

Abstract
Clostridioides difficile (C. difficile) is the leading cause of healthcare-associated infection in the U.S. and considered an urgent threat by the Centers for Disease Control and Prevention (CDC). Only two antibiotics, vancomycin and fidaxomicin, are FDA-approved for the treatment of C. difficile infection (CDI) but these therapies still suffer from high treatment failure and recurrence. Therefore, new chemical entities to treat CDI are needed. Trifluoromethylthio containing N-(1,3,4-oxadiazol-2-yl)benzamides displayed very potent activities (sub-μg/mL minimum inhibitory concentration (MIC) values) against Gram-positive bacteria. Here, we report remarkable antibacterial activity enhancement via halogen substitutions, which afforded new anti-C. difficile agents with ultrapotent activities (MICs as low as 0.003 μg/mL (0.007 μM)) that surpassed the activity of vancomycin against C. difficile clinical isolates. The most promising compound in the series, HSGN-218, was non-toxic to mammalian colon cells and is gut restrictive. In addition, HSGN-218 protected mice from CDI recurrence. Not only does this work provide a potential clinical lead for the development of C. difficile therapeutics but also it highlights dramatic drug potency enhancement via halogen substitution.
Introduction:
Clostridioides difficile (C. difficile) is a spore-forming Gram-positive anaerobic bacterium and the leading cause of nosocomial infections as well as antibiotic-associated diarrhea in the United States1. In 2017, the Centers for Disease Control and Prevention (CDC) determined that in the U.S., 223,900 patients were hospitalized with C. difficile infection (CDI), resulting in 12,800 deaths and more than $1 billion in healthcare costs2. CDI causes severe diarrhea along with life-threatening complications such as toxic megacolon, pseudomembranous colitis, and systemic inflammatory response syndrome3. Manifestations of the disease are credited to the toxin-mediated damage produced by two major toxins: toxin A (TcdA/enterotoxin) and toxin B (TcdB/cytotoxin), which catalyze the inactivation of Rho GTPases, ultimately causing intense inflammation of the gut, accompanied by necrosis and apoptosis of colonic mucosal cells4–5. Furthermore, C. difficile’s ability to produce spores hinders the clinical management of CDI because these spores are very resistant to environmental conditions, antibiotics, and disinfection processes. C. difficile spores can spread throughout the environment and once ingested by vulnerable hosts, they develop into vegetative cells that colonize the intestines, thereby producing toxins and establishing infection6–7. Therefore, C. difficile spores serve as the major cause of CDI circulation and recurrence.
CDI is typically caused from the use of antibiotics, which disrupts the reproduction of normal and protective gut microbiota, ultimately allowing C. difficile to grow in the colon and produce infectious toxins8. Although the overuse of antibiotics is one of the main reasons contributing to CDI, the management of CDI requires antibiotic treatment. Currently, there are only three drugs used to treat CDI: metronidazole, vancomycin, and fidaxomicin. Yet only vancomycin and fidaxomicin are approved by the FDA for treatment of CDI. Although, metronidazole was previously recommended as a first-line therapy for CDI, its use is now only limited to non-severe CDI cases when patients are unable to be treated with vancomycin or fidaxomicin9. Moreover, other limitations with metronidazole treatment are its potent activity against a wide spectrum of protective normal microbiota, as well as its high absorption (100% bioavailable) from the intestinal tract, restricting its concentrations in the colon10–11. Although oral vancomycin is minimally absorbed into the systemic circulation12, it has broad spectrum activity against Gram-positive bacteria, leading to a reduction in microbiome diversity13. Furthermore, both vancomycin and metronidazole treatments are inadequate due to high treatment failure (14% with vancomycin and 22% with metronidazole) and high recurrence rates (25% to 30%). This is because both antibiotics are ineffective against spores and also they cause disruption of the beneficial gut microbiota14–15. Fidaxomicin is the only new drug approved for CDI in the last 30 years. Fidaxomicin has lower recurrence rates compared to vancomycin and metronidazole because of its selectivity towards C. difficile; however, its high cost limits its use16–18. Even though vancomycin and fidaxomicin are FDA-approved therapies for CDI, emerging resistance or reduced susceptibility are evident to these antibiotics17, 19. In addition, one emerging alternative non-antibiotic therapy for CDI is fecal microbiota transplant (FMT), which restores the disrupted normal microbiome, leading to renovation of the colonization resistance to C. difficile20. While FMT appeared to be successful in the treatment of some CDI cases, it has many restrictions and poses a serious risk of transmitting infectious pathogens to the patients; especially immunocompromised and elderly patients21–22. Therefore, due to the increase in treatment failure and recurrence rates with the commonly used anti-CDI drugs, along with growths of CDI, efforts to develop novel anti-CDI therapeutics have intensified23.
Our program focuses on the discovery of new N-(1,3,4-oxadiazol-2-yl)benzamides to combat the urgent threats of antibiotic-resistant bacteria24–25. We previously reported the trifluoromethylthio-containing (1,3,4-oxadiazol-2-yl)benzamide, compound 12, as a potent anti-MRSA agent26. Compound 12 was found to have bactericidal activity as well as being non-toxic to mammalian cells26. Compound 12 was however not evaluated in vivo as it was not deemed an ideal lead due to the presence of a potential thiophene toxicophore (Figure 1). In this report, we describe the generation of a new series of trifluoromethylthio containing (1,3,4-oxadiazol-2-yl)benzamides, which leads to the identification of N-(5-(3,5-dichlorophenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (HSGN-218), which does not contain a thiophene (Figure 1). HSGN-218 was tested for its activity against a panel of clinical pathogenic C. difficile strains. Cytotoxicity against mammalian cells, bi-directional Caco-2 permeability and activity against normal gut microbiota were also investigated. Moreover, the activity of HSGN-218 treatment was evaluated in an in vivo CDI mouse model and its ability to prevent C. difficile recurrence in vivo was also investigated.
Figure 1.

Compound 12 contains a potential thiophene toxicophore but was found to be potent against C. difficile. Utilization of halogen substitution led to the discovery of an ultrapotent anti-C. difficile agent (HSGN-218) with a 70-times imporvement in potency (from 0.5 μg/mL (1.4 μM) to 0.007 μg/mL (0.02 μM).
Results and Discussion:
Halogenation, a High-Level Medicinal Chemistry Design Strategy
Halogens (X = F, Cl, Br, and I) are commonly used substituents in medicinal chemistry and drug discovery27–30. For instance, around 40% of the drugs currently FDA-approved or in clinical trials are halogenated and about 25% of the published medicinal chemistry papers and patents contain the late stage addition of halogen atoms28. Likewise, 35% of the top-15 selling drugs from 2010 to 2016 are halogenated31 (Figure 2A). Of the halogenated drugs, 57% contain fluorine, 38% contain chlorine, 4% contain bromine, and only 1% contain iodine28. The addition of halogen substitutents has been shown to have a major effect on a drug’s potency and pharmacological properties. Regarding pharmacological properties, addition of halogen substituents to lead compounds has been shown to increase lipophilicity, permeability, membrane binding and metabolic stability32–33. Likewise, insertion of halogen atoms into lead-like compounds also showed enhanced drug metabolism because the carbon-halogen bond is not easily metabolized by cytochrome P45028. Concerning potency, halogen atom substitution’s effect has been documented. For example, L86–8276, a cyclin-dependent kinase 2 (CDK2) inhibitor was shown to have an IC50 value of 2.4 μM (Figure 2B)34. Yet, the addition of a chlorophenyl group to give Flavopiridol showed a six-fold improvement in potency to give an IC50 of 0.4 μM against CDK2 (Figure 2B)34.
Figure 2.

Importance of the addition of halogen substituents to lead compounds. A. Examples of the top-15 selling drugs that are halogenated. B. Addition of chlorophenyl to CDK2 inhibitors led to a six-fold enhancement in potency.
Synthesis and Anti-C. difficile Activity of Trifluoromethylthio Containing (1,3,4-oxadiazol-2-yl)Benzamides
We previously reported that compound 12 was potent against a panel of clinically important Gram-positive bacteria26. Based on its broad-spectrum Gram-positive activity, we wondered if it would be active against C. difficile. Compound 12 inhibited C. difficile ATCC BAA 1801 with an MIC of 0.5 μg/mL (1.4 μM) (see Figure 1 and Table 1), which is comparable to vancomycin. However, compound 12 contains an unsubstituted thiophene moiety, which can lead to toxicity concerns (Figure 1). For instance, thiophene metabolism, caused by cytochrome P450 mediated oxidation, can lead to the formation of reactive metabolites, thiophene-S oxides35–36, thiophene epoxides36, and sulphenic acids37, which have a high propensity to react with nucleophiles such as water and glutathione38. We were however encouraged that compound 12 showed good activity against C. difficile, so we proceeded to make new analogs, which did not contain thiophene but instead substituted phenyl groups.
