

Abstract

A mild light-driven protocol for the direct alkylation of phenols is reported. The process is driven by the photochemical activity of a halogen-bonded complex formed upon complexation of the in situ generated electron-rich phenolate anion with the α-iodosulfone. The reaction proceeds rapidly (10 min) under microfluidic conditions, delivering a wide variety of ortho-alkylated products (27 examples, up to 97% yield, >20:1 regioselectivity, on a gram scale), including densely functionalized bioactive phenol derivatives
Phenols are highly relevant functional groups in synthetic and industrial chemistry. As a matter of fact, these aromatic systems are present in a wide number of biologically relevant compounds, such as tyramine, dopamine, paracetamol, among others (Figure 1a).1 Furthermore, phenol derivatives are key components of functional materials, including biopolymers, melanin,2 and lignin.3 Given their ubiquitous presence in different research areas, the development of methods allowing the chemical diversification of phenolic substrates, through a direct site-selective C–H functionalization process, is of great importance.5 Progress in this field has been spurred through the identification of transition-metal or organic catalysts capable to induce ortho (o)- or para (p)- selectivity in either electrophilic aromatic substitution (EArS) or cross-coupling reactions (Figure 1b).5,6 However, these methods are limited to tailored substituted phenol derivatives, while requiring high temperatures or potentially toxic catalysts. On the other hand, the classical homolytic aromatic substitution (HAS) provides a sustainable alternative method that enables the direct C–H functionalization of aromatic substrates through a radical pathway.
Figure 1.

(a) Biologically relevant phenols. (b) Reported approaches for the functionalization of phenols. (c) This work.
Nevertheless, the generation of open-shell intermediates engaging in HAS conventionally requires harsh reaction conditions, thus limiting a widespread use. Interestingly, the deprotonation of phenols 1 gives access to phenolate anions 2, which are photochemically active electron-rich intermediates that can be used to generate reactive open-shell species (Figure 1b).8
In fact, an alternative approach to the use of transition-metal catalysts is the direct excitation of the phenolate, which has been used toward the perfluoroalkylation of electron-poor derivatives.9 Nevertheless, also this approach requires prefunctionalized substrates,10 while leading to polyperfluoroalkylated products. We thus questioned if general phenolates 6 (R = H, EDG, or EWG) can be involved in the formation of photoactive halogen-bonded complexes with suitable partners.8,11 One important feature of this ground-state molecular aggregate is that, typically, its absorption profile exhibits a bathochromic shift.8,12 The excitation of the halogen-bonded complex can then occur at red-shifted wavelengths, triggering a single-electron-transfer (SET) event within the aggregate, while leading to reactive radical species.
Herein, we report a general microfluidic light-driven method for the C–H alkylation of phenols 4 using α-iodosulfones 5 (Figure 1c).13,14 The chemistry exploits the ability of the phenolate anion 6 to form a halogen-bonded complex with 5. Upon light irradiation of the resulting complex I, an electrophilic (phenylsulfonyl)alkyl radical is produced, which subsequently engages in a HAS process leading to the alkylated phenols 7. It is noteworthy that this unprecedented microfluidic halogen-complex-based strategy does not require the use of external photoredox or metal catalysts. Importantly, the implementation of this photochemical process in flow allows very short reaction times (10 min) with a high productivity rate (up to 0.6 mmol·h–1) and good to excellent o-selectivity (up to >20:1). Finally, the sulfonyl group removal under simple reductive treatment led access to important methylated bioactive phenol derivatives, extremely challenging to obtain under conventional synthetic methods.
Our exploratory studies began by testing the feasibility of a halogen-bonded association between the phenolate 6a and the α-iodosulfone 5a (Figure 2).
Figure 2.

Optical absorption spectra acquired in acetonitrile in 1 mm path quartz cuvettes: [4a] = 1.5 M; [5a] = 0.5 M; [DBU] = 1.5 M. DBU: 1,8-diazabiciclo[5.4.0]undec-7-ene.
The optical absorption spectra showed a clear, red-shifted charge-transfer (CT) band, confirming the formation of a halogen-bonded complex and the viability of our approach (blue line in Figure 2). Interestingly, the isolated chemical species (4a, 5a, and 6a) showed a negligible absorption at λ > 370 nm.
