

Abstract
Therapeutic combinations to alter immunosuppressive, solid tumor microenvironments (TMEs), such as in breast cancer, are essential to improve responses to immune checkpoint inhibitors (ICIs). Entinostat, an oral histone deacetylase inhibitor (HDACi), has been shown to improve responses to ICIs in various tumor models with immunosuppressive TMEs. The precise and comprehensive alterations to the TME induced by entinostat remain unknown. Here, we employed single-cell RNA-sequencing on HER2-overexpressing breast tumors from mice treated with entinostat and ICIs in order to fully characterize changes across multiple cell types within the TME. This analysis demonstrates that treatment with entinostat induced a shift from a pro-tumor to an anti-tumor TME signature, characterized predominantly by changes in myeloid cells. We confirmed myeloid-derived suppressor cells (MDSCs) within entinostat-treated tumors associated with a less suppressive granulocytic (G)-MDSC phenotype and exhibited altered suppressive signaling that involved the NFkB and STAT3 pathways. In addition to MDSCs, tumor-associated macrophages were epigenetically reprogrammed from a pro-tumor M2-like phenotype toward an anti-tumor M1-like phenotype, which may be contributing to a more sensitized TME. Overall, our in-depth analysis suggests that entinostat-induced changes on multiple myeloid cell types reduce immunosuppression and increase anti-tumor responses, which, in turn, improve sensitivity to ICIs. Sensitization of the TME by entinostat could ultimately broaden the population of patients with breast cancer who could benefit from ICIs.
Keywords: Epigenetics, Immunotherapy, Breast Cancer, Single Cell RNA sequencing, Immune Checkpoint Inhibitors, Entinostat, Myeloid Immunosuppression
INTRODUCTION
Immune checkpoint inhibitors (ICIs) against programmed death receptor 1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) enable immunogenic tumors with intrinsic presence of cytotoxic T cells to mount a robust immune response. ICIs prevent inhibitory signaling that dampens cytotoxic T-cell activity to permit a durable anti-tumor response in previously incurable cancers (1,2). Nonetheless, tumors with a tumor microenvironment (TME) dominated by immunosuppressive cell types often do not respond to ICIs (1). The TME across all histologic subtypes of breast cancer have been shown to include many cell types that prevent infiltration and activation of anti-tumor immune cells, including immunosuppressive cells such as T regulatory cells (Tregs), myeloid-derived suppressor cells (MDSCs), and cancer-associated fibroblasts (CAFs)(3–6). These cells secrete suppressive cytokines and upregulate the expression of immune checkpoints on T cells that bind to ligands present in the TME and halt cytotoxic T-cell activity (7).
Although single-agent ICIs have been generally unsuccessful in the treatment of most breast tumor types, additional drugs have been explored in combination with ICIs, many aiming to convert suppressive TMEs into immune-permissive environments (8–10). Epigenetic modulatory drugs, such as entinostat, have been shown to enhance the efficacy of ICIs in murine models of breast and pancreatic cancer (11–12). However, the specific mechanisms of modulation by entinostat are not fully understood. Our previous publication of phase I results of combination entinostat, nivolumab, and ipilimumab treatment in patients with advanced solid tumors reports an objective response rate of 16% and includes 10 patients with hormone receptor-positive and triple-negative breast cancer (13). Given the potential for future clinical trials in breast cancer, there is an additional need to determine the efficacy and mechanisms driving response of this promising treatment combination.
Due to the heterogeneity of cell types in the TME, single-cell RNA-sequencing (scRNAseq) is uniquely positioned to characterize the TME and identify both the molecular and cellular changes induced by entinostat in the NeuN HER2-tolerized NT2.5 syngeneic model of breast cancer (14–15). This tolerized HER2 (Erbb2)-overexpressing model of breast cancer mimics the highly suppressed, non-immunogenic TME observed in most patients with breast cancer, making it an ideal model to study poor responders to currently available ICIs. A non-immunogenic TME is defined as one consumed by immunosuppressive cells, with little to no cytotoxic T cells, creating an anti-tumor immune-excluded contexture. Here, our novel experimental design elucidated how scRNAseq could be used to analyze changes in a mouse model treated with entinostat, and with entinostat in combination with ICIs, to delineate potential mechanisms of response, while also controlling for biological variation. We characterized signatures of cell types within the NeuN TME. We found that MDSCs and anti-tumor M1-like and pro-tumor M2-like tumor-associated macrophages (TAMs) were reprogrammed by treatment with entinostat and ICIs, contributing to transformation of the TME. These in vitro and ex vivo studies further uncovered that entinostat reprogramed MDSCs through alterations in STAT3 and NFκB phosphorylation and downstream effector expression. These findings support a novel mechanism by which inhibition of HDACs orchestrate changes predominantly through two myeloid-derived cell types, MDSCs and TAMs, to promote the effects of ICIs that lead to tumor growth inhibition and promote survival.
METHODS
Mice and cell lines
Animals were kept in pathogen-free conditions and were treated in accordance with institutional and American Association of Laboratory Animal Committee policies. Animal studies have been conducted in accordance with, and with the approval of, the Johns Hopkins Animal Care and Use Program. Female NeuN mice were originally from W. Muller McMaster University (Hamilton, Ontario, Canada) and overexpress HER2 via the mouse mammary tumor virus (MMTV) promotor. Colonies were renewed yearly from Jackson labs [FVB/N-Tg(MMTVneu) 202Mul/J, Strain #002376] and bred in-house by brother/sister mating. Female Balb/c mice were obtained from Jackson labs (BALB/cJ, Strain #000651) at 8-10 weeks of age for spleen harvest used for in vitro MDSC suppression assays in which T cells were co-cultured with J774M cells. All mice for all experiments were used at 8-10 weeks of age.
NT2.5 is a rat HER-2/neu–expressing mouse mammary tumor cell line established from spontaneous mammary tumors in female NeuN mice (14). Culture conditions for NT2.5 cells were as follows: 37°C, 5% CO2 in RPMI 1640 (Gibco, cat. 11875-093) supplemented with 20% fetal bovine serum (FBS; Gemini, cat. 100-106), 1.2% HEPES buffer (Gibco, cat. 15630-080), 1% L-glutamine (Gibco, cat. 25030-081), 1% MEM non-essential amino acids (Gibco, cat. 11140-050), 0.5% penicillin-streptomycin (Gibco, cat. 15140-122), 1% sodium pyruvate (Sigma, cat. S8636), 0.2% insulin (NovoLog, cat. U-100), 0.02% gentamicin (Sigma, cat. G1397). J774M cells were previously established as a MDSC cell line (16) and were kindly provided for use in this investigation by Dr. Kebin Liu (Augusta University, USA). The J774M cell line was developed by sorting CD11b+Gr1+ cells from the ATCC macrophage line J774A.1 and have previously been shown to exhibit immunosuppressive functions like MDSCs (11,16). Culture conditions for J774M cells were as follows: 37°C, 5% CO2 in RPMI 1640 supplemented with 10% FBS, 1.5% HEPES buffer, 1% L-glutamine, 1% MEM non-essential amino acids, 1% penicillin-streptomycin, 1% sodium pyruvate, 0.0004% beta-mercaptoethanol (Sigma, cat. M3148). All cell lines were stored in liquid nitrogen and frozen down between 5-10 passages from original cell line obtained from ATCC. No cell lines were used beyond 15 passages. Cell lines were authenticated by ATCC and were regularly tested for Mycoplasma every three months in accordance with laboratory policy. All cell lines thawed from liquid nitrogen were passed once prior to use in experiments and were kept in culture no longer than 15 passages.
Tumor model, treatment dosing scheme, and tumor dissociation
Primary tumors were established in NeuN mice following injection of 5x104 NT2.5 into the mammary fat pad. Entinostat was dosed 5 days/week for 3 weeks by oral gavage at 5mg/kg in methylcellulose 2,3 (Syndax Pharmaceuticals), and given alone, with single or dual agent anti-PD-1 and anti-CTLA-4 (both by BioXCell) dosed at 100 μg/mouse via i.p. injection 2x/week, or with methylcellulose vehicle and Syrian hamster IgG (100 μg/mouse BioXCell) as appropriate controls, respectively. Tumors were seeded for 3 days prior to beginning treatment.
To obtain single-cell suspensions from breast tumors, tumors were harvested after three weeks of treatment, which is prior to any significant differences in tumor growth (11). Tumors were then weighed, diced, and dissociated using a tumor dissociation kit (Miltenyi Biotec, cat. 130-096-730) and the OctoDissociator (Miltenyi Biotec) per the manufacturer’s instructions. The 37C_m_TDK_2 program was used to dissociate tumors per the manufacturer’s instructions. Samples were filtered using a 40-mm cell strainer and red blood cells were lysed using ACK lysis buffer (Quality Biological, cat. 118-156-721). The resulting single-cell suspensions were used for subsequent isolation of specific immune cell types or flow cytometry as described below, or for scRNAseq as described below following an additional step of dead cell removal using the MACS Dead Cell Removal Kit (Miltenyi Biotec).
