

Abstract
Glutamine metabolism is reprogrammed during tumorigenesis and has been investigated as a promising target for cancer therapy. However, efforts to drug this process are confounded by the intrinsic metabolic heterogeneity and flexibility of tumors, as well as the risk of adverse effects on the anticancer immune response. Recent research has yielded important insights into the mechanisms that determine the tumor and host immune responses to pharmacological perturbation of glutamine metabolism. Here, we discuss these findings and suggest that, collectively, they point toward patient stratification and drug combination strategies to maximize the efficacy of glutamine metabolism inhibitors as cancer therapeutics.
Keywords: Cancer metabolism, Glutamine, Glutaminase (GLS), CB-839, JHU083, Immunometabolism
Metabolic reprogramming
Proliferating cells must acquire and process nutrients from their environment to meet the biosynthetic, bioenergetic, and redox demands of biomass accumulation. Almost a century ago, Otto Warburg described rapid glucose uptake coupled to lactic acid fermentation in tumor tissues, which persists even under aerobic conditions (aerobic glycolysis, or the Warburg effect) [1]. Metabolic reprogramming is now known to be a hallmark of tumorigenesis and is influenced by diverse factors, including the tissue of origin, the underlying oncogenic and epigenetic lesions, nutrient availability, and cell-cell and cell-matrix interactions within the tumor microenvironment (TME) (see Glossary). Despite remarkable heterogeneity in cancer metabolism, certain characteristics, such as increased glucose and glutamine consumption, are broadly conserved, a phenomenon that indicates fundamental biochemical necessities and that is exploited for clinical imaging of tumors with labeled metabolite analogs [2].
Because sustained anabolic metabolism is essential for tumor growth, it presents opportunities for therapeutic intervention. Antimetabolite chemotherapies, including antifolates, antipurines, and anti-pyrimidines, have been in clinical use since the 1950s, but their efficacy is restricted by dose-limiting toxicity arising from activity against other proliferative cell types, including lymphocytes. Indeed, metabolic reprogramming is also a hallmark of immune cell activation and is required for a robust anticancer immune response. The challenge, therefore, is to develop metabolism-targeted therapies that are tumor-selective, sparing the host immune system. This will likely require a personalized therapy approach based on understanding the determinants of tumor and immune response to specific metabolic perturbations. Due to its frequent dysregulation in tumors, glutamine metabolism (Figure 1) has received particular attention in this context. Several classes of glutamine metabolism inhibitors have now been developed; since 2014, an allosteric inhibitor of kidney-type glutaminase (GLS), CB-839 (Telaglenastat), has been in phase I-II clinical trials for cancer therapy (Table 1).
Figure 1. Glutamine metabolism in quiescent and proliferating cells.
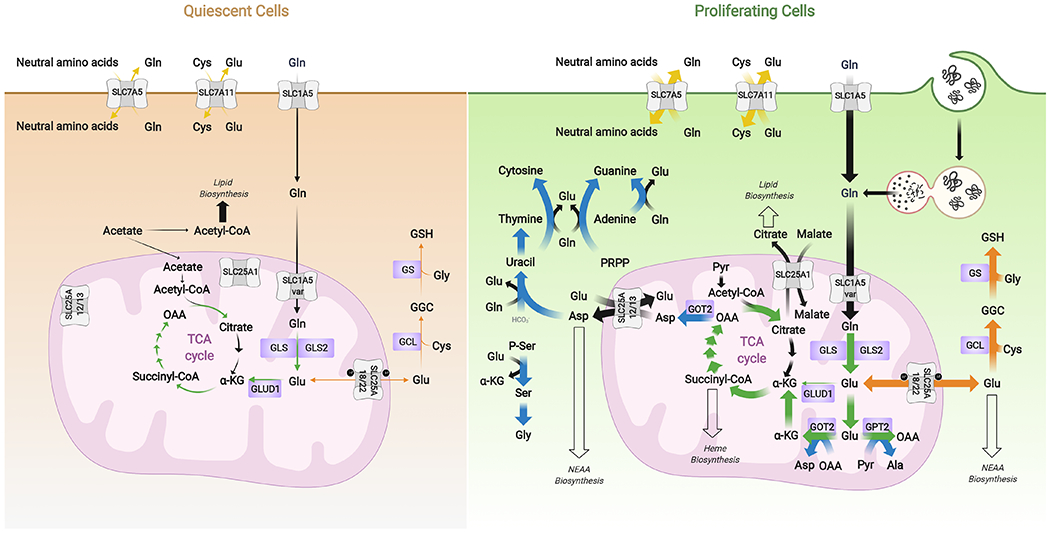
Proliferative metabolism is characterized by a broad upregulation of biosynthetic pathways. Glutamine is acquired either through uptake transporters such as SLC1A5 or, in nutrient-poor microenvironments, from extracellular protein via macropinocytosis followed by lysosomal degradation. Glutamine is an obligate nitrogen donor for nucleotide and asparagine biosynthesis and an exchange factor for some less abundant amino acids. Its catabolite glutamate is a precursor of α-KG for TCA cycle anaplerosis, a substrate for glutathione biosynthesis, a carbon and nitrogen source for NEAA biosynthesis, and also an exchange factor for other amino acids. Blue arrows indicate pathways in which glutamine/glutamate serve as a nitrogen source, green arrows indicate use of glutamine-derived carbon for anaplerosis, orange arrows represent direct incorporation of glutamate into biosynthetic pathways, and yellow arrows show exchange factor functions.
Abbreviations: α-KG, α-ketoglutarate; Ala, alanine, Asp, aspartate; Cys, cystine or cysteine; GCL, glutamate-cysteine ligase; GGC, γ-glutamylcysteine; Gln, glutamine; Glu, glutamate; GLS, glutaminase; GLS2, glutaminase 2; GLUD1, glutamate dehydrogenase 1; Gly, glycine; GOT2, mitochondrial aspartate aminotransferase; GPT2, mitochondrial alanine transaminase; GS, glutathione synthetase; GSH, reduced glutathione; NEAA, non-essential amino acids; OAA, oxaloacetate; PRPP, 5-phosphoribosyl-1-pyrophosphate; P-Ser, phosphoserine; Pyr, pyruvate; Ser, serine; SLC1A5 var, SLC1A5 variant; TCA, tricarboxylic acid.
Table 1.
