

Abstract
Protecting group-controlled annulations of tetra-substituted oxindole olefins and sulfur ylides have been achieved for the synthesis of multifunctional cyclopropane- and dihydrofuran-fused spirooxindoles. Under precise annulation regulation, a variety of cyclopropane- and dihydrofuran-fused spirooxindoles containing vicinal quaternary carbon centers were produced in up to 90% yield with up to 20 : 1 dr. This reaction demonstrates high regio-, chemo- and diastereoselectivity, broad functional group tolerance and gram-scale capacity.
Protecting group-controlled regio- and chemoselective synthesis of cyclopropane- and dihydrofuran-fused spirooxindoles containing vicinal quaternary carbon centers.
Introduction
The important scaffold spirooxindole exists in numerous natural products, biologically active molecules and lead compounds.1 In particular, three- and five-membered spirooxindoles have attracted substantial interest because of their unique skeletal diversity and biological importance (Fig. 1a).2 Various powerful strategies have been developed to construct these two skeletons, which can diversify molecular libraries in spirooxindole-related medicinal chemistry.3 One of the greatest challenges to diversifying libraries is building vicinal quaternary carbon centers on the ring with multiple functional groups; such carbon centers are important for biological activity (Fig. 1b).4 These centers are difficult to build and modify with functional groups because steric hindrance and substrate reactivity strongly affect formation of continuous quaternary carbon centers.5
Fig. 1. (a) Natural products and drugs contain a three- and five-membered spirooxindole scaffold and (b) drug molecules contain vicinal quaternary carbon centers.

We used sulfur ylide chemistry to construct structurally complex three- and five-membered spirooxindoles containing vicinal quaternary carbon centers. As a versatile and efficient synthetic tool, sulfur ylide can facilitate formation of cyclic fragments.6 Mono- or di-substituted double bond substrates tend to generate small ring compounds via the [2 + 1] pathway, such as cyclopropane, epoxy and aziridine (Scheme 1a, left column).7 Tri- or tetra-substituted double bond substrates can generate five-membered heterocyclic compounds via the [4 + 1] pathway, such as dihydrofuran, dihydropyrrole and indolin (Scheme 1a, right column).8 Tri- or tetra-substituted double bond substrates can also undergo the [2 + 1] pathway to construct a three-membered ring, albeit with different regioselectivity: the tri-substituted substrate participates in the [2 + 1] pathway with ipso or α,β selectivity;9 the tetra-substituted substrate, with α,β-selectivity (Scheme 1a, right column, lower panel).10 The tetra-substituted substrate can directly establish quaternary carbon centers in one step, as well as control the chemoselectivity between [2 + 1] and [4 + 1] pathways. In addition, tetra-substituted double bond substrates can be used to construct the spiro ring. Nevertheless, their low reactivity and steric hindrance have limited their synthetic use.
Scheme 1. (a) Annulations between various substrates and sulfur ylides and (b) construction of cyclopropane- and dihydrofuran-fused spirooxindoles containing vicinal quaternary stereocenters.

Despite the advances in using sulfur ylide as a synthetic tool, few reports have focused on using it to form rings of desired size in a chemo- and regioselective manner.11 This reflects, at least in part, uncertainty in how to control the cycloaddition pathway in a one-step reaction. Achieving chemo- and regioselective ring formation would enrich molecular libraries and accelerate new drug development. Therefore, we chose a tetra-substituted oxindole olefin as substrate to synthesize multi-substituted, three- and five-membered spirooxindoles containing vicinal quaternary carbon centers via sulfur ylide, as part of our continuing interest in synthesis of drug-like scaffolds.12 Installing different protecting groups on the tetra-substituted substrate makes the synthesis chemoselective (Scheme 1b). This reaction allows the production of desirable cyclopropane- or dihydrofuran-fused spiro-oxoindoles in high yield with high chemo-, regio- and diastereoselectivities.
Results and discussion
First, we took a tetra-substituted oxindole olefin without protecting group 1a and sulfur ylide 4a to conduct a model reaction in MeCN at ambient temperature. Two products with different ring sizes formed in one step with similar yields (5a : 8a = 1.1 : 1, Table 1, entry 1). Therefore, we set out to control the chemo- and regioselectivity of the reaction and thereby influence their relative abundance. First of all, a broad type of protecting groups were tested, including methyl (-Me), allyl, acetyl (-Ac), benzyl (-Bn) and tert-butyloxycarbonyl (-Boc) groups (see Table S1 in ESI†). To our delight, changing the N–H (1a) into N-Boc (2a) generated 6a as a major product and 9a as a minor one (Table 1, entry 2). Using N-Bn (3a) produced 10a as a major product and 7a as a minor one (entry 7). Since yields were relatively low in MeCN, these initial results encouraged us to screen solvents and temperature (entries 3–6 and 8–12). The best conditions for generation of 6a as the major product was in DCM at ambient temperature for about 2 h (entry 5). The best conditions for generation of 10a as the major product was in DCM at 50 °C for nearly 4 h (entry 11). In other words, when the reaction with 4a was conducted in DCM at ambient temperature generating cyclopropane-fused spirooxindole 6a in 82% yield. When the reaction was conducted with 4a in DCM at 50 °C, 3a preferred the [4 + 1] pathway, giving the dihydrofuran-fused spirooxindole 10a in 74% yield.
Optimization of the reaction conditionsa.

| |||||
|---|---|---|---|---|---|
| Entry | PG | Solvent | Temperature (°C) | Yieldb (%) (6a/9a) | Yieldc (%) (7a/10a) |
| 1 | H | MeCN | 25 | 45 (5a) | 48 (8a) |
| 2 | Boc | MeCN | 25 | 48/21 | — |
| 3 | Boc | THF | 25 | Trace | — |
| 4 | Boc | Tol | 25 | 68/17 | — |
| 5 | Boc | DCM | 25 | 82/9 | — |
| 6 | Boc | DCM | 50 | 63/— | — |
| 7 | Bn | MeCN | 25 | — | 19/48 |
| 8 | Bn | THF | 25 | — | Trace |
| 9 | Bn | Tol | 25 | — | 14/58 |
| 10 | Bn | DCM | 25 | — | 11/67 |
| 11 | Bn | DCM | 50 | — | 15/74 |
| 12 | Bn | DCM | 70 | — | 9/61 |
All reactions were carried out with 0.15 mmol of the substrate 1a/2a/3a, 0.165 mmol of 4a in 2.0 mL of solvent, unless otherwise stated; d.r. was determined to be > 8 : 1 by 1H-NMR analysis of the crude reaction mixture.
Yields of the isolated products 6a and 9a.
Yields of the isolated products 7a and 10a.
Having established optimal reaction conditions (Table 1, entry 5), we examined the scope of the reaction by varying the R1 and R3 groups of 2, as well as the R2 moiety of 4. Various sulfur ylides with a wide range of ortho-, meta-, and para-substituents with different electronic properties were explored (Table 2, entries 2–10). The compounds were well tolerated and afforded 6b–6g in modest to high yields, and they all demonstrated good to excellent chemo- and diastereoselectivity. This indicated that the Boc protecting group favors formation of the cyclopropane-fused spirooxindole 6. Moreover, sulfur ylide reacted well with linear and heterocycle substrates (entries 8–10). Different substituents on the aryl ring of 2 gave compound 6 in reasonable yields (entries 11–17); yield was better with an electron-donating substituent than with an electron-withdrawing one (entry 17). Replacing R3 with an OEt group led exclusively to 6r (entry 18). The relative configuration of 6r was determined by X-ray crystallographic analysis (Fig. 2), and the relative configurations of other products 6 were tentatively assigned by analogy.13 Finally, to evaluate the synthetic potential of this methodology, a gram scale reaction of 6a was carried out. 2a (3.04 mmol) and 4a (3.34 mmol) went smoothly in DCM at ambient temperature, affording the desired product 6a in 80% yield and 18 : 1 dr (entry 19).
Synthesis of cyclopropane-fused spirooxindolea.

| |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yieldb (%) (6/9) | d.r.c |
| 1 | H | Ph | CH3 | 82/9 | 20 : 1 (6a) |
| 2 | H | 2-FC6H4 | CH3 | 74/10 | 18 : 1 (6b) |
| 3 | H | 4-FC6H4 | CH3 | 70/8 | 20 : 1 (6c) |
| 4 | H | 3,4-Cl2C6H3 | CH3 | 63/10 | 20 : 1 (6d) |
| 5 | H | 4-BrC6H4 | CH3 | 75/12 | 18 : 1 (6e) |
| 6 | H | 2-CH3C6H4 | CH3 | 80/9 | 20 : 1 (6f) |
| 7 | H | 4-OCH3C6H4 | CH3 | 81/7 | 20 : 1 (6g) |
| 8 | H | Thienyl | CH3 | 68/Trace | 20 : 1 (6h) |
| 9 | H | Naphthyl | CH3 | 65/Trace | 10 : 1 (6i) |
| 10 | H | OEt | CH3 | 79/8 | 20 : 1 (6j) |
| 11 | 5-F | Ph | CH3 | 68/13 | 13 : 1 (6k) |
| 12 | 7-F | Ph | CH3 | 70/10 | 20 : 1 (6l) |
| 13 | 5-Cl | Ph | CH3 | 72/10 | 20 : 1 (6m) |
| 14 | 6-Cl | Ph | CH3 | 79/12 | 20 : 1 (6n) |
| 15 | 5-Br | Ph | CH3 | 80/5 | 15 : 1 (6o) |
| 16 | 6-Br | Ph | CH3 | 81/8 | 9 : 1 (6p) |
| 17 | 5-CH3 | Ph | CH3 | 85/Trace | 15 : 1 (6q) |
| 18d | H | Ph | OEt | 90/Trace | 18 : 1 (6r) |
| 19e | H | Ph | CH3 | 80/10 | 18 : 1 (6a) |
Unless otherwise noted, all reactions were performed with 2 (0.15 mmol), 4 (0.165 mmol) in 2 mL DCM at 25 °C for 2 h.
Isolated yields of the major compound 6 and minor 9.
The diastereoselective ratio of compounds 6 were calculated based on 1H-NMR analysis of the crude reaction mixture.
