

Abstract
The discovery of anti-neutrophil cytoplasmic antibodies (ANCA) and their antigenic targets, myeloperoxidase (MPO) and proteinase 3 (PR3), has led to further understanding as to the pathophysiologic processes that underlie vascular and tissue damage in ANCA vasculitis. ANCA trigger neutrophil activation leading to vascular damage in ANCA vasculitis. However, decades of study have determined that neutrophil activation alone is not sufficient to cause disease. Inflammatory stimuli are drivers of ANCA autoantigen expression and ANCA production. Certain infections or bacterial peptides may be crucial players in the initial steps of ANCA immunopathogenesis. Genetic and epigenetic alterations of gene encoding for MPO and PR3 provide additional disturbances to the immune homeostasis which provide a substrate for pathogenic ANCA formation from an adaptive immune system predisposed to autoreactivity. Promoted by inflammatory cytokines, ANCA binding leads to neutrophil activation, a process characterized by conformational changes, production and release of cytotoxic substances, and alternative complement pathway activation, thus creating an intense inflammatory milieu. This cascade of events perpetuates a vicious cycle of further inflammatory cell recruitment and activation, culminating in tissue necrosis. Our understanding of the pathogenic process in ANCA vasculitis paves the way for the development of therapies targeting crucial steps in this process. The greater appreciation of the role for complement, monocytes, and the adaptive immune system has already led to novel complement blockers and is poised to lead to further innovations which will allow for tailored antigen- or cell-specific immunotherapy targeting the autoimmune process without exposure to undue risks or toxicities.
Keywords: ANCA vasculitis, Neutrophil, T cell, B cell, Autoantibody, Glomerulonephritis
Introduction to ANCA vasculitis
Anti-neutrophil cytoplasmic autoantibodies (ANCA) vasculitis is characterized by small-vessel vasculitis associated with ANCA specific for proteinase 3 (PR3-ANCA) and myeloperoxidase (MPO-ANCA). Systemic vasculitis can affect any body tissue resulting in many injury patterns, including pauci-immune necrotizing crescentic glomerulonephritis, pulmonary capillaritis with hemorrhage, cutaneous leukocytoclastic angiitis causing purpura, and upper respiratory vasculitis causing sinusitis. We discuss what is known about the pathogenesis and immunopathogenesis of ANCA vasculitis leading to activation of neutrophils resulting in vascular inflammation. While the commonly used terminology has been “ANCA-associated vasculitis,” the preferred terminology is now shifting to simply “ANCA vasculitis.” This is to highlight the integral role ANCA are now known to play in initiating and perpetuating the vasculitis process as detailed below. ANCA are no longer considered possibly linked, as suggested by the term “ANCA-associated vasculitis” but rather drivers of the disease process. We therefore refer to it as ANCA vasculitis and propose this now to be the preferred nomenclature.
Pathogenic role of ANCA
ANCA targeting myeloperoxidase (MPO-ANCA) and proteinase 3 (PR3-ANCA) play a major role in the pathogenesis of ANCA vasculitis and is the rationale for antibody reduction therapies such as plasmapheresis and anti-CD20 medications. Experimental studies support the pathogenic role of ANCA in ANCA vasculitis. Little et al. showed that rats immunized with human MPO developed anti-MPO antibodies leading to leukocyte adhesion to vascular walls and small vessel vasculitis [1]. Xiao et al. demonstrated that mice injected with anti-MPO immunoglobulin, with or without functional lymphocytes, developed necrotizing crescentic glomerulonephritis and vasculitis [2]. An anecdotal example of the pathogenic potential of ANCA is the report of a pregnant woman with ANCA vasculitis whose disease flared during pregnancy. Her new-born baby developed pulmonary-renal syndrome and had circulating anti-MPO immunoglobulins similar to those in the mother [3]. No other example of this event has been published. All ANCA are not pathogenic because some individuals with ANCA vasculitis have long-lasting remission despite persistence of circulating ANCA. Rather, epitope specificity influences ANCA pathogenicity. MPO-ANCA and PR3-ANCA occur in plasma obtained from healthy blood donors [4]. Up to 20% of pauci-immune necrotizing small-vessel vasculitis cases have negative ANCA testing possibly due to an inability to detect the putative ANCA because of a circulating masking factor [5], or specificity for a neutrophil antigen that has not yet been identified. ANCA are required in the pathogenesis of ANCA vasculitis, but many other events influence the induction of the ANCA autoimmune response, and the mediation of inflammation participate in disease induction.