Immune cell isolation
Granulocytic (G)-MDSCs were isolated from single-cell suspensions from tumors following tumor dissociation using Miltenyi Biotec’s Myeloid-Derived Suppressor Cell Isolation Kit (cat. 130-094-538) according to the manufacturer’s protocol. Ly6G+ cells were positively selected to isolate G-MDSCs from Ly6G− monocytic (M)-MDSCs and were passed through LS columns (Miltenyi Biotec, cat. 130-042-401) twice to increase purity. Eluted G-MDSCs were then measured via flow cytometry for purity by staining with CD11b and Ly6G and were found to be 90-100% pure; subsequently, isolated cells were then used for the downstream assays described below.
CD8+ T cells were negatively isolated from spleens of 8-10 week–old Balb/c mice by mashing spleens through 100 mm cell strainers. Red blood cells were lysed using ACK lysis buffer, and CD8+ T cells were isolated via the EasySep Mouse CD8+ Isolation Kit (StemCell, cat. 19853) per manufacturer’s instructions. CD8+ T cells were then used for the in vitro suppression assays described below, purity of CD8+ T cells were tested after the first round of isolation using anti-CD8 and flow cytometry analysis indicated 90-100% purity.
For ex vivo experiments using intratumoral G-MDSCs, all isolated G-MDSCs were plated in tumor conditioned media after being isolated as described above (200,000-1 million G-MDSCs isolated per tumor). The number of G-MDSCs plated for suppression assays, as well as the duration of cell culture, is indicated in each assay described. Tumor-conditioned media was derived by culturing NT2.5 for 48 hours and subsequent filtering to remove tumor cells.
Flow cytometry
Isolated single-cell suspensions of breast tumors, were washed with 1X PBS (Sigma-Aldrich, VWR), counted manually using a hemocytometer, and plated in a 96 well plate with 200,000-1,000,000 cells per well. Cells were incubated for 30 minutes with Live/Dead Near-IR (ThermoFisher, cat. L10119) according to the manufacturer’s protocol. Cell were then washed with 1X PBS, followed by a 30-minute incubation with 50 μL of Fc block solution per well (BD Bioscience #553141). Surface antibodies were diluted 1:200 (Supplemental File S1) in FACS buffer (1X PBS with 2% FBS). Subsequently, samples were fixed and permeabilized with Foxp3/Transcription Factor Staining Buffer Set (eBioscience, cat. 00-5523-00) and stained with appropriate intracellular flow cytometry antibodies (Supplemental File S1). Samples were then fixed in 2% PFA and resuspended in FACS buffer to be run on a CytoFLEX (Beckman Coulter) cytometer within 2 days of fixation, and analyzed using FlowJo software version 10.5.0 (BC Biosciences) or Kaluza C software version 1.1 (Beckman Coulter). All antibodies used are listed in Supplemental File S1. Isotype control antibodies were used for all antibody targets and are listed by respective vendors; all samples were stained with target antibodies and istotype antibodies in separate wells. Gating strategies for M-MDSCs, G-MDSCs, and M1-like and M2-like Macrophages can also be found in Supplemental File S1.
J774M culturing and Luminex cytokine assays
1x105 J774M cells were seeded into 96-well plates and treated with entinostat or DMSO (Sigma-Aldrich, VWR) +/− lipopolysaccharide (LPS; 1μg/mL, Sigma-Aldrich,VWR) in serum-reduced media (2.5% FBS) at 37°C and 5% CO2. Total treatment time with entinostat, DMSO, or tumor-conditioned media was 24 hours, with LPS added during the last 6 hours. Supernatants were then collected and stored at −80°C. Supernatants were submitted to the University of Southern California (USC) Molecular Genomics Core (Los Angeles, CA) for analysis using the MILLIPLEX® standard assay for IL6, IL10, IL12p70, IFNγ, TNFα, and MIP-2 (Millipore). Samples were run on the Bio-Rad BioPlex 200 System using Luminex xMAP® technology technology and analyzed and calculated using BioPlex software. Samples treated with LPS were diluted by a factor of 20 prior analysis and 100μL submitted.
Arginase assays
Arginase activity was measured calorimetrically using Abcam’s Arginase Activity Assay Kit (cat. ab180877). For ex vivo studies, G-MDSCs were isolated from tumors as described, plated in tumor-conditioned media with either 50 or 100 μM entinostat or 1% DMSO vehicle for 16 hours, and subsequently harvested. For in vitro studies, J774M cells were cultured with (i) 0.5, 1, 5, 50, or 100 μM entinostat for 16 hours, (ii) 0.01% DMSO control for entinostat concentrations below 5 μM for 16 hours, (iii) 1% DMSO control for concentrations of entinostat 50 μM and above for 16 hours, or (iv) control with an anti-sense oligonucleotide (ASO, AstraZeneca) or a STAT3 antisense oligonucleotide inhibitor (STAT3i ASO, AstraZeneca) at 1 or 3 μM for 72 hours and subsequently harvested. Prior to analysis, dead cell removal was performed using the MACS Dead Cell Removal Kit (Miltenyi Biotec). In short, cells were lysed with the kit’s Arginase Assay lysis buffer at 1x106 cells/100uL and plated at 5x105 cells per well in two wells of a flat-bottom, low-retention plate per replicate, and topped to 40 μL with Assay Buffer. Target samples were incubated for 20 minutes at 37°C with 10 μL of included H2O2 substrate solution, while background wells were incubated with simply an additional 10 μL of buffer. Only after incubating were standards prepared and plated in duplicate per kit instructions, followed by the enzymatic reaction mixture which was added to all wells. Raw absorbance values were immediately obtained every 2 minutes over a 30-minute period using a plate reader (Molecular Devices SpectraMax M3) at OD=570 nm at 37°C. Arginase Activity Units were then calculated from raw absorbance values. ΔOD (ΔOD= (OD2-ODbg2)-(OD1-ODbg1)) was used to obtain the nmol of H2O2 generated by arginase, collected from a standard curve of known H2O2 concentrations. Arginase activity was calculated as (B/ΔT*V)*D in units/mL, where B is amount of H2O2 from standard curve (nmol), V is the sample volume added into reaction well (mL), D is sample dilution factor. One unit of Arginase activity refers to the amount of arginase that will generate 1.0 nmol of H2O2 per minute at pH 8 at 37°C.
In vitro suppression assay
J774M cells treated with entinostat or DMSO vehicle for 16 hours were co-cultured with stimulated T cells, and T-cell proliferation was measured via CFSE dilution. T cells were isolated from spleens of BALB/c mice as described above and subsequently labeled with CFSE (ThermoFisher, cat. C34554) per the manufacturer’s instructions. 2.5 x105 CFSE-labeled CD8+ T cells were co-cultured with J774M cells at varying ratios (1:1, 1:2, 1:4, and 1:8 J774M:T cells) and anti-CD3/CD28 beads (ThermoFisher, cat. 11453D) at a bead-to-T cell ratio of 1:1, per the manufacturer’s instructions, for T-cell activation. T cells were allowed to proliferate for 52 hours. Subsequently, the cultures were harvested, stained with Live/Dead NIR (ThermoFisher, cat. L10119), as well as anti-CD8, and analyzed via flow cytometric analysis as described above. Dilutions of initial CFSE were indications of T-cell division, where fewer divisions indicated greater suppressive activity. All antibodies used are listed in Supplemental File S1.
Western blots
For J774M Western blots, 2x106 J774M cells were treated with varying concentrations of entinostat (0-100 μM) for 16 hours and then either control ASO or 1 or 3 μM STAT3i ASO for 72 hours and stimulated with 1 μg/mL LPS (Sigma) for 2 hours. Whole cell extracts from J774M cells were isolated using RIPA Buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, Abcam #153034) supplemented with 1:100 protease/phosphatase inhibitor cocktail (Cell Signaling; cat. 5872). Protein concentration was determined using BCA Protein Assay Reagent (Pierce; cat. 23225). 30-100 μg of protein was loaded on a 4-15% Mini-PROTEAN TGX gel (Biorad) and transferred to PVDF membranes (Millipore). Membranes were blocked with 5% or 7% non-fat dry milk (Sigma-Aldrich, VWR) in TBS plus 0.1% Tween 20 (TBST, Sigma-Aldrich, VWR) for 1 hour at room temperature with gentle shaking. Membranes were then incubated with primary antibodies diluted in 5% or 7% non-fat dry milk in TBST at specific concentrations overnight at 4°C with gentle shaking: Phospho-Stat3 (Tyr705) Rabbit monoclonal antibody (mAb)(1:1000; Cell Signaling), Stat3 (D3Z2G) Rabbit mAb (1:1000; Cell Signaling), Phospho-NFkB p65 (Ser536)(93H1) Rabbit mAb (1:500; Cell Signaling), NFkB p65 (D14E12) Rabbit mAb (1:1000; Cell Signaling), beta-actin polyclonal antibody (1:1000; Invitrogen). Membranes were washed with TBST and incubated with secondary HRP-linked anti-rabbit IgG (1:10,000 and 1:20,000; Cell Signaling) in 1% non-fat dry milk in TBST for 1 hour at room temperature with gentle shaking. Membranes were washed with TBST, and proteins were visualized with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific). Membranes were stripped up to three times using Restore PLUS Western Blot Stripping Buffer (Thermo Scientific) and re-probed for other protein targets.
