

Abstract
Objective
The subsarcolemmal accumulation of p62 aggregates in myofibres has been proposed to be characteristic of s poradic inclusion body myositis (sIBM). The objective of this study was to analyse the patterns and prevalence of p62 immunostaining and to quantitate p62 gene expression in muscle biopsies from a large number of patients with different types of myopathic and neurogenic disorders.
Methods
For the p62 immunostaining analysis, all patients with a muscle biopsy immunostained for p62 at the Johns Hopkins Neuromuscular Pathology Laboratory from 2013 to 2017 were included (n=303). The prevalence and pattern of p62 immunostaining were compared between patients with histologically normal muscle (n=29), inflammatory myopathies (n=136), non-inflammatory myopathies (n=53), and neurogenic disorders (n=85). p62 expression levels were analysed using an existing RNAseq dataset including data from dermatomyositis (DM; n=39), immune-mediated necrotising myopathy (IMNM; n=49), antisynthetase syndrome (AS; n=18), and sIBM (n=23) patients as well as 20 histologically normal muscle biopsies.
Results
p62 staining was absent in normal biopsies, but present in biopsies from those with polymyositis (29%), non-inflammatory myopathies (all <31%), neurogenic disorders (31%), dermatomyositis (57%), sIBM (92%) and IMNM (87%). In all diseases studied, p62 accumulation was more prevalent in biopsies with more severe muscle damage. sIBM biopsies had decreased p62 expression levels compared to the other groups (corrected 0.04).
Conclusion
p62 accumulation is a general response to muscle injury and not a specific marker for sIBM. Also, in sIBM, p62 RNA levels are decreased, suggesting that, in this disease, p62 aggregation is not due to overexpression.
Keywords: myositis, dermatomyositis, inclusion body myositis, polymyositis, immune-mediated necrotising myositis, p62, muscle biopsy
Introduction
The idiopathic inflammatory myopathies are a heterogeneous family of acquired diseases that include sporadic inclusion body myositis (sIBM), dermatomositis (DM), immune-mediated necrotising myopathy (IMNM), and polymyositis (PM) (1, 2). sIBM is a slowly progressive disease that has no known effective treatment (3). Indeed, corticosteroid administration may even accelerate the progression of disability in sIBM patients (4). In its early stages, sIBM can be misdiagnosed as another type of myositis, such as PM, which are responsive to immunosuppressant medication (1). Given the radically different management of patients with sIBM and other types of myositis, it is of great importance to accurately diagnose patients with sIBM.
Anti-NT5c1a autoantibodies (5), muscle MRI (6), and muscle [18]Florbetapir positron emission tomography (7) have been proposed to help diagnose patients with sIBM. In addition, muscle biopsy features, especially the presence of rimmed vacuoles, are used as diagnostic markers for sIBM (8). However, since not all muscle biopsies from sIBM patients include rimmed vacuoles (9), other sIBM-specific histologic features could potentially improve the diagnostic utility of muscle biopsy (10).
p62 is an autophagosome cargo protein that targets other proteins that bind to it for selective autophagy (11). The presence of large p62-positive subsarcolemmal aggregates has been proposed to be a marker that could help distinguish sIBM from other types of myopathy, including PM and DM (3, 8, 10, 12, 13). Moreover, it has been reported that p62 RNA expression levels are increased in sIBM (13). Nonetheless, p62 staining and expression levels have been studied only in relatively small numbers of muscle biopsies from patients with IBM, non-sIBM inflammatory myositis, or other myopathic and neurogenic disorders. Indeed, the largest study to date included biopsies from just 53 subjects, only 12 of whom had definite sIBM (8). In the current study, we have assessed the prevalence and distribution of p62 staining in a large number of muscle biopsies from patients with sIBM, other inflammatory myopathies, non-inflammatory myopathies, and neurogenic disorders. We also quantified p62 expression levels in muscle biopsies from patients with sIBM and other types of inflammatory myopathies using RNAseq.