Clinical trials of glutaminase inhibitor CB-839 (Telaglenastat) as cancer therapeuticsa
| Trial identifier | Study | Statusb | Outcome | Refs |
|---|---|---|---|---|
| NCT02071862 i | CB-839 monotherapy or in combination with paclitaxel, everolimus, erlotinib, docetaxel, or cabozantinib in solid tumors | Phase I, 210, completed | CB-839 was well-tolerated, had clinical efficacyc with cabozantinib (ORR = 50%, DCR = 100%) or with everolimus (DCR = 92%, PFS = 7.1 months) in metastatic RCC. | [80–83] |
| NCT02071888 ii | CB-839 monotherapy, in combination with low dose dexamethasone or pomalidomide and low dose dexamethasone in hematological tumors | Phase I, 25, completed | CB-839 monotherapy was well-tolerated in patients with multiple myeloma and lymphoma. | [84] |
| NCT02071927 iii | CB-839 monotherapy or in combination with azacitidine in leukemia | Phase I, 43, completed | Platelets and PBMCs showed GLS inhibition. | [85] |
| NCT02771626 iv | CB-839 in combination with PD-1 blocking antibody, nivolumab, in patients with melanoma, ccRCC, and NSCLC | Phase I/II, 118, completed | CB-839 with nivolumab was well-tolerated in melanoma (ORR = 19%) and RCC (ORR = 21%, DCR = 74%). | [86] |
| NCT02861300 v | CB-839 with capecitabine in solid tumors and fluoropyrimidine-resistant PIK3CA mutant colorectal cancer | Phase I/II, 53, recruiting | The median PFS was 16.5 weeks for all patients and 29.5 weeks for PIK3CA mutant CRC patients. | [87] |
| NCT02944435 vi | A pharmacokinetic study of CB-839 capsule and tablet formulations in healthy adults | Phase I, 14, completed | N/A | N/A |
| NCT03047993 vii | CB-839 in combination with azacitidine in patients with advanced MDS | Phase I/II, 40, recruiting | CB-839 with azacitidine was well-tolerated in MDS patients (ORR = 63%). | [88] |
| NCT03057600 viii | CB-839 in combination with paclitaxel in patients of African ancestry and non-African ancestry with advanced TNBC | Phase II, 52, completed | CB-839 with paclitaxel was well-tolerated in advanced TNBC patients (ORR = 43%, DCR = 79%). | [89] |
|
NCT03163667ix ENTRATA |
Everolimus in combination with placebo or CB-839 in patients with RCC | Phase II, 63, completed | ENTRATA met the primary end point: improved PFS when everolimus was used in combination with CB-839 vs. placebo (3.8 months vs. 1.9 months). | [90] |
| NCT03263429 x | Novel PET/CT imaging biomarkers of CB-839 in combination with panitumumab and irinotecan in patients with metastatic and refractory RAS wildtype colorectal cancer | Phase I/II, 40, recruiting | This trial showed tolerable triple combination at full dose of each drug. | [91] |
|
NCT03428217xi CANTATA |
Cabozantinib in combination with placebo or CB-839 in patients with metastatic RCC | Phase II, 445, active, not recruiting | The combination of CB-839 and cabozantinib did not improve PFS, the primary end point. | [92]xxii |
| NCT03528642 xii | CB-839 with radiation therapy and temozolomide in patients with IDH-mutated diffuse astrocytoma or anaplastic astrocytoma | Phase I, 40, recruiting | Ongoing trial. | [93] |
| NCT03798678 xiii | CB-839 in combination with carfilzomib and dexamethasone in patients with recurrent or refractory multiple myeloma | Phase I, 36, recruiting | Ongoing trial. | [94] |
| NCT03831932 xiv | CB-839 and osimertinib in patients with EGFR-mutated stage IV NSCLC | Phase I/II, 53, recruiting | Ongoing trial. | N/A |
| NCT03872427 xv | Basket Trial of CB-839 in patients with NF1 aberrations, NF1 mutant malignant peripheral nerve sheath tumors, KEAP1/NRF2 and LKB1 aberrant tumors | Phase II, 108, recruiting | Ongoing trial. | N/A |
| NCT03875313 xvi | CB-839 in combination with the PARP inhibitor talazoparib in patients with solid tumors | Phase I/II, 33, terminated | Slow enrollment. | N/A |
| NCT03944902 xvii | CB-839 in combination with niraparib in platinum-resistant BRCA-wildtype ovarian cancer patients | Phase I, 33, not yet recruiting | N/A | N/A |
| NCT03965845 xviii | CB-839 in combination with the CDK4/6 inhibitor palbociclib in patients with solid tumors | Phase I/II, 85, recruiting | Ongoing trial. | N/A |
| NCT04250545 xix | CB-839 in combination with MLN0128 (sapanisertib) in advanced stage NSCLC | Phase I, 85, recruiting | Ongoing trial. | N/A |
|
NCT04265534xx KEAPSAKE |
CB-839 with standard-of-care chemoimmunotherapy (pembrolizumab, carboplatin, and pemetrexed) in KEAP1/NRF2-mutated, nonsquamous NSCLC | Phase II, 120, recruiting | Ongoing trial. | N/A |
| NCT04540965 xxi | Impact of the histamine H2 receptor antagonist famotidine on the pharmacokinetics of CB-839 in healthy adults | Phase I, 22, recruiting | Ongoing trial. | N/A |
Abbreviations: BRCA, breast cancer gene; ccRCC, clear cell renal cell carcinoma; CRC, colorectal cancers; DCR, disease control rate; EGFR, epidermal growth factor receptor; IDH, isocitrate dehydrogenase; MDS, myelodysplastic syndrome; NF1, neurofibromatosis type 1; NSCLC, non-small cell lung cancer; ORR, overall response rate; PARP, poly (ADP-ribose) polymerase; PBMC, peripheral blood mononuclear cell; PD-1, programmed death receptor-1; PET/CT, positron emission tomography computed tomography; PFS, progression-free survival; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; RAS, rat sarcoma gene; RCC, renal cell carcinoma; TNBC, triple negative breast cancer.
Phase stages, enrollment numbers, current status.
Calithera Biosciences initiated phase 2 ENTRATA (NCT03163667) and CANTATA (NCT03428217) trials based on the encouraging clinical efficacy and safety data.
Tumor glutamine addiction and GLS dependence are triggered by specific molecular cues and are not universal across all cancers. Some tumors even produce and secrete glutamine into the TME [3], while a subset of cancer cell lines that are ‘glutamine addicted’ in standard culture media switch to alternative fuels in vivo [4, 5]. To maximize the efficacy of glutamine metabolism inhibitors, it is critical to understand the factors that drive sensitivity to these drugs. Recent advances have been made in this field, pointing toward biomarkers and drug combination strategies to enhance the efficacy of this therapeutic approach.
Glutamine metabolism in proliferating cells
At a concentration of ~500 μM, glutamine is the most abundant amino acid in blood plasma and plays a unique nutritional role in proliferating cells. Although a classical non-essential amino acid (NEAA) that can be produced de novo by glutamine synthetase (GLUL), glutamine is considered ‘conditionally essential’ since elevated demand renders many proliferative cells dependent on an exogenous supply. There are at least 14 glutamine transporters in the solute carrier (SLC) family, with the high-affinity exchanger SLC1A5 widely upregulated in tumors [6]. Besides its function as a proteinogenic amino acid, intracellular glutamine serves as an exchange factor for less abundant nutrients or undergoes deamidation to glutamate. Amidotransferases move the amide group to a co-substrate, and through this mechanism glutamine is the obligate nitrogen donor for asparagine, hexosamine, and nucleotide biosynthesis. Alternatively, mitochondrial glutaminases (GLS and GLS2) (Box 1) release the amide group as an ammonium ion, allowing for rapid glutamate production in the absence of nitrogen acceptors.
Box 1: GLS and GLS2.
Two genes encode glutaminase enzymes in mammals, GLS and GLS2, located on chromosomes 2 and 12, respectively, in humans. In the case of GLS, two primary splice variants termed kidney-type glutaminase (KGA) and glutaminase C (GAC) share the first 14 exons, with exons 16–19 present in KGA and exon 15 in GAC [76]. The GLS gene is ubiquitously expressed in mammalian tissues, with kidney and brain having particularly high transcript levels. GLS expression is further upregulated in many cancers, especially GAC, which encodes an enzyme with a higher kcat and lower KM than other glutaminase variants [67]. In contrast, GLS2 expression is restricted to the liver, pancreas, and brain [77]. For GLS2, the two alternative transcripts are liver-type glutaminase (LGA) and glutaminase B (GAB), which arise from an alternative transcriptional start site mechanism. GAB is the longer transcript with all 18 exons, while the shorter LGA lacks part of exon 1 [78]. It is possible that the resulting difference in the N-terminal region results in distinct subcellular localization of GAB and LGA, potentially explaining reports of nuclear localization of GLS2 in brain tissue. Both GLS and GLS2 are inactive as monomers, and an allosteric modulator such as inorganic phosphate is required to drive formation of the active tetramer [79].
Glutamate itself is a critical metabolite for proliferation, but is maintained at relatively low levels (~100 μM) in plasma and is often derived primarily by glutamine deamidation intracellularly. Glutamate can be directly incorporated into the proline and glutathione biosynthetic pathways or deaminated to α-ketoglutarate (α-KG). Analogous to glutamine deamidation, this reaction is catalyzed either by transaminases, which transfer the amine group to an α-keto acid to generate another NEAA, or by mitochondrial glutamate dehydrogenases, which release ammonium [7]. Through its two-reaction conversion to α-KG, glutamine is a major anaplerotic carbon source for the tricarboxylic acid (TCA) cycle, the central metabolic hub for biosynthesis and bioenergetics (Figure 1). In addition, α-KG is the obligate co-substrate for dioxygenases, including histone and DNA demethylases, thereby connecting glutamine catabolism to regulation of the epigenome [8].
Strategies for targeting tumor glutamine metabolism
Systemic glutamine depletion
Bacterial L-asparaginases are a standard component of therapy regimens for acute lymphoblastic leukemia (ALL). In addition to asparaginase activity, these enzymes possess glutaminase activity and deplete both asparagine and glutamine in plasma [9]. Although the asparaginase activity alone is initially effective at treating ALL, the glutaminase activity is required for durable efficacy [10]. Bacterial L-glutaminases have also been investigated as possible therapeutics [11], and ongoing studies are evaluating both enzymes for treating hematological malignancies and solid tumors. Another approach for systemic glutamine depletion uses phenylbutyrate, an aromatic fatty acid approved to treat hyperammonemia in patients with urea cycle dysfunction. In the liver and kidneys, phenylbutyrate undergoes β-oxidation to generate its active form, phenylacetate. This then spontaneously conjugates with glutamine to form phenylacetylglutamine, which is excreted in urine as an alternative route for nitrogen disposal [12]. Preclinical studies have found anticancer activity for phenylbutyrate compounds, but whether this is due to glutamine depletion or another mechanism is not known [13, 14]. A major limitation of any cancer therapy approach that lowers circulating glutamine levels is that, in principle, resistance only requires activation of de novo glutamine synthesis in cancer cells or stromal cells within the TME.
