to infection), global complement activation is increased [19,20], and most notably for the present study, the capillaries of the lungs fill with neutrophils, which then avidly take up circulating microbes and protein-containing nanoparticles via a C3-dependent mechanism[21–23]. Thus, during sepsis, the lungs are a dominant RES organ in humans and rodents. In primates, rodents, and a few other mammals, the lungs are not dominant RES organs in healthy individuals (the liver dominates), but in acute inflammatory states, such as sepsis and pneumonia, neutrophils and other leukocytes appear in enormous numbers in the lung capillaries, making the lung temporarily the dominant RES organ, as it is at all times in pigs, sheep, and most other mammals[24–28]. Therefore, we chose to study Factor I’s ability to change lung uptake (a proxy for complement-dependent RES uptake) in a mouse model of sepsis, in which mice are IV-injected with lipopolysaccharides (LPS) 5 hours before nanoparticle injections.
Into these sepsis-model mice, we IV-injected 125I-labeled IgG-liposomes, plus or minus surface-conjugated Factor I. As shown in the biodistribution (obtained via gamma counter) 30 minutes after nanoparticle injection (Figure 2a), the nanoparticles with Factor I are taken up in the lungs at lower concentrations (reduction of 5.0-fold, p<0.0001). Decreased organ uptake in the dominant RES organ (lungs) should lead to higher blood concentrations, which indeed is the case (Figure 2a). Notably, these biodistribution changes induced by nanoparticle-conjugated Factor I were not observed when co-injecting nanoparticles with free Factor I (not conjugated to nanoparticles; Supplementary Figure 9). This trend of higher blood concentrations and lower lung/RES concentrations of Factor I nanoparticles persists at 6 hours, but is mostly gone by 24 hours (Figure 2b; Supplementary Figures 10 & 11), which may be because the lung’s neutrophils migrate out of the pulmonary capillaries after the sepsis stimulus (LPS) clears. Thus, at least in this major model of disease, Factor I conjugation can powerfully prevent RES uptake of nanoparticles.
Figure 2.
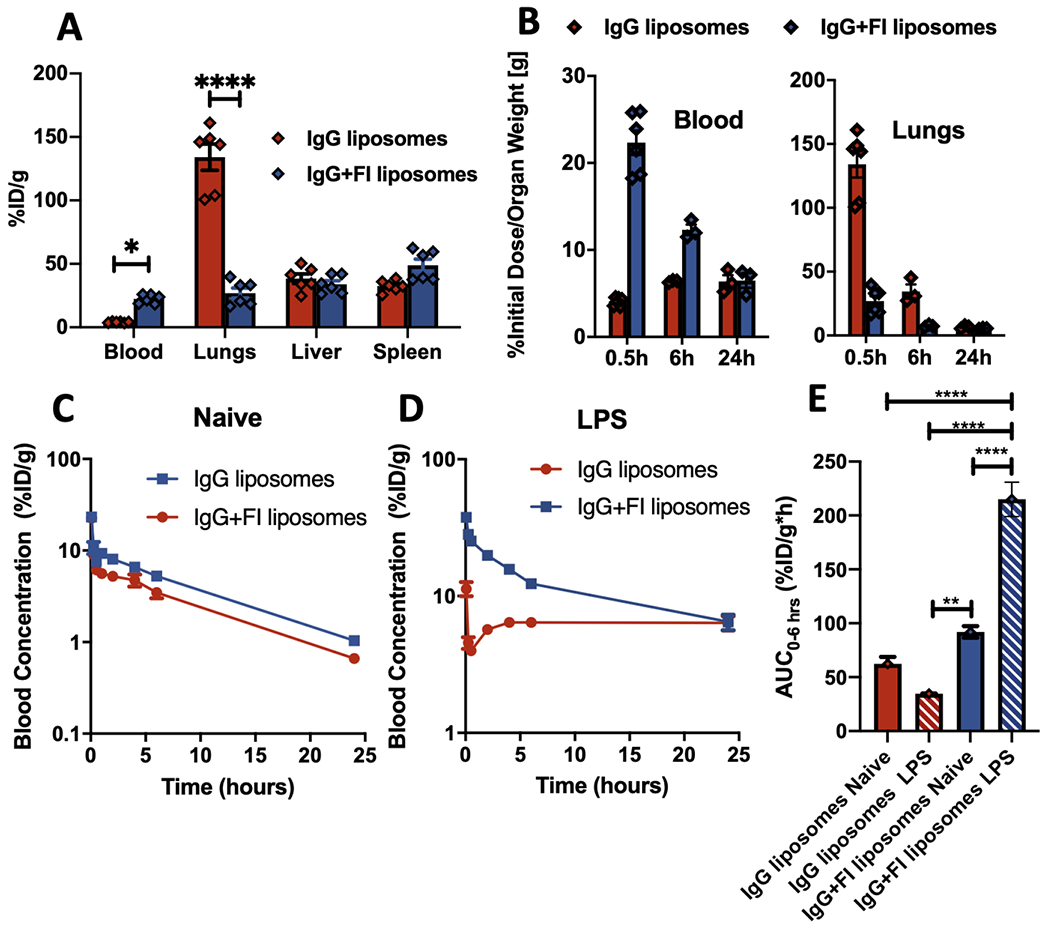
Factor I conjugation decreases nanoparticle uptake by RES organs and prolongs nanoparticle circulation in the blood in mice with acute inflammation. We employed a mouse model of sepsis, IV injection of LPS, which causes neutrophils to accumulate in the lung capillaries, making the lungs become the dominant RES organ. a) Five hours after IV LPS, mice were IV-injected with 125I-labeled IgG-liposomes, +/− Factor I, and mice were sacrificed 30 minutes later to measure the biodistribution (shown as % of the injected dose per gram of tissue [%ID/g]). Factor I conjugation decreased lung uptake of the nanoparticles, while increasing blood concentration. b) The same experiment as in (a), but also showing time points 6 & 24 hours after nanoparticle injection. c & d) Pharmacokinetics of the experiment in (a-b), measuring blood concentration over time after nanoparticle injection. Factor I may slightly increase blood concentration in naive mice (c), but the increase is much bigger in sepsis-model (IV-LPS) mice (d). e) Quantification of the area-under-the-curve (AUC) of the blood concentration vs time traces, showing Factor I (striped bars) increases the AUC (for hours 0 to 6) for both naive (blue) and sepsis-model (red) mice. Statistics: For (a, b): n=6; two-way ANOVA with Sidak’s correction for comparison between IgG +/− FI, and *=p<0.05, ***=p<0.001, ****=p<0.0001. For (e): n=3; comparisons between groups were made using 1-way ANOVA with Tukey’s post-hoc test, all comparisons with asterisks p<0.05.
