

Abstract
This work evaluated the inhibition effect of low molecular weight alcohol (ethanol) on naproxen (NAP) degradation by conventional and catalytic ozonation. The reaction system considered the ethanol as complementary organic matter in water. The conventional ozonation and in the presence of nickel oxide (O3–NiO) achieved 98% NAP degradation during the first 15 min of reaction despite the presence of ethanol. However, NAP degradation presented a delaying effect during the first minutes of treatment with this alcohol. The latter phenomenon indicates that ethanol concentration played a meaningful role in ozonation effectiveness in comparison with the presence of NiO catalyst. The presence of NiO did not generate differences in the byproducts in comparison with conventional ozonation. The intermediates were detected using the Electrospray Ionization Mass Spectrometry technique and have only one aromatic ring in their chemical structure. In samples without ethanol, these byproducts appeared only in the first 5 min of reaction. The TOC study demonstrated the increment of 25% in the mineralization degree with the presence of NiO due to the formation of ·OH species.
Ethanol concentration as organic matter delayed the NAP elimination and O3–NiO increased the mineralization degree and oxidation state of byproducts.
1. Introduction
A wide variety of anti-inflammatory drugs are consumed in human and veterinary medicine around the world. These drugs are metabolized incompletely, being eliminated from the bodies of human patients in fecal and urine excretions which end up in the sewer system.1 The irrational consumption of such pharmaceutical products has contributed to the environmental problems because these compounds can be toxic and recalcitrant to conventional wastewater treatments. Furthermore, pharmaceutical products may be able to change the reproduction cycles in aquatic species.2 The presence of these kinds of compounds is a potential danger to water bodies.3
Hospitals are a continuous and uncontrolled source of medicaments (anesthetics, analgesic, anti-inflammatories, etc.) in sewer effluents. According to Carraro et al.1 hospital effluents depend on the size of the hospital, the average number of patients, and the number of medical specialties, among others. Usually, disinfectants, detergents, biological liquids, toxic or hazardous chemicals, pharmaceutical residues, fixer solutions from photographic film and/or infections agents can be also found in these effluents. Hospital wastewater is discharged into the municipal sewer network. Due to the aforementioned characteristics, the hospital wastewater should not be treated as domestic discharge. Instead, these residuals must be considered as industrial wastewater.
The inappropriate disposal of pharmaceutical industry wastewater also contributes to this environmental problem. Because their wastewaters contain drugs with different chemical nature.4 Hence, the possible treatment of such kind of wastewaters is more complicated due to the diversity of compounds. The inadequate treatment of effluents containing several medicaments may have relevant health consequences for plants, animals, and the human beings.5
Gibson et al.6 reported that agricultural soil irrigated with wastewater in the Tula Valley (Mexico) introduced various pollutants into the terrestrial environment (including pharmaceuticals and personal care products). The bioremediation technologies applied to eliminate pharmaceutical compounds have not been successful.7,8
On the other hand, the advanced oxidation processes produced reactive species which attack different pollutants present in water.9 The common characteristic of these kinds of processes is the production of hydroxyl radicals (·OH, 2.80 eV) which have low reaction selectivity with organic compounds. The combination of the catalyst with ozone is one of the most successful methods to produce ·OH10,11 because to reduce the drawbacks of conventional ozonation for instance, the formation of toxic bromate12 and the accumulation of intermediates of low reaction constant with ozone. The presence of the catalyst can accelerate the decomposition and mineralization of pollutants in water.13–15
Several catalysts have been evaluated in catalytic ozonation (TiO2, SiO2, CeO2, etc.). Among them, NiO has shown a superior mineralization capacity over TiO2 in the elimination of the herbicide 2,4-D (2,4-dichlorophenoxyacetic acid). In this case, NiO improved the TOC removal in around 30% in comparison to conventional ozonation (O3-conv) and other catalysts (TiO2, SiO2, Al2O3).16 The degradation of benzoic and phthalic acids in aqueous solution by ozonation with NiO confirmed the activity catalyst yielding the 98% of TOC removal (52% more than O3-conv). In these works, the authors attributed the effect of NiO at three mechanisms: (1) ·OH generation, (2) direct reaction with ozone and (3) complex formation in the surface of NiO.17 The published results demonstrated that NiO is an effective catalyst for the ozonation process.
Catalytic ozonation has been evaluated as a promising technology that may degrade pharmaceutical products.18 Nowadays, the number of studies discussing the pharmaceuticals degradation is growing. However, only a few of them have studied the effect of the water matrix. The composition of the water matrix is an important parameter that must be considered in the contaminants elimination. For example, in hospitals and pharmaceutical industry effluents, ethanol is one of the components in wastewater. Chen et al.19 characterized a water effluent coming out from the main residual stream (a mixture of solvents and cleaning products) of the Pharmacy Company Manufacturing Co., a bigger amount of ethanol was detected in such residuals. To the authors' knowledge, there is a lack of information about the ethanol effect on the elimination of diverse pharmaceutical compounds.
Among pharmaceutical compounds, anti-inflammatory drugs are detected in the effluents of the municipal wastewater treatment plants.7 NAP is a non-steroidal anti-inflammatory drug. This medicament is usually prescribed in the treatment of rheumatoid arthritis and inflammation of diverse articulations and muscles. Moreover, this anti-inflammatory can be purchased without a prescription. This is the main reason to justify its common presence in municipal wastewaters.
The degradation of NAP is ineffective by the biological treatments.8 Researches on wastewater treatments such as photolysis,20 the photocatalytic process21,22 and catalytic ozonation,23 some of these works did not consider the effect of the water matrix.
The aim of this work is to evaluate O3–NiO in NAP degradation when it is dissolved in ethanol/water mixture (0 : 100, 10 : 90, 30 : 70 and 50 : 50 v/v). Besides, the influence of NiO in the ozonation effectiveness. The study also analyzed the inhibitor effect of ethanol over the NAP decomposition efficiency by O3–NiO. Details reaction pathways towards catalytic ozonation of NAP elimination has been further proposed and discussed.
2. Experimental conditions
2.1. Materials and reagents
All the reagents were analytic grade. The catalyst was commercial nickel(ii) oxide nanoparticles with diameters of less than 50 nm (Sigma-Aldrich, 99%). The NAP solution was 20 mg L−1 (Sigma-Aldrich, 99.8%) in 0 : 100, 10 : 90, 30 : 70 and 50 : 50 v/v ethanol/distillate water mixture (Chemical Meyer, 99.5%). The preparation of the model solutions included a vigorously stirring process applied during 5 h for the ethanol/water mixtures and 48 h for the solutions without ethanol. The experimental temperature was 22.0 ± 2.0 °C. The initial pH of the model solution was 5.0 ± 0.5, and it was not controlled during the ozonation.
