

Abstract
Global knockout of the nonmuscle isoform of myosin light‐chain kinase (nmMLCK), a primary cellular regulator of cytoskeletal machinery, is strongly protective in preclinical murine models of inflammatory lung injury. The current study was designed to assess the specific contribution of endothelial cell (EC) nmMLCK to the severity of murine inflammatory lung injury produced by lipopolysaccharide (LPS) and mechanical ventilation ventilator‐induced lung injury or ventilation (VILI). Responses to combined LPS/VILI exposure were assessed in: (i) wild‐type (WT) C57BL/6J mice; (ii) transgenic mice with global deletion of nmMLCK (nmMylk −/−); (iii) transgenic nmMylk −/− mice with overexpression of nmMLCK restricted to the endothelium (nmMylk −/−/ec‐tg+). Lung inflammation indices included lung histology, bronchoalveolar lavage (BAL) polymorphonuclear leukocytes (PMNs), lung protein biochemistry, tissue albumin levels, Evans blue dye (EBD) lung extravasation, and plasma cytokines (interleukin‐6 [IL‐6], keratinocyte chemoattractant [KC]/IL‐8, IL‐1bβ, extracellular nicotinamide phosphoribosyltransferase, tumor necrosis factor‐α). Compared to WT C57BL/6J mice, the severity of LPS/VILI‐induced lung injury was markedly reduced in mice with global nmMLCK deletion reflected by reductions in histologic inflammatory lung injury, BAL PMN counts, mitogen‐activated protein kinase, and NF‐kB pathway activation in lung homogenates, plasma cytokine levels, and parameters of lung permeability (increased BAL protein, tissue albumin levels, EBD lung extravasation). In contrast, mice with restricted overexpression of nmMLCK in EC (nmMylk −/−/ec‐tg+) showed significant persistence of LPS/VILI‐induced lung injury severity compared to WT mice. In conclusion, these studies strongly endorse the role of EC nmMLCK in driving the severity of preclinical inflammatory lung injury. Precise targeting of EC nmMLCK may represent an attractive therapeutic strategy to reduce lung inflammation and both lung and systemic vascular permeability.
Keywords: ARDS, MLCK
INTRODUCTION
The excessive mortality of the severe inflammatory disorder known as the acute respiratory distress syndrome (ARDS), 1 , 2 , 3 whether caused by sepsis, trauma, or either bacterial/viral pneumonias, 4 is directly attributable to unchecked vascular inflammation, and the profound loss of lung EC barrier integrity leading to alveolar flooding. The respiratory failure that ensues often requires mechanical ventilation, another potent contributor to inflammatory lung permeability and ARDS mortality, a process known as ventilator‐induced lung injury or ventilation (VILI). 1 , 3 The uncontrolled lung inflammatory response results in sustained cytokine release and systemic increases in vascular permeability, and the development of multiorgan failure, a primary cause of ARDS mortality. 4
Current concepts of vascular permeability and alveolar edema formation/resolution involves highly choreographed EC cytoskeletal regulation of the integrity of both EC and alveolar epithelial cellular barriers. Inflammation‐mediated activation of the cytoskeletal contractile apparatus results in the loss of alveolar and vascular barrier integrity, increased plasma protein influx, and the diapedesis of inflammatory cells into the lung parenchyma, particularly polymorphonuclear leukocytes (PMNs). Our system biology approaches, involving genomics, proteomics, biophysical imaging, engineered mice, and genetic epidemiology, have confirmed the critical role of the multifunctional nonmuscle myosin light chain (MLC) kinase isoform (nmMLCK1, 210 kDa, 1914 amino acids), 5 as a major regulator of vascular EC barrier function, 6 , 7 , 8 , 9 angiogenesis, 10 , 11 EC apoptosis, 12 leukocytic trafficking, 13 and vascular responses to mechanical ventilator‐derived mechanical stress. 9 Increased EC nmMLCK enzymatic activities in response to inflammatory agonists results in enhanced MLC phosphorylation at critical Ser 14 and Thr 15 amino acids and spatially specific cellular contraction, paracellular gap formation, and disruption of the vascular barrier, 16 , 17 as well as alveolar and gastrointestinal epithelial barriers. 18 We have convincingly shown nmMLCK to drive spatially localized EC cytoskeletal rearrangements in both lung vascular barrier disruption (nmMLCK‐catalyzed EC stress fiber formation) as well as in EC barrier‐restorative processes involving nmMLCK translocation to paracellular lamellipodia with gap closure. 6 , 14 , 15 , 19 The intimate involvement of nmMLCK in the regulation of sepsis‐ and ventilator‐induced inflammatory lung permeability observed in ARDS/VILI 9 , 20 is supported by genetic epidemiologic studies analyzing genetic variants (single‐nucleotide polymorphisms) identified in the gene encoding nmMLCK, MYLK, that confer increased risk of sepsis/trauma‐induced ARDS in Blacks, 21 , 22 ARDS severity in Blacks, 21 , 22 , 23 and increased risk of severe asthma in Blacks. 21 , 24 , 25
The nmMLCK isoform would appear to be an attractive ARDS/VILI therapeutic target as our in vivo studies demonstrated that nmMLCK inhibitory approaches (inhibitory oligopeptides, small interfering RNAs) reduce alveolar and vascular permeability and lung inflammation. 9 Furthermore, genetically engineered nmMLCK KO mice (nmMylk −/−) with targeted global deletion of the nmMLCK isoform exhibited strong in vivo protection from lipopolysaccharide (LPS)‐induced lung injury and VILI‐mediated lung edema, 9 , 26 confirming nmMLCK as a contributor to ARDS/VILI severity. 22 , 27 However, as nmMylk −/− mice exhibit global loss of nmMLCK, it remains entirely unclear as to which cellular sources of nmMLCK, that is, alveolar epithelium, lung endothelium, PMNs, or T‐lymphocytes, are the primary contributors to the protection from inflammatory injury observed in nmMylk −/− mice. Speculating that EC expression of nmMLCK was a major contributor to inflammatory lung injury severity, we generated transgenic mice, nmMylk ec/+, that overexpress the major nmMLCK splice variant, nmMLCK2, restricted to the vascular endothelium. We observed a strong inflammatory phenotype with marked exaggeration of LPS‐ and ventilator‐induced alveolar flooding and lung injury in a sex‐ and age‐specific manner. 20 , 28
The present study seeks to extend these prior studies by fully establishing EC nmMLCK as an attractive molecular target driving the severity of acute inflammatory lung injury in a clinically relevant murine model of ARDS/VILI. We generated genetically engineered mice, nmMylk −/−/ec‐tg+, that selectively express EC nmMLCK on the nmMylk −/− background. Our results indicate that compared to wild‐type (WT) C57BL/6J mice with combined exposure to LPS‐induced pneumonia and to VILI, nmMylk −/− mice, with global nmMLCK deletion, showed marked attenuation of lung injury severity as previously reported 9 with substantially reduced levels of inflammatory lung injury indices: histologic inflammatory lung injury, bronchoalveolar lavage (BAL) PMN counts, mitogen‐activated protein (MAP) kinase, and NF‐kB pathway activation in lung homogenates, plasma cytokine levels, and parameters of lung permeability (increased BAL protein, tissue albumin levels, and EBD lung extravasation). In contrast, mice with restricted overexpression of nmMLCK2 in EC, nmMylk −/−/ec‐tg+, showed significant preservation of LPS/VILI lung injury severity compared to WT mice. These studies highlight the critical role of the nonmuscle isoform of MLCK (nmMLCK) expressed in vascular endothelium in regulating the severity of inflammatory lung injury in preclinical murine models of ARDS/VILI. Precise targeting of EC nmMLCK may represent a highly attractive therapeutic strategy to reduce both lung inflammation and both lung and systemic vascular permeability.