The relative configuration of 6r was determined by X-ray crystallographic analysis (Fig. 2), and the relative configurations of other products 6 were tentatively assigned by analogy.
A gram scale reaction of 2a (3.04 mmol) and 4a (3.34 mmol) in DCM at 25 °C was carried out.
Fig. 2. Determination of relative configuration of products 6r and 10a by single-crystal X-ray analysis.

Next we studied the reaction's generality and limitations when generating product 10. Under the optimal conditions (Table 1, entry 11), numerous sulfur ylides reacted smoothly with compounds 3 to deliver good to high yields (Table 3, entries 2–11), although a 2-F group led to slight loss of diastereoselectivity (entry 2). Sulfur ylides with linear or heterocycle substitutions performed well (entries 8–11). In all cases, the reaction showed chemo- and diastereoselectivity in forming dihydrofuran-fused spirooxindole 10. These results support Bn as a protecting group that favors formation of compound 10. Diverse substituents on the aryl ring of 3 led to smooth reaction with 4, giving the corresponding compounds 10l–10r in 56–80% yields (entries 12–18). At last, we scaled up the reaction to gram synthesis, result proved that 3a (3.13 mmol) and 4a (3.44 mmol) were well tolerated in DCM at 50 °C, generating the target compound 10a in 80% yield and 9 : 1 dr (entry 19). The relative configuration of 10a was determined by X-ray crystallographic analysis (Fig. 2), and the relative configurations of other products 10 were tentatively assigned by analogy.14
Synthesis of dihydrofuran-fused spirooxindolea.
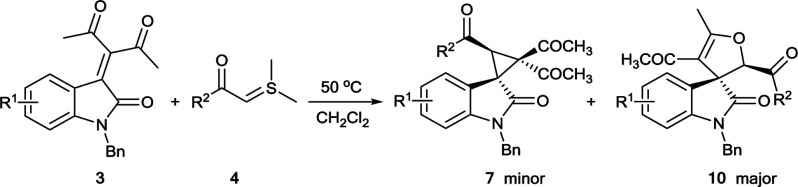
| ||||
|---|---|---|---|---|
| Entry | R1 | R2 | Yieldb (%) (7/10) | d.r.c |
| 1d | H | Ph | 15/74 | 10 : 1 (10a) |
| 2 | H | 2-FC6H4 | 13/66 | 16 : 1 (10b) |
| 3 | H | 4-FC6H4 | 11/68 | 16 : 1 (10c) |
| 4 | H | 3,4-Cl2C6H3 | 12/71 | 8 : 1 (10d) |
| 5 | H | 4-BrC6H4 | 10/75 | 10 : 1 (10e) |
| 6 | H | 2-CH3C6H4 | 8/78 | 20 : 1 (10f) |
| 7 | H | 4-OCH3C6H4 | Trace/81 | 18 : 1 (10g) |
| 8 | H | Thienyl | 15/60 | 5 : 1 (10h) |
| 9 | H | Naphthyl | 15/62 | 5 : 1 (10i) |
| 10 | H | CH3 | 14/74 | 6 : 1 (10j) |
| 11 | H | OEt | 13/76 | 5 : 1 (10k) |
| 12 | 5-F | Ph | 8/70 | 10 : 1 (10l) |
| 13 | 7-F | Ph | Trace/56 | 16 : 1 (10m) |
| 14 | 5-Cl | Ph | 14/66 | 5 : 1 (10n) |
| 15 | 6-Cl | Ph | 12/67 | 15 : 1 (10o) |
| 16 | 5-Br | Ph | 10/70 | 13 : 1 (10p) |
| 17 | 6-Br | Ph | 10/69 | 10 : 1 (10q) |
| 18 | 5-CH3 | Ph | 8/80 | 18 : 1 (10r) |
| 19e | H | Ph | 12/80 | 9 : 1 (10a) |
Unless otherwise noted, all reactions were performed with 3 (0.15 mmol), 4 (0.165 mmol) in 2 mL DCM at 50 °C for 4 h.
Isolated yields of the compound 7 and 10.
The diastereoselective ratio of compounds 10 were determined by 1H-NMR analysis of the crude reaction mixture.
The relative configuration of 10a was determined by X-ray crystallographic analysis (Fig. 2), and the relative configurations of other products 10 were tentatively assigned by analogy.
A gram scale reaction of 3a (3.13 mmol) and 4a (3.44 mmol) in DCM at 50 °C was carried out.
We turned to developing an asymmetric catalytic version of this reaction using chiral sulfur ylides and hydrogen bond catalysts. We screened a variety of chiral sulfur ylides for their ability to generate the chiral product 6a, but unfortunately, the best results we obtained were 61% yield and 18% ee (Table 4). Adding hydrogen-bonding catalysts improved yield to 77% and ee to 39% (entries 7–9). Similarly, we screened chiral sulfur ylides to afford the chiral product 10a, and obtained initial results of 53% yield and 19% ee. Screening of hydrogen-bonding catalysts identified Cat. 3 as the best, affording product 10a in 72% yield with 32% ee (entries 16–18).
Attempt to asymmetric catalytic synthesis of chiral productsa.

| |||||||
|---|---|---|---|---|---|---|---|
| Entry | PG | Sulfur ylide | Cat. | Yieldb (%) (6a/9a) | e.e.d (%) | Yieldc (%). (7a/10a) | e.e.e (%) |
| 1 | Boc | Chiral 4a | — | 61/13 | 18 | — | — |
| 2 | Boc | Chiral 4b | — | — | — | — | — |
| 3 | Boc | Chiral 4c | — | — | — | — | — |
| 4 | Boc | Chiral 4d | — | Trace | — | — | — |
| 5 | Boc | Chiral 4e | — | 35/16 | — | — | — |
| 6 | Boc | Chiral 4f | — | 56/12 | 10 | — | — |
| 7 | Boc | Chiral 4a | C1 | 78/10 | 25 | — | — |
| 8 | Boc | Chiral 4a | C2 | 68/17 | 20 | — | — |
| 9 | Boc | Chiral 4a | C3 | 77/16 | 39 | — | — |
| 10 | Bn | Chiral 4a | — | — | — | 16/53 | 19 |
| 11 | Bn | Chiral 4b | — | — | — | — | — |
| 12 | Bn | Chiral 4c | — | — | — | — | — |
| 13 | Bn | Chiral 4d | — | — | — | Trace | — |
| 14 | Bn | Chiral 4e | — | — | — | 13/41 | — |
| 15 | Bn | Chiral 4f | — | — | — | 17/59 | 8 |
| 16 | Bn | Chiral 4f | C1 | — | — | 12/76 | 17 |
| 17 | Bn | Chiral 4f | C2 | — | — | 18/65 | 13 |
| 18 | Bn | Chiral 4f | C3 | — | — | 13/72 | 32 |
Reactions were performed with 2a or 3a (0.1 mmol), 4 (0.1 mmol), or Cat. (20 mol%) in 2 mL DCM at ambient temperature for 8 h.
Yields were calculated from the isolated compound 6a or 9a respectively.
Yields were calculated from the isolated compound 7a or 10a respectively.
ee values were calculated from chiral HPLC analysis of major isomer 6.
ee values were calculated from chiral HPLC analysis of major isomer 10.
Conclusions
In summary, we set up a protecting group-controlled strategy to regulate ring size via sulfur ylide. This powerful method allows access to structurally important cyclopropane- and dihydrofuran-fused spirooxindoles containing vicinal quaternary carbon centers. This approach exhibits good functional group tolerance as well as excellent regio-, chemo- and diastereoselectivity. It can be scaled up to gram synthesis. Further studies on the bioactivity of promising spirooxindoles will be reported in due course.
Experimental
General method for the synthesis
NMR data were obtained for 1H at 400 MHz or 600 MHz, and for 13C at 100 MHz or 150 MHz. Chemical shifts are reported in ppm from tetramethylsilane with the solvent resonance in CDCl3 solution as the internal standard. ESI HRMS was performed on a Waters SYNAPT G2. Column chromatography was performed on silica gel (400–500 mesh) eluting with ethyl acetate and petroleum ether. TLC was performed on glass-backed silica plates. UV light and I2 were used to visualize products.
General procedure for the synthesis of compounds 6
To a solution of tetra-substituted oxindole olefins 2 (0.15 mmol) in CH2Cl2 (2.0 mL) was added sulfur ylides 4 (0.165 mmol) at 25 °C. The reaction mixture was stirred until the reaction completed (monitored by TLC). Then the reaction mixture was concentrated and the residue was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate = 8 : 1 to 5 : 1) to give the compounds 6 which were dried under vacuum and further analyzed by 1H-NMR, 13C-HMR, HRMS, etc.
Compound 5a was obtained according to the similar procedure. White solid, 45% yield (23.4 mg). The diastereomeric ratio was determined to be 18 : 1 by crude 1H-NMR analysis. Mp 165–166 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.37 (s, 1H), 7.89 (dd, J = 8.0, 0.8 Hz, 2H), 7.58–7.52 (m, 1H), 7.48–7.39 (m, 2H), 7.22 (td, J = 7.6, 1.2 Hz, 1H), 7.03 (d, J = 7.2 Hz, 1H), 6.96 (td, J = 7.6, 1.2 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H), 4.26 (s, 1H), 2.36 (s, 3H), 2.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 198.0, 196.1, 192.0, 173.5, 141.2, 136.5, 134.1, 128.9, 128.7, 128.5, 126.7, 122.5, 121.1, 110.1, 62.5, 43.6, 42.9, 29.8, 28.4; ESI HRMS: calcd for C21H17NO4Na+ 370.1055, found 370.1056.
Compound 6a was obtained as white solid in 82% yield (55.0 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 170–171 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.90 (dd, J = 8.0, 0.8 Hz, 2H), 7.85 (d, J = 8.0 Hz, 1H), 7.59–7.55 (m, 1H), 7.43 (t, J = 7.6 Hz, 2H), 7.30–7.27 (m, 1H), 7.09–7.02 (m, 2H), 4.28 (s, 1H), 2.35 (s, 3H), 2.26 (s, 3H), 1.64 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm) 197.7, 195.9, 191.7, 170.9, 148.5, 140.5, 136.4, 134.2, 128.9, 128.6, 126.0, 124.1, 119.8, 114.6, 85.3, 63.0, 44.5, 43.1, 29.8, 28.4, 28.1; ESI HRMS: calcd for C26H25NO6Na+ 470.1580, found 470.1581.