Capitalizing on these observations, we next evaluated the feasibility of the photochemical alkylation process between 4a and 5a, using a microfluidic photoreactor (MFP) equipped with a modular light source (370–456 nm, Table 1). The optimization process was directly performed under microfluidic conditions, since this provides a more efficient irradiation of the reaction mixture, allowing an easy scaling-up, and shorter reaction times that reduces the generation of overalkylation products.15 Specifically, the use of DBU as base, a Kessil lamp at 405 nm, and setting a residence time (tR) of 10 min led the formation of the alkylated product 7a as a mixture of the ortho- (o-) and ortho,ortho′- (o,o′-) adducts in a 3.1:1 ratio with a total yield of 10% (entry 1, Table 1). Changing the wavelength to 370 nm, where the absorption of the halogen-bonded complex is higher (blue line in Figure 2), resulted in 26% yield and a 3.3:1 ratio (entry 2, Table 1). Interestingly, increasing the initial concentration to 0.5 M, thus favoring the halogen-bonded complex formation, resulted in further improvements with a yield of 33% (entry 3, Table 1). Finally, using 1,1,3,3-tetramethylguanidine (TMG) as base,16 we obtained product 7a in a promising 10:3:1 ratio for the o:o,o′:p adducts with an overall yield of 63% (entry 4, Table 1).17 It is worth noting that the observed regioselectivity value is highly promising, also in light of the radical nature of this transformation.7,9
Table 1. Optimization of the Reaction Conditions and Control Experiments (Selected Results)a.

| entry | light source (nm) | [4a]0 (M) | base | yield (%)b | o:o,o′:p ratiob |
|---|---|---|---|---|---|
| 1 | 405 | 0.25 | DBU | 10 | 3.1:1:n.d. |
| 2 | 370 | 0.25 | DBU | 26 | 3.3:1:n.d. |
| 3 | 370 | 0.5 | DBU | 33 | 3.3:1:n.d. |
| 4 | 370 | 0.5 | TMG | 63 | 10:3:1 |
| 5c | 370 | 0.5 | TMG | 47 | 10:3:1 |
| 6d | 370 | 0.5 | TMG | 43 | 10:3:1 |
| 7 | light off | 0.5 | TMG | 0 |
Reactions were performed at ambient temperature in a microfluidic photoreactor (internal diameter: 0.8 mm).
The yield and the regioisomeric ratio were determined by 1H NMR analysis, using trichloroethylene as internal standard.
2 equiv of TMG were used.
Reaction under batch-setup in 18h.
Additionally, all the products (o-7a, o,o′-7a, and p-7a) were easily separated by flash chromatography, thus increasing the synthetic utility of this photochemical process. Control experiments confirmed that an inferior amount of base and a normal batch setup highly reduce the productivity of the system (entries 5 and 6).18
In order to gain insights on the nature of the complex between the phenolate 6a and the α-iodosulfone 5a, we performed DFT calculations at the M06-2x/Def2TZVP level of theory including polarizable continuum solvation model (MeCN). As shown in Figure 3a, natural charge distribution and the related plot of electrostatic potential indicate that the electron density in 6a is mostly localized at the oxygen atom while a positive area is present at the iodine atom (σ-hole) in 5a. Upon searching for minima structures between 6a and 5a (see the SI), we found that the two most stable complexes are characterized by (i) a hydrogen bond between a methylene proton of the α-iodosulfone 5a and the oxygen of 6a (O···H = 1.93 Å) in addition to a π–π interaction between the aromatic rings of 5a and 6a (C···C = 3.46 Å, Figure 3b) and (ii) a halogen-bonding interaction involving the oxygen and the iodine atom of 5a and 6a (O···I = 2.60 Å, Figure 3b). Although both complexes display very similar binding energy (ΔE), −7.8 and −7.2 kcal mol–1, for (i) and (ii), respectively, the computed UV–vis absorption spectrum of the halogen-bonded complex better resembles the experimental one, indicating the possible activity of this type of complex under the reaction conditions (Figure 3b).19,20 This hypothesis is also corroborated by the experiments performed using α-iodosulfones lacking the aromatic ring (vide infraScheme 1).
Figure 3.