Glutamine depletion within the TME
An alternative to systemic glutamine depletion is to target local sources of glutamine within the TME. Tumors contain a heterogeneous population of reactive stromal cells, such as cancer-associated fibroblasts (CAFs), which exist in a state of metabolic symbiosis with cancer cells. Upregulation of glutamine synthesis, accompanied by glutamine secretion, occurs in CAFs relative to normal fibroblasts [15]. Correspondingly, co-culture with CAFs but not with normal fibroblasts rescues cancer cell growth in glutamine-deficient environments, and selectively abrogating CAF glutamine anabolism in vivo suppresses ovarian tumor growth in murine models [15]. Stromal GLUL is therefore considered a potential target for cancer therapy, but to date GLUL lacks potent and selective inhibitors.
An alternative glutamine source within the TME is extracellular protein, which is a major supply route for amino acids in poorly vascularized tumors, such as pancreatic ductal adenocarcinoma (PDAC) [16]. Increased macropinocytic flux coincides with amino acid deprivation in PDAC, and consumed proteins are degraded into free amino acids in lysosomes (Figure 1). Although this process supports tumor growth in nutrient-poor environments, the feasibility of inhibiting macropinocytosis for cancer therapy remains unclear, as pathways such as coat-mediated endocytosis can potentially compensate for its loss [17].
Glutamine uptake inhibitors
The large number of glutamine uptake transporters encoded in the human genome exhibit substantial redundancy, which presents challenges for selective blockade of tumor glutamine uptake. However, frequent upregulation of the c-Myc transcriptional target SLC1A5 in tumors indicates a special role for this glutamine transporter, and its overexpression is widely associated with poor prognosis [18, 19]. These observations have motivated efforts to develop selective pharmacological agents, building on the first-generation low-potency glutamine transport antagonist L-γ-glutamyl-p-nitroanilide (GPNA) [20]. The GPNA derivative V-9302 inhibits cellular glutamine uptake with ~100-fold improved potency, shows in vitro efficacy against cancer cell lines, and suppresses tumor growth in murine models [21]. However, the proposed mechanism of action of V-9302 has been called into question by reports of SLC1A5 knockout having no effect on sensitivity to this molecule [22]. Instead, V-9302 was found to inhibit several other proteins, including the glutamine transporter SLC38A2 (SNAT2). Thus, while there is evidence that glutamine transport blockade can impact tumor growth, the current lead compounds must still be optimized, a process that might be facilitated by recent structural studies of SLC1A5 [23].
Glutamine antimetabolites
6-diazo-5-oxo-L-norleucine (L-DON), azaserine, and acivicin are secreted as antibiotics by Streptomyces bacteria and act as irreversible glutamine-competitive inhibitors of amidotransferases and glutaminases [24] (Figure 2). All of these glutamine mimetics are cytotoxic and have shown anticancer efficacy in mice. However, early clinical trials using glutamine antimetabolites were discontinued because of dose-limiting neurotoxicity, gastrointestinal toxicity, and myelosuppression, all attributed to a lack of selectivity for tumors over other glutamine-dependent cells [25, 26]. To address this issue, prodrug forms of L-DON that are inert in circulation but selectively activated within the TME have recently been developed [27]. The lead compound JHU083 causes tumor regression in multiple syngeneic murine models and has a major impact on the anticancer immune response, as discussed further below.
Figure 2. Timeline and chemical structures of representative glutamine metabolism inhibitors.
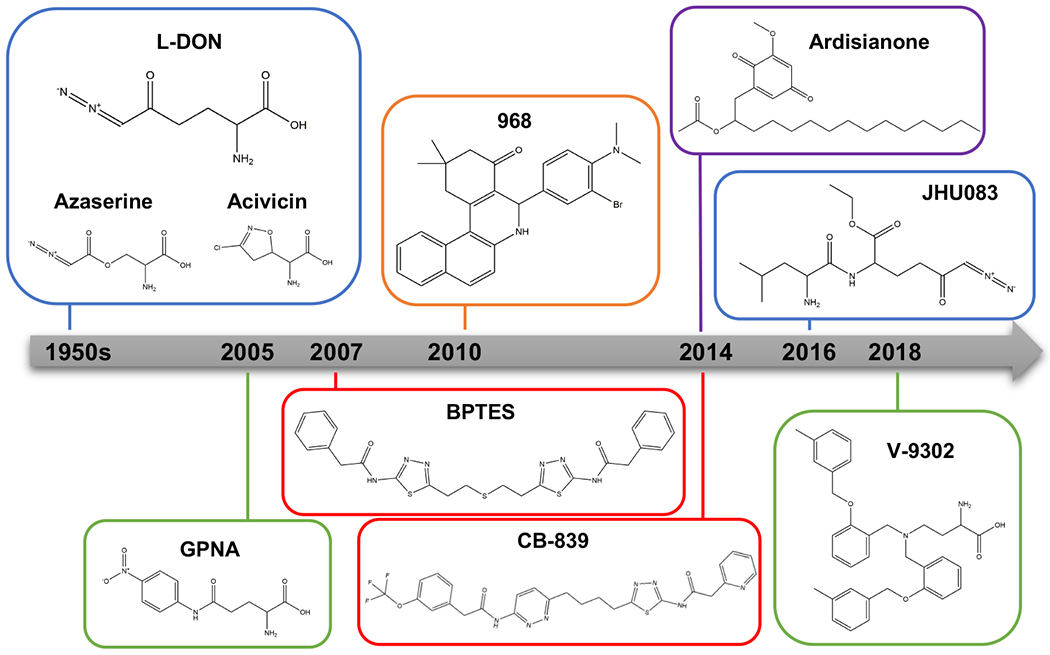
Glutamine metabolism inhibitors include glutamine antimetabolites and their prodrug forms and allosteric glutaminase inhibitors. L-DON, azaserine, and acivicin are glutamine antimetabolites, and JHU083 is a prodrug form of L-DON that is selectively activated in the tumor microenvironment. These antimetabolite-based inhibitors are marked by a blue box. The two major classes of allosteric glutaminase inhibitors are based on the lead compounds BPTES and 968, marked by red and orange boxes, respectively. Currently, CB-839 is the only GLS inhibitor to have entered clinical trials. 968 is a pan-glutaminase inhibitor, with four-fold higher potency against GLS2 than GLS. Ardisianone (AV-1), is a natural alkyl benzoquinone, the only known molecular scaffold that potently and selectively targets GLS2, marked in a purple box. In addition to GLS inhibitors, the glutamine uptake inhibitors GPNA and V-9302 are marked by green box.
Abbreviations: BPTES, bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide; GPNA, L-γ-glutamyl-p-nitroanilide; JHU083, ethyl 2-(2-Amino-4-methylpentanamido)-DON; L-DON, 6-diazo-5-oxo-L-norleucine.
Glutaminase inhibitors
GLS is highly expressed and essential in diverse malignancies and has been extensively investigated as a drug target for cancer therapy. As an alternative to broad-spectrum glutamine antagonists, highly selective GLS inhibitors have been developed over the last two decades. The two major classes of allosteric GLS inhibitors are based on the lead compounds bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) and 968 [28, 29] (Figure 2). BPTES is a symmetrical molecule that binds at the interface of two GLS dimers, where it stabilizes an inactive tetrameric conformation [28]. Docking and mutagenesis studies indicate that 968 binds between two GLS monomers to prevent formation of the active tetramer. Recently, 968 was found to be a pan-glutaminase inhibitor, with four-fold selectivity for GLS2 over GLS [30]. To date, the only known highly-selective (>10-fold) and potent (IC50 <1 μM) GLS2 inhibitors are alkyl benzoquinones isolated from Ardisia virens [31].
Co-crystal structures of BPTES and GLS have facilitated drug optimization efforts [32], leading to the development of the potent and orally bioavailable BPTES derivative CB-839 [33]. Numerous additional inhibitors based on the BPTES scaffold have been described [34–37], but only CB-839 has advanced into clinical trials (Table 1). To date, CB-839 has yielded few objective responses when used as a monotherapy. Although some phase I trials indicated promising activity for CB-839 as a component of combination therapies, the recent phase II CANTATA trial (NCT03428217) of CB-839 with the tyrosine kinase inhibitor cabozantinib in renal cell carcinoma did not meet the study’s primary end point of improved progression-free survival relative to cabozantinib alonexxii. Despite these disappointing results, new discoveries have identified biological contexts in which tumor cells become locked into GLS dependence, suggesting that carefully targeted use of GLS inhibitors might still yield clinical benefits.