Compound 6b was obtained as white solid, 74% yield (51.7 mg). The diastereomeric ratio was determined to be 18 : 1 by crude 1H-NMR analysis. Mp 143–144 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.88 (d, J = 7.8 Hz, 1H), 7.75 (td, J = 7.2, 1.8 Hz, 1H), 7.54–7.49 (m, 1H), 7.32 (dt, J = 7.8, 1.2 Hz, 1H), 7.21 (t, J = 7.2 Hz, 1H), 7.08–7.03 (m, 2H), 6.93 (dd, J = 7.8, 1.2 Hz, 1H), 4.17 (d, J = 3.0 Hz, 1H), 2.37 (s, 3H), 2.29 (s, 3H), 1.65 (s, 9H); 13C NMR (150 MHz, CDCl3) δ (ppm) 198.0, 195.9, 190.1 (d, JCF = 3.0 Hz), 170.9, 161.9 (d, JCF = 255.0 Hz), 148.7, 140.7, 135.8 (d, JCF = 9.0 Hz), 130.7 (d, JCF = 1.5 Hz), 128.9, 125.6, 124.8 (d, JCF = 4.5 Hz), 124.0, 119.9, 117.0, 116.9, 114.7, 85.2, 63.2, 48.6 (d, JCF = 7.5 Hz), 43.3 (d, JCF = 3.0 Hz), 29.9, 28.6, 28.2; ESI HRMS: calcd for C26H24FNO6Na+ 488.1485, found 488.1487.
Compound 6c was obtained as white solid, 70% yield (48.9 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 145–146 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.96–7.93 (m, 2H), 7.86 (d, J = 8.2 Hz, 1H), 7.31 (dt, J = 9.0, 1.8 Hz, 1H), 7.12–7.09 (m, 2H), 7.07 (td, J = 7.8, 1.2 Hz, 1H), 7.01 (dd, J = 7.8, 1.2 Hz, 1H), 4.22 (s, 1H), 2.35 (s, 3H), 2.26 (s, 3H), 1.65 (s, 9H); 13C NMR (150 MHz, CDCl3) δ (ppm) 197.7, 195.9, 190.1, 171.0, 166.4 (d, JCF = 255.0 Hz), 148.6, 140.5, 132.9 (d, JCF = 4.5 Hz), 131.4 (d, JCF = 10.5 Hz), 129.1, 126.0, 124.2, 119.7, 116.2 (d, JCF = 22.5 Hz), 114.7, 85.4, 63.1, 44.3, 43.1, 29.9, 28.5, 28.1; ESI HRMS: calcd for C26H24FNO6Na+ 488.1485, found 488.1487.
Compound 6d was obtained as yellow solid, 63% yield (48.8 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 144–145 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.01 (d, J = 2.0 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.75 (dd, J = 8.4, 2.4 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.33 (td, J = 8.8, 1.6 Hz, 1H), 7.08 (td, J = 8.0, 1.2 Hz, 1H), 6.98 (dd. J = 8.0, 1.2 Hz, 1H), 4.13 (s, 1H), 2.32 (s, 3H), 2.28 (s, 3H), 1.65 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm) 197.5, 195.6, 189.7, 170.7, 148.4, 140.5, 139.0, 135.9, 133.8, 131.0, 130.3, 129.2, 127.5, 125.8, 124.2, 119.4, 114.8, 85.4, 63.3, 43.8, 43.3, 29.8, 28.5, 28.1; ESI HRMS: calcd for C26H23Cl2NO6Na+ 538.0800, found 538.0802.
Compound 6e was obtained as yellow solid, 75% yield (59.2 mg). The diastereomeric ratio was determined to be 18 : 1 by crude 1H-NMR analysis. Mp 147–148 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.86 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.31 (t, J = 7.6 Hz, 1H), 7.07 (t, J = 8.0 Hz, 1H), 6.98 (d, J = 8.0 Hz, 1H), 4.20 (s, 1H), 2.34 (s, 3H), 2.26 (s, 3H), 1.65 (s, 9H); 13C NMR (150 MHz, CDCl3) δ (ppm) 197.5, 195.7, 190.7, 170.8, 148.4, 140.4, 135.0, 132.2, 129.9, 129.0, 125.8, 124.0, 119.5, 114.7, 85.32, 63.0, 44.1, 43.0, 29.8, 28.4, 28.0; ESI HRMS: calcd for C26H24BrNO6Na+ 548.0685, found 548.0686.
Compound 6f was obtained as white solid, 80% yield (55.4 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 142–143 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.88 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 7.2 Hz, 1H), 7.38 (td, J = 7.8, 1.2 Hz, 1H), 7.34–7.31 (m, 1H), 7.22 (dd, J = 15.6, 9.0 Hz, 2H), 7.09 (dd, J = 6.0, 2.4 Hz, 2H), 4.12 (s, 1H), 2.42 (s, 3H), 2.36 (s, 3H), 2.25 (s, 3H), 1.64 (s, 9H); 13C NMR (150 MHz, CDCl3) δ (ppm) 195.9, 194.7, 170.9, 148.5, 140.5, 132.6, 132.1, 129.5, 128.9, 126.2, 126.1, 124.0, 119.8, 114.6, 85.3, 63.2, 47.3, 43.5, 29.8, 28.4, 28.1, 21.2; ESI HRMS: calcd for C27H27NO6Na+ 484.1736, found 484.1734.
Compound 6g was obtained as white solid, 81% yield (58.0 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 145–146 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.88 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 8.4 Hz, 1H), 7.30–7.26 (m, 1H), 7.06–7.04 (m, 2H), 6.88 (d, J = 8.8 Hz, 2H), 4.25 (s, 1H), 3.83 (s, 3H), 2.36 (s, 3H), 2.25 (s, 3H), 1.64 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm) 197.9, 196.0, 189.7, 171.1, 164.5, 148.6, 140.4, 131.1, 129.5, 128.7, 126.1, 124.0, 120.0, 114.5, 114.1, 85.2, 62.8, 55.6, 44.6, 42.8, 29.9, 28.3, 28.1; ESI HRMS: calcd for C27H27NO7H+ 500.1685, found 500.1688.
Compound 6h was obtained as white solid, 68% yield (46.3 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 137–138 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.86 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 3.0 Hz, 1H), 7.69 (d, J = 4.8 Hz, 1H), 7.34–7.31 (m, 1H), 7.14–7.10 (m, 3H), 4.15 (s, 1H), 2.35 (s, 3H), 2.25 (s, 3H), 1.64 (s, 9H); 13C NMR (150 MHz, CDCl3) δ (ppm) 197.5, 195.6, 183.9, 170.9, 148.6, 143.7, 140.6, 135.9, 134.1, 129.0, 128.9, 126.5, 124.2, 119.8, 114.7, 85.4, 62.9, 60.5, 45.0, 43.2, 29.8, 28.1; ESI HRMS: calcd for C24H23NO6SNa+ 476.1144, found 476.1145.
Compound 6i was obtained as white solid, 65% yield (48.5 mg). The diastereomeric ratio was determined to be 10 : 1 by crude 1H-NMR analysis. Mp 153–154 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.47 (s, 1H), 7.97–7.92 (m, 2H), 7.85–7.82 (m, 3H), 7.60–7.55 (m, 2H), 7.30–7.26 (m, 1H), 7.13–7.05 (m, 2H), 4.46 (s, 1H), 2.38 (s, 3H), 2.30 (s, 3H), 1.65 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm) 197.8, 195.9, 191.4, 171.1, 148.5, 140.5, 135.9, 133.8, 132.3, 130.9, 129.9, 129.3, 128.9, 128.8, 127.8, 127.2, 126.2, 124.1, 123.5, 119.8, 114.6, 85.3, 63.1, 44.6, 43.3, 29.9, 28.4, 28.1; ESI HRMS: calcd for C30H27NO6Na+ 520.1736, found 520.1738.
Compound 6j was obtained as white solid, 79% yield (49.2 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 149–150 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.91 (d, J = 8.4 Hz, 1H), 7.36 (dt, J = 8.4, 2.4 Hz, 1H), 7.14–7.12 (m, 2H), 4.21–4.18 (m, 1H), 4.14–4.11 (m, 1H), 3.33 (s, 1H), 2.30 (s, 3H), 2.27 (s, 3H), 1.63 (s, 9H), 1.22 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 197.4, 195.5, 170.6, 166.0, 148.6, 140.8, 129.1, 125.9, 124.0, 119.9, 114.8, 85.3, 62.2, 62.1, 41.6, 40.5, 29.6, 28.6, 28.1, 14.13; ESI HRMS: calcd for C22H25NO7Na+ 438.1529, found 438.1531.
Compound 6k was obtained as white solid, 68% yield (47.5 mg). The diastereomeric ratio was determined to be 13 : 1 by crude 1H-NMR analysis. Mp 172–173 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.92 (dd, J = 8.4, 1.2 Hz, 2H), 7.84 (dd, J = 9.0, 4.8 Hz, 1H), 7.61–7.58 (m, 1H), 7.46 (dd, J = 7.8, 1.8 Hz, 2H), 7.00 (td, J = 9.0, 3.0 Hz, 1H), 6.82 (dd, J = 9.6, 3.0 Hz, 1H), 4.30 (s, 1H), 2.35 (s, 3H), 2.26 (s, 3H), 1.64 (s, 9H); 13C NMR (150 MHz, CDCl3) δ (ppm) 197.4, 195.8, 191.5, 170.7, 159.3 (d, JCF = 240 Hz), 148.5, 136.4 (d, JCF = 36 Hz), 134.5, 129.1, 128.7, 121.7 (d, JCF = 10.5 Hz), 115.7 (d, JCF = 9.0 Hz), 115.6, 114.1, 113.9, 85.6, 63.2, 44.7, 43.0, 29.8, 28.4, 28.1; ESI HRMS: calcd for C26H24FNO6Na+ 488.1485, found 488.1483.