Having observed these changes in biodistribution, we next wanted to precisely quantify the effects of Factor I conjugation on nanoparticle pharmacokinetics. As shown in Figure 2c, even in naive mice, Factor I-conjugated nanoparticles display higher blood concentrations over time (see also Supplementary Figure 12). In sepsis-model mice (LPS), the improvement in the blood-concentration vs time curve is much greater (Figure 2d). In the sepsis-model mice, the area-under-the-curve (AUC; for hours 0-6) of the blood-concentration vs time curve is 6.2-fold higher (p<0.0001) in Factor I-conjugated nanoparticles than in the same particles without Factor I (Figure 2e; see Supplementary Figure 12–13 for non-compartmental analysis and an explanation of why the concentration of nanoparticles is not monotonically decreasing over time). Thus, Factor I conjugation is able to markedly improve the pharmacokinetics of nanoparticles, in situations in which the goal is to maintain the nanoparticle circulating for a long time.
2.3. Factor I surface-conjugation prevents nanoparticle uptake by local phagocytes, thus improving targeting to the cell type of interest
We next sought to determine if Factor I conjugation could aid with targeting a cell type of interest. We hypothesized that Factor I conjugation would prevent uptake of the targeted nanoparticles by local phagocytes in the target organ, thus encouraging uptake in other cell types. We tested this hypothesis using a nanoparticle-targeting strategy that has been used to target the endothelial cells of the lungs for decades: anti-ICAM antibodies conjugated to nanoparticles[29–31].
We began by first confirming that, in vitro, Factor I conjugation could prevent uptake of nanoparticles by phagocytosis. We incubated fluorescently-tagged IgG-liposomes, plus or minus Factor I conjugation, with mouse serum for 1 hour to allow for maximal C3 deposition. We then incubated the serum-exposed nanoparticles with freshly isolated mouse neutrophils rotating at 37°C for 15 minutes. We next ran the neutrophils in a flow cytometer, assaying for the amount of liposome fluorescence associated with each neutrophil. As shown in Figure 3a, with Factor I, there were far fewer neutrophils that displayed very high amounts of nanoparticle fluorescence. Additionally, the median nanoparticle fluorescence in/on neutrophils was decreased by 2.0-fold (p<1x10−10) in the Factor I conjugated nanoparticles (Figure 3b; also Supplementary Figure 14). Thus, even under stringent in vitro conditions, Factor I managed to inhibit complement-mediated association with phagocytes.
Figure 3.
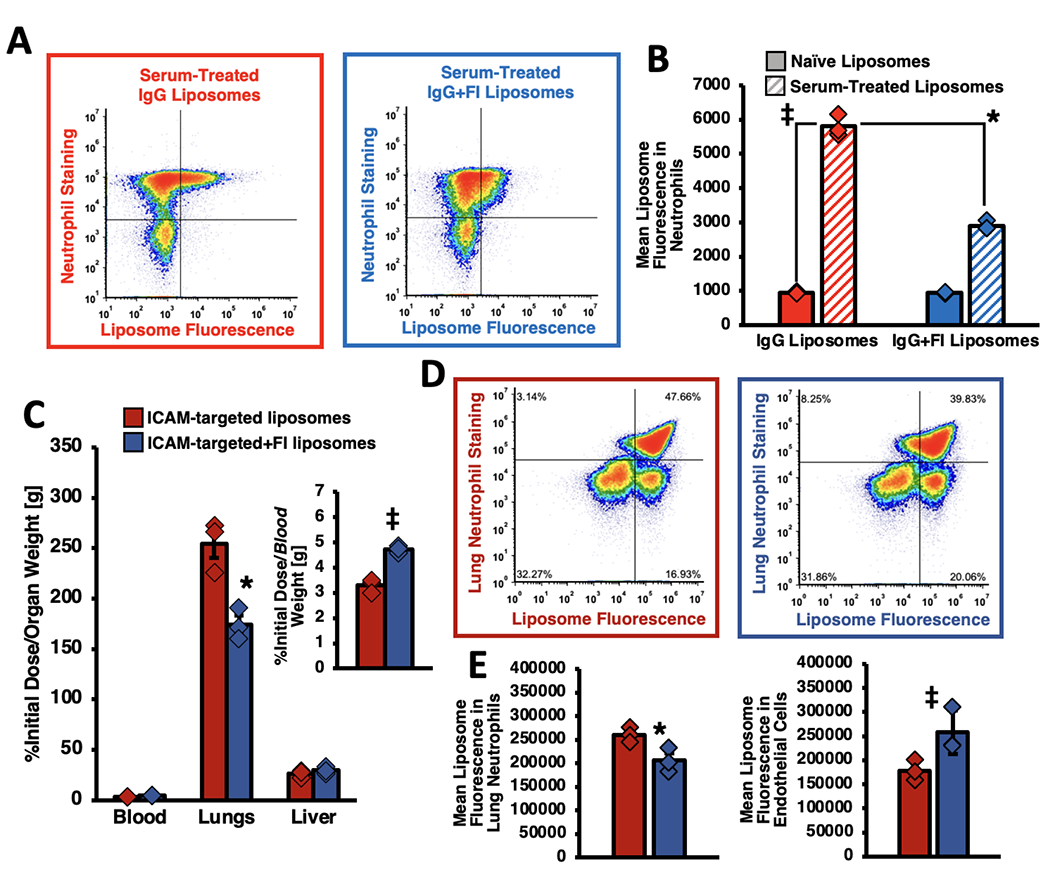
Factor I conjugation prevents phagocyte association with nanoparticles, including local phagocytes in the target organ of antibody-targeted nanoparticles. a, b) In vitro, conjugation to Factor I reduces serum-treated nanoparticle interactions with neutrophils 2.0-fold. Fluorescent-IgG-liposomes +/− Factor I were incubated with serum for 1 hour, then incubated with neutrophils for 15 minutes. Neutrophils were pelleted and washed to remove unbound nanoparticles. Neutrophils were stained with the specific marker Ly6G (y-axis) and then subjected to flow cytometry. The dot-plots show Factor I changes the relative amount of liposome fluorescence seen in/on the neutrophils, which is quantified as a mean liposome-fluorescence signal (b) in the neutrophils that is half as much when Factor I is conjugated on (blue striped bar vs red striped). c) IV-LPS mice were IV-injected with 125I-liposomes conjugated to antibodies that bind to the pulmonary endothelial marker ICAM. As shown for decades, the ICAM-targeted nanoparticles homed to the lungs, but the amount of lung targeting decreased by 31.6% and the blood concentration increased (inset) by 43.7% when the liposomes were conjugated to Factor I. d) Flow cytometry of the lungs of mice similar to (c), but with liposomes traced with fluorescence instead of radioactivity. The left panel (red outline) is from mice treated with ICAM-targeted liposomes, while the right panel (blue outline) received ICAM-targeted liposomes conjugated to Factor I. (e) Quantification of the dot plots in (d) show that Factor I led to a 21.0% lower median liposome-fluorescence in/on the lungs’ local neutrophils (left panel) but a 44.6% increase in endothelial cell-associated liposome fluorescence (right panel). This decrease in neutrophil association with nanoparticles but increase in endothelial-associated nanoparticle fluorescence shows that Factor I improved the targeting specificity of ICAM-targeted liposomes. Statistics: For (b): n=6; *p<1x10−10 via two-way ANOVA with Tukey correction; ‡ = p<1x10−10 via two-way ANOVA with Sidak’s correction. For (c): n=3; * = p=0.0001 by two-way ANOVA with Tukey; ‡ = p=0.04 by one-way ANOVA with Tukey. For (d): n=3; * = p=0.006 by two-way ANOVA with Sidak’s correction; ‡ = p=0.025 by two-way ANOVA with Sidak. For (e): n=3; * = p=0.006 by two-way ANOVA with Sidak’s correction; ‡ = p=0.005 by two-way ANOVA with Sidak’s correction.