Determinants of tumor sensitivity to glutamine blockade
The tumor oncogenotype
Reprogrammed glutamine metabolism in cancer is not a passive adaptation to the proliferative state, but instead is tightly regulated by diverse cell-intrinsic and -extrinsic factors, including the oncogenic signals that underlie tumorigenesis. Most frequently occurring oncogenic lesions in human tumors drive changes in cellular glutamine metabolism and/or sensitivity to GLS inhibitors. These changes include aberrant activation of the PI3K/mTOR axis [38], hyperactivating mutations of KRAS [39, 40], hyperactivated receptor tyrosine kinase signaling [41], overexpression of MYC or JUN [18, 42], neomorphic isocitrate dehydrogenase 1 or 2 (IDH1/2) mutations [43], changes in TGFβ or Hippo pathway signaling [44, 45], and loss-of-function mutations of the tumor suppressors TP53 (p53) [46], RB1 [47], KEAP1 [40], and STK11 (LKB1) [48].
The mechanisms by which these events regulate glutamine metabolism are varied. In the case of c-Jun and c-Myc, the expression of glutamine metabolism genes is increased either by direct transcriptional activation or by repression of microRNAs that normally prevent the translation of these transcripts (Figure 3, Key Figure). Ectopic overexpression of MYC is sufficient to drive cellular glutamine addiction, but it is possible that endogenous c-Myc does not mirror this effect, as the 3’-untranslated region (3’-UTR) of the transcript is glutamine-responsive, allowing endogenous c-Myc levels to decline when glutamine is limited [49]. Oncogenic KRAS also signals to increase expression of glutamine metabolism genes, and in KRAS-driven PDAC, glutamine-derived carbon maintains the cellular NADPH pool by supplying malic enzyme 1 (ME1) with oxidizable substrate [39].
Figure 3. Determinants of tumor sensitivity to GLS inhibitors.
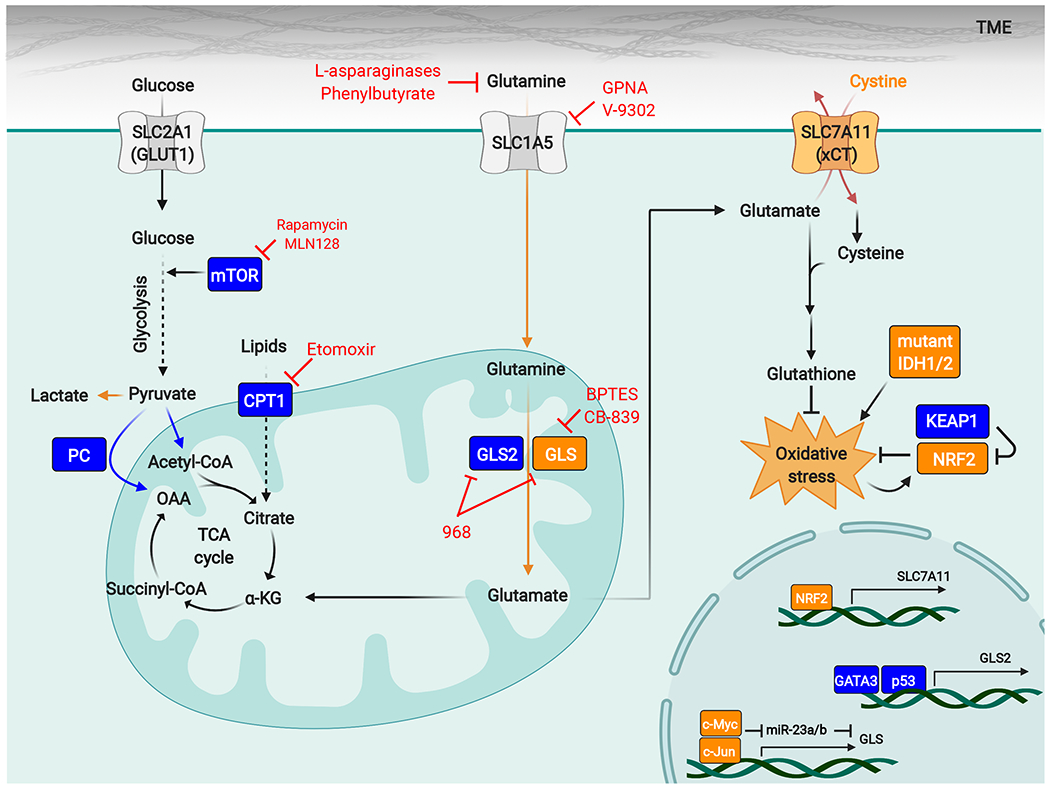
Diverse factors affect the sensitivity of tumors to GLS inhibitors, and specific oncogenotypes and metabolic stresses (shown in orange) can lock cancer cells into a GLS-dependent state. High activity of the NRF2-antioxidant response pathway has emerged as a conserved biomarker for tumor GLS dependence and can occur through genetic activation of the pathway (for example by loss-of-function mutations in KEAP1) or through factors that impose sustained oxidative stress, such as IDH1/2 mutations that lead to neomorphic 2-hydroxyglutarate-producing enzymes. Expression of GLS is regulated by c-Myc and c-Jun, and high levels of these transcription factors also favor cellular dependence on GLS. The nutrient environment also influences sensitivity to GLS inhibitors, and excessive levels of extracellular cystine can drive glutamine addiction and GLS dependence by forcing glutamate efflux through the cystine/glutamate antiporter xCT/SLC7A11. Other factors promote resistance to GLS inhibitors (shown in blue). These factors include compensatory metabolic pathways such as pyruvate carboxylation, fatty acid oxidation, and glutamine hydrolysis catalyzed by GLS2. mTORC1 can enhance metabolic flexibility by regulating a shift to glucose metabolism, and mTORC1 inhibitors synergize with GLS inhibitors.
Abbreviations: α-KG, α-ketoglutarate; CPT1, carnitine palmitoyltransferase I; GCL, glutamate-cysteine ligase; GLS, glutaminase; GPNA, L-γ-glutamyl-p-nitroanilide; IDH, isocitrate dehydrogenase; KEAP1, Kelch-like ECH-associated protein 1; mTOR, mechanistic target of rapamycin; NRF2, nuclear factor erythroid 2-related factor 2; OAA, oxaloacetate; PC, pyruvate carboxylase; ROS, reactive oxygen species; TME, tumor microenvironment.
Oncogenic mutations in IDH1/2 occur frequently in low-grade gliomas and acute myeloid leukemia and yield neomorphic enzymes that catalyze the conversion of α-KG to 2-hydroxyglutarate (2-HG) [50]. This new reaction imposes oxidative stress, partly because 2-HG production consumes NADPH, but also because the transaminases that provide glutamate for glutathione biosynthesis are inhibited by 2-HG [51]. This process leads to cellular dependence on GLS for glutamate production, such that oncogenic IDH mutations are synthetically lethal with GLS inhibition [51]. Another lesion that shows synthetic lethality with GLS blockade is loss-of-function mutation of KEAP1, which occurs in 20–30% of lung adenocarcinomas and results in sustained activation of the NRF2-antioxidant response element pathway. The NRF2 transcriptional target SLC7A11 has emerged as a major determinant of cellular glutamine addiction and sensitivity to GLS inhibitors [5, 52–54]. The gene product SLC7A11 (xCT) mediates the chloride-dependent exchange of intracellular glutamate for extracellular cystine, the limiting nutrient for glutathione biosynthesis (Figure 3). Although glutamate/cystine exchange by xCT supports redox homeostasis, excessive flux through this pathway generates an imbalance between antioxidant capacity and central carbon metabolism, and renders cells addicted to glutamine and GLS for replenishing the exported glutamate [5, 52, 54]. This phenomenon is exacerbated by loss of the STK11 (LKB1) tumor suppressor, which increases energetic and redox stress by deregulating metabolic homeostasis [48].
Oxidative stress
An emerging theme in the previous section is that oncogenotypes that increase demand on the cellular antioxidant program are key drivers of cancer cell glutamine addiction and GLS dependence. The underlying mechanism applies more broadly, such that oxidative stress appears to be a conserved biomarker for tumor sensitivity to GLS inhibitors. This is an important point, and means that GLS essentiality does not necessarily arise from its role in TCA cycle anaplerosis, but rather from its contribution to the intracellular glutamate pool for cellular redox homeostasis (Figure 3). Supporting this concept, pan-cancer analysis shows a strong co-dependency on GLS and the glutathione biosynthesis pathway [55]. A key question still to be answered is if, and how, tumors with high levels of oxidative stress or constitutive NRF2 activation can eventually acquire resistance to glutamine metabolism inhibitors, for example by downregulating SLC7A11 expression and/or by increased recycling of oxidized glutathione.