Methods
Study populations
Two groups of patients were included. For the histological analysis, biopsies from all patients with a definitive clinical diagnosis who had their biopsy processed and immunostained for p62 at the Johns Hopkins Neuromuscular Pathology Laboratory from August 2013 to January 2017 were included.
For the RNAseq analysis, all muscle biopsies with a frozen sample available for RNA extraction from patients enrolled in the longitudinal cohorts of the National Institutes of Health (Bethesda), the Johns Hopkins Myositis Center (Baltimore) the Clinic Hospital (Barcelona), and the Vall d’Hebron Hospital (Barcelona) were included if the patients had one of the following myositis-specific autoantibodies (MSAs): anti-HMGCR, anti-SRP, anti-Jo1, anti-NXP2, anti-Mi2, anti-TIF1γ or anti-MDA5, as previously described (14, 15). The DM group included biopsies from patients with anti-Mi2 (n=11), anti-NXP2 (n=12), anti-TIF1γ (n=11), and anti-MDA5 (n=5) autoantibodies, the IMNM group included biopsies from patients with anti-HMGCR (n=40) or anti-SRP patients (n=9), and the AS group included muscle biopsies from 18 patients with anti-Jo1 autoanti-bodies. 13 patients with sIBM fulfilled both the Griggs and Lloyd-Greenberg criteria (14–17) were included as well as 20 normal muscle biopsies.
For the histologic analysis, patients were classified into one of four mutually exclusive clinical subgroups. Patients were classified as having IMNM if they met the 2003 European Neuromuscular Centre (ENMC) criteria (18) and sIBM if they fulfilled Griggs’ criteria (16), If none of these criteria were met, patients were evaluated for Bohan and Peter’s criteria and classified as possible, probable or definite DM or PM (19). The control group of non-myositis was classified as “neurogenic disorders” (including radiculopathies, polyneuropathies or post-polio neuropathy) or “other myopathies” (toxic myopathy, metabolic myopathy, myasthenic syndromes, hereditary myopathies, vasculitis, and amyloid myopathy). Finally, a muscle biopsy was considered “histo-logically normal” when it did not have any pathological findings. Patients with ongoing immunosuppressant treatment at the time of the biopsy were not excluded from the study.
Muscle acquisition and staining techniques
Open muscle biopsies were obtained from the vastus lateralis, deltoid, biceps or gastrocnemius muscles. The muscle tissue was routinely frozen in isopentane cooled on dry ice and stored at −80°C. Routine staining techniques included haematoxylin and eosin (H&E), modified Gomori trichrome, periodic acid-Schiff technique, Sudan black, Congo red, reduced nicotinamide adenine dinucleotide-tetrazolium reductase, combined cytochrome C oxidase and succinate dehydrogenase, adeno-sine triphosphatase 9.4, acid phosphatase, alkaline phosphatase, non-specific esterase, myophosphorylase, and myoadenylate deaminase. Also, longitudinal paraffin sections stained with H&E were evaluated.
During the study period from August 2013 to January 2017, immunostaining for p62 was routinely performed on all muscle biopsies processed at the Johns Hopkins Neuromuscular Pathology Laboratory. Eight μm acetone-fixed cryostat sections were stained for p62 using the streptavidin-biotin complex technique with diaminobenzidine as a colour indicator. The antibody used was a 1:100 diluted mouse monoclonal anti-p62/SQSTM1 (D-3, sc-28359 from Santa Cruz Biotechnology, Santa Cruz, CA).
