

Abstract
Copy number variations (CNV) involving multiple genes are ideal models to study polygenic neuropsychiatric disorders. Since 22q11.2 deletion is regarded as the most important single genetic risk factor for developing schizophrenia, characterizing the effects of this CNV on neural networks offers a unique avenue towards delineating polygenic interactions conferring risk for the disorder. We used a Df(h22q11)/+ mouse model of human 22q11.2 deletion to dissect gene expression patterns that would spatially overlap with differential resting-state functional connectivity (FC) patterns in this model (N=12 Df(h22q11)/+ mice, N=10 littermate controls). To confirm the translational relevance of our findings, we analyzed tissue samples from schizophrenia patients and healthy controls using machine learning to explore whether identified genes were co-expressed in humans. Additionally, we employed the STRING protein-protein interaction database to identify potential interactions between genes spatially associated with hypo- or hyper-FC. We found significant associations between differential resting-state connectivity and spatial gene expression patterns for both hypo- and hyper-FC. Two genes, Comt and Trmt2a, were consistently over-expressed across all networks. An analysis of human datasets pointed to a disrupted co-expression of these two genes in the brain in schizophrenia patients, but not in healthy controls. Our findings suggest that COMT and TRMT2A form a core genetic component implicated in differential resting-state connectivity patterns in the 22q11.2 deletion. A disruption of their co-expression in schizophrenia patients points out a prospective cause for the aberrance of brain networks communication in 22q11.2 deletion syndrome on a molecular level.
Keywords: 22q11.2 deletion, schizophrenia, Comt, Trmt2a, functional connectivity, mouse
INTRODUCTION
Copy number variations (CNVs), termed as deletions or duplications of large DNA segments, provide a major avenue to unravel the molecular mechanisms conferring vulnerability for neuropsychiatric disorders. As CNVs oftentimes span over more than 20 genes, they are ideal candidates to study polygenic interactions, presumably a key mechanism in the emergence of severe disorders, such as schizophrenia (International Schizophrenia Consortium, 2009). The 22q11.2 deletion is principal in this regard. It represents one of the most important single genetic risk factors for schizophrenia (penetrance ~20-30%) (Schneider et al., 2014). Besides that, it has been shown to be associated with other neuropsychiatric conditions, among them prevalently with autism spectrum disorder (ASD) (15-50%) (Ousley et al., 2017), but also with obsessive-compulsive disorder and attention deficit hyperactivity disorder (Sebat et al., 2009). This great phenotypic variability corresponds to a considerable genotypic variance. Although the deletion spans 3 Mb in 80-90% of the carriers (Packham and Brooks, 2003), it can vary considerably, affecting up to 50 genes (Maynard et al., 2008). Besides variation in the size of the deletion itself, additional factors might influence the phenotype, including genetic variance outside the deletion acting as modifier (Michaelovsky et al., 2019), possible unknown environmental exposures during pregnancy, epigenetic variations and somatic mutations (Morrow et al., 2018).
Mouse models of 22q11.2 deletion are capable of addressing several of the scientific challenges that go along with this phenotypic variability. First, deletions in these models do not vary across animals. Second, in-bred strains offer a rather homogeneous genetic background, and standardized housing limits the effects from gene-environment interactions. Third, in contrast to human studies, mouse models offer samples unbiased by diagnosis-based pre-selection. Finally, given the extremely rare prevalence of the deletion in human populations, animal models provide the option to study sufficiently sized cohorts.
Of the six mouse models existing for the human 22q11.2 deletion (Kimber et al., 1999; Merscher et al., 2001; Sivagnanasundaram et al., 2007; Stark et al., 2008; Earls et al., 2010; Didriksen et al., 2017), Df(h22q11)/+ represents it best, containing almost all functional genes of the human 1.5 Mb segment and being congenic, i.e. bred to be essentially isogenic with a wild type group except for a selected differential chromosomal segment, and, thus, possessing an unbiased genetic background, unlike non-congenic LgDel, Df1/+ and Df(16)A+/− models (Didriksen et al., 2017). At the behavioral and electrophysiological levels this model captures features that are relevant for schizophrenia pathophysiology and characteristic for the human deletion, among which are reduced prepulse inhibition, increased acoustic startle response and impaired working memory (Didriksen et al., 2017).
Our previous study has used resting-state fMRI to identify changes in whole-brain connectivity in this mouse model (Reinwald et al., 2020). However, while the impact of the entire CNV on brain function is comparatively straightforward to study, contributions of individual genes within the deletion to these changes of neural connectivity patterns are much harder to identify. Nevertheless, such an approach appears as a major pathway towards understanding how psychiatric disorders emerge from their genetic underpinnings. One potential approach towards narrowing down the number of candidate genes is to study their expression patterns in the brain. This approach is based upon the hypothesis that genes with expression patterns that spatially overlap with changes of brain resting-state connectivity are the most likely ones to play a causal role in these brain functional alterations (Fakhry and Ji, 2015). A novel tool that has been recently introduced by our lab based on modern gene expression atlases provides a powerful opportunity to explore such a spatial overlap between transcriptomic patterns and changes of brain structure and function (Kumar et al., 2018). For our actual study, we relied upon high-resolution in situ hybridization expression maps of the genes involved in the deletion that were extracted from the Allen Mouse Brain Atlas (Lein et al., 2007). We, then, identified genes that spatially overlap with hyper- or hypoconnectivity patterns in our mouse model. The main goal of our study was to dissect the contributions of individual genes within the 22q11.2 deletion to changes of neural connectivity. In other words we aimed to clarify which of the genes involved in 22q11.2 deletion are the most likely candidates for a causal role in the emergence of changes in neural connectivity. Given this major aim, we focused our analysis on the expression maps of 24 genes located within the 22q11.2 region. To identify potential interactions between the products of genes associated with either or both of the contrasts, we used the STRING protein-protein interaction database, which allows a delineation of functional protein networks and their impact on cellular processes (Szklarcyk et al., 2019). To confirm the translational relevance of our findings, we analyzed tissue samples from patients with schizophrenia and healthy controls using machine learning to explore whether identified genes would be co-expressed in humans.
METHODS
Animals
We investigated 10-week-old male mice modeling 22q11.2 deletion (N=12, 22-26 g) (Figure 1) and its wild type (WT) littermate controls (N=10, 22-27 g). TaconicArtemis (Köln, Germany) bred the animals by mating WT C57BL/6N females with hemizygotic Df(h22q11)/+ males.
Figure 1. A schematic representation of deleted genes aligned for human (22q11.2) and mouse (16qA3) chromosomal regions.
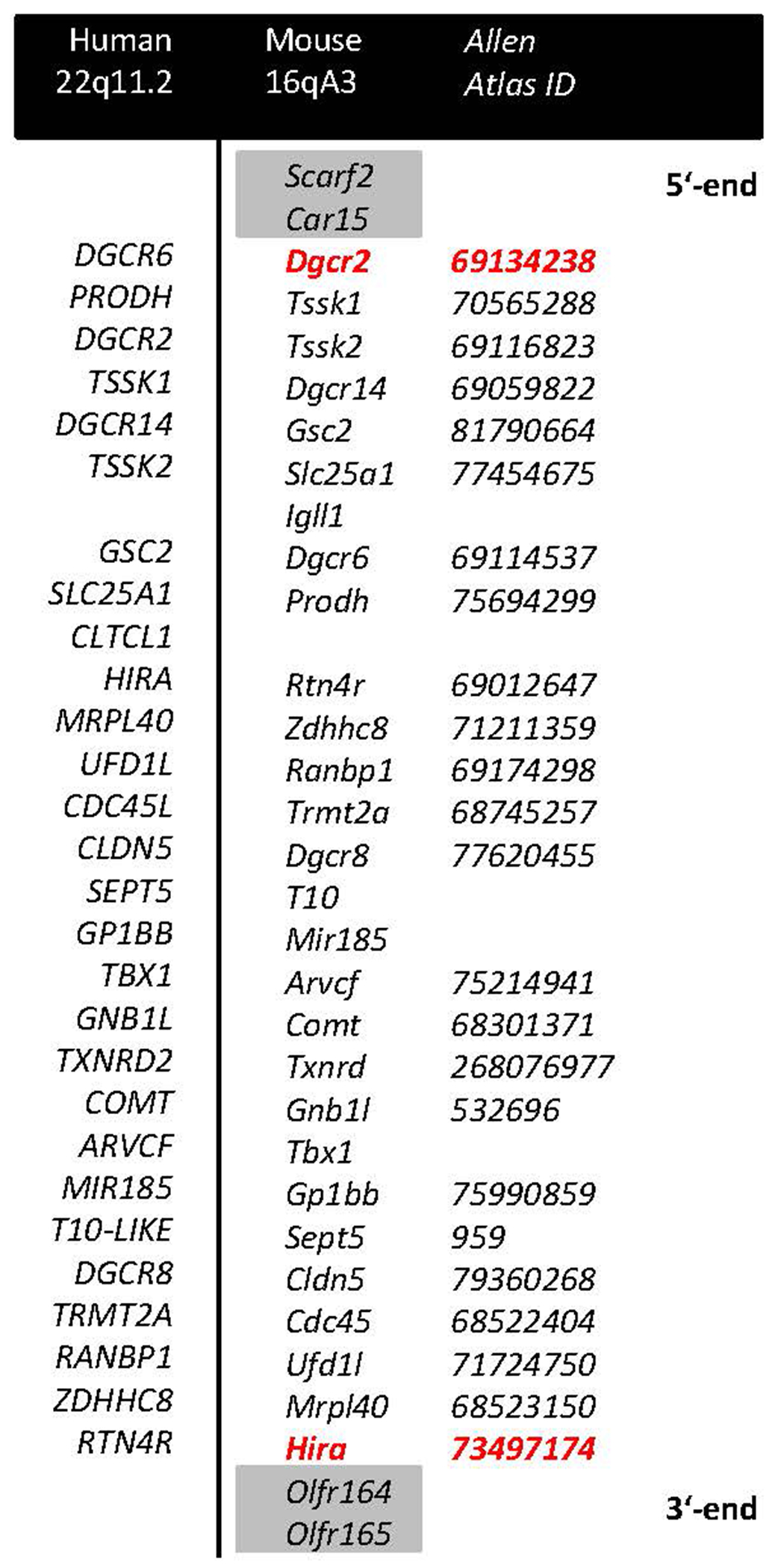
Red color denotes genes targeted with a LoxP site, grey shade denotes genes at the border not comprising part of the deletion. IDs of the expression maps for the respective genes in the Allen Mouse Brain Atlas are reported in italics.