The nutrient environment
Tumor metabolism is affected by a multitude of microenvironmental factors, including nutrient availability [56]. Metabolic regulation by these variables in turn influences tumor sensitivity to metabolism-targeted drugs [57]. Illustrating this effect of the nutrient environment, high extracellular cystine concentrations are sufficient to induce cancer cell glutamine addiction and GLS dependence when it otherwise would not exist by forcing excessive activity of xCT, resulting in glutamate efflux [5]. Whereas environmental cystine can drive glutamine addiction, extracellular pyruvate can bypass it in specific contexts by sustaining TCA cycle anaplerosis through pyruvate carboxylase-mediated production of oxaloacetate (OAA) [58]. Pyruvate is a relatively abundant nutrient in the interstitial fluid of some tissues, including the lungs, and lung metastases show a shift from glutamine to pyruvate anaplerosis relative to the parental tumor [59]. However, pyruvate cannot substitute for all of the metabolic roles of glutamine and cannot overcome glutamine addiction when demand for glutathione biosynthesis is high. In a subset of PDAC tumors, nutrient starvation triggers increased macropinocytic uptake and lysosomal degradation of extracellular protein, which yields free amino acids including glutamine [16]. The ability to activate macropinocytosis can therefore render tumors resistant to therapies that deplete extracellular glutamine or block its cellular uptake, but does not affect sensitivity to glutamine antimetabolites or GLS inhibitors, which target enzymes directly involved in glutamine metabolism.
Resistance mechanisms to GLS inhibitors
Although glutamine is the primary carbon source for the TCA cycle in some malignant cells, we suggest that this anaplerotic role can usually be substituted by other pathways, and that true glutamine addiction instead arises when rapid glutamate production is required for glutathione biosynthesis. Consistent with this proposal, in cancer cells that are not genetically or environmentally locked into an enhanced antioxidant program, GLS inhibition can be rescued by exogenous supply of TCA cycle metabolites such as α-KG or OAA. Similarly, in this class of cancer cells, intrinsic or acquired resistance to GLS inhibitors develops through upregulation of alternative supply pathways for the TCA cycle [60]. As described above, pyruvate carboxylation to OAA can bypass glutamine anaplerosis, and this pathway is linked to CB-839 resistance across multiple cancers [4, 58]. In a murine model of c-Myc-induced liver tumorigenesis, although knockout of Gls suppresses the flux of glutamine-derived carbon into the TCA cycle, increased contribution from glucose means that absolute levels of TCA cycle metabolites are unaffected [60]. Simultaneous knockout of Gls and hexokinase 2 (Hk2) in this model, to disrupt both glutamine and glucose catabolism, potently impairs tumorigenesis with almost 40% of mice failing to develop tumors after 1 year [60].
Fatty acid oxidation (FAO) provides an additional supply route for the TCA cycle, and breast cancer and PDAC with intrinsic or acquired CB-839 resistance carry out increased FAO relative to CB-839-sensitive cells [61, 62]. Branched-chain and odd-chain FAO generate both acetyl-CoA and propionyl-CoA, thus supplying the citrate synthase step of the TCA cycle as well as anaplerosis via conversion of propionyl-CoA to succinyl-CoA. There is evidence that mTORC1 signaling is involved in the adaptive switch to alternative fuel sources when one anaplerotic pathway is inhibited, and several studies have found synergism between CB-839 and mTOR inhibitors [63, 64]. This evidence has provided the scientific rationale for an ongoing phase I clinical trial (NCT04250545) of the mTOR inhibitor MLN0128 with CB-839 for treating non-small cell lung cancer (NSCLC).
An often-overlooked player in tumor glutamine metabolism is liver-type glutaminase (GLS2). The function of GLS2 in cancer metabolism appears to be context-dependent, and it is transcriptionally regulated not only by p53 but also by oncoproteins such as N-myc [46, 65]. Nevertheless, like GLS, GLS2 is primarily localized to mitochondria and catalyzes glutamine hydrolysis, thus providing the most direct resistance mechanism to GLS-selective inhibitors. Three recent studies have found that acquired resistance to GLS blockade in breast cancer is associated with increased expression of GLS2 and that ectopic GLS2 expression is sufficient for CB-839 resistance [30, 60, 66]. However, it is possible that this resistance mechanism is not universal. GLS and GLS2 exhibit markedly different enzymatic kinetics, with GLS2 having a higher KM for glutamine and lower catalytic efficiency than GLS [67]. Thus, the inherently low activity of GLS2 might be insufficient for maintaining glutamate reserves when demand is very high.
The heterogeneous immune response to GLS inhibition
Heterogeneity in glutamine metabolism and in the response to glutamine blockade occurs across other cell types within the TME, including those of the immune system. To maximize the efficacy of glutamine metabolism inhibitors for cancer therapy, it will be necessary to identify approaches that harm cancer cells while sparing or enhancing the anticancer immune response (Figure 4). Activated T cells differentiate into functional subsets with distinct metabolic programs and differing dependencies on glutamine catabolism. Although increased glutamine hydrolysis is a conserved phenotype of T cell activation, inhibiting GLS has opposing effects on the differentiation of different subsets. For example, CB-839 treatment promotes the differentiation of CD4+ T-helper 1 (Th1) cells and CD8+ cytotoxic T lymphocytes (CTLs), resulting in increased cytokine production in both cases [68]. In contrast, the same treatment impedes CD4+ T-helper 17 (Th17) cell differentiation and function, leading to decreased cytokine production and suppressed expansion of this subset. These opposing outcomes appear to be due to differences in the epigenetic response of each T cell subset to α-KG depletion following GLS blockade. Importantly, there is also temporal heterogeneity in the T cell response to GLS deficiency, which has implications for cancer immunotherapy. Knockout of the Gls gene or chronic GLS inhibition render T cells unable to sustain a long-term effector response in vivo. However, when T cells are transiently exposed to CB-839 during an initial ex vivo differentiation period, the subsequent in vivo function of Th1 cells and CD8+ CTLs is enhanced, including when chimeric antigen receptor (CAR) T cells are used to eliminate B cell leukemia [68].
Figure 4. Glutamine metabolism impacts the anticancer immune response.
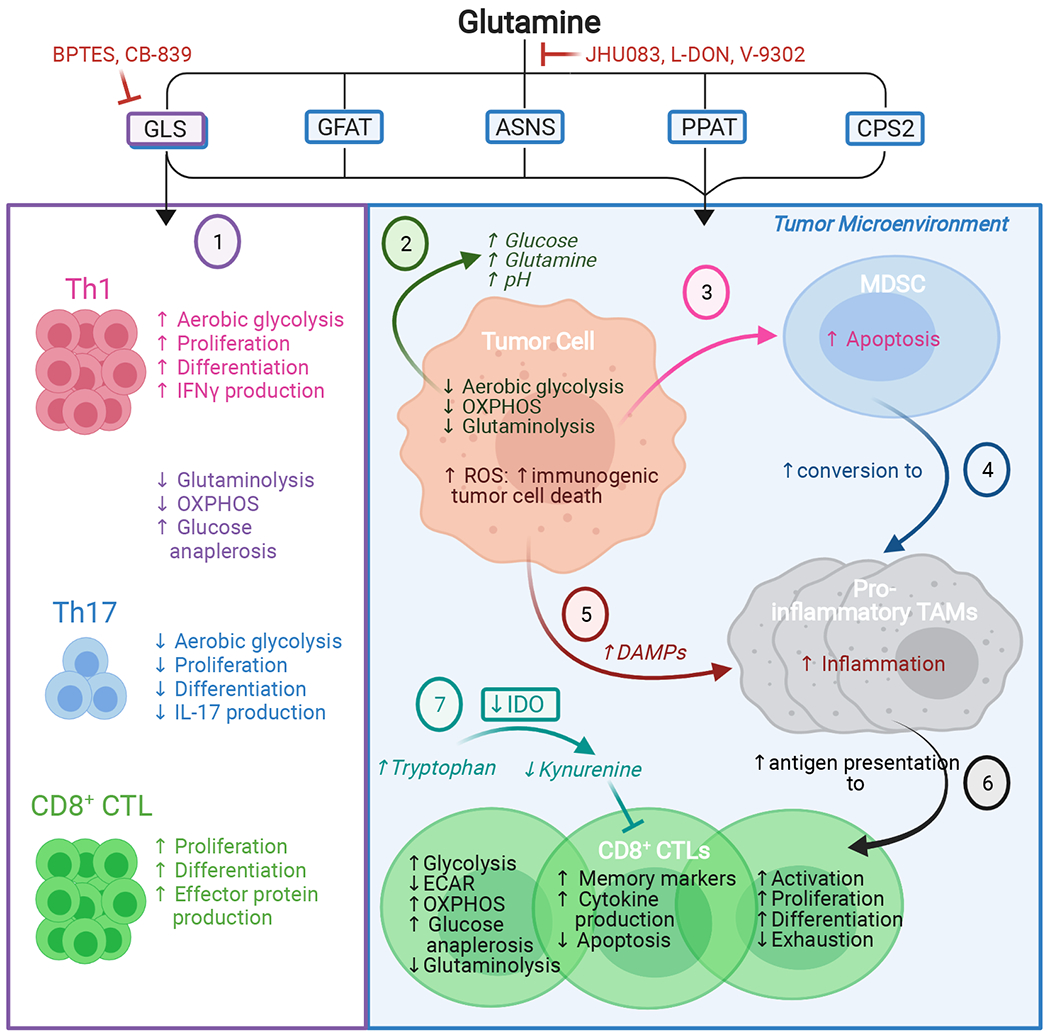
Graphical representation of the effects of allosteric GLS inhibitors (1) and non-selective glutamine antagonists (2-7). Selective GLS inhibition has distinct effects on the differentiation of CD4+ and CD8+ immune cell subsets, favoring CD4+ Th1 and CD8+ CTLs but suppressing CD4+ Th17 differentiation (1). Broader-spectrum glutamine antagonism alters the nutrient composition of the TME (2) and blocks MDSC generation and recruitment (3). Instead, glutamine antagonism favors conversion of MDSCs to pro-inflammatory TAMs, which inhibit tumor growth (4) and further activates pro-inflammatory TAMs via tumor cell-generated DAMPs (5). Inhibiting glutamine metabolism also directly modulates CD8+ CTL metabolism to promote a long-lasting, activated, memory-like phenotype; enhances antigen presentation by pro-inflammatory TAMs to CD8+ CTLs (6); decreases production of the immunosuppressive metabolite kynurenine in the TME (7); and increases CD8+ CTL tumor infiltration. These cumulative effects promote the anticancer immune response.