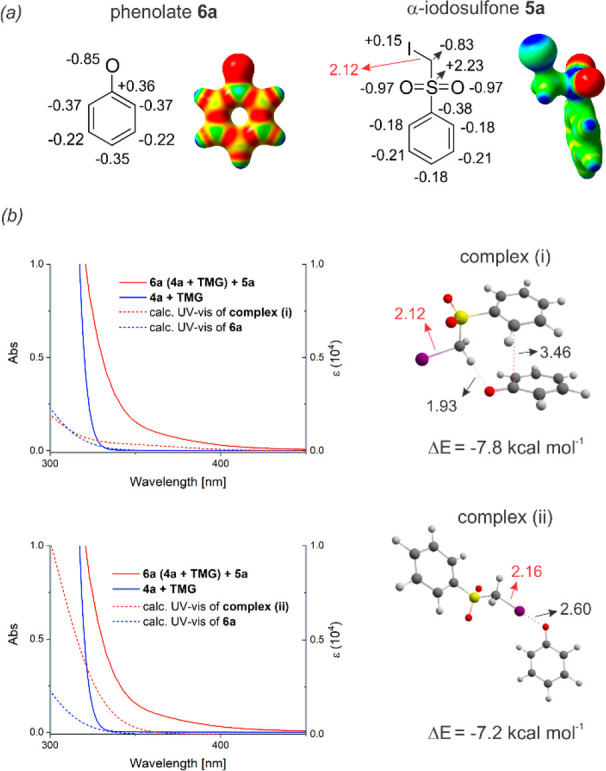
(a) Natural charge analysis and electrostatic potential (blue, positive potential; red, negative). (b) Absorption spectra for the reaction mixture (0.5 M) in the absence of 5a (blue solid line) and in the presence of 5a (red solid line). Dashed lines represent the theoretical absorption spectra computed at the M06-2X/Def2TZVP, SCRF = (IEFPCM, MeCN) level of theory of complexes (i) and (ii). ΔE represents the binding energies corrected with zero-point vibrational energy.
Scheme 1. Scope of Phenols 4 and α-Iodosulfones 5 That Can Participate in the Microfluidic Photoalkylation Process.

Reactions performed on a 1 mmol scale (see the SI for details). The reported yields refer to the total yield of the diverse regioisomers.
These substrates, lacking the phenyl ring, lose the ability to form relevant π–π interactions, while maintaining a strong ability in generating halogen-bonded complexes.21 In this regard, the calculations also indicate that the formation of a halogen-bonded adduct between 5a and 6a results in a sensible weakening of the C–I bond (C–I = 2.16 Å) of the α-iodosulfone. On the other hand, within complex (i) the length of this chemical bond remains virtually unvaried. This suggests that the halogen-bonding interaction is a key factor to favor the mesolytic cleavage of 5a and consequently the radical initiation step.22 We next performed quantum yield (QY) measurements to get information on the mechanism of this radical transformation.
For the reaction of phenol 4a and 5a, we determined a QY value of 2 mol of 7a per mole of photons, indicating the activity of a radical chain process.23 On these grounds, we propose the mechanism depicted in Figure 4. After the formation of the phenolate 6a, the halogen-bonding complex Ia is formed with the α-iodosulfone 5a. The direct excitation promotes a SET event within Ia, leading to the radicals IIa and IIIa along with the release of I–. The open-shell intermediate IIa is rapidly trapped in a reversible fashion by the ground state phenolate 6a, forming the radical anion Va. Inspection of the natural charge distribution of the phenolate 6a (Figure 3a) suggests that the sulfonyl radical IIa, behaving as a highly electrophilic species,24 should almost equally add to the electron-richer ortho and para positions of 6a. Thus, the high o-selectivity observed can be rather ascribed to the relative stability of the transient radical anion Va versus the para-adduct,25 which entails that the reaction is under thermodynamic control.26 In addition to this, the o-selectivity can also be rationalized by considering the statistically favored ortho attack (2:1 versus the para position). Considering the relatively low redox potential of the α-iodosulfone 5a [Ered (5a/5a•–) = −1.4 V vs SCE in MeCN)14 the propagation of the chain could occur by another SET event from Va to 5a, generating another molecule of the reactive radical intermediate IIa and IVa. By keto–enolic tautomerism of IVa the final alkylated phenol o-7a is formed. The chain process is terminated by a radical–radical coupling between the intermediates IIIa and IIa, ultimately leading to the formation of a molecule of product o-7a. Also, the product o-7a can potentially take part in the alkylation process, leading to the formation of bis-alkylated products.