Compound 6l was obtained as white solid, 70% yield (48.9 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 157–158 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.90 (d, J = 7.2 Hz, 2H), 7.60–7.58 (m, 1H), 7.45 (t, J = 7.8 Hz, 2H), 7.07–7.01 (m, 2H), 6.84 (dd, J = 7.8, 1.2 Hz, 1H), 4.30 (s, 1H), 2.35 (s, 3H), 2.27 (s, 3H), 1.61 (s, 9H); 13C NMR (150 MHz, CDCl3) δ (ppm) 197.2, 195.8, 191.5, 170.4, 148.4 (d, JCF = 249.1 Hz), 147.0, 136.3, 134.5, 129.1, 128.7, 127.6 (d, JCF = 10.4 Hz), 124.9 (d, JCF = 7.4 Hz), 122.9 (d, JCF = 2.2 Hz), 122.1 (d, JCF = 4.0 Hz), 117.1 (d, JCF = 20.0 Hz), 86.0, 63.2, 44.5, 43.0, 29.9, 28.4, 27.7; ESI HRMS: calcd for C26H24FNO6Na+ 488.1485, found 488.1483.
Compound 6m was obtained as yellow solid, 72% yield (41.9 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 136–137 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.93 (d, J = 7.6 Hz, 2H), 7.82 (d, J = 8.8 Hz, 1H), 7.60 (t, J = 7.2 Hz, 1H), 7.46 (t, J = 7.6 Hz, 2H), 7.29 (d, J = 1.2 Hz, 1H), 7.05 (s, 1H), 4.29 (s, 1H), 2.34 (s, 3H), 2.26 (s, 3H), 1.64 (s, 9H); 13C NMR (150 MHz, CDCl3) δ (ppm) 197.2, 195.2, 191.3, 170.3, 148.3, 138.9, 136.2, 134.4, 129.7, 128.9, 128.8, 128.6, 126.2, 121.5, 115.6, 85.6, 63.1, 44.6, 42.7, 29.6, 28.3, 28.0; ESI HRMS: calcd for C26H24ClNO6Na+ 504.1190, found 504.1189.
Compound 6n was obtained as yellow solid, 79% yield (57.1 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 145–146 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.93 (d, J = 2.0 Hz, 1H), 7.90 (d, J = 7.6 Hz, 2H), 7.59 (t, J = 7.6 Hz, 1H), 7.45 (t, J = 8.0 Hz, 2H), 7.05 (dd, J = 8.4, 1.6 Hz, 1H), 6.96 (d, J = 8.8 Hz, 1H), 4.28 (s, 1H), 2.34 (s, 3H), 2.25 (s, 3H), 1.64 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm) 197.3, 195.8, 191.6, 170.5, 148.3, 141.3, 136.3, 135.0, 134.4, 129.0, 128.6, 127.0, 124.2, 118.2, 115.4, 85.8, 62.9, 44.6, 42.8, 29.8, 28.3, 28.0; ESI HRMS: calcd for C26H24ClNO6Na+ 504.1190, found 504.1191.
Compound 6o was obtained as yellow solid, 80% yield (63.2 mg). The diastereomeric ratio was determined to be 15 : 1 by crude 1H-NMR analysis. Mp 142–143 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.93 (d, J = 7.2 Hz, 2H), 7.76 (d, J = 9.0 Hz, 1H), 7.60 (t, J = 7.8 Hz, 1H), 7.446 (t, J = 7.8 Hz, 2H), 7.43 (dd, J = 9.0, 1.8 Hz, 1H), 7.19 (d, J = 1.8 Hz, 1H), 4.28 (s, 1H), 2.34 (s, 3H), 2.26 (s, 3H), 1.63 (s, 9H); 13C NMR (150 MHz, CDCl3) δ (ppm) 197.2, 195.5, 191.3, 170.1, 148.2, 139.4, 136.2, 134.4, 131.8, 129.0, 128.9, 128.6, 121.8, 117.3, 115.9, 85.6, 63.2, 44.6, 42.6, 29.6, 28.3, 27.9; ESI HRMS: calcd for C26H24BrNO6Na+ 548.0685, found 548.0687.
Compound 6p was obtained as yellow solid, 81% yield (63.9 mg). The diastereomeric ratio was determined to be 9 : 1 by crude 1H-NMR analysis. Mp 149–150 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.09 (d, J = 0.8 Hz, 1H), 7.90 (d, J = 4.8 Hz, 2H), 7.61–7.58 (m, 1H), 7.45 (t, J = 5.2 Hz, 2H), 7.20 (dd, J = 5.6, 0.8 Hz, 1H), 6.90 (d, J = 5.6 Hz, 1H), 4.29 (s, 1H), 2.34 (s, 3H), 2.25 (s, 3H), 1.64 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm) 197.3, 195.9, 191.6, 170.5, 148.3, 141.4, 136.3, 134.5, 129.1, 128.7, 127.3, 127.2, 123.1, 118.8, 118.3, 85.9, 63.0, 44.6, 29.9, 28.4, 28.1, 28.0; ESI HRMS: calcd for C26H24BrNO6Na+ 548.0685, found 548.0683.
Compound 6q was obtained as white solid, 85% yield (58.8 mg). The diastereomeric ratio was determined to be 15 : 1 by crude 1H-NMR analysis. Mp 168–169 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.90 (d, J = 7.2 Hz, 2H), 7.72 (d, J = 8.4 Hz, 1H), 7.59–7.55 (m, 1H), 7.44 (t, J = 8.0 Hz, 2H), 7.10 (dd, J = 8.4, 0.8 Hz, 1H), 6.84 (s, 1H), 4.25 (s, 1H), 2.35 (s, 3H), 2.29 (s, 3H), 2.26 (s, 3H), 1.64 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm) 197.8, 195.9, 191.7, 171.1, 148.6, 138.2, 136.5, 134.2, 133.7, 129.5, 128.9, 128.6, 126.5, 119.7, 114.4, 85.1, 63.0, 44.5, 43.2, 29.8, 28.4, 28.1, 21.0; ESI HRMS: calcd for C27H27NO6Na+ 484.1736, found 484.1740.
Compound 6r was obtained as white solid, 90% yield (64.5 mg). The diastereomeric ratio was determined to be 18 : 1 by crude 1H-NMR analysis. Mp 147–148 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 8.10 (dd, J = 8.4, 1.2 Hz, 2H), 7.90 (d, J = 7.8 Hz, 1H), 7.59–7.56 (m, 1H), 7.47 (dt, J = 7.2, 1.2 Hz, 2H), 7.34–7.31 (m, 1H), 7.08 (dt, J = 7.8, 1.8 Hz, 1H), 7.01 (dd, J = 7.8, 1.2 Hz, 1H), 4.43–4.39 (m, 1H), 4.32–4.29 (m, 1H), 4.10 (s, 1H), 2.29 (s, 3H), 1.65 (s, 9H), 1.35 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 195.3, 191.0, 170.8, 165.5, 148.7, 140.7, 136.6, 133.9, 129.1, 128.9, 128.5, 125.7, 124.1, 119.7, 114.8, 85.0, 63.2, 56.7, 42.1, 42.0, 29.7, 28.2, 14.1; ESI HRMS: calcd for C27H27NO7Na+ 500.1685, found 500.1687.
General procedure for the synthesis of compounds 10
To a solution of Bn-protected tetra-substituted oxindole olefins 3 (0.15 mmol) in CH2Cl2 (2.0 mL) was added sulfur ylides 4 (0.165 mmol) at 50 °C. The reaction mixture was stirred until the reaction completed (monitored by TLC). Then the reaction mixture was concentrated and the residue was purified by flash chromatography on silica gel to give the compounds 10 which were dried under vacuum and further analyzed by 1H-NMR, 13C-HMR, HRMS, etc.
Compound 8a was obtained according to the similar procedure. White solid, 48% yield (25.0 mg). The diastereomeric ratio was determined to be 10 : 1 by crude 1H-NMR analysis. Mp 164–165 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.84 (s, 1H), 7.43–7.38 (m, 1H), 7.35 (dd, J = 8.4, 1.2 Hz, 2H), 7.22 (td, J = 7.6, 1.6 Hz, 2H), 6.99–6.95 (m, 1H), 6.91–6.86 (m, 2H), 6.40 (d, J = 7.6 Hz, 1H), 6.27 (s, 1H), 2.55 (s, 3H), 2.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 192.8, 191.3, 178.8, 170.2, 139.9, 134.8, 133.6, 129.0, 128.4, 127.5, 127.4, 124.9, 122.8, 118.3, 109.3, 88.8, 62.2, 28.9, 15.8; ESI HRMS: calcd for C21H17NO4Na+ 370.1055, found 370.1054.
Compound 10a was obtained as white solid in 74% yield (48.6 mg). The diastereomeric ratio was determined to be 10 : 1 by crude 1H-NMR analysis. Mp 168–169 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.42 (t, J = 7.2 Hz, 1H), 7.30 (d, J = 4.4 Hz, 4H), 7.25 (d, J = 4.8 Hz, 3H), 7.16 (t, J = 7.6 Hz, 2H), 6.93–6.89 (m, 2H), 6.86 (t, J = 7.2 Hz, 1H), 6.30 (s, 1H), 6.17 (d, J = 7.6 Hz, 1H), 5.08 (d, J = 15.6 Hz, 1H), 4.20 (d, J = 16.0 Hz, 1H), 2.56 (s, 3H), 2.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 192.9, 191.0, 177.3, 169.9, 142.2, 135.3, 135.0, 133.4, 128.9, 128.7, 128.3, 127.5, 127.3, 126.9, 124.7, 122.8, 118.4, 108.8, 89.0, 62.0, 44.6, 29.0, 15.9; ESI HRMS: calcd for C28H23NO4Na+ 460.1525, found 460.1526.