Abbreviations: ASNS, asparagine synthetase; BPTES, bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide; CPS2, carbamoyl phosphate synthetase II; CTL, cytotoxic T lymphocyte; DAMPs, damage/danger-associated molecular patterns; ECAR, extracellular acidification rate; GFAT, glutamine-fructose-6-phosphate transaminase; GLS, glutaminase; IDO, indoleamine-2,3-dioxygenase; IFNγ, interferon gamma; IL-17, interleukin 17; L-DON, 6-diazo-5-oxo-L-norleucine; MDSC, myeloid-derived suppressor cell; OXPHOS, oxidative phosphorylation; PPAT, phosphoribosyl pyrophosphate amidotransferase; TAMs, tumor-associated macrophages; Th1, T-helper 1; Th17, T-helper 17.
Glutamine blockade restores anti-tumor immunity
In contrast to GLS-selective inhibitors such as CB-839, glutamine antimetabolites inhibit all glutamine-utilizing enzymes, leading to major nutritional changes in the TME. In syngeneic murine models, treatment with the L-DON prodrug JHU083 causes broad suppression of tumor metabolic activity, mitigating hypoxia and increasing both glutamine and glucose levels in the TME [69]. Given the importance of glutamine metabolism in activated lymphocytes, a reasonable concern is whether broad-spectrum glutamine antagonism might also hamper the anticancer immune response. Remarkably, however, durable responses to JHU083 treatment occur only in immune-competent mice, and combining JHU083 with checkpoint immunotherapy yields complete response rates approaching 100%, versus 0% with immunotherapy alone. Under baseline conditions, the TME favors the residency of myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs), populations that inhibit the anticancer immune response. Treating tumor-bearing mice with JHU083 induces the differentiation of MDSCs and TAMs into pro-inflammatory TAMs, which inhibit tumor growth and cross-present tumor antigens to CD8+ T cells [70]. Ex vivo treatment with L-DON skews CD8+ T cells toward an activated, long-lived, memory-like state, analogous to the effect of CB-839 treatment or glutamine withdrawal during T cell differentiation [70, 71]. Correspondingly, JHU083-treated tumors in vivo have increased CD8+ tumor infiltrating lymphocytes (TILs), with a transcriptional program indicating a highly activated state along with expression of immunological memory markers. Consistent with glutamine antagonism supporting immunologic memory, in the small proportion of mice that are initially cured by JHU083 monotherapy, subsequent allograft tumors are completely rejected >30 days after administration of the final dose of JHU083 [69].
A ‘glutamine steal’ hypothesis has been proposed, in which cancer cell-selective glutamine blockade eliminates metabolic competition in the TME and frees up glutamine for use by immune cells [72]. However, the inhibitors used in the studies described above are not selective for cancer cells, raising the question of how glutamine antagonism enhances immune cell function while simultaneously disabling tumor cells. The explanation likely involves both cell-intrinsic factors and indirect effects from TME reconditioning. Metabolic flux analyses comparing colorectal cancer cells and T cells treated with L-DON show conserved suppression of aerobic glycolysis, but opposing effects on oxidative phosphorylation, which is suppressed in cancer cells but upregulated in T cells. These changes reflect greater metabolic flexibility in the T cells, which efficiently divert glucose-derived carbon into the TCA cycle and increase their use of extracellular acetate as an alternative fuel source [69]. Glutamine antagonism by L-DON can further enhance the anticancer immune response by impacting the mechanical properties of the tumor extracellular matrix (ECM). This effect is due to the inhibition of glutamine-fructose amidotransferase 1 (GFPT1), the rate-limiting enzyme of the hexosamine biosynthetic pathway (HBP). The HBP generates precursors for hyaluronan, a glycosaminoglycan that serves as a major component of the ECM in highly immunosuppressive tumors such as PDAC. Depletion of hyaluronan in the TME following L-DON treatment, or targeted knockdown of GFPT1, allows enhanced CD8+ T cell infiltration into PDAC tumors, and likely contributes to the observed synergism between L-DON and checkpoint immunotherapy [73].
Concluding Remarks and Future Perspectives
Recent insights into the determinants of tumor and immune responses to glutamine antagonism suggest biomarkers and strategies to maximize the efficacy of this line of therapy. Use of glutamine as a major carbon supply for the TCA cycle does not in itself predict tumor sensitivity to glutamine blockade, as many cancer cells are able to switch to alternative anaplerotic substrates such as pyruvate. Instead, glutamine addiction and cellular dependence on GLS show a robust pan-cancer correlation with the increased glutamate/glutathione demand that follows activation of the cellular antioxidant response program. This process can be driven by mutations that directly impact the pathway, such as KEAP1 loss; those that drive oxidative stress, such as neomorphic IDH1/2 mutations; cell-extrinsic factors, including the TME; and therapeutic interventions, such as radiation. Collectively, recent work suggests that oxidative stress is a conserved biomarker for tumor dependence on GLS, and tumors with a genetic or pharmacological addiction to the NRF2-antioxidant response pathway are the best candidates for treatment with GLS inhibitors. Supporting this idea, recent preclinical studies have shown that CB-839 treatment synergizes potently with radiation therapy and drugs that induce redox stress [74, 75]. The ongoing phase II KEAPSAKE trial (NCT04265534) for CB-839 treatment of KEAP1-mutant NSCLC is a key test of how well the advances made in this field extend into the clinic.
A parallel approach to improve the efficacy of glutamine metabolism inhibitors is to harness their surprising enhancement of anticancer immunity. An open question is which glutamine blockade strategy is most beneficial in this context, with glutamine antimetabolites, transport antagonists, and GLS inhibitors all reported to synergize with immune checkpoint blockade [69, 70, 72] (see Outstanding Questions). It will also be valuable to define further the temporal effects of glutamine antagonism on the immune response, given that transient CB-839 treatment modulates T cell differentiation to enhance function, but chronic treatment ultimately hampers the effector response. Despite largely disappointing results to date from clinical trials of glutamine metabolism inhibitors, insights from the last 5 years provide a scientific rationale for further evaluating carefully targeted use of these molecules and point toward rational combination therapies to maximize their efficacy in the clinic.
Outstanding Questions Box.
In tumors with genetic biomarkers for GLS dependence, such as the loss-of-function mutation of KEAP1, can resistance to GLS inhibitors still develop and, if so, through what mechanisms?
Following promising preclinical results, will ongoing clinical trials of CB-839 in tumors with KEAP1 or IDH mutations show efficacy in human patients?
Can glutamine antimetabolite prodrugs such as JHU083 be safely and effectively used to treat cancer in human patients?
Do glutamine antimetabolite prodrugs show broader anticancer activity than selective GLS inhibitors, and are there biomarkers for tumor sensitivity to these molecules?
