

Abstract
Osteoclasts are the principal bone resorption cells crucial for homeostatic bone remodeling and pathological bone destruction. Increasing data demonstrate a vital role of histone methylation in osteoclastogenesis. As an integral core subunit of H3K4 methyltransferases, Dpy30 is notal as a key chromatin regulator for cell growth and differentiation and stem cell fate determination, particularly in the hematopoietic system. However, its role in osteoclastogenesis is currently unknown. Herein, we generated Dpy30F/F; LysM-Cre+/+ mice, which deletes Dpy30 in myeloid cells, to characterize its involvement in osteoclast differentiation and function. Dpy30F/F; LysM-Cre+/+ mice showed increased bone mass, evident by impaired osteoclastogenesis and defective osteoclast activity, but no alteration of osteoblast numbers and bone formation. Additionally, our ex vivo analysis showed that the loss of Dpy30 significantly impedes osteoclast differentiation and suppresses osteoclast-related gene expression. Moreover, Dpy30 deficiency significantly decreased the enrichment of H3K4me3 on the promoter region of NFATc1. Thus, we revealed a novel role for Dpy30 in osteoclastogenesis through epigenetic mechanisms, and that it could potentially be a therapeutic target for bone destruction diseases.
Keywords: Dpy30, osteoclast, H3K4 methylation, bone resorption
1. Introduction
Osteoclasts derived from monocytic precursors of the hematopoietic cell lineage, are the sole bone resorption cells indispensable for mineral homeostasis and bone remodeling and repair (1,2). Osteoclast growth and differentiation requires macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB ligand (RANKL) (3). The formation of osteoclasts is initiated by a cascade of RANKL-induced signaling events, which results in the activation of the master transcription factor, nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) (4). Then it stimulates the expression of a series of osteoclast-specific genes such as cathepsin K (Ctsk) (5). Dysregulation of osteoclasts, such as abnormal formation or activity, can lead to numerous bone destruction diseases, from osteoporosis, rheumatoid arthritis to periodontitis (6,7). Thus, understanding the molecular mechanisms underlying osteoclastogenesis will reveal novel targets for the treatment and prevention of bone disorders.
The differentiation and function of osteoclasts are tightly controlled by gene transcription and greatly affected by the overall and local chromatin state. Although the signal pathway induced by RANKL in osteoclast differentiation is well known, increasing data demonstrate that epigenetics is also an important factor regulating gene expression in bone remodeling (8,9). In particular, histone modifications are believed to be key to controlling expression of osteoclast-related genes (10–14). H3K4 methylation, one of the predominant histone modifications, typically activates gene expression (15). Several studies suggest the influence of H3K4 methylation regulators on RANKL-induced osteoclastogenesis. Following treatment with RANKL, bone marrow macrophages (BMMs) differentiate into osteoclasts and undergo dynamic changes of chromatin structure from H3K4me3/H3K27me3 to H3K4me3, in the promoter region of several important transcription factors (13). Deletion of Pax interaction with transcription-activation domain protein-1 (PTIP), a subunit of the H3K4 methylation methyltransferases complex, reduces the enrichment of H3K4me3 signal on the promoter of peroxisome proliferator-activated receptor (PPARγ), an essential transcription factor during osteoclastogenesis (16). Although changes in H3K4 methylation during osteoclastogenesis have been noted, how the histone-modifying enzyme of H3K4 methylation regulates osteoclast differentiation remains unclear. The Set1/Mll complex is the primary histone H3K4 methylation enzyme in mammals. Their full methylation activity is determined by the relationship between the catalytic subunits and their integral common core subunits Wdr5, Rbbp5, Ash2l and Dpy30 (17,18). Dpy30 promotes H3K4 methylation at all three levels, and its optimal action is on H3K4me3 at the transcription start site (19). Although Dpy30 showed negligible contribution to the activity of the Set1/Mll complex in reconstituted systems (20), it is capable of profoundly regulating genome-wide H3K4 methylation in cells (21,22). An unbiased biochemical approach reveals components of the Set1/Mll complex to be the main proteins related to Dpy30 (23). These results allow genetic manipulation of Dpy30 to study the role of effective H3K4 methylation in numerous biological processes. In addition, Dpy30-dependent H3K4 methylation has a critical role in different processes including mammalian embryonic development (24), embryonic stem cell (ESC) differentiation (21), hematopoiesis and hematopoietic stem cell (HSC)/hematopoietic progenitor cell (HPC) differentiation (22,25,26), neural progenitor cells differentiation (27), as well as lymphomagenesis (28). These observations raise the possibility that Dpy30 also regulates osteoclast differentiation. However, the function of Dpy30 in osteoclast biology remains undefined.
In this current work, in vitro and in vivo approaches were used to ascertain the involvement of Dpy30 and H3K4 methylation in the differentiation and function of osteoclasts. The work has not only uncovered a decisive role for Dpy30 in RANKL-induced osteoclast biology, but also elucidated the epigenetic mechanisms of Dpy30 in regulating osteoclastogenesis.
2. Materials and methods
2.1. Generation of Dpy30 conditional knockout mice
LysM-Cre mice (C57BL/6 background, JAX no. 004781, The Jackson Laboratory) have been shown to be reliable in gene deletion in myeloid lineage cells and osteoclast precursors (29,30). To conditionally delete the Dpy30 gene specific to the myeloid lineage and osteoclast precursors, Dpy30F/F mice (C57BL/6 background), produced as previously described (22), were bred with LysM-Cre mice to obtain Dpy30F/F; LysM-Cre+/+ mice(Dpy30CKO). In all experiments, Dpy30CKO mice were compared with Dpy30F/F mice (Ctrl). All animal protocols were approved by the UAB Institutional Animal Care and Use Committee.
2.2. Microcomputed tomography (μCT) analysis
Dissected mouse femurs were scanned with a μCT40 scanner as described previously (Scanco Medical AG, Brüttisellen, Switzerland) (31). For trabecular bone analysis, the scan started from the growth plate and comprised 315 slices, and 200 slices in the region of interest were analyzed. For cortical bone analysis, the scan was performed at the femoral midshaft and comprised 25 slices (31). The 3-D reconstruction was performed (μCT Ray v3.8) and microarchitectural parameters were calculated with the CT Evaluation Program (v.6.5-2, Scanco Medical).
2.3. Histomorphometric analysis
All isolated femurs of 12-week-old mice were fixed in 70% ethanol and embedded in polymethylmethacrylate. For static histomorphometric analysis, 5 μm thickness sections were stained with Goldner’s Trichrome (32). For dynamic histomorphometric analysis, mice were intraperitoneally given calcein (20 mg/kg) and Alizarin Red S (40 mg/kg) 7 days and 2 days before euthanasia (32). Ten μm thickness sections were obtained and fluorescence-labeled images were taken by an Olympus BX51 microscope. All histomorphometric parameters were measured by the Bioquant Osteo (Nashville, TN, US).
For TRAP staining, after fixation in 4% PFA and decalcification in 10% EDTA, 5 μm thickness paraffin sections of dissected femurs were stained by a Leukocyte Acid Phosphatase kit (387A-1KT, Sigma) (33). Pictures were taken by a fluorescence microscope (BZ-X800, KEYENCE, Itasca, IL, USA) and osteoclast parameter was quantified.
2.4. Flow cytometry analysis
Single-cell suspensions from bone marrow were obtained as previously described (33). Then the cells were stained with CD11b-FITC, CD115 (c-fms)-APC, RANK-PE and Ly-6C-PE-Cyanine7 (eBioscience, Waltham, MA, USA) at 4 °C for 30 mins. FACS was done on the LSR II or Symphony flow cytometer (BD Biosciences), and then FlowJo (FlowJo, LLC, Ashland, OR) was used for data analysis.