Compound 10b was obtained as white solid, 66% yield (45.1 mg). The diastereomeric ratio was determined to be 16 : 1 by crude 1H-NMR analysis. Mp 154–155 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.35–7.31 (m, 4H), 7.31–7.24 (m, 3H), 6.95 (tt, J = 7.2, 1.8 Hz, 2H), 6.90 (t, J = 6.6 Hz, 2H), 6.86 (t, J = 7.8 Hz, 1H), 6.83–6.79 (m, 1H), 6.26 (s, 1H), 5.02 (d, J = 15.6 Hz, 1H), 4.33 (d, J = 15.6 Hz, 1H), 2.55 (s, 3H), 2.13 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 191.3 (d, JCF = 4.5 Hz), 191.0, 177.1, 169.9, 160.4 (d, JCF = 253.5 Hz), 142.8, 135.5, 134.7 (d, JCF = 9.0 Hz), 130.3 (d, JCF = 3.0 Hz), 129.0, 128.8, 127.6, 127.5, 127.1, 124.6, 124.3 (d, JCF = 3.0 Hz), 122.8, 115.9, 115.8, 108.9, 90.9 (d, JCF = 4.5 Hz), 61.6, 44.6, 29.1, 15.9; ESI HRMS: calcd for C28H22FNO4Na+ 478.1431, found 478.1430.
Compound 10c was obtained as white solid, 68% yield (46.5 mg). The diastereomeric ratio was determined to be 16 : 1 by crude 1H-NMR analysis. Mp 151–152 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.33–7.32 (m, 4H), 7.31–7.28 (m, 3H), 6.94 (td, J = 7.8, 1.2 Hz, 1H), 6.91 (dd, J = 7.2, 1.2 Hz, 1H), 6.86 (td, J = 7.2, 0.6 Hz, 1H), 6.81–6.76 (m, 1H), 6.28 (d, J = 7.8 Hz, 1H), 6.25 (s, 1H), 5.04 (d, J = 15.6 Hz, 1H), 4.38 (d, J = 15.6 Hz, 1H), 2.56 (s, 3H), 2.16 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 191.4, 191.0, 177.3, 169.9, 165.8 (d, JCF = 255.0 Hz), 142.3, 135.3, 131.4 (d, JCF = 3.0 Hz), 130.2 (d, JCF = 9.0 Hz), 129.1, 128.9, 127.7, 127.5, 126.9, 124.8, 122.9, 118.6, 115.6 (d, JCF = 22.5 Hz), 108.9, 88.7, 62.1, 44.7, 29.1, 15.9; ESI HRMS: calcd for C28H22FNO4Na+ 478.1431, found 478.1429.
Compound 10d was obtained as yellow solid, 71% yield (53.9 mg). The diastereomeric ratio was determined to be 8 : 1 by crude 1H-NMR analysis. Mp 196–197 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.37–7.34 (m, 3H), 7.33 (d, J = 1.8 Hz, 2H), 7.28 (dt, J = 6.0, 1.2 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.08 (dd, J = 8.4, 1.8 Hz, 1H), 6.98 (dt, J = 9.0, 4.8 Hz, 1H), 6.88 (d, J = 4.2 Hz, 2H), 6.31 (d, J = 7.8 Hz, 1H), 6.18 (s, 1H), 5.12 (d, J = 15.6 Hz, 1H), 4.36 (d, J = 15.6 Hz, 1H), 2.55 (s, 3H), 2.16 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 191.0, 190.9, 177.1, 169.7, 142.3, 138.2, 135.2, 134.4, 133.3, 130.4, 129.5, 129.3, 128.9, 127.8, 127.5, 126.7, 126.3, 124.7, 123.0, 118.5, 109.0, 88.8, 62.0, 44.8, 29.1, 15.9; ESI HRMS: calcd for C28H21Cl2NO4Na+ 528.0745, found 528.0748.
Compound 10e was obtained as white solid, 75% yield (58.1 mg). The diastereomeric ratio was determined to be 10 : 1 by crude 1H-NMR analysis. Mp 152–153 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.35–7.31 (m, 4H), 7.25 (t, J = 4.0 Hz, 3H), 7.12 (d, J = 8.4 Hz, 2H), 6.96 (td, J = 7.6, 2.0 Hz, 1H), 6.90 (dd, J = 7.2, 1.4 Hz, 1H), 6.86 (t, J = 7.6 Hz, 1H), 6.30 (d, J = 8.0 Hz, 1H), 6.23 (s, 1H), 5.00 (d, J = 15.6 Hz, 1H), 4.41 (d, J = 15.6 Hz, 1H), 2.55 (s, 3H), 2.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 191.9, 190.9, 177.2, 169.7, 142.3, 135.2, 133.6, 131.6, 129.1, 128.9, 128.8, 128.7, 127.7, 127.5, 127.4, 126.8, 124.7, 122.8, 118.5, 108.9, 88.7, 61.9, 44.8, 29.0, 15.8; ESI HRMS: calcd for C28H22BrNO4Na+ 538.0630, found 538.0632.
Compound 10f was obtained as white solid, 78% yield (52.8 mg). The diastereomeric ratio was determined to be 20 : 1 by crude 1H-NMR analysis. Mp 178–179 °C; 1H NMR (600 MHz) δ (ppm) 7.31 (td, J = 7.8, 1.2 Hz, 1H), 7.28–7.26 (m, 3H), 7.22 (t, J = 6.0 Hz, 3H), 7.12 (t, J = 7.2 Hz, 1H), 7.01 (dd, J = 7.2, 1.2 Hz, 1H), 6.97 (td, J = 7.2, 2.4 Hz, 2H), 6.89 (td, J = 7.8, 0.6 Hz, 1H), 6.30 (s, 1H), 6.20 (d, J = 7.8 Hz, 1H), 4.97 (d, J = 16.2 Hz, 1H), 4.02 (d, J = 16.2 Hz, 1H), 2.56 (s, 3H), 2.13 (s, 3H), 1.81 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 194.3, 190.9, 176.9, 169.6, 142.3, 140.1, 140.0, 135.2, 133.9, 132.3, 131.7, 129.1, 128.7, 128.5, 127.4, 127.0, 125.1, 124.5, 122.8, 118.9, 109.1, 89.4, 61.8, 44.5, 28.9, 20.0, 15.8; ESI HRMS: calcd for C29H25NO4Na+ 474.1681, found 474.1683.
Compound 10g was obtained as white solid, 81% yield (56.8 mg). The diastereomeric ratio was determined to be 18 : 1 by crude 1H-NMR analysis. Mp 188–189 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.33 (t, J = 3.0 Hz, 1H), 7.31 (t, J = 1.8 Hz, 1H), 7.29–7.28 (m, 4H), 7.26–7.24 (m, 1H), 6.94 (d, J = 7.2 Hz, 1H), 6.92 (dd, J = 7.2, 1.2 Hz, 1H), 6.85 (td, J = 7.2, 0.6 Hz, 1H), 6.63 (dt, J = 9.6, 3.0 Hz, 2H), 6.26 (d, J = 7.2 Hz, 2H), 5.03 (d, J = 15.6 Hz, 1H), 4.46 (d, J = 16.2 Hz, 1H), 3.78 (s, 3H), 2.55 (s, 3H), 2.15 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 191.1, 191.0, 177.5, 170.1, 163.9, 142.3, 135.5, 130.0, 128.9, 128.8, 128.0, 127.5, 127.4, 127.0, 124.9, 122.8, 118.6, 113.7, 108.8, 88.5, 62.2, 55.6, 44.7, 29.1, 16.0; ESI HRMS: calcd for C29H25NO5Na+ 490.1630, found 490.1631.
Compound 10h was obtained as white solid, 60% yield (39.9 mg). The diastereomeric ratio was determined to be 5 : 1 by crude 1H-NMR analysis. Mp 172–173 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.51 (dd, J = 4.8, 1.2 Hz, 1H), 7.36 (dd, J = 4.2, 1.2 Hz, 3H), 7.34–7.31 (m, 2H), 7.28 (dd, J = 6.6, 1.2 Hz, 1H), 6.98 (td, J = 7.8, 1.2 Hz, 1H), 6.95 (dd, J = 7.2, 0.6 Hz, 1H), 6.88 (dd, J = 7.2, 0.6 Hz, 1H), 6.83–6.82 (m, 1H), 6.40 (d, J = 7.8 Hz, 1H), 6.08 (s, 1H), 5.06 (d, J = 15.6 Hz, 1H), 4.64 (d, J = 15.6 Hz, 1H), 2.56 (s, 3H), 2.16 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 191.2, 184.9, 177.5, 169.9, 142.4, 140.9, 135.5, 135.4, 132.2, 129.1, 128.9, 128.8, 127.9, 127.7, 127.5, 124.8, 123.0, 118.6, 108.9, 89.2, 62.6, 44.7, 29.1, 15.9; ESI HRMS: calcd for C26H21NO4SNa+ 466.1089, found 466.1090.
Compound 10i was obtained as white solid, 62% yield (45.3 mg). The diastereomeric ratio was determined to be 5 : 1 by crude 1H-NMR analysis. Mp 170–171 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.90–7.86 (m, 2H), 7.80 (d, J = 8.0 Hz, 1H), 7.63–7.53 (m, 3H), 7.27 (d, J = 7.2 Hz, 2H), 7.09–7.08 (m, 4H), 6.97 (d, J = 7.2 Hz, 1H), 6.87–6.83 (m, 2H), 6.48 (s, 1H), 5.95 (d, J = 7.2 Hz, 1H), 5.00 (d, J = 16.0 Hz, 1H), 3.76 (d, J = 16.0 Hz, 1H), 2.59 (s, 3H), 2.16 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 192.7, 190.9, 177.3, 169.9, 142.0, 135.4, 134.9, 132.3, 131.8, 129.5, 129.1, 128.9, 128.8, 128.5, 128.3, 127.7, 127.3, 127.0, 126.9, 124.7, 123.0, 122.7, 118.4, 108.8, 89.0, 62.1, 44.4, 29.7, 29.0, 15.9; ESI HRMS: calcd for C32H25NO4Na+ 510.1681, found 510.1682.
