

Abstract
In the field of metabolomics, mass spectrometry (MS) is the method most commonly used for identifying and annotating metabolites. As this typically involves matching a given MS spectrum against an experimentally acquired reference spectral library, this approach is limited by the coverage and size of such libraries (which typically number in the thousands). These experimental libraries can be greatly extended by predicting the MS spectra of known chemical structures (which number in the millions) to create computational reference spectral libraries. To facilitate the generation of predicted spectral reference libraries we developed CFM-ID, a computer program that can accurately predict ESI-MS/MS spectrum for a given compound structure. CFM-ID is one of the best-performing methods for compound-to-mass-spectrum prediction, and also one of the top tools for in silico mass-spectrum-to-compound identification. This work improves CFM-ID’s ability to predict ESI-MS/MS spectra from compounds by: (1) learning parameters from features based on the molecular topology, (2) adding a new approach to ring cleavage that models such cleavage as a sequence of simple chemical bond dissociations and (3) expanding its hand-written rule-based predictor to cover more chemical classes, including acylcarnitines, acylcholines, flavonols, flavones, flavanones, and flavonoid glycosides. We demonstrate that this new version of CFM-ID (version 4.0) is significantly more accurate than previous CFM-ID versions, in terms of both EI-MS/MS spectral prediction and compound identification. CFM-ID 4.0 is available at http://cfmid4.wishartlab.com/ as a webservice and docker images can be downloaded at https://hub.docker.com/r/wishartlab/cfmid
Introduction
Mass spectrometry (MS) is the most widely used method for identifying or annotating metabolites in metabolomics. Many MS-based metabolite identification systems use an experimentally acquired MS spectrum (either electron impact [EI-MS] or electrospray ionization [ESI-MS/MS]) of the target compound, then attempt to find a matching MS spectrum in an experimentally acquired reference spectral library (RSL) that contains a large number of MS spectra of known metabolites1–6. However, this only works when that RSL includes the matching experimental MS spectrum and its associated molecule. Given that most RSLs have MS spectra for only a few thousand relevant compounds, their limited coverage is a major bottleneck to this approach7–11. Moreover, since many important metabolites are not included in any MS RSL, it is often not possible to identify even well-known metabolites using this approach12.
To overcome this bottleneck, researchers have developed in silico tools to identify the chemical structure of an unknown compound based on its experimentally acquired MS spectrum, without having to query an experimentally measured RSL directly. These methods can be loosely divided into two categories.
The first category of in silico methods is called MS-to-compound matching (MS2C). These methods use the observed MS spectrum to extract likely chemical and physical characteristics (the molecular formula, chemical fingerprints, subfragment structures, etc.) of the unidentified compound and then finds the corresponding chemical structure from a database of known compounds via chemical feature matching5,13–19. The most well-known MS2C approach is Heinonen et al.’s FingerID13 program, which uses a set of learned support vector machines (SVMs) to predict a bit vector (~500-bit) known as a chemical fingerprint (each bit corresponding to a specific pre-defined subregion of a molecule) for each compound, and then performs compound identification through fingerprint matching. FingerID was later extended to CSI:FingerID18, which learned more accurate SVMs that predicted a longer (1415-bit) chemical fingerprint for each given MS spectrum. CSI:FingerID is later been integrated into SIRIUS 416, one of the state-of-the-art molecular structure identification program to-date. In addition, Brouard et al.15 further extended CSI:FingerID with a single Input Output Kernel Regression (IOKR)20 method. This approach not only outperforms CSI:FingerID in terms of metabolite identification accuracy but also greatly improves both the training and inference speed compared to earlier works. IOKR and CSI:FingerID came in first and second, respectively, in the in silico fragmentation-only category (category 2) of the CASMI 2016 challenge21.
The second category of in silico methods is called compound-to-MS matching (C2MS). These methods use hand-made rules and/or computer-learned rules to predict MS spectra from millions of known chemical structures, thereby producing a comprehensive reference spectral library or RSL of in silico compound+MS pairs. The method also allows one to identify unknowns (i.e., MS2C) by comparing an experimentally measured MS spectrum against that in silico predicted reference spectral library22–28. The focus for these methods is to generate accurate in silico MS spectra (EI-MS or ESI-MS/MS) from any given molecular structure. Typically, the compound identification process (MS2C) shares the same spectral matching algorithms with standard experimental MS spectral matching tools.
Over the years, a wide range of MS spectral prediction algorithms have been developed, including LipidBlast23, MetFrag22, MIDAS29, and MAGMa30. Currently the best method for predicting both EI-MS and ESI-MS/MS spectra of a given compound, is the machine learning based competitive fragmentation modeling method called CFM-ID 2.024,25. A more recent version, called CFM-ID 3.026, extended this idea for ESI-MS/MS prediction by including (hand-coded) fragmentation rules to better handle lipids. The CFM-ID method treats a molecule fragmentation process inside a mass spectrometer as a stochastic, homogeneous Markov process. Given a molecular structure, CFM-ID first generates all its theoretically possible fragments combinatorially, structured as a fragmentation graph (see Figure 1). Each node in a fragmentation graph represents a theoretically possible ion fragment, and each directed edge between nodes encodes the possibility that one ion fragment directly produces another through a single fragmentation event. The self-loop for each node encodes the chance of the persistence transition for the given fragment. CFM-ID then estimates the probabilities of each transition (the edge in Figure 1), using parameters that it learned from a training dataset of known molecules and their associated MS spectra. Finally, CFM-ID constructs the corresponding MS spectrum for the input molecule from the fragmentation graph and associated probability estimations.
Figure 1.
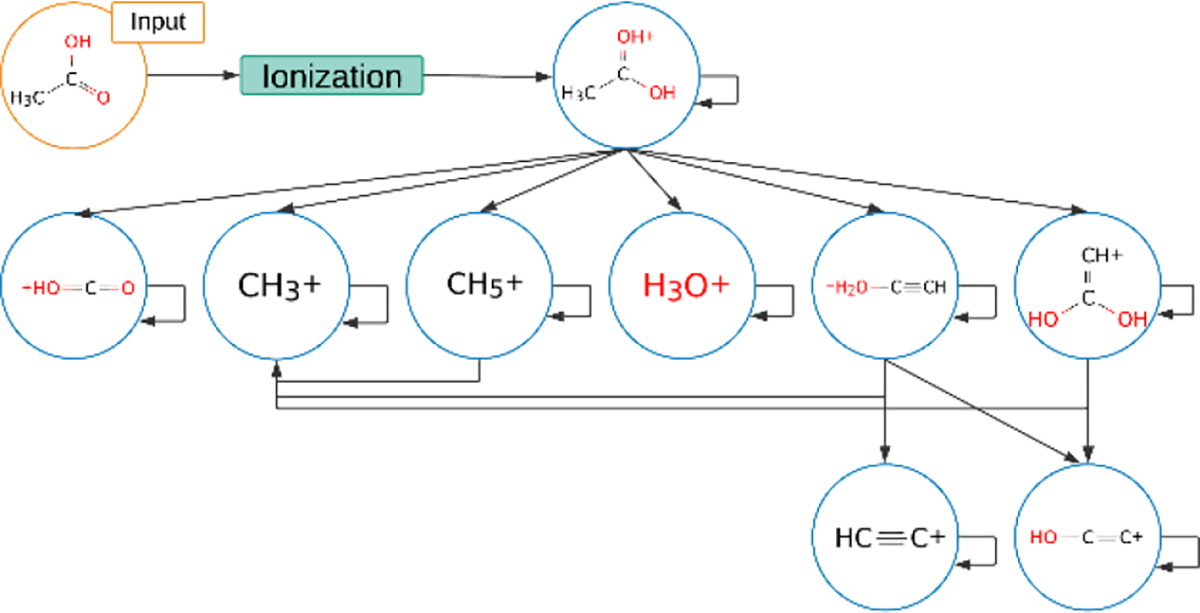
A fragmentation graph for the acetic acid [M+H]+ ion. Here each node denotes an ion fragment and each edge denotes the transition between a pair of nodes.