All animals were group-housed (2 WT and 2 Df(h22q11)/+ per cage) under controlled conditions (19-23°C, 40-60% humidity) on a 12 h light-dark cycle (lights on at 07:00) and underwent a two-week adaptation period between their arrival and the start of the MRI experiments. All procedures were approved by the German animal welfare authorities (Regierungspräsidium Karlsruhe) and were performed according to the regulations covering animal experimentation within the European Union (European Communities Council Directive 86/609/EEC) and within the German Animal Welfare Act.
MRI acquisition
Experiments were performed at a 9.4 Tesla MRI scanner (94/20 Bruker Biospec, Ettlingen, Germany) with a two-element anatomically-shaped cryogenic mouse surface coil. Anesthetic regime using medetomidine and recording of the physiological data were performed as previously (Gass et al., 2016) (for all details see Supplement). The MRI acquisition protocol for each animal comprised a FieldMap, a resting-state functional magnetic resonance imaging (rs-fMRI) time series and a 3D structural dataset. The rs-fMRI time series were acquired using an echo-planar imaging (EPI) sequence (repetition time (TR)/echo time (TE) 1300/18 ms, flip angle 50°, 21 slices, 96 x 64 matrix, field of view (17.28 x 11.52) mm2, slice thickness 0.4 mm, 400 acquisitions, acquisition time ~8.5 min). Magnetic field (B0) inhomogeneity was measured with Bruker FieldMap sequence acquired before each EPI. Structural data were acquired using rapid acquisition with refocused echoes (RARE) sequence (for sequences details see Supplement). Breathing and cardiac rates were monitored using a respiration pad placed beneath the chest (Small Animal Instruments Inc., NY, USA) and a pulse oximeter attached to the tail. Signals were recorded (10-ms resolution) during EPI acquisition using a signal breakout module (Small Animal Instruments Inc., NY, USA) and a 4-channel recorder (Velleman N.V., Gavere, Belgium) together with the scanner trigger pulses for each measured brain volume. Here we note that physiological parameters (heart and respiration rates) under medetomidine sedation demonstrated no significant differences in Df(h22q11)/+ mice compared to wild type (WT) mice (p>0.05, two-sided Wilcoxon test, Figure S1).
Pre-processing of rs-fMRI data
Pre-processing was performed, as previously (Reinwald et al., 2020), and included correction for magnetic field inhomogeneity (SPM12, http://www.fil.ion.ucl.ac.uk/spm/software/spm12), filtering of respiratory and cardiac signals (Aztec software (van Buuren et al., 2009)), slice timing correction (SPM12), spatial normalization and co-registration to a mouse brain template in the Paxinos stereotactic coordinate system (Dorr et al., 2008), regression of movement parameters and cerebrospinal fluid signal (FSL, version 4.1. http://www.fmrib.ox.ac.uk/fs (Power et al., 2012; Ayfouni and Nichols, 2018)). An additional method identifying motion-affected frames based on a decomposition of DVARS (Power et al., 2012; Ayfouni and Nichols, 2018) was integrated to further remove motion artifacts (for details see Supplement). Framewise displacement displayed relatively low levels (median values < 0.02 mm) with no significant difference between the groups; also, the number of scrubbed frames was on a low level, not exceeding the threshold of 10% for any animal and not showing significant differences between the groups (Figure S2). Next, band-pass filtering (0.01-0.1 Hz) was performed (Analysis of Functional NeuroImages software (Cox, 1996). Finally, we performed spatial smoothing using a Gaussian kernel with a 0.6 mm full width at half maximum (SPM12). One subject (control group) was classified as an outlier by comparing the overall mean functional connectivity of the correlation matrices (more than twice the standard deviation from the mean) and was excluded from the analysis.
Seed-based analysis
Using the Allen Mouse Brain Atlas (Lein et al., 2007), we defined nine seed regions which included cortical (anterior cingulate (ACC), infralimbic (IL), orbitofrontal (OC) and retrosplenial cortex (RS)), hippocampus (Ammon’s horn (CA) and dentate gyrus (DG)), and subcortical components (nucleus accumbens (Acb), substantia nigra (SN) and ventral tegmental area (VTA)). The prefrontal, retrosplenial and hippocampal regions were selected based, firstly, on their involvement in cognitive processing which is severely affected in the 22q11.1 carriers, and, secondly, on their robustly differential functional connectivity patterns, as compared to controls (Reinwald et al., 2020). As some seeds located at the edge of the brain might be prone to air-tissue interface artefacts, such as SN and VTA, we verified their coverage by overlaying them onto original EPI image. We detected a good coverage for both seeds (Figure S3), thus, ensuring that no air-tissue artifacts were present in this data. For seed-based analysis, non-eroded seed regions were used, as erosion might lead to unequal volume loss highly dependent on the original surface of the region. Nevertheless, additional analyses with eroded regions demonstrated high comparability to our original results (Figures S4, S5), underlining robustness of the connectivity findings. Mean timecourses of these regions were extracted for each animal using the unsmoothed pre-processed EPIs in order to avoid partial volume effect by e.g. cerebrospinal fluid signal from the adjacent ventricles. Then, voxel-wise functional connectivity maps were created by calculating correlation coefficients between these extracted regional timecourses and voxels covering the whole brain using the smoothed EPIs. Finally, correlation coefficients were transformed to Fisher z-scores to create z-score maps.
These z-score maps were fed into a second-level analysis, using two-sample T-tests and contrasts [1 −1] and [−1 1] to calculate differences from the control group (significance level at p<0.01, uncorrected). Here, we used uncorrected thresholded maps, as they serve as inputs to the next second-level statistical analysis and thus do not require correction for multiple testing. At this step we utilize a hypothesis-generating approach to predefine cluster regions for which we calculate different spatial associations with gene expression in the next step. We have compared these functional connectivity maps with the results of a network-based statistic (NBS) method obtained for the same dataset of Df(h22q11)/+ mice which we have recently published (Reinwald et al., 2020). The NBS relying on principles of cluster-based thresholding offers gain in power compared to mass-univariate testing with independent correction of p-values by generic procedures controlling family-wise error rate and detects subnetworks that might otherwise not be discovered (Zalesky et al., 2010). In our current seed-based approach we have found consistently similar patterns of connectivity as in NBS for all given brain regions, which strengthens the biological validity of our results.
Joint analysis of differential resting-state connectivity and spatial gene expression patterns
Besides an anatomical brain parcellation, the Allen Mouse Brain Atlas also provides high-resolution in situ hybridization gene expression maps (Lein et al., 2007). We followed our previously established protocol (Kumar et al., 2018) to analyse the expression patterns of genes belonging to the deleted chromosomal region in our mouse models to identify which gene expression patterns spatially overlapped with differential connectivity patterns (hyper- or hypoconnectivity with the seed) in these nine networks.
We downloaded all available 24 gene expression maps for genes belonging to the mouse 22q11.2 deletion. Of note, maps for three genes for the 22q11.2 deletion (Igll1, T10 and Mir185) were not available in the Allen Mouse Brain Atlas database.