2.5. Serum CTX assay
Serum samples were obtained from 12-week-old mice after fasting for 6-hours. The concentration of carboxy-terminal collagen cross-links (CTX) in the serum was measured using RatLaps™ (CTX-I) EIA (ImmunoDiagnostic Systems, Boldon Business Park, UK).
2.6. In vitro osteoclast differentiation assay
Murine bone marrow cells were obtained from long bones of 12-week-old Ctrl or Dpy30CKO mice. They were cultured overnight in tissue culture dishes containing α-MEM supplemented with 5% M-CSF. Non-attached cells were obtained and further cultured in 5% M-CSF for three days. After removing floating cells, the attached cells were collected as BMMs (12,34). Then they were induced into osteoclasts for indicated periods of time under the treatment of 5% M-CSF and different concentrations of RANKL (100 ng/ml or 10 ng/ml). To evaluate osteoclast differentiation, TRAP activity was stained using a Leukocyte Acid Phosphatase kit (Sigma), and osteoclasts were counted as TRAP-positive multinucleated cells (TRAP+ MNCs, more than 3 nuclei) (33). To assess the F-actin ring formation, cells were stained by Rhodamine Phalloidin (Invitrogen, Waltham, MA, USA) after fixation as previously described (33). For bone resorption assay, osteoclast differentiation was induced in bovine cortical bone slices as previously described (33). The bone resorption pits were stained with wheat germ agglutinin (WGA) (Sigma) and 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA, USA) and analyzed by Image J software (NIH) (33).
2.7. Quantitative PCR (qPCR) analysis
Total RNA was extracted using miRNeasy Mini (Qiagen, Germantown, MD, USA). cDNA was reverse transcribed using a SuperScript™ III First-Strand Synthesis System (Invitrogen) as described previously (33). qPCR was performed on a Real-Time PCR System (Roche, Basel, Switzerland) using TB Green Advantage qPCR Premix (Takara Bio, Mountain View, CA, USA). Primers are listed in Supplementary Table 1. Gene expressions were normalized to β-actin.
2.8. Western Blot analysis
Cells were lysed in lysis buffer (1% SDS, 10 mM EDTA, and 50 mM Tris-Cl, pH 8.0) containing freshly added protease inhibitor cocktail (MilliporeSigma, Burlington, MA, USA) (22). Proteins were separated on SDS-PAGE gel, electro-transferred to PVDF membranes, and then incubated with the following primary antibodies: NFATc1, Ctsk (Both from Santa Cruz Biotechnology); Dpy30 (Bethyl Laboratories), H3, H3K4me1, H3K4me2, H3K4me3 (All from MilliporeSigma); Erk, phospho-Erk (Thr202/Tyr204), p38, phospho-p38 (Thr180/Tyr182), JNK, phospho-JNK (Thr183/Tyr185), p65, phospho-p65 and β-actin (All from Cell Signaling Technology). After incubation with IRDye 800CW secondary antibodies (LICOR, Lincoln, NE, USA), the membranes were visualized using the Odyssey Imaging System (LICOR).
2.9. Chromatin immunoprecipitation (ChIP) and qPCR
Cells were cross-linked by treatment with 1% formaldehyde, and then terminated with 0.125 M glycine (35). After scraping off the plates in PBS containing protease inhibitors cocktail, the cells were centrifuged (7000 rpm, 10 minutes) and resuspended in lysis buffer as aforementioned for 10 minutes on ice. Cells were aliquoted at 1×106 cells/mL, and the chromatin was sheared to obtain 100–500bp DNA fragments using Bioruptor (Diagenode, Denville, NJ, USA). After centrifuging, two percent of the cell supernatants were used as input controls. Immunoprecipitation was performed with H3K4me3 or negative control IgG antibody using an Enzymatic Chromatin Immunoprecipitation kit (EZ ChIP™, MilliporeSigama). After elution, samples and inputs DNAs were de-crosslinked and purified using the phenol-chloroform extraction method. Then they were detected by qPCR using the primers on the promoter region of NFATc1 listed in the Supplementary Table 1.
2.10. Statistical analysis
Statistical difference was evaluated by Student’s t-test or one-way ANOVA analysis using GraphPad Prism 8 (San Diego, CA, USA). Data are expressed as mean ± SD. The results are representative of three independent experiments. P value less than 0.05 was considered significant.
3. Results
3.1. Expression of Dpy30 during RANKL-induced osteoclast differentiation
To determine the role of Dpy30 in osteoclast differentiation and function, we first evaluated Dpy30 expression pattern during RANKL-induced osteoclast differentiation in vitro. The gene expression of Dpy30 mainly increased on day 1, and then decreased as the osteoclast differentiation proceeded (Fig. 1A). And the protein level of Dpy30 gradually increased in this process (Fig. 1B). Meanwhile, the gene expression of osteoclast-specific gene Ctsk significantly increased with the progress of osteoclast differentiation, which confirmed the normal osteoclast differentiation (Fig. 1A). Furthermore, the global levels of H3K4me3, H3K4me2 and H3k4me1 were increased during osteoclast differentiation as detected by western blot (Fig. 1B). These results indicate the possible involvement of Dpy30, as well as its related H3K4 methylation, in RANKL-induced osteoclast differentiation.
Fig. 1.
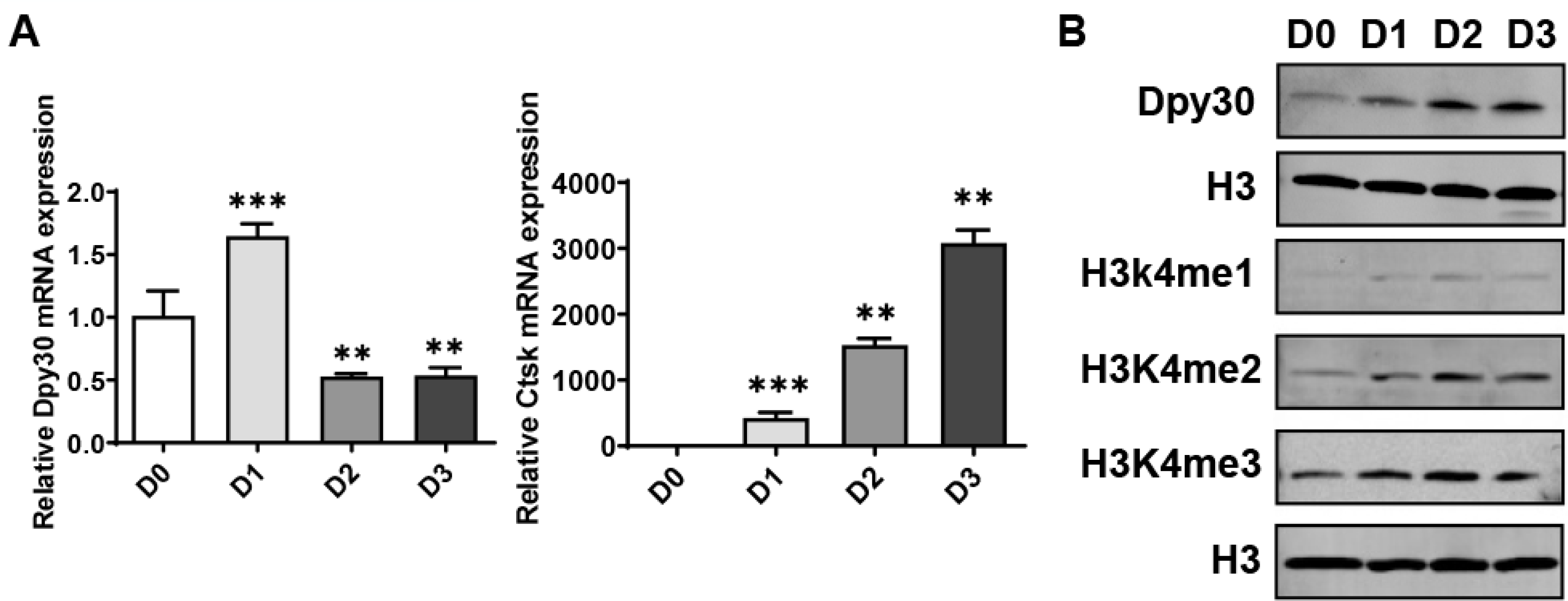
Dpy30 expression during RANKL-induced osteoclastogenesis. 12-week-old wild type bone marrow macrophages (BMMs) were exposed to RANKL (100 ng/mL) for 3 days. (A) Dpy30 and Ctsk gene expression determined by qPCR (n=3). Data are mean ± SD.*P < 0.05; **P < 0.01 relative to D0. (B) Western blotting for Dpy30, H3, H3K4me1, H3Kme2 and H4K4me3.