Figure 4.

Proposed mechanism of the photoalkylation of phenols 4 with the α-iodosulfone 5a.
With a clear mechanistic picture of the developed light-driven process, we evaluated the generality of the microfluidic photochemical alkylation process for a variety of structurally diverse phenols (Scheme 1). Phenols bearing different types of m-substituents were competent substrates, delivering the alkylated products 7b–d in yields up to 77% and selectivity up to 9:1:1 for o:p:o,p. In addition, the p-substitution was well tolerated, accessing 7e–g, bearing a t-butyl, cyano, and boronic acid pinacol ester moiety, in yields up to 67%, and up to >20:1 regioselectivity. When using phenols bearing EWGs, the corresponding complexes I showed a strong, red-shifted absorption (see the SI), thus allowing the use of a visible-light source (465 nm).8 Disubstituted precursors, derived from natural lignin biopolymer, effectively participated in the developed light-driven alkylation, furnishing the alkylated products 7h–j in up to 97% yield and >20:1 regioselectivity. Remarkably, the o-substituted products 7k and 7l, deriving from biologically relevant salicylates, were obtained in excellent yield (up to 93%) and regioselectivity. Furthermore, other bioactive electron-rich phenols, such as tyramine, tyrosine, and paracetamol were also successfully alkylated (up to 94% yield and 7:1 o:o,o′), thus demonstrating the synthetic potential of this method for the late-stage alkylation of bioactive phenols.
Importantly, the reactions giving products 7a, 7i, and 7l were easily scaled up to 10 mmol, with a high productivity (up to 0.6 mmol·h–1) and delivering up to 3.38 g of the alkylated product. We next assessed the scope of the α-iodosulfone. As shown in Scheme 1, various derivatives efficiently reacted with up to 85% yield and >20:1 regioselectivity (7p–r and 7v–y). Noteworthy, also secondary carbons were readily installed onto the phenol scaffolds (7s and 7z, up to 30% yield and >20:1 regioselectivity). In addition, this photochemical method allows the use of α-iodoalkyl sulfones as radical precursors. Remarkably, products 7t–u and 7aa were isolated in up to 50% yield and excellent regioselectivity (up to >20:1). To further demonstrate the synthetic potential of the developed photochemical alkylation process, we performed a reductive desulfonylation of the alkylated products o-7a and o,o′-7a, which are easily separated by flash column chromatography after the microfluidic photochemical alkylation process. As shown in Figure 5a, we accessed the corresponding methylated targets 8a and 8b in high isolated yields.27 While classical methylation reactions often lead to complex mixture of regioisomers,4 this two-step methylation process allows the selective generation of the single methylated regioisomers (8a–d), without the need of HPLC separation. Likewise, the reductive desulfonylation of the phenol 7d, and the paracetamol derivative 7o, afforded the methylated products 8c and 8d in 75% and 81% yields, respectively (Figure 5b). It is worth noting that there are no other existing methodologies enabling the simple methylation of 4o to access the paracetamol derivative 8d.
Figure 5.

(a) Two-step methylation process of the unsubstituted phenol 4a. (b) Two step ortho-methylation process of the substituted phenol 4d and paracetamol 4n.
In conclusion, we have developed a new photochemical strategy that enables the microfluidic C–H alkylation of phenols without the use of any external photoredox or metal catalyst. The developed method relies on the formation of a photoactive halogen-bonded complex between the phenolate anion and the α-iodosulfone. The excitation of the halogen-bonded complexes enables the formation of reactive alkyl radicals, engaging in a HAS processes with the ground state phenolates. Remarkably, this halogen-complex-based strategy enables the alkylation of a wide variety of substrates, including biologically relevant tyrosine, paracetamol, and tyramine. Furthermore, the sulfonyl moiety can be easily removed upon reductive cleavage, establishing a new straightforward strategy to access elusive methylated phenols.
Acknowledgments
Davide Bettio is acknowledged for preliminary experiments. Tommaso Bortolato is acknowledged for technical support during the quantum yields measurements. Prof. Paolo Melchiorre is acknowledged for fruitful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c00604.