2.2. Ozonation procedure
The experiments were carried out in a semi-batch type reactor (0.5 L). The ceramic diffuser was installed at the bottom of the selected Pyrex glass reactor. An ozone–oxygen mixture bubbled through a ceramic porous filter induced the agitation in the reactor. The HTU500 G ozone generator (corona discharge type) from AZCO Industries Limited – Canada produced the ozone. An ultra-dried oxygen stream was fed into the ozone generator. This ozone/oxygen stream entered in the semi-batch reactor with the flow of 0.5 L min−1. The ozone concentration was 5.5 ± 0.5 mg L−1. The BMT 946BT ozone analyzer measured the ozone concentration in the gaseous phase (BMT Messtechnik, Berlin). The NiO concentration was constant in all the catalytic ozonation experiments (100 mg L−1). In the slurry mode, the NiO was previously dried for 1 h at 100 °C.
2.3. Adsorption studies
The adsorption experiments considered the mechanic agitation of the sample during 60 min with 100 mg L−1 NiO for all the ethanol/water mixtures (0 : 100, 10 : 90, 30 : 70 and 50 : 50 v/v). The pH and temperature were kept constant along the adsorption study.
2.4. Analytical procedure for NAP degradation and byproducts identification
The PerkinElmer Flexar device (with DAD detector) was used by the HPLC analysis. The HPLC conditions for the NAP analysis were: (a) Platinum C18 column (250 × 4.6 mm, 5 μm), (b) the mobile phase was acetonitrile/water 50 : 50 adjusted to pH 2.5 with H3PO4, (c) UV-lamp wavelength of 240 nm and (d) the flow was 0.3 mL min−1.
The analysis conditions for byproducts (organic acids) were: (a) prevail organic acid column (150 mm × 4.6 mm, 5 μm), (b) the mobile phase was KH2PO4 buffer at 25 mM adjusted to pH 2.3 with H3PO4, (c) UV-lamp wavelength of 210 nm, and (d) flow of 1 mL min−1. The samples were previously filtered with 0.2 μm acrodisc syringe filter. This study reports only the mean of triplicate concentration for each sample.
The Electrospray Ionization Mass Spectrometry (ESI-MS) technique was used to identify the NAP byproducts from O3–NiO and O3-conv treatments with and without ethanol. The spectra were acquired in the negative ion scan mode using a Bruker MICROTOF-Q II 10392. The analysis was carried out by direct injection in the ESI-TOF-MS system. The studied m/z range was 50 to 3000 (n = 2). The analysis conditions were: (a) 8 μL min−1 of constant volumetric flow rate which was achieved with 100 μL of sample using a Cole Parmer syringe pump (74900-00-05), (b) the capillary temperature was 180 °C, (c) the ionization voltage was 2700 V and (d) the nitrogen spray gas flow rate was 4 L min−1 (0.4 bar). The analysis carried out for 1 min with scan and interscan times of 10 s and 0.1 s, respectively. The m/z ions (detected in the tandem analysis of ESI-MS) were evaluated by the ESI-MS-MS technique with the same analysis conditions with appropriate set mass. The fragments were analyzed by a Bruker Compass Data Analysis 4.0 (Bruker Daltonics). The widely accepted accuracy threshold for confirmation of elemental compositions was established at 5 ppm.
2.5. Chemical oxygen demand (COD) and total organic carbon (TOC)
The COD analysis was made only in the samples without ethanol with 5 and 60 min of ozonation time by chemical digestion according to method 410.4 (USEPA). The standard COD analysis kit applies the ready-to-use vials. The low range Hanna (HI94754A) reagent vials were used to digest the samples. The samples were filtered with a 0.2 μm pore filter previous digestion for 2 h at 150 °C. After digestion, the samples were measured in the equipment UV-Vis spectroscopy PerkinElmer Lambda 25 at 600 nm.
On the other hand, the TOC analysis was determined by direct injection in the Torch IQOQ equipment (Teledyne Tekmar). The solutions were previously filtered by 0.2 μm membrane. The TOC study only evaluated the samples without ethanol. These experiments used tert-butanol (TBA) as ·OH scavenger with a 200 mg L−1. The TOC technique measured the amount of organic carbon in the sample. In consequence, the TBA presence modifies the measured TOC. A reference experiment with TBA only defined the offsetting of additional organic matter in the system. Furthermore, this method determines the effect of ·OH on the ozonation efficiency (measured as mineralization), due to the scavenging of all available formed ·OH.
2.6. Analysis of NiO by XPS
The X-ray photoelectron spectroscopy (XPS) system (SPECS model) determined the photoelectron core-level spectra of ozonation samples. The XPS system has the Al Kα X-ray monochromatic source (1487 eV). The constant reference pressure was 6 × 10−10 mTorr. A double-sided carbon tape supported the powder samples. The 400 μm spot size (50 eV analyzer pass energy) generated the high-resolution scans.
The internal reference for charge correction was C 1s (284.6 eV). The fit parameters for the raw data were similar to the ones proposed by Payne et al., 2009. All the fitting procedures considered the application of Gaussian–Lorentzian (70–30%) curve shapes and Shirley-type background. Considering that NiO was the main analyzed component, the parameters suggested in Biesinger et al. (2009) were selected. The CasaXPS 2.3.16 software package processes the raw data. Furthermore, NiO commercial were characterized by other techniques, Fig. S1 and S2.†
2.7. Kinetic analysis
The kinetics study of NAP ozonation is complex because there are several oxidant species that cannot be measured on line. Then, any feasible mathematical model of such a reaction must consider a large number of intermediaries. In this study, a different kinetics analysis served to compare the catalyst and the ethanol concentration effect. It is usual to consider the pseudo-monomolecular reaction to characterize the kinetics of oxidative reactions if the radical's species are not available. The NAP ozonation is characterized by the first order kinetic reaction with the reaction rate constant kNAP.
The proposed model considers the formation and decomposition of byproducts. The mathematical form used to characterize this dynamic is:
 |
1 |
In eqn (1), the parameter kaj is the reaction rate constant (RRC) for the accumulation of the byproduct cj generated by the NAP elimination [s−1]. The parameter kdj is the RRC that characterizes the byproduct decomposition [s−1]. The model can be modified if the final compounds are studied by choosing the value of the constant kdj as zero. The term δj represents the effect of the reactions that may contribute to the byproducts dynamics (i.e. the OH radical's reaction with cj and some others). A more complex representation of the byproducts dynamics is obtained considering the interaction between the intermediary and final accumulated products (oxalic acid for example). The differential equation that represents these dynamics is the following:
 |
2 |
where kaj,k is the pseudo-monomolecular RRC characterizing the accumulation process of the final compounds k, c̄k is the accumulated byproduct concentration (no evidence of its decomposition is gotten during the ozonation period) produced by the decomposition of cj and kdj,k is the decomposition RRC of this byproduct. In this case, the term j characterizes all the reactions that affect the c̄k dynamics, such as the ones considered for eqn (2). The first-order reaction dynamics in (1, 2) have linear parameters.