We next tested whether Factor I could similarly prevent cell-targeted nanoparticles from being taken up by local phagocytes. We first showed that, in vitro, anti-ICAM-liposomes specifically bind ICAM on the surface of target cells, and that Factor I conjugation does not affect the binding avidity (Supplementary Figure 15). We next injected anti-ICAM-liposomes into sepsis-model mice. As with IgG-liposomes, Factor I decreased ICAM-targeted nanoparticle uptake in the lungs (Figure 3c; Supplementary Figure 16) and increased nanoparticle retention in the blood (Figure 3c inset). Notably, while the absolute ICAM-targeted nanoparticle uptake in the lungs decreased for the anti-ICAM+Factor I liposomes, the ICAM-targeted:IgG lung uptake ratio increased to 6.5, compared to 1.9 for liposomes without Factor I. We next disaggregated the lungs into a single-cell suspension, and used flow cytometry to check if the above-observed decreased lung uptake was due to decreased uptake by phagocytes. Indeed, the mean nanoparticle fluorescence in neutrophils (the main local, intravascular leukocyte) decreased by 21.0% (Figure 3d, upper quadrants, Figure 3e left panel, Supplementary Figure 17 left panels). Similar analysis of all lung leukocytes showed that Factor I reduced uptake in the total population of leukocytes, though specific reduction of uptake in Ly6G-negative leukocytes (non-neutrophil leukocytes) was not significant (Supplementary Figure 18). By contrast, the mean nanoparticle fluorescence increased 44.6% in endothelial cells, which are the intended target cell for anti-ICAM liposomes (Figure 3e right panel, Supplementary Figure 17 right panels). Thus, as hypothesized, Factor I surface conjugation prevented nanoparticle association with local phagocytes, and improved targeting to the cells of interest.
2.4. Factor I surface-conjugation prevents the severe anaphylactoid responses induced by nanoparticles
All of the above investigations related to Factor I conjugation improving the beneficial aspects of nanoparticles by decreasing C3b-adduct formation, decreasing RES and local phagocyte uptake, and improving biodistribution and pharmacokinetics. However, complement is also known to cause severe side effects, so we next investigated Factor I’s ability to prevent those. For decades, infusion of nanoparticles in animals and humans has been known to cause the anaphylactoid syndrome of CARPA[8,32,33]. CARPA is characterized by rapid production of two complement pathway-produced anaphylatoxins: C3a, a weak anaphylatoxin; and C5a, among the strongest known anaphylatoxins. These products, and likely other complement products, lead to urticaria (rash) and rapid (within 10 minutes) systemic capillary leak, which leads to systemic hypotension, shock, and occasionally death. These issues have been partially addressed in clinical practice, with infusion of IV fluids and for some nanomedicines, such as patisiran (the first FDA-approved lipid nanoparticle to deliver nucleic acids), the requirement for pretreatment hours ahead with steroids. However, in patient populations which cannot tolerate even slight hypotensive stimuli, such as critically ill patients with sepsis or stroke, CARPA could make nanomedicine far too risky.
To study whether Factor I conjugation could prevent CARPA in such vulnerable, critically ill populations, we tested Factor I’s ability to prevent CARPA and its attendant hypotension in the IV LPS mouse model of sepsis. Note that the complement cascade is already partially activated in IV LPS mice, as shown in Supplementary Figure 19, and prior studies[19,20]. Into such IV LPS mice, we injected IgG-liposomes +/− Factor I, and sacrificed them 10 minutes later. Assaying C3a in the blood of these mice showed that Factor I decreased the C3a concentration by 45% (Figure 4a, left panel). Even more impressively, Factor I decreased the most potent anaphylatoxin, C5a, to undetectable levels (Figure 4a, right panel). Thus, in vivo, Factor I powerfully prevents production of complement’s main anaphylatoxins.
Figure 4.
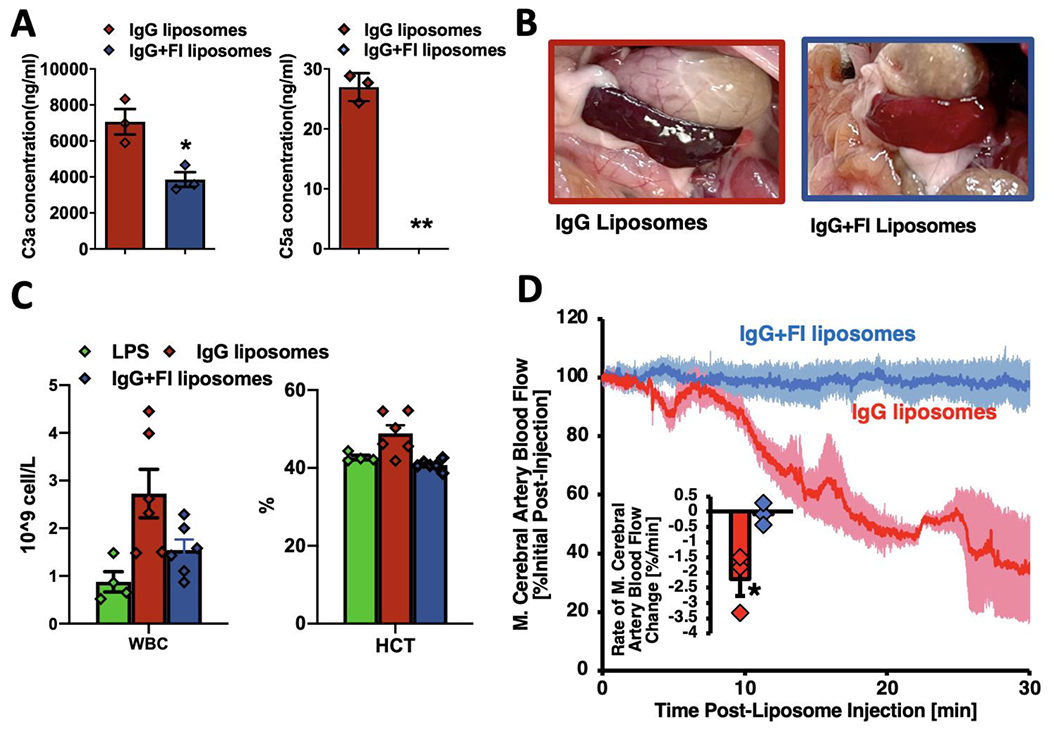
Factor I conjugation to nanoparticles prevents CARPA-associated side effects. a) Sepsis-model mice (IV-LPS) were IV-injected with IgG-liposomes +/− surface-conjugated Factor I, and sacrificed 10 minutes later for blood draw. ELISAs showed Factor I led to a nearly 50% decrease in the C3a blood concentration provoked by liposomes (left panel), and completely eliminated the production of the complement pathway’s most potent anaphylatoxin C5a (right panel). b) Factor I conjugation prevented visceral organ hypoperfusion. In the same mice from (a), we observed mice receiving IgG-liposomes had very dark spleens, indicating hypoperfusion, while Factor I conjugated liposomes did not change the spleen color from its normal light burgundy. c) Treating mice the same as in (a) & (b), we measured the CBC (complete blood count) and found that IgG-liposomes markedly increase the concentration of all circulating white blood cells (WBCs; left panel), but this was largely prevented by Factor I conjugation. IgG-liposomes also increased the hematocrit (right panel), which is the most direct measure of CARPA-related systemic capillary leak. Once again, Factor I prevented this side effect of nanoparticles. d) Factor I also prevented CARPA-related cerebral hypoperfusion. Five hours after IV-LPS, mice were injected with IgG-liposomes +/− surface-conjugated Factor I, while their cerebral blood flow was measured via Doppler ultrasound of the middle cerebral artery. IgG-liposomes (red trace) led to decreased blood flow, while Factor I conjugated liposomes (blue) did not (traces are mean +/− SEM; n = 3 condition). The inset shows the average rate of blood flow change per minute, which stayed fairly constant and negative for IgG-liposomes, but was consistently near zero for Factor I conjugated liposomes, showing that Factor I prevented liposome-provoked CARPA-related cerebral hypoperfusion. The average rate of blood flow change decreased by 2675% for IgG-liposomes vs IgG-liposomes conjugated to Factor I. Statistics: For (a): n=3; *p=.0255, **p=0.0014 by Welch’s t-test. For (d): n=3; *p=.0497 by Welch’s t-test.