Despite continuing advances in the field of computational mass spectrometry, a recent study28 using data from the 2016 Critical Assessment of Small Molecule Identification (CASMI) challenge18 showed that these pure in silico algorithmic approaches were not as good as many believed. They could only correctly identify about 25% of the testing compounds without the help of metadata, such as retention times, customized candidate databases, or compound citation counts. Hence, there is still significant room for improvement for both categories these in silico methods.
With regard to improving C2MS methods, it appears that encoding more information about chemical bond energies could help model ESI-MS/MS fragmentation more accurately. Over the years, several studies have used a range of different approaches to approximate the strength of chemical bonds. Some methods22,31,32 estimated chemical bond energies via chemical or physical properties while others32–35 estimated bond strength from the compound’s structural or topological information. Inspired by these efforts, we developed an improved version of CFM-ID (version 4.0) that learns its parameters from a detailed representation of the molecule’s chemical bond topology. We also trained and tested the program on a much larger set of quadrupole time-of-flight (QTOF) ESI-MS/MS spectra and enhanced its hand-written rule-based predictor to cover more chemical classes, including acylcarnitines, acylcholines, flavonols, flavones, flavanones, and flavonoid glycosides. Compared to the earlier versions of CFM-ID, our new version of CFM-ID can more accurately predict the ESI-MS/MS spectra of many more chemical compounds. It also performs much better on the MS2C task than any other in silico tool published to date. For the remainder of this paper we will refer to the old CFM-ID methods as CFM-ID 2.024,25 and CFM-ID 3.026 and the new method as CFM-ID 4.0.
Sequential Ring Cleavage Modeling
The first modification to earlier versions of CFM-ID was to remodel the ring cleavage in ESI-MS/MS as a two-step sequential process. During fragment graph generation, CFM-ID breaks each fragment to smaller fragments to create fragment transitions, where the ion fragment becomes a child node on the graph while the other neutral fragment is discarded. There are three types of fragment transitions: (#1) a regular fragmentation that cleaves only one chemical bond (Figure 2(a)); (#2) a ring fragmentation that cleaves two chemical bonds on the ring structure simultaneously (Figure 2(b)); and (#3) a special transition to allow a fragment remain as itself. To calculate an MS spectrum CFM-ID then needs to compute the weight of each transition. The weight of each self-transition (case #3) is fixed to 1. To compute the other two fragmentation cases, CFM-ID first describes each fragment transition as a feature vector. In particular, it defines a set of features associated with each break. As ring fragmentation (case #2) corresponds to two breaks, earlier versions of CFM-ID would produce two individual bond-break-feature-vectors FV0 and FV1 and concatenate them to produce a single vector (Figure 2(b)). In the common one-chemical-bond-cleavage case #1, CFM-ID fills the FV0 with meaningful features corresponding to that one break and FV1 with zeros -- see Figure 2(a). Returning to the ring cleavage (case #2), note, the order of the two feature vectors is arbitrary. Therefore, for the same single ring break transition, earlier versions of CFM-ID could produce two distinct feature representations. Such behavior is problematic for several reasons. First, this same input has multiple possible features, which brings on unnecessary difficulties for the learning method. Second, since the second feature vector FV2 in case #1 (non-ring cleavage) is a zero vector, the parameters learned by those cleavages cannot be effectively shared with its ring break counterparts. For instance, earlier CFM-ID models would learn that a single carbon-carbon bond is more likely to break than a double carbon-carbon bond from one-bond cleavages, yet, such knowledge is only shared by the first bond cleavage in a ring-structure cleavage case
Figure 2.
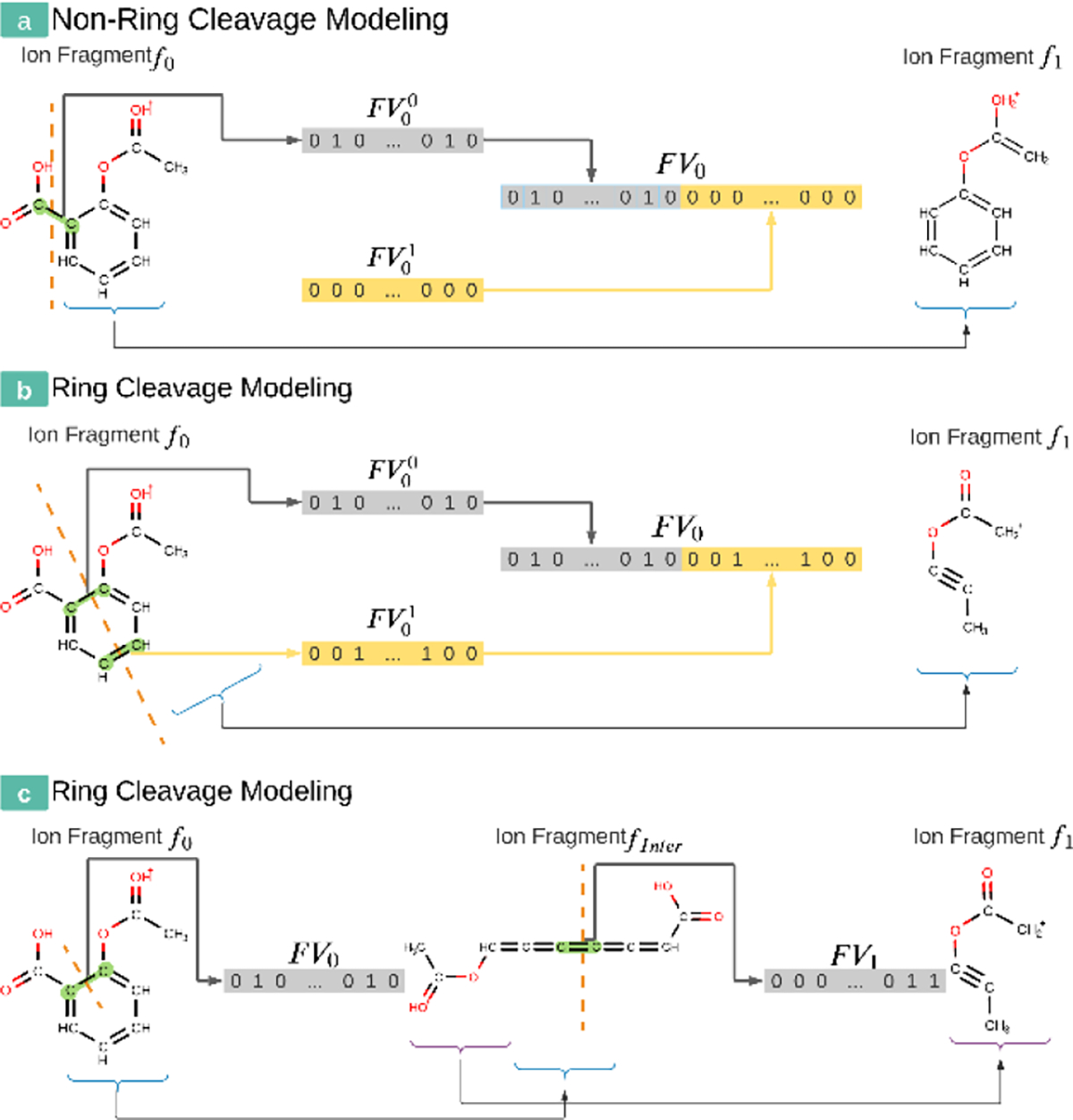
(a) An example of how earlier versions of CFM-ID model a fragmentation that only involves one chemical bond (highlighted in green). (b) An example of how earlier versions of CFM-ID model a fragmentation that involves a chemical ring structure. Note that the order of FV0 and FV1, is arbitrary. (c) An example of how the new version of CFM-ID models a fragmentation that involves the same chemical ring structure. Note that this method does not need the “double” feature vector design used by earlier versions of CFM-ID.