MATERIALS AND METHODS
Reagents
Unless otherwise noted, all reagents including LPS (Escherichia coli 0127:B8 strain) were purchased from Sigma‐Aldrich.
Animals
In‐house bred nmMylk −/−, 9 , 29 nmMylk −/−/ec‐tg+, and WT littermate controls were used for all in vivo experiments in this study. All mice were housed under standard conditions (12 h light–dark cycle, 25–27°C, ~40% humidity) in autoclaved microisolator cages with free access to food and water throughout the duration of the experiments. All animal care procedures and experiments were approved by the Institutional Animal Care and Use Committee (IACUC, University of Arizona) and were performed in accordance with IACUC and ARRIVE guidelines. The nmMylk −/−/ec‐tg+ strain of mice was created to expand upon previous findings of nmMLCK's more specific role in EC barrier integrity. To create the nmMylk −/−/ec‐tg+ line, we intercrossed these two strains, creating mice heterozygous for both loci. Next, the double heterozygotes were backcrossed to the nmMylk −/− parental strain, resulting in a colony of nmMylk −/−/ec‐tg+ mice. The double transgenic nmMylk −/−/ec‐tg+ strain did not exhibit phenotypic, fecundity, or survivability differences compared with the parental strains. All polymerase chain reaction protocols and reagents used to confirm zygosity and genotypes of the strains were used as previously described. 20 , 29 Gel electrophoresis was used to visually confirm genotypes (Figure S1).
Murine model of LPS/ventilator‐induced lung injury
Mice were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (5 mg/kg) given by intraperitoneal injection with additional doses given as needed during the VILI protocol to ensure adequate anesthetic depth. The animals were intubated with a 20‐G angiocatheter. Animals received LPS (intratracheal, 0.1 mg/kg), allowed to recover for 18 h, and then reintubated and connected to a mechanical ventilator (Advanced Ventilator System For Rodents, SAR‐1000; CWE Incorporated) as we have previously described. 26 , 28 , 30 , 31 Mice were ventilated with room air for 4 h using the following parameters: tidal volume 10 ml/kg, respiratory rate 90 breaths/min, and positive‐end expiratory pressure 0 cm H2O. Spontaneously breathing control animals received intratracheal PBS and were intubated, but allowed to breathe spontaneously on room air during the duration of the experiment.
Tissue albumin enzyme‐linked immunoassay (ELISA)
Mouse albumin ELISA Kit was purchased from Bethyl Labs and tissues were homogenized in 0.5 ml KPO4 buffer with 1.0% hexadecyl trimethyl ammonium bromide (HTAB) with the TissueLyser LT using a 5‐mm stainless steel bead, at 50 mHz for two 5 min intervals. After tissues were homogenized, an additional 0.5 ml of KPO4 buffer was added for a final volume of 1.0 ml. Tissue samples were diluted according to kit specifications to fit within the readout four‐parametric standard curve. 9
BAL analysis
At the termination of each animal experiment, mice were euthanized by approved IACUC methods. BAL of the entire lung was performed with 1 ml of cold Hank's buffered saline solution (HBSS) (Invitrogen) delivered intratracheally followed by a slow recovery of the fluid as we have previously described. 31 , 32 Cells were recovered from the collected BAL fluid by centrifugation (500 g, 20 min, 4°C) and counted using an automated cell counter (TC20; Bio‐Rad). The cell pellet was resuspended in cold 200 µl HBSS and 1 ml of RBC Lysis Solution for 5 min for the determination of total cell count. The BAL supernatant was then recentrifuged (16,5000g, 10 min, 4°C), and the supernatant collected was used to measure total protein concentration determined by a protein assay (Pierce BCA Protein Assay Kit; Thermo Scientific). After measuring, the BAL supernatant was stored at −80°C for further analysis.
Quantitative lung histology and H&E image analysis
To verify lung vascular leak damage and cell infiltration, lungs were collected from mice in experiment groups and processed for histological analysis. Mouse lung tissue was fixed in 10% neutral‐buffered formalin for a minimum of 48 h, embedded in paraffin, sectioned, mounted onto slides, and stained with Richard–Allan hematoxylin–eosin (H&E) reagents (Fisher Scientific). Specimens were then imaged using an Olympus Photomicroscope (Olympus) using both ×20 and ×40 objectives. Multiple images were obtained from each slide and slides of each group were randomly selected for quantification of H&E staining using ImageJ software as previously described. 31 , 33
Western blot analysis of proteins in lung tissues
Western blot analysis of lung homogenates was performed according to standard protocols as previously reported 31 , 32 with densitometric quantification of lung tissue expression of specific proteins, and β‐actin (total protein control).
Plasma biomarker measurements
A Meso Scale ELISA‐based Discovery Platform was utilized (Meso Scale Diagnostics) for measurements of plasma levels of IL‐6, KC/IL‐8, IL‐1β, TNF‐α, and extracellular nicotinamide phosphoribosyltransferase (eNAMPT), as we have previously described. 31 , 34 Briefly, for each cytokine, a specific biotinylated antibody was incubated (30 min, 25°C) with an analyte‐specific linker. The reaction was terminated by the addition of a free biotin solution and a second identical incubation. All cytokine‐specific‐linked antibodies are then pooled and aliquoted into a 96‐well plate and incubated (60 min, 700 rpm) and washed (three times, Tris‐buffered saline). Calibrator, sample, or standard with an equal volume of diluent was added to specific wells and incubated and washed as above. The detection antibody solution was added to each well and then incubated and washed as above. Finally, MSD Read Buffer was added to each well and the plate was read on the MSD platform, and the cytokine concentration was calculated.
Statistical analysis
All data were analyzed using Student's t test and the statistically significant threshold was at p < 0.05.
RESULTS
Histologic and inflammatory responses in LPS/VILI‐challenged WT and MYLK murine strains
H&E staining of lung tissues sections demonstrated that WT C57BL/6J mice exposed to LPS pneumonia (22 h) and VILI (4 h) exhibit substantial histologic inflammatory lung injury characterized by marked inflammatory cell infiltration, alveolar edema, and alveolar wall thickening (Figure 1a) with Image software quantification (Figure 1b). Consistent with this histologic evidence of inflammatory lung injury, LPS/VILI‐exposed WT mice also exhibited marked increases in BAL PMNs (Figure 1c) and elevated plasma levels of IL‐6, KC/IL‐8, IL‐1β, and TNF‐α and eNAMPT (Figure 2a–e). In contrast to WT mice, nmMylk −/− mice with global nmMLCK deletion were markedly protected from LPS/VILI‐induced lung injury with quantifiable reductions in histologic inflammatory lung tissue injury (Figure 1a/b), in BAL PMNs (Figure 1c), and plasma levels of IL‐6, KC/IL‐8, and TNF‐α but not IL‐1β (Figure 2a–e). The substantially attenuated severity of inflammatory lung injury observed in nmMylk −/− mice was significantly abolished in nmMylk −/−/ec‐tg+ mice expressing nmMLCK2 only in EC on a global nmMLCK KO background with a significant restoration of LPS/VILI‐induced lung inflammation (Figure 1a/b), BAL PMNs (Figure 1c), and plasma levels of IL‐6 and TNF‐α (Figure 2). This significant restoration of inflammatory lung injury in nmMylk −/−/ec‐tg+ mice suggests that EC nmMLCK is a key driver of the severity of LPS/VILI‐mediated inflammatory lung injury.