Exposure of MPO and PR3 autoantigens
Earlier studies established that most ANCA target one of two proteins, MPO and PR3, found in the granules of neutrophils and monocytes [6, 7]. Under normal homeostatic conditions, MPO and PR3 are formed and trafficked to granules during hematopoiesis. Myeloperoxidase plays a role in innate cellular defense against pathogens by forming hypochlorous acid and various other bactericidal and cytotoxic substances [8]. PR3, an intracellular serine protease, has an important role in the post-transcriptional formation of antimicrobial peptides and pro-inflammatory cytokines through its protein-cleaving abilities [9, 10]. MPO and PR3 are most abundant in neutrophils and monocytes stored in azurophilic granules. The canonical theory is that granule proteins such as MPO and PR3 are not produced within mature, circulating cells but are already stored in granules. Alterations at the genetic, epigenetic, or protein levels for MPO and PR3 may be present in ANCA vasculitis.
Studies have demonstrated that patients with ANCA vasculitis have increased expression of MPO or PRTN3, the genes that encode for MPO and PR3, respectively, compared to healthy controls [11] and that quiescent neutrophils may constitutively express ANCA antigens [12, 13]. Many patients demonstrate dynamic changes in the expression of MPO or PRTN3 with high expression during disease relapse and lower expression during remission [13, 14]. The level of expression of ANCA antigens may be genetically determined for a given individual [15]. This suggests that greater constitutive surface expression of ANCA antigen may be relevant to the pathogenesis of ANCA vasculitis but may not be so. Trauma from centrifugation during neutrophil isolation may be enough to cause PR3 surface expression and must be considered when evaluating experimental in vitro data that show an increased surface expression [16]. Individuals with MPO-ANCA or ANCA-negative vasculitis, and non-vasculitic inflammatory disorders such as rheumatoid arthritis, have a higher frequency of high PR3 membrane expression compared to healthy individuals [17]. Surface expression of ANCA antigens may be a manifestation of occult systemic inflammation or an inherent tendency for neutrophils to produce more ANCA antigens due to a greater expression of MPO and PRTN3 genes and thus release more antigens upon stimulation.
Although MPO and PRTN3 expression levels may be increased in patients with ANCA vasculitis, these expression levels are derived from mRNA of circulating leukocytes that theoretically consist of mature neutrophils. What is driving the mRNA expression in what should be transcriptionally “silent” peripheral cells? Epigenetic modifications that include DNA methylation, histone methylation, histone acetylation, and other mechanisms have the capacity to alter gene activity and expression. Several epigenetic alterations have been discovered in ANCA vasculitis.
Histone modifications in the MPO and PRTN3 gene loci may be involved in the pathogenesis of ANCA vasculitis. Ciavatta et al. found that chromatin modification of H3K27me3 levels, associated with gene silencing, was depleted at MPO and PR3 loci in neutrophils from ANCA vasculitis patients [11], indicating that epigenetic modifications associated with gene silencing may contribute to aberrant expression of MPO and PR3. Another study found that ANCA vasculitis patients with decreased DNA methylation, a gene silencing process, at the PRTN3 promoter had a greater risk of relapse [18]. Thus, DNA methylation of PRTN3 may predict disease remission and relapse. These studies suggest that epigenetics may be important in the development of ANCA vasculitis and perpetuation of aberrant expression of autoantigen genes.
Increased transcription and inferred translation of MPO and PR3 alone are not enough to drive the disease. For ANCA to form against MPO or PR3, they need exposure to the adaptive immune system either via mobilization from the intracellular space to the cell surface or a release into the extracellular space after activation of the neutrophils. During priming and activation, neutrophils go from a quiescent state to one that is optimized for anti-microbial activity via the expression of adhesion molecules, the generation of reactive oxygen species (ROS), and the release of granule contents [19]. Neutrophils are the first cellular line of defense against pathogens, and inflammatory cytokines such as TNF-α and bacterial components such as lipopolysaccharides have been shown to be major stimulants for the expression and mobilization of ANCA antigens [20–23]. Another important mediator is the complement component anaphylatoxin C5a through binding to its receptor on neutrophils [24], discussed below. The release of MPO and PR3 from neutrophils can be triggered by many different stimuli because of the important role they play in innate immune defense, for example against pathogens. Even events with low morbidity and mortality such as transient infections, or minor inflammatory stimuli such as surgical procedures or cigarette smoking, release MPO and PR3 from neutrophils [25].