To address these issues in CFM-ID 4.0, a given ring cleavage transition is modelled as a sequence of two individual one-bond fragment transitions – i.e., as two “case #1’s”. The new approach does not need a second bond feature vector. First, Figure 2(b) shows how the earlier CFM-ID approach would cleave two bonds (highlighted in green) of the ion fragment f0 to create the subsequent fragment f1. In contrast, our new sequential ring cleavage model represents the same sequence in two steps. Figure 2(c) shows how it cleaves a single bond on the ring of f0 and creates an intermediate fragment finter. Next, it produces a fragment f1 by cleaving another chemical bond that was on the same ring. This design also introduces a minor issue: intermediate fragments are chemically unstable. To address such a problem the persistence transitional probability of the intermediate fragment is fixed to zero. As a result, an unstable intermediate fragment will not result in a peak in the predicted MS spectrum. This sequential ring cleavage modeling can be further extended to handle fragment transitions involving more than two bonds. However, ring cleavages are already an unusual event in ESI-MS/MS. The chance of three or more bonds cleaving simultaneously is even rarer. Since the computational cost of CFM-ID scales poorly with the number of transitions in each fragmentation graph, we decided not to cover those uncommon cases to simplify our model and reduce its required computations.
Connectivity Matrix Features
In MS, the probability of a fragment transition occurring depends on two factors: 1) whether the associated chemical bond will cleave, and 2) which fragment will have a charge after the bond cleavage. For the first question, Tanaka et al32 recently developed a method to predict whether a chemical bond would cleave by checking its bonding patterns, the location of the chemical bond, the character of the two associated atoms, and the chemical groups attached to it. Tanaka’s analysis provides a physicochemical basis for our hypothesis that the chance of a bond cleavage during MS/MS fragmentation depends largely on the local structure surrounding a chemical bond. For the second question, our observations from many annotated MS data shows that atoms such as sulphur and nitrogen have a higher tendency to acquire an unpaired valence electron than other atom types such as carbon. This phenomenon indicates that atom types contribute to the position of the charge after bond cleavage. To simplify the problem, we assume that the atoms near the potential cleavage site can provide sufficient information to answer the question of charge acquisition. While this assumption may not be entirely correct from a pure chemical or physical perspective, from a computational point of view, it provides a reason for a useful compromise.
Because CFM-ID computes transitional probabilities with respect to a given source ion, it describes a bond cleavage event through the following three components: (1) the chemical bond that is cleaved, (2) the ion fragment, and (3) the neutral loss fragment. As products of fragmentation, the ion fragment and the neutral loss fragment can each be viewed as the root of a rooted graph G = (V, E, VL, EL, R), where V is a set of vertices whose elements correspond to an atom in the fragment and E is a set of chemical bonds between each pair of atoms in the fragment. VL and EL are the sets of labels assigned to each vertex and edge, respectively. (In step (b) of Figure 3, G is the graph shown, V is the set of 10 atoms, E is the set of 10 edges.) In this example, features stored in VL are atom types, which are presented as the label assigned to each vertex. Similarly, features stored in EL indicate chemical bond type, which are shown as the numbers associated with each edge. R denotes the root vertex of the graph, which is one of the two atoms connected to the bond before it cleaves. To include more topological information about each molecule, we developed an adjacency matrix based chemical structure representation for CFM-ID 4.0 that extracts topological features of fragments that are MS relevant. This feature representation method has two objectives. First, it is well known that a rooted graph could have multiple adjacency matrices depending on the indexing of each vertex. By carefully designing our indexing method, this method will output an identical feature representation for rooted chemical structures sharing the same underlying structure and the same root atom. Second, this feature representation needs to reflect the topological relationship between each atom and the root atom – i.e., atoms with larger index numbers should be farther away from the root atom. This implies that features for closer atoms should also be closer to the start of the feature representation.
Figure 3.

(a) An ion fragment with its root atom highlighted by the blue circle. (b) Extracting a graph G from the ion fragment. (c) Labelling every vertex in the graph G. (d) Indexing each vertex is determined based on its label. (e) Selecting a subgraph SG from graph G. (f) Computing adjacency matrix from graph SG. (g) Creating two tensors from the adjacency matrix. (h) Flattening two tensors into vectors. (i) Joining two vectors into the output feature vector.
