

Abstract
The mitochondrial unfolded protein response (UPRmt) has emerged as a predominant mechanism that preserves mitochondrial function. Consequently, multiple pathways likely exist to modulate UPRmt. We discovered that the tRNA processing enzyme, homolog of ELAC2 (HOE-1), is key to UPRmt regulation in Caenorhabditis elegans. We find that nuclear HOE-1 is necessary and sufficient to robustly activate UPRmt. We show that HOE-1 acts via transcription factors ATFS-1 and DVE-1 that are crucial for UPRmt. Mechanistically, we show that HOE-1 likely mediates its effects via tRNAs, as blocking tRNA export prevents HOE-1-induced UPRmt. Interestingly, we find that HOE-1 does not act via the integrated stress response, which can be activated by uncharged tRNAs, pointing toward its reliance on a new mechanism. Finally, we show that the subcellular localization of HOE-1 is responsive to mitochondrial stress and is subject to negative regulation via ATFS-1. Together, we have discovered a novel RNA-based cellular pathway that modulates UPRmt.
Research organism: C. elegans
Introduction
Mitochondria are central to a myriad of cellular processes including energy production, cellular signaling, biogenesis of small molecules, and regulation of cell death via apoptosis (Nunnari and Suomalainen, 2012). Mitochondrial dysfunction can lead to metabolic and neurological disorders, cardiovascular disease, and cancers (Vafai and Mootha, 2012). To maintain proper mitochondrial function cellular mechanisms have evolved that respond to, and mitigate, mitochondrial stress (Baker et al., 2012; Wang and Chen, 2015; Wrobel et al., 2015; Munkácsy et al., 2016; Tjahjono and Kirienko, 2017; Weidberg and Amon, 2018; Naresh and Haynes, 2019; Fessler et al., 2020; Guo et al., 2020).
One of the predominant mitochondrial stress response mechanisms is the mitochondrial unfolded protein response (UPRmt). Although first discovered in mammals (Zhao et al., 2002), UPRmt has been best characterized in Caenorhabditis elegans (Naresh and Haynes, 2019). UPRmt is primarily characterized by transcriptional upregulation of genes whose products respond to and ameliorate mitochondrial stress (Yoneda et al., 2004; Nargund et al., 2012).
In C. elegans, activation of UPRmt relies on the transcription factor ATFS-1 that primarily localizes to mitochondria, but under mitochondrial-stress conditions is trafficked to the nucleus where it drives the expression of mitochondrial stress response genes (Haynes et al., 2010; Nargund et al., 2012; Nargund et al., 2015). However, it has become increasingly apparent that UPRmt is under multiple levels of control: Mitochondrial stress in neurons can activate intestinal UPRmt non-cell-autonomously via retromer-dependent Wnt signaling (Durieux et al., 2011; Berendzen et al., 2016; Zhang et al., 2018); overexpression of two conserved histone demethylases are independently sufficient to activate UPRmt (Merkwirth et al., 2016); and ATFS-1 is post-translationally modified to facilitate its stability and subsequent UPRmt activation (Gao et al., 2019). Given mitochondrial integration into many diverse cellular signaling and metabolic pathways, there are likely yet-to-be identified pathways regulating UPRmt.
In conducting a small-scale RNAi screen to interrogate the effects of perturbing mitochondrial RNA processing we discovered that the 3’-tRNA zinc phosphodiesterase, homolog of ELAC2 (HOE-1), is a key regulator of UPRmt in C. elegans. ELAC2 is an essential endonuclease that cleaves 3’-trailer sequences from nascent tRNAs—a necessary step of tRNA maturation—in both nuclei and mitochondria (Nashimoto et al., 1999; Mayer et al., 2000; Schiffer et al., 2002; Takaku et al., 2003; Dubrovsky et al., 2004; Brzezniak et al., 2011; Sanchez et al., 2011; Siira et al., 2018). ELAC2 has also been reported to cleave other structured RNAs yielding tRNA fragments, small nucleolar RNAs (snoRNAs) and micro RNAs (miRNAs) (Kruszka et al., 2003; Lee et al., 2009; Bogerd et al., 2010; Siira et al., 2018). In humans, mutations in ELAC2 are associated with hypertrophic cardiomyopathy (Haack et al., 2013; Shinwari et al., 2017; Saoura et al., 2019) and prostate cancer (Tavtigian et al., 2001; Korver et al., 2003; Noda et al., 2006) while in C. elegans, loss of HOE-1 has been shown to compromise fertility (Smith and Levitan, 2004).
Surprisingly, we find that it is not the mitochondrial, but rather the nuclear activity of HOE-1 that is required for activation of UPRmt. Remarkably, compromising nuclear export of HOE-1 is sufficient to specifically and robustly activate UPRmt. Blocking tRNA export from the nucleus suppresses this HOE-1-dependent UPRmt induction, suggesting that HOE-1 generates RNA species required in the cytosol to trigger UPRmt. Finally, we show that HOE-1 nuclear levels are dynamically regulated under conditions of mitochondrial stress, supporting a physiological role for HOE-1 in mitochondrial stress response. Taken together, our results provide a novel mechanism by which UPRmt is regulated as well as provide critical insight into the biological role of the conserved tRNA processing enzyme, HOE-1.
Results
hoe-1 is required for maximal UPRmt activation
We discovered that RNAi against hoe-1, a gene encoding a 3’-tRNA phosphodiesterase, attenuates hsp-6p::GFP induction—a fluorescence based transcriptional reporter of UPRmt activation (Yoneda et al., 2004). Knockdown of hoe-1 by RNAi is sufficient to attenuate UPRmt reporter activation induced by a loss-of-function mutation in the mitochondrial electron transport chain (ETC) complex I subunit NUO-6 (nuo-6(qm200)) (Figure 1A and B).
Figure 1. hoe-1 is required for maximal UPRmt activation.
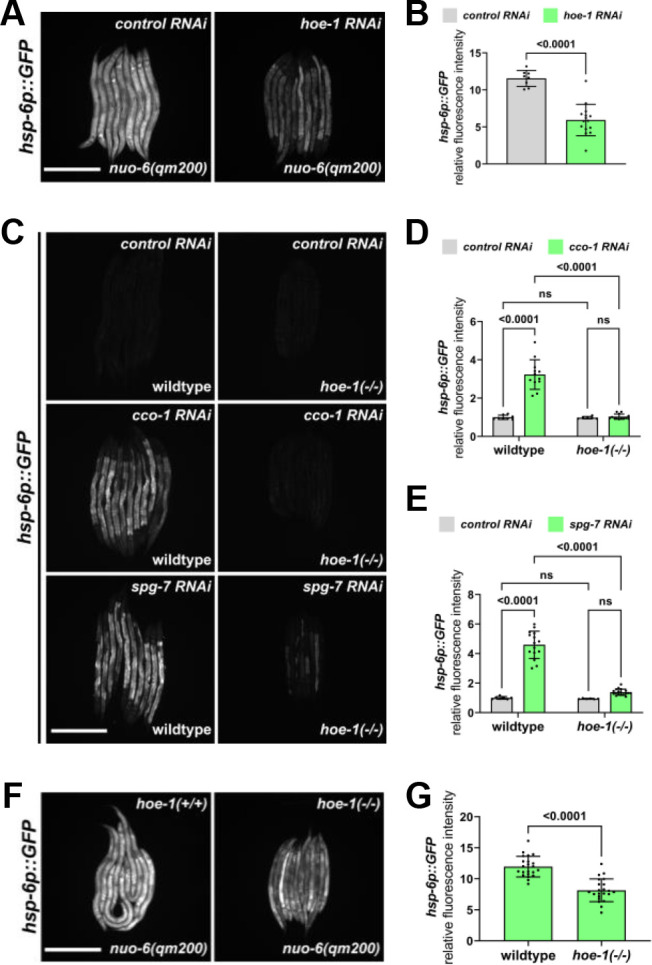
(A) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in L4 nuo-6(qm200) animals on control and hoe-1 RNAi. Scale bar 200 μm. (B) Fluorescence intensity quantification of hsp-6p::GFP in individual L4 nuo-6(qm200) animals on control and hoe-1 RNAi normalized to hsp-6p::GFP in a wildtype background on control RNAi (n = 8 and 15 respectively, mean and SD shown, unpaired t-test). (C) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in L3/L4 wildtype and hoe-1 null (hoe-1(-/-)) animals on control, cco-1, and spg-7 RNAi. Scale bar 200 μm. (D) Fluorescence intensity quantification of hsp-6p::GFP in individual L3/L4 wildtype and hoe-1(-/-) animals on control and cco-1 RNAi (n = 8,12,6 and 13 respectively, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (E) Fluorescence intensity quantification of hsp-6p::GFP in individual L3/L4 wildtype and hoe-1(-/-) animals on control and spg-7 RNAi (n = 7,15,6 and 18 respectively, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (F) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in L3/L4 nuo-6(qm200) animals with (hoe-1(+/+)) and without (hoe-1(-/-)) hoe-1. Scale bar 200 μm. (G) Fluorescence intensity quantification of hsp-6p::GFP in individual L3/L4 nuo-6(qm200) animals with (hoe-1(+/+)) and without (hoe-1(-/-)) hoe-1 normalized to hsp-6p::GFP in a wildtype background (n = 22 for each condition, mean and SD shown, unpaired t-test).
To further interrogate the potential role of hoe-1 in UPRmt regulation, we used CRISPR/Cas9 to generate a hoe-1 null mutant (hoe-1(-/-)) by deleting the open reading frame of hoe-1 (Dokshin et al., 2018). The hoe-1 null mutants do not develop past late larval stage 3, thus the allele is maintained over a balancer chromosome, tmC25 (Dejima et al., 2018). UPRmt induced by the knockdown of both the mitochondrial protease, spg-7, and ETC complex IV subunit, cco-1, is robustly attenuated in hoe-1 null animals (Figure 1C–E). Furthermore, UPRmt induced by nuo-6(qm200) is attenuated in hoe-1 null animals similarly to what is seen in nuo-6(qm200) animals on hoe-1 RNAi (Figure 1F and G). Taken together, these findings suggest that HOE-1 is generally required for maximal UPRmt activation.
HOE-1 is dual-targeted to nuclei and mitochondria
To better understand the role of HOE-1 in UPRmt regulation, we sought to identify where HOE-1 functions in the cell. HOE-1 is predicted to localize to both nuclei and mitochondria and this dual-localization has been shown for HOE-1 orthologs in Drosophila, mice, and human cell lines (Dubrovsky et al., 2004; Brzezniak et al., 2011; Rossmanith, 2011; Siira et al., 2018). To determine the subcellular localization of HOE-1 in C. elegans, we C-terminally tagged HOE-1 with GFP at its endogenous locus (HOE-1::GFP). Both hoe-1::GFP homozygous and hoe-1::GFP/hoe-1(-/-) trans-heterozygous animals grow and develop indistinguishably from wildtype animals suggesting that GFP-tagging HOE-1 does not compromise its essential functions (Figure 2—figure supplement 1A). We found that HOE-1 localizes to both mitochondria and nuclei (Figure 2A).
Figure 2. Nuclear HOE-1 is required for maximal UPRmt activation.
(A) Fluorescence images of a terminal intestinal cell in a wildtype animal expressing HOE-1::GFP (green) stained with TMRE (magenta) to visualize mitochondria. GFP and TMRE co-localization shown in white in merged image. Arrow indicates nuclei. Scale bar 20 μm. Representative line segment analysis of individual mitochondrion. (B) Schematic of HOE-1 protein showing the mitochondrial targeting sequence (MTS) and nuclear localization signals (NLS). ΔMTS allele created by replacing START codon with an alanine (M1A). Transcription begins at M74 for nuclear localized HOE-1. ΔNLS allele created by compromising the most N-terminal NLS (636KRPR > AAPA). (C) Fluorescence images of UPRmt reporter (hsp-6p::GFP) in L4 wildtype and hoe-1(ΔMTS) animals on control and spg-7 RNAi. Scale bar 200 μm. (D) Fluorescence intensity quantification of hsp-6p::GFP in individual L4 wildtype and hoe-1(ΔMTS) animals on control and spg-7 RNAi (n = 15,20,17, and 19 respectively, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (E) Fluorescence images of UPRmt reporter (hsp-6p::GFP) in L4 wildtype and hoe-1(ΔNLS) animals on control and spg-7 RNAi. Scale bar 200 μm. (F) Fluorescence intensity quantification of hsp-6p::GFP in individual L4 wildtype and hoe-1(ΔNLS) animals on control and spg-7 RNAi (n = 15 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (G) Fluorescence images of UPRmt reporter in L4 nuo-6(qm200) animals in wildtype and hoe-1(ΔNLS) backgrounds. Scale bar 200 μm. (H) Fluorescence intensity of hsp-6p::GFP in individual L4 nuo-6(qm200) animals in wildtype and hoe-1(ΔNLS) backgrounds (n = 30 for each condition, mean and SD shown, unpaired t-test). (I) mRNA transcript quantification of hsp-6 in L4 wildtype and hoe-1(ΔNLS) animals on control and spg-7 RNAi normalized to ama-1 (n = 4 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test).