Quantitative (q)PCR
Reverse transcription of mRNA and subsequent qPCR was performed on RNA isolates from J774M cells treated with entinostat (16 hours) or STAT3i (72 hours) and then stimulated with LPS for 2 hours. Treated J774M cells were harvested, and RNA was subsequently extracted using Quick-RNA Miniprep or Microprep (Zymo Research; cat. R1050, R1054). RNA was reverse transcribed into cDNA using Maxima First Strand cDNA Synthesis Kit for RT-qPCR (ThermoFisher; cat. K1642). qPCR of subsequent cDNA was performed using PowerUp™ SYBR™ Green Master Mix (ThermoScientific; cat. A25742) and primer sets from Integrated DNA Technologies as follows: Actb (5’-GACTCATCGTACTCCTGCTTG-3’ and 5’-GATTACTGCTCTGGCTCCTAG-3’), Polr2a (5’-CAGGGTCATATCTGTCAGCATG-3’ and 5’-GGTCCTTCGAATCCGCATC-3’), Il6 (5’-TCCTTAG0CCACTCCTTCTGT-3’ and 5’-AGCCAGAGTCCTTCAGAGA-3’), Il10 (5’-ATGGCCTTGTAGACACCTTG-3’ and 5’-GTCATCGATTTCTCCCCTGTG-3’), Nox2/Cybb (5’-TGTTCCTGTACCTTTGTGAGAG-3’ and 5’-CACCTCCATCTTGAATCCCTT-3’), S100a9 (5’-CATCAGCATCATACACTCCTCA-3’ and 5’-GGAATTCAGACAAATGGTGGAAG-3’), and Stat3 (5’-GTTCAAGCACCTGACCCTTAG-3’ and 5’-AGTCTCGAAGGTGATCAGGT-3’). . The real-time PCR cycling conditions used were 50 °C for 2 min, 95 °C for 2 min, 40 cycles of 95 °C for 15 s, 52°C for 15 s, and 72°C for 1 min. Gene expression was quantified using the delta-delta CT method normalized to two reference genes, Actb and Polr2a (17). Samples were run in triplicate for each run, and each run was repeated 3 times. Approximately 10 ng of template was used per reaction. Samples were run on the QuantStudio(TM) 6 Flex System (ThermoScientific).
scRNAseq, quality control, and analysis
For library preparation, 10× Genomics Chromium Single Cell 3′ RNA-seq kits v2 were used. Gene expression libraries were prepared according to the manufacturer’s protocol. RNA was extracted from 20 whole tumors from the following groups: Vehicle control (V), entinostat-treated (E), entinostat and anti-PD-1 (EP), entinostat and anti-CTLA-4 (EC), entinsotat with anti-PD-1 and anti-CTLA-4 (EPC), with 4 biological replicates from each of the 5 experimental groups. Tumors were sequenced in 4 batches: RunA (8 tumors; 1 E, 2 EP, 2 EC, 2 EPC, 1 V), RunB (8 tumors; 1 E, 2 EP, 2 EC, 2 EPC, 1 V), Pilot1 (2 tumors, 1 E and 1 V) and Pilot2 (2 tumors, 1 E and 1 V). Each batch had an equal assortment of samples from each treatment group to reduce technical biases. Illumina HiSeqX Ten or NovaSeq were used to generate ~6.5 billion total reads (see Supplementary File S2 for quality metrics). Paired-end reads were processed using CellRanger v3.0.2 and mapped to the mm10 transcriptome v1.2.0 by 10x Genomics with default settings. ScanPy v1.4 was used for quality control and basic filtering. For gene filtering, all genes expressed in less than 3 cells were removed. Cells expressing less than 200 genes or more than 8000 genes or had more than 15% mitochondrial gene expression were also removed. Cell type proportions were normalized to total cells isolated per treatment group. CoGAPS v3.58 was used for non-negative matrix factorization (NMF) with the following parameters: distributed = “single-cell”, sparseOptimization = TRUE, nPatterns = 50 patterns for broad cell types, nPatterns = 20 for myeloid cell types and defaults for the remaining parameters (18). We implemented the Monocle3 v1.2.2.0 pipeline, including Batchelor for batch correction of library runs (Supplementary Figure S1A; 19), UMAP low dimensional embedding, and Leidenbase 0.1.0 for community detection (implemented in Monocle; 20). Doublets were removed from final UMAP. For each cell type, entinostat-treated cells were compared to vehicle-treated cells, and each independent combination treatment was compared to entinostat. To do this, we created cell data set (CDS) subsets for each cell type and each treatment group comparison. fit_models function in monocle3 was then applied to those subsets for logistic regression analysis with library batches, cell cycle theta estimate (for cell cycle correction on cancer cells only), and treatment groups used as covariates in model (model_formula_str = “~treatment + run” or model_formula_str = “~treatment + run + theta”). The estimate outputs were used to rank genes for pathway analysis, and the p value statistics were FDR (false discovery rate) adjusted. Volcano plots were created using the enhanced volcano plot package v.1.7. SAVER v. 1.1.2 was used to impute missing data due to drop out. Heatmaps were created using ComplexHeatmap v2.80 and Pheatmap v1.0.12. The geneSetTest and barcodeplot functions from limma package v.3.44.1 were implemented for pathway analysis. Further pseudotime analyses were also performed with Monocle3 upon the Batchelor batch corrected UMAP embedding, and statistics from pseudotime were computed similarly to the treatment effects described above. InferCNV (of the Trinity CTAT Project. https://github.com/broadinstitute/inferCNV) package v1.4.0 generated a heatmap that illustrates inferred copy number variation (CNV) across the cancer population in reference to fibroblasts, using pooled cells, the mm10 genome reference for chromosome mapping, and default parameters. This confirmed that the CNV profile of the NT2.5 cell line that was used to inoculate the mice, did not diverge across different tumors and treatments (Supplementary Figure S1B). Of note, the consistent CNV observed in the tumor cells across treatments served as an internal control, indicating that no expected differences in overall tumor cell identity between mice. We also observed a small cluster of cells we called “Cancer 2,” which was consistent across treatments and had a cancer and CAF intermediate CNV profile. These cells formed their own partition and were deemed to be either a technical artifact or an uncharacterizable epithelial cell type and, thus, removed from final UMAP (Supplementary Figure S1B). Cell cycle estimation was performed using Tricycle v1.0 to identify topological differences driven by cell cycle stage, such as in cancer cells (21, Supplementary Figure S2) The full code used in the analysis is made available on the following github repository: Dimitri-Sid/Entinostat_ICI_scRNAseq.
RNAseq of J774M cells
Cells were cultured as described above and treated overnight with 0.5 μM entinostat or 0.01% DMSO in biological triplicates. RNA-sequencing was performed by Novogene Corporation Inc. In brief, Qubit and Bioanalyzer instruments were used to perform quality control check for RNA samples. The NEBNext Ultra II non-directional RNA Library Prep kit was used to prepare libraries for sequencing. Labchip and qPCR assays were used to assess library quality and concentration. Novoseq6000 instrument was used to perform PE150 sequencing of the libraries. RNA Integrity Number (RIN) values of the samples ranged from 9.1 to 9.8. Preprocessing of the data was performed using Salmon, version 0.8.2 for alignment to the gencode vm10 reference index, and TXImport version 1.2.0 for transcript level summarization. Differential expression analysis was performed using the ebayes function from the limma R package version 3.46.0. The 3 entinostat samples were contrasted with the 3 DMSO control samples, and Benjamini-Hochberg was used to adjust the resulting p values for multiple testing correction. Heatmaps used to visualize the data were created using the heatmap.2 function from the gplots R package version 3.1.1.
Statistical analyses
For differential gene expression, monocle3 fit_models function was used, which implements a generalized linear model using a quasipoisson distribution. Coefficient_table() function was used to test if each coefficient differed from zero using a Wald test, and p-values were adjusted by applying a Benjamini-Hochberg FDR correction. Wilcoxon rank sum tests were used in gene set enrichment pathway analyses using Limma (wilcoxGST and geneSetTest). Pseudotime and NMF pattern weight distribution comparisons were tested using rank sum Wilcoxon and Kruskal-Wallis one way ANOVA on ranks tests. For bar graphs and box plots, Welch Two Sample t-test or one-way ANOVA with adjustments for multiple comparisons (Sidak’s test if pre-specified comparisons otherwise Tukey’s test) were conducted. All in vitro and ex vivo experiments were repeated at least 3 times. Statistical analyses were performed using GraphPad Prism v7.00 and R v4.1.1. Statistically significant p values are abbreviated as follows: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Availability of data and materials
Raw and processed sequencing data files are available under NCBI BioProject accession number PRJNA683665 and NCBI Gene Expression Omnibus accession number GSE166321. Code used in the analysis is made available on GitHub (Dimitri-Sid/Entinostat_ICI_scRNAseq).