To achieve these two objectives, our proposed approach first selects from G, a subgraph SG that contains the most relevant atoms, and then encodes this selected subgraph as an adjacency matrix-based method. The selection phase of this method consists of two graph traversals. The first traversal assigns a string heuristic value to each vertex in the graph, and the second traversal is responsible for selecting and indexing vertices using those heuristic values. For a given molecular graph, in the first step, a heuristic string is created and assigned to each vertex through a breadth-first traversal (Figure 3). First, we define a bond-order-atom-type pair as a 3-character string. The first character uses a number to indicate the order of the chemical bond, which leads to the current vertex, and the remaining two characters define atom types of the current vertex using atomic symbols. For the one-character atom symbols, an empty string is inserted between the atom type and bond order. For instance, a single bond leading to a carbon atom will have “1 C” as its bond-order-atom-type pair while the string for a double bond leading to chlorine is “2Cl”. For any non-root vertex in the graph, its heuristic string consists of the following two parts: (1) a bond-order-atom-type pair to describe the type of chemical bond leading to the current vertex and the atom type associated to the current vertex. (2) A set of alphanumeric heuristic strings from its child vertices, each placed within a bracket. For instance, the green vertex in Figure 3 (c) has a carbon atom associated to it, and it is connected to its parent vertex via a single bond. Therefore, its bond-order-atom-type pair is “1 C”, which forms the first part of the heuristic label. Its child vertex has a double-bond-carbon pair and a single-bond-oxygen pair. If we sort those two labels alphanumerically, the second part of the current heuristic label is “[1 O][2 C]”. Once joined together, the heuristic label of the green vertex is “1 C[1 O][2 C]”. In the situation where multiple edges lead to the same vertex, our method chooses the edge on the shortest path to the root vertex to create the heuristic label. If there are still multiple edges, we use the edge with the lowest bond order to break the tie.
The second graph traversal assigns a new index to each vertex based on its labels. It also follows the rules of breadth-first, with additional steps to determine the visiting order of the child vertices at each step. As shown in Figure 3, starting from the root vertex, at each traversing step, the heuristic label created in the previous traversal will be used to determine which child vertex to visit next. For any given vertex, the visiting order of its child vertices is based on the label of the child vertices, with an alphanumerically smaller label being visited first, and vice versa. For example, after visiting the green vertex in Figure 2, our traversal process will visit Vertex8 and Vertex9 because “1 C” is alphanumerically smaller than “2 O”.
Once the input graph is indexed, our method extracts the closest N vertices to the root based on their index to create an adjacency matrix. (In Fig. 3, this N is 6.) Because the index number also reflects the distance between the current vertex to the root, the first N vertices are also the closest N vertices to the root. Our implementation uses 10 as the threshold since it is large enough to cover all atoms in our dataset with a distance of 2 bonds. Compared to the distance-based filtering that selects all neighboring vertices within a given distance, this nearest N-neighborhood approach not only has a better feature space utilization but also can provide a better representation for a more linear chemical structure. For example, when using distance-based selection, long-chain structures with various lengths will have a l ower chance of obtaining a unique feature vector. While using a fixed-range filter with up to 3 atoms or 2 chemical bonds, we see that some fragments produced by fragmenting, for example propane and n-butylamine, will share the same feature representation while they are structurally different. In comparison, the same problem is less severe if the threshold is based on the number of neighborhood atoms instead of their distance. Through this indexing and selection process, the importance of atoms with respect to the current bond cleavage is captured by the topological distance between the atom pairs in the chemical space and labeling order inside the feature space. More proximal atoms will be represented on the one end of the feature representation, and more distal ones will be represented on the other end.
Our method uses tensors derived from an adjacency matrix (Figure 3(f)) of a fragment to create this feature representation. The adjacency matrix is a square matrix where each (i, j) entry is 1 if there is an atom at vertexi connected to an atom at vertexj. Such a representation method can be easily extended to handle more complicated labels via additional vectors or tensors. In our case, we represent a fragment graph G through a combination of two tensors. Let Nv represent the number of vertices, and Dv represent the length of the one-hot encoded feature vector per bond. Therefore, a Nv × Nv × Dv tensor Tadj holds the connectivity information of the given graph as well as the label of each chemical bond. To represent each atom, the second tensor, Tvertex, has the size Ne × De where Ne is the number of vertices, and De is the size of the one-hot encoded labels for each atom. Tensor Tadj is created from the adjacency matrix Madj. Its first two axes resemble the Madj, and the third axis stores the features per edge. For every pair of vertices Vi and Vj, the feature vector Tadj(i, j) is a zero vector if there is no edge. Otherwise, vector Tadj(i, j) stores its associated chemical bond type as a one-hot encoded feature vector. Bond types can belong to one of the following categories, single, double, triple, and higher bond orders, aromatic, or conjugated. Tensor Tvertex is used to store feature vectors for atoms. For the ith vertex and its associated atom in Tadj, its feature vector is stored by the ith vector of Tvertex. This vertex feature vector consists of two components. The first component describes the vertex’s atom type as being one of the following: {carbon, nitrogen, oxygen, phosphorus, sulfur, other}. The second component indicates the current vertex’s neighborhood atom counts in one of the following scales: {1, 2, 3, 4, 5 or more}. Before feeding this information into the CFM-ID training algorithm, the un-duplicated part of Tadj and all of Tvertex are flattened into a 1-dimensional vector and then concatenated. Note that, since G is an undirected graph, Tvertex needs to include Element(i,j) where i < j.
Please note that our proposed method requires more computing resources than the old CFM-ID. We have also developed a set of sampling methods to ensure that the training model is computationally feasible. Details of these sampling methods can be found in supplementary materials.
Extending Fragmentation Rules
Similar to earlier versions of CFM-ID, for a given chemical structure, CFM-ID 4.0 will first predict MS/MS spectra through its MS rule-based (MSRB) fragmentation module. If that rule-base does not produce a prediction, CFM-ID then predicts spectra via its machine-learned module (MSML). For instance, CFM-ID 3.0 used more than a dozen hand-made MSRBs for predicting lipid MS spectra. This not only improved the quality of the predicted MS/MS spectra but also sped up the prediction process by a factor of 100 or more. Lipids have a modular chemical structure consisting of different combinations of head groups and tail groups (acyl or fatty acid) that fragment in predictable ways in most mass spectrometers. These fragmentation reactions can be described by SMIRKS36 generic reaction strings. Many other chemicals commonly encountered in metabolomic studies also exhibit modular structures with predictable modular fragmentation tendencies. These include acylcarnitines, acylcholines, flavonols, flavones, flavanones, and flavonoid glycosides. CFM-ID 4.0 now includes hand-made MSRB fragmentation code for these modular compounds. Just as with the lipid rules in CFM-ID 3.0, the new version of CFM-ID uses ClassyFire37 to identify the specific chemical classes that are suitable for MSRB fragmentation. Detailed manual analysis of experimental MS spectra collected on QTOF instruments from various vendors enabled us to define an extended set of SMIRKS-based fragmentation rules covering 11 chemical classes and 3 adduct types (Supplemental Table S1). This led to the generation of a total of 313 rules. Each rule uses SMIRKS strings to describe a chemical reaction that fragments the precursor molecule to generate a specific fragment. The rules also incorporate average scaled relative intensities from the experimental spectra. The structure and the mass-to-charge ratio of each predicted fragment can be easily computed based on the encoded pattern. For each compound class covered in the rule-based module, an ESI-MS/MS spectrum (QTOF) can be simulated by CFM-ID 4.0 at collision energies of 10, 20, and 40 eV.