3.2. Dpy30 deficiency in myeloid cells results in increased bone mass
To further study the importance of Dpy30 in osteoclast development, we generated Dpy30CKO mice and assessed the bone phenotype of Dpy30CKO mice at 12-weeks and 22-weeks old using μCT analysis. The images showed an osteopetrotic phenotype at 12-weeks old age in Dpy30CKO mice, indicated by a more microstructurally disconnected trabecular bone network, compared with Ctrl mice (Fig. 2A). Moreover, there was a significant higher trabecular bone density, as confirmed by the significant augmentation in bone volume, trabecular number, and bone mineral density, while an according reduction in trabecular separation in the Dpy30CKO mice in comparison to Ctrl mice (Fig. 2B). Furthermore, a marked elevation of bone mass was seen at 22-weeks old Dpy30CKO mice (Fig. 2C), represented by a significant augmentation in trabecular bone density, and even a significantly higher cortical bone density in comparison to Ctrl mice (Fig. 2D). This data indicates that Dpy30 controls bone mass in vivo.
Fig. 2.
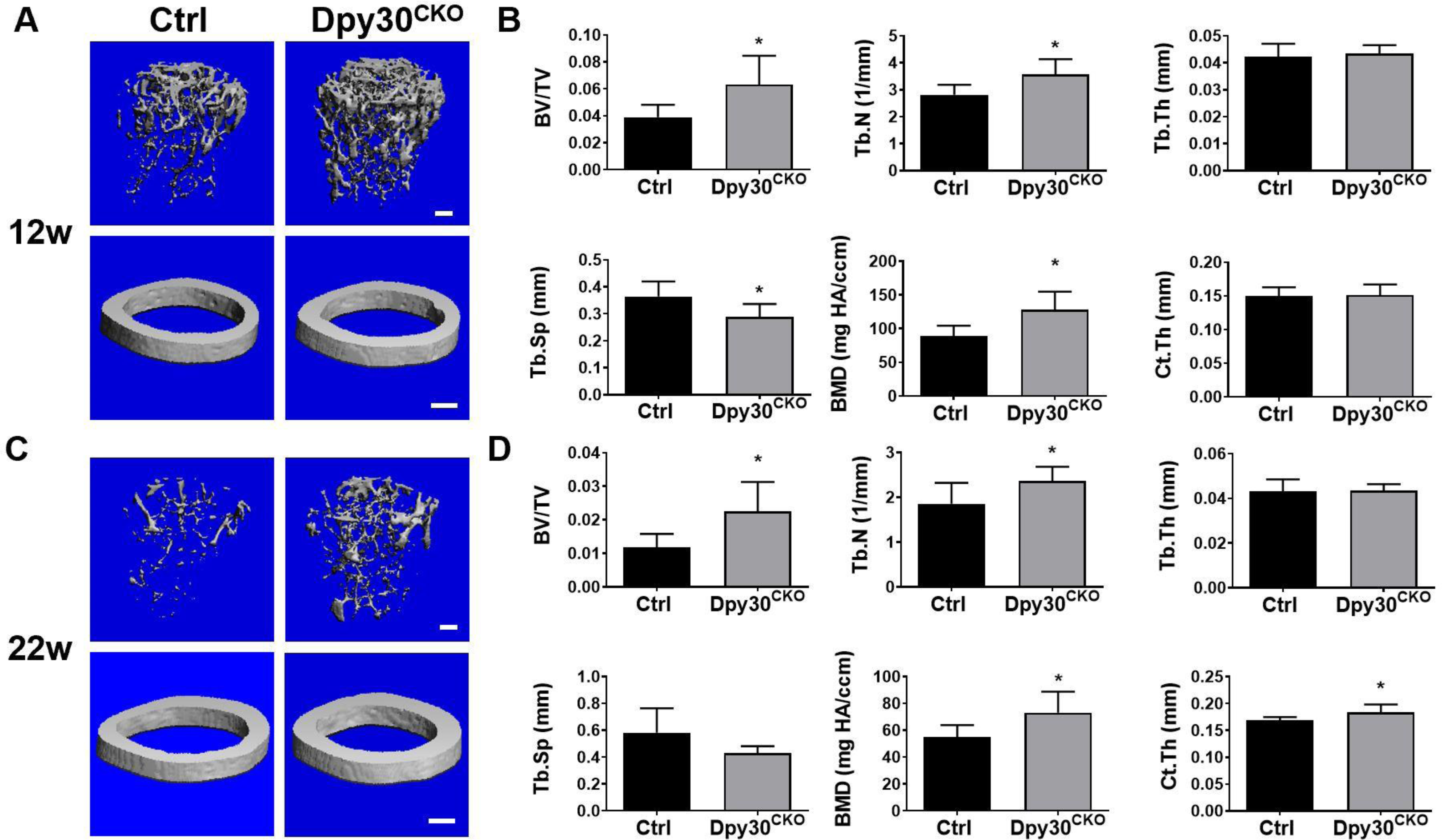
Dpy30 deficient mice resulted in increased bone mass. (A) Representative μCT images of femurs from Ctrl and Dpy30CKO mice (12-week-old). (B) Bone morphometric analysis and quantification from A (n=6); (C) Representative μCT images of femurs from Ctrl and Dpy30CKO mice (22-week-old). (D) Bone morphometric analysis and quantification from C (n=7); Data are mean ± SD. *P < 0.05; **P < 0.01. BV/TV, bone volume per tissue volume; Tb.N, trabecular bone number; Tb.Th, trabecular bone thickness; Tb.Sp, trabecular bone space; BMD, bone mineral density; Ct.Th, cortical thickness. Scale bar: 100 μm.
3.3. Dpy30 deficiency in myeloid cells results in decreased osteoclasts in trabecular bone
Bone mass is maintained by the balanced activity of osteoclasts and osteoblasts (36). To further clarify whether the elevated bone mass of Dpy30CKO mice results from decreased osteoclast activity or may involve alterations in the osteoblast function, we next analyzed bone histomorphometry. Static histomorphometric analysis revealed a significant difference of osteoclast parameters between Ctrl and Dpy30CKO mice, as shown by a significantly decreased osteoclast surface per bone surface (OcS/BS) and number of osteoclast per bone surface (N.Oc/BS) seen in the Dpy30CKO mice (Fig. 3A, B). These results demonstrate that Dpy30 is important in osteoclast differentiation in vivo. However, unlike the results seen in the osteoclast parameters, no significant changes were observed regarding the osteoblast parameters between Ctrl and Dpy30CKO mice (Fig. 3A, B). Consistent with the static histomorphometric analysis of osteoblast parameters, no significant difference of bone formation parameters between Ctrl and Dpy30CKO mice was observed by dynamic histomorphometric analysis (Fig. 3C, D). These findings indicate that osteoblast differentiation or survival was not impacted by Dpy30 deletion.
Fig. 3.