Table 1.
MICs in μg/mL (μM) of HSGN-218, analogs, and control antibiotics, against C. difficle ATCC BAA 1801.
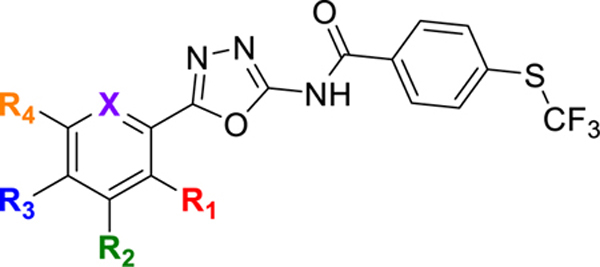
|
| Compound/Control Antibiotic | -R1 | -R2 | -R3 | -R4 | X | MICs in μg/mL (μM) |
|---|---|---|---|---|---|---|
| Compound 12 | - | - | - | - | - | 0.5 (1.4) |
| 1 | H | H | H | H | CH | 2 (5.5) |
| 2 | F | H | H | H | CH | 4 (10.4) |
| 3 | Cl | H | H | H | CH | 4 (10.0) |
| 4 | H | F | H | H | CH | 0.03 (0.08) |
| 5 | H | Cl | H | H | CH | 0.03 (0.08) |
| 6 | H | OMe | H | H | CH | 4 (10.1) |
| 7 | H | CF3 | H | H | CH | 0.015 (0.04) |
| 8 | H | H | F | H | CH | 4 (10.4) |
| 9 | H | H | Cl | H | CH | 2 (5.0) |
| 10 | H | H | CF3 | H | CH | 0.125 (0.29) |
| 11 | H | H | OMe | H | CH | 4 (10.1) |
| 12 | H | H | CH3 | H | CH | 4 (10.5) |
| 13 | H | H | i-Propyl | H | CH | 128 (314.2) |
| 14 | Cl | H | Cl | H | CH | 0.03 (0.07) |
| 15; HSGN-218 | H | Cl | H | Cl | CH | 0.007 (0.02) |
| 16 | OMe | H | H | Cl | CH | 2 (4.7) |
| 17 | H | H | H | H | N | 8 (21.8) |
| Vancomycin | - | - | - | - | - | 1 (0.7) |
| Metronidazole | - | - | - | - | - | 0.25 (1.46) |
| Fidaxomicin | - | - | - | - | - | 0.06 (0.06) |
In our previous report26, we determined that the 4-(trifluoromethylthio)phenyl group is vital for optimal activity so we kept this constant. The synthesis of the compounds began with a substituted benzaldehyde followed by the addition of semicarbazide and sodium acetate to give the corresponding semicarbazone. Then, using bromine and sodium acetate, the semicarbazone was cyclized into the subsequent 1,3,4-oxadizol-2-amine (Scheme 1). Amide coupling between the 1,3,4-oxadiazol-2-amine and 4-trifluoromethylthio benzoic acid using benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP) reagent gave the desired trifluoromethylthio containing (1,3,4-oxadiazol-2-yl)benzamides (Scheme 1).
Scheme 1: General Route for the Synthesis of trifluoromethylthio-containing N-(1,3,4-oxadiazol-2-yl)benzamidesa.

aReagents and Conditions: (a) Semicarbazide hydrochloride, NaOAc, MeOH:H2O (1:1), rt, 30 min, 95% (b) Bromine, NaOAc, AcOH, 60 °C, 1 h, 40% – 70% (c) BOP Reagent, DIPEA, DMF, rt, 12 h, 16% – 33%.
With the compounds in hand (see Table 1), we proceeded to evaluate them against C. difficile. Halogen substitutions (especially the Cl, F or CF3 groups) resulted in the most active compounds. Substitution with OMe, Me, and i-propy groups showed only moderate to no actvity (see Table 1 for MICs of compounds 6, 11, 12, and 13 against C. difficile ATCC BAA 1801). For halogen substituents, the position on the phenyl ring was also important. For example, the MIC for meta-Cl (5) was 0.03 μg/mL (0.08 μM), whereas that for the ortho- (3) and para- (9) analogs were 4 μg/mL (10.0 μM) and 2 μg/mL (5.0 μM) respectively against C. difficile ATCC BAA 1801 (Table 1). Additionally, for di-substituted halogen containing compounds, the position of the halogens affected actvity. For instance, the 3,5-dichlorophenyl analog (15, HSGN-218) was more than four times more potent than the 2,4-dichlorophenyl (14) analog (MIC = 0.06 μg/mL and 0.007 μg/mL for 14 and 15 respectively). We also proceeded to investigate subsitution of the phenyl group with heteroaromatics, such as pyridinyl (17) which had only moderate activity (Table 1) allowing us to conclude that the phenyl ring is needed for optimal activity.
Comprehensive antibacterial profile of HSGN-218 against various C. difficile clinical isolates
After the initial screening of HSGN-218, we assessed its antibacterial profile against a panel of C. difficile clinical isolates. As depicted in Table 2, HSGN-218 exhibited exceptional actvity against C. difficile clinical isolates with MICs ranging from 0.003 μg/mL (0.007 μM) to 0.03 μg/mL (0.07 μM). Vancomycin displayed MICs ranging from 0.25 μg/mL (0.2 μM) to 1 μg/mL (0.7 μM) against all the tested strains (Table 2). With regard to micromolar concentrations, HSGN-218 is between 2.5 to 100 times more potent than vancomycin in inhibiting clinically relevant C. difficile growth in vitro. Metronidazole inhibited the growth of the tested C. difficile strains at concentrations ranging from 0.125 μg/mL (0.7 μM) to 0.25 μg/mL (1.46 μM). Fidaxomicin displayed MIC values ranging from 0.015 μg/mL (0.01 μM) to 0.06 μg/mL (0.06 μM).
Table 2.
MICs in μg/mL (μM) of HSGN-218 and control antibiotics against various C. difficile clinical isolates.
| Compound/Control Antibiotic | C. difficile NR-13432 (isolate 6) | C. difficile NR-13435 (isolate 9) | C. difficile NR-32883 (P2) | C. difficile NR-32891 (P13) | C. difficile NR-32895 (P19) | C. difficile NR-32904 (P30) | C. difficile ATCC 43255 |
|---|---|---|---|---|---|---|---|
| HSGN-218 | 0.03 (0.07) | 0.003 (0.007) | 0.007 (0.02) | 0.007 (0.02) | 0.007 (0.02) | 0.007 (0.02) | 0.015 (0.04) |
| Vancomycin | 0.25 (0.2) | 1 (0.7) | 0.5 (0.4) | 0.5 (0.4) | 1 (0.7) | 1 (0.7) | 1 (0.7) |
| Metronidazole | 0.25 (1.46) | 0.125 (0.7) | 0.125 (0.7) | 0.125 (0.7) | 0.25 (1.46) | 0.25 (1.46) | 0.25 (1.46) |
| Fidaxomicin | 0.06 (0.06) | 0.06 (0.06) | 0.03 (0.03) | 0.015 (0.01) | 0.03 (0.03) | 0.015 (0.01) | 0.015 (0.01) |
Antibacterial profile of HSGN-218 against vancomycin-resistant enterococci and Gram-negative bacteria
Next, the antibacterial activity of HSGN-218 was assessed against vancomycin-resistant enterococci (VRE) and Escherichia coli that are highly common bacteria in the gut. The overgrowth of VRE and colonization of the gut are one of the major issues associated with the vancomycin and metronidazole treatment of CDI39–40. Thus, anticlostridial agents capable of inhibiting the growth of VRE are highly desirable. On the other hand, E. coli is the predominant aerobic bacteria colonizing in the gut which remains resident throughout the life of the host41. As depicted in Table 3, HSGN-218 exhibited potent actvity against VRE clinical isolates with MICs ranging from 0.06 μg/mL (0.14 μM) to 0.125 μg/mL (0.29 μM) outperforming vancomycin and metronidazole. When tested against E. Coli, HSGN-218 was found to be inactive against E. coli BW25113 (wild-type strain). Conversely, the compound showed moderate activity (MIC = 4 μg/mL (9.2 μM)) against E. coli JW55031 which is deficient in AcrAB-TolC efflux pump. Thus, the lack of activity against the wild-type E. coli could be attributed to that HSGN-218 may be a substrate for AcrAB-TolC efflux pump.