Figure 1.
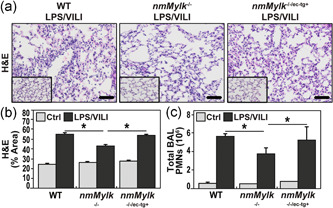
Endothelial‐specific overexpression of nmMLCK abolishes the protection against LPS/VILI lung injury observed innmMYLK −/−mice. (a/b) Compared to PBS‐challenged wild‐type (WT) C57BL6 mice (inset), H&E staining WT mice exposed to a “two‐hit” injury model of LPS (18 h) and ventilator‐induced lung injury (4 h) (LPS/VILI) reveals significant increases in alveolar edema, leukocyte infiltration, and inflammatory lung injury (first panel in a). In contrast, LPS/VILI‐challenged nmMylk −/− mice were protected with significantly reduced PMN infiltration and decreases in vascular leakage (second panel in a). This reduction in lung injury severity was nearly entirely eliminated in nmMylk −/−/ec‐tg+ mice with global nmMLCK KO, except for overexpression of nmMLCK restricted to the endothelium (third panel in a). (b) Image J quantification of H&E staining expressed as the % area of staining. * p < 0.05 comparisons. (c) Total BAL PMN counts corroborate H&E staining findings, showing that compared to either the LPS/VILI‐challenged WT or nmMylk −/−/ec‐tg+ mice, the nmMylk −/− mice have a significant reduction in PMN's found in the alveolar space, indicating a higher level of EC barrier integrity. There is no significant difference in barrier integrity via PMN infiltration between the wild‐type mice and the nmMylk −/−/ec‐tg+ mice. *p < 0.05 comparisons. Ctrl, control; H&E, hematoxylin and eosin; KO, knockout; LPS, lipopolysaccharide; nmMLCK, nonmuscle isoform of myosin light‐chain kinase; PMN, polymorphonuclear leukocyte; VILI, ventilator‐induced lung injury or ventilation; WT, wild type.
Figure 2.

Inflammatory cytokine plasma levels are reduced or differentially expressed in nmMylk –/− compared to the nmMylk −/−/ec‐tg+ . The Meso Scale ELISA‐based Discovery platform was utilized to assess plasma cytokine levels in the LPS/VILI injury model. Depicted are the mouse plasma levels of inflammatory cytokines of IL‐6 (a), IL‐1β (b), KC (c), TNF‐α (d), and eNAMPT (e) obtained in control spontaneous breathing mice (n = 3) for each genotype/group and in mice exposed to LPS/VILI (n = 3) for each genotype/group. Cytokine levels in all animals exposed to LPS/VILI show a drastic increase compared to their control counterparts. nmMylk −/− mice exhibited significantly lower levels of cytokines (a, c, d, e) compared to WT mice, except for plasma levels of IL‐1β (b) were unaffected by nmMylk genotypes. The reduced plasma cytokine levels observed in LPS/VILI‐challenged nmMylk −/− mice were restored in nmMylk −/−/ec‐tg+ mice to levels approximating WT mice. Baseline levels of all five cytokines in each genotype group were comparable, except for the levels of IL‐1β in nmMylk −/−/ec‐tg+ mice, which exhibited an elevated baseline level compared to either the WT or nmMylk −/− control mice. *p < 0.05. Ctrl, control; ELISA, enzyme‐linked immunoassay; eNAMPT, extracellular nicotinamide phosphoribosyltransferase; IL, interleukin; KC, keratinocyte‐derived cytokine; LPS, lipopolysaccharide; VILI, ventilator‐induced lung injury or ventilation; WT, wild type.
Inflammation‐induced lung vascular barrier responses in WT and MYLK murine strains
We next assessed the role of EC nmMLCK in regulating lung vascular barrier responses in response to LPS/VILI‐induced lung inflammation and vascular leak in WT and engineered mice. Images from excised whole lungs obtained from mice injected with EBD 60 min before harvesting after 20 h exposure to intratracheal LPS/4 h VILI demonstrate that exposure to LPS/VILI produced prominent EBD vascular extravasation compared to PBS‐challenged WT mice (Figure 3a/b). In contrast, the magnitude of EBD vascular leak is clearly reduced in the nmMylk −/− mice with global nmMLCK KO (Figure 3c). Importantly, LPS/VILI‐challenged nmMylk −/−/ec‐tg+ mice, with global nmMLCK KO, but restricted nmMLCK expression only in EC, demonstrate increased EBD leakage compared to nmMylk −/− mice approximating the level of EBD extravasation in LPS/VILI‐exposed WT mice (Figure 3d).
Figure 3.

Effect of nmMylk genotypes on murine lung vascular integrity. (a–d) Representative photos of murine lungs exposed to PBS or LPS/VILI via an intratracheal instillation (1 mg/kg, harvest timepoint 20 h post LPS instillation, VILI 20 ml/kg, 4 h). Sixty minutes before harvest, mice (n = 3/group) were injected intravenously with 30 mg/kg of Evans blue dye (EBD) conjugated to albumin as we have previously described. 31 Lungs were flushed with PBS before removal. Comparisons of EBD extravasation in each group were compared with EBD leakage minimal in PBS‐exposed mice. All genotypes exhibited similar EBD levels for the PBS‐exposed mice (a). EBD leakage was markedly increased in each LPS/VILI‐exposed group with greatest increases in EBD extravasation observed in nmMylk −/−/ec‐tg+ mice (b) with marked reductions in nmMylk −/− mice (c). EBD leakage in nmMylk −/−/ec‐tg+ mice showed increased EBD extravasation approximating WT mice (d). (e/f) Consistent with the findings of EC barrier integrity disruption shown in EBD pictures, LPS/VILI‐exposed WT, and nmMylk −/−/ec‐tg+ mice had similar values for BAL protein levels and lung tissue albumin levels compared to PBS‐exposed animals. In contrast, by comparison, LPS/VILI‐exposed nmMylk −/− mice had significantly lower values in both readouts of vascular permeability (* p < 0.05 comparisons). Ctrl, control; LPS, lipopolysaccharide; PBS, phosphate‐buffered saline; VILI, ventilator‐induced lung injury or ventilation; WT, wild type.