We next examined whether Factor I can prevent the visceral organ hypoperfusion predicted to occur with CARPA. To do this, in the same mice in which we assayed C5a blood levels, we photographed the spleen, which can become dark during states of visceral organ hypoperfusion or hypoxia. As shown in Figure 4b and Supplementary Figure 20, the spleens of mice receiving IgG-liposomes were very dark, almost black, whereas the spleens of mice receiving Factor I-conjugated IgG-liposomes were the normal light burgundy color of naive mice, suggesting Factor I protected from abdominal organ hypoperfusion.
Next we measured whether the transient leukocytosis previously described for CARPA is also observed in these IV LPS mice after injection of IgG-liposomes. Figure 4c and Supplementary Figure 21 show that IgG-liposomes increase leukocyte concentration by 4.1-fold. Conjugation of Factor I reduced that nanoparticle-induced leukocytosis by 57.7%.
We next investigated if nanoparticles injected into IV-LPS mice also induce the massive capillary leak phenomenon of CARPA, previously shown in pigs. To measure capillary leak, we measured the hematocrit, which is a measure of the fraction of blood’s volume that is occupied by red blood cells. In states of rapid capillary leak, the hematocrit increases (called “hemoconcentration”), as plasma leaks into the tissues. Indeed, we found that IgG-liposomes caused a 14.4% increase in the hematocrit 10 minutes after nanoparticle injection. Importantly, this hemoconcentration was completely abrogated by Factor I conjugation, suggesting that Factor I can indeed prevent capillary leak.
Finally, we finished by determining if Factor I can prevent hypoperfusion to the brain, the most sensitive organ during acute critical illnesses. Many groups have for decades pursued nanoparticle-based treatments for acute ischemic stroke. However, we hypothesized that CARPA from nanoparticles could induce cerebral hypoperfusion, which could make cerebral infarcts enlarge, as the at-risk, partially perfused “penumbra” region around a stroke’s core is very sensitive to hypoperfusion. As hypothesized, IgG-liposomes decreased perfusion to the mouse brain (measured by Doppler ultrasound of the middle cerebral artery) by more than 60%, starting within 5 minutes after nanoparticle injection, and lasting at least 30 minutes (Figure 4d; Supplementary Figures 22–23). As a comparator, in the transient middle cerebral artery occlusion (tMCAO) mouse model of ischemic stroke, a 70% reduction in middle cerebral artery blood flow is standardly considered sufficient to induce a large volume cerebral infarct (stroke)[34]. Importantly, this cerebral hypoperfusion was completely prevented by Factor I conjugation. Analysis of middle cerebral blood flow by both average rate of blood flow reduction and area under curve showed significant improvement with Factor I (Figure 4d, inset, Supplementary Figure 22). Conjugation to Factor I eliminated IgG liposome effects on middle cerebral artery blood flow for two densities of IgG on the liposomes, 200 or 100 IgG per liposome (Supplementary Figure 23). Thus, while nanoparticles could have outsized side effects in critically ill patients like those with strokes, Factor I may be able to prevent these and enable nanomedicine to move into the intensive care unit (ICU).
3. Discussion & Conclusions
Nanomedicine has faced several challenges on its quest to safely shuttle drugs to their desired location for a specified length of time. Among the greatest of these challenges has been created by the complement system, which has had a half-billion-year head start in designing a defensive system to prevent nano-scale particles, meaning microbes and later engineered nanoparticles, from going where they want in the body. Nanomaterials engineers have made significant strides against this defense, most notably with the introduction of hydrophilic polymer brushes, which have served as partial blockers of C3 reaching the nanoparticle surface. But clearly, more is needed, as most nanoparticles, with or without PEG or other brush polymers, end up in the RES organs rather than in their intended tissue[7].
However, nanoengineers have started to borrow from nature’s armamentarium to fight complement. The first example was co-injecting complement inhibitors into the blood at the same time or right before nanoparticle injection[14,15]. These elegant studies showed in vitro that regulators of complement activity (RCAs), such as Factor H, decreased complement activation when nanoparticles were added to serum. However, clinically, co-injection of long-acting complement inhibitors with nanoparticles comes with the significant danger of causing systemic complement inhibition, which is a powerful way to suppress the immune system. Such immunosuppression could be very dangerous, especially in hospitalized patients, who are already at greatly increased risk of acquiring new infections.
Therefore, we sought to build on the clever technological advances thus far, by conjugating RCAs directly onto the nanoparticle surface. Surface conjugation greatly increases the local concentration of the RCAs, and thus, at least theoretically, should require far lower numbers of RCA molecules to be introduced into patients, avoiding immunosuppression. As predicted, we found that appending RCAs to the nanoparticle surface efficiently prevented C3 opsonization (Figure 1), leading to numerous other benefits: decreased RES organ uptake and thus prolonged nanoparticle circulation in the blood (Figure 2); less nanoparticle uptake by phagocytes, including local phagocytes in the target organ (Figure 3); and decreased CARPA-associated side effects, such as systemic capillary leak, visceral hypoperfusion, and most importantly, cerebral hypoperfusion (Figure 4). Thus, RCA surface conjugation, especially of Factor I, appears to be a straightforward way of overcoming many of the hurdles that have tripped up nanomedicine for years.