Table 3.
MICs in μg/mL (μM) of HSGN-218 and control antibiotics against vancomycin-resistant enterococci (VRE) and Escherichia coli isolates.
| Compound/Control Antibiotic | E. faecium ATCC 700221 | E. faecalis ATCC 51299 | E. coli JW55031 (TolC Mutant | E. coli BW25113 (wild-type strain) |
|---|---|---|---|---|
| HSGN-218 | 0.125 (0.29) | 0.06 (0.14) | 4 (9.2) | >16 (>36.8) |
| Vancomycin | 32 (22.1) | >64 (>44.2) | >64 (>44.2) | >64 (>44.2) |
| Metronidazole | >64 (>373.9) | >64 (>373.9) | NT1 | NT |
| Linezolid | 1 (3.0) | 1 (3.0) | 16 (47.4) | >64 (>189,7) |
| Gentamicin | NT | NT | 0.25 (0.52) | 0.25 (0.52) |
NT1, not tested
HSGN-218 is highly tolerable to human cell lines
Prokaryotic cell selectivity is a vital attribute for any antibiotic candidate. Thus, HSGN-218 was assessed for toxicity to mammalian cells. HSGN-218 showed an excellent safety profile against human colorectal cells (Caco-2) (Figure 3). It was highly tolerable to Caco-2 cells at concentrations higher than 64 μg/mL. This concentration is more than 9,000-times higher than the compound’s corresponding MIC value against C. difficile ATCC BAA 1801 used in the initial screening.
Figure 3.

In vitro cytotoxicity assessment of HSGN-218 (tested in triplicate) against human colorectal cells (Caco-2) using the MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay. Results are presented as percent viable cells relative to DMSO (negative control). Error bars represent standard deviation values. A one-way ANOVA, with post hoc Dunnet’s multiple comparisons test, determined no statistical difference between the values obtained for the compound and DMSO.
HSGN-218 demonstrates low Caco-2 permeability:
In order to treat CDI, it’s vital that a compound does not cross the gastrointestinal tract but instead stays localized in the gut. Thus, we assessed whether HSGN-218 would permeate across the gastrointestinal tract via a Caco-2 bidirectional permeability assay42. The assay (performed as a service at Eurofins Panlabs (MO, USA) demonstrated that HSGN-218 showed limited ability to permeate across Caco-2 bilayers (Papp = 0.2 × 10−6 cm s−1 from the apical to basolateral and Papp = 0.1 × 10−6 cm s−1 from the basolateral to apical, see Table 4). This permeability is comparable to rinitidine (Papp = 0.5 × 10−6 cm s−1 from the apical to basolateral and Papp = 1.3 × 10−6 cm s−1 from the basolateral to apical, see Table 4), a drug that is known to have low permeability across Caco-2 bilayers. Propranolol was used as a high permeability control as its Papp = 37.2 × 10−6 cm s−1 from the apical to basolateral and Papp = 22.7 × 10−6 cm s−1 from the basolateral to apical (Table 4). Therefore, the Caco-2 permeability results indicate that HSGN-218 will not cross the gastrointestinal tract and instead concentrate in the gut, the site for C. difficile infections.
Table 4.
Caco-2 Permeability Analysis for HSGN-218 and Control Drugs.
| Compound/Control Drug | Mean A → B Papp (cm s−1) | Mean B → A Papp (cm s−1) | Notes |
|---|---|---|---|
| HSGN-218 | 0.2 × 10−6 | 0.1 × 10−6 | Low Permeability |
| Ranitidine | 0.5 × 10−6 | 1.3 × 10−6 | Low Permeability Control |
| Propranolol | 37.2 × 10−6 | 22.7 × 10−6 | High Permeability Control |
In vitro antibacterial evaluation of HSGN-218 against normal microflora.
Antibiotics administration (especially broad-spectrum ones) causes alteration of the normal intestinal microbial composition, resulting in gut colonization by opportunistic pathogens like C. difficile43. Consequently, we investigated whether HSGN-218 has a deleterious effect on important representative members of the normal gut microbiota such as Lactobacillus spp and Bacteroides spp. Bacteroides spp comprise a large proportion of the intestinal microbiota, which were reported to contribute to bile acid-mediated inhibition of C. difficile and prevent CDI in mouse model44–45. Additionally, lactobacilli were reported to interfere with C. difficile both in vitro and in vivo46–47. As depicted in Table 5, HSGN-218 exhibited weak antibacterial activity against Lactobacillus strains (MIC = 16 μg/mL (36.8 μM)) and inhibited growth of species of Bacteroides (MIC=1–2 μg/mL (2.3–4.6 μM)). Similarly, vancomycin inhibited Lactobacillus strains (MICs = ≤1–2 μg/mL (0.7–1.4 μM)) and exhibited weak activity against Bacteroides spp (MICs = 32–64 μg/mL (22.1–44.2 μM)). Although HSGN-218 was similar to vancomycin, the anti-CDI drug of choice, in inhibiting the growth of certain species of the normal microbiota, it must be noted that HSGN-218 inhibits C. difficile at concentrations that are 100-times less than what is needed to inhibit Bacteroides (compare Table 2 with Table 5). On the other hand, vancomycin inhibited both C. difficile and Lactobacillus strains with comparable MIC values of 1–2 μg/mL. Metronidazole and fidaxomicin (to a lesser extent) also inhibit certain members of the normal intestinal microbiota48–50.
Table 5.
MICs in μg/mL (μM) of HSGN-218 and control antibiotics against human normal gut microbiota.
| Bacterial strains | HSGN-218 | Vancomycin | Metronidazole | Fidaxomicin |
|---|---|---|---|---|
| Lactobacillus gasseri HM-400 | 16 (36.8) | ≤1 (≤0.7) | >64 (>373.9) | >64 (>60.5) |
| Lactobacillus crispatus HM-103 | 16 (36.8) | 2 (1.4) | >64 (>373.9) | >64 (>60.5) |
| Lactobacillus crispatus HM-371 | 16 (36.8) | 2 (1.4) | >64 (>373.9) | >64 (>60.5) |
| Bacteroides fragilis HM-711 | 2 (4.6) | 64 (44.2) | ≤1 (5.84) | >64 (>60.5) |
| Bacteroides fragilis HM-709 | 1 (2.3) | 32 (22.1) | 2 (11.68) | >64 (>60.5) |
| Bacteroides dorei HM-719 | 2 (4.6) | 64 (44.2) | ≤1 (5.84) | >64 (>60.5) |
Frequency of mutation.
The promising results of HSGN-218 led us to investigate the likelihood of C. difficile to develop resistance to HSGN-218. No resistant mutants were isolated at a concentration of 15 × MIC and 20 × MIC in the presence of a high inoculum of C. difficile (Table 6), indicating that C. difficle is unlikely to form rapid resistance to HSGN-218. Likewise, vancomycin exhibited low frequency of mutation (<1.1× 10−9) and no resistant mutants were isolated, in agreement with a previous report51.
Table 6.
Frequency of mutation of HSGN-218 against C. difficile ATCC 43255
| Test agent | Frequency of mutation | |
|---|---|---|
| 15 × MIC | 20 × MIC | |
| HSGN-218 | <1.1× 10−9 | <1.1× 10−9 |
| Vancomycin | <1.1× 10−9 | <1.1× 10−9 |
In vivo efficacy of HSGN-218 in a CDI mouse model52
The potent antibacterial activities of HSGN-218 against C. difficile prompted us to investigate its efficacy in a CDI mouse model and its potential to protect mice from CDI recurrence, as described before. As shown in Figure 4, vancomycin (10 mg/kg) protected 100% of mice up to 5 days, as previously reported53–54. HSGN-218 (50 mg/kg), was able to significantly protect 66.7% of the mice against C. difficile during the 5-days treatment period.
Figure 4.

In vivo efficacy of HSGN-218 in a CDI mouse model. Kaplan–Meier survival curves were analyzed using a log-rank (Mantel–Cox) test. Asterisks (*) denote statistically significant difference between mice treated with either HSGN-218, or vancomycin in comparison with the vehicle-treated mice.