Training and Testing
To test and train our new CFM-ID model, we performed two sets of tasks. The first evaluated CFM-ID’s ability to generate spectra (the MS2C task). The second focused on CFM-ID’s ability to identify compounds from MS spectra (the C2MS task) using the CASMI 2016 challenge set21. We used experimental ESI-MS/MS data obtained from the Scripps Research Institute’s Metlin9,38 MS database to train CFM-ID 4.0. These MS/MS spectra were all collected by a Quadrupole Time-of-Flight (QTOF) instrument in positive [M+H]+ ionization or negative [M-H]− ionization modes. All spectra had an associated chemical structure. Each training/testing sample consisted of molecules with a molecular mass ≤ 1000 Da and QTOF ESI-MS/MS spectra measured at three different collision energy levels: 10, 20, and 40 eV. Note that only the most common isotope peaks are presented in these spectra. In total, 4 data sets were used in this study which are from Metlin database: (1) Metlin Metabolites 2015 ([M+H]+) 4473[M+H]+ spectra for 1491 non-peptide metabolites. (2) Metlin Metabolites 2015 ([M-H]−) 2928 [M-H]− spectra for 976 non-peptide metabolites. (3) Metlin Metabolites 2019 ([M+H]+) 12,165 [M+H]+ spectra for 4055 non-peptide metabolites,1483 metabolites in this set are also included in the 2015 set. (4) Metlin Metabolites 2019 ([M-H]−) 6120 [M-H]− spectra for 2040 non-peptide metabolites. 913 metabolites in this set are also included in the 2015 set. Note that earlier versions of CFM-ID used only the first two data sets.
All experiments were carried out under a 10-fold cross-validation framework. To enable direct comparisons between CFM-ID 4.0 and earlier versions of CFM-ID, we purposely set the hyperparameters of CFM-ID 4.0 to be the same as earlier versions of CFM-ID. This involved setting the maximum depth of the fragmentation graphs, the maximum number of allowed ring breaks, and the model depth to two. We only trained the neural network variant of CFM-ID with a fully connected structure consisting of two hidden layers, each with 128 nodes. Our datasets included the following chemical features: (1) Break Atom Pair: Indicators for the atom type pairs formed by the Ion Root and the NL Root (Ion Root and NL Root denotes the root atoms for the ion fragment and the neutral loss fragment, respectively); (2) BrokenOrigBondType: Indicators for the type of cleaved bond, which is one of {single, double, triple, aromatic, conjugated, ionic, hydrogen bond}; (3) Gasteiger Charges: Features for Gasteiger charges for the Ion Root and the NL Root in the original unbroken molecule24; (4) Hydrogen Movement: Features for how hydrogen atoms switch sites24; and (5) Ion Root and NL Root Connectivity Matrix Features: our proposed feature presentation capped the number of atoms at 10.
For the spectral prediction (C2MS) task, a post-processing step similar to the original CFM-ID study21 was applied. For a predicted ESI-MS/MS spectrum, this post-processing first sorts all peaks by their intensity, then iteratively selects the most significant peaks by intensity. The selection process is stopped once the output spectrum meets one of the following criteria: (1) the sum of peak intensities ≥80% of the total input intensity or (2) the number of peaks equals to 30.
Results: Performance of Spectra Prediction
We evaluated the C2MS task by assessing each version of CFM-ID to predict ESI-MS/MS spectra based on an input chemical structure. For this performance evaluation, we computed the metrics using the experimental MS/MS spectrum SM and the predicted MS/MS spectrum SP. Given a pair of spectra, SM and SP, we consider a pair of peaks to match (e.g., for intersection) if their mass-to-charge-ratio difference is smaller than 0.01 Da or 10 ppm whichever occurs first. While we used Dice coefficient (This measurement was incorrectly called the “Jaccard Index” in the original CFM-ID papers) between presents of matching peaks to evaluate our models, our full list of metrics also includes Precision, Recall and a re-weighted version of Stein’s Dot Product24,39. The first three metrics, Dice, Precision, and Recall only considered the presence of peaks, while the Dot Product also includes the intensity of the peaks (mathematical details of each metric can be found in the Supplemental Material).
First, we compared the performance of CFM-ID 4.0 vs. CFM-ID 2.0 and 3.0 on the Metlin 2015 [M+H]+ and [M-H]− spectral dataset. As the rule-based extensions introduced in CFM-ID 3.026 do not cover any compounds in these two datasets, therefore CFM-ID 2.024,25 and CFM-ID 3.026 produced the same predicted outputs. Table 1 provides the 10-fold cross-validation results between CFM-ID 4.0 and earlier versions of CFM-ID using [M+H]+ and [M-H]− spectra. In contrast to the baseline model, CFM-ID 4.0 achieved better performance in both the Dice coefficient and Dot Product measurements. Considering the averaged results of all energy level, CFM-ID 4.0’s average Dice coefficients improved over CFM-ID 2.0/3.0 by 31.8% on [M+H]+ spectra and by 8% on [M-H]− spectra (2-sided t-test p-values 1E-43 and 3E-29 ). In terms of the Dot Product, the improvements over CFM-ID 2.0/3.0 were 26.7% on [M+H]+ spectra and 20.6% on [M-H]− spectra (p-values 6E-05 and 8E-12). The performance improvements in the Dice and Dot Product scores suggest that the spectra predicted by our model not only have more accurately predicted m/z values but also more accurate intensities for each peak. In terms of the overall contribution of each component (MSRB vs. topological features vs. ring cleavage) to CFM-ID 4.0’s performance, we found that the most significant contributor to CFM-ID 4.0’s improvement was from the addition of molecular topology features (~65%); followed by the addition of ring cleavage features (35%); followed the addition of rule-based features (~1%). Note that MSRB predicted less than 3% of all MS/MS spectra (see Supplemental Tables S2–5)”.Those performance improvements in the Dice score and Dot Product suggested that spectra predicted by our model not only have more accurately predicted m/z values but also more accurate intensities for each peak.
Table 1.
Spectrum prediction results for the Metlin 2015 [M+H]+ and [M-H]−.