Dpy30 deficient mice showed decreased osteoclasts cell numbers. (A) Representative Goldner’s Trichrome staining images of femoral static histomorphometric analysis. Scale bar: 100 μm. (B) Quantification of femoral static histomorphometric analysis from A (n=7). (C) Representative fluorescence-labeled images of femoral dynamic histomorphometric analysis. Scale bar: 100 μm. (D) Quantification of femoral dynamic histomorphometric analysis from C (n=7). (E) Representative TRAP staining image of femurs. Scale bar: 50 μm. (F) Quantification of N.Oc/BS from E (n=6). (G) Serum analysis of CTX-1 (n = 10). N.Oc/BS, number of osteoclast per bone surface; N.Ob/BS, number of osteoblast per bone surface; OcS/BS, osteoclast surface per bone surface; ObS/BS, osteoblast surface per bone surface. MS/BS, mineralizing surface; MAR, mineral apposition rate; BFR/BS, bone form rate per bone surface; BFR/BV, bone form rate per bone volume. Data are mean ± SD. **P < 0.01.
To further confirm the impaired osteoclastogenesis, TRAP staining of decalcified distal femurs were also analyzed, and the results also showed reduced TRAP activity in the femur of Dpy30CKO mice in comparison with Ctrl mice (Fig. 3E). Additionally, the histomorphometric analysis showed a significant decrease of N.Oc/BS in Dpy30CKO mice in comparison with Ctrl mice (Fig. 3F). Consistently, serum levels of CTX-1, a hallmark related to bone resorption activity, markedly decreased in Dpy30CKO mice (Fig. 3G). In sum, this data demonstrates that Dpy30CKO mice acquired higher bone mass owing to the impaired osteoclast development and bone resorption activity.
3.4. Dpy30 deficiency inhibits osteoclast formation and function
Given that osteoclasts originate from osteoclast precursors of the myeloid cell lineage as we previously reported (33,37), we evaluated if Dpy30 deficiency will affect the development of osteoclast precursors. As shown by the flow cytometric analysis, the percentages of osteoclast precursors CD11b+c-fms+ and CD11b+c-fms+Ly6Chi do not differ between Dpy30CKO and Ctrl mice (Fig. 4A, B). Expression level of RANK in these osteoclast precursors are similar between Ctrl and Dpy30CKO mice (Supplemental Fig 2A). The data indicate that the defect in osteoclast formation in Dpy30CKO does not stem from the change in the number of osteoclast progenitor cells nor down-regulation of RANK expression on them.
Fig. 4.
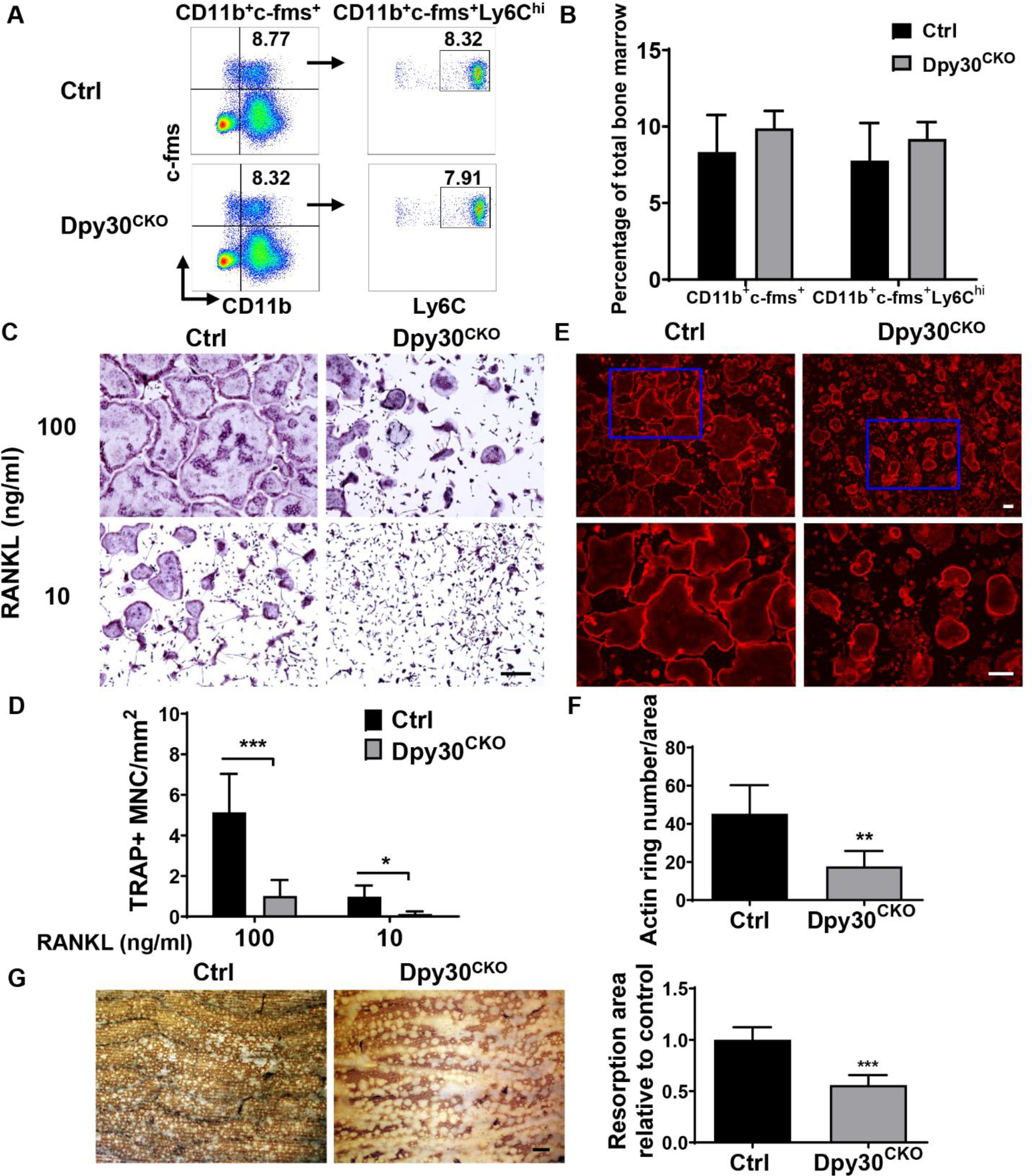
Dpy30 deficiency inhibits osteoclast formation and function. (A) Flow cytometry plot of osteoclast precursor population from bone marrow of 12-week-old Ctrl and Dpy30CKO mice. (B) Percentage of osteoclast precursor population as aforementioned (n = 6). (C) TRAP staining image of mature osteoclast from 12-week-old Ctrl and Dpy30CKO mice BMMs stimulated with RANKL (10 ng/ml or 100 ng/ml) at day 3. Scale bar: 50 μm. (D) Quantification of TRAP+ cell number in C (n=7). (E) F-actin ring fluorescence staining image of mature osteoclasts at day 3. Scale bar: 100 μm. (F) Quantification of F-actin ring+ number in E (n=7). (G) Bone resorption area by WGA staining in the cultures of mature osteoclasts at day 8. Scale bar: 100 μm. Data are mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
To evaluate whether the loss of Dpy30 influences the formation and function of osteoclasts, BMMs from Ctrl and Dpy30CKO mice were induced into osteoclasts on exposure of RANKL. The TRAP staining results showed a significantly lower number and smaller size of TRAP+ osteoclasts in Dpy30CKO BMMs in comparison to Ctrl BMMs with both optimal (100 ng/ml) and permissive (10 ng/ml) RANKL concentrations (Fig. 4C, D). Additionally, the actin ring formation was assessed by immunostaining. As a morphological feature of mature osteoclasts, actin ring is a sealing zone composed of actin with clear edges, which determines resorption activity of osteoclasts (1,38). Consistent with the results of TRAP staining, we observed a significant reduction in F-actin ring+ cells and remarkable smaller F-actin rings for the Dpy30CKO BMMs in comparison to Ctrl BMMs (Fig. 4E, F). Further bone resorption assays were analyzed by WGA staining of bone slices, and the results showed that Dpy30 deficiency inhibited osteoclastic bone resorption (Fig. 4G). Besides, there is similar osteoclast survive rate between Ctrl and Dpy30CKO mice when the osteoclasts are extensively cultured for 11 days. (Supplemental Fig. 2B, C). This data demonstrates that lack of Dpy30 severely affects osteoclast formation and function, but not alter osteoclast survival.