We, then, used our Spatial Gene Expression Analysis Tool (https://github.com/UniversityOfIowaHealthCare/gene-expression-tool) to align all 24 gene expression maps into the Waxholm space (Johnson et al., 2010), as a common anatomical reference space.
To identify potential associations of differential resting-state connectivity with expression patterns of genes involved in the deletion, we extracted the gene expression (“energy”) values of all 24 genes overlapping with the clusters that stemmed from our seed-based analyses. In brief, significant raw gene expression data was converted into a 3D volume and thresholded and binarized to eliminate any noise effects from the conversion. Likewise, the clusters representing connectivity were thresholded and binarized to account for any potential spatial uncertainties from the transformation. From this point the masks were overlaid and the gene expression maps were checked for regional overlap with the connectivity clusters (Figure 2). This value was then used to calculate the relation between the two.
Gene expression values were pooled over all clusters from one distinct contrast (hypo- or hyperconnectivity) and distribution of gene expression values within the voxels of the clusters was compared to the distribution of gene expression values of all voxels over the entire brain using a rank sum test (http://de.mathworks.com/help/stats/ranksum.html) at a threshold of p<0.05 to identify genes that were significantly enriched in the clusters. The results are presented as fold changes, in which fold change=[(mean cluster expression – mean brain expression)/mean brain expression].
Figure 2. Overview over the pipeline for the joint analysis of gene expression patterns and differential resting state connectivity.

The top left image contains layered 2D expression images of the COMT gene which gets converted into the 3D image below. Please note that the lateral edges of the brain are not fully covered by the sagittal slices. Images then get binarized and aligned to the Waxholm space, which serves as a standard reference space for both gene expression maps and clusters indicating differential resting-state connectivity (top right image). After this alignment, a direct comparison between differential resting-state connectivity and gene expression patterns can be achieved.
Protein-protein-interaction network analysis of enriched genes
We used the STRING protein-protein interaction database to identify functional networks between enriched genes of a contrast (see also Supplement). This is a database that collects, scores and integrates publicly available information on interactions between proteins (termed in the database “functional associations” (Szklarczyk et al., 2019)). We used the web-based user interface of the database to upload the genes that were identified in previous analyses to significantly overlap with brain functional changes into the STRING database. This was done separately for every rs-fMRI network studied. The database then assesses whether a set of given genes or proteins is enriched in functional associations. This enrichment is assessed by comparing the observed number of functional associations between the proteins from the list to a null model. In other words, a question has been posed: is the number of edges (functional associations) between nodes (proteins) higher than can be expected by chance? If there were indeed more functional associations between proteins than can be expected by chance, we regarded that as indicative of a functional network formed by these proteins.
Analysis of co-expression patterns of COMT and TRMT2A in humans
COMT and TRMT2A expressions were explored in two human lymphoblastoid cell line (LCL) datasets (LCLBATCH1: N=407, with 187 schizophrenia patients and 220 HC; LCLBATCH2: N=449, with 225 schizophrenia patients and 224 healthy controls, respectively). Co-expression was subsequently explored in three human post-mortem brain expression datasets (combined N=151).
Expression microarray datasets were retrieved from the GEO database and dbGaP (Table S1) and pre-processed (for all details see Supplement). Surrogate variables were determined to account for the effect of unobserved confounders using the R library sva (Leek et al., 2012), specifying diagnostic group, and known confounders (age, age2, sex) as variables of interest. Multiple regression was applied to residualize the expression levels of each gene for a given dataset with respect to age, age2, sex and the first 10 surrogate variables. The residualized expression levels were ranked prior to downstream analysis. Co-expression between COMT and TRMT2A and interactions with diagnosis were assessed using multiple linear regression. Post-hoc analyses of diagnosis-specific co-expression were performed using Pearson correlation analysis (p<0.05). As we used a discovery-validation approach, the p-values related to the significance of the COMT-TRMT2A co-expression, were not corrected for multiple testing.
RESULTS
Functional connectivity analysis
Seed-based analyses detected predominantly higher connectivity for the VTA (Figure 3A) and SN (Figure 3B) covering the prefrontal and somatosensory cortices, caudate putamen (CPu), hippocampal areas, periaqueductal gray and superior colliculus (p<0.01) in the Df(h22q11)/+ mice, as compared to WT group. Contrary, CA (Figure 3C) and DG (Figure 3D) exhibited overall reduced connectivity (p<0.01), except for VTA and SN. The Acb manifested both reduced (with IL, somatosensory cortex, presubiculum) and increased (with insula, cortical amygdaloid area, Cpu) correlations (p<0.01) (Figure 3E).
Figure 3. Between-group comparisons of seed-based resting-state functional connectivity for the subcortical brain regions between Df(h22q11)/+ and wild type mice.
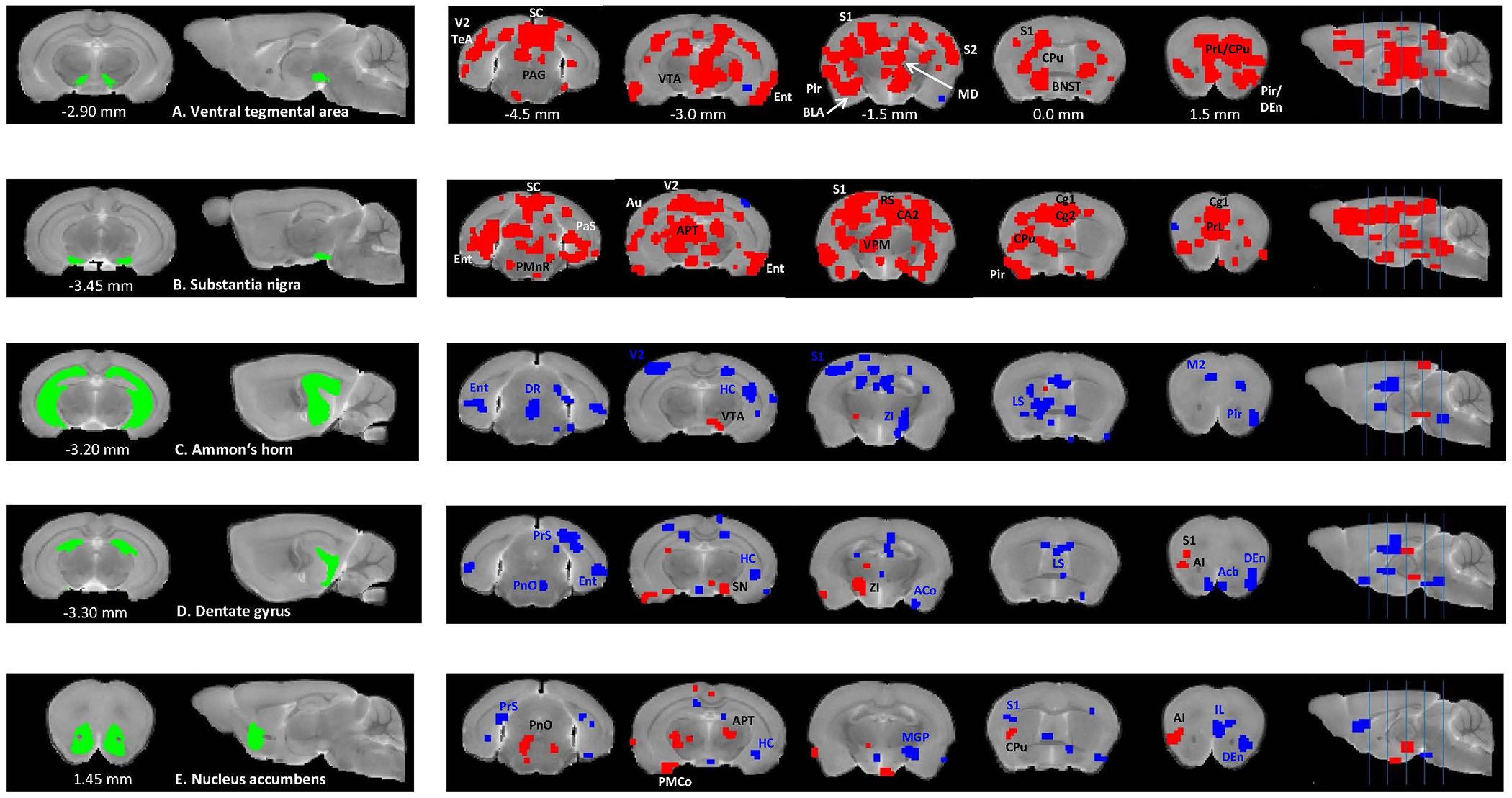
Two-sample t-tests (p <0.01) show significantly higher (red) and lower (blue) connectivity between ventral tegmental area (A), substantia nigra (B), Ammon’s horn (C), dentate gyrus (D), nucleus accumbens (E), and multiple cortical and subcortical brain regions in Df(h22q11)/+ mice, compared to the control group. Coordinates are in mm to Bregma. Abbreviations: ACo – anterior cortical amygdaloid nucleus, AI – agranular insular cortex, APT – anterior pretectal nucleus, Au – auditory cortex, BLA – basolateral amygdaloid nucleus, BNST – bed nucleus of stria terminalis, CA2 – field CA2 of the hippocampus, Cg1 – cingulate cortex area 1, Cg2 - cingulate cortex area 2, CPu – caudate putamen, DEn – dorsal endopiriform nucleus, DR – dorsal raphe nucleus, Ent – entorhinal cortex, HC - hippocampus, LS – lateral septal nucleus, M2 – secondary motor cortex, MD – mediodorsal thalamic nucleus, MGP – medial globus pallidus, PAG – periaqueductal gray, PaS - parasubiculum, Pir – piriform cortex, PMnR – paramedian raphe nucleus, PMCo – posteromedial cortical amygdaloid area, PnO – pontine reticular nucleus oral part, PRh – perirhinal cortex, PrL – prelimbic cortex, PrS - presubiculum, S1 – primary somatosensory cortex, S2 - secondary somatosensory cortex, SC – superior colliculus, TeA – temporal association cortex, V2 – secondary visual cortex, VPM – ventral posteromedial thalamic nucleus, ZI – zona incerta.