Figure 2—figure supplement 1. hoe-1::GFP does not compromise growth or development and is sufficient to rescue the developmental arrest of hoe-1(-/-) animals.
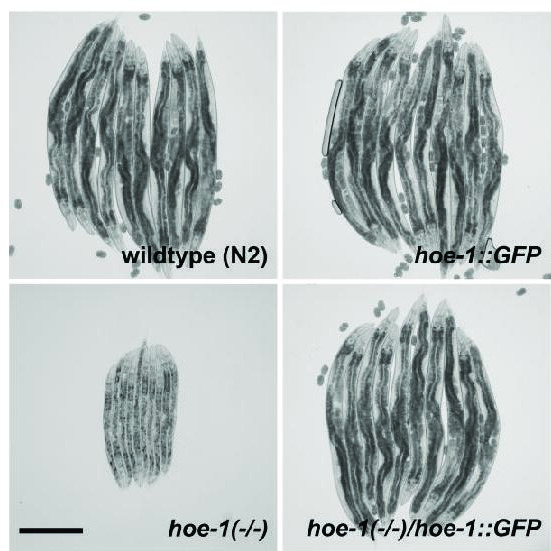
Figure 2—figure supplement 2. hoe-1(ΔMTS) allele attenuates HOE-1 mitochondrial localization.
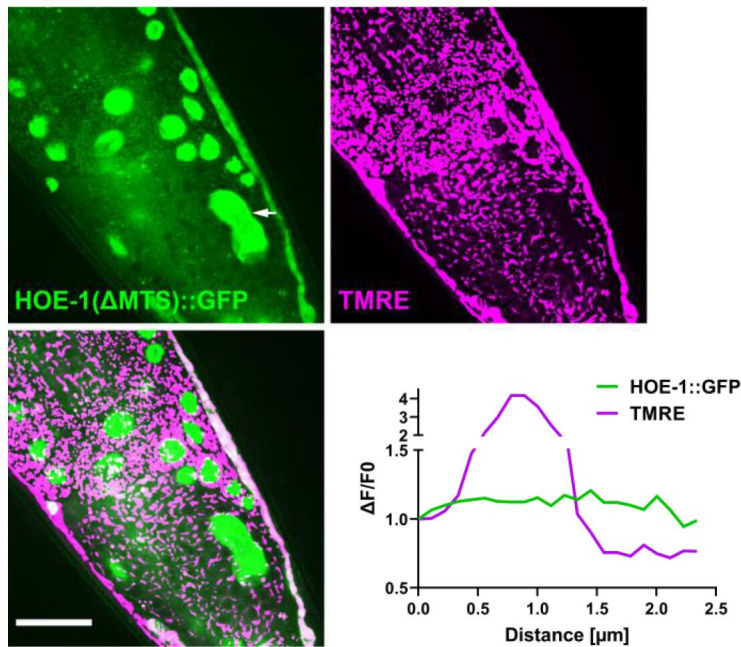
Figure 2—figure supplement 3. hoe-1(ΔMTS) does not attenuate cco-1 RNAi-induced UPRmt.

Figure 2—figure supplement 4. hoe-1(ΔNLS) allele attenuates nuclear HOE-1 localization.

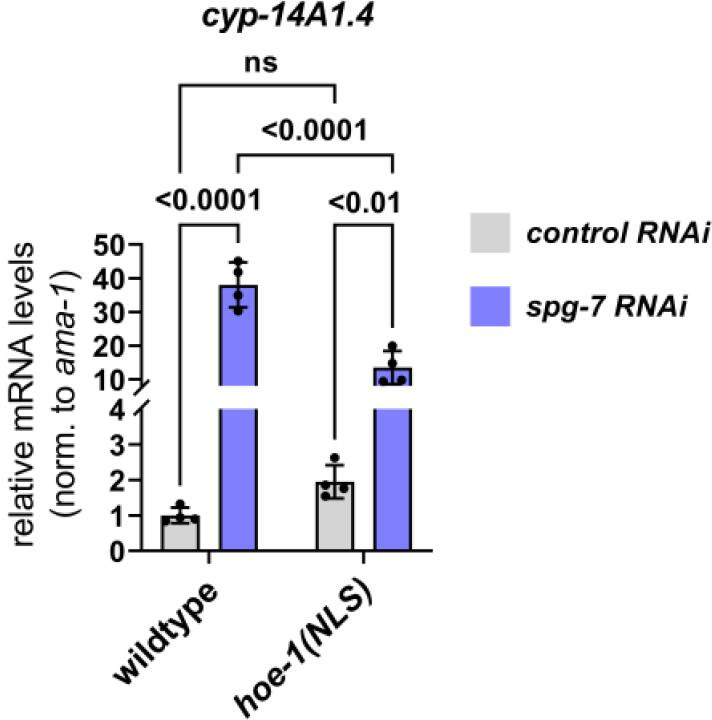
Figure 2—figure supplement 5. UPRmt-responsive gene cyp-14A1.4 is downregulated under mitochondrial stress conditions in hoe-1(ΔNLS) animals relative to wildtype.
Mitochondrial HOE-1 is not required for UPRmt activation
Given the dual-localization of HOE-1, we questioned whether it is mitochondrial or nuclear HOE-1 that is required for UPRmt activation. To address this question, we created mitochondrial and nuclear compartment-specific loss-of-function mutants of HOE-1 (Figure 2B). hoe-1 contains two functional start codons. Translation beginning from the first start codon (encoding methionine 1 (M1)) produces a protein containing a mitochondrial targeting sequence (MTS). Translation beginning from the second start codon (encoding methionine 74 (M74)), which is 3’ to the MTS, produces a nuclear specific protein. This feature is conserved in human ELAC2 and it has been shown that mutating M1 to an alanine produces a mitochondrial-specific knockout (Brzezniak et al., 2011). Thus, we used the same approach to create a mitochondrial-specific knockout of C. elegans HOE-1 (hoe-1(ΔMTS)). This mutation is sufficient to strongly attenuate mitochondrial targeting without impacting nuclear localization (Figure 2—figure supplement 2A).
UPRmt reporter activation by spg-7 and cco-1 RNAi is not attenuated in hoe-1(ΔMTS) animals (Figure 2C and D and Figure 2—figure supplement 3A and B). In fact, UPRmt reporter activation is slightly elevated in hoe-1(ΔMTS) animals relative to wildtype. These data suggest that mitochondrial HOE-1 is not required for UPRmt activation.
Nuclear HOE-1 is required for UPRmt activation
HOE-1 is predicted to contain two nuclear localization signals (NLS). Given that hoe-1 null mutant animals are developmentally arrested and hoe-1(ΔMTS) animals are superficially wildtype we reasoned that completely ablating nuclear localization of HOE-1 may result in recapitulation of the null phenotype. In effort to disentangle the developmental effects from the effect on UPRmt we ablated only one of the nuclear localization signals of HOE-1. To compromise nuclear localization, we mutated the positively charged residues of the most N-terminal NLS to alanines (hoe-1(ΔNLS)). These mutations are sufficient to strongly attenuate, but not completely ablate, HOE-1 nuclear localization whilst still allowing animals to develop to adulthood (Figure 2—figure supplement 4A–C).
In contrast to loss of mitochondrial HOE-1, loss of nuclear HOE-1 robustly attenuates UPRmt reporter activation induced by spg-7 RNAi (Figure 2E and F) and attenuates UPRmt reporter activation induced by nuo-6(qm200) (Figure 2G and H). Furthermore, loss of nuclear HOE-1 attenuates the transcriptional upregulation of UPRmt target genes hsp-6 (Figure 2I) and cyp-14A1.4 (Figure 2—figure supplement 5A) under conditions of mitochondrial stress. Together these data suggest that HOE-1 is required in the nucleus to facilitate UPRmt activation.
Compromising HOE-1 nuclear export is sufficient to activate UPRmt
Like many nuclear localized proteins (la Cour et al., 2004), HOE-1 has both nuclear localization signals and a nuclear export signal (NES). Given that loss of nuclear HOE-1 results in UPRmt attenuation we questioned if compromising HOE-1 nuclear export, by ablating the NES of HOE-1, is sufficient to activate UPRmt. We created a HOE-1 NES knockout mutant (hoe-1(ΔNES)) by replacing the strong hydrophobic residues of the predicted NES with alanines (Figure 3—figure supplement 1A). hoe-1(ΔNES) animals are superficially wildtype in their development but are sterile. Thus, the allele is balanced with tmC25. Homozygous hoe-1(ΔNES) animals have elevated nuclear HOE-1 accumulation relative to wildtype (Figure 3—figure supplement 1B, Figure 2—figure supplement 4B and C).
Strikingly, the UPRmt reporter hsp-6p::GFP is robustly activated in adult hoe-1(ΔNES) animals similarly to that seen in mitochondrial stressor nuo-6(qm200) and constitutive UPRmt activation in atfs-1 gain-of-function (atfs-1(et15)) mutant animals (Figure 3A and B). hoe-1(ΔNES) also mildly induces the less sensitive UPRmt reporter hsp-60p::GFP (Figure 3—figure supplement 2A and B).
Figure 3. Nuclear export defective HOE-1 is sufficient to specifically activate UPRmt.
(A) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in day 2 adult wildtype, nuo-6(qm200), atfs-1(et15), and hoe-1(ΔNES) animals. Scale bar 200 μm. (B) Fluorescence intensity quantification of hsp-6p::GFP in individual day 2 adult wildtype, nuo-6(qm200), atfs-1(et15), and hoe-1(ΔNES) animals (n = 10 for each condition, mean and SD shown, ordinary one-way ANOVA with Tukey’s multiple comparisons test). (C–E) mRNA transcript quantification of hsp-6, clec-47, and cyp-14A1.4, respectively, in day 2 adult wildtype and hoe-1(ΔNES) animals normalized to ama-1 mRNA levels (n = 4 for each condition, mean and SD shown, unpaired t-test). (F) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in day 2 adult hoe-1(ΔNES) animals on control and atfs-1 RNAi. Scale bar 200 μm. (G) Fluorescence intensity quantification of hsp-6p::GFP in individual day 2 adult wildtype and hoe-1(ΔNES) animals on control and atfs-1 RNAi (n = 10 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (H) Fluorescence images of UPRER reporter (hsp-4p::GFP) activation in day 2 adult wildtype and hoe-1(ΔNES) animals. Scale bar 200 μm. (I) Fluorescence intensity quantification of hsp-4p::GFP in individual day 2 adult wildtype and hoe-1(ΔNES) animals (n = 10 for each condition, mean and SD shown, unpaired t-test). (J) Fluorescence images of intestinal-specific basal protein reporter (ges-1p::GFPcyto) activation in day 2 adult wildtype and hoe-1(ΔNES) animals. Scale bar 200 μm. (K) Fluorescence intensity quantification of ges-1p::GFPcyto in individual day 2 adult wildtype and hoe-1(ΔNES) animals (n = 10 for each condition, mean and SD shown, unpaired t-test).