These results indicating key regulation of vascular barrier integrity by nmMLCK in EC were confirmed by BAL protein measurements in WT and nmMylk‐engineered mice with Figure 3e showing the significant increase in BAL protein in LPS/VILI‐challenged WT mice which is significantly reduced in nmMylk −/− mice. Consistent with the restored indices of lung inflammation and EBD leakage, LPS/VILI‐challenged nmMylk −/−/ec‐tg+ mice expressing nmMLCK only in EC exhibit BAL protein levels that are significantly greater than nmMylk −/− mice and comparable to WT mice. Lung tissue albumin measurements also show significantly greater tissue albumin content in LPS/VILI‐challenged WT mice compared to unchallenged control mice (Figure 3f) and significantly reduced in nmMylk −/− mice. In contrast, nmMylk −/−/ec‐tg+ mice exhibited levels of tissue albumin content that is significantly greater than nmMylk −/− mice and again comparable to LPS/VILI‐challenged WT mice. Together, these results are consistent with EC nmMLCK serving as a major regulator of lung vascular barrier integrity and lung edema formation.
Lung signaling responses in LPS/VILI‐challenged WT and MYLK murine strains
Finally, we assessed NF‐kB and MAP kinase pathway signaling in lung homogenates from LPS/VILI‐challenged WT and Mylk‐engineered murine strains. As expected and previously reported, 31 LPS/VILI‐challenged WT mice exhibit robust NF‐kB phosphorylation (Figure 4a/b) and MAP kinase activation (p42/44 ERK) (Figure 4a/c). A key effector of the MAP kinase family pathways is the Ser/Thr ribosomal S6 kinase (RSK) or p90rsk as p90rsk is activated by ERK MAPK and signals to the contractile machinery via phosphorylation of the myosin phosphatase‐targeting subunit 1 (MYPT1). 35 Like ERK phosphorylation, p‐pS90RSK phosphorylation was also highly induced in lung tissues from LPS/VILI‐ exposed WT mice (Figure 4a/d). In contrast to WT mice, LPS/VILI‐exposed nmMylk −/− mice showed dramatically reduced levels of NF‐kB, ERK MAPK, and pS90RSK phosphorylation with nmMylk −/−/ec‐tg+ mice exhibiting levels of NF‐kB, ERK MAPK, and p‐pS90RSK phosphorylation that were significantly greater than nmMylk −/− mice and comparable to LPS/VILI‐challenged WT mice (Figure 4). Figure 4 also depicts the prominent LPS/VILI‐induced reductions in NRF2 protein expression in concert with increases in the expression of the reactive oxygen species (ROS)‐generating protein NOX‐4 in WT mice. In contrast to NF‐kB, ERK MAPK, and pS90RSK, biochemical studies of lung homogenates failed to identify significant alterations in either NRF2 or NOX4 in either nmMylk −/− mice or nmMylk −/−/ec‐tg+ with global nmMLCK deletion, but restricted EC nmMLCK expression.
Figure 4.

(a) Western blots in mouse lung tissues exposed to LPS/VILI showing significantly increased phosphorylation/expression of p‐NF‐kB, p‐ERK, NOX4, NRF2, and p‐p90RSK compared to PBS‐exposed control mice. (b–d) Quantifications of panel (a) western blots showing significant reductions in levels of p‐NF‐ƘB, p‐ERK and p‐p90RSK in LPS/VILI nmMylk −/− mice, approximating levels of PBS‐exposed controls. In contrast, LPS/VILI‐exposed nmMylk −/−/ec‐tg+ mice exhibited significantly greater phosphorylation/expression levels, comparable to LPS/VILI‐exposed WT mice. *p < 0.05 vs control; **p <0.05 vs WT; ***p<0.05 vs nmMylk‐/‐. Ctrl, control; LPS, lipopolysaccharide; NF‐kB, nuclear factor‐kB; VILI, ventilator‐induced lung injury or ventilation; WT, wild type.
DISCUSSION
A cardinal pathophysiologic feature of all inflammatory disorders is the disruption of the continuous, semipermeable EC barrier in intact blood vessels, resulting in increased vascular permeability and leakage of luminal contents into the interstitium of vital organs, including the lung. In addition to ARDS, the MLCK‐regulated cytoskeleton driven by specific and distinct MLCK isoforms is an essential participant in multiple lung inflammatory diseases, including asthma. 36 , 37 , 38 For example, we have shown that nmMylk −/− mice exhibit significant reductions in ovalbumin‐mediated allergic lung and airway inflammation. 36 Conversely, mice overexpressing nmMLCK only in the endothelium (nmMylk ec/+) exhibit elevated susceptibility and severity in asthmatic inflammation. 36 Extrapulmonary inflammatory disorders involving the MCLK pathway and MLC phosphorylation include heart failure, 39 inflammatory bowel disease, 40 severe burn injury, 41 irritable bowel syndrome, 42 radiation injury, 43 diabetes, 44 and cancer. 45 In addition to direct endothelial and intestinal epithelial barrier regulation, 40 nmMLCK influences the expression of key genes involved in the severity of cancer and asthma with increased mortality. 38 , 46
Mortality in the critically ill is directly attributable to unchecked inflammation‐induced vascular permeability and development of multiorgan failure, 4 events governed by the dynamic contractile function of the actin‐based EC cytoskeleton 15 with nmMLCK a critical actin‐binding protein involved in both tensile force generation and linkage of the actin cytoskeleton to adhesive membrane components. nmMLCK is intimately involved in key aspects of the inflammatory response including PMN transmigration with LPS‐induced vascular leakage and alveolar flooding in vivo 7 , 12 , 13 , 15 accompanied by increases in MLC phosphorylation 16 , 17 and a reorganization of the actomyosin cytoskeleton. 6 , 15 , 47 Studies utilizing various nmMLCK inhibitory approaches or genetically engineered mice nmMLCK KO mice (nmMylk −/−) with targeted global deletion of the nmMLCK isoform confirmed targeting of nmMLCK as a viable approach to reduce lung inflammation and alveolar and vascular permeability. 9 , 29 While confirming nmMLCK as a contributor to ARDS/VILI severity, 22 , 27 studies demonstrating nmMylk −/− mice with global nmMLCK knockout (KO) are protected in murine inflammatory lung injury models (LPS, VILI, and lung irradiation) 9 , 43 failed to identify the primary cellular inflammatory effector most adversely affected by the loss of nmMLCK expression. Despite multiple biologically plausible cellular candidates such as the alveolar epithelium or inflammatory and immune effector cells including PMNs and T‐cell lymphocytes, we speculated EC expression of nmMLCK to be a major if not primary contributor to the development of inflammatory lung permeability and subsequent to the magnitude of lung injury severity. Our earlier studies utilizing transgenic nmMylk ec/+ mice overexpressing nmMLCK2 in the vascular endothelium supported this hypothesis with these mice exhibiting a strong inflammatory phenotype that exacerbated inflammatory lung injury. To extend these studies, we created nmMylk −/−/ec‐tg+ mice selectively expressing EC nmMLCK on the nmMylk −/− background to fully establish the critical importance of EC nmMLCK in driving the severity of acute inflammatory lung injury.