Muscle biopsy evaluation
All biopsies from the Neuromuscular Pathology Laboratory were interpreted for clinical purposes by an experienced myopathologist (A.M.C.), who was not blinded to clinical information. The positivity, subcellular pattern, and location of p62 staining were determined by a myology specialist (J.C.M.) who was masked to disease activity and subgroup. Biopsies were evaluated for round, polygonal or angular atrophy, perifascicular atrophy, degeneration or necrosis, regeneration, ragged red fibres, COX-negative cells, glycogen deposits, increased of lipid, inflammatory infiltrate (partial invasion of non-necrotic muscle fibres, endomysial, perimysial, perivascular, granulomatous), rimmed vacuoles or non-rimmed vacuoles, endomysial fibrosis, protein aggregates, amyloid deposits, and fibre type grouping. p62/SQSTM1 immunostaining was classified as “negative” if all muscle fibres were negative, and positive if any fibres exhibited any of the six patterns described in Figure 1.
Fig. 1.
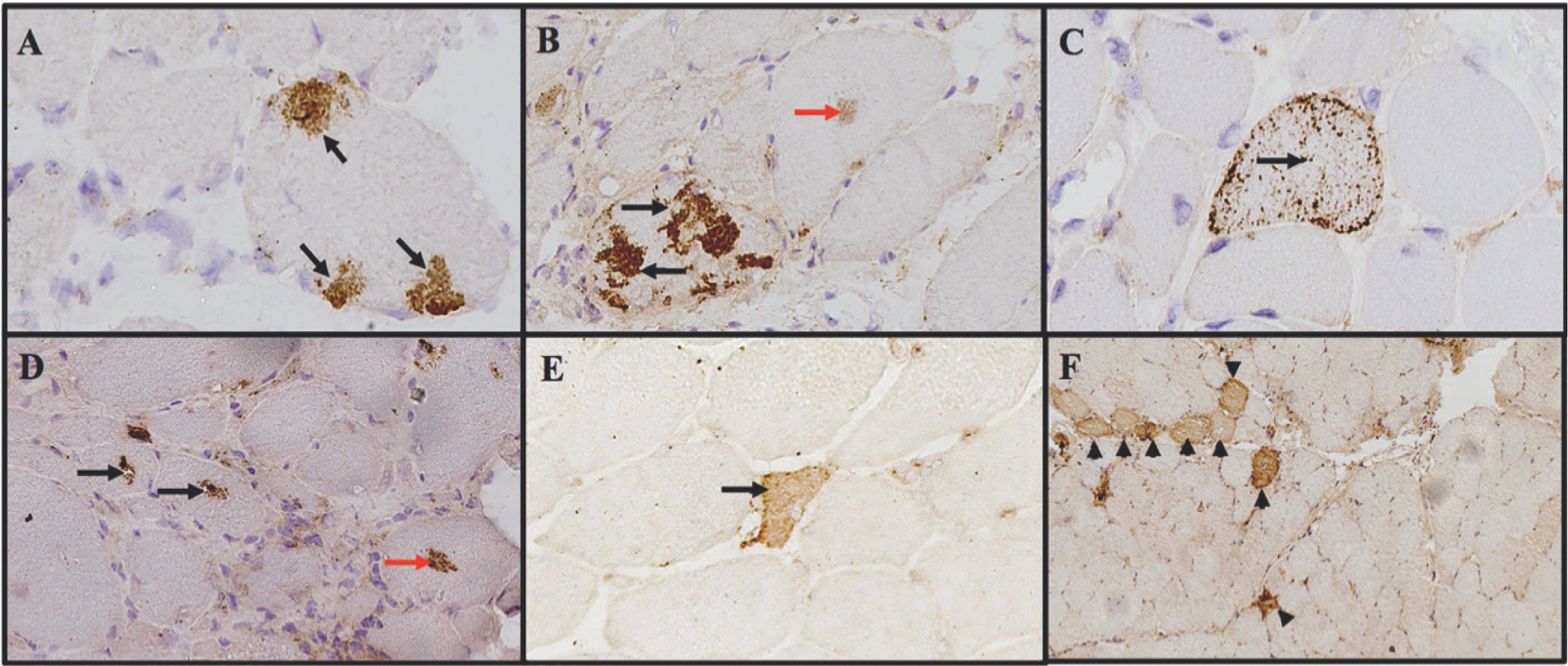
Examples of different patterns of p62 immunostaining in patients with myositis.