The parametric identification solution used in this study (estimate the values of kNAP, kaj, kdj, kaj,k and kdj,k) uses a robust exact differentiator based on the super-twisting algorithm or STA for short:24
 |
3 |
where ĉα is the estimation for the concentration of cα. Notice here that cα may represent each of the compounds considered in the model of the catalytic ozonation (NAP and byproducts), ĉβ is an auxiliary variable needed to execute the differentiator, dα is the estimation of the time derivative of cα which is used further to calculate the uncertain RRCs. The constants ρα and ρβ are positive and should be selected according to the theoretical result presented by Moreno and Osorio.25 For instance, in the case of the model proposed in (4), it can be represented as:
| yk(t) = kaj,kcj(t) − kdj,kc̄k(t) + j(t) | 4 |
where 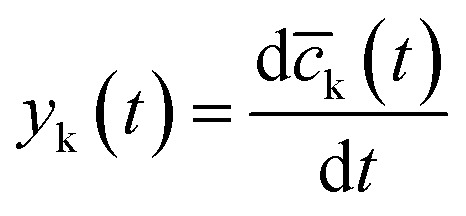 .
.
If the STA is applied considering that cα = c̄k. Therefore, after a finite time T, dα(t) = yk(t) for all t ≥ T. The estimation of kaj,k and kdj,k can be solved by the least mean squares method considering the sampling process of the experimental data.
The model in (5) is (conspiring the sample data at specific time t = ts):
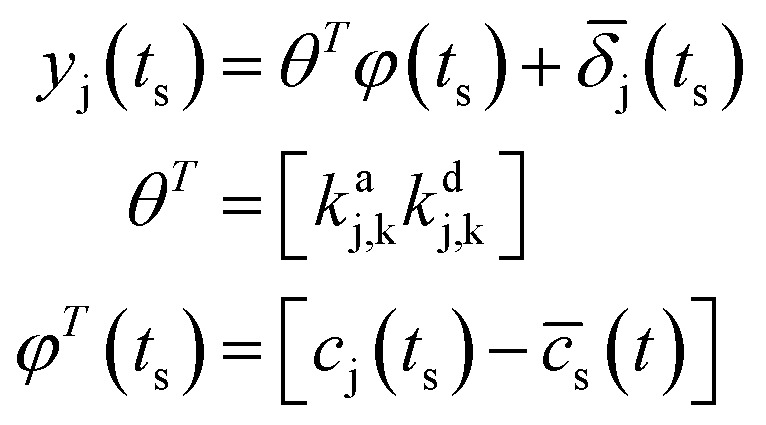 |
5 |
The solution of the parametric modelling strategy is:
 |
6 |
where S is the number of samples taken during ozonation. The similar procedure can be used to estimate the remainder RRC for cNAP and cj.
3. Results and discussion
3.1. Adsorption of NAP on NiO
Prior to the ozonation treatments, NAP adsorption experiments on NiO showed the pollutant removal from the solution by mass transfer of the contaminant to the catalyst. The results from these experiments after 60 min of NAP contact with NiO are shown in Fig. 1. At the pH 5.0 ± 0.5, the maximum adsorption of NAP after 60 min with 50% of ethanol was 7.4% while without ethanol was of 4.5%. At the pH of 5.0, the NiO surface is positively charged due to the formation of NiO–OH specie.
Fig. 1. NAP adsorption study on NiO in presence of diverse ethanol/water ratios.

On the other hand, NiO zero potential is at pH 10.8 which is bigger than the experimental pH.26 In our adsorption experiments, the charge in the NiO surface might promote an electrostatic interaction with the deprotonated molecules of NAP. The NAP deprotonation was incomplete as a consequence of the NAP pKa of 4.16.27 Therefore, charge differences (positive for the NiO and slightly negative for the NAP) may explain the low adsorption obtained in this study. On the other hand, Pacheco and Martínez28 reported that the solubility of NAP is governed by the solvation of the pharmaceutical with 30% of ethanol in the solution. Based on these results, NAP removal by adsorption has not a significant difference regarding the ethanol/water ratio. It was found that the adsorption contribution in NAP removal is less than 7.5% of the NAP concentration.
3.2. Ozonation of NAP in water with and without NiO
This section reports the influence of different ethanol/water ratios on the NAP elimination with or without NiO (Fig. 2). Both, O3-conv and O3–NiO removed 98% of initial NAP concentration after 15 min of treatment independently of ethanol/water ratios (Fig. 2). The latter results demonstrated the effectiveness of O3-conv to removed NAP.
Fig. 2. NAP concentration profiles with 0, 10, 30 and 50% v of ethanol. (a) Conventional and (b) catalytic ozonation with NiO.

The reported NAP and other drugs concentrations are underneath other components concentrations in wastewater.29,30 These uneven distributions (pharmaceutical/organic compounds in the wastewater) difficult pharmaceutical compounds degradation. In the case of drugs, they are dissolved in complex mixtures of organic compounds. Pharmaceutical products from hospitals, manufacturing industry, among others release pharmaceuticals into the water. These effluents feature is the ethanol content because this organic compound is widely used as a raw material and disinfectant agent.31 Therefore, we use ethanol as an artificial organic matter. High proportions of ethanol in the solution allows studying the effect of organic matter on the degradation of recalcitrant molecules at trace levels. So far, few studies have analyzed the effect of interfering compounds on the ozonation kinetics and on the target compound decomposition efficiency. Here, the organic matter influence was evaluated with the mixtures ethanol/water (0 : 100, 10 : 90, 30 : 70, 50 : 50 v/v).
Fig. 2a depicts the variation of NAP concentrations as a function of the ethanol/water ratio in O3 treatment. Higher ethanol concentrations (50% v/v) delayed NAP elimination to 60% after 3 min of treatment. In the experiments without ethanol, the NAP elimination by O3 was 97.3% at the same time of reaction. According to the results shown in Fig. 2, NAP degradation was slower if the ethanol proportion was larger, independently of the catalyst presence. Despite ethanol nature as a radical scavenger32 and its high concentrations (10, 30 and 50% v), the NAP degradation was above 98% after 15 min of treatment.
The interference effect of ethanol in naphthalene elimination was reported by Aguilar et al.33 In the case of naphthalene, the presence of 30% v/v of ethanol after 60 min of O3-conv degraded 99%, however, 50% v/v of ethanol reached only 66% of degradation. On the other hand, Ghauch et al.32 studied the interference of ethanol (as a ·OH scavenger) on the NAP degradation treated by thermally active persulfate. The persulfate treatment reported 32% of NAP elimination with ethanol compared to 82% without ethanol after 60 min. Therefore, ethanol presence affects contaminants degradation efficiency as a result of ethanol scavenger activity attributed to its reaction with ·OH (k = 1.9 × 109 M−1 s−1).34
The catalytic effect of NiO in the NAP degradation appears in the Fig. 2b. Both O3-conv and O3–NiO degraded NAP with a similar degradation rate despite the ethanol concentration. The latter indicates that direct ozonation mechanism governs NAP degradation.
In summary, the presence of ethanol showed two effects:
(a) The presence of ethanol inhibited the NAP elimination, which increased if high (30 and 50%) proportions of alcohol are in the ozonation system.
(b) The presence of catalyst did not change the NAP concentration profiles in comparison with conventional ozonation. Recall that if the reaction rate of organic compound with ozone did not change under the presence of catalyst, it is feasible that ozone reacts intensively with the contaminants (direct reaction mechanism). By this reason, there was not an evident effect of catalyst on the NAP ozonation. However, the reaction of ozone and catalyst produces ·OH which promote the byproducts elimination. Diverse publications demonstrated the effect of catalyst in combination with ozone on the intermediate.17,35,36
3.3. Identification of byproducts obtained during NAP ozonation
In all the degradation experiments (Fig. 2), O3–NiO decomposes the initial contaminant equally faster than O3-conv. This may indicate that radicals (including ·OH which are provided by O3–NiO) have limited contribution efficiency of the model compound elimination. However, the radicals might be involved in NAP byproducts degradation.