While Factor I conjugation to nanoparticles appears to have numerous advantages, it is prudent to consider possible side effects. In particular, we must consider whether conjugated Factor I would cause immunosuppression and consequent infections, as a very high systemic concentration of Factor I might inhibit C3 so much that microbes escape. However, it is very unlikely that the amount of Factor I present on our nanoparticles would have this immunosuppressive effect. We are proposing the injection of very small masses of Factor I, as shown in the following dose calculations: In humans, most IV liposomes (e.g., Doxil) are dosed at, maximally, 10 mg/kg[35]. Converting that maximal dose to a 10 mg/kg mouse dose, for our average targeted liposomes in this study, that is 3 x 1011 liposomes injected per mouse. In Figure 4D, we showed that the optimal # of Factor I molecules per liposomes is 20. Thus we are injecting 6 x 1012 Factor I molecules into the 2 mL of blood that a mouse has, which, given the 88 kDa weight of Factor I, means we are injecting 0.4 ug of Factor I mL of blood. By comparison, Factor I is present at 35 ug/mL in the blood[12]). Thus, the Factor I technology proposed here will only increase the amount of Factor I in the blood by ~1%, which is unlikely to cause immunosuppression.
While Factor I and other RCA conjugation appears to have many benefits, there are still many remaining scientific questions and required engineering. For example, it is somewhat surprising that Factor I worked as well as Factor H in our experiments, while in contrast, multiple species of bacteria evolved to coat themselves with Factor H-binding proteins, not Factor I. We conducted most of our experiments with Factor I rather than H for practical engineering reasons: Factor H is a very large protein whose 40-disulfide bonds make it very sensitive to redox conditions, and thus we found that it occasionally aggregated unexpectedly, while Factor I was easy to conjugate and never aggregated or unfolded. Indeed, there was a previous report in which Factor H was physisorbed (non-covalent adsoprtion) onto silicon nanoparticles[17], and this adsorption did not significantly reduce C3a activation in whole serum, and failed to reduce phagocyte uptake, suggesting that Factor H is very sensitive to the method of adsorption onto nanoparticles.
Several other intriguing questions remain for understanding how Factor-I-conjugation provides benefits and how to optimize these advantages. It is unclear how Factor I works so efficiently appended to surfaces without appended cofactors, since it usually must bind Factor H or a surface-bound RCA to function. Further work is clearly warranted to test if Factor I conjugation can be made even more efficient by addition of Factor I cofactors. This may be particularly challenging, since the efficiency of complement protection afforded by Factor I varied non-monotonically with the number of Factor I molecules per liposomes (Fig 1D). It will be important to investigate the mechanism underlying this surprising quantitative relationship, as the mechanism may provide insight into further optimizing Factor I conjugation.
Lastly, and most importantly, it will be interesting to port this system to other species, especially humans. Factor I is well conserved in mammals, but the complement system itself varies considerably in how easy it is to trigger, with mice usually considered as having a high-threshold for C3 activation, while humans have lower thresholds for triggered C3-adduct formation[6,8,36]. Additionally, human neutrophils may interact with nanoparticles differently than mouse neutrophils, so further engineering might be required to not just modulate complement, but also downstream neutrophil behavior [37]. Thus, while the present studies suggest conjugation of RCAs like Factor I to the nanoparticle surface can be a powerful tool for nanomedicine, there is still work to be done to optimize this and other approaches, so we can finally conquer the defenses of complement and allow nanomedicine to reach its full potential.
4. Methods
4.1. Nanoparticle preparation
Liposome were prepared from 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[azido(polyethylene gly-col)-2000 (azide PEG 2000 DSPE) and cholesterol (54:40:6 mol%) using the classical lipid thin film extrusion method as described previously[18]. In brief, lipids were dissolved in chloroform, then added to together in the above molar ratios to a final lipid molarity of 20 nM, and the cholorform was evaporated by blowing nitrogen onto the liquid, resulting in a thin film at the bottom of a borosilicate tube. We hydrated the films by adding 500 uL of PBS, followed by bath sonication at 55C. We then extruded the lipids through a 200-nanometer filter using an Avanti Polar Lipids syringe extruder. For fluorescent liposomes, an additional 1% of TopFluor PC was included in the formulation. Liposomes were then sized via dynamic light scattering (DLS; Malvern Zetasizer).
We next conjugated liposomes to specific proteins using the common “click chemistry” of strain-promoted alkyne-azide cycloaddition (SPAAC)g, as described previously.[18] The method of modifying the specific proteins for SPAAC was is described in the section below, “Modification of Proteins.” The DBCO-modified proteins were covalently conjugated to the azide-functionalized liposomes by mixing the protein and liposomes in PBS and incubated at 4 °C overnight. The ratios of protein-to-liposoome were determined by the desired # of proteins per liposome (see Results), assuming a conjugation efficiency of 60-90%. We then measured the # of actual proteins per liposome via Sepharose column (see below), and adjusted reaction ratios to achieve our goal # of proteins per liposome. For further details on conjugation of proteins to liposomes, see the section below, 4.4. Characterization of protein conjugation to liposomes
Immunoliposomes were purified from residual antibodies using a 20 mL Sepharose 4B-Cl column (GE Healthcare, Pittsburg, PA). DLS measurement of hydrodynamic particle size and polydispersity index using a Zetasizer Nano ZSP (Malvern Panalytical, Malvern UK). The resulting liposome concentration in number per ml was measured using NanoSight NS300 (NanoSight, Salisbury, United Kingdom) using a 4x104 dilution into high purity DI water.
4.2. Modification of Proteins
Proteins were conjugated to azide functionalized liposomes via copper-free click chemistry[18], antibodies (whole molecule IgG, Thermofisher; anti-ICAM mAb YN1/1.7.4 grown from hybridoma per ATCC) and complement factors (Factor H and I, Complement Technology, Inc. Tyler, TX) were modified using DBCO-PEG4-NHS ester (Jena Bioscience, Jena, Germany) according to the manufacturer’s protocol. Briefly, antibodies were mixed with NHS ester in DMSO at 1:20 molar ratio, and Factor H or Factor I at 1:5 molar ratio. After reaction for 30 min at room temperature, modified antibodies were purified from residual DBCO reagent and free NHS ester using Amicon Ultracel-50kDA membrane filter (Millipore, Burlington, MA) to remove unreacted NHS ester PEG4 DBCO. The efficiency of DBCO-IgG reaction was determined optically, with absorbance at 280 nm indicating IgG concentration and absorbance at 309 nm indicating DBCO concentration. Spectral overlap of DBCO and IgG absorbance was noted by correcting absorbance at 280 nm. Molar protein concentration was determined using Beer’s Law calculation. The number of DBCO per IgG was determined as the ratio. Secondary fluorescent labeling of proteins using Alexafluor 488 NHS ester (Thermofisher) followed manufacturer’s instructions.