In the ACC, the seed-based analysis detected reduced connectivity with the superior and inferior colliculi, visual and temporal association cortices, hippocampus and increased connectivity with the substantia nigra (p<0.01) in the Df(h22q11)/+ mice, as compared to WT group (Figure 4A). The IL and OC presented similar patterns and, in addition to the SN, an increased connectivity with the amygdala (p<0.01) (Figures 4B, 4C). Finally, the RS also exhibited reduced correlations with the inferior colliculus, hippocampus (DG and CA1), but additionally increased connectivity with the amygdalohippocampal area, bed nucleus of stria terminalis and Acb (p<0.01) (Figure 4D).
Figure 4. Between-group comparisons of seed-based rs-FC for the cortical brain regions between Df(h22q11)/+ and wild type mice.
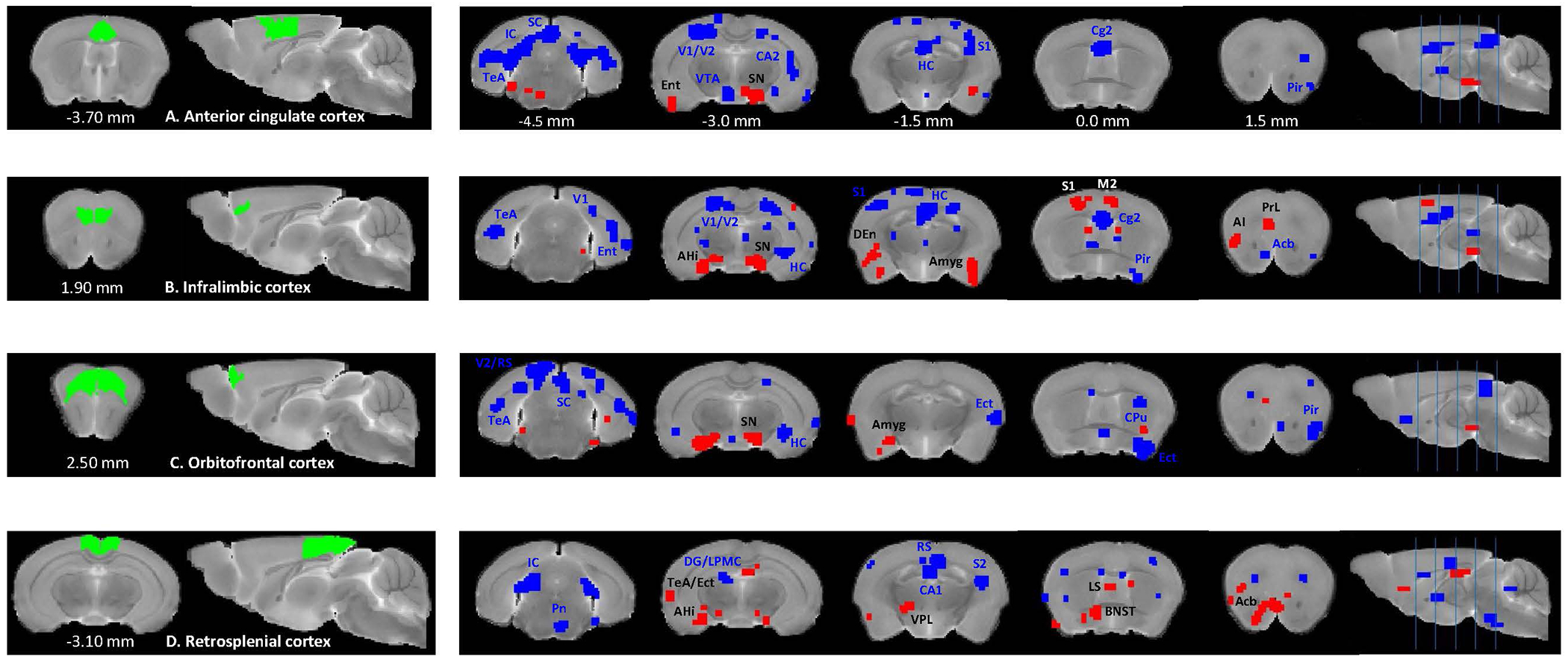
Two-sample t-tests (p <0.01) show significantly higher (red) and lower (blue) connectivity between anterior cingulate cortex (A), infralimbic cortex (B), orbitofrontal cortex (C) and retrosplenial cortex (D) and multiple cortical and subcortical brain regions in Df(h22q11)/+ mice, compared to the control group. Coordinates are in mm to Bregma. Abbreviations: AHi – amygdalohippocampal area, Amyg - amygdala, Ect – ectorhinal cortex, LPMC – lateral posterior thalamic nucleus mediocaudal part, Pn – pontine nuclei, V1 – primary visual cortex, VPL – ventral posterolateral thalamic nucleus. Other abbreviations are the same as in Figure 3.
Identification of genes with expression patterns that are spatially associated with differential connectivity patterns
In a next step, we aimed to explore which of the deleted genes showed expression patterns that were spatially associated with differential resting-state connectivity in the Df(h22q11)/+ mice. The results of our analyses are summarized in Figure 5. We found significant associations between differential resting-state connectivity and spatial gene expression patterns for all contrasts. The number of individual genes implicated ranged from 7 (seed: CA) to 19 (seed: IL) for regions showing hyperconnectivity, and 4 (seed: DG) to 18 (seed: ACC) for regions with hypoconnectivity to the seeds.
Figure 5. Genes overexpressed in networks indicating hyper- and hypoconnectivity for seed regions.
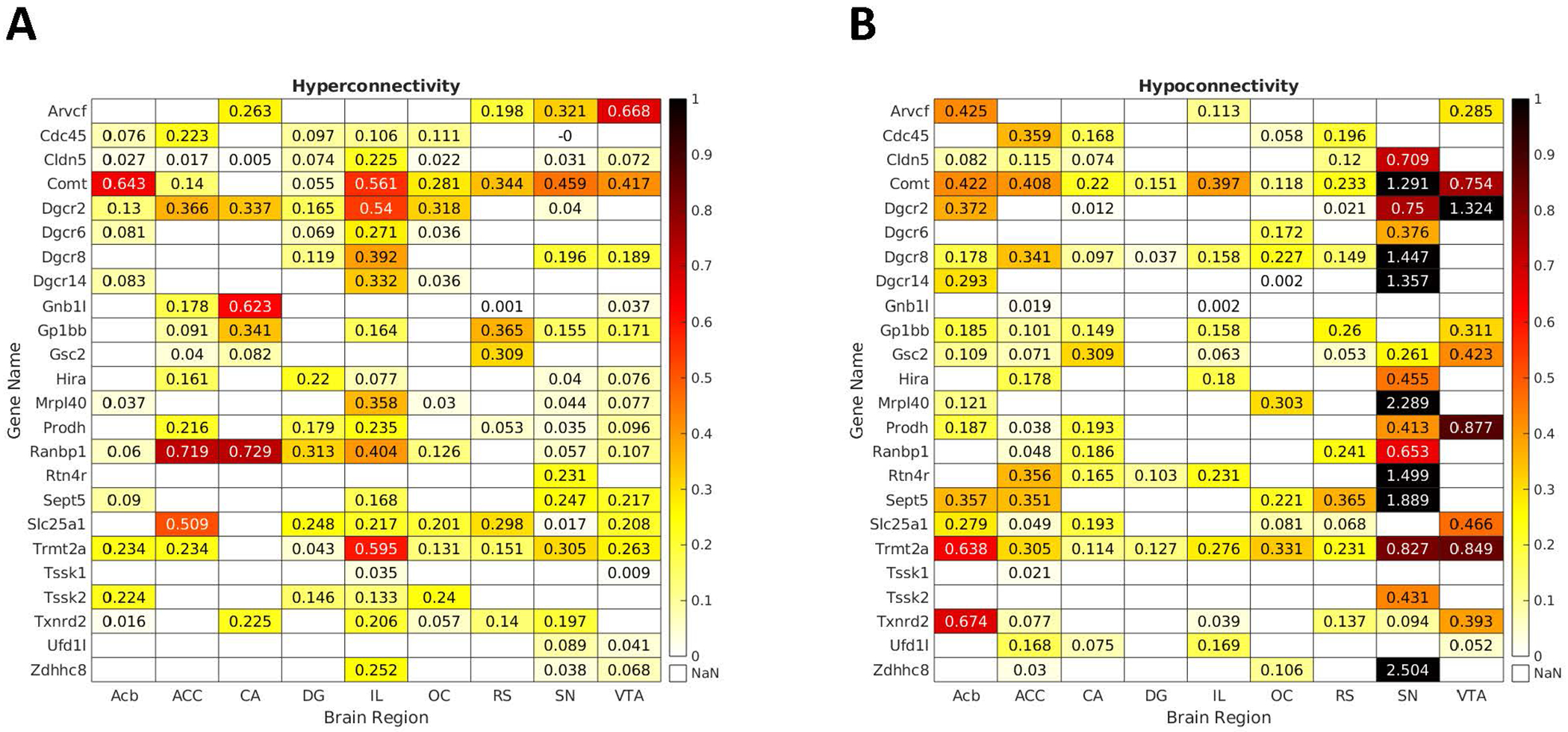
Depicted are the matrices presenting fold changes between neural networks hyper- (left) and hypoconnected (right) to the seed regions and spatial expression patterns of the hemi-deleted genes in these networks. Only values for significant overexpression are provided. Comt and Trmt2a were the most consistently implicated genes across the studied networks. Other genes showed expression patterns that were associated either with hyper- or hypoconnected networks.