Figure 3—figure supplement 1. Nuclear export defective HOE-1 has increased nuclear accumulation relative to wildtype.
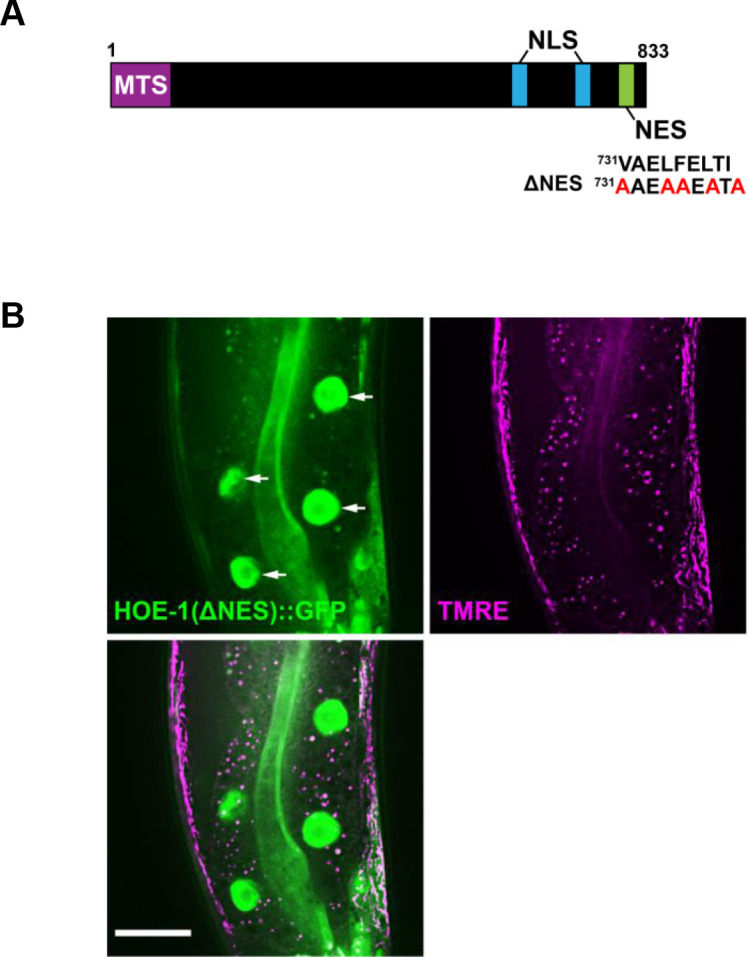
Figure 3—figure supplement 2. Nuclear export defective HOE-1 activates UPRmt.

Figure 3—figure supplement 3. Compromised nuclear import of HOE-1 completely attenuates hoe-1(ΔNES)-induced UPRmt.

Figure 3—figure supplement 4. Compromised mitochondrial import of HOE-1 exacerbates hoe-1(ΔNES)-induced UPRmt.

Figure 3—figure supplement 5. Nuclear export defective HOE-1 activates UPRmt in the intestine cell autonomously.

UPRmt activation is characterized by the transcriptional upregulation of a suite of mitochondrial stress response genes that encode chaperone proteins, proteases, and detoxification enzymes that function to restore mitochondrial homeostasis (Nargund et al., 2012). To interrogate the extent of UPRmt induction in hoe-1(ΔNES) animals, we measured transcript levels of a diverse set of UPRmt associated genes. We found that the UPRmt genes encoding a chaperone protein (hsp-6), stress response involved C-type lectin (clec-47), and P450 enzyme (cyp-14A4.1) are all upregulated in hoe-1(ΔNES) animals (Figure 3C, D and E). These data support hoe-1(ΔNES) being sufficient to activate the UPRmt transcriptional response.
UPRmt activation is dependent upon the transcription factor ATFS-1 (Haynes et al., 2010; Nargund et al., 2012). Thus, we tested if UPRmt reporter activation in hoe-1(ΔNES) animals is ATFS-1 dependent. Knockdown of atfs-1 is sufficient to completely attenuate UPRmt reporter activation in hoe-1(ΔNES) animals (Figure 3F and G), showing that UPRmt induction by hoe-1(ΔNES) is ATFS-1 dependent.
Elevated nuclear HOE-1 levels in hoe-1(ΔNES) animals is likely responsible for UPRmt activation
To further interrogate how UPRmt is activated in hoe-1(ΔNES) animals, we made double localization mutants of hoe-1. If UPRmt is activated in hoe-1(ΔNES) animals due to elevated nuclear HOE-1 levels we reasoned that introducing a hoe-1(ΔNLS) mutation in the hoe-1(ΔNES) background (hoe-1(ΔNLS+ΔNES)) should be sufficient to attenuate UPRmt activation. Indeed, hoe-1(ΔNLS+ΔNES) animals have UPRmt reporter activation comparable to wildtype animals (Figure 3—figure supplement 3A and B). Furthermore, we reasoned that compromising mitochondrial localization of HOE-1 in a hoe-1(ΔNES) background (hoe-1(ΔMTS+ΔNES)) may further enhance hoe-1(ΔNES)-induced UPRmt activation as what would be the mitochondrial targeted HOE-1 pool should be diverted to the nucleus as well. Consistent with our hypothesis, hoe-1(ΔMTS+ΔNES) animals have even higher activation of UPRmt than hoe-1(ΔNES) alone (Figure 3—figure supplement 4A and B). Taken together, these data strongly suggest that hoe-1(ΔNES) triggers UPRmt activation due to elevated nuclear HOE-1 levels.
Compromising HOE-1 nuclear export activates UPRmt cell-autonomously in the intestine
Contrary to UPRmt induced by nuo-6(qm200) and atfs-1(et15), hoe-1(ΔNES) animals appear to have UPRmt activated specifically in the intestine (Figure 3A). We questioned if this UPRmt activation is occurring cell autonomously or non-cell autonomously as UPRmt has been shown to be able to be signaled across tissues, particularly from neurons to intestine (Durieux et al., 2011; Berendzen et al., 2016; Zhang et al., 2018). To determine which tissue HOE-1 is required in for UPRmt activation we took advantage of the auxin-inducible degradation (AID) system that allows for tissue-specific protein degradation (Zhang et al., 2015). Briefly, degron-tagged proteins will be degraded in the presence of the plant hormone auxin but only in tissues wherein E3 ubiquitin ligase subunit, TIR1, is expressed. We C-terminally degron-tagged hoe-1(ΔNES) (hoe-1(ΔNES)::degron) and crossed this allele into backgrounds in which TIR1 is driven under an intestinal-specific (ges-1p::TIR1) or a neuronal-specific (rgef-1p::TIR) promoter (Ashley et al., 2021). hoe-1(ΔNES)-induced UPRmt is only attenuated when HOE-1 is selectively degraded in the intestine (Figure 3—figure supplement 5A and B). This data strongly suggests that compromised nuclear export of HOE-1 activates UPRmt cell-autonomously in the intestine.
Compromising HOE-1 nuclear export specifically activates UPRmt
Changes in protein synthesis rates and associated protein folding capacity can broadly activate cellular stress response mechanisms (Wang and Kaufman, 2016; Das et al., 2017; Boos et al., 2019). Given the role of hoe-1 in tRNA maturation, we questioned if the robust upregulation of UPRmt in hoe-1(ΔNES) animals may be the result of compromised cellular proteostasis in general rather than specific activation of UPRmt. One stress response mechanism that is sensitive to global proteotoxic stress is the endoplasmic reticulum unfolded protein response (UPRER) (Preissler and Ron, 2019). We find that the UPRER reporter hsp-4p::GFP is not activated in hoe-1(ΔNES) animals (Figure 3H and I), suggesting that hoe-1(ΔNES) does not cause ER stress nor cellular proteotoxic stress. Additionally, a basal reporter of GFP that has been used to proxy general protein expression (Gitschlag et al., 2016), ges-1p::GFPcyto, is only mildly upregulated in hoe-1(ΔNES) animals relative to wildtype (Figure 3J and K). Together these findings support that impaired nuclear export of HOE-1 specifically activates UPRmt.
Compromising HOE-1 nuclear export reduces mitochondrial membrane potential
UPRmt is known to be triggered when mitochondrial membrane potential is compromised (Rolland et al., 2019; Shpilka et al., 2021). Thus, we assessed mitochondrial membrane potential, using TMRE staining, in adult hoe-1(ΔNES) animals where UPRmt is robustly activated. Consistent with UPRmt activation, we found that mitochondrial membrane potential is severely depleted in adult hoe-1(ΔNES) animals (Figure 4A and B). However, hoe-1(ΔNLS) animals also exhibit compromised mitochondrial membrane potential without UPRmt activation suggesting that decreased membrane potential does not guarantee UPRmt induction (Figure 4A and B). Compromised mitochondrial membrane potential can be both a cause and consequence of UPRmt activation (Rolland et al., 2019; Shpilka et al., 2021). Thus, we assessed whether or not compromised membrane potential in hoe-1(ΔNES) animals is atfs-1-dependent. Mitochondrial membrane potential is not rescued in hoe-1(ΔNES) animals on atfs-1 RNAi (Figure 4C and D) suggesting that reduced mitochondrial membrane potential in hoe-1(ΔNES) animals is not a result of UPRmt activation. Taken together, these data show that compromised nuclear export of HOE-1 results in depletion of mitochondrial membrane potential. Furthermore, this depletion in membrane potential correlates with UPRmt activation, consistent with the possibility that hoe-1(ΔNES) activates UPRmt via depletion of mitochondrial membrane potential.
Figure 4. Nuclear export defective HOE-1 activates UPRmt, correlating with reduced mitochondrial membrane potential.