4.3. Radiolabeling of Antibodies
Antibodies were radioiodinated with [125I]Na (Perkin Elmer, Waltham, MA) using Pierce lodgen radiolabeling reagent and purified using Zeba desalting spin columns (ThermoFisher Scientific). Radiochemical purity was assessed via TLC using a mobile phase of 75% methanol : 25% NH4 acetate, and confirmed > 90% in all cases. To radiolabel immunoliposomes, 2% radiolabeled untargeted (random IgG) antibodies were added for conjugation.
4.4. Characterization of protein conjugation to liposomes
Proteins, including Factor H or I, IgG or mAb, modified with DBCO were combined in designated amounts and incubated with azide functionalized liposomes. Combining the measured Nanosight NTA liposomes concentration values in #/ml, and spectrophotometric protein concentrations, we combined mixtures of the DBCO functionalized proteins (including <10% by volume relative to total protein of labeled tracer protein), with azide liposomes for simultaneous conjugation at 4C overnight. Calculations include an efficiency estimate from 60-90% of protein binding. For example, if IgG DBCO is estimated to have 90% binding efficiency and the target coating density is 100 IgG/liposome then we will calculate an addition of 110 IgG per particle. After conjugation incubation, the reaction mixtures were characterized and purified by size exclusion chromatography (SEC) using Sepharose 4B-CL (Sigma Aldrich) as previously described[18]. Protein conjugation was quantified by tracing ligand fluorescence or radioactivity (each fraction read on a plate reader or a gamma counter). Efficiency of conjugation reaction is quantitatively defined as the ratio of the area under the curve of the ligand signal in the liposome peak (7-9 mL) over the sum of that peak combined with the free protein peak. Supplement Figure 2 shows chromatography data of a titration of Factor I coating on liposomes, and 2 IgG coating densities, all indicating characteristic elution peaks. Fractions containing protein bearing liposomes are collected and re-concentrated using Amicon filtration devices (Millipore), then measured again for size and concentration using DLS and NTA as described above.
4.5. C3a and C5a ELISA
ELISA testing was conducted to measure the activated C3a and C5a levels in vitro and in vivo, per manufacturer protocol. Briefly, for in vitro measurement, 20 μL of fresh serum was incubated with 20 μL of immunoliposomes (2x1012 liposomes/mL, conjugated with designated DBCO modified proteins) for 15 minutes, EDTA was added to a final concentration of 20mM, to inhibit further complement activation. For in vivo measurement, plasma was collected from mouse inferior vena cava with EDTA coated syringes, and then chelated with 20mM EDTA and the pan-complement inhibitor Futhan (0.05mg/ml, BD Pharmingen) to inhibit further complement activation. Serum/plasma C3a and C5a levels were measured by using sandwich ELISA kits from BD Biosciences Company.
4.6. Nanoparticle tracking analysis for C3 adhesion to liposomes
To prepare fluorophore-labeled C3, human complement protein C3 (Complement Technology) was incubated with NHS ester Alexa Fluor 488 (ThermoFisher) at 1:5 mol:mol ratio in PBS with 0.1 M NaHCO3 on ice for two hours. Afterwards, excess fluorophore was removed from C3 by 3-fold passage against molecular weight cutoff 10 kDa centrifugal filter (Amicon) with PBS washing between passages. After C3 recovery from the centrifugal filter, spectrophotometer measurement of optical density at 280 nm determined fluorescent C3 concentration and optical density measurement at 488 nm determined the number of fluorophores per C3.
Immediately before experiments, liposome concentrations were determined by nanoparticle tracking analysis (Nanosight, Malvern). In a total reaction volume of 40 μL, 4x1010 liposomes were combined with 20 μL mouse serum and fluorescent C3 was doped into the solution at a final concentration of 0.3 mg/mL. Fluorescent C3, serum, and liposomes were incubated in the dark at room temperature for 20 minutes. Fluorescent C3 was also added to serum solutions at identical concentration, without liposomes, verifying that the fluorescent C3 did not adhere to endogenous serum components at detectable concentrations. The C3-serum-liposomes reactions were terminated by 1:250 dilution in PBS and the diluted suspensions were used for nanoparticle tracking analysis. Nanoparticle tracking analysis was conducted with a 488 nm excitation laser and a 500 nm long pass filter to image and track Alexa Fluor 488 signal from fluorescent C3 on nanoparticles. Automated analysis of fluorescence nanoparticle tracking data in Malvern Nanosight software used a uniform detection threshold of 5 for all samples. The same samples were immediately analyzed with an open filter to assess light scattering species, rather than just fluorescent-tagged species, therefore imaging and tracking all serum components and unlabeled liposomes in the sample and verifying that the fluorescent population was distinct from the total population of serum components in its size distribution and concentration. Scattering-based nanoparticle tracking data was analyzed in Malvern Nanosight software with a detection threshold of 12. For both fluorescence data and scattering data, five technical replicates were obtained for each sample and an average of those replicates was taken as representative of the size-concentration profile for each sample.
4.7. Animal studies: Pharmacokinetics and biodistribution of immunoliposomes in naive and inflamed mice
Immunoliposomes (3 mg/kg, ~2x1012 liposomes/mL) were intravenously injected in naive or lipopolysaccharide (LPS) treated groups. For LPS groups, mice were anesthetized with 3% isoflurane, LPS from E. coli strain B4 (Sigma) was administered at 2 mg/kg in 100 μL PBS 5 hours prior to liposome injection. After five hours, mice were anesthetized with ketamine-xylazine (10 mg/kg ketamine, 100 mg/kg xylazine, via intramuscular administration) and were injected intravascularly with 3 mg/kg immunoliposomes conjugated with designated DBCO modified proteins (IgG, anti ICAM YN1, Factor I ). The animals were euthanized at designated times after injections (30 minutes after nanoparticle injection, unless otherwise stated), and the organs of interest were harvested, rinsed with saline, blotted dry, and weighed. Blood samples (~200 ul) were spun down at 500 rcf in a microcentrifuge tube with RBCs separated from plasma. Biodistribution quantification was determined by measuring the radioactivity in the blood and other tissues using a Wallac 2470 Wizard gamma counter (PerkinElmer Life and Analytical Sciences-Wallac Oy, Turku, Finland). The gamma data of the 125I measurements and organ weights were used to calculate the tissue biodistribution injected dose per gram of tissue. The total injected dose was measured prior to injections, corrected for tube and syringe residuals, and verified to be ≥75% of the sum of the individual measures.
Blood pharmacokinetic data was analyzed via standard noncompartmental analysis (NCA) in order to derive the area under the concentration vs. time curve (AUC), which was calculated using the linear trapezoidal rule. This can be done using MATLAB’s Simbiology NCA system.