Comt and Trmt2a were the most consistently implicated genes across the studied networks. Comt expression patterns were associated both with networks hyperconnected (except the CA) and hypoconnected to all seeds. Trmt2a expression patterns showed similar association patterns as Comt for hyperconnected networks (also except the CA), and were significantly associated with all hypoconnected networks.
Other genes showed expression patterns that were associated either with hyper- or hypoconnected networks. Expression patterns of Ranbp1 were associated with all networks hyperconnected to their seeds, except for RS. Dgcr8 expression patterns showed the opposite direction, as they were associated with all networks hypoconnected to their seeds, except for VTA.
Genes implicated as associated with differential connectivity form networks above chance level
For all networks, genes formed protein-protein-interaction networks above chance level. This indicates that the proteins, encoded by the respective genes, are functionally related and linked to common biological tasks.
We, next, wanted to explore the specific biological properties, which these protein-protein-interaction networks would mediate. A data-driven search identified an axonal localization of their gene products as the main characteristic of the genes implicated across all contrasts (both hyper- and hypoconnectivity). Across regions with hyperconnectivity to the seeds, our analyses identified the terms “methyltransferase”, “regulation of chromatin silencing” and “response to drug” as most enriched. For regions with hypoconnectivity to the seeds, a slightly different picture emerged, with “transit peptide” and “leucine rich repeat C-terminal and N-terminal domains” being enriched, besides “methyltransferase activity” (Figure 6).
Figure 6. Protein-protein interaction networks overexpressed in networks indicating hyper- and hypoconnectivity.
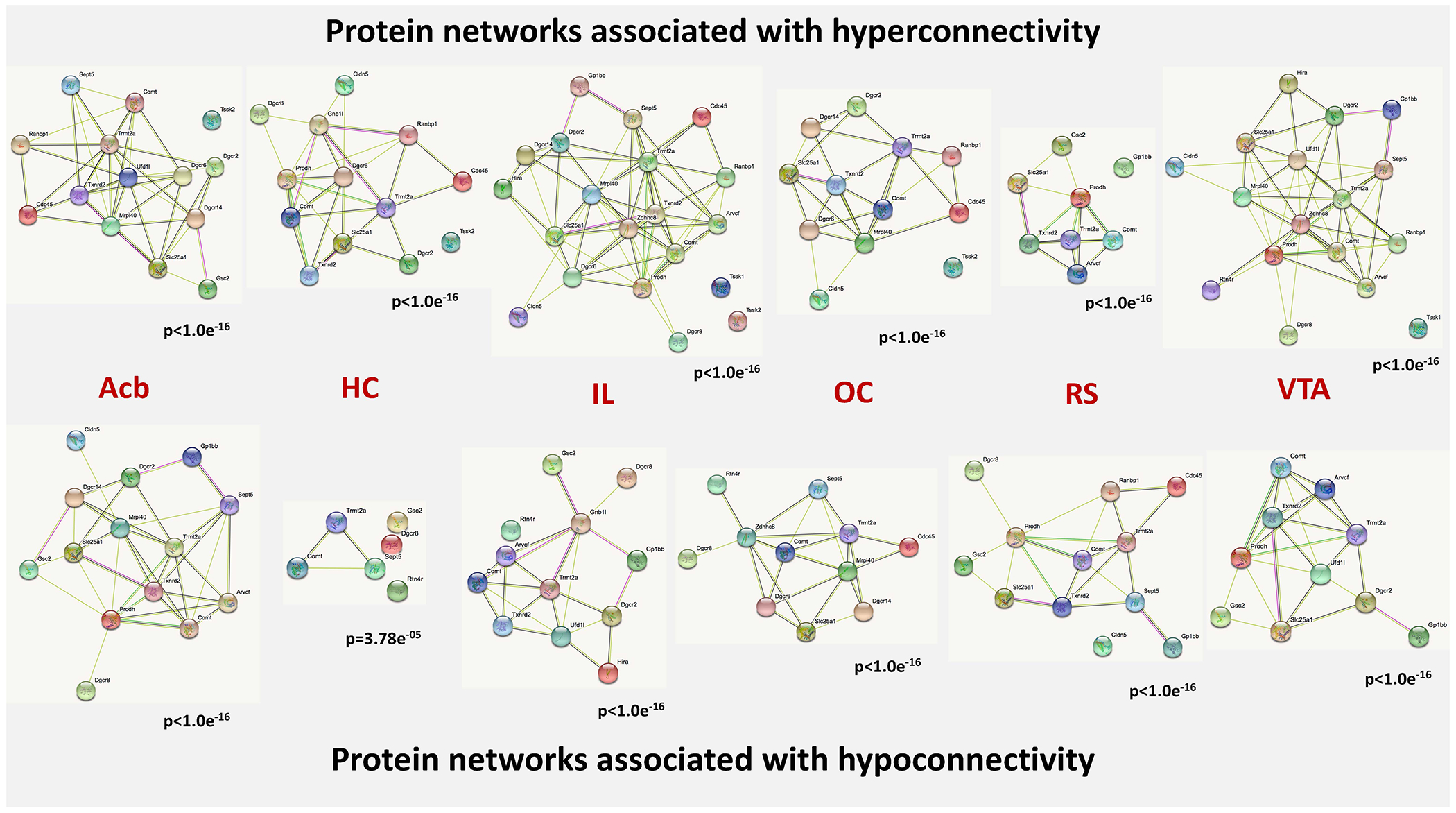
The nodes of the network represent the proteins, which are encoded by the genes entered into our analysis. The content of the node indicates, whether the 3D structure of the gene product is known or not. The colors of the edges indicate the kind of evidence for protein-protein interactions. Nodes (proteins) without edges indicate that a protein has no functional associations with other proteins of that contrast. The p-values under the networks indicate the likelihood that the involved genes form a protein-protein network above chance level. Known interactions: light blue – from curated databases, pink – experimentally determined; predicted interactions: green – gene neighborhood, red – gene fusions, dark blue – gene co-occurrence; other interactions: light green – text mining, black – co-expression, violet – protein homology. Abbreviations: Acb - nucleus accumbens, HC - hippocampus, IL - infralimbic cortex, OC - orbitofrontal cortex, RS - retrosplenial cortex, VTA - ventral tegmental area.
COMT and TRMT2A are co-expressed in the CNS in healthy controls, but not in schizophrenia patients
As our analyses indicated an association of Comt and Trmt2a expression patterns with aberrant brain function in our mouse model of 22q11.2 del, we wanted to investigate potential co-expression patterns of these two genes in specimens of actual human schizophrenia patients to explore a translational relevance. We found that in two lymphoblastoid cell line datasets, COMT and TRMT2A were significantly, but weakly, co-expressed independent of diagnosis (β=0.17, p=4·10−4 and β=0.10, p=0.04, respectively).