| Dice | Dot Prod | Recall | Precision | Dice | Dot Prod | Recall | Precision | |||
|---|---|---|---|---|---|---|---|---|---|---|
| CFM-ID 2.0/3.0 [M+H]+ | Overall | 0.30 | 0.30 | 0.44 | 0.26 | CFM-ID 2.0/3.0 [M-H]− |
0.23 | 0.29 | 0.27 | 0.29 |
| 10 eV | 0.37 | 0.53 | 0.53 | 0.34 | 0.31 | 0.55 | 0.31 | 0.40 | ||
| 20 eV | 0.29 | 0.27 | 0.44 | 0.26 | 0.20 | 0.18 | 0.24 | 0.23 | ||
| 40 eV | 0.22 | 0.12 | 0.36 | 0.19 | 0.20 | 0.14 | 0.25 | 0.24 | ||
| CFM-ID 4.0 [M+H]+ | Overall | 0.37 | 0.38 | 0.37 | 0.50 | CFM-ID 4.0 [M-H]− |
0.25 | 0.35 | 0.23 | 0.48 |
| 10 eV | 0.46 | 0.60 | 0.42 | 0.71 | 0.30 | 0.58 | 0.24 | 0.70 | ||
| 20 eV | 0.35 | 0.35 | 0.37 | 0.46 | 0.26 | 0.33 | 0.24 | 0.47 | ||
| 40 eV | 0.29 | 0.20 | 0.33 | 0.32 | 0.19 | 0.13 | 0.21 | 0.29 |
In the context of in silico spectral prediction, high Recall or high Precision can be easily achieved separately -- the former can be done by simply enumerating all possible peaks, while the latter can be achieved by only predicting the peak corresponding to molecular ions. Note that the earlier CFM-ID versions have a noticeable difference between their Precision and Recall. In contrast, difference between Precision and Recall for CFM-ID 4.0 are much smaller. Furthermore, CFM-ID achieved a 90.6% improvement in terms of Precision while its Recall performance dropped slightly with a 15.8% decrease, relative to CFM-ID 2.0/3.0. On average, the Dice coefficient and the Dot Product values increased by 14.3% and 19.6% for CFM-ID 4.0. To further investigate the performance of CFM-ID 4.0, we then trained and e valuated it using larger and more chemically diverse datasets, namely Metlin Metabolites 2019 [M+H]+ and Metlin Metabolites 2019 [M-H]−. Despite having more chemical diversity in these two datasets, CFM-ID 4.0 still maintained excellent MS spectral predictive ability. We also carried out an in-depth analysis of CFM-ID 4.0’s spectral prediction performance for each chemical class, as determined by ClassyFire37. Our results suggest that CFM-ID 4.0’s spectral prediction performance works well for a wide range of chemicals, with no significant negative bias towards any specific class of chemicals (Supplemental Table S6).
To understand the predictive performance of CFM-ID in different metabolomics research fields, we evaluated CFM-ID on three data sets containing molecules commonly found in exposomics, foodomics, and human metabolomics studies. Each molecule is sampled from the Metlin 2019 dataset compounds according to the superclass distribution of compounds in the HMDB40, FooDB41, and ContaminantDB39. Table 2 presents the results of CFM-ID 4.0’s spectra prediction performance on these three domains, as well a comparison between purely machine-learned classification (MSML) versus a model that uses both expert-generated rules and a machine-learned classifier (MSML+MSRB). The rule-based module enhances the Dice score for molecules that include many modular components such as lipids and polyphenols. Our results showed that the combination of two methods outperforms the machine learning-only method in the foodomics and metabolomics datasets (which have large numbers of lipids, carnitines and polyphenols). On the other hand, the rule-based module does not improve the prediction quality for the exposomics dataset (that covers mostly man-made, synthetic compounds, which did not have any MSRB fragmentation data). Note that the rule-based module is usually 5–7 times faster than the machine-learned module. However, the machine-learned module can obviously predict spectra for a far wider range of molecules compared to the rule-based module. More details can be found in Supplemental Figures S1–S6.
Table 2.
Comparison of two versions of CFM-ID 4.0 for different analyte datasets.
| Expos-ome | Food-ome | HMDB | ||
|---|---|---|---|---|
| MSML [M+H]+ | Dice | 0.38 | 0.32 | 0.29 |
| Dot Product | 0.38 | 0.36 | 0.38 | |
| MSML& MSRB [M+H]+ | Dice | 0.38 | 0.38 | 0.36 |
| Dot Product | 0.38 | 0.37 | 0.38 | |
| MSML [M-H]− | Dice | 0.32 | 0.30 | 0.30 |
| Dot Product | 0.33 | 0.31 | 0.32 | |
| MSML& MSRB [M-H]− | Dice | 0.33 | 0.32 | 0.30 |
| Dot Product | 0.34 | 0.33 | 0.33 |
Compound ID with the CASMI 2016 Datasets
To evaluate CFM-ID 4.0’s performance on the spectral identification task (MS2C), we used the CASMI 2016 competition (category 3) dataset, which contained 208 experimentally collected Orbitrap MS/MS spectra: 127 measured in the positive [M+H]+ ion mode, and 81 in the negative [M-H]− mode. While this dataset is nearly 5 years old, it is widely used as a “gold standard” for comparing the performance of in silico MS2C and C2MS systems. This is because both historic and current performance data exist for many in silico predictors using this dataset. Since the MS spectra in the CASMI dataset were collected on an OrbiTrap mass spectrometer using higher-energy collisional dissociation (HCD), and not the collision-induced dissociation (CID) used by our QTOF training data, we used a pre-processing step to determine the corresponding QTOF collision energy level for each spectrum with a conversion equation provided by Thermo Fisher Scientific. This equation maps each Orbitrap HCD spectrum to a QTOF spectrum with 10, 20, 40 eV collision energies. Note that the experimental spectra used in this challenge are merged spectra collected in the stepped mode18, where CFM-ID is predicting its MS spectra based on individual CID levels. Note also that we retrieved candidate structures from CFM-ID 3.0’s spectral database26 with a mass tolerance of 10 ppm. Only a MSML model is used in this evaluation task, it was trained on all samples in the Metlin 2019 [M+H]+ set and the Metlin 2019 [M-H]− set for their respective adduct types,.
To evaluate each of the predictors, we used the modified Dot Product function introduced by CFM-ID 2.024. Table 3 compares the MS2C classification results between CFM-ID 2.0, CFM-ID 3.0, and CFM-ID 4.0. The CFM-ID 4.0, without CFM-ID 3.0’s metadata scoring function, managed to correctly identify 147 out of 208 compounds with in silico spectra only. That is only 2 fewer than CFM-ID 3.0even though the latter not only used metadata but also exploited experimentally collected MS/MS spectra in its prediction. CFM-ID 4.0 also exhibited a 22.5% improvement in its identification accuracy over the original CFM-ID 2.0. Furthermore, this result also surpassed the result achieved by MS-Finder42,43, and SIRIUS 416. Using the same meta-data scoring method from CFM-ID 3.0, we found that CFM-ID 4.0-metadata (which includes metadata such as each compound’s citation frequency) was able to correctly identify 162 MS/MS spectra out of 208 total input spectra, a significant improvement over that reported by all existing in silico predictors. It is also notable that even though CFM-ID (all versions) has been trained using only QTOF MS/MS data, it is able to perform very well with Orbitrap MS/MS data. This is despite the well-known differences in ion intensity and fragmentation behavior between these two instrument types. Our result suggests that if CFM-ID 4.0 were to be trained specifically on Orbitrap MS/MS data, it may perform somewhat better on the CASMI data than what was reported here.
Table 3.
Results of CASMI 2016 Category 321.
Conclusion
In this work, we have introduced a new and improved version of CFM-ID, called CFM-ID 4.0. CFM-ID 4.0 incorporates a more robust machine learning process that uses a novel tensor representation for describing the topology of chemical structures. It also uses a new approach for handling ring cleavage and new rule-based methods that enhance MS/MS spectral prediction for specific classes of chemicals where the machine-learned model does poorly. We empirical compared CFM-ID 4.0’s in silico spectra prediction performance on multiple ESI-MS/MS data sets, spanning a wide range of chemical classes, in both positive and negative ionization modes. While still imperfect, CFM-ID 4.0 outperformed the earlier versions of CFM-ID by a significant margin across all data sets. In addition, we assessed CFM-ID 4.0’s in silico MS2C compound identification ability via the CASMI 2016 data set (category 3) and found that CFM-ID 4.0 achieved significantly better identification results than all existing approaches.