3.5. Dpy30 deficiency inhibits osteoclast-related genes and proteins
To determine the molecular basis of Dpy30 deletion on osteoclastogenesis, osteoclast-related genes expression was examined in BMMs from Ctrl and Dpy30CKO mice after RANKL stimulation. Deletion of Dpy30 at the cell level was confirmed at the gene and protein level during the process of osteoclast differentiation (Fig. 5A, 5B). As revealed by gene expression analysis, a significantly lower level of expression of osteoclast-related genes Ctsk, Trap, Car2, Atpv60d2 and Oscar, and transcription factor NFATc1 were observed in Dpy30CKO BMMs in comparison to Ctrl BMMs at different time points (Fig. 5A). Likewise, NFATc1 and Ctsk were also markedly reduced at the protein levels in Dpy30CKO BMMs compared to Ctrl BMMs (Fig. 5B). This data indicates that Dpy30 deficiency impedes osteoclast differentiation through inhibition of osteoclast-related genes at the transcription and protein level.
Fig. 5.
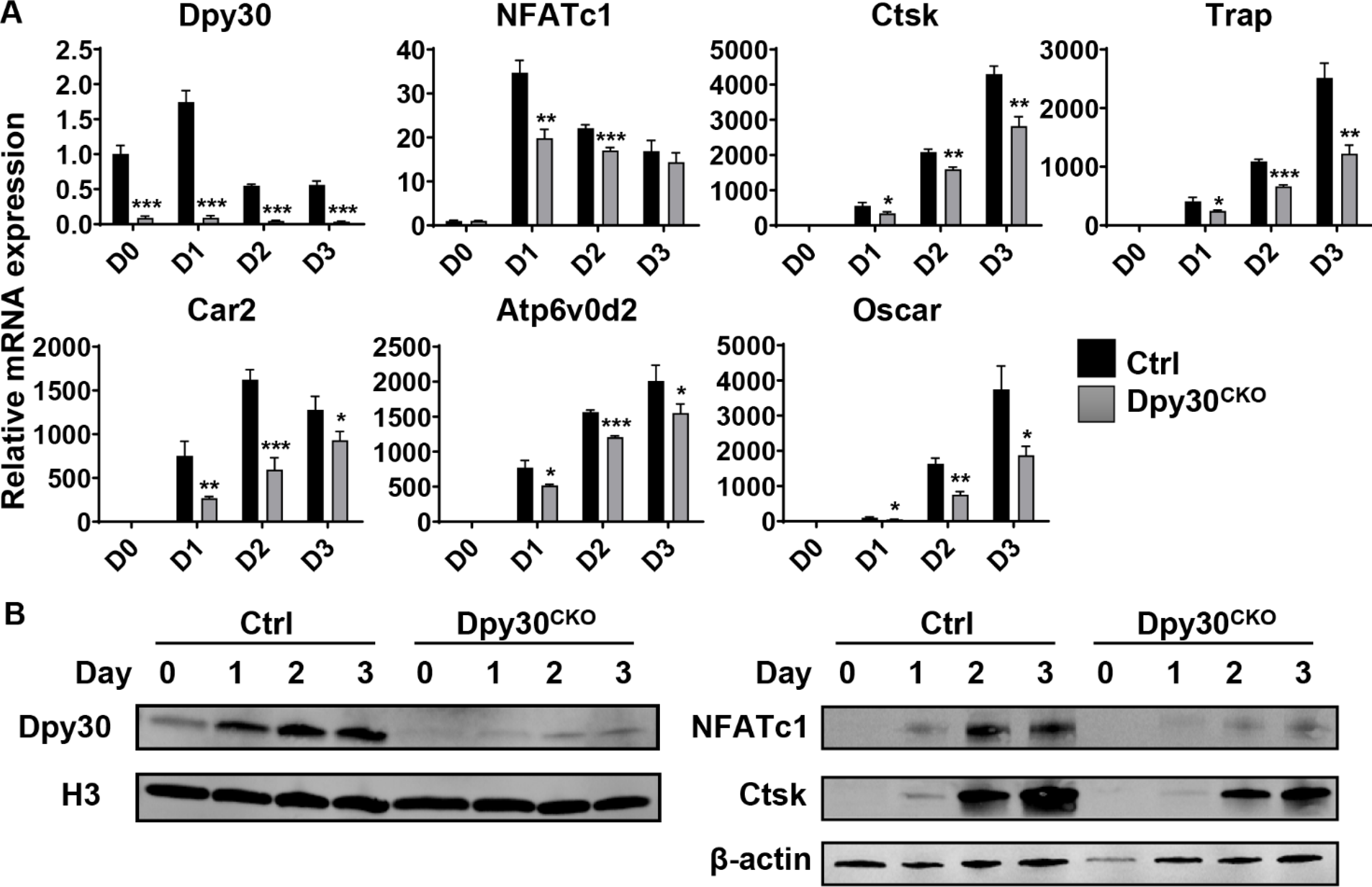
Dpy30 downregulates the expression of osteoclast-related genes at the transcription and protein level. (A) qPCR analysis of mRNA expression of Dpy30, NFATc1, Ctsk, Trap, Car2, Atp6v0d2 and Oscar during osteoclastogenesis using BMMs from the 12-week-old Ctrl and Dpy30CKO mice stimulated with RANKL (100 ng/ml) at different days (n=3). Data are mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001. (B) Western blotting of Dpy30, NFATc1 and Ctsk expression of cells as aforementioned.
3.6. Dpy30 deficiency inhibits H3K4 methylation at the NFATc1 promoter
It is reported that signal pathways, such as MAPKs and NF-κB, are vital in the differentiation and survival of osteoclasts (39). Thus, we assessed the impact of Dpy30 deficiency on these signal pathways in BMMs on exposure of RANKL. Our data showed that conditional deletion of Dpy30 have no influence on the activation of RANKL-induced MAPKs or NF-κB pathways (Fig. 6A). H3K4 methylation is typically related to gene activation. Dpy30 can directly and preferentially control H3K4 methylation, as well as the expression of various hematopoietic development-related genes (22). We therefore assessed if the H3K4 methylation activity was altered in Dpy30CKO BMMs during osteoclast differentiation. We found that during this process, lack of Dpy30 significantly decreased the levels of H3K4me1, H3K4me2, especially H3K4me3 (Fig. 6B). To investigate how Dpy30 regulates gene expression during osteoclastogenesis, we evaluated H3K4me3 at the NFATc1 regulatory regions. Results of the ChIP-qPCR using H3K4me3 antibody showed that H3K4me3 enrichment on the NFATc1 promoter was notably reduced in Dpy30CKO osteoclasts as compared to Ctrl osteoclasts (Fig. 6C). This observation indicates that Dpy30 regulates NFATc1 expression through H3K4me3 on the NFATc1 promoter, thus establishing it as a critical chromatin modulator for osteoclast differentiation and function through transcriptional regulation.
Fig. 6.