3.3.1. Byproduct identified by ESI-MS-MS
All the NAP treated samples were evaluated by ESI-MS-MS. The analysis strategy considered samples with and without ethanol obtained at 5 and 60 min of O3-conv (the first time was selected for the fastest NAP elimination without ethanol and the later time corresponding to the end of the reaction). In all samples, the NAP ion (229.09 m/z) was not detected. However, ten ion signals appeared in ESI-MS analysis in negative mode.
The absence of the NAP ion and the detection of some other ions demonstrated that NAP molecule has been transformed into its byproducts without reaching complete mineralization in all treatments. In the absence of ethanol, the ESI-MS technique identified the signals corresponding to four specific ions that appeared in both treatments with and without the catalyst: 133.06, 149.06, 177.06, and 237.11 m/z. Furthermore, the ion with 221.04 m/z was only detected in O3-conv. In the case of the O3–NiO, it was the 217.08 m/z. All these ions corresponded to the byproducts formed along the first 5 min of NAP degradation.
The ESI-MS technique did not detect any byproduct in the samples taken at 60 min without ethanol. If ethanol was in the reaction, the analysis of ESI-MS after 60 min of treatment detected similar peaks observed in samples without alcohol at 5 min (133.06, 149.05, 159.05, 177.05, 193.05, 205.09, 217.08, 221.04, 237.11 and 249.11 m/z). The latter indicates that the same reaction pathway might regulate the O3-conv and O3–NiO systems.
Table 1 shows the ions detected by the ESI-MS-MS during the NAP degradation (Fig. S3 and S4†). Compound with 217.08 m/z is attributed to 1-(6-methoxynaphthalen-2-yl)ethylhydroperoxide. This compound appears as an intermediary of NAP degradation by photocatalytic21,37,38 and photolysis systems.20 The 217.08 m/z byproduct is generated by the loss of –COO− from the propionic acid substituent with a subsequent attack by O2−˙.21,37 The compounds 237.11, 221.11, 205.09, 177.06 and 149.06 m/z suggested molecules with a single aromatic ring. The presence of these ions demonstrated the oxidation capacity of O3-based treatments for breaking down one of the aromatic rings of NAP. These single aromatic ring structures have not been reported before by any other oxidation processes.
Ions m/z detected in ESI-MS-MS analysis (negative mode) in samples of NAP degradationa.
| Compound | Structure | Formula | [M–H]− | Error (ppm) | mSigma | Abundance (%) | |
|---|---|---|---|---|---|---|---|
| 1 | 2-(3-(Hydroxymethyl)-4-(2-methoxyethyl)phenyl)propanoic acid |
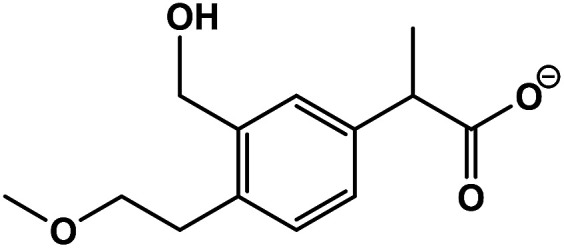
|
C13H18O4 | 237.1125 | 3.1 | 2.9 | 3.6 |
| 2 | 2-(4-(2-Mmethoxyethyl)-3-methylphenyl)propanoic acid |
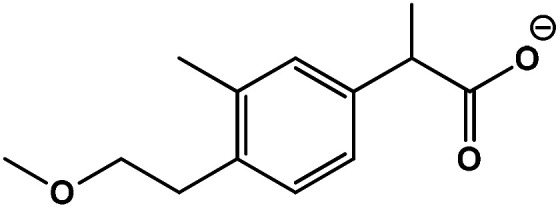
|
C13H18O4 | 221.1153 | 15.9 | 1.2 | 2.5 |
| 3 | 1-(6-Methoxynaphthalene-2-yl)ethylhydroperoxide |
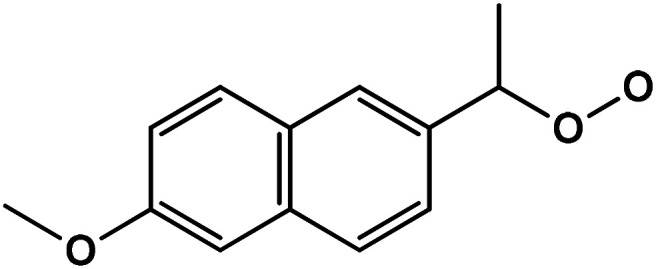
|
C13H14O3 | 217.0852 | 8.2 | 9.4 | 1.6 |
| 4 | 2-(4-(2-Hydroxyvinyl)phenyl)acetic acid |
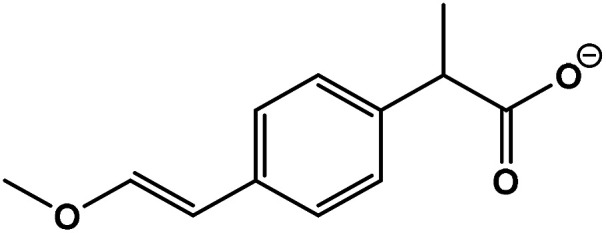
|
C12H14O3 | 205.0833 | −18.0 | 9.8 | 2.0 |
| 5 | 2-(4-(2-Hydroxyvinyl)phenyl)acetic acid |
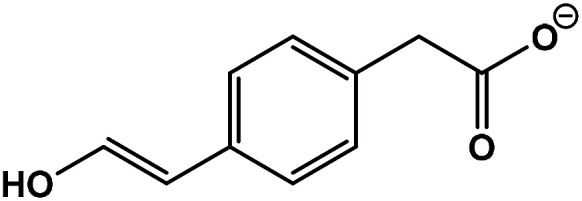
|
C10H10O3 | 177.0554 | 1.7 | 12.4 | 3.2 |
| 6 | 2-(p-Tolyl)acetic acid |
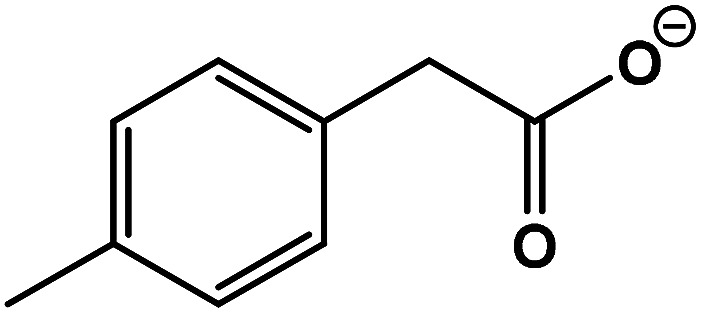
|
C9H10O2 | 149.0592 | −10.8 | 8.5 | 1.8 |
Error [ppm]: absolute value of the deviation between measured mass and theoretical mass of the selected peak in [ppm]. mSigma: combined value for the standard deviation of the masses and intensities for all peaks, given in [milliSigma].