A) subsarcolemmal aggregates, B) sarcoplasmic aggregates, C) punctate pattern, D) perivacuolar pattern, E) necrotic fibre and F) perifascicular pattern. Red arrows indicated fine grouped aggregates.
Statistical analysis
Dichotomous variables were expressed as percentages, and absolute frequencies and groups were compared using a chi-squared test or Fisher’s exact test, as appropriate. Multivariate logistic regression analysis was used to compare these groups adjusting for the type of disease. The levels of CK among groups were compared using the Wilcoxon rank-sum test. Statistical analyses were performed using Stata/MP v. 14.1. A two-sided p-value of 0.05 or less was considered statistically significant with no correction for multiple comparisons.
RNA-sequencing
RNA-sequencing (RNA-seq) was performed as previously described (14, 15). Briefly, RNA was prepared using TRIzol. Libraries were made using the NeoPrepTM system according to the TruSeqM Stranded mRNA Library Prep protocol (Illumina) and sequenced using the Illumina HiSeq 2500 or 3000. Reads were aligned using the STAR v. 2.5 (20), the abundance of each gene was quantified using StringTie v. 1.3.3 (21), and the differential gene expression was performed using DESeq2 v. 1.20.0 (22). The Benjamini-Hochberg correction was used to adjust for multiple comparisons and a corrected p-value (q-value) of 0.05 or less was considered statistically significant.
Results
Patients and groups
Three hundred and three muscle biopsies that had p62 immunostaining performed were available for the study. From these, 45% (n=136) were from patients with inflammatory myopathies (37 sIBM, 31 IMNM, 40 DM, and 28 PM), 46% (n=138) were from disease comparators (85 had neurogenic disease and 53 had other types of myopathy), and 10% (n=29) were histologically normal (Supplementary Fig. S1). The average disease duration was longer in IBM (5.9 years [SD 4.3]) than in DM (1.4 years [SD 2.4] or IMNM (0.5 years [SD 0.2]). The difference between p62-positive and p62-negative biopsies was not significant in terms of disease duration at the time of the biopsy (p=0.9). The creatine kinase was higher in patients with a positive-p62 biopsy (p62+ median[Q1–Q3]: 1074 [473–3340] vs. p62-: 794 [166–1891], p=0.04).
Prevalence and pattern of p62 immunostaining
Six major types of p62 staining patterns were observed (Fig. 1). Within myofibres, large subsarcolemmal aggregates, large sarcoplasmic aggregates, small scattered punctate aggregates, and perivacuolar aggregates were identified as distinct patterns (Fig. 1). The presence of diffuse p62-positive staining was also noted in necrotic fibres (Fig. 1). Finally, diffuse p62-positive muscle cells were sometimes present in a perifascicular distribution (Fig. 1). The presence or absence of each p62 staining pattern was recorded for each muscle biopsy included in the study (Table I).
Table I.
Prevalence of p62 positivity and the pattern of staining among different clinical subgroups.