Although CFM-ID 4.0 has surpassed its predecessors in terms of performance, there is still room for further improvement, both in terms of accuracy and runtime. One future direction we intend to pursue is to use deeper fragmentation graphs and more efficient ways to explore them. A deeper fragmentation graph may be beneficial when handling more complicated chemical structures, however, it will also introduce extra computational complexity. Another potential improvement is to extend CFM-ID with even better molecular modelling methods using graph neural networks. Finally, we plan to adapt CFM-ID to handle MS spectra collected using instruments other than QTOF instruments, such as Orbitrap mass spectrometers.
Availability
CFM-ID 4.0 is available as webservice at http://cfmid4.wishartlab.com/ and docker image at https://hub.docker.com/r/wishartlab/cfmid. Source code can be found at https://bitbucket.org/wishartlab/cfm-id-code/ and https://bitbucket.org/wishartlab/msrb-fragmenter for MSML and MSRB respectively. Details of CFM-ID 4.0 web server spectra library is available at Supplemental Table S7.
Supplementary Material
Acknowledgements
This research was supported by Genome Canada, Genome British Columbia and Genome Alberta (project 284MBO, 264PRO), the Canada Foundation for Innovation, the National Institutes of Health, National Institute of Environmental Health Sciences grant U2CES030170 and by the Estonian Research Council grant (PUTJD903). This research was also supported by the Natural Sciences and Engineering Research Council, the Canadian Institutes of Health Research, the Canadian Institute for Advanced Research and Alberta Machine Intelligence Institute.JChem was used for drawing and displaying chemical structures, JChem 17.21.0, ChemAxon (https://www.chemaxon.com)
References
- (1).Dunn WB; Ellis DI Metabolomics: Current Analytical Platforms and Methodologies. Trends Anal. Chem 2005, 24 (4), 285–294. [Google Scholar]
- (2).Kind T; Wohlgemuth G; Lee DY; Lu Y; Palazoglu M; Shahbaz S; Fiehn O FiehnLib: Mass Spectral and Retention Index Libraries for Metabolomics Based on Quadrupole and Time-of-Flight Gas Chromatography/Mass Spectrometry. Anal. Chem 2009, 81 (24), 10038–10048. 10.1021/ac9019522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Stein S Mass Spectral Reference Libraries: An Ever-Expanding Resource for Chemical Identification. Anal. Chem 2012, 84 (17), 7274–7282. 10.1021/ac301205z. [DOI] [PubMed] [Google Scholar]
- (4).Tautenhahn R; Cho K; Uritboonthai W; Zhu Z; Patti GJ; Siuzdak G An Accelerated Workflow for Untargeted Metabolomics Using the METLIN Database. Nat. Biotechnol 2012, 30 (9), 826–828. 10.1038/nbt.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hufsky F; Scheubert K; Böcker S Computational Mass Spectrometry for Small-Molecule Fragmentation. TrAC Trends Anal. Chem 2014, 53, 41–48. 10.1016/j.trac.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kind T; Tsugawa H; Cajka T; Ma Y; Lai Z; Mehta SS; Wohlgemuth G; Barupal DK; Showalter MR; Arita M; Fiehn O Identification of Small Molecules Using Accurate Mass MS/MS Search. Mass Spectrom. Rev 2018, 37 (4), 513–532. 10.1002/mas.21535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dunn WB; Erban A; Weber RJM; Creek DJ; Brown M; Breitling R; Hankemeier T; Goodacre R; Neumann S; Kopka J; Viant MR Mass Appeal: Metabolite Identification in Mass Spectrometry-Focused Untargeted Metabolomics. Metabolomics 2013, 9 (S1), 44–66. 10.1007/s11306-012-0434-4. [DOI] [Google Scholar]
- (8).Uppal K; Walker DI; Liu K; Li S; Go Y-M; Jones DP Computational Metabolomics: A Framework for the Million Metabolome. Chem. Res. Toxicol 2016, 29 (12), 1956–1975. 10.1021/acs.chemrestox.6b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Guijas C; Montenegro-Burke JR; Domingo-Almenara X; Palermo A; Warth B; Hermann G; Koellensperger G; Huan T; Uritboonthai W; Aisporna AE; Wolan DW; Spilker ME; Benton HP; Siuzdak G METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem 2018, 90 (5), 3156–3164. 10.1021/acs.analchem.7b04424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Stephen S NIST/EPA/NIH Mass Spectral Library with Search Program Data Version: NIST V20; Mass Spectrometry Data Center National Institute of Standards and Technology, 2020. [Google Scholar]
- (11).MzCloud; HighChem LLC, 2021. [Google Scholar]
- (12).Tsugawa H Advances in Computational Metabolomics and Databases Deepen the Understanding of Metabolisms. Curr. Opin. Biotechnol 2018, 54, 10–17. 10.1016/j.copbio.2018.01.008. [DOI] [PubMed] [Google Scholar]
- (13).Heinonen M; Shen H; Zamboni N; Rousu J Metabolite Identification and Molecular Fingerprint Prediction through Machine Learning. Bioinformatics 2012, 28 (18), 2333–2341. 10.1093/bioinformatics/bts437. [DOI] [PubMed] [Google Scholar]
- (14).Shen H; Dührkop K; Böcker S; Rousu J Metabolite Identification through Multiple Kernel Learning on Fragmentation Trees. Bioinformatics 2014, 30 (12), i157–i164. 10.1093/bioinformatics/btu275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Brouard C; Shen H; Dührkop K; d’Alché-Buc F; Böcker S; Rousu J Fast Metabolite Identification with Input Output Kernel Regression. Bioinformatics 2016, 32 (12), i28–i36. 10.1093/bioinformatics/btw246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dührkop K; Fleischauer M; Ludwig M; Aksenov AA; Melnik AV; Meusel M; Dorrestein PC; Rousu J; Böcker S SIRIUS 4: A Rapid Tool for Turning Tandem Mass Spectra into Metabolite Structure Information. Nat. Methods 2019, 16 (4), 299–302. 10.1038/s41592-019-0344-8. [DOI] [PubMed] [Google Scholar]
- (17).Shen H; Zamboni N; Heinonen M; Rousu J Metabolite Identification through Machine Learning— Tackling CASMI Challenge Using FingerID. Metabolites 2013, 3 (2), 484–505. https://doi.org/10/gchm59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dührkop K; Shen H; Meusel M; Rousu J; Böcker S Searching Molecular Structure Databases with Tandem Mass Spectra Using CSI:FingerID. Proc. Natl. Acad. Sci 2015, 112 (41), 12580–12585. 10.1073/pnas.1509788112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ludwig M; Dührkop K; Böcker S Bayesian Networks for Mass Spectrometric Metabolite Identification via Molecular Fingerprints. Bioinformatics 2018, 34 (13), i333–i340. 10.1093/bioinformatics/bty245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Brouard C; Szafranski M Input Output Kernel Regression: Supervised and Semi-Supervised Structured Output Prediction with Operator-Valued Kernels. 48.