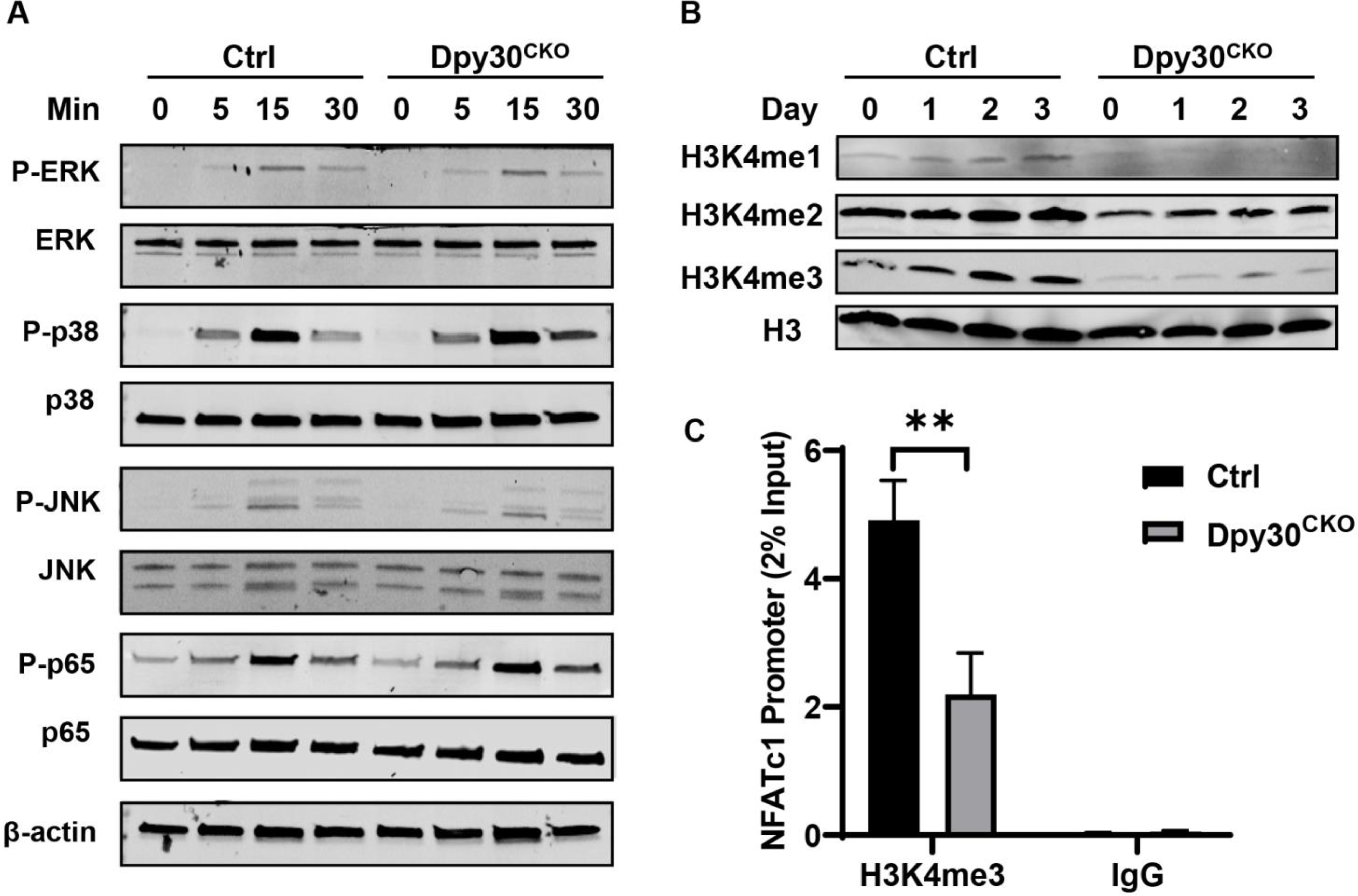
Dpy30 deficiency inhibits H3K4 methylation at the NFATc1 promoter. (A) Total protein levels and phosphorylation of MAPKs and NF-ĸB were assessed in Ctrl and DPY30CKO BMMs stimulated by RANKL (100 ng/ml) for 0, 5, 15 or 30 minutes. (B) Western blotting of H3, H3K4me1, H3K4me2 and H3K4me3 in Ctrl and Dpy30CKO BMMs stimulated with RANKL (100 ng/ml) at different days. (C) Ctrl and Dpy30CKO BMMs were stimulated by RANKL (100 ng/ml) for 1 day. The enrichment of H3K4me3 on the NFATc1 promoter region was evaluated by ChIP assay (n=3). Data are mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
4. Discussion
Osteoclasts are the unique cells primarily responsible for bone resorption, and their differentiation and activation are tightly regulated. Recent evidence has shown that epigenetic regulation, such as histone methylation, is important in controlling osteoclast differentiation (9). However, little is known about the regulation of osteoclastogenesis via histone-modifying enzymes involved in H3K4 methylation. Herein, we have uncovered an unprecedented role for Dpy30, a critical chromatin regulator of H3K4 methylation, in osteoclast differentiation, skeletal development, and bone homeostasis. By characterizing the bone phenotype of mice with Dpy30 deletion in myeloid cells, we found that Dpy30CKO mice formed fewer osteoclasts than the control mice, resulting in elevated bone mass. We also found that in vitro culture of osteoclasts derived from BMMs from Dpy30CKO mice showed reduced osteoclast formation and impaired function. Our findings, from in vitro and in vivo approaches, strongly demonstrate a critical role for Dpy30 in osteoclast differentiation and function.
Osteoclasts originate from osteoclast precursors of HSC origin (33,37). After differentiating from their hematopoietic precursors, osteoclast precursors undergo lineage commitment and cell maturation, and terminate into osteoclasts under the control of a series of molecular events (40). Studies have shown that genes important for hematopoiesis, such as PU.1, C/BBPα and ASXL1, have critical roles in osteoclast differentiation (35,41–43). Dpy30 is a crucial chromatin regulator of hematopoiesis, which acts through controlling H3K4 methylation and affecting the transcription of numerous genes (22,25,26). Dpy30 knockdown in human HPCs impaired its myelomonocytic differentiation (25). The crucial role Dpy30 plays in generating myelomonocytic cells (which are also osteoclast precursors) raises the possibility that failure to express Dpy30 might deter development of osteoclasts, resulting in arrested bone resorption and osteopetrosis. In the present study, Dpy30 was deleted in myeloid lineage using LysM-Cre. Our results showed that Dpy30CKO mice possess elevated bone mass because of the reduction in the number of osteoclasts without affecting osteoblasts, demonstrating a regulatory role of Dpy30 on osteoclast differentiation. However, no alteration in the number of osteoclast precursors was seen in Dpy30 deficient mice, indicating that regulation of osteoclast formation by Dpy30 was not due to its effect on the generation of osteoclast precursors.
The activation of a series of osteoclast-related genes including Ctsk, Oscar and Atp6v0d2, is required for RANKL-induced osteoclastogenesis (3). Our results showed that deletion of Dpy30 downregulates osteoclast-related genes (e.g., Ctsk, Trap, Oscar, Car2 and Atp6v0d2). These genes play important roles in osteoclast function. For example, Ctsk, a cysteine proteinase highly expressed in osteoclasts, participates dominantly in the degradation of bone matrix (44). These gene changes contribute to osteoclast-induced bone resorption involved with actin ring formation and matrix protein degradation, which is consistent with the impaired actin ring formation and repressed osteoclast formation that we observed. On the other hand, osteoclastogenesis is regulated by the activation or inhibition of osteoclast marker genes at the transcription level by various transcription factors (40). NFATc1, a critical early transcription factor of osteoclast-macrophage lineage determination (45), is also significantly downregulated in Dpy30CKO mice, suggesting that Dpy30 cooperates with NFATc1 to regulate osteoclast differentiation and activity. Hence, Dpy30 deficiency can suppress RANKL-induced expression of osteoclast-specific genes and transcription factor, thus resulting in arrested osteoclast differentiation and activity.