After testing the efficacy of HSGN-218 in the CDI mouse model, we sought to investigate this promising activity of HSGN-218 in preventing C. difficile recurrence. C. difficile recurrence is challenging to treat. In addition to the subsequent prolongation of C. difficile shedding and transmission, 1 out of every 5 patients experienced C. difficile recurrence episode died within 30 days of diagnosis55. Therefore, we sought to investigate this promising activity of HSGN-218 in preventing C. difficile recurrence. Mice were infected and treated for 5 days and then they were monitored for survival and possible C. difficile recurrence until the 21st day. Vancomycin-treated mice survived the first 5 days (similar to prior reports)53, but in accordance with previous studies52, 54, mice treated with vancomycin were susceptible to C. difficile recurrence and 83.3% of vancomycin-treated mice died after stopping vancomycin treatment. In contrast, HSGN-218 (50 mg/kg), significantly protected mice from CDI recurrence with 100% survival after 5- days treatment period (Figure 5).
Figure 5.

In vivo efficacy of HSGN-218 against CDI recurrence. Mice were treated with HSGN-218 (50 mg/kg), vancomycin (10 mg/kg) or the vehicle for 5 days and treatments were stopped thereafter. Kaplan–Meier survival curves were analyzed using a log-rank (Mantel–Cox) test. Asterisks (*) denote statistically significant difference between mice treated with either HSGN-218, or vancomycin in comparison with the vehicle-treated mice. Pound (#) denotes statistically significant difference between mice treated with compound HSGN-218 in comparison with vancomycin-treated mice
Conclusion:
In conclusion, we have identified HSGN-218 as a highly potent small molecule inhibitor of C. difficile growth. HSGN-218 is up to 100-times more active (MICs ranging from 0.003 μg/mL (0.007 μM) to 0.03 μg/mL (0.07 μM)) against C. difficile clinical isolates than vancomycin, the drug of choice for CDI. The compound is also non-toxic to mammalian cells as well as demonstrates low Caco-2 bidirectional permeability, indicating that HSGN-218 would have minimal systemic absorption. Even though HSGN-218 inhibited the growth of certain representative members of normal microbiota, excitingly, HSGN-218 protected mice from CDI as well as it showed significant efficacy against C. difficile recurrence. Therefore, compound HSGN-218 is considered as a lead compound to develop as anti- C. difficile therapeutic and deserves serious consideration.
Experimental Section:
Chemistry:
General Information: unless noted otherwise, all reagents and solvents were purchased from commercial sources and used as received. The 1H, 13C, and 19F NMR spectra were obtained in DMSO-d6 as solvent using a 500 MHz spectrometer with Me4Si as an internal standard. Chemical shifts are reported in parts per million (δ) and are calibrated using residual undeuterated solvent as an internal reference. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, or combinations thereof. High resolution mass spectra (HRMS) were obtained using electron spray ionization (ESI) technique and as TOF mass analyzer. Compounds were characterized by 1H NMR, 13C NMR, 19F NMR, and HRMS data. The purity of compounds was determined to be greater than 95% by measuring the absorbance at 260 nm with high performance liquid chromatography (HPLC) (See supprting information). HPLC spectra were recorded on an Agilent 1260 Infinity system using a ZORBAX RR Eclipse Plus C18 column. The mobile phase gradient went from 50% H2O : 50% MeOH over 5 minutes and then 40% H2O : 60% MeOH for 5 minutes, followed by 10% H2O : 90% MeOH for 2 minues and lastly 50% H2O : 50% MeOH for 3 minutes at a 1 mL/min flow rate.
Synthesis of 1,3,4-oxadiazol-2-amines [I.1 – I.17]:
The synthesis of I.1-I.17 was performed using a literature reported procedure56. Obtained 1H, 13C, and 19F spectra were in agreement with literature reported data.
Amide Coupling Procedure for the Synthesis of Compounds 1–17:
A 20 mL screw caped vial, charged with the corresponding acid (1 eq.), amine (1 eq.), BOP reagent (2.7 eq.) and diisopropylethylamine (1.5 mL) in DMF solvent (5 mL) was stirred at room temperature for 16 h. After completion, the reaction mixture was concentrated under reduced pressure, followed by flash column chromatography (hexanes:ethyl acetate 90:10 to 70:30) to give the desired product.
N-(5-Phenyl-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (1):
Off-white solid (46 mg, 28%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 8.0 – 7.9 (m, 2H), 7.9 (m, 2H), 7.6 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.6, 161.1, 158.6, 136.0, 135.8, 132.2, 131.1 (q, J = 308.7 Hz), 130.1, 129.8, 128.6, 126.6, 123.8. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C16H11F3N3O2S [M + H]+ 366.0524, found 366.0522. Purity by HPLC was found to be 96%.
N-(5-(2-Fluorophenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (2):
Off-white solid (38 mg, 22%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 8.0 (td, J = 7.6, 1.8 Hz, 1H), 7.9 (m, 2H), 7.7 (tdd, J = 7.4, 5.1, 1.8 Hz, 1H), 7.5 – 7.4 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.6, 160.6 (d, J = 257.0 Hz), 158.7, 157.7, 136.1, 135.7, 134.5 (d, J = 8.82 Hz), 131.1 (q, J = 308.7 Hz), 130.1, 129.7, 128.6, 125.8, 117.6 (d, J = 20.2 Hz), 112.2 (d, J = 11.3 Hz). 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F), −112.0 (d, J = 5.7 Hz, 1F). HRMS (ESI) m/z calcd for C16H10F4N3O2S [M + H]+ 384.0430, found 384.0429. Purity by HPLC was found to be 96%.
N-(5-(2-Chlorophenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (3):
Off-white solid (41 mg, 23%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 8.0 – 7.9 (m, 1H), 7.9 (m, 2H), 7.7 (m, 1H), 7.6 (td, J = 7.8, 1.6 Hz, 1H), 7.6 (t, J = 7.6 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 165.2, 159.2, 158.8, 136.1, 135.6, 133.5, 132.3, 131.6, 131.5, 131.1 (q, J = 308.7 Hz), 130.1, 128.7, 128.3, 123.0. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C16H10ClF3N3O2S [M + H]+ 400.0134, found 400.0135. Purity by HPLC was found to be 96%.
N-(5-(3-Fluorophenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (4):
Off-white solid (55 mg, 32%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 7.9 (m, 2H), 7.8 (m, 1H), 7.7 – 7.6 (m, 2H), 7.5 (td, J = 8.5, 2.7 Hz, 1H).13C NMR (126 MHz, DMSO-d6) δ 165.3, 163.7 (d, J = 245.7 Hz), 160.2, 158.7, 136.0, 135.5, 132.4 (d, J = 8.82 Hz), 131.1 (q, J = 308.7 Hz), 130.2, 128.7, 125.9 (d, J = 8.82 Hz), 122.8, 119.3 (d, J = 21.4 Hz), 113.3 (d, J = 25.2 Hz). 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F), −112.5 (q, J = 8.5 Hz, 1F). HRMS (ESI) m/z calcd for C16H10F4N3O2S [M + H]+ 384.0430, found 384.0429. Purity by HPLC was found to be 98%.
N-(5-(3-Chlorophenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (5):
Off-white solid (35 mg, 19%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 7.9 – 7.8 (m, 4H), 7.7 (dd, J = 23.9, 7.9 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.4, 159.8, 159.0, 136.1, 134.6, 132.0, 132.0, 131.1 (q, J = 308.7 Hz), 130.2, 128.9, 128.5, 126.0, 125.8, 125.2. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C16H10ClF3N3O2S [M + H]+ 400.0134, found 400.0135. Purity by HPLC was found to be 96%.
N-(5-(3-Methoxyphenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (6):
Off-white solid (42 mg, 24%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 7.9 (m, 2H), 7.6 – 7.5 (m, 2H), 7.4 (s, 1H), 7.2 (d, J = 7.9 Hz, 1H), 3.8 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.5, 160.9, 160.2, 158.6, 136.0, 135.7, 131.2 (q, J = 308.7 Hz), 130.1, 128.7, 128.6, 125.0, 118.9, 118.3, 111.5, 55.9. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C17H13F3N3O2S [M + H]+ 396.0630, found 396.0632. Purity by HPLC was found to be 98%.