RESULTS
ScRNAseq reveals myeloid-derived immune cells predominate the breast TME
To evaluate the molecular and cellular determinants of entinostat’s impact on immunosuppressive cell populations in the murine breast TME, we analyzed tumors derived from NeuN mice. We performed scRNAseq on 20 whole tumors from mice treated with entinostat alone or combined with ICIs (anti-PD-1 and/or anti-CTLA-4) to characterize the cell types and pathways impacted by entinostat that sensitize the TME for treatment with ICIs (Figure 1A). Tumors were implanted, and mice were treated for 3 weeks with vehicle control (V), entinostat alone (E), entinostat in combination with anti-PD-1 (EP) or anti-CTLA-4 (EC), or the triple combination (EPC). After 3 weeks, 20 whole tumors were harvested (4 from each group), processed into single cell suspension, and scRNAseq was performed using the 10x Genomics Chromium platform. A total of 56,731 individual cells were annotated into cell types by clustering the RNA expression profiles and assessing the expression of canonical and previously reported cell type markers (Figures 1B–C, Supplementary Table S1), and differentially expressed genes between clusters were identified (Figure 1D). Our analysis characterized cells into 5 cell type populations broadly defined by canonical gene markers. These included 24,798 cancer cells (Erbb2+, Cdh1+), 6,645 CAFs (Col12a1+, Mmp2+), 1,419 lymphoid cells (Cd3e+), 13,301 monocytes/macrophages (Adgre1+, Itgam+, Cd14+), and 10,568 MDSCs (S100a8/9+, Itgam+, Cd14+).
Figure 1. Single-cell transcriptional data of NeuN tumors treated with HDACi entinostat alone or in combination with ICIs.

A. Summary of cell numbers obtained for each treatment group in the NT2.5 tumor model (see Methods). Whole tumors from 20 mice, 4 in each experimental group, were used to perform scRNAseq. B. UMAP with each cell partition annotated by its corresponding cell type. C. UMAPS colored by canonical marker genes used for cell type identification. D. Dot plot showing top differentially expressed markers between partitions computed using monocle3. E. Cell weights for the CoGAPS NMF patterns found to correspond to partitions; boxplot lines correspond to medians. F. Gene weights for the genes most associated with each of the CoGAPS patterns that corresponded with cellular partitions. G. Proportion of cells in each group by treatment condition. H. Evaluation of Cd274 (PDL1) and Pdcd1lg2 (PDL2) ligand expression across cellular groups quantified in violin plot and overlayed with UMAP. White dots correspond to medians and black dots to means.
To further characterize cellular compartments in the TME, we also performed unsupervised learning using CoGAPS non-negative matrix factorization to identify patterns of biological activity in our dataset. To corroborate our cellular annotations from clustering, we compared whether any cell-level features (called patterns) corresponded to the single-cell clusters and their respective top-ranked genes directly from CoGAPS (Figures 1E–F). In total, we observed a total of 36 CoGAPS patterns that distinguished immune cells, myeloid cells, epithelial/fibroblast cells, and technical noise (Supplementary Figure S3). Several patterns corresponded directly with UMAP annotated clusters: two epithelial-associated patterns (Pattern 4 and Pattern 33), a lymphoid-associated pattern (Pattern 25), and a myeloid-associated pattern (Patterns 14). Altogether, these patterns combined with the clustering analysis yielded five broad cell types in the breast cancer TME. These cellular groupings did not vary significantly between treatment groups (Figure 1G). In all cases, the immunosuppressive myeloid cell types predominated the non-tumor cell population (Figure 1G–H).
To first determine the impact of entinostat on tumor cells, we performed differential expression and pathway analysis of the cancer cells, comparing entinostat treatment to vehicle. Firstly, we estimated cell cycle stage in each cell and found topology driven by cell cycle in the cancer partition (Supplementary Figure S2). To isolate treatment effects independent of the cell cycle changes, we performed further differential expression analysis in the cancer cells correcting for cell cycle and identified 179 differentially expressed genes. Gene set analysis identified a significant increase in antigen processing and presentation in entinostat-treated cells at a pathway level (Supplementary Figure S4A–B). To test the hypothesis that upregulation of these pathways corresponded to increased T-cell infiltration, we conducted a clustering analysis on the lymphoid cells alone and identified 812 regulatory (Treg), 112 cytotoxic (Tc), 455 helper (Th) T cells, as well as 40 B cells (Supplementary Figure S5A–B, Figure 5C). Tregs and Th cells were the most abundant types of T cells observed, consistent with the immunosuppressive landscape of most breast cancers and specifically this HER2+ mouse model (Supplementary Figure S5 C–D; 22). The greatest increase in T-cell content following treatment with entinostat, however, was observed in Treg and Th cell subpopulations. Previous functional studies of infiltrating CD8+ T cells after entinostat treatment demonstrates an increase in proliferation and a trend toward increased granzyme B and IFNγ production in response to combination ICIs with entinostat (11); however, these changes were not observed at the mRNA level, likely due to the small number of T cells captured with scRNAseq, limiting the statistical power of additional analyses with these data.
Figure 5. Proposed model of molecular mechanisms affected by treatment in the myeloid lineage of the TME.
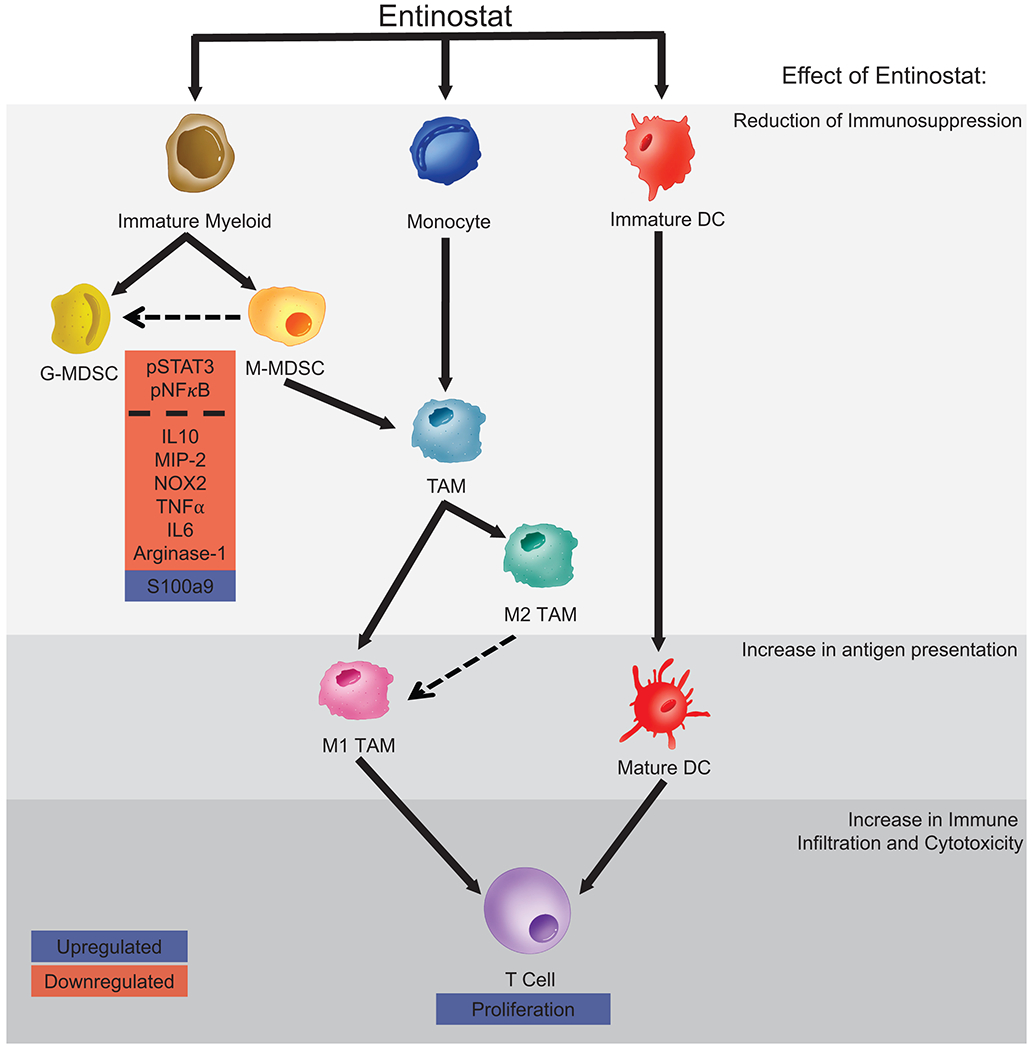
Entinostat promotes an anti-tumor signature in the TME and is likely accomplished by reducing immunosuppression via polarizing MDSCs to a more G-MDSC state (dotted arrow) and decreasing their suppressive capabilities through altered STAT3 and NFκB signaling. Intratumoral maturation of Myeloid DCs, in combination with TAMs polarizing from an M2-like towards an M1-like phenotype (dotted arrow), likely contribute to increased antigen presentation. Lastly, treatment effects on the T-cell compartment promote T-cell proliferation and also support increased anti-tumor responses. Transcription factors and cytokines in pink boxes are downregulated, and those in blue are upregulated.
Entinostat + ICIs modulate the phenotype of immature myeloid cells within the breast TME
The predominantly myeloid TME signature and expression of Cd274 and Pdcd1lg2 (PD-L1/2 checkpoint ligands) in MDSCs and macrophages prompted us to focus our analysis on the molecular changes induced by entinostat treatment. Many studies support an abundance of MDSC aggregation and/or change in suppressive function in tumors and suggest that these immature myeloid cell populations are key in preventing cytotoxic T-cell infiltration and restraining efficacy of ICIs (23). A better understanding of how treatments may affect the functional plasticity of these cells will contribute to determining what is driving improved sensitivity of the TME to ICIs. We first gathered a representative list of canonical markers and differentially expressed genes used to identify specific myeloid cell subclusters (Figure 2A–B, Supplementary File S2). We cross-validated these subclusters with additional patterns derived by a secondary CoGAPS implementation performed solely on the myeloid cells (Supplementary Figure S6). This analysis identified 18 myeloid cell patterns, in which 7 corresponded to myeloid sub-clusters, and the remainder corresponded to broader groups of cells with shared function or technical noise (Supplementary Figure S6, Figure 2C–D). Specifically, our analysis revealed 5,069 M-MDSCs (Pattern 14), 5,499 G-MDSCs (Pattern 5), 368 dendritic cells (DCs, Pattern 11), 2,471 monocytes (Pattern 16), 430 proliferating myeloid cells (Pattern 6), 8,079 M1-like (Pattern 9) and 1,953 M2-like (Pattern 3) TAMs (Figures 2E–F).