Our results indicate that compared to WT C57BL/6J mice, nmMylk −/− mice exposed to LPS/VILI‐induced murine lung injury model showed marked attenuation of inflammatory lung permeability and injury as we previously reported. 9 In contrast, a similar challenge of nmMylk −/−/ec‐tg+ mice, selectively expressing EC nmMLCK on the nmMylk −/− background, showed near‐complete loss of the significant protection observed in LPS/VILI‐challenged nmMylk −/− mice, with significantly increased histologic lung inflammation, increased BAL PMNs, and increased loss of vascular barrier integrity reflected by EBD leakage, BAL protein, and tissue albumin measurements. These studies strongly endorse a primary contributory role of EC nmMLCK in driving the severity of acute inflammatory lung injury and loss of EC barrier integrity.
In addition to the obvious contribution of EC‐expressed nmMLCK protein to increases in EC paracellular gap formation and permeability elicited by inflammatory stimuli such as mechanical stress, 9 thrombin, 7 and inflammatory cytokines such as TNF 12 and eNAMPT, 48 we strongly speculate the nmMLCK protein to exert regulatory proinflammatory gene and protein expression in EC, which potentially serve to enhance the severity of the lung inflammatory response. This premise is supported by our prior report demonstrating that compared with wild‐type mice, nmMLCK KO mice exhibited significant reductions in VILI‐induced gene expression in biological pathways such as NF‐kB signaling pathway, NRF2‐mediated oxidative stress, coagulation, p53 signaling, leukocyte extravasation, and IL‐6 signaling. The finding that NF‐kB genes were significantly affected by deletion of the nmMLCK allele supports our previous finding that nmMLCK activation is directly involved in NF‐kB activation and NF‐kB‐dependent increases in the transcription of inflammatory and barrier‐regulatory genes, including the TNF signaling pathway. 49 In the current study, we again noted dramatic LPS/VILI‐induced increases in NF‐kB phosphorylation, which were abrogated in nmMylk −/− mice. In addition, LPS/VILI‐induced elevations in the plasma levels of IL‐6, IL‐8/KC, TNF‐α, and eNAMPT, observed in WT mice, were all significantly reduced in nmMylk −/− mice, whereas IL‐1β was unaffected, findings consistent with our earlier reports of LPS/VILI‐induced gene expression in nmMylk −/− mice. 9 Consistent with the capacity for nmMLCK expression in EC to restore the fully integrated inflammatory response and histologic evidence of inflammatory lung injury, LPS/VILI‐exposed nmMylk −/− mice with global nmMLCK deletion but restricted EC nmMLCK expression demonstrated significant elevations in plasma levels of IL‐6 and TNF‐α approaching levels in LPS/VILI‐exposed WT mice, whereas levels of IL‐1β IL‐8/KC and eNAMPT were unaffected.
Levels of ROS are substantially elevated in every preclinical model of sepsis, trauma, and mechanical stress/VILI, are strongly linked to TLR4/NF‐kB signaling, 31 , 50 and are reduced in nmMylk −/− mice exposed to LPS, VILI, or hyperoxia. 9 , 51 We have previously shown a very strong and inverse relationship between the expression of the antioxidant transcription factor, NRF2, and Mylk transcription via a highly novel, negative ARE‐dependent effect of NRF2 on Mylk promoter activity. 52 Reductions in NRF2 expression or the NRF2 inhibitor, brusatol, significantly increase nmMLCK protein levels, actin stress fiber formation, and augment loss of EC barrier integrity, 52 whereas pharmacologic NRF2 activators reduced nmMLCK protein levels. In the current in vivo studies, we noted prominent LPS/VILI‐induced reductions in NRF2 protein expression in concert with increases in the expression of the ROS‐generating protein NOX‐4. Biochemical studies of lung homogenates failed to identify significant alterations in these responses in either nmMylk −/− mice or nmMylk −/−/ec‐tg+ with global nmMLCK deletion, but restricted EC nmMLCK expression.
We have also previously identified a 45 nmMLCK‐influenced gene signature that successfully predicted human asthma severity and exacerbation status. 46 Pathway analysis of these nmMLCK‐influenced genes highlight the epidermal growth factor receptor (ErbB) signaling pathway known to involve in the participation of the JNK and ERK MAP kinase signaling pathways with several MAP kinase family genes such as Map3k7, (encoding mitogen‐activated protein kinase kinase kinase 7) and Map3k12 (mitogen‐activated protein kinase kinase kinase 12) among nmMLCK‐influenced differentially expressed genes. 9 , 46 We and others have shown the involvement of the activated MAP kinase signaling pathway in LPS‐ and VILI‐induced increases in actin stress fiber formation and paracellular gap formation in vitro and in vivo. 31 , 53 Anti‐inflammatory NF‐kB inhibitory strategies 31 , 53 attenuate both LPS/VILI‐induced robust MAP kinase family activation as well as inflammatory lung injury. We recently focused on a key MAP kinase family effector, the Ser/Thr RSK or p90rsk that is activated by ERK MAPK and signals to the contractile machinery via phosphorylation of the MYPT1. 54 p90rsk is prominently phosphorylated in LPS/VILI‐exposed mice, rats, and pigs. 50 In human lung EC, p90rsk is involved in eNAMPT‐ and LPS‐induced increases, MLC phosphorylation, EC contraction, and permeability with p90rsk inhibition reducing eNAMPT‐ and LPS‐induced TER. 18 In the current study, comparisons of p42/44 ERK activation in lung homogenates from LPS/VILI‐challenged WT and MYLK‐engineered murine strains showed mice with global nmMLCK KO with marked blunting of ERK MAPK and p‐pS90rsk phosphorylation/activation, two key determinants of EC permeability, consistent with the observed decreases in the level of p90rsk activation evoked by TLR4/NFkB inhibition. In contrast to the reduced p90rsk activation and EC permeability in global nmMLCK KO, these responses were preserved in nmMylk −/−/ec‐tg+ mice and comparable to LPS/VILI‐challenged WT mice.
The current study does exhibit several limitations. In generating nmMylk −/−/ec‐tg+ mice, we inserted the expression of the nmMLCK2 splice variant in an EC‐specific manner similarly as we had performed in generating the nmMylk ec/+ transgenic mouse line. 20 While this mouse line has proven useful, important differences between nmMLCK1 and nmMLCK2 constitute a limitation of the current study. nmMLCK2 is structurally identical to WT nmMLCK1, except for the absence of amino acids 437–506 encoded by the spliced out exon 11 and lacks the immunoglobulin‐like cell‐adhesion molecule domain, IgCAM3. 18 , 19 , 55 , 56 Two key sites of tyrosine phosphorylation Y464 and Y471 present in the nmMLCK1 IgCAM3 domain 6 , 19 , 57 are absent in the nmMLCK2 protein. Compared with nmMLCK1, nmMLCK2 is selectively induced by TNF‐α and mechanical stress (18% cyclic stretch), 19 and exhibits reduced peripheral translocation during EC barrier recovery. 19 nmMLCK2 silencing dramatically reduces the magnitude of thrombin‐induced barrier dysfunction and promotes more rapid EC barrier recovery. 58 Thus, insertion of the proinflammatory nmMLCK2 splice variant and not nmMLCK1 on a global nmMLCK KO background does not allow for a true evaluation of the exact role of nmMLCK1 in dynamic, spatially localized EC barrier‐regulatory responses to injurious inflammatory stimuli. A final limitation is that we have shown that nmMylk is differentially regulated by DNA methylation in Black subjects with sepsis‐induced ARDS; 27 , 59 however, nmMylk methylation in nmMylk −/− KO mice expressing EC nmMLCK was not assessed in the current study.
