

Abstract
Identifying contexts in which cancer cells become addicted to specific nutrients is critical for developing targeted metabolic therapies. In this issue of Cancer Cell, Momcilovic et al. report that suppressed glycolysis following mTOR inhibition is countered by adaptive glutamine catabolism in lung squamous cell carcinoma, sensitizing tumors to glutaminase inhibition.
Lung squamous cell carcinoma (SCC), a subtype of non-small cell lung cancer (NSCLC), is responsible for 40,000–50,000 deaths per year in the US and has a 5-year survival rate of only 15%–20% (Liao et al., 2012). Druggable oncogenic lesions such as epidermal growth factor receptor (EGFR) mutations are rare in SCC, and treatment options are therefore normally limited to surgery, radiation therapy, cytotoxic chemotherapy, and, more recently, immunotherapy. Thus, there is a continued need to develop new strategies to treat SCC. In this issue of Cancer Cell, Momcilovic et al. (2018) describe a novel therapeutic approach aimed at targeting the metabolic requirements of SCC.
Metabolic reprogramming in cancer cells enables the opportunistic use of nutrients to satisfy the biosynthetic and redox requirements of cell proliferation. Although a small number of metabolic oncoproteins and tumor suppressors have been identified, in most cancers normal metabolic pathways become rewired as a consequence of oncogenic signaling or microenvironmental conditions such as hypoxia. The concept of selectively targeting cancer cell metabolism for therapy has been intensively explored in recent years, leading to rapid advances in our understanding of how metabolism can support or drive tumorigenesis. In 2017, the first-in-class inhibitor of mutant isocitrate dehydrogenase 2 (IDH2) was approved by the FDA for treatment of acute myeloid leukemia. Several other metabolism inhibitors, including the glutaminase (GLS)-selective molecule CB-839, are currently in clinical trials for cancer treatment. However, the inherent plasticity of cellular metabolism and the high degree of metabolic heterogeneity in human tumors pose great challenges for developing effective patient stratification and drug combination strategies.
By carrying out in vivo metabolic profiling, Momcilovic et al. set out to identify druggable metabolic nodes in SCC (Momcilovic et al., 2018). Using the KrasG12D;Lkb1−/− (KL) mouse model, which simultaneously develops lung SCC and adenocarcinoma (ADC) tumors, the authors first established that SCC tumors have a higher proliferative index than ADC in vivo. Correspondingly, uptake of 18F-fluoro-2-deoxyglucose (18F-FDG) and 11C-glutamine is highly elevated in SCC relative to ADC, as is expression of the uptake transporters for glucose and glutamine, GLUT1 and SLC1A5, respectively.
Previously, the same group found that SCC tumors escape chronic treatment with the mTOR kinase inhibitor MLN128 (Momcilovic et al., 2015). Although MLN128 potently inhibits mTORC1 signaling and glucose metabolism in SCC, the proliferative index of these tumors is unaffected. Momcilovic et al. therefore reasoned that an alternative carbon source must be fueling tumor growth. In the current study, by perfusing mice with stable isotope-labeled glucose or glutamine, they found that, whereas glucose catabolism is downregulated in SCC following MLN128 treatment, glutamine catabolism becomes upregulated. Thus, SCC tumors avidly consume both glucose and glutamine in vivo and rapidly adapt to suppression of glycolysis by further upregulating glutamine catabolism.
Through an elaborate series of signaling experiments, the authors then established the mechanism driving adaptive glutamine metabolism. Upon mTOR inhibition, a feedback signaling loop activates AKT, and AKT in turn phosphorylates and inhibits glycogen synthase kinase 3 (GSK3). This results in stabilization of the transcription factors c-Myc and c-Jun, positive regulators of GLS expression (Gao et al., 2009; Lukey et al., 2016), which are normally targeted for degradation by GSK3 (Figures 1A and 1B). Consistent with earlier reports that the effects of c-Myc on cancer metabolism are context dependent (Yuneva et al., 2012), the authors found that c-Jun is a conserved regulator of GLS in lung SCC, whereas the role of c-Myc varies between cell lines. Levels of inhibited (phospho)-GSK3 and activated c-Jun consistently delineated tumors that resisted mTOR inhibition via adaptive glutaminolysis.
Figure 1. Adaptive Glutamine Catabolism following mTOR Inhibition in Lung SCC.