Experimental procedures, characterization data, and optical absorption, UV, and NMR spectra, CV, coordinates (PDF)
Author Contributions
# S.C. and C.R. contributed equally.
Author Contributions
All authors have given approval to the final version of the manuscript.
This work was supported by the University of Padova P-DiSC#11BIRD2020-UNIPD (L.D.), CariParo Foundation, Synergy – Progetti di Eccellenza 2018 (L.D.), and MIUR (PRIN Nanoredox, Prot. 2017PBXPN4, M.P., M.B.). G.F. kindly acknowledges FRA2021 funded by the University of Trieste and Microgrants 2021 founded by Region FVG (LR 2/2011, ART. 4). P.C. thanks the Seal of Excellence@UNIPD, QuantaCOF, for a postdoctoral fellowship. S.C. acknowledges the DIMED of UniPD for a postdoctoral fellowship.
The authors declare no competing financial interest.
Supplementary Material
References
- a Quideau S.; Deffieux D.; Douat-Casassus C.; Pouységu L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chem., Int. Ed. 2011, 50, 586–621. 10.1002/anie.201000044. [DOI] [PubMed] [Google Scholar]; b McGrath N. A.; Brichacek M.; Njardarson J. T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87, 1348–1349. 10.1021/ed1003806. [DOI] [Google Scholar]
- d’Ischia M.; Napolitano A.; Ball V.; Chen C.-T.; Buehler M. J. Polydopamine and Eumelanin: From Structure–Property Relationships to a Unified Tailoring Strategy. Acc. Chem. Res. 2014, 47, 3541–3550. 10.1021/ar500273y. [DOI] [PubMed] [Google Scholar]
- Davin L. B.; Jourdes M.; Patten A. M.; Kim K.-W.; Vassão D. G.; Lewis N. G. Dissection of Lignin Macromolecular Configuration and Assembly: Comparison to Related Biochemical Processes in Allyl/Propenyl Phenol and Lignan Biosynthesis. Nat. Prod. Rep. 2008, 25, 1015–1090. 10.1039/b510386j. [DOI] [PubMed] [Google Scholar]
- Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2003. [Google Scholar]
- Huang Z.; Lumb J. P. Phenol-Directed C-H Functionalization. ACS Catal. 2019, 9, 521–555. 10.1021/acscatal.8b04098. [DOI] [Google Scholar]
- a Youn S. W.; Cho C.-G. Transition-Metal-Catalyzed Ortho-Selective C–H Functionalization Reactions of Free Phenols. Org. Biomol. Chem. 2021, 19, 5028–5047. 10.1039/D1OB00506E. [DOI] [PubMed] [Google Scholar]; b Dai J.-L.; Shao N.-Q.; Zhang J.; Jia R.-P.; Wang D.-H. Cu (II)-Catalyzed ortho-Selective Aminomethylation of Phenols. J. Am. Chem. Soc. 2017, 139, 12390–12393. 10.1021/jacs.7b06785. [DOI] [PubMed] [Google Scholar]
- Bowman W. R.; Storey J. M. D. Synthesis Using Aromatic Homolytic Substitution—Recent Advances. Chem. Soc. Rev. 2007, 36, 1803–1822. 10.1039/b605183a. [DOI] [PubMed] [Google Scholar]
- a Bartolomei B.; Gentile G.; Rosso C.; Filippini G.; Prato M. Turning the Light on Phenols: New Opportunities in Organic Synthesis. Chem.—Eur. J. 2021, 27, 16062–16070. 10.1002/chem.202102276. [DOI] [PubMed] [Google Scholar]; b Liang K.; Li T.; Li N.; Zhang Y.; Shen L.; Ma Z.; Xia C. Redox-neutral photochemical Heck-type arylation of vinylphenols activated by visible light. Chem. Sci. 2020, 11, 2130–2135. 10.1039/C9SC06184C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini G.; Nappi M.; Melchiorre P. Photochemical Direct Perfluoroalkylation of Phenols. Tetrahedron 2015, 71, 4535–4542. 10.1016/j.tet.2015.02.034. [DOI] [Google Scholar]
- a Slama-Schwok A.; Blanchard-Desce M.; Lehn J.-M. Intramolecular charge transfer in donor-acceptor molecules. J. Phys. Chem. 1990, 94, 3894–3902. 10.1021/j100373a007. [DOI] [Google Scholar]; b Ahn M.; Kim M.-J.; Cho D. W.; Wee K.-R. Electron Push–Pull Effects on Intramolecular Charge Transfer in Perylene-Based Donor–Acceptor Compounds. J. Org. Chem. 2021, 86, 403–413. 10.1021/acs.joc.0c02149. [DOI] [PubMed] [Google Scholar]
- Guo Q.; Wang M.; Liu H.; Wang R.; Xu Z. Visible-Light-Promoted Dearomative Fluoroalkylation of β-Naphthols through Intermolecular Charge Transfer. Angew. Chem., Int. Ed. 2018, 57, 4747–4751. 10.1002/anie.201800767. [DOI] [PubMed] [Google Scholar]