Despite these limitations, the current study highlights the use of genetically engineered mice to define the critical role of the nmMLCK expressed in vascular endothelium in regulating the severity of inflammatory lung injury in preclinical murine models of ARDS. Utilizing genetically engineered mice, nmMylk −/−/ec‐tg+, which selectively express EC nmMLCK on the nmMylk −/− background, have successfully confirmed EC nmMLCK as a potentially attractive molecular target in ARDS/VILI and have provided direct support for EC nmMLCK driving the severity of acute inflammatory lung injury and loss of the vascular endothelial barrier. Precise targeting of EC nmMLCK represents a highly attractive therapeutic strategy to reduce lung inflammation and both lung and systemic vascular permeability.
AUTHOR CONTRIBUTIONS
Conception and design of the work and the analysis and interpretation of data for the work: Joe G. N. Garcia, Carrie L. Kempf, Saad Sammani, Sara M. Camp, and Jin H. Song. Drafting and revision of the manuscript: Joe G. N. Garcia. Approval of the final version to be published: Joe G. N. Garcia, Carrie L. Kempf, Saad Sammani, Sara M. Camp, Jin H. Song, Carrie L. Kempf, Saad Sammani, Tadeo Bermudez, Vivian Reyes Hernon, Matthew K. Hufford, Jessica Burt, and Jin H. Song. Critical revision of content: Carrie L. Kempf, Saad Sammani, Sara M. Camp, and Jin H. Song. Collection and analysis of data: Carrie L. Kempf, Saad Sammani, Tadeo Bermudez, Vivian Reyes Hernon, Matthew K. Hufford, Jessica Burt, and Jin H. Song. Revision of the manuscript: Carrie L. Kempf, Saad Sammani, Tadeo Bermudez, Vivian Reyes Hernon, Matthew K. Hufford, Jessica Burt, and Jin H. Song.
CONFLICTS OF INTEREST
Joe G. N. Garcia is CEO and founder of Aqualung Therapeutics Corporation. The remaining authors declare no conflicts of interest.
ETHICS STATEMENT
The ethics statement is not available.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This study was supported by the NIH/NHLBI grant P01 HL126609 (to J. G. N. G.).
Kempf CL, Sammani S, Bermudez T, Song JH, Hernon VR, Hufford MK, Burt J, Camp SM, Dudek SM, Garcia JGN. Critical role for the lung endothelial nonmuscle myosin light‐chain kinase isoform in the severity of inflammatory murine lung injury. Pulmonary Circulation. 2022;12:e12061. 10.1002/pul2.12061
DATA AVAILABILITY STATEMENT
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–93. 10.1056/NEJMoa050333 [DOI] [PubMed] [Google Scholar]
- 2. Sedhai YR, Yuan M, Ketcham SW, Co I, Claar DD, McSparron JI, Prescott HC, Sjoding MW. Validating measures of disease severity in acute respiratory distress syndrome. Ann Am Thorac Soc. 2021;18:1211–18. 10.1513/AnnalsATS.202007-772OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zambon M, Vincent JL. Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest. 2008;133:1120–27. 10.1378/chest.07-2134 [DOI] [PubMed] [Google Scholar]
- 4. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–49. 10.1056/NEJM200005043421806 [DOI] [PubMed] [Google Scholar]
- 5. Garcia JG, Lazar V, Gilbert‐McClain LI, Gallagher PJ, Verin AD. Myosin light chain kinase in endothelium: molecular cloning and regulation. Am J Respir Cell Mol Biol. 1997;16:489–94. [DOI] [PubMed] [Google Scholar]
- 6. Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JGN. Pulmonary endothelial cell barrier enhancement by sphingosine 1‐phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279:24692–700. [DOI] [PubMed] [Google Scholar]
- 7. Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163:510–22. 10.1002/jcp.1041630311 [DOI] [PubMed] [Google Scholar]
- 8. Garcia JG, Verin AD, Schaphorst K, Siddiqui R, Patterson CE, Csortos C, Natarajan V. Regulation of endothelial cell myosin light chain kinase by Rho, cortactin, and p60(src). Am J Physiol. 1999;276:L989–98. [DOI] [PubMed] [Google Scholar]
- 9. Mirzapoiazova T, Moitra J, Moreno‐Vinasco L, Sammani S, Turner JR, Chiang ET, Evenoski C, Wang T, Singleton PA, Huang Y, Lussier YA, Watterson DM, Dudek SM, Garcia JGN. Non‐muscle myosin light chain kinase isoform is a viable molecular target in acute inflammatory lung injury. Am J Respir Cell Mol Biol. 2011;44:40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Linz‐McGillem LA, Moitra J, Garcia JG. Cytoskeletal rearrangement and caspase activation in sphingosine 1‐phosphate‐induced lung capillary tube formation. Stem Cells Dev. 2004;13:496–508. 10.1089/scd.2004.13.496 [DOI] [PubMed] [Google Scholar]
- 11. Wadgaonkar R, Dudek SM, Zaiman AL, Linz‐McGillem L, Verin AD, Nurmukhambetova S, Romer LH, Garcia JGN. Intracellular interaction of myosin light chain kinase with macrophage migration inhibition factor (MIF) in endothelium. J Cell Biochem. 2005;95:849–58. 10.1002/jcb.20472 [DOI] [PubMed] [Google Scholar]
- 12. Petrache I, Birukov K, Zaiman AL, Crow MT, Deng H, Wadgaonkar R, Romer LH, Garcia JG. Caspase‐dependent cleavage of myosin light chain kinase (MLCK) is involved in TNF‐alpha‐mediated bovine pulmonary endothelial cell apoptosis. FASEB J. 2003;17:407–16. 10.1096/fj.02-0672com [DOI] [PubMed] [Google Scholar]
- 13. Garcia JGN, Verin AD, Herenyiova M, English D. Adherent neutrophils activate endothelial myosin light chain kinase: role in transendothelial migration. J Appl Physiol. 1998;84:1817–21. [DOI] [PubMed] [Google Scholar]
- 14. Wang T, Brown ME, Kelly GT, Camp SM, Mascarenhas JB, Sun X, Dudek SM, Garcia JGN. Myosin light chain kinase (MYLK) coding polymorphisms modulate human lung endothelial cell barrier responses via altered tyrosine phosphorylation, spatial localization, and lamellipodial protrusions. Pulm Circ. 2018;8(2):1–7. 10.1177/2045894018764171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. [DOI] [PubMed] [Google Scholar]
- 16. Bogatcheva NV, Verin AD, Wang P, Birukova AA, Birukov KG, Mirzopoyazova T, Adyshev DM, Chiang ET, Crow MT, Garcia JG. Phorbol esters increase MLC phosphorylation and actin remodeling in bovine lung endothelium without increased contraction. Am J Physiol. 2003;285:L415–26. 10.1152/ajplung.00364.2001 [DOI] [PubMed] [Google Scholar]
- 17. Shi S, Verin AD, Schaphorst KL, Gilbert‐McClain LI, Patterson CE, Irwin RP, Natarajan V, Garcia JG. Role of tyrosine phosphorylation in thrombin‐induced endothelial cell contraction and barrier function. Endothelium. 1998;6:153–71. 10.3109/10623329809072202 [DOI] [PubMed] [Google Scholar]