ANCA vasculitis is a rare disease with an incidence of 1–2 cases and a prevalence of 5–20 cases per 100,000 individuals [26], despite frequent if not constant exposure to ANCA antigens released from neutrophils. This supports the role of dysregulation at the level of the adaptive immune system that can turn ANCA antigen exposures to immunogenic ones that cause ANCA disease, as discussed in the next section.
The theory of molecular mimicry and complementary proteins is proposed as mechanisms that initiate a pathogenic ANCA response. In PR3-ANCA, patients have antibodies recognizing a peptide translated from anti-sense PR3 (thereby complementary PR3, cPR3). Through a series of serendipitous experiments, it was determined that a subset of PR3-ANCA vasculitis patients also harbors autoantibodies that react to cPR3 [27]. To determine cause and effect, mice injected with the cPR3 peptide and monitored for both antibodies to PR3 and cPR3 developed antibodies to both proteins, supporting the theory that complementary proteins can promote the development of antibodies to both sense and anti-sense proteins [27]. However, where would anti-sense or complementary protein sources emerge in the human population? Through sequence analysis and Basic Local Alignment Search Tool of peptide sequences, the cPR3 sequence shares high levels of homology with numerous microbial sources including Staphylococcus aureus and E. histiolytica [27]. This underscores the relevance of molecular mimicry in the development of autoimmune disease and ANCA vasculitis, especially as many patients report symptoms of infection during the weeks leading up to disease onset and diagnosis.
Apoptosis may provide another path for exposure of ANCA antigens to become immunogenic. Apoptotic neutrophils display ANCA antigens, allowing immune cells to interact with them [28]. Dysregulated apoptosis, through the display of autoantigens to the immune system, has been hypothesized as a major pathway in the pathogenesis of some autoimmune diseases, particularly systemic lupus erythematosus [29]. Neutrophil extracellular traps (NETs), discussed below, contain MPO and PR3 antigens, DNA, and the immunostimulatory antimicrobial protein LL37 [30]. Exteriorized DNA with LL37 from NETs trigger self-recognition of DNA by dendritic cells in other autoimmune conditions such as psoriasis [31]. Exteriorized MPO and PR3, along with LL37, from NETs could play a role in the aberrant self-recognition of antigens by the immune system leading to the formation of ANCA.
Targeting of ANCA antigens by the adaptive immune system
With any immune response to antigen, whether a foreign pathogen or self-antigen, the interaction between innate and adaptive immunity rests with MHC and peptide presentation. To prevent autoimmune disease, the immune system contains checks and balances that inform the discrimination between self and non-self antigens, termed tolerance. Tolerance is broken in autoimmunity, permitting the immune system to target, recognize, and activate in response to self-antigen.
Autoimmune disease results from multiple hits that break tolerance. One hit is genetic predisposition, often involving HLA antigen binding capabilities. Genome-wide association studies (GWAS) have been performed in patient cohorts with various autoimmune diseases. The top genetic association with autoimmune disease is human leukocyte antigen (HLA) [32]. GWAS performed in two cohorts of patients with ANCA vasculitis, one a European cohort [33] and another North American cohort [34], determined that HLA-DPB1 was associated with ANCA vasculitis and notably enriched in the PR3-ANCA vasculitis population. HLA-DQ (specifically DQA1 and DQB1) has been associated with MPO-ANCA vasculitis, although both the European and North American cohorts were underpowered for detection within the MPO-ANCA vasculitis subgroup.
Each HLA gene encodes for a variety of alleles that determine the HLA antigen-binding groove. Therefore, different HLA proteins (encoded for by different alleles) have a lower or higher affinity for certain antigenic peptides. Several HLAs associated with MPO- or PR3-ANCA vasculitis have a strong affinity for binding peptides from both autoantigens. Through in silico predictions that were confirmed with in vitro HLA-binding assays, HLA-DPB1*04:01 has strong affinity for several MPO and PR3 peptides [35, 36].