3.3.2. Final product identified by HPLC
In both O3-conv and O3–NiO, the non-identified compound (with a retention time (RT) of 12.9 min HPLC) showed the effect of ethanol (Fig. 3a). In the presence of 50% v/v of ethanol, the higher intensity of the non-identified compound appeared around 10 min. However, in the ethanol concentration of 30% v/v, the time for the higher intensity value changed to 5 min. In absence of ethanol, the greater accumulation of this compound occurred after 3 min of reaction. It was concluded that larger ethanol concentrations increase the time when the maximum byproduct accumulation happens. The byproduct elimination is also influenced by the ethanol concentration. In reactions with higher ethanol concentrations (30 and 50% v/v), complete elimination of this byproduct was not observed even after 60 min of treatment. The samples collected at 60 min of O3-conv and O3–NiO treatments achieved around 50, 70 and 95% elimination for this byproduct with 50, 30 and 10% v/v of ethanol, respectively. When the solution did not contain the ethanol, the complete removal of this byproduct was achieved after 10 min of ozonation (six times lower than the time obtained with 10% v/v of ethanol). The observed inhibitory effect was a consequence of the reaction between ozone and ethanol. The presence of NiO did not modify the byproduct profile in comparison with O3-conv. This fact was confirmed independently of the ethanol concentration.
Fig. 3. Dynamics of (a) non-identified compound (12.9 min of RT) and (b) oxalic acid a different ethanol/water ratio by conventional and catalytic ozonation. Inset: oxalic acid concentration without ethanol in the first five min of reaction.

It is known that short chain organic acids are the main products of conventional ozonation treatments. These acids accumulated due to their low constants reaction rates with ozone.39 In this study, the oxalic acid was the main product in all the studies systems (Fig. 3b). The profiles of oxalic acid concentration had an accumulative tendency in the presence of ethanol. In the higher ethanol concentration (50% v/v), the oxalic acid duplicates its presence in comparison with treatments with 30% v/v of ethanol after 60 min. The experiments with 10% v/v of ethanol produced 20 mg L−1 of oxalic acid, which is about the half of the obtained with 30% v/v of ethanol. The proportional increments of oxalic acid with respect to ethanol concentrations indicate that ethanol degradation contributed to this organic acid accumulation. In absence of ethanol, the oxalic acid contribution was from the byproducts generated from the NAP degradation. Several studies suggest that the oxalic acid is the product of the attack to NAP′ propionic acid. Jallouli et al.4 proposed that the oxalic acid formation is due to the breakout of the 3-carbon fragment in the NAP molecule which transforms the carboxylic link into malic acid (4-atom carbon acid). In consequence, the decomposition of malic acid leads to the formation of oxalic acid.
The O3–NiO treatment produced higher concentrations of oxalic acid than the O3-conv. The most important difference between oxalic acid concentration in O3-conv and O3–NiO treatments appeared in mixtures with 30 and 50% v/v of ethanol in the first minutes of reaction (Fig. 3b). The treatments without ethanol produced the maximum oxalic acid concentrations. In the case of the O3–NiO treatments, oxalic acid was approximately 10.8 mg L−1, meanwhile, O3-conv generated 3.6 mg L−1 both in the 3 min of reaction (inset in Fig. 3b). The difference in the oxalic acid concentration was attributed to the presence of NiO. The dissimilarities of organic acids production as a function of the catalysts in advanced oxidations processes was also reported by Fuentes et al.40 in the terephthalic acid degradation by the photocatalytic ozonation with VxOy/TiO2.
Considering the byproducts identification (HPLC and ESI-MS-MS) and TOC analysis, the NAP degradation pathway was proposed (Fig. 4). The ESI-MS-MS ions analysis (Table 1) leads to propose some byproducts having only one aromatic ring. The NAP ring-cleavage corresponds to the first general stage in the identified degradation pathway. Such NAP degradation pathway has not been described by any other authors. On the other hand, the ion 218.08 m/z (detected by ESI-MS-MS analysis) has been previously reported.37,38 The similarity of molecular weights between NAP and this byproduct justified the modification of the carboxylic acid substituent. In this degradation way, no changes in the aromatic ring can be assumed. This byproduct was introduced as part of a secondary pathway in the degradation process of NAP. In both treatments (O3-conv and O3–NiO) the oxalic and formic acid detection demonstrated the breakdown of the NAP molecule. The TOC study confirmed partial mineralization in the system (Section 3.4).
Fig. 4. Reaction pathway proposed for the degradation of NAP in samples with ethanol by conventional and catalytic ozonation.

3.4. Chemical oxygen demand and total organic carbon removals
Fig. 5a shows the COD analysis of NAP samples without ethanol in the O3-conv and O3–NiO. The addition of the NiO diminished the COD at 84% in comparison with the 27% obtained with O3-conv. These results confirmed that NiO contributed to eliminating the NAP through radical species generation. The samples collected at 60 min without ethanol did not present significant COD differences with the results of samples at 5 min due to recalcitrant byproducts in the reaction.
Fig. 5. (a) COD concentration, (b) TOC analysis ([NAP + TBA]0 = 319.84 mg C L−1, [NAP + TBA]t=60 min = 259.22 mg C L−1) and (c) AOSC results (at 60 min) of the degradation of NAP in samples without ethanol by conventional and catalytic ozonation.

The TOC analysis, for NAP degradation without ethanol, showed a 12.3% of TOC removal in O3-conv at 60 min (Fig. 5b). The percentage removal increased with the use of NiO (34.1%). These results proved the effectiveness of the catalyst in the NAP and its intermediaries' elimination. The increment of the ozonation time up to 120 min forced the mineralization increment by the effect of the catalyst. The use of NiO increased the TOC removal from 23.5% by O3-conv to 48.1% at 120 min. According to Zhang et al.41 the NiO in ozonation induces ·OH production. The ·OH production obtained from NiO is justified by the interaction of the actives sites on the catalyst with ozone as Ni(OH)2–O–H which is produced by the interaction with the adsorbed water. The ·OH generated by the catalyst contributed to the decomposition of NAP, its intermediaries, and the accumulated products. This contribution depends on the ·OH concentration obtained in presence of the catalyst during the ozonation. One common manner to evaluate the ·OH contribution is adding a strong radical scavenger.32,42 The presence of ·OH in catalytic ozonation was demonstrated by the use of TBA as a scavenger.
Fig. 5b shows the TOC analysis of NAP catalytic ozonation (60 min of reaction) in samples without ethanol and in presence of TBA (200 mg L−1). The TOC removal was 19% at the end of the ozonation. This removal was 6.64% higher than the conventional treatment and 15% smaller than the TOC removed for the catalytic system. The TOC reduction in samples with TBA demonstrates the ·OH generation by NiO. This production contributed to the NAP degradation as well as the byproducts mainly.