Consistent with findings from LCL data, COMT and TRMT2A expression levels were significantly associated in three human post-mortem brain gene expression datasets (β=0.23, p=0.004). In contrast to the peripheral blood, however, there was a significant effect of diagnosis on co-expression (p=0.01), and post-hoc analysis demonstrated that the two genes were co-expressed in controls (rho=0.46, p=4.1·10−5), but not in patients (p>0.75). Expression did not significantly differ between patients and controls.
DISCUSSION
22q11.2 deletion is the most important single genetic risk factor associated with schizophrenia, but also with other neuropsychiatric disorders, such as ASD (Ousley et al., 2017; Bassett and Chow, 2008). While the impact of the entire CNV on brain structure (Sun et al., 2020; Villalon-Reina et al., 2020) and function (Zoller et al., 2019) has been studied in humans and animals (Reinwald et al., 2020), there still is a critical gap in understanding, how these neural changes mechanistically emerge and which genes within the deletion are involved in mediating these effects. In our current study, we linked changes of resting-state connectivity in a mouse model of 22q11.2 deletion to spatial expression patterns of genes involved in the deletion. The two most consistently implicated genes by this approach across all networks, COMT and TRMT2A, showed co-expression in the brain of healthy human subjects, but not in schizophrenia patients. In the peripheral tissue, the two genes were co-expressed both in patients and controls. Since our mouse model captures schizophrenia features and the human samples were extracted from schizophrenia patients, we discuss our findings in the context of schizophrenia.
The major changes in functional connectivity comprised hyperconnectivity for the dopaminergic pathways of the ventral tegmental area and substantia nigra, and opposing predominant hypoconnectivity for the rest of the investigated cortical and subcortical areas, particularly reduced hippocampal-prefrontal connectivity and hypoconnectivity for the auditory, visual and fronto-temporal structures. These patterns consistently replicate connectivity abnormalities in schizophrenia (Brandl et al., 2019) and findings from mouse models of the 22q11.2 deletion (Sigurdsson et al., 2010; Chun et al., 2017; Hamm et al., 2017) and from human carriers (Mattiaccio et al., 2016; Larsen et al., 2018). Hippocampal-prefrontal hypoconnectivity is a central feature of many neuropsychiatric syndromes hypothesized to underlie cognitive impairment (Godsil et al., 2013), while the aberrant connectivity within the sensory systems could serve as a marker of risk for developing psychotic symptoms (Chun et al., 2017; Hamm et al., 2017; Ottet et al., 2013).
Of note, genes associated with hyper- or hypoconnected networks across seeds encode proteins that interacted with each other above chance level. An enrichment of genes with axonal function and/or localization across all regions with hyper- and hypoconnectivity appear as plausible molecular underpinning of changes in resting-state networks (Richiardi et al., 2015).
Although some regions (e.g., the superior colliculus, secondary visual or primary somatosensory cortex) showed changes of connectivity for several seeds, there was no common spatial signature across all networks. Despite this heterogeneity, the expression patterns of Comt and Trmt2a demonstrated consistent spatial association with changes in functional connectivity. We would interpret this constellation as supportive of a plausible biological involvement of these genes in the emergence of brain functional changes in our model, rather than an artifact driven by the involvement of identical brain regions across networks.
As the length of the 22q11.2 deletion doesn’t affect schizophrenia risk, it is possible that only several genes are critical. Among genes present in different types of deletion is COMT (Weksberg et al., 2007). However, as COMT is also present in 22q11.2 carriers with no history of psychosis (Weksberg et al., 2007), it appears more plausible that gene-gene and gene-environment interactions, rather than a single gene, would play a causal role in inducing schizophrenia. COMT is one of the best studied genes in the context of psychiatric disorders (Hiroi et al., 2019) and cognition. COMT genotype (Val108/158Met) affects prefrontal (Dennis et al., 2010), prefrontal-hippocampal (Bertolino et al., 2006) and default-mode network connectivity (Liu et al., 2010) during cognitive processing and at rest in healthy individuals (Tunbridge et al., 2013) (however, also see (Nickl-Jockschat et al., 2015)). Interestingly, it affects neural pathways and connectivity related to cognitive performance in patients with schizophrenia differentially, than in healthy individuals (Wang et al., 2018). Given its role in the regulation of catecholaminergic, particularly, dopaminergic signaling (Mannisto and Kaakkola, 1999), COMT has been hypothesized to be a major player in the manifestation of psychosis (Tunbridge et al., 2006). COMT dose alterations due to a varying number of copies of 22q11.2 segment affect development of working memory capacity, a critical component of executive function compromised in neuropsychiatric disorders (Boku et al., 2018). Our own data demonstrating a consistent association between its expression and patterns of hypo- and hyperconnectivity across seeds fits the view of COMT as a gene with central pathophysiological relevance within the deletion. It is not surprising that both connectivity directions (hyper- and hypo-) were associated with Comt expression, as catecholamines produce both excitatory and inhibitory effects acting on different types of receptors. The monoallelic deletion of Comt also provides a mechanistic explanation for the observed hyperconnectivity of the dopaminergic ventral tegmental area and substantia nigra in the Df(h22q11)/+ mice that parallels the observed hyperdopaminergic state in patients with 22q11.2 deletion syndrome (Butcher et al., 2017). However, it should be noted that Comt expression patterns weren’t exclusively associated with classical dopaminergic networks, which could indicate a pathomechanism beyond a simple up-regulation of dopamine levels due to reduced degradation.
Trmt2a, the second implicated gene, is much less extensively studied. It encodes a transfer RNA (tRNA) methyltransferase, which catalyzes the post-transcriptional chemical modification of tRNA, stabilizing its structure and increasing the fidelity and efficiency of protein synthesis (Chang et al., 2019). Additionally, this gene has been linked to deficits in sustained attention in schizophrenia patients, which might represent a disease endophenotype (Liu et al., 2007). Of note, a potential epistasis between COMT and TRMT2A has not been described yet. However, our translational analyses in humans support the idea of a potential connection between the two, namely our finding of a co-expression of TRMT2A and COMT in the healthy human brain, which was disrupted in schizophrenia patients. This finding further supports the notion of COMT and TRMT2A as being relevant for brain connectivity and, hence, behavior in 22q11.2 deletion syndrome and should be regarded as an important translational validation marker. There is an intriguing possibility of an epistatic interaction between TRMT2A and COMT accomplished via a cross-talk between protein biosynthesis and neurotransmission or vice versa, which could be disrupted in schizophrenia.
Two other genes had a consistent association either with hyper- or hyponnectivity. The first one was Ranbp1 whose expression patterns were associated with all networks hyperconnected to their seeds, except for the retrosplenial cortex. Its product is RNA-binding G-protein participating in cell cycle (Clements et al., 2017) and involved in the metabotropic glutamate receptor gene network (Wenger et al., 2016). A homozygous knockout of this gene results in microcephaly and selective disruption of layer 2/3 cortical projection neuron generation (Paronett et al., 2015). The association between Ranbp1 expression and hyperconnectivity in the Df(h22q11)/+ mice might result from its involvement in excitatory glutamatergic signaling and the generation of cortical projections.
In contrast to Ranbp1, the Dgcr8 expression patterns showed the opposite association, as they were associated with all networks hypoconnected to their seeds, except for the ventral tegmental area. A conditional deletion of Dgcr8 in cortical pyramidal neurons leads to reduction of interneurons in the prefrontal cortex, accompanied by an inhibitory synaptic transmission deficit (Hsu et al., 2012), which could serve as a mechanistic explanation of the observed association between Dgcr8 expression and hypoconnectivity in the Df(h22q11)/+ mice.