Studies to elucidate the role of HLA and binding autoantigenic peptides were key to understanding the formation of autoreactive T and B cells that leads to the production of ANCA. By understanding the interaction of certain HLA with autoantigen peptides, tetramers (HLA multimers loaded with selected peptides and conjugated to a fluorophore) can be utilized to further profile the immune system specificity. Tetramers will bind to T cell receptors that recognize the presented antigen. This tool permitted quantification and characterization of MPO-reactive CD4 + T cells in the periphery of patients with ANCA vasculitis [35]. Autoreactive, MPO-specific T cells are detectable in the peripheral blood of ANCA vasculitis, and these T cells are pro-inflammatory and clonally expanded [35]. The detection of these T cells indicates that ANCA are high-affinity, class-switched antibodies that require antigen-specific T cell help to B cells and highlight the role T cells play in pathogenesis.
Current clinical assays detect ANCA that are a polyclonal pool of autoantibodies with multiple different epitope specificities. Epitope mapping to determine where ANCA bind to MPO or PR3 may lead to clinical epitope-specific ANCA assays that provide more useful information for patient care than assays for polyclonal ANCA. Studies using recombinant mutants, human/mouse chimeric proteins, or epitope-excision mass spectrometry have elucidated a key region of MPO that is targeted by MPO-ANCA [5, 37–39]. Certain ANCA epitope pairings correlate with disease activity and may be better markers of relapse compared to the traditional measurement of total polyclonal ANCA [5]. Studies elucidating the precise targeting of autoantigens by the immune system are key to developing personalized therapies.
ANCA binding resulting in neutrophil activation
Immunopathogenesis leading to the formation of autoreactive T cells and autoantibodies is critical for the formation of ANCA (see Fig. 1 for a summary of the antigen recognition and immune recognition process detailed above), but the essential interaction that leads to vasculitis is the binding of ANCA to their cognate antigen on neutrophils and monocytes, inducing activation, degranulation, and release of injurious factors on the vascular endothelium.
Fig. 1.
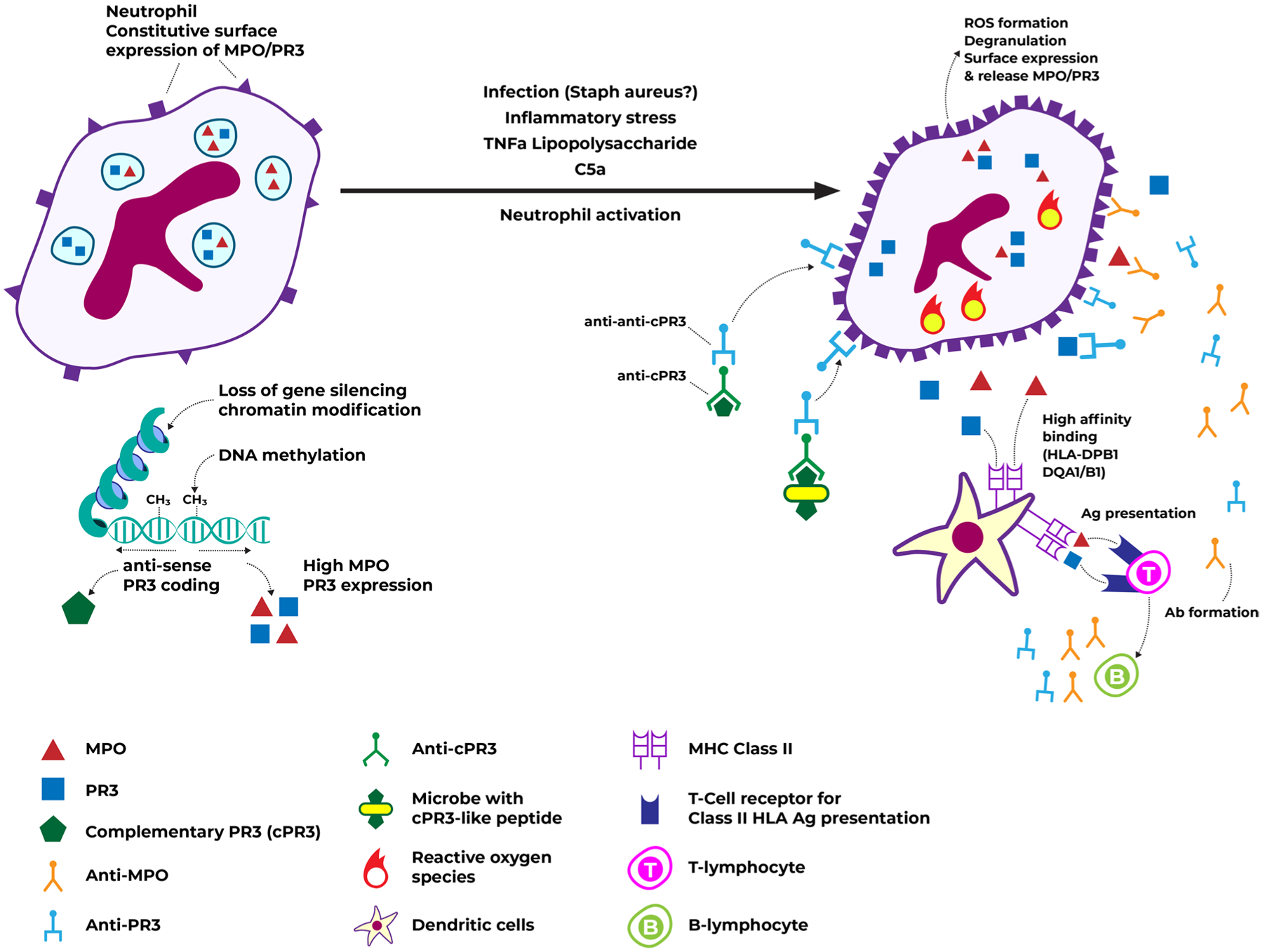