Osteoclast development is tightly regulated by various molecular mechanisms. Signaling pathways, such as MAPKs and NF-κB, exert an important function in osteoclast differentiation and survival (39,46). However, no change was observed in RANKL-induced expression or activation of these classic osteoclast signaling molecules in BMMs in the absence of Dpy30, indicating that the decreased differentiation of osteoclasts caused by the loss of Dpy30 involves other mechanisms. Increased evidence indicates that mechanisms based on epigenetics also have pivotal roles in coordinating gene expression during osteoclast differentiation (8,9). Previous studies showed that RANKL-stimulated osteoclastogenesis occurs along with a change of chromatin architecture from H3K4me3/H3K27me3 to H3K4me3, on the promoter of NFATc1, suggesting that H3K4me3 is important in maintaining osteoclast differentiation (13). Dpy30 is an important chromatin modulator on H3K4 methylation, especially on H3K4me3 (17,22). Here, we observed that Dpy30 deletion downregulated the global levels of H3K4 methylation during osteoclast differentiation process and Dpy30 deficiency significantly decreased H3K4me3 on the promoter of NFATc1. By directly controlling the expression of transcription factor NFATc1, Dpy30 may exert an indispensable effect in regulating the expression of crucial osteoclast-related genes, thereby promoting the differentiation, activation, and function of osteoclasts, and regulating bone remodeling. In line with our results, it was shown that PTIP decreases H3K4me3 on the promoter of PPARγ, leading to the impairment of osteoclast differentiation and function (16). In addition, ARTD1 controls osteoclastogenesis and bone homeostasis by regulating H3K4me3 on the promoter of IL-1β (47). Similarly, loss of ASXL1 in myeloid cells causes loss of transcription-repressive H3K27me3 and gain of transcription-active H3K4me3 at NFATc1, leading to an elevated accumulation of osteoclasts and thus a lower bone mass (35). Therefore, instead of affecting the classical MAPKs and NF-κB pathways, Dpy30 regulates osteoclast differentiation by affecting the activation of NFATc1 via changing active histone methylation, which links the prominent H3K4 methylation mechanism to osteoclastogenesis. However, it is also possible that Dpy30 may bind directly to the NFATc1 promoter region, which would indicate that Dpy30 could also directly regulates NFATc1 expression. In addition, Dpy30 may regulate H3K4 methylation via its association with proteins other than components of the Set1/Mll complexes (19).
The Set1/Mll complex is the primary histone H3K4 methylation enzyme in mammals. Their full methylation activity is determined by the relationship between the catalytic subunits and their integral common core subunits Wdr5, Rbbp5, Ash2l and Dpy30 (17). We found that the core subunit and Set1/Mll complex show the similar expression trend as Dpy30 in osteoclast differentiation process (Supplementary Fig.1), which suggest these genes may also participate the osteoclast differentiation process, which needs further investigation. In addition to the Set1/Mll complex components as the major associated proteins, Dpy30 also associates with a few other proteins (23), thus we cannot rule out a role for Dpy30 in regulating osteoclast differentiation by interacting with other factors.
In summary, by using in vitro and in vivo methodologies, we revealed a novel epigenetic regulatory mechanism of Dpy30 and its associated H3K4 methylation on osteoclastogenesis. Our study showed that loss of Dpy30 can inhibit osteoclast differentiation and function, as well as increases bone mass, mainly by its effect on osteoclasts without affecting osteoblasts. Moreover, Dpy30 deficiency regulates transcription factor NFATc1 through decreasing H3K4me3 on the promoter of NFATc1 and thus inhibits osteoclastogenesis (Fig. 7). Insights gained from this study will not only contribute to our knowledge of epigenetic regulation in osteoclast biology, but will also lay a foundation for the new and effective therapeutic approaches targeting epigenetic modulators of osteolytic diseases.
Fig. 7. Schematic diagram of the role of Dpy30 in osteoclastogenesis.
Dpy30 regulates H3K4me3 on the promoter of transcription factor NFATc1 and thus regulates RANKL-induced osteoclastogenesis.
Supplementary Material
Highlights.
Dpy30 deficiency in myeloid cells inhibits osteoclast differentiation and function in vitro and in vivo.
Dpy30 deficiency in myeloid cells affects bone mass by regulating osteoclast-mediated bone resorption without altering osteoblast-mediated bone formation.
Dpy30 deficiency decreases H3K4me3 on the promoter of NFATc1.
Acknowledgements
We thank Greg Harber for his technical assistance. This research was supported by grants from the National Institute of Dental and Craniofacial Research R01 DE026465 (PZ), R90 DE023056 (YZ, ZL) and UAB GC-CODED Pilot & Feasibility Award (YZ). The University of Alabama at Birmingham Comprehensive Flow Cytometry Core is supported by the National Institutes of Health grants P30AI27667 and P30AR048311.
Footnotes
Declaration of competing interest
The authors declare no conflicts of interest.
The following are the supplementary data related to this article.
Supplementary Fig. 1. Wdr5, Rbbp5 and Ash21 expression during RANKL-induced osteoclastogenesis. 12-week-old wild type bone marrow macrophages (BMMs) were exposed to RANKL (100 ng/mL) for 3 days. Wdr5, Rbbp5 and Ash21 gene expression are determined by qPCR (n=3). Data are mean ± SD.*P < 0.05; **P < 0.01 relative to D0
Supplementary Fig. 2 Dpy30 deficiency does not alter RANK expression in osteoclast precursors and osteoclasts survival. (A) Flow cytometry analysis of RANK expression in osteoclast precursor CD11b+c-fms+ and CD11b+c-fms+Ly6Chi population from bone marrow of 12-week-old Ctrl and Dpy30CKO mice. (B) TRAP staining image of extensive culture of osteoclasts from 12-week-old Ctrl and Dpy30CKO mice at day 5 and day 11, without media change. Scale bar: 100 μm. (C) The relative survive rate analyzed from B, and D5 was analyzed as baseline (n=3). Data are mean ± SD
Supplement Table 1. Mouse primer sequences for qPCR
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boyle WJ, Simonet WS, and Lacey DL (2003) Osteoclast differentiation and activation. Nature 423, 337–342 [DOI] [PubMed] [Google Scholar]
- 2.Novack DV, and Teitelbaum SL (2008) The osteoclast: friend or foe? Annu Rev Pathol 3, 457–484 [DOI] [PubMed] [Google Scholar]
- 3.Teitelbaum SL, and Ross FP (2003) Genetic regulation of osteoclast development and function. Nat Rev Genet 4, 638–649 [DOI] [PubMed] [Google Scholar]
- 4.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, and Taniguchi T (2002) Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3, 889–901 [DOI] [PubMed] [Google Scholar]
- 5.Takeshita S, Kaji K, and Kudo A (2000) Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res 15, 1477–1488 [DOI] [PubMed] [Google Scholar]
- 6.Feng X, and McDonald JM (2011) Disorders of bone remodeling. Annu Rev Pathol 6, 121–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graves DT, Li J, and Cochran DL (2011) Inflammation and uncoupling as mechanisms of periodontal bone loss. J Dent Res 90, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurotaki D, Yoshida H, and Tamura T (2020) Epigenetic and transcriptional regulation of osteoclast differentiation. Bone 138, 115471. [DOI] [PubMed] [Google Scholar]
- 9.Astleford K, Campbell E, Norton A, and Mansky KC (2020) Epigenetic Regulators Involved in Osteoclast Differentiation. Int J Mol Sci 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang C, Qiao Y, Mun SH, Lee MJ, Murata K, Bae S, Zhao B, Park-Min KH, and Ivashkiv LB (2016) Cutting Edge: EZH2 Promotes Osteoclastogenesis by Epigenetic Silencing of the Negative Regulator IRF8. J Immunol 196, 4452–4456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Y, and Ge W (2018) The histone methyltransferase DOT1L inhibits osteoclastogenesis and protects against osteoporosis. Cell Death Dis 9, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yi SJ, Jang YJ, Kim HJ, Lee K, Lee H, Kim Y, Kim J, Hwang SY, Song JS, Okada H, Park JI, Kang K, and Kim K (2021) The KDM4B-CCAR1-MED1 axis is a critical regulator of osteoclast differentiation and bone homeostasis. Bone Res 9, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasui T, Hirose J, Tsutsumi S, Nakamura K, Aburatani H, and Tanaka S (2011) Epigenetic regulation of osteoclast differentiation: possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J Bone Miner Res 26, 2665–2671 [DOI] [PubMed] [Google Scholar]