(A) Fluorescence images of TMRE stained day 1 adult wildtype, hoe-1(ΔNES), and hoe-1(ΔNLS) individuals. Scale bar 20 μm. (B) Fluorescence intensity quantification of TMRE staining in individual day 1 adult wildtype, hoe-1(ΔNES), and hoe-1(ΔNLS) animals (n = 57, 60, and 63 respectively, mean and SD shown, ordinary one-way ANOVA with Tukey’s multiple comparisons test). (C) Fluorescence images of TMRE stained day 1 adult wildtype and hoe-1(ΔNES) animals on control and atfs-1 RNAi. Scale bar 20 μm. (D) Fluorescence intensity quantification of TMRE staining in individual day 1 adult wildtype and hoe-1(ΔNES) animals on control and atfs-1 RNAi (n = 65, 62, 65, and 61 respectively, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test).
Compromising HOE-1 nuclear export elevates nuclear levels of UPRmt transcription factors ATFS-1 and DVE-1
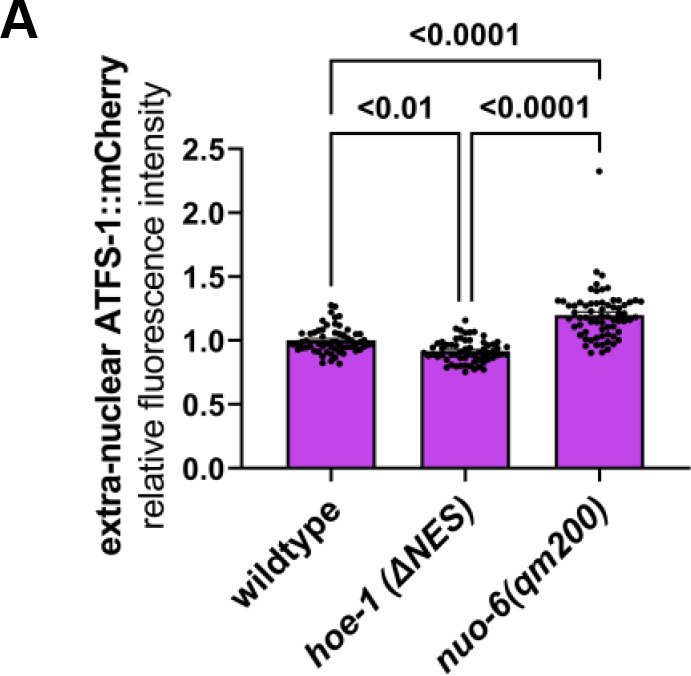
UPRmt activation is dependent upon nuclear accumulation of the transcription factor ATFS-1 (Nargund et al., 2012; Nargund et al., 2015). Thus, we tested if ATFS-1 accumulates in nuclei of hoe-1(ΔNES) animals by assessing the fluorescence intensity of ectopically expressed mCherry-tagged ATFS-1 (atfs-1p::ATFS-1::mCherry) in wildtype, hoe-1(ΔNES), and mitochondrial-stressed nuo-6(qm200) animals. Both hoe-1(ΔNES) and nuo-6(qm200) animals have elevated nuclear accumulation of ATFS-1 relative to wildtype (Figure 5A and B). However, while nuo-6(qm200) animals exhibit elevated levels of total cellular and extranuclear levels of ATFS-1::mCherry relative to wildtype, hoe-1(ΔNES) animals do not (Figure 5C and Figure 5—figure supplement 1A). We find that atfs-1 mRNA levels are also elevated in hoe-1(ΔNES) animals relative to wildtype comparable to that seen in nuo-6(qm200) animals (Figure 5D).
Figure 5. Nuclear export defective HOE-1 animals have increased nuclear accumulation of UPRmt transcription factors ATFS-1 and DVE-1.
(A), Fluorescence images of ATFS-1::mCherry in the terminal intestine of day 2 adult wildtype hoe-1(ΔNES), and nuo-6(qm200) individuals (tip of the tail is in the bottom of each panel). Intestinal nuclei outlined with dashed white line. Scale bar 20 μm. (B) Fluorescence intensity quantification of nuclear ATFS-1::mCherry in wildtype, hoe-1(ΔNES), and nuo-6(qm200) individuals (n = 65, 74, and 72 respectively, mean and SD shown, ordinary one-way ANOVA with Tukey’s multiple comparisons test). (C) Fluorescence intensity quantification of total cellular ATFS-1::mCherry in wildtype, hoe-1(ΔNES), and nuo-6(qm200) individuals (n = 61, 62, and 67 respectively, mean and SD shown, ordinary one-way ANOVA with Tukey’s multiple comparisons test). (D) mRNA transcript quantification of atfs-1 in day 2 adult wildtype, nuo-6(qm200), and hoe-1(ΔNES) animals normalized to ama-1 (n = 4 for each condition, mean and SD shown, ordinary one-way ANOVA with Tukey’s multiple comparisons test). (E) Fluorescence images of dve-1p::DVE-1::GFP in day 2 adult wildtype and hoe-1(ΔNES) animals. Scale bar 200 μm. (F) Number of intestinal cell nuclei with DVE-1::GFP puncta above brightness threshold of 25 in day 2 adult wildtype and hoe-1(ΔNES) animals (n = 33 and 41 respectively, unpaired t-test). (G) Western blot for DVE-1::GFP and actin from day 1 adult wildtype and hoe-1(ΔNES) animals. (H) Quantification of DVE-1::GFP western blot band intensity from day 1 adult wildtype and hoe-1(ΔNES) animals normalized to total protein (n = 4 for each condition, mean and SD shown, unpaired t-test).

Figure 5—figure supplement 1. Nuclear export defective HOE-1 does not elevate extra-nuclear ATFS-1::mCherry levels.
The transcription factor DVE-1 is required for full UPRmt activation (Haynes et al., 2007; Tian et al., 2016). Thus, we asked if DVE-1::GFP nuclear expression is higher in hoe-1(ΔNES) than in wildtype animals. We found that accumulation of DVE-1::GFP in intestinal cell nuclei is significantly higher in hoe-1(ΔNES) than in wildtype animals (Figure 5E and F). Qualitatively, cellular DVE-1::GFP levels appear mildly elevated in hoe-1(ΔNES) animals based on actin (Figure 5G, Figure 5—source data 1), though the difference in DVE-1::GFP levels is not significant when normalized to total protein (Figure 5H). Thus, while we cannot rule out the possibility of a slight increase in the cellular levels of DVE-1, elevation in the nuclear localization of DVE-1 in hoe-1(ΔNES) animals is the more robust phenotype. Together, these data suggest that UPRmt induction in hoe-1(ΔNES) animals is a result of increased nuclear accumulation of UPRmt transcription factors ATFS-1 and DVE-1.
UPRmt is activated by altered tRNA processing in animals with compromised HOE-1 nuclear export
The canonical role of HOE-1 is to cleave 3’-trailer sequences from nascent tRNAs (Nashimoto et al., 1999; Mayer et al., 2000; Schiffer et al., 2002; Takaku et al., 2003; Dubrovsky et al., 2004; Brzezniak et al., 2011; Sanchez et al., 2011; Siira et al., 2018). This enzymatic function is dependent upon zinc binding (Ma et al., 2017; Bienert et al., 2017). Thus, we queried if UPRmt activation by hoe-1(ΔNES) is dependent upon the catalytic activity of HOE-1. To test this, we generated a catalytically-dead HOE-1 mutant by changing an essential aspartate of the zinc-binding pocket of HOE-1 to alanine in both a wildtype (hoe-1(D624A)) and hoe-1(ΔNES) (hoe-1(D624A+ΔNES)) background. Animals homozygous for D624A recapitulate the growth arrest phenotype of the hoe-1 null mutant precluding us from assessing the impact of D624A on UPRmt induction in adult hoe-1(ΔNES) animals. To overcome this constraint, we assessed UPRmt activation in hoe-1(ΔNES) versus hoe-1(ΔNES)/hoe-1(D624A+ΔNES) trans-heterozygous animals. A single copy of catalytically dead hoe-1 is sufficient to attenuate hoe-1(ΔNES)-induced UPRmt (Figure 6—figure supplement 1A and B). These data suggest that hoe-1(ΔNES)-induced UPRmt requires the catalytic activity of HOE-1.
Given that HOE-1 catalytic activity is required for UPRmt, we further interrogated the potential role of tRNA processing as a mechanism by which HOE-1 may modulate UPRmt induction. Production of mature tRNAs begins with transcription of tRNA gene loci by RNA polymerase III followed by sequential cleavage of 5’-leader and 3’-trailer sequences from immature tRNA transcripts by RNase P and HOE-1, respectively. Following cleavage of 3’-trailer sequences, tRNAs can be transported to the cytosol by tRNA exportin (Hopper and Nostramo, 2019).
Given that HOE-1 nuclear levels are elevated in hoe-1(ΔNES) animals, we reasoned that 3’-tRNA processing should be elevated due to increased nuclear activity of HOE-1. Thus, we questioned if UPRmt induction in hoe-1(ΔNES) animals is a result of elevated 3’-tRNA processing. First, we knocked-down RNA pol III subunit rpc-1 to attenuate the production of total RNA pol III-dependent transcripts in hoe-1(ΔNES) animals. If hoe-1(ΔNES)-induced UPRmt is due to elevated processing of tRNAs we hypothesized that restriction of nascent tRNA production should attenuate UPRmt activation. Indeed, we found that rpc-1 RNAi robustly attenuates hoe-1(ΔNES)-induced UPRmt (Figure 6—figure supplement 2A and B). Interestingly, rpc-1 RNAi has little impact on mitochondrial stress-induced UPRmt (nuo-6(qm200)) (Figure 6—figure supplement 2C and D). These data show that rpc-1 is required for hoe-1(ΔNES)-induced UPRmt and support our hypothesis that increased 3’-tRNA processing by HOE-1 activates UPRmt.
For the majority of tRNAs 5’-end processing by the RNase P complex is a prerequisite for 3’-end processing by HOE-1 (Frendewey et al., 1985; Yoo and Wolin, 1997). Thus, if increased 3’-tRNA end processing is responsible for UPRmt activation, compromising 5’-end processing by RNAi against RNAse P should attenuate hoe-1(ΔNES)-induced UPRmt. RNAi against a subunit of the RNase P complex, popl-1, attenuates UPRmt induction in hoe-1(ΔNES) animals (Figure 6A and B). popl-1 RNAi also attenuates both UPRmt induced by nuo-6(qm200) (Figure 6C and D) as well as basal induction of ges-1p::GFPcyto (Figure 6E and F), albeit to a lesser extent than the attenuation seen in hoe-1(ΔNES) animals. These data suggest that popl-1 RNAi may have a broad impact on protein expression but supports that elevated 3’-tRNA processing in hoe-1(ΔNES) animals is responsible for UPRmt activation given that popl-1 RNAi strongly attenuates hoe-1(ΔNES)-induced UPRmt.
Figure 6. Nuclear export defective HOE-1 activates UPRmt via altered tRNA processing.
(A) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in day 2 adult wildtype and hoe-1(ΔNES) animals on control and popl-1 RNAi. Scale bar 200 μm. (B) Fluorescence intensity quantification of hsp-6p::GFP in individual day 2 adult wildtype and hoe-1(ΔNES) animals on control and popl-1 RNAi (n = 24 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (C) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in day 2 adult wildtype and nuo-6(qm200) animals on control and popl-1 RNAi. Scale bar 200 μm. (D) Fluorescence intensity quantification of hsp-6p::GFP in individual day 2 adult wildtype and nuo-6(qm200) animals on control and popl-1 RNAi (n = 24 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (E) Fluorescence images of intestinal-specific basal protein reporter (ges-1p::GFPcyto) activation in day 2 adult wildtype animals on control and popl-1 RNAi. Scale bar 200 μm. (F) Fluorescence intensity quantification of ges-1p::GFPcyto in individual day 2 adult wildtype animals on control and popl-1 RNAi (n = 24 for each condition, mean and SD shown, unpaired t-test). (G) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in day 2 adult wildtype and hoe-1(ΔNES) animals on control and xpo-3 RNAi. Scale bar 200 μm. (H) Fluorescence intensity quantification of hsp-6p::GFP in individual day 2 adult wildtype and hoe-1(ΔNES) animals on control and xpo-3 RNAi (n = 24 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (I) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in day 2 adult wildtype and nuo-6(qm200) animals on control and xpo-3 RNAi. Scale bar 200 μm. (J) Fluorescence intensity quantification of hsp-6p::GFP in individual day 2 adult wildtype and nuo-6(qm200) animals on control and xpo-3 RNAi (n = 24 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (K) Fluorescence images of intestinal-specific basal protein reporter (ges-1p::GFPcyto) activation in day 2 adult wildtype animals on control and xpo-3 RNAi. Scale bar 200 μm. (L) Fluorescence intensity quantification of ges-1p::GFPcyto in individual day 2 adult wildtype animals on control and xpo-3 RNAi (n = 24 for each condition, mean and SD shown, unpaired t-test).

Figure 6—figure supplement 1. Nuclear export defective HOE-1 induced UPRmt is dependent upon the catalytic activity of HOE-1.

Figure 6—figure supplement 2. RNAi against RNA polymerase III subunit, rpc-1, preferentially attenuates hoe-1(ΔNES)-induced UPRmt.

Figure 6—figure supplement 3. RNAi against tRNA nucleotidyl transferase, hpo-31, and tRNA ligase, rtcb-1, mildly attenuate both hoe-1(ΔNES)- and nuo-6(qm200)-induced UPRmt.