| Myositis | Comparators | Histologically normal (n=29) | |||||
|---|---|---|---|---|---|---|---|
| sIBM (n=37) | IMNM (n=31) | DM (n=40) | PM (n=28) | Neurogenic disorders (n=85) | Other myopathies (n=53) | ||
| p62 positive | 92% (34) | 87% (27) | 57% (23) | 29% (8) | 31% (26) | 26% (14) | 0% (0) |
| p62 subsarcolemmal aggregates | 76% (28) | 55% (17) | 28% (11) | 7% (2) | 14% (12) | 13% (7) | 0% (0) |
| p62 sarcoplasmic aggregates | 73% (27) | 68% (21) | 32% (13) | 14% (4) | 19% (16) | 15% (8) | 0% (0) |
| p62 punctate pattern | 51% (19) | 61% (19) | 40% (16) | 25% (7) | 14% (12) | 21% (11) | 0% (0) |
| p62 circumscribed punctuation | 3% (1) | 0% (0) | 0% (0) | 4% (1) | 4% (3) | 0% (0) | 0% (0) |
| p62 perivacuolar | 19% (7) | 0% (0) | 0% (0) | 0% (0) | 4% (3) | 6% (3) | 0% (0) |
| p62 necrotic cells | 3% (1) | 55% (17) | 2% (1) | 4% (1) | 2% (2) | 2% (1) | 0% (0) |
| p62 perifascicular | 0% (0) | 0% (0) | 32% (13) | 11% (3) | 0% (0) | 0% (0) | 0% (0) |
Overall, 62% of the inflammatory myopathy muscle biopsies had some type of positive staining for p62. This was most prevalent in muscle biopsies from those with sIBM (92%), IMNM (87%), and DM (57%). Staining for p62 was also present in PM (29%), neurogenic disorders (31%) and other types of myositis (26%).
Subsarcolemmal p62 aggregates, which have been proposed to be helpful in the diagnosis of sIBM, were present in 76% of sIBM cases. In addition, p62-positive subsarcolemmal aggregates were often present in IMNM (55%) and DM (28%) muscle biopsies. Perivacuolar p62 staining was very specific for sIBM, but only occurred in 19% of patients with this diagnosis. The presence of p62-positive necrotic fibres was common only in patients with IMNM (55%). Other patterns of p62 staining were varied and not specifically associated with a particular type of myositis (Table I).
Histopathological patterns associated with p62
To investigate whether p62 aggregation is specific for a type of disease process or is non-specifically associated with myofibre damage, we assessed the relationship between the presence of p62 aggregates with various types of pathological histological findings (Table II). Independent of the type of disease, p62 accumulation was more prevalent in biopsies with endomysial and perivascular inflammatory infiltrates, degeneration, regeneration, and/or rimmed vacuoles. Biopsies with a partial invasion of non-necrotic muscle fibres and in biopsies with abundant COX-negative fibres had a non-significant trend towards staining positive for p62.
Table II.
Prevalence of different pathological features in muscle biopsies according to the p62 status.
| p62 positive (n=132) | p62 negative (n=171) | univariate p-value | multivariate p-value | Total (n=303) | |
|---|---|---|---|---|---|
| Inflammatory infiltrate | 57% (75) | 23% (39) | <0.001 | 0.002 | 38% (114) |
| Endomysial inflammation | 41% (54) | 9% (16) | <0.001 | <0.001 | 23% (70) |
| Perimysial inflammation | 14% (19) | 10% (17) | 0.2 | 0.8 | 12% (36) |
| Perivascular inflammation | 31% (41) | 17% (29) | 0.004 | 0.04 | 23% (70) |
| Partial invasion of non-necrotic muscle fibres | 27% (36) | 5% (9) | <0.001 | 0.1 | 15% (45) |
| Degeneration | 83% (110) | 27% (46) | <0.001 | <0.001 | 51% (156) |
| Regeneration | 79% (104) | 22% (38) | <0.001 | <0.001 | 47% (142) |
| Perifascicular atrophy | 11% (15) | 7% (12) | 0.2 | 0.7 | 9% (27) |
| COX negative | 35% (46) | 15% (26) | <0.001 | 0.1 | 24% (72) |
| Rimmed vacuoles | 25% (33) | 1% (2) | <0.001 | 0.003 | 12% (35) |
Univariate analysis was performed using chi-square or Fisher’s exact test as appropriate.