Given the temporal variation in the response of T cells to GLS inhibition, and the markedly different biological half-lives of CB-839 versus immune checkpoint inhibitors, what dosing regimens are most effective in combination therapies?
Highlights.
Genetic and microenvironmental factors can ‘lock’ tumor cells into a state of glutamine addiction and dependence on glutaminase (GLS).
Oxidative stress and activation of the NRF2-antioxidant response pathway are conserved biomarkers for tumor sensitivity to GLS inhibitors.
Glutamine antagonism reconditions the tumor microenvironment and can enhance the anticancer immune response through both direct and indirect mechanisms.
Clinical trial results to date have shown tolerability of the GLS inhibitor CB-839, but mixed results with regard to efficacy, indicating the need to continue mechanistic, pharmacological, and translational research.
Targeting conserved biomarkers and developing rational drug synergisms will enhance the efficacy of glutamine metabolism inhibitors in cancer therapy.
Acknowledgements
MJL gratefully acknowledges support from the Breast Cancer Coalition of Rochester, METAvivor, The Elsa U. Pardee Foundation, and the Department of Defense Breast Cancer Research Program (BC200599). TJ gratefully acknowledges funding from The Mark Foundation for Cancer Research (Endeavor Award).
Glossary Box
- Anaplerosis
The process of replenishing the intermediates of a metabolic pathway. In proliferating cells, the tricarboxylic acid (TCA) cycle serves as the source of biosynthetic precursors and thereby loses carbon (cataplerosis). To maintain metabolic homeostasis, anaplerotic flux must match cataplerotic flux.
- Anticancer immune response
Innate and adaptive immune responses that contribute to the control of tumor growth. The immune system interacts with cancer cells in three phases, elimination, equilibrium, and escape. In the elimination phase, the immune system destroys cancer cells, and in the equilibrium phase, the tumor remains stable. In the escape phase, cancer cells evade the immune system and an immunosuppressive environment is established. Cancer immunotherapy approaches aim to restore immune control of cancer.
- Antimetabolite
A molecule that is chemically similar to a natural metabolite, but which inhibits the normal processing of that metabolite. Antimetabolites can interfere with the functions of one or more of the enzymes that normally interact with the natural metabolite, thereby affecting the cell’s normal metabolic processes.
- Glutaminase (GLS)
An amidohydrolase enzyme that catalyzes the conversion of glutamine to glutamate and an ammonium ion.
- NRF2-antioxidant response element pathway
A cellular mechanism that coordinates the response to oxidative and electrophilic stress by upregulating the transcription factor NRF2. The transcriptional targets of NRF2 include genes encoding key mediators of the detoxification and elimination of reactive oxidants and electrophiles. NRF2 is negatively regulated by KEAP1, a tumor suppressor whose function is frequently lost in non-small cell lung cancer.
- Tumor microenvironment (TME)
Tumors consist of a heterogeneous population of cancer cells and also contain both resident and infiltrating host cells, such as cancer-associated fibroblasts, endothelial cells, and lymphocytes. Together with the extracellular matrix, the tumor interstitial fluid, and secreted factors, these components constitute the TME.
- xCT (SLC7A11)
The gene product SLC7A11 (xCT) heterodimerizes with SLC3A2 to form the plasma membrane cystine/glutamate antiporter (system xc-), which plays a role in the antioxidant response by supplying cysteine, the rate-limiting substrate for glutathione biosynthesis. The SLC7A11 gene is a transcriptional target of NRF2.
Resources
References
- 1.Warburg O (1925) The metabolism of carcinoma cells. The Journal of Cancer Research 9, 148–163. [Google Scholar]
- 2.Dunphy MP et al. (2018) In vivo PET assay of tumor glutamine flux and metabolism: in-human trial of 18F-(2 S, 4 R)-4-fluoroglutamine. Radiology 287, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuneva MO et al. (2012) The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell metabolism 15, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson SM et al. (2016) Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muir A et al. (2017) Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6, e27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhutia YD and Ganapathy V (2016) Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1863, 2531–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mates J et al. (2013) Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Current molecular medicine 13, 514–534. [DOI] [PubMed] [Google Scholar]
- 8.Chang S et al. (2019) The cancer driver genes IDH1/2, JARID1C/KDM5C, and UTX/KDM6A: crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Experimental & Molecular Medicine 51, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avramis VI (2012) Asparaginases: Biochemical Pharmacology and Modes of Drug Resistance. Anticancer Research 32, 2423–2437. [PubMed] [Google Scholar]
- 10.Chan W-K et al. (2019) Glutaminase activity of L-asparaginase contributes to durable preclinical activity against acute lymphoblastic leukemia. Molecular cancer therapeutics 18, 1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller C et al. (2008) A phase IIa study of PEGylated glutaminase (PEG-PGA) plus 6-diazo-5-oxo-L-norleucine (DON) in patients with advanced refractory solid tumors. Journal of Clinical Oncology 26, 2533–2533. [Google Scholar]
- 12.Darmaun D et al. (1998) Phenylbutyrate-induced glutamine depletion in humans: effect on leucine metabolism. American Journal of Physiology-Endocrinology and Metabolism 274, E801–E807. [DOI] [PubMed] [Google Scholar]
- 13.Thibault A et al. (1994) A phase I and pharmacokinetic study of intravenous phenylacetate in patients with cancer. Cancer research 54, 1690–1694. [PubMed] [Google Scholar]
- 14.Carducci MA et al. (1996) Phenylbutyrate induces apoptosis in human prostate cancer and is more potent than phenylacetate. Clin. Cancer. Res. 2, 379–387. [PubMed] [Google Scholar]
- 15.Yang L et al. (2016) Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metabolism 24, 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Commisso C et al. (2013) Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ha KD et al. (2016) Macropinocytosis Exploitation by Cancers and Cancer Therapeutics. Frontiers in physiology 7, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao P et al. (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hassanein M et al. (2013) SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clinical cancer research 19, 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esslinger CS et al. (2005) Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorganic & medicinal chemistry 13, 1111–1118. [DOI] [PubMed] [Google Scholar]
- 21.Schulte ML et al. (2018) Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nature Medicine 24, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bröer A et al. (2018) Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Frontiers in Pharmacology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scopelliti AJ et al. (2018) Structural characterisation reveals insights into substrate recognition by the glutamine transporter ASCT2/SLC1A5. Nature Communications 9, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magill G et al. (1957) Pharmacological and initial therapeutic observations on 6-Diazo-5-Oxo-L-Norleucine (Don) in human neoplastic disease. Cancer 10, 1138–1150. [DOI] [PubMed] [Google Scholar]
- 25.Lynch G et al. (1982) Phase II evaluation of DON (6-diazo-5-oxo-L-norleucine) in patients with advanced colorectal carcinoma. American journal of clinical oncology 5, 541–543. [PubMed] [Google Scholar]
- 26.Earhart RH et al. (1990) Phase II trial of 6-diazo-5-oxo-L-norleucine versus aclacinomycin-A in advanced sarcomas and mesotheliomas. Investigational new drugs 8, 113–119. [DOI] [PubMed] [Google Scholar]
- 27.Rais R et al. (2016) Discovery of 6-diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. Journal of medicinal chemistry 59, 8621–8633. [DOI] [PubMed] [Google Scholar]
- 28.Robinson MM et al. (2007) Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). The Biochemical journal 406, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang JB et al. (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer cell 18, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lukey MJ et al. (2019) Liver-Type Glutaminase GLS2 Is a Druggable Metabolic Node in Luminal-Subtype Breast Cancer. Cell Rep 29, 76–88 e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu CC et al. (2013) Ardisianone, a natural benzoquinone, efficiently induces apoptosis in human hormone-refractory prostate cancers through mitochondrial damage stress and survivin downregulation. The Prostate 73, 133–145. [DOI] [PubMed] [Google Scholar]
- 32.Thangavelu K et al. (2012) Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proceedings of the National Academy of Sciences 109, 7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gross MI et al. (2014) Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Molecular Cancer Therapeutics 13, 890–901. [DOI] [PubMed] [Google Scholar]
- 34.Zimmermann SC et al. (2016) Allosteric Glutaminase Inhibitors Based on a 1,4-Di(5-amino-1,3,4-thiadiazol-2-yl)butane Scaffold. ACS medicinal chemistry letters 7, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Q et al. (2018) Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. The Journal of biological chemistry 293, 3535–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Z et al. (2018) Novel 1, 3, 4-selenadiazole-containing kidney-type glutaminase inhibitors showed improved cellular uptake and antitumor activity. Journal of medicinal chemistry 62, 589–603. [DOI] [PubMed] [Google Scholar]