4.8. In vitro neutrophil uptake of liposomes
Neutrophils were purified from ~8-week-old C57BL/6 mouse femur bone marrow. Femurs were harvested after euthanasia, their ends were cut off, and the marrow was extracted by flushing media through the cut end. The marrow cells were then subjected to magnetic bead pull down of non-neutrophils using RoboSep Mouse Neutrophil Enrichment Kit (StemCell Technologies), exactly according to manufacturer instructions. The neutrophils were placed into 500uL media at a concentration of approximately 2x106 cells mL.
Within 1 hour of neutrophil isolation, 1x106 neutrophils were rotated with 5x109 liposomes in 20μL PBS for 15 minutes at 37°C. Alternatively, serum-treated liposomes were prepared by incubating 5x109 fluorescent liposomes in 10 μL PBS with 10 μL of mouse serum for one hour at 37°C prior to addition to 1x106 neutrophils. The mouse serum was prepared by drawing blood from wild-type mice, allowing the blood to coagulate in a 1.5 mL Eppendorf centrifuge tube for 30 min at room temperature, and then centrifuging at 1500g x 10 minutes at 4C. For flow cytometry (BD Accuri C6), neutrophils were washed and stained with PerCP/Cy5.5 Ly6G antibodies (BD Biosciences, 1:100 dilution) and non-neutrophils were excluded from analysis via Ly6G staining (see Supplementary Figure 14 for gating). Liposome fluorescence in neutrophils was quantified by mean fluorescence intensity, gated on Ly6G-positive populations.
4.9. Flow cytometry analysis of ICAM-targeted liposome distribution among cell types in the lungs
Mice were injected with LPS (2mg/kg, IV) 5 hours prior to IV-injection of fluorescent (1% of TopFluor PC) anti-ICAM-liposomes +/− surface-conjugated Factor I. Mice were anesthetized with ketamine/xylazine (10 mg/kg ketamine, 100 mg/kg xylazine, intramuscular administration) in order to place a tracheal catheter secured by suture. Thirty minutes after liposome administration, mice were sacrificed by terminal exsanguination via the vena cava and lungs were perfused by right ventricle injection of ~10 mL of cold PBS. The lungs were then infused via the tracheal catheter with 1 mL of a digestive enzyme solution consisting of 5 U/mL dispase, 2.5 mg/mL collagenase type I, and 1 mg/mL of DNAse I in cold PBS. Immediately after infusion, the trachea was sutured shut while removing the tracheal catheter. The lungs with intact trachea were removed via thoracotomy and kept on ice prior to manual disaggregation by vigorous chopping with scissors and razors.
Disaggregated lung was aspirated in an additional 2 mL of digestive enzyme solution and incubated at 37°C for 45 minutes, with vortexing every 10 minutes. After addition of 1 mL of fetal calf serum, tissue suspensions were strained through 100 μm filters and centrifuged at 500 xg for 5 minutes. After removal of supernatant, the pelleted material was resuspended in 10 mL of cold ACK lysing buffer. The resulting suspensions were strained through a 40 μm filter and incubated for 10 minutes on ice. The suspensions were centrifuged at 500 xg for 5 minutes and the resulting pellets were rinsed in 10 mL of FACS buffer (2% fetal calf serum and 1 mM EDTA in PBS). After centrifugation at 500xg for 5 minutes, the rinsed cell pellets were resuspended in 2% PFA in 1 mL FACS buffer for 10 minutes incubation at room temperature in the dark. The fixed cell suspensions were centrifuged at 500 xg for 5 minutes and resuspended in 1 mL of FACS buffer.
To stain fixed cells, 100 μL aliquots of cell suspensions were pelleted 500xg for 5 minutes, then resuspended in labeled antibody diluted in FACS buffer (1:150 dilution for Alexa Fluor 647 anti-Ly6G or APC-anti-CD31 and 1:500 dilution for PerCP/Cy5.5 anti-CD45). Samples were incubated with staining antibodies for 20 minutes at room temperature in the dark, diluted with 1 mL of FACS buffer, and pelleted at 500xg for 5 minutes. Stained pellets were resuspended in 200 μL of FACS buffer immediately prior to flow cytometry analysis (BD Accuri). Data was gated on FSC vs. SSC and FSC(height) vs. FSC(area) to exclude debris and doublets. Controls with no stain, obtained from IV-LPS-injured mice not receiving fluorescent nanoparticles, established gates for negative/positive staining with TopFluor PC liposomes, Alexa Fluor 647-or APC-labeled antibodies, or PerCP/Cy5.5-labeled antibodies. Single stain controls allowed automatic generation of compensation matrices in FCS Express software during final analysis of the data. Association of liposomes with cell types was identified by coincidence of green fluorescent signal with anti-CD45, anti-Ly6G, or anti-CD31 signal.
4.10. Brain doppler blood flow measurements of inflamed mice treated with liposomes
Mice were injected with LPS (2mg/kg, IV) 5 hours prior to measuring the blood flow, using a moorVMS-LSD laser doppler perfusion system. Anesthetized mice (2% isoflurane) were placed on a heating pad with a rectal probe to keep the temperature at 37+/−0.5 C. An incision was made between the ear and the eye, the masseter muscle was separated from the skull to find the middle cerebral artery (MCA). The doppler probe was fixed over the MCA and the blood flow monitored for 2 minutes prior to the injection of the liposomes (3 mg/kg). Blood flow was followed 30 minutes after the injection of liposomes. Isoflurane was reduced to 1% if the blood flow was reduced by a 50% of the baseline (which only happened in the mice that received liposomes without Factor I) to prevent animal death. Finally, mice were ethically euthanized and the blood collected for complete blood count (CBC).
4.11. Animal protocols
All animal studies were carried out in strict accordance with Guide for the Care and Use of Laboratory Animals as adopted by National Institute of Health and approved by University of Pennsylvania Institutional Animal Care and Use Committee (IACUC) Male C57BL/6J mice, 6-8 weeks old, were purchased from Jackson Laboratories. Mice were maintained at 22–26°C and on a 12/12 hour dark/light cycle with food and water ad libitum.
4.12. Statistics and software
All statistics were analyzed with GraphPad Prism, with the tests used indicated in the figure legends.
Supplementary Material
Acknowledgements
Funding for this work came from: NIH K08-HL-138269 (Brenner); NIH R01-HL-153510 (Brenner); NIH K99-HL-153696 (Glassman); AHA Career Development Award (Marcos Contreras); NIH R01-HL-157189,-HL-155106,-HL-157189 (Muzykantov); NIH R01-AI-146162,-AI-148797.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library as a separate file.
Contributor Information
Zhicheng Wang, Departments of Medicine and Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA.
Elizabeth D. Hood, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA.
Jia Nong, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA.
Jing Ding, Department of Pediatrics, Peking University People’s Hospital, Beijing, 100044, China.
Oscar A. Marcos-Contreras, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA
Patrick M. Glassman, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA
Kathryn M. Rubey, Department of Pediatrics, Children’s Hospital of Philadelphia, Philadelphia, PA, 19104 USA
Michael Zaleski, Departments of Medicine and Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA.
Carolann L. Espy, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA
Damodara Gullipali, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA.