Following 3’-end processing in the nuclei, tRNAs can be exported to the cytosol by tRNA exportin (Hopper and Nostramo, 2019). To test if elevated levels of 3’-processed tRNAs are required in the cytosol to activate UPRmt, we asked if restricting tRNA nuclear export via RNAi against tRNA exportin, xpo-3, attenuates hoe-1(ΔNES)-induced UPRmt. Strikingly, xpo-3 RNAi robustly attenuates hoe-1(ΔNES)-induced UPRmt(Figure 6G and H). However, xpo-3 RNAi does not attenuate nuo-6(qm200) induced UPRmt (Figure 6I and J) nor basal ges-1p::GFP levels (Figure 6K and L). These data suggest that in hoe-1(ΔNES) animals 3’-processed tRNAs are required in the cytosol to activate UPRmt.
While 5’- and 3’-tRNA processing are the only steps known to be required for tRNA export from the nucleus, there are other downstream tRNA maturation processes that occur (Hopper and Nostramo, 2019). Some nascent tRNAs include introns that need to be removed and then ligated by a tRNA ligase (Englert and Beier, 2005; Popow et al., 2012). For tRNAs to be charged with corresponding amino acids, nascent tRNAs must contain a CCA sequence as part of the 3’ acceptor stem. This can be achieved by a CCA-adding tRNA nucleotidyl transferase (Hou, 2010). Knockdown of both tRNA ligase, rtcb-1, and tRNA nucleotidyl transferase, hpo-31 mildly attenuate hoe-1(ΔNES)-induced UPRmt (Figure 6—figure supplement 3A–C). However, rtcb-1 RNAi also mildly attenuates nuo-6(qm200)-induced UPRmt (Figure 6—figure supplement 3D and E). Knockdown of hpo-31 severely compromised growth of nuo-6(qm200) animals and thus the impact on UPRmt could not accurately be assessed. These data suggest that tRNA ligation and CCA addition have limited involvement in hoe-1(ΔNES)-induced UPRmt.
Taken together, these data suggest that UPRmt induction by nuclear export deficient HOE-1 is the result of increased 3’-tRNA processing and that these tRNA species are required in the cytosol to trigger UPRmt.
Compromised HOE-1 nuclear export does not activate UPRmt via GCN2 or eIF2α
Alteration to tRNA processing can activate cellular signaling pathways (Raina and Ibba, 2014). One such pathway is the integrated stress response (ISR) in which uncharged tRNAs activate the kinase GCN2 which, in turn, phosphorylates the eukaryotic translation initiation factor, eIF2α. This inhibitory phosphorylation of eIF2α leads to upregulation of a select number of proteins including the transcription factor ATF4 (Pakos-Zebrucka et al., 2016; Costa-Mattioli and Walter, 2020). Interestingly, ATF4 and one of its targets, ATF5, are orthologs of ATFS-1 (Fiorese et al., 2016). Moreover, GCN2 and ISR in general have been shown to be responsive to mitochondrial stress (Baker et al., 2012; Fessler et al., 2020; Guo et al., 2020; Koncha et al., 2021). Thus, we questioned if UPRmt activation by hoe-1(ΔNES) is mediated via GCN2 and eIF2α phosphorylation. We found that hoe-1(ΔNES)-induced UPRmt is only slightly reduced in both a gcn-2 null (gcn-2(ok871)) and an eIF2α non-phosphorylatable mutant (eIF2α(S46A,S49A)) background (Figure 7A and B). These data suggest that a mechanism independent of ISR must largely be responsible for UPRmt activation by hoe-1(ΔNES) animals.
Figure 7. Nuclear export defective HOE-1 induced UPRmt is not gcn-2 or eIF2α dependent.

(A) Fluorescence images of UPRmt reporter (hsp-6p::GFP) activation in day 2 adult wildtype, gcn-2(ok871), eIF2α(S46A,S49A), hoe-1(ΔNES), hoe-1(ΔNES);gcn-2(ok871), and hoe-1(ΔNES);eIF2α(S46A,S49A) animals. Scale bar 200 μm. (B) Fluorescence intensity quantification of hsp-6p::GFP in individual day 2 adult wildtype, gcn-2(ok871), eIF2α(S46A,S49A), hoe-1(ΔNES), hoe-1(ΔNES);gcn-2(ok871), and hoe-1(ΔNES);eIF2α(S46A,S49A) animals (n = 24 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test).
Nuclear HOE-1 is dynamically responsive to mitochondrial stress and negatively regulated by ATFS-1
To better understand the potential physiological implications of HOE-1 in UPRmt, we assessed hoe-1 expression and subcellular dynamics of HOE-1 during mitochondrial stress. It is predicted that two major transcripts are produced from the hoe-1 gene locus: one that contains an MTS and one that does not, which are translated into mitochondrial- and nuclear-targeted HOE-1, respectively. However, it has been shown in other systems that hoe-1 orthologs produce a single transcript that encodes both a mitochondrial targeted and nuclear targeted HOE-1 isoform via alternative translation initiation (Rossmanith, 2011). Thus, we first sought to determine which mechanism is used for hoe-1 expression. To do so, we designed two sets of primers complementary to hoe-1 mRNA one of which amplifies only mRNA that includes the MTS and the other which amplifies all hoe-1 mRNA (spans a sequence that is included in all predicted HOE-1 isoforms). If there are two independent hoe-1 transcripts, we expected there to be higher levels of hoe-1 mRNA measured by the primer pair for total transcripts than for the mitochondrial specific pair. However, we found that both primer pairs measured similar levels of hoe-1 mRNA (Figure 8—figure supplement 1A) suggesting that, like in other systems, there is a single hoe-1 transcript. Next, we assessed hoe-1 mRNA levels in non-stress versus mitochondrial stress conditions. We found, using both primer pairs, that hoe-1 mRNA levels are modestly elevated in nuo-6(qm200) animals relative to wildtype (Figure 8—figure supplement 1B and C) suggesting that hoe-1 may be transcriptionally upregulated under conditions of mitochondrial stress.
Next, we assessed the subcellular dynamics of HOE-1 in response to mitochondrial stress. We found that HOE-1::GFP nuclear levels are markedly diminished under mitochondrial stress induced by nuo-6(qm200), cco-1 RNAi, and spg-7 RNAi (Figure 8A and B and Figure 8—figure supplement 2A and B). This observation was unexpected given that hoe-1 transcript levels are elevated during mitochondrial stress and it runs contrary to the fact that compromising HOE-1 nuclear export is sufficient to induce UPRmt (Figure 3A and B). A common feature of signaling pathways is negative regulation. Thus, we questioned if reduced nuclear HOE-1 is a result of negative feedback rather than a direct result of mitochondrial stress. Given that mitochondrial stress activates UPRmt, we assessed HOE-1::GFP status in a mitochondrial stress background wherein atfs-1 is knocked down by RNAi. HOE-1 levels are significantly upregulated in nuclei of nuo-6(qm200) animals on atfs-1 RNAi relative to nuo-6(qm200) animals on control RNAi, as well as both wildtype animals on control and atfs-1 RNAi (Figure 8A and B and Figure 8—figure supplement 3A–C). Moreover, total cellular HOE-1 levels are elevated under mitochondrial stress in an atfs-1 RNAi background (Figure 8C and D and Figure 8—source data 1A–E and Figure 8—source data 2A–E). Additionally, mitochondrial HOE-1 levels are elevated under mitochondrial stress conditions irrespective of RNAi treatment (Figure 8—figure supplement 3D). Together these data suggest that HOE-1 is upregulated and accumulates in nuclei upon mitochondrial stress. Then, nuclear HOE-1 is negatively regulated by ATFS-1 once UPRmt is activated.
Figure 8. Nuclear HOE-1 levels are elevated during mitochondrial stress in the absence of ATFS-1 but decreased in the presence of ATFS-1.
(A) Fluorescence images of HOE-1::GFP in day 1 adult wildtype and nuo-6(qm200) animals on control and atfs-1 RNAi. Scale bar 200 μm. (B) Fluorescence intensity quantification of intestinal nuclei relative to extranuclear signal in day 1 adult wildtype and nuo-6(qm200) animals on control and atfs-1 RNAi (n = 40 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (C) Western blot for HOE-1::GFP and actin from day 1 adult wildtype and nuo-6(qm200) animals on control and atfs-1 RNAi. (D) Quantification of HOE-1::GFP western blot band intensity from day 1 adult wildtype and nuo-6(qm200) animals on control and atfs-1 RNAi normalized to total protein (n = 4 for each condition, mean and SD shown, ordinary two-way ANOVA with Tukey’s multiple comparisons test). (E) Fluorescence images of HOE-1::GFP in day 1 adult wildtype and atfs-1(et15) animals. Scale bar 200 μm. (F) Fluorescence intensity quantification of intestinal nuclei relative to extranuclear signal in day 1 adult wildtype and atfs-1(et15) animals (n = 40 for each condition, mean and SD shown, unpaired t-test). (G) Western blot for HOE-1::GFP and actin from day 1 adult wildtype and atfs-1(et15) animals. (H) Quantification of HOE-1::GFP western blot band intensity from day 1 adult wildtype and atfs-1(et15) animals normalized to total protein (n = 4 for each condition, mean and SD shown, unpaired t-test). (I) Mitochondrial stress triggers activation of HOE-1 resulting in altered RNA processing that facilitates UPRmt via ATFS-1. Activation of UPRmt negatively regulates HOE-1.

Figure 8—figure supplement 1. hoe-1 mRNA levels are upregulated under conditions of mitochondrial stress.

Figure 8—figure supplement 2. UPRmt-inducing cco-1 and spg-7 RNAi both attenuate HOE-1 nuclear levels.

Figure 8—figure supplement 3. Nuclear HOE-1 levels are elevated during mitochondrial stress in the absence of ATFS-1 but decreased in the presence of ATFS-1.

Figure 8—figure supplement 4. Constitutive activation of UPRmt by atfs-1 gain-of-function (atfs-1(et15)) depletes nuclear HOE-1 levels.