ANCA antigen expression and immune recognition. Caption: under normal circumstances, neutrophils constitutively express some level of MPO/PR3 on their surface, but most are stored in granules. Chromatin modifications, loss of gene silencing, and decreased DNA methylation may contribute to higher MPO/PR3 expression. Neutrophil activation, via inflammatory stimuli, leads to degranulation, greater surface expression, and release of MPO/PR3. Peptides from these antigens bind with high affinity to HLA, leading to antigen presentation and auto-antibody formation. Auto-antibodies may also occur from the formation of antibodies against anti-cPR3 or against antibodies targeting cPR3-like microbial peptides, which then cross react with regular PR3. These auto-antibodies against MPO/PR3 constitute ANCA. Abbreviations: ANCA, anti-neutrophil cytoplasmic antibodies; cPR3, complementary PR3; DNA, deoxyribonucleic acid; HLA, human leukocyte antigen; MPO, myeloperoxidase; PR3, proteinase 3
The interaction between ANCA and its antigenic target on neutrophils is key in the pathogenesis of ANCA vasculitis. Binding of ANCA to antigens occurs through interaction of the Fab′2 segment with its corresponding antigen. The Fc regions of ANCA bound to antigen engage Fc receptor on the surface of neutrophils [40, 41], initiating a series of intracellular signals culminating in neutrophil activation [42]. Neutrophil activation in response to ANCA is present to a much greater extent in individuals with ANCA vasculitis compared to healthy controls [43], suggesting an inherent predisposition for a greater inflammatory response in affected patients. ANCA-activated neutrophils increase the production of ROS and release the contents of their intracellular granules [22, 44]. This process is amplified with prior exposure to neutrophil-activating factors [22, 45] that can lead to a self-amplifying loop of ANCA antigen expression on the cell surface, causing further activation by ANCA and perpetuating the inflammatory response. The ability of MPO- and PR3-ANCA to activate neutrophils may differ, possibly accounting for differences in disease phenotypes. PR3-ANCA may induce more neutrophil degranulation and ROS formation than MPO-ANCA [44], which may explain why the former has more active lesions (e.g., crescents and fibrinoid necrosis) and the latter more often manifests with greater chronicity and sclerosis on biopsy [46, 47]. The reasons underlying these differences in presentation between MPO-ANCA and PR3-ANCA vasculitis are unknown.