Figure 2. The NeuN TME is characterized by a dynamic population of immunosuppressive cells of myeloid origin.
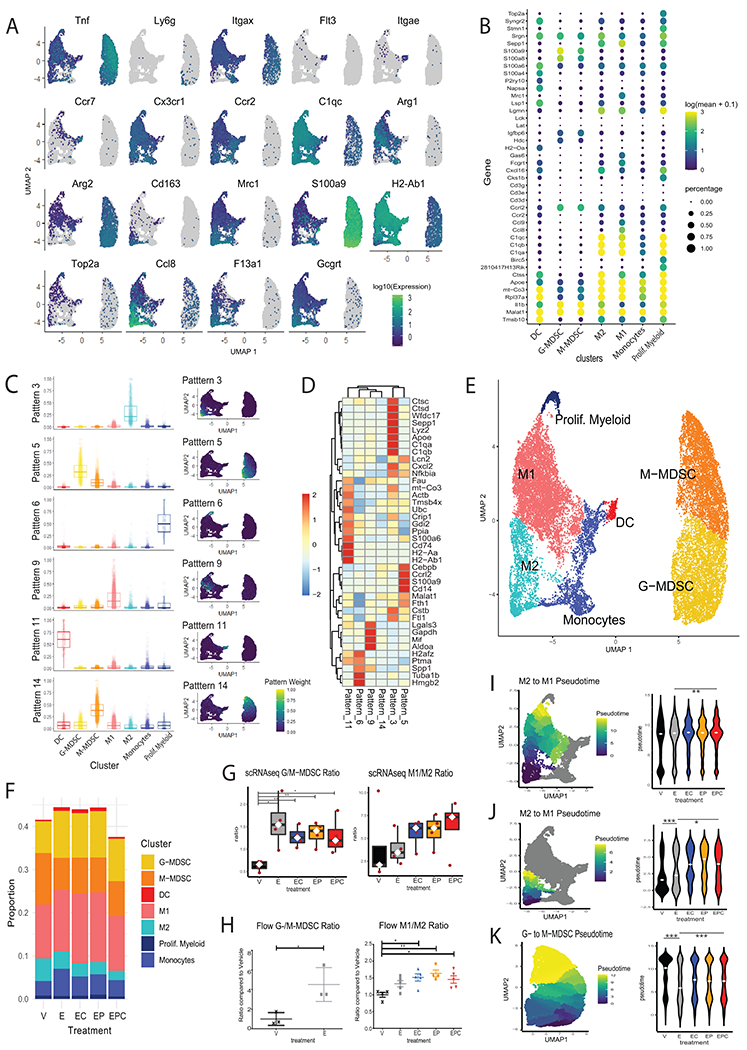
Myeloid cellsfrom mice in Fig. 1 were subseted from the total TME population and analyzed further. All data shown in panels A-E were analyzed from all 20 mice in all treatment groups combined. Data shown in panels F-K represent 4 mice/ treatment group. A. UMAPs of canonical myeloid cell subtype markers. B. Dot plot showing the top differentially expressed genes in each cluster. C. Cell weights for the CoGAPS NMF patterns obtained from the cells in the myeloid partition that corresponded to the cell clusters in boxplots by cell type and overlaid in UMAPs, boxplot lines correspond to medians. D. Gene weights for the top 3 genes most associated with each of the CoGAPS patterns that correspond with cellular partitions. E. Annotated clusters projected onto the UMAP of the myeloid subset population. F. Proportion of myeloid cells in each treatment condition. G. G-MDSC:M-MDSC ratio (left) and M1:M2 ratio (right) in tumors estimated from the single-cell RNAseq data. Each red dot corresponds to one tumor; significance calculated using Welch Two Sample t-test. H. Flow cytometry analysis showing G:M-MDSC ratios (left) and M1:M2 macrophage ratios (right) of breast tumors from entinostat- and ICI-treated NeuN mice. Each dot represents one mouse, and each bar represents mean +/− SEM; n=3–5 mice/group. Significance was determined by a one-way ANOVA with Tukey multiple comparisons test. I. M2 to M1 transition pseudotime weights overlayed in UMAP and graphed by treatment in violin plot for all TAMS. J. Analysis for the M2 population only performed as in (I). K. G-MDSC to M-MDSC transition pseudotime overlayed in UMAP and plotted by treatment. White spots on boxplots, and violin plots correspond to medians. Significance was determined by Wilcoxon signed-rank test. Statistically significant P values are shown as follows: *P< 0.05; **P<0.01; ***P<0.001.
Ratios of G/M-MDSCs and M1/M2 macrophages reflect suppressive function and pro- vs. anti-tumor responses, respectively (24–25). Using the clusters defined from the single-cell data, we found a significantly elevated G/M-MDSC ratio with entinostat treatment (p<0.05) and an increasing M1/M2 macrophage ratio with entinostat and combination treatments, although not statistically significant (Figures 2G). To corroborate these changes at the protein level, we conducted flow cytometry analysis of tumors from treated NeuN mice. The cytometry results showed a significantly elevated G-/M-MDSC ratio in mice treated with entinostat compared to vehicle-treated mice (p<0.05) and a significantly elevated M1/M2 ratio in mice treated with entinostat and ICI combinations (p<0.05, Figure 2H).
To evaluate the impact of the combination treatment on functional plasticity within these myeloid cells, we performed pseudotime analysis in the TAM and MDSC clusters. Specifically, pseudotime analysis was conducted to quantify the transition from M2-like to M1-like across the entire TAM cluster (Figure 2I). Consistent with the analysis of the M1/M2 ratio, we did not observe statistically significant differences in pseudotime from M2-like to M1-like macrophages between the treatment groups, although there was a trend towards increased pseudotime. Therefore, we performed additional pseudotime analysis within the M2-like cluster to further quantify the transition of the gene expression profiles in these cells towards the M1-like cluster (Figure 2J). We observed that entinostat treatment associated with significantly higher M2- to M1-like pseudotime (p<0.001), and combination treatments associated with significantly increased pseudotime compared to both entinostat alone and vehicle (p<0.05, Figure 2I). Finally, pseudotime was used to quantify the transition of cells from the G-MDSC to M-MDSC phenotypein the MDSC cluster. In this analysis, entinostat-treated groups associated with significantly lower G-MDSC to M-MDSC pseudotime (p<0.001, Figure 2K). Because G-MDSCs are considered less suppressive than M-MDSCs and are less likely to further differentiate into more specialized myeloid cells (26–27) and M1-like macrophages are considered to contribute to a more robust myeloid anti-tumor response (28), both changes in cell phenotypes suggest promotion of a less suppressed TME, which likely contributes to sensitization to treatment with ICIs.
Differential expression analysis within the MDSC and monocyte/macrophage cell clusters in mice treated with entinostat vs. vehicle revealed 182 differentially expressed genes in M1-like, 76 in M2-like, 15 in M-MDSC, and 30 in G-MDSCs (Supplementary Figure S7, Supplementary File S3). We ordered macrophages and MDSCs based on the respective pseudotimes and overlayed cluster identities, CoGAPS NMF pattern weights, and expression of representative differentially expressed genes. We observed that phenotypic transitions between M1/M2 and G-MDSC/M-MDSC associated with changes in the expression of genes involved with immune cytokine and chemokine signaling, such as Tnf, Ccl9, Il1r2, Ifitm1, MHC genes H2-Aa, H2-Ab1, H2-K1, H2−DMa, and alarmins S100a9 and S100a8 (Figure 3A–B, Supplementary File S3).
Figure 3. Treatment with entinostat modifies the transcriptional landscape of myeloid cell types.