On the other hand, Pocostales et al.43 reported a manner to know the average oxidation stated of the carbon atoms (AOSC) from the results of TOC and COD. The AOSC value satisfies the expression (7).
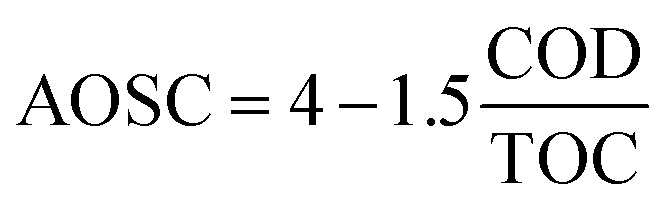 |
7 |
The lower AOSC values determine the lower oxidation state of organic matter present in the solution. The AOSC results are in the interval of +4 to −4 (examples of carbon dioxide and methane AOSC values, respectively).43Fig. 5c presents the AOSC from the samples of O3-conv and O3–NiO at 60 min of treatment. The single ozonation had AOSC values of 2.86, which are like the values of initial samples, while the reaction in presence of NiO increased this parameter to 3.68. These results demonstrated that the initial compound was removed by O3-conv. On the other hand, the refractory compounds produced by ozone were eliminated by the catalyst action (·OH or/and surface oxidation reactions).
3.5. XPS analysis
To investigate the effect of the reaction medium, 30 : 70 and 50 : 50 ethanol/water mixtures were selected because of their interference effect in the elimination of NAP's intermediaries. First, the ozonation was carried out in a water rich mixture of 30 : 70 volume ratio. After 30 min, a catalyst sample was analyzed as showed in Fig. 6a–c.
Fig. 6. High resolution XPS spectra for catalyst samples recovered from ozonation reactions of NAP. The first reaction was carried out in a 30 : 70 mixture volume ratio: (a–c) 30 and (d–f) 60 min reaction time. For the second reaction a 50 : 50 mixture was used: (g–i) 30 and (j–l) 60 min reaction time.

Spectrum (a) shows the entire Ni 2p region. The main structure, 2p3/2, was fitted according to the model proposed by Biesigner et al.44 For sake of clarity, satellite peaks are the dashed line style while the main photoelectron peaks have continuous line style. The most intense peak shows up at 553.6 eV correspond to Ni2+ in NiO. The second peak at 855.7 eV (and satellite) was necessary to fit correctly the raw data, it correlates very close to the binding energy for nickel hydroxide, Ni(OH)2.45 The intensity ratio of NiO/Ni(OH)2 is 2.0.
The O 1s region is fitted with peaks for oxygen species associated with carbon. The main peak at 529.1 eV corresponds to O2− in NiO, alongside with the known peak for oxygen in defective sites of nickel oxide, NiOdef, at 530.9 eV.44 A further peak at 531.0 eV for oxygen in Ni(OH)2 was included. The intensity ratio of NiO and NiOdef to Ni(OH)2, was intentionally fixed to the same value seen for the same species in the Ni 2p region; in this case is 2 with a good fitting (Fig. 6b). This result mutually supports each other. In the C 1s region are the carbon associated specie incorporated in the O 1s fittings (Fig. 6c).
After 60 min reaction time, Fig. 6d–f, the overall nickel to carbon intensity ratio is very close to that after 30 min. Also, the intensity ratio NiO/Ni(OH)2 is now 3.3 in the Ni 2p3/2 region, as well as in the corresponding O 1s region. In the C 1s, there is the distribution of carbon-associated species similar to the one observed after 30 min. Next, the reaction medium was enriched in ethanol up to a 50 : 50 v/v mixture ratio with water. After 30 min reaction time, Fig. 6g and h, the intensity ratio NiO/Ni(OH)2 reached the lowest value at 1.6. In the 60 min of treatment, it increased up to 2.3 Fig. 6j and k.
The distribution of carbon species was scarcely influenced by the reaction time, as seen after 60 min in Fig. 6l. Similarly, the richer ethanol medium did not influence carbon species. However, the overall nickel to carbon intensity ratio is the highest at 30; compared to a ratio of 15 for 30 min reaction time. It was observed that the added ethanol helped to keep the nickel catalyst surface from carbonaceous accumulation.
XPS results show that ethanol presence did not change the chemical environment on catalyst when the NAP elimination took place. A preliminary study demonstrated that the naphthalene ozonation with ethanol as cosolvent generated Ni-carbonaceous on the catalyst (confirmed by XPS). In consequence, the initial compound is a key factor to explain the catalyst (NiO) surface modifications during the ozonation.
3.6. Kinetic study of NAP decomposition and byproducts dynamics
The solution of the method proposed in (5) yields to characterize the decomposition of NAP. The same method was also used to describe the decomposition and accumulation of byproducts formed through out ozonation. Table 2 incorporates a summary of all RRC and correlation coefficients. The higher values of all the correlation factors prove that the simplified model proposed in this study can be used to characterize the kinetic reactions.
Reaction rate constants and statistical analysis determined for the conventional and catalytic ozonation processes under different ethanol concentrationsa.
| Ethanol (% v) | Treatment | Naproxen kNAP (s−1) | Oxalic acid kOA (s−1) | Byproduct compound (12.9 min of RT) kIC (s−1) | |
|---|---|---|---|---|---|
| Accumulation | Degradation | ||||
| 0 | O3 conv | 0.144 (0.986) | 0.025 (0.933) | 0.990 (0.914) | 0.091 (0.946) |
| O3–NiO | 0.167 (0.952) | 0.013 (0.946) | 0.976 (0.966) | 0.086 (0.937) | |
| 10 | O3 conv | 0.126 (0.979) | 1.251 (0.966) | 0.895 (0.975) | 0.060 (0.966) |
| O3–NiO | 0.143 (0.926) | 1.569 (0.915) | 0.886 (0.974) | 0.053 (0.957) | |
| 30 | O3 conv | 0.085 (0.936) | 4.414 (0.956) | 0.770 (0.984) | 0.025 (0.967) |
| O3–NiO | 0.085 (0.925) | 4.907 (0.932) | 0.813 (0.921) | 0.022 (0.896) | |
| 50 | O3 conv | 0.005 (0.956) | 7.696 (0.966) | 0.976 (0.924) | 0.015 (0.934) |
| O3–NiO | 0.010 (0.977) | 6.166 (0.923) | 0.305 (0.890) | 0.014 (0.915) | |
The correlation data are presented in parentheses.
The mathematical structure of the reactions proposed in eqn (1) and (2) was suitable to compare the relative efficiency of each ozonation system (including the effect of ethanol) considered in this study. The first result of the kinetic analysis was the decomposition dynamics of NAP. It was determined (constant kinetic reaction decreased 87.5%) by the presence of 10% v/v ethanol while the catalyst showed a less important effect on the ozonation reaction rate. In the ethanol concentration of 50% v/v, the NAP decomposition reaction rate was the smallest (considering the ethanol concentration as the comparison variable) despite the presence of the catalysts. The reduction of ethanol concentration to 30% v/v increased the constant kinetic reaction 8 times for the O3–NiO and 16 times for the O3-conv. This difference is a consequence of the ·OH scavenger nature of ethanol which affects the catalytic process, (Fig. S5 and S6†). However, the reaction rates were similar (0.0851 and 0.0845 s−1) despite the presence of the catalytic. The similarity of reaction rates between the O3-conv and O3–NiO keep if the ethanol concentration decreased to 10% and the 0%. These results confirmed that the kinetic rate constants depend on ethanol concentration.