If interaction of ANCA with MPO or PR3 is at the core of ANCA vasculitis pathogenesis, why are immune complexes and immunofluorescence staining for immunoglobulin so rarely seen in ANCA vasculitis (pauci-immune disease)? Immune complexes are likely degraded during the pathogenic process and become undetectable during testing. Animal models and human studies of ANCA vasculitis have shown that positive immunofluorescence staining for immunoglobulins and complement can be seen [48, 49] but then disappear with infiltration of immune cells and ensuing inflammatory damage [50]. A similar process is seen in Arthus reaction, a type III immune complex hypersensitivity reaction, induced in the skin of Guinea pigs where immunoglobulin and complement staining on histological exam of lesions were detectable up to 18 h after the initial reaction and then could not be detected, suggesting they were degraded or eliminated by the inflammatory reaction over time [51]. Despite the paucity of immune complexes found on histopathologic examination of biopsy specimens, ANCA vasculitis is driven by the interaction of ANCA antibodies with their target antigens, which is therefore a variant of immune complex-mediated disease.
Cascade of neutrophil activation in ANCA vasculitis
The binding of ANCA to their antigens leads to neutrophil activation and initiates a cascade of events that culminates in vascular inflammation. There may be dysregulation of multiple steps along a series of events, and not just ANCA production and binding to antigens that lead to end-organ damage in ANCA vasculitis.
Cytoskeleton changes
Neutrophils are 12–14 μm diameters and require some degree of deformability to pass through the glomerular capillary network where segments may have a diameter smaller than 10 μm [52]. Binding of ANCA to their antigenic targets on neutrophils not only causes morphologic changes allowing it to get to its site of action but also contributes to small-vessel damage characteristic of ANCA vasculitis. Tse et al. demonstrated that ANCA-activated neutrophils had polymerization of their actin via interaction with the Fc receptor, which took on a filamentous form [53]. Neutrophils developed a more rigid structure, lending it resistant to deformation, demonstrated by slower transfer into a micropipette of 5 μm [53]. This may account for ANCA vasculitis predilection for small vascular beds such as those in the kidneys, lungs, and skin, all of which have capillaries smaller than the diameter of a normal neutrophil [52, 54, 55]. A non-deformable neutrophil is unable to pass through the capillary bed, leading to obstruction of flow and allowing for adhesion to the endothelium with subsequent release of effectors of local damage. This may explain why the liver and spleen, having sinusoidal capillaries of larger diameter than regular capillaries, are not classically affected by ANCA vasculitis. It is unclear why this occurs less frequently in other organ micro-circulation such as the gastrointestinal tract or musculoskeletal system. Cases of ANCA vasculitis leading to muscle necrosis have been reported [56]. Overall, ANCA vasculitis predilection for certain organs is incompletely understood and unlikely to be explained solely by neutrophil cytoskeleton changes.
Vascular adherence
A neutrophil that is physiologically activated mediates an inflammatory response, adheres to the endothelium, and then undergoes a series of structural changes that allow it to roll across the endothelium and transmigrate through to gain access to the stimulus of the inflammation, for example, tissue trauma or a microbial pathogen. In ANCA vasculitis, the inflammatory response is inappropriately directed against normal vessels. Inflammatory cytokines involved in the activation of neutrophils promote adherence of neutrophils to the endothelium. TNF-α activates neutrophil integrins, adhesive membrane receptors that will bind to endothelial ligands, causing endothelial adherence [57]. Hu et al. demonstrated that both TNF-α and IL-8 induced a downregulation of neutrophil chemokine receptors CXCR1 and CXCR2 in patients with ANCA vasculitis compared to controls, regardless of disease activity [58]. Normally, CXCR1 and CXCR2 serve as signals to indicate where neutrophils transmigrate across the endothelium towards injured tissue. If these receptors are downregulated in patients with ANCA vasculitis, their activated neutrophils remain adherent to the endothelium. Circulating ANCA then have the liberty to bind to their antigens expressed on the surface of neutrophils adherent to endothelium. Moreover, when ANCA bind to their Fcγ receptor, they activate integrins so that neutrophils become aberrantly fixed to endothelium, effectively halting the normal rolling process [59]. Therefore, activated neutrophils from patients with ANCA vasculitis seem predisposed to remain within the vascular space. The ensuing massive inflammatory response will be focused along the endothelium and within the vascular space.