To further test if nuclear HOE-1 is negatively regulated by UPRmt activation rather than by mitochondrial stress, we assessed HOE-1 localization in ATFS-1 gain-of-function animals (atfs-1(et15)). atfs-1(et15) constitutively activates UPRmt in the absence of mitochondrial stress (Rauthan et al., 2013). Thus, we asked if atfs-1(et15) is sufficient to reduce nuclear HOE-1 levels. Indeed, nuclear HOE-1 levels are markedly reduced in atfs-1(et15) animals relative to wildtype (Figure 8E and F and Figure 8—figure supplement 4A–C) while total and mitochondrial HOE-1 protein levels are largely unperturbed (Figure 8G and H, Figure 8—figure supplement 4D, Figure 8—source data 3A–E and Figure 8—source data 4A–E). These data further support that UPRmt activation negatively regulates nuclear HOE-1.
Discussion
Regulation of UPRmt is not completely understood and elucidating this mechanism has broad implications for understanding cellular response to mitochondrial dysfunction. Here, we describe a novel mechanism by which mitochondrial stress is transduced to activate UPRmt and how that response is regulated through a feedback mechanism (Figure 8I).
Multiple factors have been identified that are required for maximal activation of UPRmt. This includes the mitochondrial localized proteins, CLPP-1 protease and peptide transmembrane transporter HAF-1 (Haynes et al., 2007; Haynes et al., 2010). Additionally, the transcription factors ATFS-1 and DVE-1 along with the co-transcriptional activator UBL-5 are required for UPRmt activation (Benedetti et al., 2006; Haynes et al., 2007; Haynes et al., 2010; Nargund et al., 2012; Nargund et al., 2015; Tian et al., 2016). Histone modifications, chromatin remodeling, and post-translational modifications of ATFS-1 are also involved in fully activating UPRmt (Tian et al., 2016; Merkwirth et al., 2016; Gao et al., 2019; Shao et al., 2020). We show for the first time that nuclear HOE-1 is required for maximal activation of UPRmt as its induction by various stressors is attenuated in hoe-1 RNAi, hoe-1 null, and hoe-1(ΔNLS) backgrounds.
We show that loss of hoe-1 results in varied attenuation of UPRmt depending on how UPRmt is activated. UPRmt induction by RNAi (cco-1 and spg-7) is robustly attenuated by loss of hoe-1 while nuo-6(qm200)-induced UPRmt is only modestly attenuated. RNAi by feeding works well in all tissues except neurons (Timmons et al., 2001; Kamath et al., 2003). Importantly, UPRmt can be activated non-cell autonomously in the intestine by mitochondrial stress in neurons (Durieux et al., 2011; Berendzen et al., 2016; Zhang et al., 2018). UPRmt induced cell-autonomously in the intestine by RNAi may be hoe-1 dependent while neuron-to-intestine UPRmt induction may work primarily in a hoe-1-independent manner. Consistent with this, increased nuclear accumulation of HOE-1 only activates UPRmt in the intestine. These results further exemplify the complexity of UPRmt signaling.
UPRmt is generally triggered via compromised mitochondrial membrane potential which facilitates the nuclear accumulation of ATFS-1 (Rolland et al., 2019; Shpilka et al., 2021). We find that UPRmt activation via hoe-1(ΔNES) correlates with a decrease in mitochondrial membrane potential providing a potential trigger for UPRmt induction. Furthermore, we show that the UPRmt transcription factors ATFS-1 and DVE-1 have increased nuclear localization in hoe-1(ΔNES) animals, thus likely facilitating the robust UPRmt activation.
HOE-1 functions in tRNA processing (Nashimoto et al., 1999; Mayer et al., 2000; Schiffer et al., 2002; Takaku et al., 2003; Dubrovsky et al., 2004; Brzezniak et al., 2011; Sanchez et al., 2011; Siira et al., 2018). Here, we show that increased 3’-tRNA processing by HOE-1 is likely responsible for UPRmt activation. Restricting HOE-1-dependent 3’-tRNA trailer sequence cleavage indirectly by RNAi against RNA polymerase III subunit, rpc-1, and RNase P subunit, popl-1, strongly attenuate hoe-1(ΔNES)-induced UPRmt. Moreover, these RNA species must be required in the cytosol to activate UPRmt as RNAi against tRNA exportin xpo-3 is sufficient to robustly attenuate hoe-1(ΔNES)-induced UPRmt. Our findings herein are the first reported connection between altered tRNA processing and UPRmt in C. elegans. Given the general requirement for tRNAs in protein translation on the one hand, and the mitochondria-specific nature of UPRmt on the other, our findings of a connection between the two are intriguing. However, besides performing their core housekeeping function in protein translation, tRNAs have also emerged as small RNAs with important regulatory roles inside cells (Avcilar-Kucukgoze and Kashina, 2020). Perhaps, the most well-characterized regulatory role for tRNAs is in the activation of the integrated stress response (ISR). In ISR, uncharged tRNAs activate the eIF2α kinase, GCN2, resulting in the upregulation of ATFS-1 orthologs ATF4 and ATF5 (Pakos-Zebrucka et al., 2016; Costa-Mattioli and Walter, 2020). However, we show that gcn-2 and eIF2α are not required for hoe-1(ΔNES)-induced UPRmt activation suggesting that a different mechanism is responsible. The lack of involvement of ISR in HOE-1’s role in UPRmt is not too surprising as there may be a greater pool of fully mature tRNAs in the cytosol in hoe-1(ΔNES) animals due to increased 3’-end processing of tRNAs above wildtype levels. This would result in an excess of charged tRNAs in the cytosol, the opposite of what is required to trigger GCN2-dependent ISR. Instead, we can speculate on several additional possibilities for the consequences of increased levels of charged tRNAs that can explain the role of HOE-1 in UPRmt regulation. For example, the use of amino acids to charge excess tRNAs in hoe-1(ΔNES) animals may limit the pool of free amino acids available for mitochondrial import, thus affecting translation of proteins encoded by the mitochondrial genome. This may result in stoichiometric imbalance between nuclear and mitochondrial-encoded components of the electron transport chain, which is known to compromise mitochondrial membrane potential and trigger UPRmt (Houtkooper et al., 2013). Alternatively, mito-nuclear imbalance in hoe-1(ΔNES) animals may result from excessive translation of nuclear-encoded mitochondrial proteins due to increased abundance of available charged tRNAs in the cytosol. In yet another scenario, UPRmt may not be the consequence of a global increase in the levels of all cytosolic tRNAs but rather, may be due to changes in the levels of specific tRNAs that preferentially impact translation of genes enriched for the corresponding codons. Such selective upregulation of tRNAs has been shown previously to have specific cellular consequences (Gingold et al., 2014; Goodarzi et al., 2016). Finally, it is possible that a tRNA-like RNA or other small RNA species such as tRNA fragments are responsible for UPRmt induction in hoe-1(ΔNES) animals (Kruszka et al., 2003; Lee et al., 2009; Bogerd et al., 2010; Siira et al., 2018). However, if this is the case, our data argue that such an RNA species would need to be transported to the cytosol by tRNA exportin. Non-tRNA transport by an ortholog of xpo-3 has not yet been reported (Hopper and Nostramo, 2019).
We show that nuclear HOE-1 is dynamically regulated by mitochondrial stress. In the presence of stress, nuclear HOE-1 levels are depleted. However, this is UPRmt dependent as HOE-1 nuclear levels under mitochondrial stress are elevated above wild-type levels when UPRmt is blocked by atfs-1 RNAi. These data, paired with the fact that compromising HOE-1 nuclear export triggers UPRmt, lead us to hypothesize that upon mitochondrial stress, nuclear HOE-1 levels are elevated. This upregulation of nuclear HOE-1 elevates 3’-tRNA processing thereby triggering a signaling cascade that results in elevated nuclear ATFS-1 and DVE-1 and subsequent UPRmt induction. Activated UPRmt then negatively regulates HOE-1 nuclear levels thus providing a feedback mechanism to tightly control mitochondrial stress response. UPRmt-negative regulation of HOE-1 is further supported by our data showing that constitutive activation of UPRmt by atfs-1(et15) is sufficient to reduce nuclear HOE-1 levels in the absence of mitochondrial stress. How it is that mitochondrial stress activates HOE-1 is still unknown. Multiple mitochondrial derived small molecules have been reported to communicate mitochondrial status including reactive oxygen species (ROS), NAD+, and acetyl-CoA (Baker et al., 2012; Mouchiroud et al., 2013; Ramachandran et al., 2019; Tjahjono et al., 2020; Zhu et al., 2020) We look forward to further investigating whether these, or other molecules, are involved in HOE-1 regulation.
In humans, mutations in the ortholog of HOE-1, ELAC2, are associated with both hypertrophic cardiomyopathy (Haack et al., 2013; Shinwari et al., 2017; Saoura et al., 2019) and prostate cancer (Tavtigian et al., 2001; Korver et al., 2003; Noda et al., 2006). Historically, it has been suggested that mutations in ELAC2 cause disease because of a loss of mature tRNA production. Our works suggests an intriguing alternative whereby ELAC2 mutations lead to altered tRNA processing that triggers aberrant stress response signaling resulting in disease state. Our system provides a convenient opportunity to interrogate these disease causing variants.
Taken together, our findings provide a novel mechanism—involving the tRNA processing enzyme HOE-1—by which mitochondrial stress is transduced to activate UPRmt thus providing important insight into the regulation of mitochondrial stress response.
Methods
Worm maintenance
Worms were grown on nematode growth media (NGM) seeded with OP50 E. coli bacteria and maintained at 20 °C.
Mutants and transgenic lines
A complete list of C. elegans strains used can be found in Supplementary file 1. All new mutant and transgenic strains generated via CRISPR/Cas9 for this study were confirmed by Sanger sequencing.
CRISPR/Cas9
CRISPR was conducted as previously described (Dokshin et al. Genetics 2018; Paix et al. Genetics 2015) using Alt-R S.p. Cas9 Nuclease V3 (IDT #1081058) and tracrRNA (IDT #1072532). A complete list of crRNA and repair template sequences purchased from IDT can be found in Supplementary file 2.
Genetic crosses
Strains resulting from genetic crosses were generated by crossing ~20 heterozygous males of a given strain to 5–8 L4 hermaphrodites of another strain (heterozygous males were generated by first crossing L4 hermaphrodites of that strain to N2 males). F1, L4 hermaphrodites were then cloned out and allowed to have self-progeny. F2 progeny were cloned out and once they had progeny were genotyped or screened (if fluorescent marker) for presence of alleles of interest. All genotyping primers were purchased from IDT and can be found in Supplementary file 2.
Fluorescence microscopy
All whole animal imaging was done using Zeiss Axio Zoom V16 stereo zoom microscope. For all whole animal imaging, worms were immobilized on 2% agar pads on microscope slides in ~1 μl of 100 mM levamisole (ThermoFisher #AC187870100) and then coverslip applied.
Fluorescence image analysis
For whole animal fluorescence intensity quantification, total pixels (determined by tracing individual animals and summing the total number of pixels within the bounds of the trace) and pixel fluorescence intensity (pixel fluorescence intensity on 1–255 scale) were quantified using imageJ and mean fluorescence intensity for each worm was calculated (sum total of fluorescence intensity divided by total number of pixels within bounds of the trace). For DVE-1::GFP image analysis (Figure 5E&F), brightness threshold was set to 25 in imageJ and then the number of gut cell nuclei that were saturated at this threshold were counted. For Figure 8A&B and Figure 8E&F, and Figure 8—figure supplement 2A&B, mean fluorescence intensity was calculated within the bounds of gut cell nuclei and outside of the bounds of gut cell nuclei and then graphed as the ratio fluorescence intensity of nuclear to extranuclear signal.
RNAi
RNAi by feeding was conducted as previously described (Gitschlag et al. Cell Met. 2016). Briefly, RNAi clones were grown overnight from single colony in 2 ml liquid culture of LB supplemented with 50 μg/ml ampicillin. To make 16 RNAi plates, 50 ml of LB supplemented with 50 μg/ml ampicillin was inoculated with 500 μl of overnight culture and then incubated while shaking at 37 °C for 4–5 hours (to an OD550-600 of about 0.8). Cultures were then induced by adding 50 ml additional LB supplemented with 50 μg/ml ampicillin and 4 mM IPTG and then continued incubating while shaking at 37 °C for 4 hours. Following incubation, bacteria were pelleted by centrifugation at 3900 rpm for 6 min. Supernatant was decanted and pellets were gently resuspended in 4 ml of LB supplemented with 8 mM IPTG. 250 μl of resuspension was seeded onto standard NGM plates containing 1 mM IPTG. Plates were left to dry overnight and then used within 1 week. Bacterial RNAi feeder strains were all from Ahringer RNAi Feeding Library, grown from single colony and identity confirmed by Sanger sequencing. atfs-1 (ZC376.7), cco-1 (F26E4.9), hoe-1 (E04A4.4), hpo-31 (F55B12.4), popl-1 (C05D11.9), rpc-1 (C42D4.8), rtcb-1 (F16A11.2), spg-7 (Y47G6A.10), xpo-3 (C49H3.10).