- a Crisenza G. E. M.; Mazzarella D.; Melchiorre P. Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lima C. G. S.; Lima T. D. M.; Duarte M.; Jurberg I. D.; Paixão M. W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6, 1389–1407. 10.1021/acscatal.5b02386. [DOI] [Google Scholar]; c Yuan Y.; Majumder S.; Yang M.; Guo S. Recent Advances in Catalyst-Free Photochemical Reactions via Electron-Donor-Acceptor (EDA) Complex Process. Tetrahedron Lett. 2020, 61, 151506. 10.1016/j.tetlet.2019.151506. [DOI] [Google Scholar]; d Fu M.-C.; Shang R.; Zhao B.; Wang B.; Fu Y. Photocatalytic decarboxylative alkylations mediated by triphen ylphosphine and sodium iodide. Science 2019, 363, 1429–1434. 10.1126/science.aav3200. [DOI] [PubMed] [Google Scholar]
- a Dell’Amico L.; Mateos J.; Cuadros S.; Vega-Peñaloza A. Unlocking the Synthetic Potential of Light-Excited Aryl Ketones: Applications in Direct Photochemistry and Photoredox Catalysis. Synlett 2022, 33, 116. 10.1055/a-1403-4613. [DOI] [Google Scholar]; b Mateos J.; Vega-Peñaloza A.; Franceschi P.; Rigodanza F.; Andreetta P.; Companyó X.; Pelosi G.; Bonchio M.; Dell’Amico L. A Visible-Light Paternò–Büchi Dearomatisation Process towards the Construction of Oxeto-Indolinic Polycycles. Chem. Sci. 2020, 11, 6532–6538. 10.1039/D0SC01569E. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Franceschi P.; Mateos J.; Vega-Peñaloza A.; Dell’Amico L. Microfluidic Visible-Light Paternò–Büchi Reaction of Oxindole Enol Ethers. Eur. J. Org. Chem. 2020, 2020, 6718–6722. 10.1002/ejoc.202001057. [DOI] [Google Scholar]
- Filippini G.; Silvi M.; Melchiorre P. Enantioselective Formal α-Methylation and α-Benzylation of Aldehydes by Means of Photo-Organocatalysis. Angew. Chem., Int. Ed. 2017, 56, 4447–4451. 10.1002/anie.201612045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Buglioni L.; Raymenants F.; Slattery A.; Zondag S. D. A.; Noël T. Technological Innovations in Photochemistry for Organic Synthesis: Flow Chemistry, High-Throughput Experimentation, Scale-up, and Photoelectrochemistry. Chem. Rev. 2022, 122, 2752–2906. 10.1021/acs.chemrev.1c00332. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cambié D.; Bottecchia C.; Straathof N. J. W.; Hessel V.; Noël T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]; c Rehm T. H. Flow Photochemistry as aTool in Organic Synthesis. Chem.—Eur. J. 2020, 26, 16952–16974. 10.1002/chem.202000381. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Su Y.; Straathof N. J. W.; Hessel V.; Noël T. Photochemical Transformations Accelerated in Continuous-Flow Reactors: Basic Concepts and Applications. Chem.—Eur. J. 2014, 20, 10562–10589. 10.1002/chem.201400283. [DOI] [PubMed] [Google Scholar]; e Noël T. A Personal Perspective on the Future of Flow Photochemistry. J. Flow Chem. 2017, 7, 87–93. 10.1556/1846.2017.00022. [DOI] [Google Scholar]; f Plutschack M. B.; Pieber B.; Gilmore K.; Seeberger P. H. The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev. 2017, 117, 11796–11893. 10.1021/acs.chemrev.7b00183. [DOI] [PubMed] [Google Scholar]
- The ability of TMG to form halogen-bonded adducts with perfluoroalkyliodides has been documented, see:; Sladojevich F.; McNeill E.; Borgel J.; Zheng S.-L.; Ritter T. Condensed-Phase, Halogen-Bonded CF3I and C2F5I Adducts for Perfluoroalkylation Reactions. Angew. Chem., Int. Ed. 2015, 54, 3712–3716. 10.1002/anie.201410954. [DOI] [PubMed] [Google Scholar]; Our optical absorption studies revealed the possible formation of a complex between 5a and TMG (see section G.1 in the SI). However, in our reaction conditions, the main photoabsorbing species is the complex formed between the phenolate 6a and the iodosulfone 5a
- Dehalogenation of the starting material 5a is a competing pathway that is minimized under the optimal reaction conditions (Table 1, entry 4).