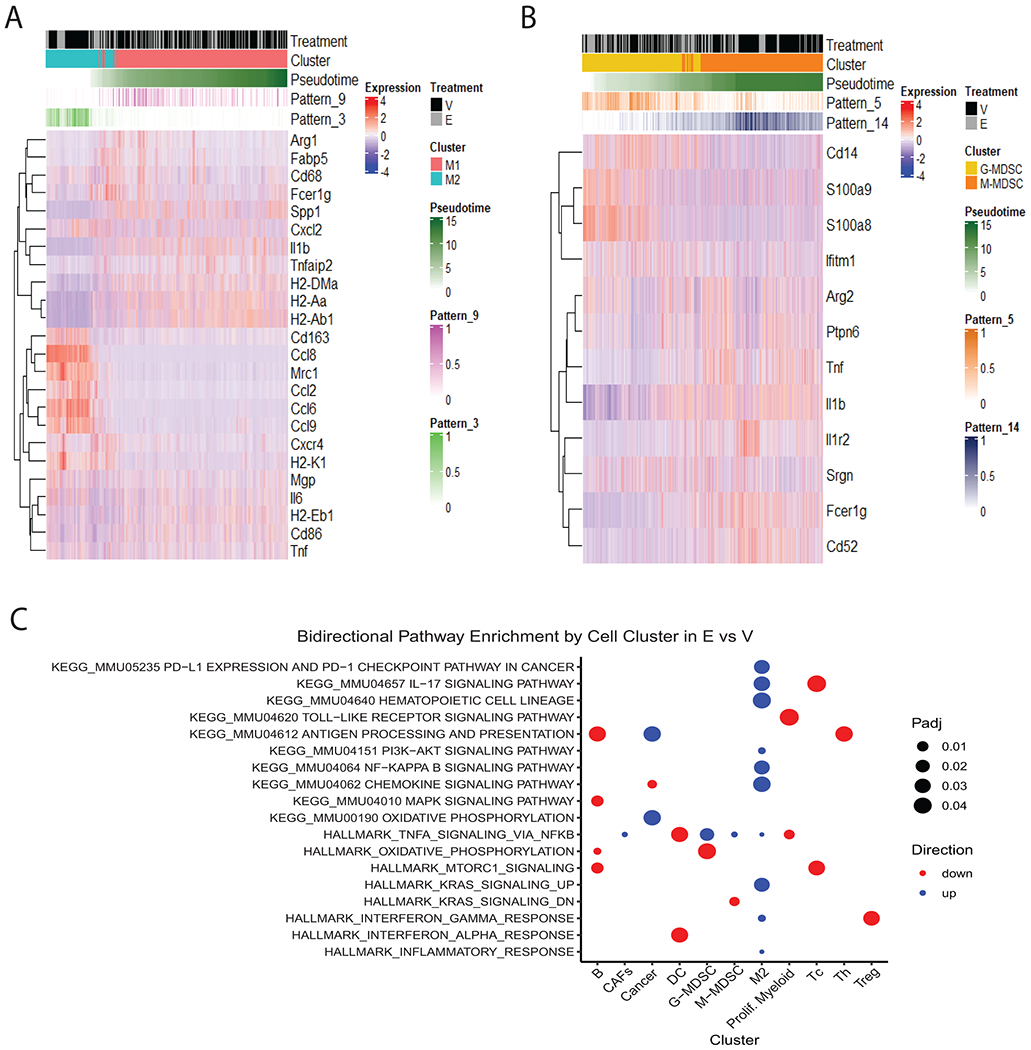
A. Heatmap of TAMs identified in the myeloid cell populations subset from the total TME population in the 20 whole tumors sequenced in this study. TAMs were ordered by pseudotime and annotated by cluster treatment and CoGAPS NMF patterns illustrating changes in expression of canonical markers and differentially expressed genes. B. Analysis for MDSCs performed as in (A). C. Enriched KEGG and Hallmark immune-related gene sets in myeloid clusters.
To further interpret the differential expression results, we performed gene set analysis on immunosuppression and immune signaling pathways annotated in KEGG and Hallmark MSigDB pathways. In MDSCs, we found that TNFα signaling via NFκB was significantly enriched by entinostat treatment in both G-MDSCs and M-MDSCs, and NFκB signaling was enriched in M2 macrophages (Figure 3C). We also found that oxidative phosphorylation was decreased in G-MDSCs with entinostat treatment relative to vehicle (Figure 3C). In Tregs, IFNγ response was significantly downregulated, whereas this pathway was upregulated in M2-like macrophages (Figure 3C). M2-like macrophages had the highest number of immune-related pathways significantly downregulated across all the cell types identified in the TME, whereas M1-like macrophages did not have any significant enrichment of the selected immune-related pathways (Figure 3C). Other important alterations in immune-related pathways between entinostat treatment and vehicle controls are summarized in Figure 3C.
STAT3 and NFκB signaling are implicated in decreased MDSC suppression by entinostat
The scRNAseq data suggested that entinostat induced a transition in myeloid cells into less immunosuppressive states (i.e., from M- to G-MDSC phenotypes) and identified significant changes in NFκB and oxidative phosphorylation (OXPHOS) pathways, all known to contribute to suppressive signaling (29). These data indicate that there are additional signaling pathways affected by treatment and support our previous functional data which demonstrates decreased suppressive function of G-MDSCs following entinostat treatment (11). This expanded our focus beyond altered phosphorylation of STAT3 in G-MDSCs (11). To investigate potential mechanisms driving the cell state transitions induced by treatment, we used an MDSC-like cell line (J774M cells)(30) to validate gene expression changes induced by entinostat. Bulk RNA-sequencing of J774M cells treated with entinostat or DMSO control identified an altered transcriptional profile of over 220 differentially expressed genes with entinostat (Supplementary Figure S8A, Supplementary File S4). Pathway enrichment analysis revealed decreased OXPHOS signaling and increased TNFα signaling via NFκB, consistent with the scRNAseq data; however, it also revealed an increase in the IL6, JAK, and STAT3 signaling (Supplementary Figure S8B), similar our previous findings in from tumor-infiltrating MDSCs (11). We subsequently examined the effect of entinostat on STAT3 and NFκB activation, as well as downstream targets of these pathways that mediate suppression. Stimulation of cells with LPS, a stimulus shown to mimic an acute inflammatory response in immune cells of myeloid origin (31) and a tool often used to help unravel underlying mechanisms of immune function (32), demonstrated entinostat decreased STAT3 and NFκB phosphorylation (Figure 4A), with subsequent decreases in transcripts of well-known downstream targets involved in immunosuppression, including Il6, Il10, and Nox2 (Figure 4B). Because these products promote the immunosuppressive phenotype of MDSCs, we evaluated secreted cytokines in the presence of entinostat. In LPS-treated cells, entinostat also significantly decreased production of cytokines, including GM-CSF, IL6, MIP-2/CXCL2, and TNFα (Figure 4C).
Figure 4. Effects of entinostat are mediated by STAT3 and NFkB pathway modulation.
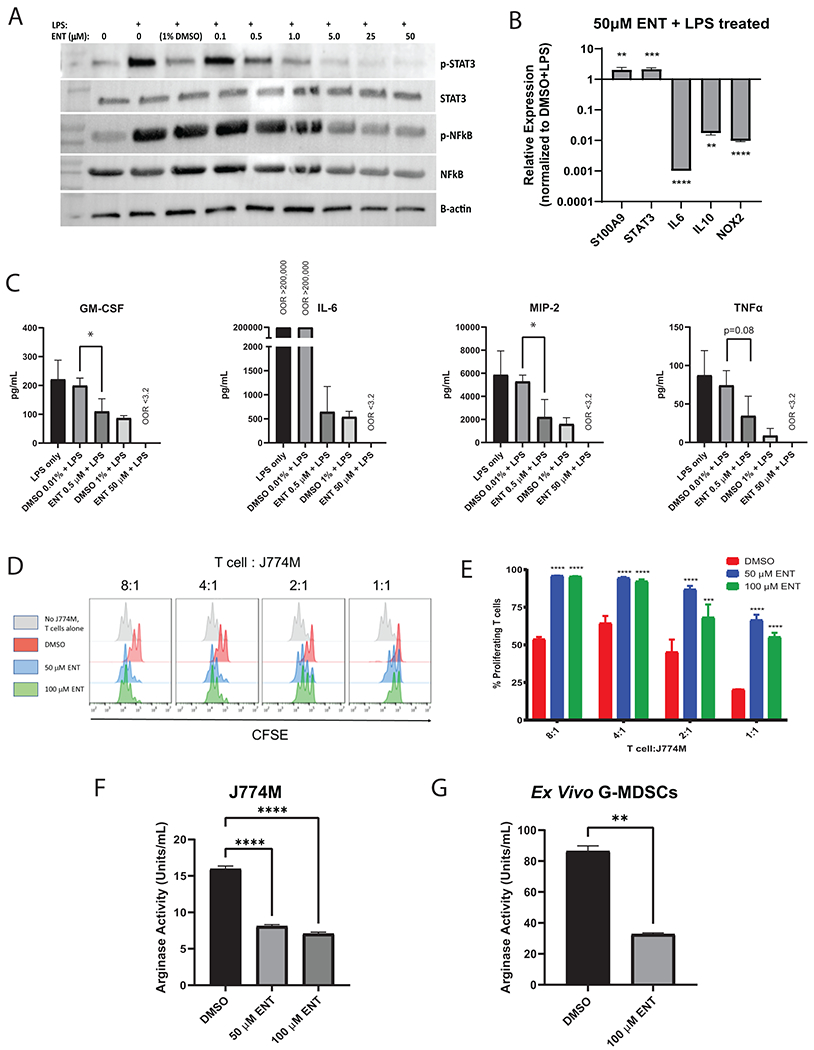
A. J774M cells pre-treated with varying concentrations of entinostat overnight were stimulated with LPS for 2 hours. Western blot depicts lysates probed for phospho-(p)STAT3, STAT3, pNFκB, NFκB, and beta-actin. B. qPCR of inflammatory genes, S100a9, Stat3, Il6, Il10, and Nox2 was performed on J774M cells treated with 50 μM entinostat (ENT) for 16 hours stimulated with LPS. Error bars represent SD from 3 replicates. Experiment was repeated 3 times. C. Multiplex analysis of cytokines released in vitro. Error bars represent SD from 3 repeated experiments. OOR: out of range. 0.01% DMSO was the control for 0.5 μM ENT and 1% DMSO was the control for 50 μM ENT. D. CFSE-labeled T cells stimulated with anti-CD3/CD28 beads for 52 hours at the indicated E:T ratio with J774M cells were analyzed by flow cytometry. Each peak represents a cell division as determined by CFSE dilution. E. Quantification of percentages from (D) of proliferating CD8+ T cells in each treatment group compared to the DMSO control. Error bars represent SD from replicates. Experiment was repeated twice. F-G, J774M cells (F) and Ly6G+ G-MDSCs (G) isolated from tumors of untreated NeuN mice were treated with entinostat ex vivo for 16 hours in tumor conditioned media before arginase activity was measured. Error bars represent SD from 3 replicates, with experiments repeated twice. Statistically significant p values: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
We then showed that these changes translated to functional differences in vitro, where entinostat reduced the suppressive function of J774M cells, as measured in T-cell proliferation and arginase activity assays (Figure 4D–F). Specifically, entinostat restored T-cell proliferation (Figure 4E) and reduced tumor cell arginase activity (Figure 4F). We further confirmed reduction of suppressive activity (via arginase activity) in Ly6G+ G-MDSCs isolated from tumors of untreated NeuN mice and treated ex vivo with entinostat for 16 hours in tumor conditioned media (Figure 4G). These results in J774M cells correlate with results from our previous study demonstrating restoration of T-cell proliferation and decreased Arg-1 production from isolated intratumoral G- and M-MDSCs (11).