There are several limitations in this study. First, we relied upon gene expression maps from the Allen Mouse Brain Atlas, which are derived from wild type mice. Expression patterns differ across mouse strains and can be reasonably expected to be different in animals modeling 22q11.2 deletion. However, it should be noted that a disruption of Comt and Trmt2a co-expression, as indicated by our analyses in mice, obviously has a translational capacity in human schizophrenia patients. Second, it should be noted that the Allen Mouse Brain Atlas did not provide an expression map for every gene in the deletion. In detail, 3 maps (Igll1, T10 and Mir185) were missing. Although we would not expect our results to change decisively with these three maps added, we do acknowledge that their lack in the analysis might distort the results to a certain degree. Third, substantia nigra and ventral tegmental area displayed the strongest results in connectivity (hyperconnectivity) and gene expression, however the latter correlated with hypoconnectity pattern, possibly due to the fact that smaller connectivity clusters had lower variance and thus possibly deviated more from the average distribution in the brain. Therefore, the statistical strength of the results should be interpreted with caution. Also, we did not correct our gene expression analyses for multiple comparisons. As the Allen Mouse Brain Atlas only provides a single expression map for most genes (Lein et al., 2007), corrections for multiple comparisons would go along with a significantly increased risk for false negative findings. To the best of our knowledge, none of the studies in the field has so far applied such corrections (Fakhry and Ji, 2015; Kumar et al., 2018; Fernandes et al., 2017). As this approach goes along with a risk for false positive findings, we would regard our approach as an option to formulate observer-independent data-driven hypotheses on the potential underpinnings of changes in brain function or structure, but have to emphasize that these hypotheses need verification by additional experiments (Kumar et al., 2018). Importantly, our findings in human datasets of schizophrenia patients and healthy controls support the idea of a potentially central role for COMT and TRMT2A in mediating the impact of 22q11.2 deletion on the brain. Finally, while the Df(h22q11)/+ mouse model of human 22q11.2 deletion shows behavioral and electrophysiological phenotypes associated with schizophrenia - and, hence, the focus of our approach and discussion has been placed accordingly, - it should be noted that this CNV is nosologically unspecific. 22q11.2 deletion is associated with a broad set of neuropsychiatric disorders, such as ASD, attention-deficit hyperactivity disorder, anxiety disorders, intellectual disability (Schneider et al., 2014; Zinkstok et al., 2019), and obsessive-compulsive disorders (Gothelf et al., 2004; Sebat et al., 2009). Our findings of disrupted co-expression patterns between COMT and Trmt2a in post-mortem brain samples of human patients point towards a translational relevance of our results at least in schizophrenia. However, given the lack of nosological specificity of 22q11.2 deletion, it remains to be clarified whether that holds up for other disorders associated with this CNV. Consequently, the results obtained in our mouse model should be rather interpreted as the effects of a given CNV on brain connectivity, than regarded as a rodent model of a particular disease.
Acknowledging these caveats, our findings suggest that COMT and TRMT2A form a core genetic component modulating psychosis risk via differential neural activity in 22q11.2 deletion. A better understanding of the interplay of these two genes, along with other genes within the deletion, yields the potential to therapeutically influence liability for psychosis in 22q11 deletion.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Felix Hörner and Claudia Falfan-Melgoza for excellent technical assistance.
FUNDING
NEWMEDS – the research leading to these results, has received support from the Innovative Medicine Initiative Joint Undertaking under Grant Agreement no.115008 of which resources are composed of European Federation of Pharmaceutical Industries and Associations (EFPIA) in-kind contribution and financial contribution from the European Union’s Seventh Framework Programme (FP7/ 2007-2013). This work was also supported by the grants from the German Research Foundation (Deutsche Forschungsgemeinschaft): DFG SA 1869/15-1 and DFG GA 2109/2-1 to N.G., DFG SA 1869/11-2 within the priority program (SPP1629) “Thyroid Trans Act” to A.S. and J.R., and EB 187/8-1 within the Clinician Scientist Program of the Medical Faculty Mannheim, Heidelberg University, to J.R. The authors would like to thank the University of Iowa for their in-house funding of this project. We would also like to thank Dr. Michael Hawrylylcz and his team at the Allen Institute for Brain Science, Seattle, WA, USA, for their continued collaboration and support.
N.G., Z.P., A.S., W.W.-F., J.R., M.S., J.C., H.C., G.K., E.S. and T.N.-J. have no conflicts of interest to report. M.D. and T.B.S. receive full-time salaries from H. Lundbeck A/S, Copenhagen, Denmark. A.J.S. is employed at Takeda Pharmaceuticals, Cambridge, Massachusetts, USA. A.M.L. has received consultant fees from Blueprint Partnership, Boehringer Ingelheim, Daimler und Benz Stiftung, Elsevier, F. Hoffmann-La Roche, ICARE Schizophrenia, K. G. Jebsen Foundation, L.E.K. Consulting, Lundbeck International Foundation (LINF), R. Adamczak, Roche Pharma, Science Foundation, Synapsis Foundation–Alzheimer Research Switzerland, and System Analytics and has received lectures including travel fees from Boehringer Ingelheim, Fama Public Relations, Institut d’investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Janssen-Cilag, Klinikum Christophsbad, Göppingen, Lilly Deutschland, Luzerner Psychiatrie, LVR Klinikum Düsseldorf, LWL Psychiatrie Verbund Westfalen-Lippe, Otsuka Pharmaceuticals, Reunions i Ciencia S. L., Spanish Society of Psychiatry, Südwestrundfunk Fernsehen, Stern TV, and Vitos Klinikum Kurhessen.
REFERENCES
- Afyouni S, Nichols TE, 2018. Insight and inference for DVARS. Neuroimage. 172, 291–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, 2008. Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep. 10(2), 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolino A, Rubino V, Sambataro F, Blasi G, Latorre V, Fazio L, et al. , 2006. Prefrontal-hippocampal coupling during memory processing is modulated by COMT val158met genotype. Biol Psychiatry. 60(11), 1250–1258. [DOI] [PubMed] [Google Scholar]
- Boku S, Izumi T, Abe S, Takahashi T, Nishi A, Nomaru H, et al. , 2018. Copy number elevation of 22q11.2 genes arrests the developmental maturation of working memory capacity and adult hippocampal neurogenesis. Mol Psychiatry. 23(4), 985–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl F, Avram M, Weise B, Shang J, Simoes B, Bertram T, et al. , 2019. Specific Substantial Dysconnectivity in Schizophrenia: A Transdiagnostic Multimodal Meta-analysis of Resting-State Functional and Structural Magnetic Resonance Imaging Studies. Biol Psychiatry. 85, 573–583. [DOI] [PubMed] [Google Scholar]
- Butcher NJ, Marras C, Pondal M, Rusjan P, Boot E, Christopher L, et al. , 2017. Neuroimaging and clinical features in adults with a 22q11.2 deletion at risk of Parkinson’s disease. Brain. 140, 1371–1383. [DOI] [PubMed] [Google Scholar]
- Chang YH, Nishimura S, Oishi H, Kelly VP, Kuno A, Takahashi S, 2019. TRMT2A is a novel cell cycle regulator that suppresses cell proliferation. Biochem Biophys Res Commun. 508, 410–415. [DOI] [PubMed] [Google Scholar]
- Chun S, Du F, Westmoreland JJ, Han SB, Wang YD, Eddins D, et al. , 2017. Thalamic miR-338-3p mediates auditory thalamocortical disruption and its late onset in models of 22q11.2 microdeletion. Nat Med. 23, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements CC, Wenger TL, Zoltowski AR, Bertollo JR, Miller JS, de Marchena AB, et al. , 2017. Critical region within 22q11.2 linked to higher rate of autism spectrum disorder. Mol Autism. 8, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RW, 1996. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res. 29, 162–173. [DOI] [PubMed] [Google Scholar]
- Dennis NA, Need AC, LaBar KS, Waters-Metenier S, Cirulli ET, Kragel J, et al. , 2010. COMT val108/158 met genotype affects neural but not cognitive processing in healthy individuals. Cereb Cortex. 20(3), 672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didriksen M, Fejgin K, Nilsson SR, Birknow MR, Grayton HM, Larsen PH, et al. , 2017. Persistent gating deficit and increased sensitivity to NMDA receptor antagonism after puberty in a new mouse model of the human 22q11.2 microdeletion syndrome: a study in male mice. J Psychiatry Neurosci. 42, 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr AE, Lerch JP, Spring S, Kabani N, Henkelman RM, 2008. High resolution three-dimensional brain atlas using an average magnetic resonance image of 40 adult C57Bl/6J mice. Neuroimage. 42, 60–69. [DOI] [PubMed] [Google Scholar]
- Earls LR, Bayazitov IT, Fricke RG, Berry RB, Illingworth E, Mittleman G, et al. , 2010. Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. J Neurosci. 30, 15843–15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhry A, Ji S, 2015. High-resolution prediction of mouse brain connectivity using gene expression patterns. Methods. 73, 71–78. [DOI] [PubMed] [Google Scholar]