The kinetic rate constants of the non-identified compound (RT = 12.9 min) was also determinate for all the studied systems. The increment of the non-identified compound RRC has the same tendency that NAP decomposition. The accumulation kinetics rate constant of the non-identified compound in presence of the catalyst showed a variation of 320% between the reaction rate with 50% v and without ethanol.
The higher concentration of ethanol in the ozonation showed the slower reaction rates to decompose the NAP and its intermediary. This reaction condition yields to the larger rate to accumulate oxalic acid. When the O3-conv was evaluated, the accumulation kinetic rate constants of oxalic acid was 0.025 s−1 without ethanol. In the case of the ethanol concentration of 50% v/v, kinetic rate constants increased around 308% (k = 7.696 s−1). The O3–NiO produced the increment in kinetic rate constants from 0.013 to 6.166 s−1 under the ethanol proportions of 0 and 50% v/v, respectively. The significant differences in accumulation rate constants for oxalic acid with respect to the decomposition rates for the non-identified compound demonstrated that ethanol was also transformed to oxalic acid by the ozonation, independently of the catalyst presence.
Taking into consideration the results obtained for NAP removal as well as the non-identified compound and oxalic acid production, ethanol strongly affects the ozonation efficiency. This condition is more evident at the higher accumulation of recalcitrant short chain organic acids. The removal of oxalic acid was not detected in the ozonation reactions considered in this study. This condition is rarely achieved in chemical oxidation processes based on the application of conventional ozone. The kinetic study was supported by the narrow correlation factors obtained for all the reaction rate constants calculated by the method proposed in this study. All these values were 0.90 or above.
In summary, the contribution of this researching study is the confirmation of the full degradation of a NAP by O3-conv and O3–NiO in presence of different concentrations of ethanol. This strategy offers an alternative manner of using ethanol as an artificial organic matter. The effect of ethanol was characterized as a retardant of the NAP and byproducts elimination. Nevertheless, the O3–NiO increased the mineralization degree and oxidation state of byproducts obtained from the NAP oxidation. These byproducts were analyzed by the ESI-MS-MS.
4. Conclusions
The final remarks of this study are:
• The NAP degradation by O3-conv and O3–NiO in solutions with (10, 30 and 50% v/v) and without ethanol was reached after 15 min of treatment. The increment of ethanol concentration (used to emulated organic matter) reduced the reaction rate of NAP elimination, only during the initial period (5 min) of the reaction. This fact serves as an indirect confirmation of the inhibitory effect by ethanol in the catalytic ozonation of NAP.
• The byproducts analysis (ESI-MS-MS) suggested the NAP degradation by two pathways. The first pathway considers the attacking and breaking of the aromatic ring in the NAP (byproducts not described by other authors). The second manner suggested changes in the propionic acid substituent without breaking the aromatic structure. Without ethanol, the detected byproducts appeared only in the first 5 min of reaction. All these results confirmed that ethanol in the samples interfered on the degradation of the byproducts formed after 60 min of ozonation. The byproducts remained in the solution after this reaction time.
• The NiO effect was scarcely observed in the NAP degradation profiles. However, the oxalic acid had a different behavior depending on the ozonation system (conventional or catalytic). In samples without ethanol, the NiO presence increased 25% the mineralization degree in comparison with conventional ozonation (2 h of reaction).
• The AOSC analysis confirmed the higher oxidation of organic matter with the catalyst. The TBA study confirmed the production of ·OH by the NiO in the treatment, promoting an indirect mechanism of the reaction.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The author thanks the Eng. David Dominguez for the XPS support, Denisse Fabiola González Ramírez, Iveete Cecilia Guzmán Rodríguez and the institutions UNAM PAPIIT 1N105114, 1N107715 and Instituto Politécnico Nacional-México (Project: SIP 20180459, SIP 20195237, SIP 20195998) for the economic support.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02133g
Reference
- Carraro E. Bonetta S. Bertino C. Lorenzi E. Gilli G. J. Environ. Manage. 2016;168:185–199. doi: 10.1016/j.jenvman.2015.11.021. [DOI] [PubMed] [Google Scholar]
- Park J. C. Han J. Lee M. C. Seo J. S. Lee J. S. Aquat. Toxicol. 2017;189:16–24. doi: 10.1016/j.aquatox.2017.05.012. [DOI] [PubMed] [Google Scholar]
- Grabicova K. Grabic R. Fedorova G. Fick J. Cerveny D. Kolarova J. Turek J. Zlabek V. Randak T. Water Res. 2017;124:654–662. doi: 10.1016/j.watres.2017.08.018. [DOI] [PubMed] [Google Scholar]
- Jallouli N. Elghniji K. Hentati O. Ribeiro A. R. Silva A. M. T. Ksibi M. J. Hazard. Mater. 2016;304:329–336. doi: 10.1016/j.jhazmat.2015.10.045. [DOI] [PubMed] [Google Scholar]
- Muir D. Simmons D. Wang X. Peart T. Villella M. Miller J. Sherry J. Sci. Rep. 2017;7:16999. doi: 10.1038/s41598-017-15462-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson R. Duran-Alvarez J. C. Estrada K. L. Chavez A. Jimenez Cisneros B. Chemosphere. 2010;81:1437–1445. doi: 10.1016/j.chemosphere.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Gavrilescu M. Demnerova K. Aamand J. Agathos S. Fava F. New Biotechnol. 2015;32:147–156. doi: 10.1016/j.nbt.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Quintana J. B. Weiss S. Reemtsma T. Water Res. 2005;39:2654–2664. doi: 10.1016/j.watres.2005.04.068. [DOI] [PubMed] [Google Scholar]