To further support the role of entinostat’s effects on the STAT3 pathway and on decreased immunosuppression, we showed that STAT3 inhibition with STAT3 antisense oligonucleotide inhibitor (STAT3i)(Supplementary Figure S9A) resulted in similar gene expression as entinostat treatment (Supplementary Figure S9B). At the functional level, STAT3i decreased arginase activity in J774M cells (Supplementary Figure S9C). The similar results between entinostat and STAT3i are congruent with the hypothesis that entinostat modifies MDSC phenotype at least partly through the STAT3 pathway.
Collectively, these data suggest that entinostat treatment promotes the transition of an immunosuppressed TME towards one that supports anti-tumor responses through its effects on immune related signaling in cell types within the TME—most predominantly those of myeloid origin such as MDSCs, monocytes, and DCs (Figure 5). We previously suggested a limited model whereby entinostat’s sensitization of the TME relied heavily on its effect on MDSC suppression via STAT3 activation and now provide additional evidence in support of a more complex mechanism driving changes in tumor-infiltrating MDSC phenotype and function via its effects not only on STAT3, but also via NFkB and OXPHOS pathways (Figure 5). Our data suggests that the observed decrease in immunosuppression by G-MDSCs is in part the result of decreased STAT3 and NFκB activity and phosphorylation by entinostat, which leads to decreased production of suppressive cytokines. Our data supports an expanded model of TME sensitization that includes entinostat’s promotion of phenotypic shifts between M2 and M1-like TAMs, and maturation of DCs, which we hypothesize also contributes to increased antigen presentation and improved T-cell proliferation and cytotoxicity (Figure 5). The changes induced by entinostat contribute to the transformation of the non-immunogenic breast TME into a more immunogenic tumor that is more likely to respond to ICIs to promote tumor killing.
DISCUSSION
A wide range of cancers accumulate immunosuppressive cell types, such as MDSCs and M2-like TAMs, in the TME, exacerbating poor anti-tumor responses and ineffective immune checkpoint inhibition (33–34). We previously showed that combination treatment of entinostat with ICIs can improve response rates to ICIs in murine breast and pancreatic cancer models by reducing MDSC suppressive function (11). Given the ubiquitous effects of entinostat on cellular components of a tumor, our comprehensive investigation of the effects of this combination therapy on components of the breast TME by scRNAseq provides new insight into the complexity of the observed anti-tumor immune response. Here, we presented an in-depth characterization of the cellular composition and transcriptional landscape of primary breast tumors from the NeuN murine model of HER2+ breast cancer treated with entinostat in combination with ICIs. We used single-cell RNA-seq data to characterize the cellular heterogeneity and identified an approximately 1:1 ratio of myeloid cells to cancer cells in all treatment groups. Our analysis included a robust annotation of cell types by utilizing a combination of canonical marker genes, clustering, and NMF analysis to account for gene drop out at the RNA level. This analysis revealed a distinct MDSC cell partition, predominantly representing G-MDSCs in an entinostat-driven manner, as indicated by pseudotime analysis of the scRNA-seq data and RNA expression of alarmins S100a8/a9, cytokines Tnf and Il1b, and immune checkpoint ligands Cd274 and Pdcd1lg2. It also revealed that gene expression changes induced by entinostat in M2-like macrophages could promote a more M1-like TAM phenotype and contributed to a sensitized TME. Lastly, Tregs and Th cells are the most abundant T-cell subtypes observed within the TME, and robust expression of Ctla4 and Pdcd1 on these cells could support decreased suppression following treatment with ICIs. Still, the limited number of T cells in our study warrants future single-cell studies with enriched populations to assess the impact of this combination therapy on the lymphoid population.
MDSCs have been described as a dynamic myeloid cell type of neutrophilic and monocytic origin that are often recruited into solid TMEs of many cancer types and then reprogrammed to inhibit anti-tumor immune responses (35). Specifically, peripheral MDSCs can be pathologically recruited intratumorally and shift from a primarily G- to M-MDSC phenotype (26). In support of this, we demonstrate that in untreated breast tumors, there is accumulation of M-MDSCs harboring a pro-tumorigenic signature. Following treatment with entinostat, our data showed a reversal of this shift from M- to G-MDSCs, suggesting this could be one mechanism by which entinostat promotes a less suppressive TME. Also, a shift from M- to G-MDSC phenotype provides fewer immature myeloid cells with the potential to differentiate into immune-suppressive TAMs and DCs in response to cancer stimuli (35–36), as supported by our pseudotime analysis. A decrease in STAT3 and NFκB activation by entinostat treatment also contributed to the decreased immunosuppression, measures reduced cytokine and chemokine production in MDSCs. Although we previously identified MDSCs as an important component of improved response to ICIs following combined treatment with entinostat (11), we now have a better understanding of how entinostat may affect MDSC plasticity and what changes in signaling cascades may be responsible for these observed changes. Further evaluation of additional functional effects on MDSCs induced by ICIs is also necessary to fully understand the contribution their modulation has to an improved anti-tumor response.
Bulk RNA-sequencing of unstimulated J774M cells following treatment with low-dose entinostat revealed increased TNFα via NFκB and IL6, JAK, STAT3 signaling, whereas Western blot, qPCR, and luminex data demonstrated decreased expression of phospho-(p)STAT3, pNFkB, IL6, IL10, and Nox2. A decrease in pSTAT3 was also previously reported from intratumoral MDSCs treated with entinostat (11). It is not surprising that under conditions of an immune stimulus there can be differential effects of immune-related signaling. It is also possible that differences in expression at the mRNA level are representative of the complex feedback loops between NFkB and STAT3 pathways (29). This could also explain why we did not observe a significant increase in IL6/JAK/STAT3 signaling in intratumoral MDSCs as we did in the J774M cells. We hypothesize that in vivo, there is likely crosstalk between STAT3 and NFκB pathways which may be independently affected by entinostat or may work in concert as a result of changes by entinostat. It is also important to consider that these signaling cascades are likely not just occurring as autocrine signaling within MDSCs but could be occurring as a complex network of paracrine signaling with other cell types identified by this study that affect the phenotypic state and suppressive function of MDSCs because of treatment with entinostat. One example of an additional mechanism that could be driving decreased suppressive function of MDSCs via paracrine signaling is via entinostat-driven alteration of T-cell signaling, which could promote a decrease in suppressive signaling by MDSCs. Future studies are necessary to evaluate the likely complex mechanism of entinostat’s modulation of these signaling pathways in MDSCs.
Altogether, our analysis leads us to propose a model that entinostat reduces immunosuppression by promotion of myeloid cell plasticity toward the less suppressive G-MDSC phenotype and when combined with ICIs, more anti-tumor M1-like TAM phenotypes. We propose that decreased immune suppression is occurring via the direct effect of entinostat on MDSC function through altered STAT3 and NFkB signaling that then leads to altered cytokine production (Figure 5). We also propose that decreased suppression is not only occurring via the direct effect of treatment on MDSC function, but also through the impact of treatment on T-cell function which could indirectly decrease myeloid cell suppression specifically through IFNγ secretion. This hypothesis is supported by our finding of upregulated IFNγ signaling in M2-like macrophages, known to support the M2 to M1-like macrophage switching, and a downregulation of IFNγ signaling in Tregs, known to support suppression (37). We previously examined IFNγ secretion by intratumoral T cells following different treatment combinations and did not observe statistically significant changes (11). The inability to discern differences in cytokine secretion in intratumoral T cells could be because the small numbers of T cells infiltrating these tumors limit detection of changes in protein secretion in response to therapy. Other groups have seen similar findings, for example the combination of tumor targeted IL12 and entinostat can eradicate tumors in EMT6 breast cancer models and in MC38 and CT26 colorectal cancer models via M1-like TAM polarization, as well as increased cytotoxic T-cell infiltration and neutrophil activation (38).