Takashi Miwa, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA.
Vladimir R. Muzykantov, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA
Wen-Chao Song, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA.
Jacob W. Myerson, Department of Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA.
Jacob S. Brenner, Departments of Medicine and Pharmacology, University of Pennsylvania, Philadelphia, PA, 19104 USA.
References
- [1].Moghimi SM, Simberg D, Skotland T, Yaghmur A, Hunter AC, J. Pharmacol. Exp. Ther 2019, 370, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Moghimi SM, Simberg D, Papini E, Farhangrazi ZS, Adv. Drug Deliv. Rev 2020, DOI 10.1016/j.addr.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].La-Beck NM, Islam MR, Markiewski MM, Front. Immunol 2020, 11, 603039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nonaka M, Curr. Opin. Immunol 2001, 13, 69. [DOI] [PubMed] [Google Scholar]
- [5].Fam SY, Chee CF, Yong CY, Ho KL, Mariatulqabtiah AR, Tan WS, Nanomaterials (Basel) 2020, 10, DOI 10.3390/nano10040787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD, Immunol. Rev 2016, 274, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Nature Reviews Materials 2016, 1, 16014. [Google Scholar]
- [8].Szebeni J, Mol. Immunol 2014, 61, 163. [DOI] [PubMed] [Google Scholar]
- [9].Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW, J. Am. Chem. Soc 2012, 134, 2139. [DOI] [PubMed] [Google Scholar]
- [10].Chen F, Wang G, Griffin JI, Brenneman B, Banda NK, Holers VM, Backos DS, Wu L, Moghimi SM, Simberg D, Nat. Nanotechnol 2017, 12, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vu VP, Gifford GB, Chen F, Benasutti H, Wang G, Groman EV, Scheinman R, Saba L, Moghimi SM, Simberg D, Nat. Nanotechnol 2019, 14, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nilsson SC, Sim RB, Lea SM, Fremeaux-Bacchi V, Blom AM, Mol. Immunol 2011, 48, 1611. [DOI] [PubMed] [Google Scholar]
- [13].Lachmann PJ, Immunobiology 2019, 224, 511. [DOI] [PubMed] [Google Scholar]
- [14].Mészáros T, Csincsi ÁI, Uzonyi B, Hebecker M, Fülöp TG, Erdei A, Szebeni J, Józsi M, Nanomedicine 2016, 12, 1023. [DOI] [PubMed] [Google Scholar]
- [15].Gifford G, Vu VP, Banda NK, Holers VM, Wang G, Groman EV, Backos D, Scheinman R, Moghimi SM, Simberg D, Control J. Release 2019, 302, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Parente R, Clark SJ, Inforzato A, Day AJ, Cell. Mol. Life Sci 2017, 74, 1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Park JH, Jackman JA, Ferhan AR, Belling JN, Mokrzecka N, Weiss PS, Cho N-J, ACS Nano 2020, 14, 11950. [DOI] [PubMed] [Google Scholar]
- [18].Hood ED, Greineder CF, Shuvaeva T, Walsh L, Villa CH, Muzykantov VR, Bioconjug. Chem 2018, 29, 3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sax HC, Talamini MA, Hasselgren PO, Rosenblum L, Ogle CK, Fischer JE, J. Surg. Res 1988, 44, 109. [DOI] [PubMed] [Google Scholar]
- [20].Ward PA, Nat. Rev. Immunol 2004, 4, 133. [DOI] [PubMed] [Google Scholar]
- [21].Doerschuk CM, Microcirculation 2001, 8, 71. [PubMed] [Google Scholar]
- [22].Kuebler WM, Goetz AE, Eur. Surg. Res 2002, 34, 92. [DOI] [PubMed] [Google Scholar]
- [23].Myerson JW, Patel PN, Habibi N, Walsh LR, Lee Y-W, Luther DC, Ferguson LT, Zaleski MH, Zamora ME, Marcos-Contreras OA, Glassman PM, Johnston I, Hood ED, Shuvaeva T, Gregory JV, Kiseleva RY, Nong J, Rubey KM, Greineder CF, Mitragotri S, Worthen GS, Rotello VM, Lahann J, Muzykantov VR, Brenner JS, bioRxiv 2020, 2020.04.15.037564. [Google Scholar]
- [24].Brain JD, Molina RM, DeCamp MM, Warner AE, Am. J. Physiol 1999, 276, L146. [DOI] [PubMed] [Google Scholar]
- [25].Schneberger D, Aharonson-Raz K, Singh B, Am. J. Physiol. Lung Cell. Mol. Physiol 2012, 302, L498. [DOI] [PubMed] [Google Scholar]
- [26].Gill SS, Suri SS, Janardhan KS, Caldwell S, Duke T, Singh B, Respir. Res 2008, 9, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yipp BG, Kim JH, Lima R, Zbytnuik LD, Petri B, Swanlund N, Ho M, Szeto VG, Tak T, Koenderman L, Pickkers P, Tool ATJ, Kuijpers TW, van den Berg TK, Looney MR, Krummel MF, Kubes P, Sci Immunol 2017, 2, DOI 10.1126/sciimmunol.aam8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Granton E, Kim JH, Podstawka J, Yipp BG, Trends Immunol 2018, 39, 890. [DOI] [PubMed] [Google Scholar]
- [29].Atochina EN, Balyasnikova IV, Danilov SM, Granger DN, Fisher AB, Muzykantov VR, Am. J. Physiol 1998, 275, L806. [DOI] [PubMed] [Google Scholar]
- [30].Ferrer MCC, Shuvaev VV, Zern BJ, Composto RJ, Muzykantov VR, Eckmann DM, PLoS One 2014, 9, e102329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brenner JS, Bhamidipati K, Glassman P, Ramakrishnan N, Jiang D, Paris AJ, Myerson JW, Pan DC, Shuvaev VV, Villa C, Hood ED, Kiseleva R, Greineder CF, Radhakrishnan R, Muzykantov VR, Nanomedicine 2017, DOI 10.1016/j.nano.2016.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fülöp T, Nemes R, Mészáros T, Urbanics R, Kok RJ, Jackman JA, Cho N-J, Storm G, Szebeni J, Control J. Release 2018, 270, 268. [DOI] [PubMed] [Google Scholar]
- [33].Zamboni WC, Szebeni J, Kozlov SV, Lucas AT, Piscitelli JA, Dobrovolskaia MA, Adv. Drug Deliv. Rev 2018, 136-137, 82. [DOI] [PubMed] [Google Scholar]
- [34].Chiang T, Messing RO, Chou W-H, J. Vis. Exp 2011, DOI 10.3791/2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gabizon A, Shmeeda H, Barenholz Y, Clin. Pharmacokinet 2003, 42, 419. [DOI] [PubMed] [Google Scholar]
- [36].Ricklin D, Mastellos DC, Reis ES, Lambris JD, Nat. Rev. Nephrol 2018, 14, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kelley WJ, Fromen CA, Lopez-Cazares G, Eniola-Adefeso O, Acta Biomater 2018, 79, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.