Compound 10j was obtained as white solid, 74% yield (41.7 mg). The diastereomeric ratio was determined to be 6 : 1 by crude 1H-NMR analysis. Mp 145–146 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.48 (d, J = 7.2 Hz, 2H), 7.36 (t, J = 7.8 Hz, 3H), 7.29 (d, J = 7.2 Hz, 1H), 7.15 (dt, J = 3.0, 1.2 Hz, 1H), 6.93 (d, J = 2.4 Hz, 1H), 6.71 (d, J = 7.8 Hz, 1H), 5.38 (s, 1H), 5.10 (d, J = 16.2 Hz, 1H), 4.99 (d, J = 15.6 Hz, 1H), 2.50 (s, 3H), 2.12 (s, 3H), 1.77 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 201.4, 191.1, 176.9, 169.3, 142.6, 135.5, 129.4, 128.8, 127.7, 127.4, 123.5, 122.9, 109.6, 91.7, 61.2, 44.7, 28.9, 27.3, 15.7; ESI HRMS: calcd for C23H21NO4Na+ 398.1368, found 398.1368.
Compound 10k was obtained as white solid, 76% yield (46.2 mg). The diastereomeric ratio was determined to be 5 : 1 by crude 1H-NMR analysis. Mp 168–169 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.44 (d, J = 7.8 Hz, 2H), 7.34 (t, J = 7.2 Hz, 2H), 7.28 (d, J = 7.2 Hz, 1H), 7.14 (td, J = 7.8, 1.2 Hz, 1H), 7.02 (dd, J = 7.8, 1.2 Hz, 1H), 6.93 (td, J = 7.8, 0.6 Hz, 1H), 6.67 (d, J = 7.8 Hz, 1H), 5.43 (s, 1H), 5.17 (d, J = 15.6 Hz, 1H), 4.86 (d, J = 15.6 Hz, 1H), 4.20 (m, 1H), 3.72 (m, 1H), 2.50 (s, 3H), 2.13 (s, 3H), 0.50 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 191.2, 176.9, 169.5, 166.1, 143.0, 135.8, 128.3, 128.8, 128.0, 127.7, 123.7, 122.7, 109.2, 85.7, 61.5, 44.6, 34.4, 31.8, 29.0, 15.8, 13.3; ESI HRMS: calcd for C24H23NO5Na+ 428.1474, found 428.1476.
Compound 10l was obtained as white solid, 70% yield (47.8 mg). The diastereomeric ratio was determined to be 10 : 1 by crude 1H-NMR analysis. Mp 158–159 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.46 (tt, J = 7.8, 1.2 Hz, 1H), 7.33 (dd, J = 8.4, 1.2 Hz, 2H), 7.31–7.28 (m, 4H), 7.20 (td, J = 7.8, 1.8 Hz, 2H), 6.69 (dd, J = 7.8, 2.4 Hz, 1H), 6.60 (td, J = 8.4, 2.4 Hz, 1H), 6.31 (s, 1H), 6.07 (dd, J = 8.4, 4.2 Hz, 1H), 5.30 (s, 1H), 5.10 (d, J = 16.0 Hz, 1H), 4.18 (d, J = 16.2 Hz, 1H), 2.57 (s, 3H), 2.22 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 192.6, 190.9, 177.2, 159.2 (d, JCF = 241.5 Hz), 138.3, 135.0, 134.9, 128.9, 128.5, 127.7, 127.6, 127.2, 118.6, 115.3 (d, JCF = 24.0 Hz), 112.7 (d, JCF = 24.0 Hz), 109.4 (d, JCF = 9.0 Hz), 88.8, 62.4, 44.8, 29.1, 15.9; ESI HRMS: calcd for C28H22FNO4Na+ 478.1431, found 478.1429.
Compound 10m was obtained as white solid, 56% yield (38.2 mg). The diastereomeric ratio was determined to be 16 : 1 by crude 1H-NMR analysis. Mp 160–161 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.43 (tt, J = 7.8, 1.2 Hz, 1H), 7.39 (d, J = 7.2 Hz, 2H), 7.32 (td, J = 7.2, 1.8 Hz, 2H), 7.28–7.25 (m, 1H), 7.23 (dd, J = 8.4, 1.2 Hz, 2H), 7.12 (t, J = 7.2 Hz, 2H), 6.82–6.79 (m, 1H), 6.72–6.68 (m, 2H), 6.27 (s, 1H), 5.09 (d, J = 15.6 Hz, 1H), 4.48 (d, J = 15.0 Hz, 1H), 2.56 (s, 3H), 2.16 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 192.7, 191.0, 177.1, 170.1, 146.9 (d, JCF = 244.9 Hz), 136.6, 134.8, 133.8, 128.6, 128.4, 127.6 (d, JCF = 2.0 Hz), 127.4, 123.5, 123.4, 120.7, 120.6, 118.6, 117.2, 117.0, 89.0, 62.2, 46.1, 29.1, 15.9; ESI HRMS: calcd for C28H22FNO4Na+ 478.1431, found 478.1432.
Compound 10n was obtained as white solid, 66% yield (46.7 mg). The diastereomeric ratio was determined to be 5 : 1 by crude 1H-NMR analysis. Mp 159–160 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.46 (t, J = 7.8 Hz, 1H), 7.31 (dd, J = 7.8, 1.2 Hz, 3H), 7.28 (d, J = 5.4 Hz, 3H), 7.24 (dd, J = 6.6, 5.4 Hz, 1H), 7.21 (t, J = 7.8 Hz, 2H), 6.91 (d, J = 1.8 Hz, 1H), 6.87 (dd, J = 8.4, 2.4 Hz, 1H), 6.29 (s, 1H), 6.06 (d, J = 8.4 Hz, 1H), 5.08 (d, J = 16.2 Hz, 1H), 4.18 (d, J = 16.2 Hz, 1H), 2.57 (s, 3H), 2.24 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 192.7, 191.0, 177.1, 170.1, 147.7, 146.1, 136.6, 134.8, 133.8, 128.6, 128.4, 127.7, 127.6, 127.5, 127.4, 123.5, 123.4, 120.6, 118.6, 117.2, 117.0, 89.0, 62.2, 46.1, 29.0, 15.9; ESI HRMS: calcd for C28H22ClNO4Na+ 494.1135, found 494.1133.
Compound 10o was obtained as yellow solid, 67% yield (47.4 mg). The diastereomeric ratio was determined to be 15 : 1 by crude 1H-NMR analysis. Mp 164–165 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.47 (tt, J = 7.2, 1.2 Hz, 1H), 7.33–7.31 (m, 1H), 7.31–7.28 (m, 5H), 7.21 (td, J = 8.4, 1.8 Hz, 2H), 6.84 (d, J = 1.8 Hz, 2H), 6.28 (s, 1H), 6.16 (s, 1H), 5.05 (d, J = 15.6 Hz, 1H), 4.18 (d, J = 16.2 Hz, 1H), 2.56 (s, 3H), 2.20 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 192.7, 190.9, 177.4, 170.0, 143.5, 134.9, 134.7, 133.8, 129.0, 128.5, 128.9, 127.8, 127.5, 125.6, 125.5, 122.7, 118.6, 109.4, 88.8, 61.8, 44.7, 29.1, 15.9; ESI HRMS: calcd for C28H22ClNO4Na+ 494.1135, found 494.1136.
Compound 10p was obtained as white solid, 70% yield (54.2 mg). The diastereomeric ratio was determined to be 13 : 1 by crude 1H-NMR analysis. Mp 200–201 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.46 (td, J = 7.2, 0.6 Hz, 1H), 7.31 (d, J = 7.8 Hz, 3H), 7.29–7.27 (m, 4H), 7.21 (t, J = 7.2 Hz, 2H), 7.04 (d, J = 1.8 Hz, 1H), 7.02 (dt, J = 8.4, 2.4 Hz, 1H), 6.29 (s, 1H), 6.02 (dd, J = 7.8, 0.6 Hz, 1H), 5.08 (d, J = 15.6 Hz, 1H), 4.18 (d, J = 16.2 Hz, 1H), 2.57 (s, 3H), 2.24 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 192.6, 190.9, 176.9, 170.1, 141.4, 135.0, 134.8, 133.7, 131.8, 128.2, 128.9, 128.5, 127.7, 127.7, 127.6, 127.2, 118.6, 115.4, 110.3, 88.8, 62.0, 44.7, 29.1, 16.0; ESI HRMS: calcd for C28H22BrNO4Na+ 538.0630, found 538.0632.
Compound 10q was obtained as yellow solid, 69% yield (54.5 mg). The diastereomeric ratio was determined to be 10 : 1 by crude 1H-NMR analysis. Mp 184–185 °C; 1H NMR (600 MHz, CDCl3) δ (ppm) 7.47 (tt, J = 7.8, 1.2 Hz, 1H), 7.32–7.31 (m, 2H), 7.30–7.26 (m, 5H), 7.21 (td, J = 7.2, 1.2 Hz, 2H), 7.00 (dd, J = 7.8, 1.8 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 6.31 (d, J = 1.8 Hz, 1H), 6.28 (s, 1H), 5.05 (d, J = 15.6 Hz, 1H), 4.18 (d, J = 16.2 Hz, 1H), 2.55 (s, 3H), 2.21 (s, 3H); 13C NMR (150 MHz, CDCl3) δ (ppm) 192.7, 190.9, 177.3, 170.0, 143.6, 134.8, 134.7, 133.8, 128.9, 128.5, 127.8, 127.5, 127.2, 126.1, 125.9, 125.7, 122.6, 118.5, 112.1, 88.7, 61.8, 44.7, 29.1, 15.9; ESI HRMS: calcd for C28H22BrNO4Na+ 538.0630, found 538.0633.
Compound 10r was obtained as white solid, 80% yield (54.1 mg). The diastereomeric ratio was determined to be 18 : 1 by crude 1H-NMR analysis. Mp 162–163 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.42 (t, J = 7.2 Hz, 1H), 7.25 (d, J = 4.4 Hz, 4H), 7.25 (d, J = 7.2 Hz, 3H), 7.16 (t, J = 7.6 Hz, 2H), 6.74 (s, 1H), 6.70 (d, J = 8.0 Hz, 1H), 6.29 (s, 1H), 6.05 (d, J = 8.0 Hz, 1H), 5.06 (d, J = 16.0 Hz, 1H), 4.16 (d, J = 16.0 Hz, 1H), 2.56 (s, 3H), 2.18 (s, 3H), 2.13 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 193.0, 191.1, 177.1, 169.9, 139.8, 135.4, 135.1, 133.3, 132.3, 129.2, 128.7, 128.5, 128.4, 128.3, 127.5, 127.4, 127.3, 126.9, 125.4, 118.4, 108.5, 89.1, 62.0, 44.6, 29.0, 20.9, 15.9; ESI HRMS: calcd for C29H25NO4Na+ 474.1681, found 474.1683.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful for financial support from the National Natural Science Foundation of China (81573588, 81773889, 21702035), the Science & Technology Department of Sichuan Province (2017JZYD0001, 2017JY0323, 2016TD0006).