Multivariate logistic regression analysis was adjusted for the type of disease.
p62 expression levels
The expression levels of p62 RNA were assessed in a separate collection of muscle biopsy specimens that underwent RNAseq analysis as previously described (14, 15). The expression of p62 was decreased in sIBM both compared to normal muscle tissue (log2FoldChange = −0.64, q-value=0.03) and to all the other types of myositis, including IMNM (log2FoldChange = −0.69, q-value=0.03), DM (log2FoldChange = −0.83, q-value=0.001), and AS (log2FoldChange = −0.83, q-value=0.01).
Discussion
It has been proposed that p62 immunohistochemical staining can help to differentiate sIBM from other types of inflammatory myopathies such as PM and DM (13). Indeed, p62 staining plays a prominent role, together with the presence of other pathologic features, in at least one proposed diagnostic algorithm for the histological diagnosis of sIBM (10). Nonetheless, the results of the current study emphasise that p62 staining is common not only in sIBM, but also in muscle biopsies from patients with IMNM and DM.
We have also demonstrated that independent of the type of muscle disease, p62-positive muscle biopsies have more inflammation, degeneration, and regeneration compared to p62-negative muscle biopsies. Moreover, p62 accumulation is common in the scattered necrotic fibres of IMNM and in the perifascicular myofibres of DM, which are the sites of injury in these two diseases. Taken together, our observations suggest that p62 accumulation is more likely to be a downstream consequence of muscle fibre injury rather than a distinctive pathological process that occurs exclusively in sIBM.
Interestingly, the RNA expression of p62 in muscle from patients with sIBM was decreased compared to normal tissue and other types of myositis. This observation suggests that the p62 immunohistochemistry staining in biopsies from sIBM patients reflects an accumulation of the p62 protein that is not due to an increased rate of synthesis. Of note, one prior study utilising qPCR with expression levels normalised to housekeeping genes reported that RNA expression of p62 was increased in sIBM (13). However, expression levels of these housekeeping genes could be altered in patients with sIBM. Unlike qPCR, RNAseq does not rely on using housekeeping as a reference, eliminating this potential bias. Additionally, RNAseq has shown to be highly accurate and reproducible for quantifying gene expression levels (23, 24).
Our study has several limitations. First, immunohistochemical and RNA expression studies were not performed in the same set of samples. However, given the large number of samples included in each section of the study, this is unlikely to have had a significant effect on the results. Also, the immunohistochemical and the RNA expression studies used different classificatory systems because some samples of the immunohistochemical analysis did not have paired sera to define the serological diagnosis. However, sIBM patients included in both the histological analysis and RNAseq analysis were both classified as such according to the same criteria. Moreover, the other myositis subsets utilised were appropriate to answer the relevant questions in each section. Finally, the p62 staining was read by a single researcher with no intrarater reliability evaluation.
In summary, even though p62 immunostaining may play a useful role (when taken along with other histologic features) in diagnosing sIBM, our results show that p62 aggregates are not a specific feature of sIBM. Rather, p62 accumulation is found in other inflammatory myopathies, other myopathic processes, and in neurogenic processes. Moreover, independent of the type of muscle disease, positive p62 staining is associated with the presence of various other histopathologic features, suggesting that p62 aggregation is a common downstream effect of muscle injury.
Supplementary Material
Key messages.
p62 accumulation is a general response to injury and not a specific marker for sIBM.
p62 levels are decreased in sIBM, suggesting that p62 aggregation is not due to overexpression.
Funding:
this work was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases and National Institute of Environmental Health Sciences and the Zhang fund.
Footnotes
Competing interests: none declared.