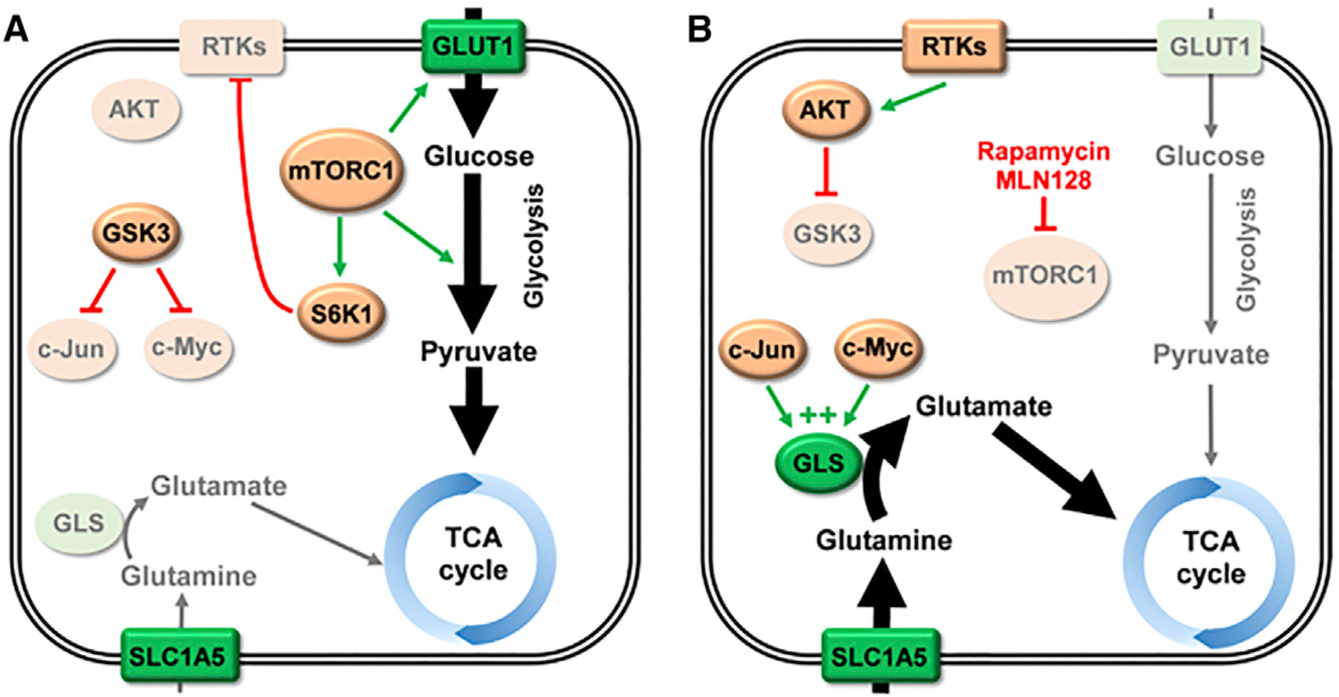
(A) In untreated lung squamous cell carcinoma (SCC), mTORC1 is often hyperactivated. One signaling axis downstream of mTORC1 leads to upregulated glucose uptake and glycolytic flux. A proportion of the glucose-derived carbon supplies the tricarboxylic acid (TCA) cycle. Another signaling axis activates S6K1, which mediates feedback inhibition of receptor tyrosine kinases (RTKs) such as the insulin receptor. Under these conditions, GSK3 is active and targets c-Myc and c-Jun for degradation, leading to decreased expression of glutaminase (GLS). (B) Treatment with the mTORC1 inhibitor rapamycin, or the mTOR kinase inhibitor MLN128, decreases glucose uptake, glycolytic enzyme expression, and supply of glucose-derived carbon to the TCA cycle. It also blocks activation of S6K1, and, consequently, feedback inhibition of RTKs is lost. Elevated RTK signaling activates AKT via phosphorylation of residue T308. Activated AKT then phosphorylates and inhibits GSK3, allowing c-Myc and c-Jun to accumulate. These transcription factors upregulate expression of GLS, increasing cellular glutaminase activity and allowing glutamine-derived carbon to supply the TCA cycle. Lighter shading indicates enzymes or pathways with suppressed expression/activity. Nutrient transporters and metabolic enzymes are shown in green. Abbreviations: GLS, glutaminase; GLUT1, glucose transporter 1; GSK3, glycogen synthase kinase 3; RTKs, receptor tyrosine kinases; TCA, tricarboxylic acid.