- 18. Clayburgh DR, Rosen S, Witkowski ED, Wang F, Blair S, Dudek S, Garcia JGN, Alverdy JC, Turner JR. A differentiation‐dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. J Biol Chem. 2004;279:55506–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown M, Adyshev D, Bindokas V, Moitra J, Garcia JGN, Dudek SM. Quantitative distribution and colocalization of non‐muscle myosin light chain kinase isoforms and cortactin in human lung endothelium. Microvasc Res. 2010;80:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moitra J, Evenoski C, Sammani S, Wadgaonkar R, Turner JR, Ma SF, Garcia JGN. A transgenic mouse with vascular endothelial over‐expression of the non‐muscle myosin light chain kinase‐2 isoform is susceptible to inflammatory lung injury: role of sexual dimorphism and age. Transl Res. 2008;151:141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao L, Grant AV, Rafaels N, Stockton‐Porter M, Watkins T, Gao P, Chi P, Munoz M, Watson H, Dunston G, Togias A, Hansel N, Sevransky J, Maloney JP, Moss M, Shanholtz C, Brower R, Garcia JGN, Grigoryev DN, Cheadle C, Beaty TH, Mathias RA, Barnes KC. Polymorphisms in the myosin light chain kinase gene that confer risk of severe sepsis are associated with a lower risk of asthma. J Allergy Clin Immunol. 2007;119:1111–8. [DOI] [PubMed] [Google Scholar]
- 22. Gao L, Grant A, Halder I, Brower R, Sevransky J, Maloney JP, Moss M, Shanholtz C, Yates CR, Meduri GU, Shriver MD, Ingersoll R, Scott AF, Beaty TH, Moitra J, Ma SF, Ye SQ, Barnes KC, Garcia JGN. Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell Mol Biol. 2006;34:487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Christie JD, Ma SF, Aplenc R, Li M, Lanken PN, Shah CV, Fuchs B, Albelda SM, Flores C, Garcia JGN. Variation in the myosin light chain kinase gene is associated with development of acute lung injury after major trauma. Crit Care Med. 2008;36:2794–800. [DOI] [PubMed] [Google Scholar]
- 24. Flores C, Ma SF, Maresso K, Ober C, Garcia JGN. A variant of the myosin light chain kinase gene is associated with severe asthma in African Americans. Genet Epidemiol. 2007;31:296–305. [DOI] [PubMed] [Google Scholar]
- 25. Acosta‐Herrera M. Fine mapping of the myosin light chain kinase (MYLK) gene replicates the association with asthma in populations of Spanish descent. J Allergy Clin Immunol. 2015;136:1116–8, 19. 10.1016/j.jaci.2015.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rizzo AN, Sammani S, Esquinca AE, Jacobson JR, Garcia JGN, Letsiou E, Dudek SM. Imatinib attenuates inflammation and vascular leak in a clinically relevant two‐hit model of acute lung injury. Am J Physiol. 2015;309:L1294–304. 10.1152/ajplung.00031.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Szilagyi KL, Liu C, Zhang X, Wang T, Fortman JD, Zhang W, Garcia JGN. Epigenetic contribution of the myosin light chain kinase gene to the risk for acute respiratory distress syndrome. Transl Res. 2017;180:12–21. 10.1016/j.trsl.2016.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goldman JL, Sammani S, Kempf C, Saadat L, Letsiou E, Wang T, Moreno‐Vinasco L, Rizzo AN, Fortman JD, Garcia JG. Pleiotropic effects of interleukin‐6 in a "two‐hit" murine model of acute respiratory distress syndrome. Pulm Circ. 2014;4:280–8. 10.1086/675991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rossi JL, Velentza AV, Steinhorn DM, Watterson DM, Wainwright MS. MLCK210 gene knockout or kinase inhibition preserves lung function following endotoxin‐induced lung injury in mice. Am J Physiol. 2007;292:L1327–34. 10.1152/ajplung.00380.2006 [DOI] [PubMed] [Google Scholar]
- 30. Letsiou E, Rizzo AN, Sammani S, Naureckas P, Jacobson JR, Garcia JGN, Dudek SM. Differential and opposing effects of imatinib on LPS‐ and ventilator‐induced lung injury. Am J Physiol. 2015;308:L259–69. 10.1152/ajplung.00323.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quijada H, Bermudez T, Kempf CL, Valera DG, Garcia AN, Camp SM, Song JH, Franco E, Burt JK, Sun B, Mascarenhas JB, Burns K, Gaber A, Oita RC, Reyes Hernon V, Barber C, Moreno‐Vinasco L, Sun X, Cress AE, Martin D, Liu Z, Desai AA, Natarajan V, Jacobson JR, Dudek SM, Bime C, Sammani S, Garcia JGN. Endothelial eNAMPT amplifies preclinical acute lung injury: efficacy of an eNAMPT‐neutralising mAb. Eur Respir J. 2021;57:2002536. 10.1183/13993003.02536-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1‐phosphate in murine endotoxin‐induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–51. 10.1164/rccm.200309-1258OC [DOI] [PubMed] [Google Scholar]
- 33. Jensen EC. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat Rec. 2013;296:378–81. 10.1002/ar.22641 [DOI] [PubMed] [Google Scholar]
- 34. Garcia AN, Casanova NG, Valera DG, Sun X, Song JH, Kempf CL, Moreno‐Vinasco L, Burns K, Bermudez T, Valdez M, Cuellar G, Gregory T, Oita RC, Hernon VR, Barber C, Camp SM, Martin D, Liu Z, Bime C, Sammani S, Cress AE, Garcia JG. Involvement of eNAMPT/TLR4 signaling in murine radiation pneumonitis: protection by eNAMPT neutralization. Transl Res. 2021;239:44–57. 10.1016/j.trsl.2021.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Borbiev T, Birukova A, Liu F, Nurmukhambetova S, Gerthoffer WT, Garcia JG, Verin AD. p38 MAP kinase‐dependent regulation of endothelial cell permeability. Am J Physiol. 2004;287:L911–8. 10.1152/ajplung.00372.2003 [DOI] [PubMed] [Google Scholar]
- 36. Wang T, Moreno‐Vinasco L, Ma SF, Zhou T, Shimizu Y, Sammani S, Epshtein Y, Watterson DM, Dudek SM, Garcia JGN. Nonmuscle myosin light chain kinase regulates murine asthmatic inflammation. Am J Respir Cell Mol Biol. 2014;50:1129–35. 10.1165/rcmb.2013-0434OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang T, Zhou T, Saadat L, Garcia JG. A MYLK variant regulates asthmatic inflammation via alterations in mRNA secondary structure. Eur J Hum Genet. 2015;23:874–6. 10.1038/ejhg.2014.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou T, Wang T, Garcia JG. Genes influenced by the non‐muscle isoform of myosin light chain kinase impact human cancer prognosis. PLoS One. 2014;9:e94325. 10.1371/journal.pone.0094325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Markandran K, Yu H, Song W, Lam D, Madathummal MC, Ferenczi MA. Functional and molecular characterisation of heart failure progression in mice and the role of myosin regulatory light chains in the recovery of cardiac muscle function. Int J Mol Sci. 2021;23:23. 10.3390/ijms23010088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF‐{alpha} modulation of Caco‐2 intestinal epithelial tight junction barrier: role of myosin light‐chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288:G422–30. 10.1152/ajpgi.00412.2004 [DOI] [PubMed] [Google Scholar]