- Andreozzi R. Caprio V. Insola A. Marotta R. Catal. Today. 1999;53:51–59. doi: 10.1016/S0920-5861(99)00102-9. [DOI] [Google Scholar]
- Xu B. Qi F. Zhang J. Li H. Sun D. Robert D. Chen Z. Chem. Eng. J. 2016;284:942–952. doi: 10.1016/j.cej.2015.09.032. [DOI] [Google Scholar]
- Song Z. Zhang Y. Liu C. Xu B. Qi F. Yuan D. Pu S. Chem. Eng. J. 2019;357:655–666. doi: 10.1016/j.cej.2018.09.182. [DOI] [Google Scholar]
- Zhang Y. Xia Y. Li Q. Qi F. Xu B. Chen Z. Sep. Purif. Technol. 2018;197:261–270. doi: 10.1016/j.seppur.2018.01.019. [DOI] [Google Scholar]
- Nawrocki J. Kasprzyk-Hordern B. Appl. Catal., B. 2010;99:27–42. doi: 10.1016/j.apcatb.2010.06.033. [DOI] [Google Scholar]
- Esplugas S. Bila D. M. Krause L. G. T. Dezotti M. J. Hazard. Mater. 2007;149:631–642. doi: 10.1016/j.jhazmat.2007.07.073. [DOI] [PubMed] [Google Scholar]
- Xu B. Qi F. Sun D. Chen Z. Robert D. Chemosphere. 2016;146:22–31. doi: 10.1016/j.chemosphere.2015.12.016. [DOI] [PubMed] [Google Scholar]
- Rodríguez J. L. Valenzuela M. A. Poznyak T. Lartundo L. Chairez I. J. Hazard. Mater. 2013;262:472–481. doi: 10.1016/j.jhazmat.2013.08.041. [DOI] [PubMed] [Google Scholar]
- Magallanes D. Rodriguez J. L. Poznyak T. Valenzuela M. A. Lartundo L. Chairez I. New J. Chem. 2015;39:7839–7848. doi: 10.1039/C5NJ01385B. [DOI] [Google Scholar]
- Kasprzyk-Hordern B. Ziółek M. Nawrocki J. Appl. Catal., B. 2003;46:639–669. doi: 10.1016/S0926-3373(03)00326-6. [DOI] [Google Scholar]
- Chen Z. Ren N. Wang A. Zhang Z.-P. Shi Y. Water Res. 2008;42:3385–3392. doi: 10.1016/j.watres.2008.04.020. [DOI] [PubMed] [Google Scholar]
- Arany E. Szabó R. K. Apáti L. Alapi T. Ilisz I. Mazellier P. Dombi A. Gajda-Schrantz K. J. Hazard. Mater. 2013;262:151–157. doi: 10.1016/j.jhazmat.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Kanakaraju D. Motti C. A. Glass B. D. Oelgemöller M. Chemosphere. 2015;139:579–588. doi: 10.1016/j.chemosphere.2015.07.070. [DOI] [PubMed] [Google Scholar]
- Méndez-Arriaga F. Esplugas S. Giménez J. Water Res. 2008;42:585–594. doi: 10.1016/j.watres.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Rosal R. Rodríguez A. Gonzalo M. S. García-Calvo E. Appl. Catal., B. 2008;84:48–57. doi: 10.1016/j.apcatb.2008.03.003. [DOI] [Google Scholar]
- Cruz-Zavala E. Moreno J. A. Fridman L. M. IFAC Proceedings Volumes. 2011;44:3039–3044. doi: 10.3182/20110828-6-IT-1002.03529. [DOI] [Google Scholar]
- Moreno J. A. and Osorio M., 2008
- Hernández N. Moreno R. Sánchez-Herencia A. J. Fierro J. L. G. J. Phys. Chem. B. 2005;109:4470–4474. doi: 10.1021/jp0448954. [DOI] [PubMed] [Google Scholar]
- Babić S. Horvat A. J. M. Mutavdžić Pavlović D. Kaštelan-Macan M. TrAC, Trends Anal. Chem. 2007;26:1043–1061. doi: 10.1016/j.trac.2007.09.004. [DOI] [Google Scholar]
- Pacheco D. P. Martínez F. Phys. Chem. Liq. 2007;45:581–595. doi: 10.1080/00319100701313862. [DOI] [Google Scholar]
- Gadipelly C. Pérez-González A. Yadav G. D. Ortiz I. Ibáñez R. Rathod V. K. Marathe K. V. Ind. Eng. Chem. Res. 2014;53:11571–11592. doi: 10.1021/ie501210j. [DOI] [Google Scholar]
- Grossberger A. Hadar Y. Borch T. Chefetz B. Environ. Pollut. 2014;185:168–177. doi: 10.1016/j.envpol.2013.10.038. [DOI] [PubMed] [Google Scholar]
- Sajedi-Amin S. Barzegar-Jalali M. Fathi-Azarbayjani A. Kebriaeezadeh A. Martínez F. Jouyban A. J. Mol. Liq. 2017;232:152–158. doi: 10.1016/j.molliq.2017.02.056. [DOI] [Google Scholar]
- Ghauch A. Tuqan A. M. Kibbi N. Chem. Eng. J. 2015;279:861–873. doi: 10.1016/j.cej.2015.05.067. [DOI] [Google Scholar]
- Aguilar C. M. Rodriguez J. L. Chairez I. Tiznado H. Poznyak T. Environ. Sci. Pollut. Res. Int. 2017;24:25550–25560. doi: 10.1007/s11356-016-6134-2. [DOI] [PubMed] [Google Scholar]
- Buxton G. V. Greenstock C. L. Helman W. P. Ross A. B. J. Phys. Chem. Ref. Data. 1988;17:513–886. doi: 10.1063/1.555805. [DOI] [Google Scholar]
- Rodriguez J. L. Valenzuela M. A. Poznyak T. Lartundo L. Chairez I. J. Hazard. Mater. 2013;262:472–481. doi: 10.1016/j.jhazmat.2013.08.041. [DOI] [PubMed] [Google Scholar]
- Xu B. Qi F. Clean: Soil, Air, Water. 2016;44:1099–1105. [Google Scholar]
- Jallouli N. Pastrana-Martínez L. M. Ribeiro A. R. Moreira N. F. F. Faria J. L. Hentati O. Silva A. M. T. Ksibi M. Chem. Eng. J. 2018;334:976–984. doi: 10.1016/j.cej.2017.10.045. [DOI] [Google Scholar]
- Wang F. Wang Y. Feng Y. Zeng Y. Xie Z. Zhang Q. Su Y. Chen P. Liu Y. Yao K. Lv W. Liu G. Appl. Catal., B. 2018;221:510–520. doi: 10.1016/j.apcatb.2017.09.055. [DOI] [Google Scholar]
- Feng J. Zhang X. M. Fu J. S. Chen H. L. Catal. Commun. 2018;110:28–32. doi: 10.1016/j.catcom.2018.03.001. [DOI] [Google Scholar]
- Fuentes I. Rodriguez J. L. Poznyak T. Chairez I. Environ. Sci. Pollut. Res. Int. 2014;21:12241–12248. doi: 10.1007/s11356-014-3176-1. [DOI] [PubMed] [Google Scholar]
- Zhang X. Li X. Qin W. Chem. Phys. Lett. 2009;479:310–315. doi: 10.1016/j.cplett.2009.08.029. [DOI] [Google Scholar]
- Nasseri S. Mahvi A. H. Seyedsalehi M. Yaghmaeian K. Nabizadeh R. Alimohammadi M. Safari G. H. J. Mol. Liq. 2017;241:704–714. doi: 10.1016/j.molliq.2017.05.137. [DOI] [Google Scholar]
- Pocostales P. Álvarez P. Beltrán F. J. Chem. Eng. J. 2011;168:1289–1295. doi: 10.1016/j.cej.2011.02.042. [DOI] [Google Scholar]
- Biesinger M. C. Payne B. P. Lau L. W. M. Gerson A. Smart R. S. C. Surf. Interface Anal. 2009;41:324–332. doi: 10.1002/sia.3026. [DOI] [Google Scholar]
- Payne B. P. Biesinger M. C. McIntyre N. S. J. Electron Spectrosc. Relat. Phenom. 2009;175:55–65. doi: 10.1016/j.elspec.2009.07.006. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.