- 14.Kim K, Shin Y, Kim J, Ulmer TS, and An W (2018) H3K27me1 is essential for MMP-9-dependent H3N-terminal tail proteolysis during osteoclastogenesis. Epigenetics Chromatin 11, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kouzarides T (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 16.Das P, Veazey KJ, Van HT, Kaushik S, Lin K, Lu Y, Ishii M, Kikuta J, Ge K, Nussenzweig A, and Santos MA (2018) Histone methylation regulator PTIP is required to maintain normal and leukemic bone marrow niches. Proc Natl Acad Sci U S A 115, E10137–E10146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, and Roeder RG (2006) Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 13, 713–719 [DOI] [PubMed] [Google Scholar]
- 18.Hyun K, Jeon J, Park K, and Kim J (2017) Writing, erasing and reading histone lysine methylations. Exp Mol Med 49, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang H (2020) The complex activities of the SET1/MLL complex core subunits in development and disease. Biochim Biophys Acta Gene Regul Mech 1863, 194560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haddad JF, Yang Y, Takahashi YH, Joshi M, Chaudhary N, Woodfin AR, Benyoucef A, Yeung S, Brunzelle JS, Skiniotis G, Brand M, Shilatifard A, and Couture JF (2018) Structural Analysis of the Ash2L/Dpy-30 Complex Reveals a Heterogeneity in H3K4 Methylation. Structure 26, 1594–1603 e1594 [DOI] [PubMed] [Google Scholar]
- 21.Jiang H, Shukla A, Wang X, Chen WY, Bernstein BE, and Roeder RG (2011) Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell 144, 513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Z, Shah K, Khodadadi-Jamayran A, and Jiang H (2016) Dpy30 is critical for maintaining the identity and function of adult hematopoietic stem cells. J Exp Med 213, 2349–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang H, Lu X, Shimada M, Dou Y, Tang Z, and Roeder RG (2013) Regulation of transcription by the MLL2 complex and MLL complex-associated AKAP95. Nat Struct Mol Biol 20, 1156–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertero A, Madrigal P, Galli A, Hubner NC, Moreno I, Burks D, Brown S, Pedersen RA, Gaffney D, Mendjan S, Pauklin S, and Vallier L (2015) Activin/nodal signaling and NANOG orchestrate human embryonic stem cell fate decisions by controlling the H3K4me3 chromatin mark. Genes Dev 29, 702–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Z, Augustin J, Chang C, Hu J, Shah K, Chang CW, Townes T, and Jiang H (2014) The DPY30 subunit in SET1/MLL complexes regulates the proliferation and differentiation of hematopoietic progenitor cells. Blood 124, 2025–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z, Shah K, Khodadadi-Jamayran A, and Jiang H (2019) Control of Hematopoietic Stem and Progenitor Cell Function through Epigenetic Regulation of Energy Metabolism and Genome Integrity. Stem Cell Reports 13, 61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah K, King GD, and Jiang H (2020) A chromatin modulator sustains self-renewal and enables differentiation of postnatal neural stem and progenitor cells. J Mol Cell Biol 12, 4–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Z, Shah K, Busby T, Giles K, Khodadadi-Jamayran A, Li W, and Jiang H (2018) Hijacking a key chromatin modulator creates epigenetic vulnerability for MYC-driven cancer. J Clin Invest 128, 3605–3618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dallas SL, Xie Y, Shiflett LA, and Ueki Y (2018) Mouse Cre Models for the Study of Bone Diseases. Curr Osteoporos Rep 16, 466–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inoue K, Deng Z, Chen Y, Giannopoulou E, Xu R, Gong S, Greenblatt MB, Mangala LS, Lopez-Berestein G, Kirsch DG, Sood AK, Zhao L, and Zhao B (2018) Bone protection by inhibition of microRNA-182. Nat Commun 9, 4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Zhao Y, Chen Z, Katz J, Michalek SM, Li Y, and Zhang P (2021) Age-related expansion and increased osteoclastogenic potential of myeloid-derived suppressor cells. Mol Immunol 137, 187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Shi Z, Jules J, Chen S, Kesterson RA, Zhao D, Zhang P, and Feng X (2019) Specific RANK Cytoplasmic Motifs Drive Osteoclastogenesis. J Bone Miner Res 34, 1938–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Y, Li Z, Su L, Ballesteros-Tato A, Katz J, Michalek SM, Feng X, and Zhang P (2020) Frontline Science: Characterization and regulation of osteoclast precursors following chronic Porphyromonas gingivalis infection. J Leukoc Biol 108, 1037–1050 [DOI] [PubMed] [Google Scholar]
- 34.Shin Y, Ghate NB, Moon B, Park K, Lu W, and An W (2019) DNMT and HDAC inhibitors modulate MMP-9-dependent H3 N-terminal tail proteolysis and osteoclastogenesis. Epigenetics Chromatin 12, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rohatgi N, Zou W, Collins PL, Brestoff JR, Chen TH, Abu-Amer Y, and Teitelbaum SL (2018) ASXL1 impairs osteoclast formation by epigenetic regulation of NFATc1. Blood Adv 2, 2467–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zaidi M (2007) Skeletal remodeling in health and disease. Nat Med 13, 791–801 [DOI] [PubMed] [Google Scholar]
- 37.Cai X, Li Z, Zhao Y, Katz J, Michalek SM, Feng X, Li Y, and Zhang P (2020) Enhanced dual function of osteoclast precursors following calvarial Porphyromonas gingivalis infection. J Periodontal Res 55, 410–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jurdic P, Saltel F, Chabadel A, and Destaing O (2006) Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol 85, 195–202 [DOI] [PubMed] [Google Scholar]
- 39.Park JH, Lee NK, and Lee SY (2017) Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol Cells 40, 706–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asagiri M, and Takayanagi H (2007) The molecular understanding of osteoclast differentiation. Bone 40, 251–264 [DOI] [PubMed] [Google Scholar]
- 41.Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, and Teitelbaum SL (1997) Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature 386, 81–84 [DOI] [PubMed] [Google Scholar]
- 42.Carey HA, Hildreth BE 3rd, Geisler JA, Nickel MC, Cabrera J, Ghosh S, Jiang Y, Yan J, Lee J, Makam S, Young NA, Valiente GR, Jarjour WN, Huang K, Rosol TJ, Toribio RE, Charles JF, Ostrowski MC, and Sharma SM (2018) Enhancer variants reveal a conserved transcription factor network governed by PU.1 during osteoclast differentiation. Bone Res 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen W, Zhu G, Jules J, Nguyen D, and Li YP (2018) Monocyte-Specific Knockout of C/ebpalpha Results in Osteopetrosis Phenotype, Blocks Bone Loss in Ovariectomized Mice, and Reveals an Important Function of C/ebpalpha in Osteoclast Differentiation and Function. J Bone Miner Res 33, 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costa AG, Cusano NE, Silva BC, Cremers S, and Bilezikian JP (2011) Cathepsin K: its skeletal actions and role as a therapeutic target in osteoporosis. Nat Rev Rheumatol 7, 447–456 [DOI] [PubMed] [Google Scholar]
- 45.Takayanagi H (2007) The role of NFAT in osteoclast formation. Ann N Y Acad Sci 1116, 227–237 [DOI] [PubMed] [Google Scholar]
- 46.Soysa NS, and Alles N (2009) NF-kappaB functions in osteoclasts. Biochem Biophys Res Commun 378, 1–5 [DOI] [PubMed] [Google Scholar]
- 47.Robaszkiewicz A, Qu C, Wisnik E, Ploszaj T, Mirsaidi A, Kunze FA, Richards PJ, Cinelli P, Mbalaviele G, and Hottiger MO (2016) ARTD1 regulates osteoclastogenesis and bone homeostasis by dampening NF-kappaB-dependent transcription of IL-1beta. Sci Rep 6, 21131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.