If SCC tumors adapt to mTOR inhibition (and suppression of glycolysis) by increasing glutamine uptake and catabolism, can resistance be overcome by combination therapy with the GLS inhibitor CB-839? Using cell lines and patient-derived xenografts, the authors show that SCC tumors are largely non-responsive to monotherapy with either MLN128 or CB-839 (Momcilovic et al., 2018). However, combination treatment almost completely abolished tumor growth. Moreover, when CB-839 was used as a second-line therapy to treat tumors progressing on MLN128 alone, rapid tumor necrosis and suppression of growth occurred. Finally, the authors identified a conserved signature in hypermetabolic cancers, including head and neck squamous cell carcinoma and osteosarcoma, which correlates with patient outcome and response to combined mTOR and GLS inhibition. This signature includes elevated phospho-GSK3S21/9 and phospho-c-JunS73, indicating that these could serve as biomarkers to stratify patients for metabolic therapy.
The findings in this study raise some interesting questions concerning the details of how adaptive glutamine catabolism rescues chronic mTOR inhibition in SCC. Inhibition of mTOR suppresses glycolysis and entry of glucose-derived carbon into the TCA cycle, but stimulates glutamine-mediated anaplerosis. It would be of interest to examine other glucose-supplied processes, such as the pentose phosphate and serine biosynthesis pathways, given the pronounced drop in 18F-FDG accumulation following MLN128 treatment. Another intriguing question is whether adaptive glutamine metabolism might drive resistance to therapies that indirectly impact mTOR signaling, or whether the resistance mechanism described here is uniquely dependent on the potent AKT feedback activation that follows direct mTORC1 inhibition.
The last 2 years have seen a number of studies shed light on the role of glutamine catabolism, and GLS in particular, in lung cancers. Using mouse models that develop lung ADC, Davidson et al. reported that tumors in vivo can be less dependent on GLS than cells cultured ex vivo (Davidson et al., 2016). This difference was attributed to abundant cystine in cell culture medium, which increases GLS dependence by driving glutamate efflux through the xCT cystine/glutamate antiporter (Muir et al., 2017). However, subsequent studies found that activation of the NRF2 antioxidant pathway, a frequent event in human lung cancers, is sufficient to upregulate xCT and cause dependence on GLS in tumors in vivo (Romero et al., 2017; Sayin et al., 2017). These studies illustrate that for tumors with high mutational burdens, such as those of the lung, individual mouse models harboring few somatic mutations may not perfectly mirror the human disease. Hypermutant mouse models for lung and colon cancers, currently under development, have the potential to address this issue.
The new work by Momcilovic et al. adds to a clear theme that has emerged from recent studies of GLS as a possible drug target. For inhibition of glutamine catabolism to be a successful strategy for cancer therapy, the precise contexts in which this metabolic pathway becomes essential need to be understood. Dependence on the glutaminase reaction may be driven by genetic lesions such as KEAP1 mutations, which lead to activation of the NRF2 antioxidant program (Romero et al., 2017; Sayin et al., 2017). Alternatively, as in the present study, dependence may be engineered through pharmacological interventions such as mTOR inhibition, with glutamine catabolism serving as a druggable resistance mechanism. By identifying the biological basis of addiction to glutaminolysis, the potential for using GLS inhibitors to block tumor growth in patients continues to increase.
REFERENCES
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, and Dang CV (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao RG, Watanabe H, Meyerson M, and Hammerman PS (2012). Targeted therapy for squamous cell lung cancer. Lung Cancer Manag. 1, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukey MJ, Greene KS, Erickson JW, Wilson KF, and Cerione RA (2016). The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat. Commun. 7, 11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momcilovic M, McMickle R, Abt E, Seki A, Simko SA, Magyar C, Stout DB, Fishbein MC, Walser TC, Dubinett SM, and Shackelford DB (2015). Heightening energetic stress selectively targets LKB1-deficient non-small cell lung cancers. Cancer Res. 75, 4910–4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, Graeber TG, Parlati F, Demo S, Li R, et al. (2018). The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell 33, this issue, 905–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, and Vander Heiden MG (2017). Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6, e27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sánchez-Rivera FJ, et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 23, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, Alvarez SW, Wu WL, Karakousi TR, Zavitsanou AM, et al. (2017). Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6, e28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva MO, Fan TWM, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Matés JM, Alonso FJ, Wang C, Seo Y, et al. (2012). The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 15, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]