Electronic supplementary information (ESI) available: Detailed experimental procedures and full spectroscopic data of all the compounds. CCDC 1895883 and 1895884. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra02192b
Notes and references
- For selective reviews on spirooxindole, see:; (a) Yu B. Zheng Y.-C. Shi X.-J. Qi P.-P. Liu H.-M. Anti-Cancer Agents Med. Chem. 2016;16:1315–1324. doi: 10.2174/1871520615666151102093825. [DOI] [PubMed] [Google Scholar]; (b) Somei M. Yamada F. Nat. Prod. Rep. 2005;22:73–103. doi: 10.1039/B316241A. [DOI] [PubMed] [Google Scholar]; (c) Kochanowska-Karamyan A. J. Hamann M. T. Chem. Rev. 2010;110:4489–4497. doi: 10.1021/cr900211p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Badillo J. J. Hanhan N. V. Franz A. K. Curr. Opin. Drug Discovery Dev. 2010;13:758–776. [PubMed] [Google Scholar]; (e) Galliford C. V. Scheidt K. A. Angew. Chem., Int. Ed. 2007;46:8748–8758. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]
- (a) Zhang Z. Wang Z. Huang K. Liu Y. Wei C. Zhou J. Zhang W. Wang Q. Liang H. Zhang A. Wang G. Zhen Y. Han L. Cancer Lett. 2019;443:91–107. doi: 10.1016/j.canlet.2018.11.034. [DOI] [PubMed] [Google Scholar]; (b) Cui C.-B. Kakeya H. Osada H. Tetrahedron. 1996;52:12651–12666. doi: 10.1016/0040-4020(96)00737-5. [DOI] [Google Scholar]; (c) Yu B. Yu D.-Q. Liu H.-M. Eur. J. Med. Chem. 2015;97:673–698. doi: 10.1016/j.ejmech.2014.06.056. [DOI] [PubMed] [Google Scholar]
- For selective reviews on the synthesis of spirooxindole, see:; (a) Singh G. S. Desta Z. Y. Chem. Rev. 2012;112:6104–6155. doi: 10.1021/cr300135y. [DOI] [PubMed] [Google Scholar]; (b) Cheng D. Ishihara Y. Tan B. Barbas, III C. F. ACS Catal. 2014;4:743–762. doi: 10.1021/cs401172r. [DOI] [Google Scholar]; (c) Mei G.-J. Shi F. Chem. Commun. 2018;54:6607–6621. doi: 10.1039/C8CC02364F. [DOI] [PubMed] [Google Scholar]; (d) Xia M. Ma R.-Z. J. Heterocycl. Chem. 2014;51:539–554. doi: 10.1002/jhet.1114. [DOI] [Google Scholar]; (e) Santos M. M. M. Tetrahedron. 2014;70:9735–9757. doi: 10.1016/j.tet.2014.08.005. [DOI] [Google Scholar]; (f) Liu Y. Wang H. Wan J. Asian J. Org. Chem. 2013;2:374–386. doi: 10.1002/ajoc.201200180. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Franz A. K. Hanhan N. V. Ball-Jones N. R. ACS Catal. 2013;3:540–553. doi: 10.1021/cs300801y. [DOI] [Google Scholar]; (h) Hong L. Wang R. Adv. Synth. Catal. 2013;355:1023–1052. doi: 10.1002/adsc.201200808. [DOI] [Google Scholar]; (i) Ball-Jones N. R. Badillo J. J. Franz A. K. Org. Biomol. Chem. 2012;10:5165–5181. doi: 10.1039/C2OB25184A. [DOI] [PubMed] [Google Scholar]; (j1) MacDonald J. P. Badillo J. J. Arevalo G. E. Silva-García A. Franz A. K. ACS Comb. Sci. 2012;14:285–293. doi: 10.1021/co300003c. [DOI] [PMC free article] [PubMed] [Google Scholar]; . For the lasted examples on the synthesis of spirooxindole, see:; (k) Wang C.-S. Li T.-Z. Cheng Y.-C. Zhou J. Mei G.-J. Shi F. J. Org. Chem. 2019;84:3214–3222. doi: 10.1021/acs.joc.8b03004. [DOI] [PubMed] [Google Scholar]; (l) Jiang F. Luo G.-Z. Zhu Z.-Q. Wang C.-S. Mei G.-J. Shi F. J. Org. Chem. 2018;83:10060–10069. doi: 10.1021/acs.joc.8b01390. [DOI] [PubMed] [Google Scholar]; (m) Jiang X.-L. Liu S.-J. Gu Y.-Q. Mei G.-J. Shi F. Adv. Synth. Catal. 2017;359:3341–3346. doi: 10.1002/adsc.201700487. [DOI] [Google Scholar]; (n) Lian X.-L. Adili A. Liu B. Tao Z.-L. Han Z.-Y. Org. Biomol. Chem. 2017;15:3670–3673. doi: 10.1039/C7OB00484B. [DOI] [PubMed] [Google Scholar]; (o) Roh H. J. Kim S. Y. Min B. K. kim J. N. Tetrahedron Lett. 2017;58:21–24. doi: 10.1016/j.tetlet.2016.11.077. [DOI] [Google Scholar]; (p) Zhan G. Shi M.-L. He Q. Lin W.-J. Ouyang Q. Du W. Chen Y.-C. Angew. Chem., Int. Ed. 2016;55:2147–2151. doi: 10.1002/anie.201510825. [DOI] [PubMed] [Google Scholar]; (q) Chan W.-L. Tang X. Zhang F. Quek G. Mei G.-J. Lu Y. Angew. Chem., Int. Ed. 2019 doi: 10.1002/anie.201900758. [DOI] [PubMed] [Google Scholar]
- (a) Kimishima A. Umihara H. Mizoguchi A. Yokoshima S. Fukuyama T. Org. Lett. 2014;16:6244–6247. doi: 10.1021/ol503175n. [DOI] [PubMed] [Google Scholar]; (b) Son B. W. Jensen P. R. Kaulfman C. A. Fenical W. Nat. Prod. Rep. 1999;13:213–222. [Google Scholar]; (c) Calverley P. M. A. Anderson J. A. Celli B. Ferguson G. T. Jenkins C. Jones P. W. Yates J. C. Vestbo J. N. Engl. J. Med. 2007;356:775–789. doi: 10.1056/NEJMoa063070. [DOI] [PubMed] [Google Scholar]
- (a) Liu Y. Han S.-J. Liu W.-B. Stoltz B. M. Acc. Chem. Res. 2015;48:740–751. doi: 10.1021/ar5004658. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Long R. Huang J. Gong J. Yang Z. Nat. Prod. Rep. 2015;32:1584–1601. doi: 10.1039/C5NP00046G. [DOI] [PubMed] [Google Scholar]; (c) Peterson E. A. Overman L. E. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11943–11948. doi: 10.1073/pnas.0402416101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selective books about sulfur ylide, see:; (a) Aggarwal V. K. and Richardson J., in Science of Synthesis : Sulfur Ylides, ed. A.Padwa and D.Bellus, George Thieme Verlag, Stuttgart, 2004, pp. 21–105 [Google Scholar]; (b) Aggarwal V. K., Richardson J. and Winn C. L., in Science of Synthesis: α-Substituted Sulfur Ylides, ed. A. B.Charette, George Thieme Verlag, Stuttgart, 2005, pp. 11–74, For selective reviews on sulur ylide, see: [Google Scholar]; (c) Lu L.-Q. Li T.-R. Wang Q. Xiao W.-J. Chem. Soc. Rev. 2017;46:4135–4149. doi: 10.1039/C6CS00276E. [DOI] [PubMed] [Google Scholar]; (d) Zhu C. Ding Y. Ye L.-W. Org. Biomol. Chem. 2015;13:2530–2536. doi: 10.1039/C4OB02556C. [DOI] [PubMed] [Google Scholar]; (e) Sun X.-L. Tang Y. Acc. Chem. Res. 2008;41:937–948. doi: 10.1021/ar800108z. [DOI] [PubMed] [Google Scholar]; (f) Aggarwal V. K. Winn C. L. Acc. Chem. Res. 2004;37:611–620. doi: 10.1021/ar030045f. [DOI] [PubMed] [Google Scholar]; (g) Li A.-H. Dai L.-X. Aggarwal V. K. Chem. Rev. 1997;97:2341–2372. doi: 10.1021/cr960411r. [DOI] [PubMed] [Google Scholar]