N-(5-(3-Trifluoromethylphenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (7):
Off-white solid (53 mg, 27%). 1H NMR (500 MHz, DMSO-d6) δ 8.2 (d, J = 7.9 Hz, 1H), 8.2 – 8.1 (m, 3H), 8.0 (m, 1H), 7.9 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.4, 159.8, 159.1, 136.1, 135.8, 131.4, 131.1 (q, J = 308.7 Hz), 130.8 (q, J = 31.5 Hz), 130.5, 130.4, 130.1, 128.6, 125.2 (q, J = 272.2 Hz), 125.0, 122.8, 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F), −62.8 (s, 3F). HRMS (ESI) m/z calcd for C17H10F6N3O2S [M + H]+ 434.0398, found 434.0399. Purity by HPLC was found to be 98%.
N-(5-(4-Fluorophenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (8):
Off-white solid (43 mg, 24%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 8.0 (dd, J = 8.7, 5.5 Hz, 2H), 7.9 (m, 2H), 7.4 (t, J = 8.7 Hz, 2H).13C NMR (126 MHz, DMSO-d6) δ 165.5 (d, J = 250.7 Hz), 160.4, 158.6, 136.1, 135.7, 131.1 (q, J = 308.7 Hz), 130.1, 129.3 (d, J = 8.82 Hz), 128.6, 120.5, 117.2 (d, J = 22.7 Hz). 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F), −108.8. (s, 1F). HRMS (ESI) m/z calcd for C16H10F4N3O2S [M + H]+ 384.0430, found 384.0431. Purity by HPLC was found to be 96%.
N-(5-(4-Chlorophenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (9):
Off-white solid (36 mg, 20%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 8.0 – 7.9 (m, 2H), 7.9 (m, 2H), 7.7 – 7.6 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 164.5, 160.4, 158.6, 137.0, 136.1, 135.6, 131.1 (q, J = 308.7 Hz), 130.1, 128.6, 128.4, 127.2, 122.7. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C16H10ClF3N3O2S [M + H]+ 400.0134, found 400.0132. Purity by HPLC was found to be 97%.
N-(5-(4-Trifluoromethylphenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (10):
Off-white solid (42 mg, 22%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (dd, J = 14.0, 8.0 Hz, 4H), 8.0 (m, 2H), 7.9 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.2, 160.0, 159.0, 136.1, 135.6, 132.0 (q, J = 31.5 Hz), 131.1 (q, J = 308.7 Hz), 130.1, 128.7, 127.6, 127.4, 126.9, 125.3 (q, J = 272.2 Hz).19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F), −62.8 (s, 3F). HRMS (ESI) m/z calcd for C17H10F6N3O2S [M + H]+ 434.0398, found 434.0397. Purity by HPLC was found to be 99%.
N-(5-(4-Methoxyphenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (11):
Off-white solid (48 mg, 27%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (d, J = 8.0 Hz, 2H), 7.9 – 7.8 (m, 4H), 7.1 (m, 2H), 3.8 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 164.0, 162.5, 161.4, 158.1, 136.1, 131.1 (q, J = 308.7 Hz), 130.1, 128.4, 127.3, 116.1, 115.4, 115.1, 56.0. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C17H13F3N3O2S [M + H]+ 396.0630, found 396.0631. Purity by HPLC was found to be 99%.
N-(5-(4-Methylphenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (12):
Off-white solid (35 mg, 21%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 7.9 (m, 4H), 7.4 (m, 2H), 2.4 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.7, 161.3, 158.4, 142.5, 136.1, 135.8, 131.1 (q, J = 308.7 Hz), 130.4, 130.1, 128.5, 126.6, 121.1, 21.6. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C17H13F3N3O2S [M + H]+ 380.0681, found 380.0682. Purity by HPLC was found to be 98%.
N-(5-(4-Isopropylphenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (13):
Off-white solid (39 mg, 21%). 1H NMR (500 MHz, DMSO-d6) δ 8.2 – 8.1 (m, 2H), 7.9 (dd, J = 8.3, 2.5 Hz, 4H), 7.5 (m, 2H), 3.0 (h, J = 6.9 Hz, 1H), 1.2 (d, J = 6.9 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 165.5, 161.2, 158.4, 153.1, 136.1, 135.8, 131.2 (q, J = 308.7 Hz), 130.1, 128.5, 127.9, 126.7, 121.4, 33.9, 23.9. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C19H17F3N3O2S [M + H]+ 408.0994, found 408.0993. Purity by HPLC was found to be 97%.
N-(5-(2,4-Dichlorophenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (14):
Off-white solid (35 mg, 18%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 8.0 (m, 1H), 7.9 – 7.8 (m, 3H), 7.7 (dd, J = 8.5, 2.1 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 165.1, 158.8, 158.5, 137.5, 136.1, 135.5, 133.3, 132.6, 131.2, 131.1 (q, J = 308.7 Hz), 130.1, 128.7, 122.0. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C16H9Cl2F3N3O2S [M + H]+ 433.9745, found 433.9747. Purity by HPLC was found to be 99%.
N-(5-(3,5-Dichlorophenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (15, HSGN-218):
Off-white solid (37 mg, 19%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 7.9 (m, 5H). 13C NMR (126 MHz, DMSO-d6) δ 165.4, 159.7, 158.7, 136.1, 135.7, 131.4, 131.2 (q, J = 308.7 Hz), 130.1, 128.4, 127.2, 126.3, 124.9. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C16H9Cl2F3N3O2S [M + H]+ 433.9745, found 433.9744. Purity by HPLC was found to be 99%.
N-(5-(5-Chloro-2-methoxyphenyl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (16):
Off-white solid (46 mg, 24%). 1H NMR (500 MHz, DMSO-d6) δ 8.1 (m, 2H), 7.9 (m, 2H), 7.8 (d, J = 2.7 Hz, 1H), 7.6 (dd, J = 9.0, 2.8 Hz, 1H), 7.3 (m, 1H), 3.9 (s, 3H).13C NMR (126 MHz, DMSO-d6) δ 165.8, 158.7, 156.8, 136.1, 135.8, 133.3, 131.2 (q, J = 308.7 Hz), 130.1, 129.4, 128.6, 126.3, 124.9, 115.4, 114.4, 57.1. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C17H12ClF3N3O3S [M + H]+ 430.0240, found 430.0241. Purity by HPLC was found to be 97%.
N-(5-(Pyridin-2-yl)-1,3,4-oxadiazol-2-yl)-4-((trifluoromethyl)thio)benzamide (17):
Off-white solid (26 mg, 16%). 1H NMR (500 MHz, DMSO-d6) δ 8.8 (d, J = 4.7 Hz, 1H), 8.1 (dd, J = 8.2, 2.7 Hz, 3H), 8.0 (td, J = 7.8, 1.8 Hz, 1H), 7.9 (m, 2H), 7.6 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 164.9, 160.8, 159.0, 150.7, 143.2, 138.3, 136.1, 135.5, 131.1 (q, J = 308.7 Hz), 130.1, 128.7, 126.6, 122.9. 19F NMR (471 MHz, DMSO-d6) δ −42.4 (s, 3F). HRMS (ESI) m/z calcd for C15H10F3N4O2S [M + H]+ 367.0477, found 367.0475. Purity by HPLC was found to be 98%.
Bacterial strains media, cell lines and reagents
Bacterial strains used in this study (Table 1S) were obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources) and the American Type Culture Collection (ATCC). E. coli BW25113 and JW25113 were obtained from the Coli Genetic Stock Center (CGSC), Yale University, USA. Brain heart infusion broth was purchased from Becton, Dickinson and Company (Cockeysville, MD, USA) and was purchased from Fisher Scientific. Yeast extract, L-cysteine, vitamin K, hemin and phosphate buffered saline (PBS) were all obtained from commercial vendors. Human colorectal adenocarcinoma epithelial cells (Caco-2) (ATCC HTB-37) was obtained from the American Type Culture Collection (ATCC) (Manassas, VA, USA). Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS) and phosphate-buffered saline (PBS) was purchased from Corning (Manassas, VA, USA). Vancomycin hydrochloride (Gold Biotechnology, St. Louis, MO, USA), linezolid and gentamicin sulfate (Chem-Impex International, Wood Dale, IL, USA), metronidazole (Alfa Aesar, Ward Hill, MA, USA), and fidaxomicin (Cayman Chemical, Ann Arbor, MI, USA) were purchased commercially. Compounds were synthesized from commercial sources in our laboratory.