- 41. Chen C, Wang P, Su Q, Wang S, Wang F, Weber CR. Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS One. 2012;7:e34946. 10.1371/journal.pone.0034946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xie Y, Zhan X, Tu J, Xu K, Sun X, Liu C, Ke C, Cao G, Zhou Z, Liu Y. Atractylodes oil alleviates diarrhea‐predominant irritable bowel syndrome by regulating intestinal inflammation and intestinal barrier via SCF/c‐kit and MLCK/MLC2 pathways. J Ethnopharmacol. 2021;272:113925. 10.1016/j.jep.2021.113925 [DOI] [PubMed] [Google Scholar]
- 43. Wang T, Mathew B, Wu X, Shimizu Y, Rizzo AN, Dudek SM, Weichselbaum RR, Jacobson JR, Hecker L, Garcia JG. Nonmuscle myosin light chain kinase activity modulates radiation‐induced lung injury. Pulm Circ. 2016;6:234–9. 10.1086/686491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hu W, Feng P. Myosin light chain kinase is involved in the mechanism of gastrointestinal dysfunction in diabetic rats. Dig Dis Sci. 2012;57:1197–202. 10.1007/s10620-012-2041-7 [DOI] [PubMed] [Google Scholar]
- 45. Cao F, Zhu L, Zhang J, Pongkorpsakol P, Kuo WT, Turner JR, Zhou Q, Wang Y, Chen F, Liu Y, Zuo L. Myosin light chain kinase is a potential target for hypopharyngeal cancer treatment. Biomed Pharmacother. 2020;131:110665. 10.1016/j.biopha.2020.110665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou T, Wang T, Garcia JG. A nonmuscle myosin light chain kinase‐dependent gene signature in peripheral blood mononuclear cells is linked to human asthma severity and exacerbation status. Pulm Circ. 2015;5:335–8. 10.1086/680357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garcia JG, Schaphorst KL. Regulation of endothelial cell gap formation and paracellular permeability. J Investig Med. 1995;43:117–26. [PubMed] [Google Scholar]
- 48. Ye SQ, Zhang LQ, Adyshev D, Usatyuk PV, Garcia AN, Lavoie TL, Verin AD, Natarajan V, Garcia JG. Pre‐B‐cell‐colony‐enhancing factor is critically involved in thrombin‐induced lung endothelial cell barrier dysregulation. Microvasc Res. 2005;70:142–51. 10.1016/j.mvr.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 49. Wadgaonkar R, Linz‐McGillem L, Zaiman AL, Garcia JG. Endothelial cell myosin light chain kinase (MLCK) regulates TNFalpha‐induced NFkappaB activity. J Cell Biochem. 2005;94:351–64. 10.1002/jcb.20250 [DOI] [PubMed] [Google Scholar]
- 50. Bermudez T, Sammani S, Song JH, Hernon VR, Kempf CL, Garcia AN, Burt J, Hufford M, Camp SM, Cress AE, Desai AA, Natarajan V, Jacobson JR, Dudek SM, Cancio LC, Alvarez J, Rafikov R, Li Y, Zhang DD, Casanova NG, Bime C, Garcia J. eNAMPT neutralization reduces preclinical ARDS severity via rectified NFkB and Akt/mTORC2 signaling. Sci Rep. 2022;12:696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Usatyuk PV, Singleton PA, Pendyala S, Kalari SK, He D, Gorshkova IA, Camp SM, Moitra J, Dudek SM, Garcia JGN, Natarajan V. Novel role for non‐muscle MLCK in hyperoxia‐induced recruitment of cytoskeletal proteins, NADPH oxidase activation and reactive oxygen species generation in lung endothelium. J Biol Chem. 2012;287:9360–75. 10.1074/jbc.M111.294546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu P, Rojo de la Vega M, Sammani S, Mascarenhas JB, Kerins M, Dodson M, Sun X, Wang T, Ooi A, Garcia J, Zhang DD. RPA1 binding to NRF2 switches ARE‐dependent transcriptional activation to ARE‐NRE‐dependent repression. Proc Natl Acad Sci USA. 2018;115:E10352–61. 10.1073/pnas.1812125115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Camp SM, Ceco E, Evenoski CL, Danilov SM, Zhou T, Chiang ET, Moreno‐Vinasco L, Mapes B, Zhao J, Gursoy G, Brown ME, Adyshev DM, Siddiqui SS, Quijada H, Sammani S, Letsiou E, Saadat L, Yousef M, Wang T, Liang J, Garcia JG. Unique Toll‐like receptor 4 activation by NAMPT/PBEF induces NFkappaB signalling and inflammatory lung injury. Sci Rep. 2015;5:13135. 10.1038/srep13135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Samson SC, Elliott A, Mueller BD, Kim Y, Carney KR, Bergman JP, Blenis J, Mendoza MC. p90 ribosomal S6 kinase (RSK) phosphorylates myosin phosphatase and thereby controls edge dynamics during cell migration. J Biol Chem. 2019;294:10846–62. 10.1074/jbc.RA119.007431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lazar V, Garcia JG. A single human myosin light chain kinase gene (MLCK; MYLK) transcribes multiple nonmuscle isoforms. Genomics. 1999;57:256–67. [DOI] [PubMed] [Google Scholar]
- 56. Verin AD, Lazar V, Torry RJ, Labarrere CA, Patterson CE, Garcia JGN. Expression of a novel high molecular‐weight myosin light chain kinase in endothelium. Am J Respir Cell Mol Biol. 1998;19:758–66. [DOI] [PubMed] [Google Scholar]
- 57. Dudek SM, Chiang ET, Camp SM, Guo Y, Zhao J, Brown ME, Singleton PA, Wang L, Desai A, Arce FT, Lal R, Van Eyk JE, Imam SZ, Garcia JGN, Nusrat A. Abl tyrosine kinase phosphorylates nonmuscle myosin light chain kinase to regulate endothelial barrier function. Mol Biol Cell. 2010;21:4042–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mascarenhas JB, Tchourbanov AY, Fan H, Danilov SM, Wang T, Garcia JG. Mechanical stress and single nucleotide variants regulate alternative splicing of the MYLK gene. Am J Respir Cell Mol Biol. 2017;56:29–37. 10.1165/rcmb.2016-0053OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun X, Sun BL, Sammani S, Bermudez T, Dudek SM, Camp SM, Garcia JGN. Genetic and epigenetic regulation of the non‐muscle myosin light chain kinase isoform by lung inflammatory factors and mechanical stress. Clin Sci. 2021;135:963–77. 10.1042/CS20201448 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.