- (a) Wang L. Cao W. Mei H. Hu L. Feng X. Adv. Synth. Catal. 2018;360:4089–4093. doi: 10.1002/adsc.201800937. [DOI] [Google Scholar]; (b) Mei H. Pan G. Zhang X. Lin L. Liu X. Feng X. Org. Lett. 2018;20:7794–7797. doi: 10.1021/acs.orglett.8b03187. [DOI] [PubMed] [Google Scholar]; (c) Li Q.-Z. Zhang X. Zeng R. Dai Q.-S. Liu Y. Shen X.-D. Leng H.-J. Yang K.-C. Li J.-L. Org. Lett. 2018;20:3700–3704. doi: 10.1021/acs.orglett.8b01537. [DOI] [PubMed] [Google Scholar]; (d) Yuan Z. Fang X. Li X. Wu J. Yao H. Lin A. J. Org. Chem. 2015;80:11123–11130. doi: 10.1021/acs.joc.5b01793. [DOI] [PubMed] [Google Scholar]; (e) Zhang X.-Z. Du J.-Y. Deng Y.-H. Chu W.-D. Yan X. Yu K.-Y. Fan C.-A. J. Org. Chem. 2016;81:2598–2606. doi: 10.1021/acs.joc.5b02725. [DOI] [PubMed] [Google Scholar]; (f) Liu L. Yuan Z. Pan R. Zeng Y. Lin A. Yao H. Huang Y. Org. Chem. Front. 2018;5:623–628. doi: 10.1039/C7QO00846E. [DOI] [Google Scholar]; (g) Ye S. Huang Z.-Z. Xia C.-A. Tang Y. Dai L.-X. J. Am. Chem. Soc. 2002;124:2432–2433. doi: 10.1021/ja0172969. [DOI] [PubMed] [Google Scholar]; (h) Deng X.-M. Cai P. Ye S. Sun X.-L. Liao W.-W. Li K. Tang Y. Wu Y.-D. Dai L.-X. J. Am. Chem. Soc. 2006;128:9730–9740. doi: 10.1021/ja056751o. [DOI] [PubMed] [Google Scholar]; (i) Marsini M. A. Reeves J. T. Desrosiers J.-N. Herbage M. A. Savoie J. Li Z. Fandrick K. R. Sader C. A. McKibben B. Gao D. A. Cui J. Gonnella N. C. Lee H. Wei X. Roschangar F. Lu B. Z. Senanayake C. H. Org. Lett. 2015;17:5614–5617. doi: 10.1021/acs.orglett.5b02838. [DOI] [PubMed] [Google Scholar]; (j) Wu H.-Y. Chang C.-W. Chein R.-J. J. Org. Chem. 2013;78:5788–5793. doi: 10.1021/jo400648f. [DOI] [PubMed] [Google Scholar]; (k) Illa O. Namutebi M. Saha C. Ostovar M. Chen C. C. Haddow M. F. Nocquet-Thibault S. Lusi M. McGarrigle E. M. Aggarwal V. K. J. Am. Chem. Soc. 2013;135:11951–11966. doi: 10.1021/ja405073w. [DOI] [PubMed] [Google Scholar]; (l) Illa O. Arshad M. Ros A. McGarrigle E. M. Aggarwal V. K. J. Am. Chem. Soc. 2010;132:1828–1830. doi: 10.1021/ja9100276. [DOI] [PubMed] [Google Scholar]; (m) Zhu B.-H. Zheng J.-C. Yu C.-B. Sun X.-L. Zhou Y.-G. Shen Q. Tang Y. Org. Lett. 2010;12:504–507. doi: 10.1021/ol9027072. [DOI] [PubMed] [Google Scholar]; (n) Aggarwal V. K. Charmant J. P. H. Fuentes D. Harvey J. N. Hynd G. Ohara D. Picoul W. Robiette R. Smith C. Vasse J.-L. Winn C. L. J. Am. Chem. Soc. 2006;128:2105–2114. doi: 10.1021/ja0568345. [DOI] [PubMed] [Google Scholar]; (o) Janardanan D. Sunoj R. B. J. Org. Chem. 2008;73:8163–8174. doi: 10.1021/jo800652c. [DOI] [PubMed] [Google Scholar]; (p) Aggarwal V. K. Vasse J.-L. Org. Lett. 2003;5:3987–3990. doi: 10.1021/ol035554w. [DOI] [PubMed] [Google Scholar]; (q) Aggarwal V. K. Ford J. G. Fonquerna S. Adams H. Jones R. V. H. Fieldhouse R. J. Am. Chem. Soc. 1998;120:8328–8339. doi: 10.1021/ja9812150. [DOI] [Google Scholar]
- (a) Zheng J.-C. Zhu C.-Y. Sun X.-L. Tang Y. Dai L.-X. J. Org. Chem. 2008;73:6909–6912. doi: 10.1021/jo801135j. [DOI] [PubMed] [Google Scholar]; (b) Lu L.-Q. Zhang J.-J. Li F. Cheng Y. An J. Chen J.-R. Xiao W.-J. Angew. Chem., Int. Ed. 2010;49:4495–4498. doi: 10.1002/anie.201000755. [DOI] [PubMed] [Google Scholar]; (c) Wang Q. Qi X. Lu L.-Q. Li T.-R. Yuan Z.-G. Zhang K. Li B.-J. Lan Y. Xiao W.-J. Angew. Chem., Int. Ed. 2016;55:2840–2844. doi: 10.1002/anie.201510413. [DOI] [PubMed] [Google Scholar]; (d) Yang Q.-Q. Wang Q. An J. Chen J.-R. Lu L.-Q. Xiao W.-J. Chem.–Eur. J. 2013;19:8401–8404. doi: 10.1002/chem.201300988. [DOI] [PubMed] [Google Scholar]; (e) Lu L.-Q. Cao Y.-J. Liu X.-P. An J. Yao C.-J. Ming Z.-H. Xiao W.-J. J. Am. Chem. Soc. 2008;130:6946–6948. doi: 10.1021/ja800746q. [DOI] [PubMed] [Google Scholar]; (f) Chen J.-R. Dong W.-R. Candy M. Pan F.-F. Jörres M. Bolm C. J. Am. Chem. Soc. 2012;134:6924–6927. doi: 10.1021/ja301196x. [DOI] [PubMed] [Google Scholar]; (g) Xie P. Wang L. Yang L. Li E. Ma J. Huang Y. Chen R. J. Org. Chem. 2011;76:7699–7705. doi: 10.1021/jo2008737. [DOI] [PubMed] [Google Scholar]
- (a) Tang X. Zhu H.-P. Zhou J. Chen Y. Pan X.-L. Guo L. Li J.-L. Peng C. Huang W. Org. Biomol. Chem. 2018;16:8169–8174. doi: 10.1039/C8OB02034E. [DOI] [PubMed] [Google Scholar]; (b) Roy S. Piradhi V. ChemistrySelect. 2017;2:6159–6162. doi: 10.1002/slct.201701221. [DOI] [Google Scholar]; (c) Li Y. Li Q.-Z. Huang L. Liang H. Yang K.-C. Leng H.-J. Liu Y. Shen X.-D. Guo X.-J. Li J.-L. Molecules. 2017;22:328. doi: 10.3390/molecules22020328. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Meng X.-S. Jiang S. Xu X.-Y. Wu Q.-X. Gu Y.-C. Shi D.-Q. Eur. J. Org. Chem. 2016;2016:4778–4781. doi: 10.1002/ejoc.201600759. [DOI] [Google Scholar]; (e) Bernard A. M. Frongia A. Piras P. P. Secci F. Spiga M. Org. Lett. 2005;7:4565–4568. doi: 10.1021/ol0514606. [DOI] [PubMed] [Google Scholar]; (f) Corey E. J. Chaykovsky M. J. Am. Chem. Soc. 1962;84:867–868. doi: 10.1021/ja00864a040. [DOI] [Google Scholar]
- (a) Volkers A. A. Mao X. S. Klunder A. J. H. Zwanenburg B. Tetrahedron. 2009;11:2364–2367. doi: 10.1016/j.tet.2009.01.002. [DOI] [Google Scholar]; (b) Sheikh S. E. Kausch N. Lex J. Neudorfl J.-M. Schmalz H.-G. Synlett. 2006;10:1527–1530. [Google Scholar]; (c) Blanchfield J. T. Chow S. Bernhardt P. V. Kennard C. H. L. Kitching W. Aust. J. Chem. 2004;57:673–676. doi: 10.1071/CH04047. [DOI] [Google Scholar]
- (a) Chen J. Jia P. Huang Y. Org. Lett. 2018;20:6715–6718. doi: 10.1021/acs.orglett.8b02810. [DOI] [PubMed] [Google Scholar]; (b) Oost R. Neuhaus J. D. Misale A. Meyrelles R. Veiros L. F. Maulide N. Chem. Sci. 2018;9:7091–7995. doi: 10.1039/C8SC02815J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li C. Jiang K. Ouyang Q. Liu T.-Y. Chen Y.-C. Org. Lett. 2016;18:2738–2741. doi: 10.1021/acs.orglett.6b01194. [DOI] [PubMed] [Google Scholar]
- (a) Xie X. Huang W. Peng C. Han B. Adv. Synth. Catal. 2018;360:194–228. doi: 10.1002/adsc.201700927. [DOI] [Google Scholar]; (b) Zhao Q. Peng C. Huang H. Liu S.-J. Zhong Y.-J. Huang W. He G. Han B. Chem. Commun. 2018;54:8359–8362. doi: 10.1039/C8CC04732D. [DOI] [PubMed] [Google Scholar]; (c) Yang M.-C. Peng C. Huang H. Yang L. He X.-H. Huang W. Cui H.-L. He G. Han B. Org. Lett. 2017;19:6752–6755. doi: 10.1021/acs.orglett.7b03516. [DOI] [PubMed] [Google Scholar]; (d) Li X. Huang W. Liu Y.-Q. Kang J.-W. Xia D. He G. Peng C. Han B. J. Org. Chem. 2017;82:397–406. doi: 10.1021/acs.joc.6b02489. [DOI] [PubMed] [Google Scholar]; (e) Han B. Huang W. Ren W. He G. Wang J.-H. Peng C. Adv. Synth. Catal. 2015;357:561–568. doi: 10.1002/adsc.201400764. [DOI] [Google Scholar]; (f) Zhou R. Wu Q.-J. Guo M.-R. Huang W. He X.-H. Yang L. Peng F. He G. Han B. Chem. Commun. 2015;51:13113–13116. doi: 10.1039/C5CC04968G. [DOI] [PubMed] [Google Scholar]; (g) Li X. Yang L. Peng C. Xie X. Leng H.-J. Wang B. Tang Z.-W. He G. Ouyang L. Huang W. Han B. Chem. Commun. 2013;49:8692–8694. doi: 10.1039/C3CC44004D. [DOI] [PubMed] [Google Scholar]; (h) Xie X. Peng C. He G. Leng H.-J. Wang B. Huang W. Han B. Chem. Commun. 2012;48:10487–10489. doi: 10.1039/C2CC36011J. [DOI] [PubMed] [Google Scholar]; (i) Wang B. Leng H.-J. Yang X.-Y. Han B. Rao C.-L. Liu L. Peng C. Huang W. RSC Adv. 2015;5:88272–88276. doi: 10.1039/C5RA15735H. [DOI] [Google Scholar]
- CCDC 1895883 (for 6r) contains the supplementary crystallographic data for this paper.†
- CCDC 1895884 (for 10a) contains the supplementary crystallographic data for this paper.†
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.