- (21).Schymanski EL; Ruttkies C; Krauss M; Brouard C; Kind T; Dührkop K; Allen F; Vaniya A; Verdegem D; Böcker S; Rousu J; Shen H; Tsugawa H; Sajed T; Fiehn O; Ghesquière B; Neumann S Critical Assessment of Small Molecule Identification 2016: Automated Methods. J. Cheminformatics 2017, 9 (1), 22. https://doi.org/10/f9z6rq. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ruttkies C; Schymanski EL; Wolf S; Hollender J; Neumann S MetFrag Relaunched: Incorporating Strategies beyond in Silico Fragmentation. J. Cheminformatics 2016, 8 (1). 10.1186/s13321-016-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kind T; Liu K-H; Lee DY; DeFelice B; Meissen JK; Fiehn O LipidBlast in Silico Tandem Mass Spectrometry Database for Lipid Identification. Nat. Methods 2013, 10 (8), 755–758. 10.1038/nmeth.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Allen F; Greiner R; Wishart D Competitive Fragmentation Modeling of ESI-MS/MS Spectra for Putative Metabolite Identification. Metabolomics 2015, 11 (1), 98–110. 10.1007/s11306-014-0676-4. [DOI] [Google Scholar]
- (25).Allen F; Pon A; Greiner R; Wishart D Computational Prediction of Electron Ionization Mass Spectra to Assist in GC/MS Compound Identification. Anal. Chem 2016, 88 (15), 7689–7697. h 10.1021/acs.analchem.6b01622. [DOI] [PubMed] [Google Scholar]
- (26).Djoumbou-Feunang Y; Pon A; Karu N; Zheng J; Li C; Arndt D; Gautam M; Allen F; Wishart DS CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites 2019, 9 (4), 72. https://doi.org/10/gf7c7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wei JN; Belanger D; Adams RP; Sculley D Rapid Prediction of Electron-Ionization Mass Spectrometry Using Neural Networks. ACS Cent. Sci 2019, 5 (4), 700–708. 10.1021/acscentsci.9b00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gasteiger J; Hanebeck W; Schulz KP Prediction of Mass Spectra from Structural Information. J. Chem. Inf. Model 1992, 32 (4), 264–271. 10.1021/ci00008a001. [DOI] [Google Scholar]
- (29).Wang Y; Kora G; Bowen BP; Pan C MIDAS: A Database-Searching Algorithm for Metabolite Identification in Metabolomics. Anal. Chem 2014, 86 (19), 9496–9503. 10.1021/ac5014783. [DOI] [PubMed] [Google Scholar]
- (30).Ridder L; van der Hooft JJJ; Verhoeven S Automatic Compound Annotation from Mass Spectrometry Data Using MAGMa. Mass Spectrom. 2014, 3 (Special_Issue_2), S0033–S0033. https://doi.org/10/gf68sm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kangas LJ; Metz TO; Isaac G; Schrom BT; Ginovska-Pangovska B; Wang L; Tan L; Lewis RR; Miller JH In Silico Identification Software (ISIS): A Machine Learning Approach to Tandem Mass Spectral Identification of Lipids. Bioinformatics 2012, 28 (13), 1705–1713. https://doi.org/10/f332qz. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tanaka W; Arita M Physicochemical Prediction of Metabolite Fragmentation in Tandem Mass Spectrometry. Mass Spectrom. 2018, 7 (1), A0066–A0066. 10.5702/massspectrometry.A0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Duvenaud D; Maclaurin D; Aguilera-Iparraguirre J; Gómez-Bombarelli R; Hirzel T; Aspuru-Guzik A; Adams RP Convolutional Networks on Graphs for Learning Molecular Fingerprints. ArXiv150909292 Cs Stat 2015. [Google Scholar]
- (34).Kearnes S; McCloskey K; Berndl M; Pande V; Riley P Molecular Graph Convolutions: Moving beyond Fingerprints. J. Comput. Aided Mol. Des 2016, 30 (8), 595–608. 10.1007/s10822-016-9938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wu Z; Ramsundar B; Feinberg EN; Gomes J; Geniesse C; Pappu AS; Leswing K; Pande V MoleculeNet: A Benchmark for Molecular Machine Learning. ArXiv170300564 Phys. Stat 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).SMIRKS (2007) A Reaction Transform Language.
- (37).Djoumbou Feunang Y; Eisner R; Knox C; Chepelev L; Hastings J; Owen G; Fahy E; Steinbeck C; Subramanian S; Bolton E; Greiner R; Wishart DS ClassyFire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J. Cheminformatics 2016, 8 (1). 10.1186/s13321-016-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Smith CA; O’maille G; Want EJ; Qin C; Trauger SA; Brandon TR; Custodio DE; Abagyan R; Siuzdak G METLIN : A Metabolite Mass Spectral Database. Ther. Drug Monit 2005, 27 (6), 747–751. [DOI] [PubMed] [Google Scholar]
- (39).Stein SE; Scott DR Optimization and Testing of Mass Spectral Library Search Algorithms for Compound Identification. J. Am. Soc. Mass Spectrom 1994, 5 (9), 859–866. 10.1016/1044-0305(94)87009-8. [DOI] [PubMed] [Google Scholar]
- (40).Wishart DS; Feunang YD; Marcu A; Guo AC; Liang K; Vázquez-Fresno R; Sajed T; Johnson D; Li C; Karu N; Sayeeda Z; Lo E; Assempour N; Berjanskii M; Singhal S; Arndt D; Liang Y; Badran H; Grant J; Serra-Cayuela A; Liu Y; Mandal R; Neveu V; Pon A; Knox C; Wilson M; Manach C; Scalbert A HMDB 4.0: The Human Metabolome Database for 2018. Nucleic Acids Res. 2018, 46 (D1), D608–D617. 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).FooDB www.foodb.ca (accessed 2020 –07 –01).
- (42).Tsugawa H; Kind T; Nakabayashi R; Yukihira D; Tanaka W; Cajka T; Saito K; Fiehn O; Arita M Hydrogen Rearrangement Rules: Computational MS/MS Fragmentation and Structure Elucidation Using MS-FINDER Software. Anal. Chem 2016, 88 (16), 7946–7958. https://doi.org/10/f8xqsb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lai Z; Tsugawa H; Wohlgemuth G; Mehta S; Mueller M; Zheng Y; Ogiwara A; Meissen J; Showalter M; Takeuchi K; Kind T; Beal P; Arita M; Fiehn O Identifying Metabolites by Integrating Metabolome Databases with Mass Spectrometry Cheminformatics. Nat. Methods 2018, 15 (1), 53–56. https://doi.org/10/ggjdgv. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.