Determination of the MICs against C. difficile clinical isolates:
The minimum inhibitory concentrations (MICs) of tested compounds and control drug; vancomycin, were determined using the broth microdilution method, as previously described57–60 against C. difficile clinical isolates. Briefly, 0.5 McFarland bacterial solution was prepared and diluted in brain heart infusion supplemented (BHIS) broth (to an inoculum size ~5 × 105 CFU/mL). Test agents were added and serially diluted before plates were incubated anaerobically at 37°C for 48 hours. MICs reported are the lowest drug concentration that completely suppressed the growth of bacteria, as observed visually.
Determination of the MICs against vancomycin-resistant enterococci (VRE) and Escherichia coli strains
The MICs of HSGN-218 and control drugs were determined using the broth microdilution method, according to guidelines outlined by the Clinical and Laboratory Standards Institute (CLSI61) against Enterococcus faecium, Enterococcus faecalis and Escherichia coli strains. Bacterial strains were grown aerobically overnight on tryptone soy agar (TSA) plates at 37° C. Afterwards, a bacterial solution equivalent to 0.5 McFarland standard was prepared and diluted in cation-adjusted Mueller-Hinton broth (CAMHB) (for E. coli) or tryptone soy broth (TSB) (for enterococcal strains), to achieve a bacterial concentration of about 5 × 105 CFU/mL. Test agents were added in the first row of the 96-well plates and serially diluted along the plates. Plates were then, incubated as previously described. MICs reported in Table 3 are the minimum concentrations of the test agents that completely inhibited the visual growth of bacteria.
In vitro cytotoxicity analysis of HSGN-218 against human colorectal cells.
Compounds were assayed for potential cytotoxicity against a human colorectal adenocarcinoma (Caco-2) cell line, as described previously62–63. Briefly, tested compounds were incubated with Caco-2 cells for 2 hours. Then, cells were incubated with MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) reagent for 4 hours before measuring absorbance values (OD490).
Caco-2 permeability assay
Assay and data analysis were performed by Eurofins Panlabs (MO, USA) according to a previously reported protocol64–65. The apparent permeability coefficient (Papp) of the tested agents was calculated using the equation below:
where VR is the volume of the receiver chamber. CR,end is the concentration of the test compound in the receiver chamber at the end time point, Δt is the incubation time and A is the surface area of the cell monolayer. CD,mid is the calculated mid-point concentration of the test compound in the donor side, which is the mean value of the donor concentration at time 0 minute and the donor concentration at the end time point. CR,mid is the mid-point concentration of the test compound in the receiver side, which is one half of the receiver concentration at the end time point. Concentrations of the test compound were expressed as peak areas of the test compound.
In vitro antibacterial evaluation of HSGN-218 against normal microflora.
The broth microdilution assay was utilized to determine the MICs of HSGN-218 against commensal organisms that compose the human gut microflora, as described elsewhere48, 61, 66. A bacterial solution equivalent to 0.5 McFarland standard was prepared and diluted in BHIS broth (for Bacteroides) or in MRS broth (for Lactobacillus) to achieve a bacterial concentration of about 5 × 105 CFU/mL. Test agents were added and serially diluted along the plates. Plates were incubated for 48 hours at 37°C before recording the MIC by visual inspection of growth.
Frequency of spontaneous mutation.
HSGN-218 was tested against C. difficile to determine the likelihood of development of spontaneous mutation as previously described51, 67. Briefly, HSGN-218 and vancomycin were added to BHIS agar to achieve a final concentration of 15 × MIC and 20 × MIC and poured in plates and left to dry out. An inoculum of ~ 109 CFU/mL of C. difficile ATCC 43255 was spread over the plates and incubated anaerobically at 37°C for 48 hours before plates were checked for the possible bacterial growth.
Preparation of C. difficile spores for mice infection
C. difficile spores were prepared as described earlier 68,52. Briefly, C. difficile ATCC 43255 was inoculated onto BHIS agar and incubated anaerobically for 5 days. Spores were collected anaerobically using PBS containing 10% bovine serum albumin, heated at 70°C for 20 minutes to get rid of vegetative cells and counted by dilution and plating onto BHIS supplemented with 0.1% taurocholic acid. Spores were then, stored at 4℃ overnight before infecting mice.
C. difficile infection (CDI) mouse model
The study was reviewed, approved and performed following the guidelines of the Purdue University Animal Care and Use Committee (PACUC) and according to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Mice were housed in individually ventilated autoclaved cages and received sterile food and water ad libitum throughout the duration of the experiment. CDI mouse model was performed as described previously52. Eight-week-old female pathogen-free C57BL/6 mice (Jackson, ME, USA) were pre-treated with an antibiotic cocktail in sterile drinking water to disrupt the mice normal intestinal microflora, reducing the colonization resistance and facilitating infection with the toxigenic strain of C. difficile. Afterwards, mice were switched to regular autoclaved water for 2 days and they received a single dose of clindamycin (10 mg/kg) intraperitoneally 1 day prior to C. difficile challenge. For infection, mice were restrained and infected via oral gavage with 1.3 × 106 spores of C. difficile ATCC 43255. Following infection, mice were randomly allocated into groups (n=6) for treatment. Two hours post-infection, one groups were treated orally with HSGN-218 (50 mg/kg), one group was treated with vancomycin (10 mg/kg) via oral gavage, and one group was treated orally with the vehicle (10% DMSO, 10% tween 80, 80% PBS). Treatments were continued once daily for five days and mice were closely monitored for disease signs (including weight loss, behavioral changes, hunched posture, decreased activity, wet tail and diarrhea).
In vivo efficacy of HSGN-218 in C. difficile recurrence
In order to investigate the activity of HSGN-218 in preventing C. difficile recurrence, mice were infected, as described above and one group was treated orally with HSGN-218 (50 mg/kg), one group was treated with vancomycin (10 mg/kg) via oral gavage, and one group was treated orally with the vehicle for 5 days. Treatments were stopped after 5 days, and mice were monitored for disease signs and recurrence of infection till the 21st day. Then, mice were humanely euthanized using CO2 asphyxiation.
Statistical analyses
The survival data were analyzed by Log-rank (Mantel-Cox) test utilizing GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla, CA, USA).
In Silico PAINS Analysis
All synthesized analogs were subjected to PAINS filters by using the SwissADME program69. Molecular formula strings of analogs were manually entered into the program, which indicated no PAINS were found.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by Purdue University, the National Institute of Allergy And Infectious Diseases of the National Institutes of Health under Award Numbers T32AI148103 and R01AI130186.
Abbreviations:
- ATCC
American type culture collection
- BHIS
brain heart infusion supplemented
- BOP
benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate
- CDI
Clostridium difficile infection
- CLSI
clinical and laboratory standards institute
- DIPEA
diisopropylethylamine
- DMEM
dulbecco’s modified eagle medium
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- H2O
water
- MeOH
methanol
- MIC
minimum inhibitory concentration
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)
- NaOAc
sodium acetate
- PAINS
pan assay interference compounds
- PBS
phosphate buffered saline
- RT
room temperature
- TSA
tryptic soy agar
- TSB
tryptic soy broth
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.xxxxxxx.
• Various bacterial strains used in this study; and 1H NMR, 13C NMR, and 19F NMR spectra of analogs
• Molecular SMILES strings and MIC values
The authors declare no competing financial interest.