Quantification of gene expression
cDNA was synthesized using Maxima H Minus First Strand cDNA Synthesis Kit, with dsDNase (ThermoFisher #K1682) according to manufacturer’s directions. Lysates for cDNA synthesis were made by transferring 10, day 2 adult worms to 10 μl of lysis buffer supplemented with 20 mg/ml proteinase K and incubating at 65 °C for 10 min, 85 °C for 1 min and 4 °C for 2 min. Quantification of gene expression was performed using droplet digital PCR (ddPCR) with Bio-Rad QX200 ddPCR EvaGreen Supermix (Bio-Rad #1864034). Primers used for ddPCR can be found in Supplementary file 2.
TMRE staining
A total of 500 μl of 1 mM TMRE (ThermoFisher #T669) solution in M9 buffer (prepared from a stock TMRE solution of 0.5 M in DMSO) was supplemented on top of standard NGM plates pre-seeded with 200 ul lawn of OP50 and allowed to dry overnight in the dark. The following day young L4 animals were transferred to TMRE plates and incubated on TMRE for 16 hr. After 16 hr, animals were transferred from TMRE plates to seeded standard NGM plates for 1 hr to remove any non-specific TMRE signal from cuticle and intestinal lumen. Animals were then imaged via confocal microscopy as described below.
Confocal fluorescence imaging
Worms were grown at 20 °C and age-synchronized by timed egg-lays on NGM plates seeded with OP50 or HT115 bacteria for RNAi experiments. Before imaging, worms were immobilized with 3 μl 0.05 µm Polybead microsphere suspension (Polysciences) on a 10% agarose pad with a coverslip (1). Images were taken in the mid- or posterior intestine using a Nikon Ti2 with CSU-W1 spinning disk and Plan-Apochromat 100 X/1.49 NA objective. HOE-1::GFP was imaged by 488 nm laser excitation and ET525/36 m emission filter. 2 X integration was applied (Nikon Elements) to increase signal strength. TMRE and ATFS-1::mCherry were imaged with 561 nm laser excitation and ET605/52 M emission filter.
Image processing and analysis was performed with Nikon Elements software. Raw images were subjected to deconvolution and rolling ball background subtraction. Mitochondrial networks were segmented using the TMRE signal after excluding dye aggregates via Bright Spot Detection. To objectively set threshold parameters across groups with different TMRE intensity levels, the low threshold for segmentation was calculated based on a linear correlation with mean TMRE intensity within each group, y = 0.6411*x + 89.71 (x = mean TMRE intensity and constants derived from an initial manual validation). Regions of interest (ROIs) were manually drawn to encompass a single intestinal cell, and nuclei were identified and segmented manually using brightfield images. Mean intensities were measured within the resulting masks.
To detect localization of HOE-1::GFP in mitochondria, images of TMRE-stained intestinal cells of control and ΔMTS worms were collected and blinded. Mitochondria were segmented by TMRE signals as above. For each cell, one representative line scan was drawn manually across the mitochondrial short axis.
Western blot
Fifty adult worms were transferred into a tube containing 20 μl of M9 Buffer. Then, 20 μl of 2 x Laemmli Buffer (BioRad #161–0737) supplemented with 2-mercaptoethanol (i.e. βME) was added to worm suspension and gently pipetted up and down 5 times to mix. Worms were lysed at 95 °C for 10 min in thermocycler followed by ramp down to room temperature (25 °C). Lysates were then pipetted up and down 10 times to complete disrupt and homogenize suspension. Samples were briefly centrifuged to pellet any worm debris. 20 μl of lysate supernatant was loaded onto precast Mini-PROTEAN TGX Stain-Free Gel (BioRad #4568045). Gel was run for 30 min at 100 V and then an additional 40–45 min at 130 V in 1 x Tris/Glycine/SDS Running Buffer (BioRad #1610732). Following electrophoresis gel was activated and imaged for total protein. Gel was equilibrated in Trans-Blot Turbo Transfer Buffer (BioRad #10026938) and transferred to activated and equilibrated Trans-Blot Turbo LF PVDF Membrane (BioRad #10026934) for 7 min at 2.5 A/25 V on Trans-Blot Turbo Transfer System. Following transfer, stain-free membrane was imaged for total protein. Membrane was then blocked in 5% milk in TBST for 2 hr rocking at room temperature. Following blocking, membrane was incubated in primary antibody overnight rocking at 4 °C. Mouse monoclonal anti-β-actin (Santa Cruz Biotechnology #sc-47778) or mouse monoclonal anti-GFP (#sc-9996) were used at a dilution of 1:2,500 in 5% milk in TBST. The following day the membrane was washed three times for 5 min each with TBST and then incubated with HRP-conjugated goat anti-mouse antibody (sc-2005) at 1:2000 in 5% milk in TBST for 2 hours at room temperature. Membrane was again washed three times for 5 min each with TBST. Membranes were then incubated for 5 min in Clarity Western ECL Substrate (BioRad #1705060) and immediately imaged on a BioRad ChemiDoc MP imager. Band intensity was quantified using imageJ.
Statistical analysis
Experiment-specific details regarding sample size and statistical test used can be found in the corresponding Figure Legends. Significant p-values under 0.05 are denoted on all graphs and p-values above 0.05 are considered non-significant (ns). All statistical analysis was performed in GraphPad Prism 9. All data points for each experiment are included (no outlier exclusion was performed). For all whole animal fluorescence analysis, a sample size of 24 animals was generally used, each animal considered a biological replicate. Statistical analysis of high resolution fluorescence confocal imaging (HOE-1::GFP, ATFS-1::mCherry, and TMRE) was conducted on sample sizes between 60 and 80 animals of which animals were collected and imaged on three independent days, each animal considered a biological replicate. For western blot analysis, four independent samples were used for each condition, each sample (containing 50 worms each) is considered a biological replicate. For ddPCR analysis, a sample size of 4 was used for each condition, each sample (containing 10 worms each) is considered a biological replicate, each biological replicate was run in technical duplicate of which the average value was used for analysis.
Acknowledgements
We thank Lantana K Grub and Cassidy A Johnson for their valuable feedback on the manuscript. We thank WormBase for invaluable tools and information used to plan and execute the research described. Worm strain itSi001 was graciously shared with us by Sasha de Henau. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was generously supported by R01 GM123260 (MRP), R35 GM145378 (MRP), R00 AG052666 (KB), and by the support provided to JPH by the Training Program in Environmental Toxicology (T32ES007028). Some confocal microscopy imaging was performed through the Vanderbilt Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126). Droplet Digital PCR to quantify transcript levels was performed through the Vanderbilt University Medical Center’s Immunogenomics, Microbial Genetics and Single Cell Technologies core.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Maulik R Patel, Email: maulik.r.patel@vanderbilt.edu.
Xiaochen Wang, Institute of Biophysics Chinese Academy of Sciences, China.
David Ron, University of Cambridge, United Kingdom.
Funding Information
This paper was supported by the following grants:
National Institute of General Medical Sciences R01GM123260 to James P Held, Benjamin R Saunders, Maulik R Patel, Claudia V Pereira.
National Institute on Aging R00AG052666 to Gaomin Feng, Kristopher Burkewitz.
National Institute of Environmental Health Sciences T32ES007028 to James P Held.
National Institute of General Medical Sciences R35GM145378 to James P Held, Benjamin R Saunders, Maulik R Patel.
Additional information
Competing interests
No competing interests declared.
No competing interests declared.
Author contributions
Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing - original draft, Writing – review and editing.
Formal analysis, Investigation, Methodology, Validation, Writing – review and editing.
Investigation.
Investigation, Writing – review and editing.
Funding acquisition, Supervision, Writing – review and editing.
Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review and editing.
Additional files
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting files. Source data files have been provided for Figures 5 and 8.
References
- Ashley GE, Duong T, Levenson MT, Martinez MAQ, Johnson LC, Hibshman JD, Saeger HN, Palmisano NJ, Doonan R, Martinez-Mendez R, Davidson BR, Zhang W, Ragle JM, Medwig-Kinney TN, Sirota SS, Goldstein B, Matus DQ, Dickinson DJ, Reiner DJ, Ward JD. An expanded auxin-inducible degron toolkit for Caenorhabditis elegans. Genetics. 2021;217:iyab006. doi: 10.1093/genetics/iyab006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avcilar-Kucukgoze I, Kashina A. Hijacking tRNAs From Translation: Regulatory Functions of tRNAs in Mammalian Cell Physiology. Frontiers in Molecular Biosciences. 2020;7:610617. doi: 10.3389/fmolb.2020.610617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Nargund AM, Sun T, Haynes CM, Larsson N-G. Protective Coupling of Mitochondrial Function and Protein Synthesis via the eIF2α Kinase GCN-2. PLOS Genetics. 2012;8:e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–239. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendzen KM, Durieux J, Shao LW, Tian Y, Kim HE, Wolff S, Liu Y, Dillin A. Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell. 2016;166:1553–1563. doi: 10.1016/j.cell.2016.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienert S, Waterhouse A, de Beer TAP, Tauriello G, Studer G, Bordoli L, Schwede T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Research. 2017;45:D313–D319. doi: 10.1093/nar/gkw1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Karnowski HW, Cai X, Shin J, Pohlers M, Cullen BR. A mammalian herpesvirus uses noncanonical expression and processing mechanisms to generate viral MicroRNAs. Molecular Cell. 2010;37:135–142. doi: 10.1016/j.molcel.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos F, Krämer L, Groh C, Jung F, Haberkant P, Stein F, Wollweber F, Gackstatter A, Zöller E, van der Laan M, Savitski MM, Benes V, Herrmann JM. Mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nature Cell Biology. 2019;21:442–451. doi: 10.1038/s41556-019-0294-5. [DOI] [PubMed] [Google Scholar]
- Brzezniak LK, Bijata M, Szczesny RJ, Stepien PP. Involvement of human ELAC2 gene product in 3’ end processing of mitochondrial tRNAs. RNA Biology. 2011;8:616–626. doi: 10.4161/rna.8.4.15393. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science (New York, N.Y.) 2020;368:eaat5314. doi: 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Melo JA, Thondamal M, Morton EA, Cornwell AB, Crick B, Kim JH, Swartz EW, Lamitina T, Douglas PM, Samuelson AV. The homeodomain-interacting protein kinase HPK-1 preserves protein homeostasis and longevity through master regulatory control of the HSF-1 chaperone network and TORC1-restricted autophagy in Caenorhabditis elegans. PLOS Genetics. 2017;13:e1007038. doi: 10.1371/journal.pgen.1007038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejima K, Hori S, Iwata S, Suehiro Y, Yoshina S, Motohashi T, Mitani S. An Aneuploidy-Free and Structurally Defined Balancer Chromosome Toolkit for Caenorhabditis elegans. Cell Reports. 2018;22:232–241. doi: 10.1016/j.celrep.2017.12.024. [DOI] [PubMed] [Google Scholar]
- Dokshin GA, Ghanta KS, Piscopo KM, Mello CC. Robust Genome Editing with Short Single-Stranded and Long, Partially Single-Stranded DNA Donors in Caenorhabditis elegans. Genetics. 2018;210:781–787. doi: 10.1534/genetics.118.301532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrovsky EB, Dubrovskaya VA, Levinger L, Schiffer S, Marchfelder A. Drosophila RNase Z processes mitochondrial and nuclear pre-tRNA 3’ ends in vivo. Nucleic Acids Research. 2004;32:255–262. doi: 10.1093/nar/gkh182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert M, Beier H. Plant tRNA ligases are multifunctional enzymes that have diverged in sequence and substrate specificity from RNA ligases of other phylogenetic origins. Nucleic Acids Research. 2005;33:388–399. doi: 10.1093/nar/gki174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, Jae LT. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature. 2020;579:433–437. doi: 10.1038/s41586-020-2076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Current Biology. 2016;26:2037–2043. doi: 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frendewey D, Dingermann T, Cooley L, Söll D. Processing of precursor tRNAs in Drosophila. Processing of the 3’ end involves an endonucleolytic cleavage and occurs after 5’ end maturation. The Journal of Biological Chemistry. 1985;260:449–454. doi: 10.1016/S0021-9258(18)89752-6. [DOI] [PubMed] [Google Scholar]