References
- 1.SELVA-O’CALLAGHAN A, PINAL-FERNANDEZ I, TRALLERO-ARAGUAS E, MILISENDA JC, GRAU-JUNYENT JM, MAMMEN AL: Classification and management of adult inflammatory myopathies. Lancet Neurol 2018; 17: 816–28. [DOI] [PubMed] [Google Scholar]
- 2.MARASCO E, CIOFFI E, COMETI L et al. : One year in review 2018: idiopathic inflammatory myopathies. Clin Exp Rheumatol 2018; 36: 937–47. [PubMed] [Google Scholar]
- 3.GREENBERG SA: Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol 2019; 15: 257–72. [DOI] [PubMed] [Google Scholar]
- 4.BENVENISTE O, GUIGUET M, FREEBODY J et al. : Long-term observational study of sporadic inclusion body myositis. Brain 2011; 134: 3176–84. [DOI] [PubMed] [Google Scholar]
- 5.LLOYD TE, CHRISTOPHER-STINE L, PINAL-FERNANDEZ I et al. : Cytosolic 5’-nucleotidase 1A as a target of circulating autoantibodies in autoimmune diseases. Arthritis Care Res (Hoboken) 2016; 68: 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.PINAL-FERNANDEZ I, CASAL-DOMINGUEZ M, CARRINO JA et al. : Thigh muscle MRI in immune-mediated necrotising myopathy: extensive oedema, early muscle damage and role of anti-SRP autoantibodies as a marker of severity. Ann Rheum Dis 2017; 76: 681–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LILLEKER JB, HODGSON R, ROBERTS M et al. : [18F]Florbetapir positron emission tomography: identification of muscle amyloid in inclusion body myositis and differentiation from polymyositis. Ann Rheum Dis 2019; 78: 657–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.HINIKER A, DANIELS BH, LEE HS, MARGETA M: Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Commun 2013; 1: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.CHAHIN N, ENGEL AG: Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 2008; 70: 418–24. [DOI] [PubMed] [Google Scholar]
- 10.BRADY S, SQUIER W, SEWRY C, HANNA M, HILTON-JONES D, HOLTON JL: A retrospective cohort study identifying the principal pathological features useful in the diagnosis of inclusion body myositis. BMJ Open 2014; 4: e004552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LIU WJ, YE L, HUANG WF et al. : p62 links the autophagy pathway and the ubiqutinproteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 2016; 21: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.NAKANO S, OKI M, KUSAKA H: The role of p62/SQSTM1 in sporadic inclusion body myositis. Neuromuscul Disord 2017; 27: 363–9. [DOI] [PubMed] [Google Scholar]
- 13.NOGALSKA A, TERRACCIANO C, D’AGOSTINO C, KING ENGEL W, ASKANAS V: p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol 2009; 118: 407–13. [DOI] [PubMed] [Google Scholar]
- 14.AMICI DR, PINAL-FERNANDEZ I, MAZALA DA et al. : Calcium dysregulation, functional calpainopathy, and endoplasmic reticulum stress in sporadic inclusion body myositis. Acta Neuropathol Commun 2017; 5: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.PINAL-FERNANDEZ I, AMICI DR, PARKS CA et al. : Myositis autoantigen expression correlates with muscle regeneration but not autoantibody specificity. Arthritis Rheumatol 2019; 71: 1371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.GRIGGS RC, ASKANAS V, DIMAURO S et al. : Inclusion body myositis and myopathies. Ann Neurol 1995; 38: 705–13. [DOI] [PubMed] [Google Scholar]
- 17.LLOYD TE, MAMMEN AL, AMATO AA, WEISS MD, NEEDHAM M, GREENBERG SA: Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology 2014; 83: 426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.HOOGENDIJK JE, AMATO AA, LECKY BR et al. : 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004; 14: 337–45. [DOI] [PubMed] [Google Scholar]
- 19.BOHAN A, PETER JB: Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975; 292: 403–7. [DOI] [PubMed] [Google Scholar]
- 20.DOBIN A, DAVIS CA, SCHLESINGER F et al. : STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.PERTEA M, PERTEA GM, ANTONESCU CM, CHANG TC, MENDELL JT, SALZBERG SL: StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 2015; 33: 290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LOVE MI, HUBER W, ANDERS S: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.NAGALAKSHMI U, WANG Z, WAERN K et al. : The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008; 320: 1344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.WANG Z, GERSTEIN M, SNYDER M: RNASeq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009; 10: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.