References:
- 1.Zhang S; Palazuelos-Munoz S; Balsells EM; Nair H; Chit A; Kyaw MH, Cost of hospital management of Clostridium difficile infection in United States-a meta-analysis and modelling study. BMC Infect. Dis 2016, 16 (1), 447. DOI: 10.1186/s12879-016-1786-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention (CDC). Antibiotic / antimicrobial resistance (AR / AMR), biggest threats and data. https://www.cdc.gov/drugresistance/biggest-threats.html
- 3.Kachrimanidou M; Malisiovas N, Clostridium difficile infection: a comprehensive review. Crit. Rev. Microbiol 2011, 37 (3), 178–187. [DOI] [PubMed] [Google Scholar]
- 4.Davies AH; Roberts AK; Shone CC; Acharya KR, Super toxins from a super bug: structure and function of Clostridium difficile toxins. Biochem. J 2011, 436 (3), 517–526. [DOI] [PubMed] [Google Scholar]
- 5.Chumbler NM; Farrow MA; Lapierre LA; Franklin JL; Haslam D; Goldenring JR; Lacy DB, Clostridium difficile toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PloS Pathog. 2012, 8 (12), DOI: 10.1371/journal.ppat.1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awad MM; Johanesen PA; Carter GP; Rose E; Lyras D, Clostridium difficile virulence factors: insights into an anaerobic spore-forming pathogen. Gut Microbes 2014, 5 (5), 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Viswanathan VK; Mallozzi MJ; Vedantam G, Clostridium difficile infection: an overview of the disease and its pathogenesis, epidemiology and interventions. Gut Microbes 2010, 1 (4), 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dethlefsen L; Huse S; Sogin ML; Relman DA, The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6 (11), e280, DOI: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald LC; Gerding DN; Johnson S; Bakken JS; Carroll KC; Coffin SE; Dubberke ER; Garey KW; Gould CV; Kelly C; Loo V; Sammons JS; Sandora TJ; Wilcox MH, Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the infectious diseases society of America (IDSA) and society for healthcare epidemiology of America (SHEA). Clin. Infect. Dis 2018, 66 (7), E1–E48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamp KC; Freeman CD; Klutman NE; Lacy MK, Pharmacokinetics and pharmacodynamics of the nitroimidazole antimicrobials. Clin. Pharmacokinet 1999, 36 (5), 353–373. [DOI] [PubMed] [Google Scholar]
- 11.Bolton RP; Culshaw MA, Faecal metronidazole concentrations during oral and intravenous therapy for antibiotic associated colitis due to Clostridium difficile. Gut 1986, 27 (10), 1169–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pepin J; Alary ME; Valiquette L; Raiche E; Ruel J; Fulop K; Godin D; Bourassa C, Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin. Infect. Dis 2005, 40 (11), 1591–1597. [DOI] [PubMed] [Google Scholar]
- 13.Louie TJ; Cannon K; Byrne B; Emery J; Ward L; Eyben M; Krulicki W, Fidaxomicin preserves the intestinal microbiome during and after treatment of Clostridium difficile infection (CDI) and reduces both toxin reexpression and recurrence of CDI. Clin. Infect. Dis 2012, 55 Suppl 2, S132–S142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vardakas KZ; Polyzos KA; Patouni K; Rafailidis PI; Samonis G; Falagas ME, Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int. J. Antimicrob. Agents 2012, 40 (1), 1–8. [DOI] [PubMed] [Google Scholar]
- 15.Smits WK; Lyras D; Lacy DB; Wilcox MH; Kuijper EJ, Clostridium difficile infection. Nat. Re.v Dis. Primers 2016, 2, 16020, DOI: 10.1038/nrdp.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orenstein R, Fidaxomicin failures in recurrent Clostridium difficile infection: a problem of timing. Clin. Infect. Dis 2012, 55 (4), 613–614. [DOI] [PubMed] [Google Scholar]
- 17.Cornely OA; Miller MA; Louie TJ; Crook DW; Gorbach SL, Treatment of first recurrence of Clostridium difficile infection: fidaxomicin versus vancomycin. Clin. Infect. Dis 2012, 55 Suppl 2, S154–S161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhanel GG; Walkty AJ; Karlowsky JA, Fidaxomicin: A novel agent for the treatment of Clostridium difficile infection. Can. J. Infect. Dis. Med 2015, 26 (6), 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baines SD; Wilcox MH, Antimicrobial resistance and reduced susceptibility in Clostridium difficile: potential consequences for induction, treatment, and recurrence of C. difficile infection. Antibiotics 2015, 4 (3), 267–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakken JS; Borody T; Brandt LJ; Brill JV; Demarco DC; Franzos MA; Kelly C; Khoruts A; Louie T; Martinelli LP; Moore TA; Russell G; Surawicz C, Treating Clostridium difficile infection with fecal microbiota transplantation. Clin. Gastroenterol. Hepatol 2011, 9 (12), 1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodworth MH; Carpentieri C; Sitchenko KL; Kraft CS, Challenges in fecal donor selection and screening for fecal microbiota transplantation: a review. Gut Microbes 2017, 8 (3), 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pamer EG, Fecal microbiota transplantation: effectiveness, complexities, and lingering concerns. Mucosal Immunol. 2014, 7 (2), 210–214. [DOI] [PubMed] [Google Scholar]
- 23.Naclerio GA; Sintim HO, Multiple ways to kill bacteria via inhibiting novel cell wall or membrane targets. Future Med. Chem 2020, 12 (13), 1253–1279. [DOI] [PubMed] [Google Scholar]
- 24.Naclerio GA; Karanja CW; Opoku-Temeng C; Sintim HO, Antibacterial small molecules that potently inhibit Staphylococcus aureus lipoteichoic acid biosynthesis. ChemMedChem 2019, 14 (10), 1000–1004. [DOI] [PubMed] [Google Scholar]
- 25.Opoku-Temeng C; Naclerio GA; Mohammad H; Dayal N; Abutaleb NS; Seleem MN; Sintim HO, N-(1,3,4-oxadiazol-2-yl)benzamide analogs, bacteriostatic agents against methicillin- and vancomycin-resistant bacteria. Eur. J. Med. Chem 2018, 155, 797–805. [DOI] [PubMed] [Google Scholar]
- 26.Naclerio GA; Abutaleb NS; Onyedibe KI; Seleem MN; Sintim HO, Potent trifluoromethoxy, trifluoromethylsulfonyl, trifluoromethylthio and pentafluorosulfanyl containing (1,3,4-oxadiazol-2-yl)benzamides against drug-resistant Gram-positive bacteria. RSC Med. Chem 2020, 11 (1), 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendez L; Henriquez G; Sirimulla S; Narayan M, Looking back, looking forward at halogen bonding in drug discovery. Molecules 2017, 22 (9), DOI: 10.3390/molecules22091397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandes MZ; Cavalcanti SM; Moreira DR; de Azevedo Junior WF; Leite AC, Halogen atoms in the modern medicinal chemistry: hints for the drug design. Curr. Drug Targets 2010, 11 (3), 303–314. [DOI] [PubMed] [Google Scholar]
- 29.Wilcken R; Zimmermann MO; Lange A; Joerger AC; Boeckler FM, Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem 2013, 56 (4), 1363–1388. [DOI] [PubMed] [Google Scholar]
- 30.Guo M; Zheng Y; Terell JL; Ad M; Opoku-Temeng C; Bentley WE; Sintim HO, Geminal dihalogen isosteric replacement in hydrated AI-2 affords potent quorum sensing modulators. Chem. Commun 2015, 51 (13), 2617–2620. [DOI] [PubMed] [Google Scholar]
- 31.Suarez-Castro A; Valle-Sanchez M; Cortes-Garcia CJ; Chacon-Garcia L, Molecular docking in halogen bonding. In Molecular Docking, IntechOpen: 2018. [Google Scholar]
- 32.Gerebtzoff G; Li-Blatter X; Fischer H; Frentzel A; Seelig A, Halogenation of drugs enhances membrane binding and permeation. Chembiochem 2004, 5 (5), 676–684. [DOI] [PubMed] [Google Scholar]
- 33.Gentry CL; Egleton RD; Gillespie T; Abbruscato TJ; Bechowski HB; Hruby VJ; Davis TP, The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides 1999, 20 (10), 1229–1238. [DOI] [PubMed] [Google Scholar]
- 34.De Azevedo WF Jr.; Mueller-Dieckmann HJ; Schulze-Gahmen U; Worland PJ; Sausville E; Kim SH, Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. USA 1996, 93 (7), 2735–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valadon P; Dansette PM; Girault JP; Amar C; Mansuy D, Thiophene sulfoxides as reactive metabolites: formation upon microsomal oxidation of a 3-aroylthiophene and fate in the presence of nucleophiles in vitro and in vivo. Chem. Res. Toxicol 1996, 9 (8), 1403–1413. [DOI] [PubMed] [Google Scholar]