Future work should evaluate the contribution of these additional immune cells with regard to altered suppression within the TME leading to a more robust anti-tumor response. Ultimately, all these changes likely work in concert to increase cytotoxicity within the TME. Our results add to the growing evidence that epigenetic modulation can prime tumors to respond to ICIs by modifying the transcriptional landscape of myeloid and T cells. It must be noted, however, that although entinostat promotes phenotypic plasticity in immune cells, it is also possible that non-immune cells in the TME may further introduce inter-cellular interactions in response to entinostat treatment that contribute to the observed transitions in the myeloid compartment. Our study exemplifies how scRNAseq of murine tumors treated with a novel therapeutic combination, entinostat + ICIs enabled a comprehensive genomic analysis of the treatment effects on the cellular players within the breast TME that are likely to contribute to the mechanism of response. This analysis highlights that the mechanism driving response is complex, not only due to the number of cell types involved, but also due to the complexity of the changes within each cell type. Future mechanistic studies should explore how treatment with entinostat and ICIs affects the functional plasticity of each of the cell types we identified to determine their contribution to response, which could potentially provide useful biomarkers and/or uncover novel therapeutic targets. Mirroring these studies to our human clinical studies of this treatment combination (13) has directed us to investigate changes in immune cell types beyond that of MDSCs and to investigate functional plasticity of these cells, which could provide a foundation for low-throughput MDSC-and TAM-based biomarkers to evaluate in future clinical studies.
Supplementary Material
Synopsis:
Epigenetic modulation using entinostat is demonstrated to sensitize HER2+ breast tumors to immune checkpoint inhibition. Entinostat promotes anti-tumor myeloid cell signatures, reduces MDSC suppression, and boosts T-cell infiltration and cytotoxicity in the tumor microenvironment.
Acknowledgments:
We would like to thank all members of the Jaffee lab for help throughout the course of these experiments. We would like to thank Syndax Pharmaceuticals for supplying the entinostat, and Ionis for supplying STAT3i used in these experiments. We would like to thank the AstraZeneca members of the Sidney Kimmel Comprehensive Cancer Center Experimental and Computational Genomics Core (ECGC), supported by NIH/NCI grant P30CA006973, for support with next-generation sequencing and data processing. We would like to thank Dr. Kebin Liu, Augusta University, for providing the J774M cells.
Funding:
This work was supported through funding from: NIH (NCI R01CA184926 for EMJ; P50CA062924 for EMJ, EJF, and LTK; NCI R01CA177669 for EJF and LTK; NCI U01 CA253403 for EJF; NCI 5T32 CA009071–34; NCI P30 CA006973; and NIDCR R01DE27809 to DAG and EJF Stand Up To Cancer – Lustgarten Foundation Pancreatic Cancer Convergence Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT14–14); Stand Up To Cancer is a division of the Entertainment Industry Foundation. The indicated SU2C grant is administered by the American Association for Cancer Research.; the Lustgarten Foundation’s Research Investigator’s Award Program; the Broccoli Foundation (EMJ and ERT); Research Scholarship Grant, RSG-21-020-01-MPC, from the American Cancer Society (DAG); The Bloomberg~Kimmel Institute for Cancer Immunotherapy; The Skip Viragh Center for Pancreas Cancer Clinical Research and Patient Care; The Commonwealth Foundation for Cancer Research (SY, DS, LTK and EJF); the Allegheny Foundation (DS, LTK, and EJF); Tower Cancer Research Foundation Career Development Award (ERT); the Emerson Foundation (EJF and EMJ); P30CA014089 from the National Cancer Institute (ERT); NIH NCI P30 CA014089 (ERT); MacMillan Pathway to Independence Fellowship (ERT); and the Maryland Cigarette Restitution Fund (SY).
Competing interests:
Through a licensing agreement with JHU, Dr. Jaffee has the potential to receive royalties from Aduro for a human GVAX vaccine. Dr. Jaffee receives research funding from Aduro Biotech and Bristol-Myers Squibb. RC has received research grants to institutions from Novartis, Puma Biotechnology, Merck, Genentech, Macrogenics, travel support from Genentech and an unrestricted educational grant from Pfizer.
REFERENCES
- 1.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Umansky V, Sevko A. Tumor microenvironment and myeloid-derived suppressor cells. Cancer Microenviron. 2013;6:169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gatti-Mays ME, Balko JM, Gameiro SR, Bear HD, Prabhakaran S, Fukui J, et al. If we build it they will come: targeting the immune response to breast cancer. NPJ Breast Cancer. 2019;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu K, Iyoda T, Okada M, Yamasaki S, Fujii S-I. Immune suppression and reversal of the suppressive tumor microenvironment. Int Immunol. 2018;30:445–54. [DOI] [PubMed] [Google Scholar]
- 6.Cha YJ, Koo JS. Role of Tumor-Associated Myeloid Cells in Breast Cancer. Cells. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paluskievicz CM, Cao X, Abdi R, Zheng P, Liu Y, Bromberg JS. T regulatory cells and priming the suppressive tumor microenvironment. Front Immunol. 2019;10:2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Cao F, Li M, Li P, Yu Y, Xiang L, et al. Hydroxychloroquine induced lung cancer suppression by enhancing chemo-sensitization and promoting the transition of M2-TAMs to M1-like macrophages. J Exp Clin Cancer Res. 2018;37:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCaw TR, Li M, Starenki D, Liu M, Cooper SJ, Arend RC, et al. Histone deacetylase inhibition promotes intratumoral CD8+ T-cell responses, sensitizing murine breast tumors to anti-PD1. Cancer Immunol Immunother. 2019;68:2081–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wall JA, Meza-Perez S, Scalise CB, Katre A, Londoño AI, Turbitt WJ, et al. Manipulating the Wnt/β-catenin signaling pathway to promote anti-tumor immune infiltration into the TME to sensitize ovarian cancer to ICB therapy. Gynecol Oncol. 2021;160:285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol Res. 2018;6:1561–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci USA. 2014;111:11774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roussos Torres ET, Rafie CI, Wang C, Lim D, Brufsky AM, LoRusso PM, et al. Phase 1 Study of Entinostat and Nivolumab with or without Ipilimumab in Advanced Solid Tumors (ETCTN-9844). Clin Cancer Res. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reilly RT, Gottlieb MB, Ercolini AM, Machiels JP, Kane CE, Okoye FI, et al. HER-2/neu is a tumor rejection target in tolerized HER-2/neu transgenic mice. Cancer Res. 2000;60:3569–76. [PubMed] [Google Scholar]
- 15.Mace K, Mayhew E, Mihich E, Ehrke MJ. Alterations in murine host defense functions by adriamycin or liposome-encapsulated adriamycin. Cancer Res. 1988;48:130–6. [PubMed] [Google Scholar]
- 16.Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. 2016;5:e1247135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fertig EJ, Ding J, Favorov AV, Parmigiani G, Ochs MF. CoGAPS: an R/C++ package to identify patterns and biological process activity in transcriptomic data. Bioinformatics. 2010;26:2792–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haghverdi L, Lun ATL, Morgan MD, Marioni JC. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol. 2018;36:421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Traag VA, Waltman L, van Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep. 2019;9:5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng SC, Stein-O’Brien G, Augustin JJ, Slosberg J, Carosso GA, Winer B, et al. Universal prediction of cell cycle position using transfer learning. BioRxiv. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foote JB, Kok M, Leatherman JM, Armstrong TD, Marcinkowski BC, Ojalvo LS, et al. A STING Agonist Given with OX40 Receptor and PD-L1 Modulators Primes Immunity and Reduces Tumor Growth in Tolerized Mice. Cancer Immunol Res. 2017;5:468–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi H, Li K, Ni Y, Liang X, Zhao X. Myeloid-Derived Suppressor Cells: Implications in the Resistance of Malignant Tumors to T Cell-Based Immunotherapy. Front Cell Dev Biol. 2021;9:707198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Youn J-I, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40:2969–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macciò A, Gramignano G, Cherchi MC, Tanca L, Melis L, Madeddu C. Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci Rep. 2020;10:6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37:208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218:1402–10. [DOI] [PubMed] [Google Scholar]
- 29.Fan Y, Mao R, Yang J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. 2013;4:176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S., et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin Cancer Res. 2017. Sep 1;23(17):5187–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996;60:8–26. [DOI] [PubMed] [Google Scholar]
- 32.Segre E, Fullerton JN. Stimulated whole blood cytokine release as a biomarker of immunosuppression in the critically ill: the need for a standardized methodology. Shock. 2016;45:490–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostrand-Rosenberg S, Fenselau C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol. 2018;200:422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Genard G, Lucas S, Michiels C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front Immunol. 2017;8:828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21:485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol. 2018;9:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hicks KC, Chariou PL, Ozawa Y, Minnar CM, Knudson KM, Meyer TJ, et al. Tumour-targeted interleukin-12 and entinostat combination therapy improves cancer survival by reprogramming the tumour immune cell landscape. Nat Commun. 2021;12:5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed sequencing data files are available under NCBI BioProject accession number PRJNA683665 and NCBI Gene Expression Omnibus accession number GSE166321. Code used in the analysis is made available on GitHub (Dimitri-Sid/Entinostat_ICI_scRNAseq).