- When the reaction was performed under a classical batch setup, we observed an increased amount of the dehalogenated α-iodosulfone byproduct (50%). This experimental outcome indicated the key role of the microfludic setup in the developed synthetic process.
- During the preparation of this manuscript, it has been reported that thiophenolate anions can engage in photoabsorbing halogen-bonded complexes with iodobenzene via a halogen-bonding interaction. See:; Li T.; Liang K.; Tang J.; Ding Y.; Tong X.; Xia C. A photoexcited halogen-bonded complex of the thiophenolate anion with iodobenzene for C(sp3)–H activation and thiolation. Chem. Sci. 2021, 12, 15655. 10.1039/D1SC03667J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K.; Yamaguchi E.; Itoh A. Halogen-bonding-promoted Photo-induced C-X Borylation of Aryl Halide using Phenol Derivatives. ChemRxiv 2022, 10.26434/chemrxiv-2022-glvkg. [DOI] [PubMed] [Google Scholar]
- DFT calculations show significant binding energies upon complexation of 6a with alkyl α-iodosulfones (−7.3 and −7.1 kcal mol–1) via halogen bonding, while displaying a significant elongation of the C–I.
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The halogen bond. Chem. Rev. 2016, 116, 2478–1601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cismesia M. A.; Yoon T. P. Characterizing chain processes in visible light photoredox catalysis. Chem. Sci. 2015, 6, 5426–5434. 10.1039/C5SC02185E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Parsaee F.; Senarathna M. C.; Kannangara P. B.; Alexander S. N.; Damien P.; Arche E.; Welin E. R. Radical philicity and its role in selective organic transformations. Nat. Rev. Chem. 2021, 5, 486–499. 10.1038/s41570-021-00284-3. [DOI] [PubMed] [Google Scholar]; b Della E. W.; Graney S. D. The regiochemistry of cyclization of α-sulfenyl, α-sulfinyl, and α-sulfonyl-5-hexenyl radicals: procedures leading to regioselective syntheses of cyclic sulfones and sulfoxides. J. Org. Chem. 2004, 69, 3824–3835. 10.1021/jo0497306. [DOI] [PubMed] [Google Scholar]
- We performed preliminary DFT calculations at the (U)M06-2x/ Def2TZVP, SCRF = (IEFPCM, MeCN) level of theory for computing the energy gaining on the formation of intermediate Va (depicted in Figure 4) for both ortho and para pathways. We found that the radical anion intermediate in the ortho-position is more stable by 8.2 kcal mol–1 compared to that in para-position (see Figure S22 of the SI).
- Studer A.; Bossart M.. Homolytic Aromatic Substitutions. Radicals in Organic Synthesis; Wiley-VCH; 2001; p 62–80. [Google Scholar]
- Gui J.; Zhou Q.; Pan C.-M.; Yabe Y.; Burns A. C.; Collins M. R.; Ornelas M. A.; Ishihara Y.; Baran P. S. C–H Methylation of Heteroarenes Inspired by Radical SAM Methyl Transferase. J. Am. Chem. Soc. 2014, 136, 4853–4856. 10.1021/ja5007838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.