- 37.Shukla K et al. (2012) Design, Synthesis, and Pharmacological Evaluation of Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl Sulfide 3 (BPTES) Analogs as Glutaminase Inhibitors. Journal of Medicinal Chemistry 55, 10551–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Csibi A et al. (2013) The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153, 840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Son J et al. (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romero R et al. (2017) Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gallipoli P et al. (2018) Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 131, 1639–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lukey MJ et al. (2016) The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat Commun 7, 11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seltzer MJ et al. (2010) Inhibition of Glutaminase Preferentially Slows Growth of Glioma Cells with Mutant IDH1. Cancer Research 70, 8981–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edwards DN et al. (2017) The receptor tyrosine kinase EphA2 promotes glutamine metabolism in tumors by activating the transcriptional coactivators YAP and TAZ. Science Signaling 10, eaan4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andratsch M et al. (2007) TGF-β signaling and its effect on glutaminase expression in LLC-PK1-FBPase+ cells. American Journal of Physiology-Renal Physiology 293, F846–F853. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki S et al. (2010) Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proceedings of the National Academy of Sciences 107, 7461–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reynolds MR et al. (2014) Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 33, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galan-Cobo A et al. (2019) LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in <em>KRAS</em>-Mutant Lung Adenocarcinoma. Cancer Research 79, 3251–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dejure FR et al. (2017) The MYC mRNA 3′-UTR couples RNA polymerase II function to glutamine and ribonucleotide levels. The EMBO journal 36, 1854–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujii T et al. (2016) Targeting isocitrate dehydrogenase (IDH) in cancer. Discovery medicine 21, 373–380. [PubMed] [Google Scholar]
- 51.McBrayer SK et al. (2018) Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 175, 101–116.e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shin CS et al. (2017) The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat Commun 8, 15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Timmerman LA et al. (2013) Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer cell 24, 450–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sayin VI et al. (2017) Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 6, e28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daemen A et al. (2018) Pan-Cancer Metabolic Signature Predicts Co-Dependency on Glutaminase and De Novo Glutathione Synthesis Linked to a High-Mesenchymal Cell State. Cell Metab 28, 383–399 e389. [DOI] [PubMed] [Google Scholar]
- 56.Muir A and Vander Heiden MG (2018) The nutrient environment affects therapy. Science 360, 962–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cantor JR et al. (2017) Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272.e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng T et al. (2011) Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proceedings of the National Academy of Sciences 108, 8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Christen S et al. (2016) Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Reports 17, 837–848. [DOI] [PubMed] [Google Scholar]
- 60.Mendez-Lucas A et al. (2020) Identifying strategies to target the metabolic flexibility of tumours. Nat Metab 2, 335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biancur DE et al. (2017) Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun 8, 15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reis LMD et al. (2019) Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. The Journal of biological chemistry 294, 9342–9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Momcilovic M et al. (2018) The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer cell 33, 905–921 e905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanaka K et al. (2015) Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. The Journal of clinical investigation 125, 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao D et al. (2015) Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget 6, 40655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dias MM et al. (2020) GLS2 is protumorigenic in breast cancers. Oncogene 39, 690–702. [DOI] [PubMed] [Google Scholar]
- 67.Cassago A et al. (2012) Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proceedings of the National Academy of Sciences 109, 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson MO et al. (2018) Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 175, 1780–1795 e1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leone RD et al. (2019) Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oh MH et al. (2020) Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest 130, 3865–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nabe S et al. (2018) Reinforce the antitumor activity of CD8(+) T cells via glutamine restriction. Cancer science 109, 3737–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edwards DN et al. (2021) Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J. Clin. Invest. 131, e140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharma NS et al. (2020) Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J Clin Invest 130, 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gregory MA et al. (2019) Targeting glutamine metabolism and redox state for leukemia therapy. Clinical Cancer Research 25, 4079–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rashmi R et al. (2020) Glutaminase Inhibitors Induce Thiol-Mediated Oxidative Stress and Radiosensitization in Treatment-Resistant Cervical Cancers. Molecular Cancer Therapeutics 19, 2465–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Katt WP et al. (2017) A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis. Future medicinal chemistry 9, 223–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Altman BJ et al. (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nature Reviews Cancer 16, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martín-Rufián M et al. (2012) Mammalian glutaminase Gls2 gene encodes two functional alternative transcripts by a surrogate promoter usage mechanism. PloS one 7, e38380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Curthoys NP and Watford M (1995) Regulation of glutaminase activity and glutamine metabolism. Annual review of nutrition 15, 133–159. [DOI] [PubMed] [Google Scholar]
- 80.Harding JJ et al. (2015) Safety and tolerability of increasing doses of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase, in solid tumors. J. Clin. Oncol. 33, 2512–2512. [Google Scholar]
- 81.DeMichele A et al. (2016) Phase 1 study of CB-839, a small molecule inhibitor of glutaminase (GLS) in combination with paclitaxel (Pac) in patients (pts) with triple negative breast cancer (TNBC). J. Clin. Oncol. 34, 1011–1011. [Google Scholar]
- 82.Tannir NM et al. (2018) Phase 1 study of glutaminase (GLS) inhibitor CB-839 combined with either everolimus (E) or cabozantinib (Cabo) in patients (pts) with clear cell (cc) and papillary (pap) metastatic renal cell cancer (mRCC). J. Clin. Oncol. 36, 603–603. [Google Scholar]
- 83.Meric-Bernstam F et al. (2019) CB-839, a glutaminase inhibitor, in combination with cabozantinib in patients with clear cell and papillary metastatic renal cell cancer (mRCC): Results of a phase I study. Journal of Clinical Oncology 37, 549–549. [Google Scholar]
- 84.Vogl DT et al. (2015) Phase 1 Study of CB-839, a First-in-Class, Glutaminase Inhibitor in Patients with Multiple Myeloma and Lymphoma. Blood 126, 3059–3059. [Google Scholar]
- 85.Wang ES et al. (2015) Phase 1 Study of CB-839, a First-in-Class, Orally Administered Small Molecule Inhibitor of Glutaminase in Patients with Relapsed/Refractory Leukemia. Blood 126, 2566–2566. [Google Scholar]
- 86.Meric-Bernstam F et al. , CX-839-004: a phase 1/2 study of CB-839, a first-in-class glutaminase inhibitor, combined with nivolumab in patients with advanced melanoma (MEL), renal cell carcinoma (RCC), or non-small cell lung cancer (NSCLC), Society for Immunotherapy of Cancer Annual Meeting, 2017. [Google Scholar]
- 87.Eads JR et al. (2018) Phase I clinical trial of the glutaminase inhibitor CB-839 plus capecitabine in patients with advanced solid tumors. J. Clin. Oncol. 36, 2562–2562. [Google Scholar]
- 88.Guerra VA et al. (2019) Interim Analysis of a Phase II Study of the Glutaminase Inhibitor Telaglenastat (CB-839) in Combination with Azacitidine in Advanced Myelodysplastic Syndrome (MDS). Blood 134, 567–567. [Google Scholar]
- 89.Vidal G et al. (2019) Abstract P6-20-07: Efficacy and safety of CB-839, a small molecule inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer (TNBC): Initial findings from a multicenter, open-label phase 2 study. Cancer Res. 79, P6-20-07-P26-20-07. [Google Scholar]
- 90.Motzer RJ et al. (2019) LBA54 - ENTRATA: Randomized, double-blind, phase II study of telaglenastat (tela; CB-839) + everolimus (E) vs placebo (pbo) + E in patients (pts) with advanced/metastatic renal cell carcinoma (mRCC). Ann. Oncol. 30, v889–v890. [Google Scholar]
- 91.Ciombor KK et al. (2019) CB-839, panitumumab, and irinotecan in RAS wildtype (WT) metastatic colorectal cancer (mCRC): Phase I results. J. Clin. Oncol. 37, 574–574. [Google Scholar]
- 92.Tannir NM et al. (2019) CANTATA: Randomized, international, double-blind study of CB-839 plus cabozantinib versus cabozantinib plus placebo in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 37, TPS682–TPS682. [Google Scholar]
- 93.Kizilbash SH et al. (2019) A phase Ib trial of CB-839 (telaglenastat) in combination with radiation therapy and temozolomide in patients with IDH-mutated diffuse astrocytoma and anaplastic astrocytoma (NCT03528642). J. Clin. Oncol. 37, TPS2075–TPS2075. [Google Scholar]
- 94.Gonsalves WI et al. (2019) Trial in Progress: Phase I Dose-Escalation and Dose-Expansion Trial of a Novel Glutaminase Inhibitor (CB-839 HCl) in Combination with Carfilzomib and Dexamethasone in Relapsed and/or Refractory Multiple Myeloma. Blood 134, 3160–3160. [Google Scholar]