- Fernandes DJ, Ellegood J, Askalan R, Blakely RD, Dicicco-Bloom E, Egan SE, et al. , 2017. Spatial gene expression analysis of neuroanatomical differences in mouse models. Neuroimage. 163, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass N, Weber-Fahr W, Sartorius A, Becker R, Didriksen M, Stensbol TB, et al. , 2016. An acetylcholine alpha7 positive allosteric modulator rescues a schizophrenia-associated brain endophenotype in the 15q13.3 microdeletion, encompassing CHRNA7. Eur Neuropsychopharmacol. 26, 1150–1160. [DOI] [PubMed] [Google Scholar]
- Godsil BP, Kiss JP, Spedding M, Jay TM, 2013. The hippocampal-prefrontal pathway: the weak link in psychiatric disorders? Eur Neuropsychopharmacol. 23, 1165–1181. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Presburger G, Zohar AH, Burg M, Nahmani A, Frydman M, et al. , 2004. Obsessive-compulsive disorder in patients with velocardiofacial (22q11 deletion) syndrome. Am J Med Genet B Neuropsychiatr Genet. 126B, 99–105. [DOI] [PubMed] [Google Scholar]
- Hamm JP, Peterka DS, Gogos JA, Yuste R, 2017. Altered Cortical Ensembles in Mouse Models of Schizophrenia. Neuron. 94, 153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi N, Yamauchi T, 2019. Modeling and Predicting Developmental Trajectories of Neuropsychiatric Dimensions Associated With Copy Number Variations. Int J Neuropsychopharmacol. 22, 488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu R, Schofield CM, Dela Cruz CG, Jones-Davis DM, Blelloch R, Ullian EM, 2012. Loss of microRNAs in pyramidal neurons leads to specific changes in inhibitory synaptic transmission in the prefrontal cortex. Mol Cell Neurosci. 50, 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, et al. , 2009. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 460, 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GA, Badea A, Brandenburg J, Cofer G, Fubara B, Liu S, et al. , 2010. Waxholm space: an image-based reference for coordinating mouse brain research. Neuroimage. 53, 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimber WL, Hsieh P, Hirotsune S, Yuva-Paylor L, Sutherland HF, Chen A, et al. , 1999. Deletion of 150 kb in the minimal DiGeorge/velocardiofacial syndrome critical region in mouse. Hum Mol Genet. 8, 2229–2237. [DOI] [PubMed] [Google Scholar]
- Kumar VJ, Grissom NM, McKee SE, Schoch H, Bowman N, Havekes R, et al. , 2018. Linking spatial gene expression patterns to sex-specific brain structural changes on a mouse model of 16p11.2 hemideletion. Transl Psychiatry. 8, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen KM, Morup M, Birknow MR, Fischer E, Hulme O, Vangkilde A, et al. , 2018. Altered auditory processing and effective connectivity in 22q11.2 deletion syndrome. Schizophr Res. 197, 328–336. [DOI] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD, 2012. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. , 2007. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Liu YL, Fann CS, Liu CM, Chang CC, Yang WC, Wu JY, et al. , 2007. HTF9C gene of 22q11.21 region associates with schizophrenia having deficit-sustained attention. Psychiatr Genet. 17, 333–338. [DOI] [PubMed] [Google Scholar]
- Liu B, Song M, Li J, Liu Y, Li K, Yu C, Jiang T, 2010. Prefrontal-related functional connectivities within the default network are modulated by COMT val158met in healthy young adults. J Neurosci. 30(1), 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannisto PT, Kaakkola S. 1999. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 51, 593–628. [PubMed] [Google Scholar]
- Mattiaccio LM, Coman IL, Schreiner MJ, Antshel KM, Fremont WP, Bearden CE, et al. , 2016. Atypical functional connectivity in resting-state networks of individuals with 22q11.2 deletion syndrome: associations with neurocognitive and psychiatric functioning. J Neurodev Disord. 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, et al. , 2008. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol Cell Neurosci. 39, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, et al. , 2001. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 104, 619–629. [DOI] [PubMed] [Google Scholar]
- Michaelovsky E, Carmel M, Frisch A, Salmon-Divon M, Pasmanik-Chor M, Weizman A, et al. , 2019. Risk gene-set and pathways in 22q11.2 deletion-related schizophrenia: a genealogical molecular approach. Transl Psychiatry. 9, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow BE, McDonald-McGinn DM, Emanuel BS, Vermeesch JR, Scambler. P.J., 2018. Molecular genetics of 22q11.2 deletion syndrome. Am J Med Genet A. 176, 2070–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickl-Jockschat T, Janouschek H, Eickhoff SB, Eickhoff CR, 2015. Lack of meta-analytic evidence for an impact of COMT Val158Met genotype on brain activation during working memory tasks. Biol Psychiatry. 78(11), e43–46. [DOI] [PubMed] [Google Scholar]
- Ottet MC, Schaer M, Cammoun L, Schneider M, Debbane M, Thiran JP, et al. , 2013. Reduced fronto-temporal and limbic connectivity in the 22q11.2 deletion syndrome: vulnerability markers for developing schizophrenia? PLoS One. 8, e58429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousley O, Evans AN, Fernandez-Carriba S, Smearman EL, Rockers K, Morrier MJ, et al. , 2017. Examining the Overlap between Autism Spectrum Disorder and 22q11.2 Deletion Syndrome. Int J Mol Sci. 18, 1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packham EA, Brook JD, 2003. T-box genes in human disorders. Hum Mol Genet. 12 Spec No 1: R37–44. [DOI] [PubMed] [Google Scholar]
- Paronett EM, Meechan DW, Karpinski BA, LaMantia AS, Maynard TM, 2015. Ranbp1, Deleted in DiGeorge/22q11.2 Deletion Syndrome, is a Microcephaly Gene That Selectively Disrupts Layer 2/3 Cortical Projection Neuron Generation. Cereb Cortex. 25, 3977–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE, 2012. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage. 59, 2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinwald JR, Sartorius A, Weber-Fahr W, Sack M, Becker R, Didriksen M, et al. , 2020. Separable neural mechanisms for the pleiotropic association of copy number variants with neuropsychiatric traits. Transl Psychiatry. 10(1), 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richiardi J, Altmann A, Milazzo AC, Chang C, Chakravarty MM, Banaschewski T, et al. , 2015. BRAIN NETWORKS. Correlated gene expression supports synchronous activity in brain networks. Science. 348, 1241–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Debbané M, Bassett AS, Chow EW, Fung WL, van den Bree M, et al. , 2014. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry. 171(6), 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Levy DL, McCarthy SE, 2009. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 25, 528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA, 2010. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 464, 763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivagnanasundaram S, Fletcher D, Hubank M, Illingworth E, Skuse D, Scambler P, 2007. Differential gene expression in the hippocampus of the Df1/+ mice: a model for 22q11.2 deletion syndrome and schizophrenia. Brain Res. 1139, 48–59. [DOI] [PubMed] [Google Scholar]
- Stark KL, Xu B, Bagchi A, Lai WS, Liu H, Hsu R, et al. , 2008. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet. 40, 751–760. [DOI] [PubMed] [Google Scholar]
- Sun D, Ching CRK, Lin A, Forsyth JK, Kushan L, Vajdi A, et al. , 2020. Large-scale mapping of cortical alterations in 22q11.2 deletion syndrome: Convergence with idiopathic psychosis and effects of deletion size. Mol Psychiatry. 25, 1822–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. , 2019. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunbridge EM, Harrison PJ, Weinberger DR, 2006. Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry. 60, 141–151. [DOI] [PubMed] [Google Scholar]
- Tunbridge EM, Farrell SM, Harrison PJ, Mackay CE, 2013. Catechol-O-methyltransferase (COMT) influences the connectivity of the prefrontal cortex at rest. Neuroimage. 68, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Buuren M, Gladwin TE, Zandbelt BB, van den Heuvel M, Ramsey NF, Kahn RS, et al. , 2009. Cardiorespiratory effects on default-mode network activity as measured with fMRI. Hum Brain Mapp. 30, 3031–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalon-Reina JE, Martinez K, Qu X, Ching CRK, Nir TM, Kothapalli D, et al. , 2020. Altered white matter microstructure in 22q11.2 deletion syndrome: a multisite diffusion tensor imaging study. Mol Psychiatry. 25, 2818–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhang B, Zeng B, Tang Y, Zhang T, Zhao S, et al. , 2018. Association between catechol-O-methyltransferase genetic variation and functional connectivity in patients with first-episode schizophrenia. Schizophr Res. 199, 214–220. [DOI] [PubMed] [Google Scholar]
- Weksberg R, Stachon AC, Squire JA, Moldovan L, Bayani J, Meyn S, et al. , 2007. Molecular characterization of deletion breakpoints in adults with 22q11 deletion syndrome. Hum Genet. 120(6), 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger TL, Kao C, McDonald-McGinn DM, Zackai EH, Bailey A, Schultz RT, et al. , 2016. The Role of mGluR Copy Number Variation in Genetic and Environmental Forms of Syndromic Autism Spectrum Disorder. Sci Rep. 6, 19372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalesky A, Fornito A, Bullmore ET, 2010. Network-based statistic: identifying differences in brain networks. Neuroimage. 53, 1197–207. [DOI] [PubMed] [Google Scholar]
- Zinkstok JR, Boot E, Bassett AS, Hiroi N, Butcher NJ, Vingerhoets C, et al. , 2019. Neurobiological perspective of 22q11.2 deletion syndrome. Lancet Psychiatry. 6, 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller D, Sandini C, Karahanoglu FI, Padula MC, Schaer M, Eliez S, et al. , 2019. Large-Scale Brain Network Dynamics Provide a Measure of Psychosis and Anxiety in 22q11.2 Deletion Syndrome. Biol Psychiatry Cogn Neurosci Neuroimaging. 4, 881–892. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.