- 36.Dansette PM; Bertho G; Mansuy D, First evidence that cytochrome P450 may catalyze both S-oxidation and epoxidation of thiophene derivatives. Biochem. Biophys. Res. Commun 2005, 338 (1), 450–455. [DOI] [PubMed] [Google Scholar]
- 37.Mansuy D; Dansette PM, Sulfenic acids as reactive intermediates in xenobiotic metabolism. Arch. Biochem. Biophys 2011, 507 (1), 174–185. [DOI] [PubMed] [Google Scholar]
- 38.Gramec D; Peterlin Masic L; Sollner Dolenc M, Bioactivation potential of thiophene-containing drugs. Chem. Res. Toxicol 2014, 27 (8), 1344–1358. [DOI] [PubMed] [Google Scholar]
- 39.Al-Nassir WN; Sethi AK; Li Y; Pultz MJ; Riggs MM; Donskey CJ, Both oral metronidazole and oral vancomycin promote persistent overgrowth of vancomycin-resistant enterococci during treatment of Clostridium difficile-associated disease. Antimicrob. Agents Chemother 2008, 52 (7), 2403–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seiler P; Enderlin-Paput M; Pfaff P; Weiss M; Ritz D; Clozel M; Locher HH, Cadazolid does not promote intestinal colonization of vancomycin-resistant enterococci in mice. Antimicrob. Agents Chemother 2016, 60 (1), 628–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delmas J; Dalmasso G; Bonnet R, Escherichia coli: The good, the bad and the ugly. Clin. Microbiol 2015, 4, DOI: 10.4172/2327-5073.1000195. [DOI] [Google Scholar]
- 42.Hidalgo IJ; Raub TJ; Borchardt RT, Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96 (3), 736–749. [PubMed] [Google Scholar]
- 43.Kim S; Covington A; Pamer EG, The intestinal microbiota: antibiotics, colonization resistance, and enteric pathogens. Immunol. Rev 2017, 279 (1), 90–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoon S; Yu J; McDowell A; Kim SH; You HJ; Ko G, Bile salt hydrolase-mediated inhibitory effect of Bacteroides ovatus on growth of Clostridium difficile. J. Microbiol 2017, 55 (11), 892–899. [DOI] [PubMed] [Google Scholar]
- 45.Deng H; Yang S; Zhang Y; Qian K; Zhang Z; Liu Y; Wang Y; Bai Y; Fan H; Zhao X; Zhi F, Bacteroides fragilis prevents Clostridium difficile infection in a mouse model by restoring gut barrier and microbiome regulation. Front. Microbiol 2018, 9, 2976, DOI: 10.3389/fmicb.2018.02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quigley L; Coakley M; Alemayehu D; Rea MC; Casey PG; O’Sullivan O; Murphy E; Kiely B; Cotter PD; Hill C; Ross RP, Lactobacillus gasseri APC 678 reduces shedding of the pathogen Clostridium difficile in a murine model. Front. Microbiol 2019, 10, 273, DOI: 10.3389/fmicb.2019.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naaber P; Smidt I; Stsepetova J; Brilene T; Annuk H; Mikelsaar M, Inhibition of Clostridium difficile strains by intestinal Lactobacillus species. J. Med. Microbiol 2004, 53 (Pt 6), 551–554. [DOI] [PubMed] [Google Scholar]
- 48.Abutaleb NS; Seleem MN, Repurposing the antiamoebic drug diiodohydroxyquinoline for treatment of Clostridioides difficile infections. Antimicrob. Agents Chemother 2020, 64 (6), DOI: 10.1128/AAC.02115-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rafii F; Sutherland JB; Cerniglia CE, Effects of treatment with antimicrobial agents on the human colonic microflora. Ther. Clin. Risk Manag 2008, 4 (6), 1343–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ajami NJ; Cope JL; Wong MC; Petrosino JF; Chesnel L, Impact of oral fidaxomicin administration on the intestinal microbiota and susceptibility to Clostridium difficile colonization in mice. Antimicrob. Agents Chemother 2018, 62 (5), DOI: 10.1128/AAC.02112-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mascio CT; Chesnel L; Thorne G; Silverman JA, Surotomycin demonstrates low in vitro frequency of resistance and rapid bactericidal activity in Clostridium difficile, Enterococcus faecalis, and Enterococcus faecium. Antimicrob Agents Chemother 2014, 58 (7), 3976–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abutaleb NS; Seleem MN, Auranofin, at clinically achievable dose, protects mice and prevents recurrence from Clostridioides difficile infection. Sci. Rep 2020, 10 (1), 7701, DOI: 0.1038/s41598–020-64882–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hutton ML; Pehlivanoglu H; Vidor CJ; James ML; Thomson MJ; Lyras D, Repurposing auranofin as a Clostridioides difficile therapeutic. J. Antimicrob. Chemother 2020, 75 (2), 409–417. [DOI] [PubMed] [Google Scholar]
- 54.Chen X; Katchar K; Goldsmith JD; Nanthakumar N; Cheknis A; Gerding DN; Kelly CP, A mouse model of Clostridium difficile-associated disease. Gastroenterology 2008, 135 (6), 1984–1992. [DOI] [PubMed] [Google Scholar]
- 55.Frieden T. Centers for disease control and prevention (CDC). Antibiotic resistance threats in the United States 2013. (accessed 02/20/2018). [Google Scholar]
- 56.Kaur J; Soto-Velasquez M; Ding Z; Ghanbarpour A; Lill MA; van Rijn RM; Watts VJ; Flaherty DP, Optimization of a 1,3,4-oxadiazole series for inhibition of Ca(2+)/calmodulin-stimulated activity of adenylyl cyclases 1 and 8 for the treatment of chronic pain. Eur. J. Med. Chem 2019, 162, 568–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.AbdelKhalek A; Abutaleb NS; Mohammad H; Seleem MN, Antibacterial and antivirulence activities of auranofin against Clostridium difficile. Int. J. Antimicrob. Agents 2019, 53 (1), 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mody D; Athamneh AIM; Seleem MN, Curcumin: a natural derivative with antibacterial activity against Clostridium difficile. J. Glob. Antimicrob. Resist 2019, 21, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pal R; Seleem MN, Screening of natural products and approved oncology drug libraries for activity against Clostridioides difficile. Sci. Rep 2020, 10 (1), 5966, DOI: 10.1038/s41598-020-63029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shao X; AbdelKhalek A; Abutaleb NS; Velagapudi UK; Yoganathan S; Seleem MN; Talele TT, Chemical space exploration around thieno[3,2-d]pyrimidin-4(3H)-one scaffold led to a novel class of highly active Clostridium difficile inhibitors. J. Med. Chem 2019, 62 (21), 9772–9791. [DOI] [PubMed] [Google Scholar]
- 61.Clinical and Laboratory Standards Institute, C., Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. 9 ed.; 2012. [Google Scholar]
- 62.Kotb A; Abutaleb NS; Seleem MA; Hagras M; Mohammad H; Bayoumi A; Ghiaty A; Seleem MN; Mayhoub AS, Phenylthiazoles with tert-Butyl side chain: metabolically stable with anti-biofilm activity. Eur. J. Med. Chem 2018, 151, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.ElAwamy M; Mohammad H; Hussien A; Abutaleb NS; Hagras M; Serya RAT; Taher AT; Abouzid KA; Seleem MN; Mayhoub AS, Alkoxyphenylthiazoles with broad-spectrum activity against multidrug-resistant gram-positive bacterial pathogens. Eur. J. Med. Chem 2018, 152, 318–328. [DOI] [PubMed] [Google Scholar]
- 64.Obach RS; Baxter JG; Liston TE; Silber BM; Jones BC; MacIntyre F; Rance DJ; Wastall P, The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther 1997, 283 (1), 46–58. [PubMed] [Google Scholar]
- 65.Hammad A; Abutaleb NS; Elsebaei MM; Norvil AB; Alswah M; Ali AO; Abdel-Aleem JA; Alattar A; Bayoumi SA; Gowher H; Seleem MN; Mayhoub AS, From phenylthiazoles to phenylpyrazoles: broadening the antibacterial spectrum toward carbapenem-resistant bacteria. J. Med. Chem 2019, 62 (17), 7998–8010. [DOI] [PubMed] [Google Scholar]
- 66.AbdelKhalek A, M. H., Mayhoub AS, Seleem MN, Screening for potent and selective anticlostridial leads among FDA-approved drugs. J. Antibiot 2020, 73, 392–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thangamani S; Mohammad H; Abushahba MF; Sobreira TJ; Hedrick VE; Paul LN; Seleem MN, Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Sci. Rep 2016, 6, 22571, DOI: 10.1038/srep22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edwards AN; McBride SM, Isolating and purifying Clostridium difficile spores. Methods Mol. Biol 2016, 1476, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Daina A; Michielin O; Zoete V, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep 2017, 7, 42717, DOI: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.