- Gao K, Li Y, Hu S, Liu Y. SUMO peptidase ULP-4 regulates mitochondrial UPR-mediated innate immunity and lifespan extension. eLife. 2019;8:e41792. doi: 10.7554/eLife.41792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, Kooistra SM, Christophersen NS, Christensen LL, Borre M, Sørensen KD, Andersen LD, Andersen CL, Hulleman E, Wurdinger T, Ralfkiær E, Helin K, Grønbæk K, Ørntoft T, Waszak SM, Dahan O, Pedersen JS, Lund AH, Pilpel Y. A dual program for translation regulation in cellular proliferation and differentiation. Cell. 2014;158:1281–1292. doi: 10.1016/j.cell.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Gitschlag BL, Kirby CS, Samuels DC, Gangula RD, Mallal SA, Patel MR. Homeostatic Responses Regulate Selfish Mitochondrial Genome Dynamics in C. elegans. Cell Metabolism. 2016;24:91–103. doi: 10.1016/j.cmet.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF. Modulated Expression of Specific tRNAs Drives Gene Expression and Cancer Progression. Cell. 2016;165:1416–1427. doi: 10.1016/j.cell.2016.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YHT, Wiita AP, Xu K, Correia MA, Kampmann M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature. 2020;579:427–432. doi: 10.1038/s41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Kopajtich R, Freisinger P, Wieland T, Rorbach J, Nicholls TJ, Baruffini E, Walther A, Danhauser K, Zimmermann FA, Husain RA, Schum J, Mundy H, Ferrero I, Strom TM, Meitinger T, Taylor RW, Minczuk M, Mayr JA, Prokisch H. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. American Journal of Human Genetics. 2013;93:211–223. doi: 10.1016/j.ajhg.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Developmental Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Molecular Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper AK, Nostramo RT. tRNA Processing and Subcellular Trafficking Proteins Multitask in Pathways for Other RNAs. Frontiers in Genetics. 2019;10:96. doi: 10.3389/fgene.2019.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YM. CCA addition to tRNA: implications for tRNA quality control. IUBMB Life. 2010;62:251–260. doi: 10.1002/iub.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Koncha RR, Ramachandran G, Sepuri NBV, Ramaiah KVA. CCCP-induced mitochondrial dysfunction - characterization and analysis of integrated stress response to cellular signaling and homeostasis. The FEBS Journal. 2021;288:5737–5754. doi: 10.1111/febs.15868. [DOI] [PubMed] [Google Scholar]
- Korver W, Guevara C, Chen Y, Neuteboom S, Bookstein R, Tavtigian S, Lees E. The product of the candidate prostate cancer susceptibility gene ELAC2 interacts with the gamma-tubulin complex. International Journal of Cancer. 2003;104:283–288. doi: 10.1002/ijc.10945. [DOI] [PubMed] [Google Scholar]
- Kruszka K, Barneche F, Guyot R, Ailhas J, Meneau I, Schiffer S, Marchfelder A, Echeverría M. Plant dicistronic tRNA-snoRNA genes: a new mode of expression of the small nucleolar RNAs processed by RNase Z. The EMBO Journal. 2003;22:621–632. doi: 10.1093/emboj/cdg040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Cour T, Kiemer L, Mølgaard A, Gupta R, Skriver K, Brunak S. Analysis and prediction of leucine-rich nuclear export signals. Protein Engineering, Design & Selection. 2004;17:527–536. doi: 10.1093/protein/gzh062. [DOI] [PubMed] [Google Scholar]
- Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs. Genes & Development. 2009;23:2639–2649. doi: 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Li de la Sierra-Gallay I, Lazar N, Pellegrini O, Durand D, Marchfelder A, Condon C, van Tilbeurgh H. The crystal structure of Trz1, the long form RNase Z from yeast. Nucleic Acids Research. 2017;45:6209–6216. doi: 10.1093/nar/gkx216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M, Schiffer S, Marchfelder A. tRNA 3’ processing in plants: nuclear and mitochondrial activities differ. Biochemistry. 2000;39:2096–2105. doi: 10.1021/bi992253e. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM, Steffen KK, Williams EG, Mouchiroud L, Tronnes SU, Murillo V, Wolff SC, Shaw RJ, Auwerx J, Dillin A. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell. 2016;165:1209–1223. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo Y-S, Viswanathan M, Schoonjans K, Guarente L, Auwerx J. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkácsy E, Khan MH, Lane RK, Borror MB, Park JH, Bokov AF, Fisher AL, Link CD, Rea SL. DLK-1, SEK-3 and PMK-3 Are Required for the Life Extension Induced by Mitochondrial Bioenergetic Disruption in C. elegans. PLOS Genetics. 2016;12:e1006133. doi: 10.1371/journal.pgen.1006133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naresh NU, Haynes CM. Signaling and Regulation of the Mitochondrial Unfolded Protein Response. Cold Spring Harbor Perspectives in Biology. 2019;11:a033944. doi: 10.1101/cshperspect.a033944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science (New York, N.Y.) 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt. Molecular Cell. 2015;58:123–133. doi: 10.1016/j.molcel.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashimoto M, Tamura M, Kaspar RL. Minimum requirements for substrates of mammalian tRNA 3’ processing endoribonuclease. Biochemistry. 1999;38:12089–12096. doi: 10.1021/bi9911942. [DOI] [PubMed] [Google Scholar]
- Noda D, Itoh S, Watanabe Y, Inamitsu M, Dennler S, Itoh F, Koike S, Danielpour D, ten Dijke P, Kato M. ELAC2, a putative prostate cancer susceptibility gene product, potentiates TGF-beta/Smad-induced growth arrest of prostate cells. Oncogene. 2006;25:5591–5600. doi: 10.1038/sj.onc.1209571. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Reports. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J, Schleiffer A, Martinez J. Diversity and roles of (t)RNA ligases. Cellular and Molecular Life Sciences. 2012;69:2657–2670. doi: 10.1007/s00018-012-0944-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S, Ron D. Early Events in the Endoplasmic Reticulum Unfolded Protein Response. Cold Spring Harbor Perspectives in Biology. 2019;11:a033894. doi: 10.1101/cshperspect.a033894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina M, Ibba M. tRNAs as regulators of biological processes. Frontiers in Genetics. 2014;5:171. doi: 10.3389/fgene.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran PV, Savini M, Folick AK, Hu K, Masand R, Graham BH, Wang MC. Lysosomal Signaling Promotes Longevity by Adjusting Mitochondrial Activity. Developmental Cell. 2019;48:685–696. doi: 10.1016/j.devcel.2018.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauthan M, Ranji P, Aguilera Pradenas N, Pitot C, Pilon M. The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. PNAS. 2013;110:5981–5986. doi: 10.1073/pnas.1218778110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland SG, Schneid S, Schwarz M, Rackles E, Fischer C, Haeussler S, Regmi SG, Yeroslaviz A, Habermann B, Mokranjac D, Lambie E, Conradt B. Compromised Mitochondrial Protein Import Acts as a Signal for UPRmt. Cell Reports. 2019;28:1659–1669. doi: 10.1016/j.celrep.2019.07.049. [DOI] [PubMed] [Google Scholar]
- Rossmanith W. Localization of human RNase Z isoforms: dual nuclear/mitochondrial targeting of the ELAC2 gene product by alternative translation initiation. PLOS ONE. 2011;6:e19152. doi: 10.1371/journal.pone.0019152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez MIGL, Mercer TR, Davies SMK, Shearwood A-MJ, Nygård KKA, Richman TR, Mattick JS, Rackham O, Filipovska A. RNA processing in human mitochondria. Cell Cycle (Georgetown, Tex.) 2011;10:2904–2916. doi: 10.4161/cc.10.17.17060. [DOI] [PubMed] [Google Scholar]
- Saoura M, Powell CA, Kopajtich R, Alahmad A, Al-Balool HH, Albash B, Alfadhel M, Alston CL, Bertini E, Bonnen PE, Bratkovic D, Carrozzo R, Donati MA, Di Nottia M, Ghezzi D, Goldstein A, Haan E, Horvath R, Hughes J, Invernizzi F, Lamantea E, Lucas B, Pinnock K-G, Pujantell M, Rahman S, Rebelo-Guiomar P, Santra S, Verrigni D, McFarland R, Prokisch H, Taylor RW, Levinger L, Minczuk M. Mutations in ELAC2 associated with hypertrophic cardiomyopathy impair mitochondrial tRNA 3’-end processing. Human Mutation. 2019;40:1731–1748. doi: 10.1002/humu.23777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer S, Rösch S, Marchfelder A. Assigning a function to a conserved group of proteins: the tRNA 3’-processing enzymes. The EMBO Journal. 2002;21:2769–2777. doi: 10.1093/emboj/21.11.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao LW, Peng Q, Dong M, Gao K, Li Y, Li Y, Li CY, Liu Y. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nature Communications. 2020;11:4639. doi: 10.1038/s41467-020-18501-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinwari ZMA, Almesned A, Alakhfash A, Al-Rashdan AM, Faqeih E, Al-Humaidi Z, Alomrani A, Alghamdi M, Colak D, Alwadai A, Rababh M, Al-Fayyadh M, Al-Hassnan ZN. The Phenotype and Outcome of Infantile Cardiomyopathy Caused by a Homozygous ELAC2 Mutation. Cardiology. 2017;137:188–192. doi: 10.1159/000465516. [DOI] [PubMed] [Google Scholar]
- Shpilka T, Du Y, Yang Q, Melber A, Uma Naresh N, Lavelle J, Kim S, Liu P, Weidberg H, Li R, Yu J, Zhu LJ, Strittmatter L, Haynes CM. UPRmt scales mitochondrial network expansion with protein synthesis via mitochondrial import in Caenorhabditis elegans. Nature Communications. 2021;12:479. doi: 10.1038/s41467-020-20784-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siira SJ, Rossetti G, Richman TR, Perks K, Ermer JA, Kuznetsova I, Hughes L, Shearwood A-MJ, Viola HM, Hool LC, Rackham O, Filipovska A. Concerted regulation of mitochondrial and nuclear non-coding RNAs by a dual-targeted RNase Z. EMBO Reports. 2018;19:e46198. doi: 10.15252/embr.201846198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MM, Levitan DJ. The Caenorhabditis elegans homolog of the putative prostate cancer susceptibility gene ELAC2, hoe-1, plays a role in germline proliferation. Developmental Biology. 2004;266:151–160. doi: 10.1016/j.ydbio.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Takaku H, Minagawa A, Takagi M, Nashimoto M. A candidate prostate cancer susceptibility gene encodes tRNA 3’ processing endoribonuclease. Nucleic Acids Research. 2003;31:2272–2278. doi: 10.1093/nar/gkg337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Simard J, Teng DHF, Abtin V, Baumgard M, Beck A, Camp NJ, Carillo AR, Chen Y, Dayananth P, Desrochers M, Dumont M, Farnham JM, Frank D, Frye C, Ghaffari S, Gupte JS, Hu R, Iliev D, Janecki T, Kort EN, Laity KE, Leavitt A, Leblanc G, McArthur-Morrison J, Pederson A, Penn B, Peterson KT, Reid JE, Richards S, Schroeder M, Smith R, Snyder SC, Swedlund B, Swensen J, Thomas A, Tranchant M, Woodland AM, Labrie F, Skolnick MH, Neuhausen S, Rommens J, Cannon-Albright LA. A candidate prostate cancer susceptibility gene at chromosome 17p. Nature Genetics. 2001;27:172–180. doi: 10.1038/84808. [DOI] [PubMed] [Google Scholar]
- Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, Meyer BJ, Dillin A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt. Cell. 2016;165:1197–1208. doi: 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- Tjahjono E, Kirienko NV. A conserved mitochondrial surveillance pathway is required for defense against Pseudomonas aeruginosa. PLOS Genetics. 2017;13:e1006876. doi: 10.1371/journal.pgen.1006876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjahjono E, McAnena AP, Kirienko NV. The evolutionarily conserved ESRE stress response network is activated by ROS and mitochondrial damage. BMC Biology. 2020;18:74. doi: 10.1186/s12915-020-00812-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- Wang X, Chen XJ. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature. 2015;524:481–484. doi: 10.1038/nature14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- Weidberg H, Amon A. MitoCPR-A surveillance pathway that protects mitochondria in response to protein import stress. Science (New York, N.Y.) 2018;360:eaan4146. doi: 10.1126/science.aan4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel L, Topf U, Bragoszewski P, Wiese S, Sztolsztener ME, Oeljeklaus S, Varabyova A, Lirski M, Chroscicki P, Mroczek S, Januszewicz E, Dziembowski A, Koblowska M, Warscheid B, Chacinska A. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature. 2015;524:485–488. doi: 10.1038/nature14951. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. Journal of Cell Science. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- Yoo CJ, Wolin SL. The yeast La protein is required for the 3’ endonucleolytic cleavage that matures tRNA precursors. Cell. 1997;89:393–402. doi: 10.1016/s0092-8674(00)80220-2. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ward JD, Cheng Z, Dernburg AF. The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development (Cambridge, England) 2015;142:4374–4384. doi: 10.1242/dev.129635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wu X, Chen P, Liu L, Xin N, Tian Y, Dillin A. The Mitochondrial Unfolded Protein Response Is Mediated Cell-Non-autonomously by Retromer-Dependent Wnt Signaling. Cell. 2018;174:870–883. doi: 10.1016/j.cell.2018.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. The EMBO Journal. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Wu X, Zhou J, Li X, Huang X, Li J, Wu J, Bian Q, Wang Y, Tian Y. NuRD mediates mitochondrial stress-induced longevity via chromatin remodeling in response to acetyl-CoA level. Science Advances. 2020;6:eabb2529. doi: 10.1126/sciadv.abb2529. [DOI] [PMC free article] [PubMed] [Google Scholar]