Reactive oxygen species production and degranulation
Major components of the neutrophil’s weapons against pathogens are ROS, highly toxic substances for any living organism, as well as the contents of their intracellular azurophilic granules, including MPO and PR3, involved in the degradation of microbes. When neutrophils adhere to the endothelium and are activated by ANCA, the damage caused by the release of ROS and granule contents will be misdirected against the endothelium. One consequence of damage to endothelial cells is detachment and endothelial cell apoptosis. An important oxygen radical produced by neutrophils is H2O2, which is toxic to endothelial cells [60]. The granule protease elastase can also damage endothelial cells causing detachment [61]. Released ANCA antigens have been shown to bind to endothelial cells, contributing to vascular damage. Indeed, internalization of PR3 into the endothelial cell leads to apoptosis [62–64]. Internalization of MPO into endothelial cells leads to release of ROS causing endothelial damage and detachment and promotes complement-mediated cytotoxicity resulting from ANCA interaction with endothelial cell-bound MPO [62, 65]. Airway endothelial cells internalize MPO but not PR3 [62]. Denudation of the endothelium exposes thrombogenic basement membranes and perivascular matrix to the circulation, initiating the coagulation cascade leading to activation of coagulation factors resulting in fibrin formation and the characteristic fibrinoid necrosis of ANCA vasculitis. These varying pathogenic interactions with the endothelium between the neutrophil granule proteins MPO and PR3 offer another hypothesis to account for the differences in disease phenotypes between MPO and PR3-ANCA vasculitis. Along with possible time-dependent degradation of immune complexes, the internalization of ANCA antigens by endothelial cells may also explain why ANCA vasculitis rarely shows intense immunoglobulin staining on biopsy.
Neutrophil extracellular trap release
NETs are a meshwork of granule proteins (including proteases such as elastase and MPO), and nuclear components (deoxyribonucleic acid [DNA], histone) that are released by neutrophils upon activation, providing another weapon in the neutrophil armamentarium against pathogens. They are released as a mesh that binds gram-positive and gram-negative bacteria; the meshwork effectively locks pathogens in place allowing proteases from granules within the NETs to degrade bacterial virulence factors and facilitate disposal by other leukocytes [66].
A potential role for NETs in the pathogenesis of ANCA vasculitis is suggested by the fact that NETs contain MPO and PR3 [30, 66]. Patients with ANCA vasculitis may have higher levels of circulating NET remnants during active disease compared to healthy controls, and activation of neutrophils by ANCA has been shown to initiate the release of NETs [30, 67]. NETs have been localized at sites of vascular injury in individuals with ANCA vasculitis [68, 69] and have been proposed as potential triggers for the immunogenic formation of ANCA by providing MPO to myeloid dendritic cells, a process that could be prevented by treatment with DNAse [70]. NETs may also be involved in the pathogenic process in ANCA vasculitis through activation of the complement cascade. Wang et al. showed that ANCA-induced NETs could activate the alternative complement pathway both locally, resulting in the deposition of C3b and C5b-9 on NETs, and systemically, resulting in higher levels of serum complement degradation products than if NETs were degraded by DNAse [71]. The role of the complement cascade in the pathogenesis of ANCA vasculitis is discussed below. Finally, NETs may have pro-coagulant properties that could account for the increased risk for thrombotic events in patients with ANCA vasculitis and for the fibrin deposition seen within fibrinoid necrosis, a classic histologic lesion. The meshwork of proteinaceous substances released from neutrophils may contain tissue factor and tissue-factor bearing microparticles, which could trigger the extrinsic pathway of the coagulation cascade [72]. Levels of tissue factor containing NETs and microparticles decrease in serum of patients with ANCA vasculitis once remission is achieved compared to active disease [73]. This may explain the thromboembolic risks in ANCA vasculitis being the greatest during active disease [74]. NETs may be a key mediator in the pathogenesis of ANCA vasculitis and potential target for future therapies; however, this requires further study.
Contribution of the complement system to pathogenesis of ANCA vasculitis
The complement cascade comprises a complex series of serum and cell-bound proteins that is an important part of the innate immune system critical for appropriate homeostatic and defense mechanisms. It also functions as a mediator of injury in many autoimmune and auto-inflammatory diseases. Complement can be activated via the classical pathway (immune complex mediated), lectin pathway (recognition of carbohydrate patterns on microbial and other surfaces), and the constitutively active alternative pathway. As part of the innate immune system, pathogens, microbial substances, and damaged or apoptotic cells are major activators of the complement cascade [75]. The most important pathway involved in the pathogenesis of ANCA vasculitis is the alternative pathway [76, 77].
Recent ANCA vasculitis research highlights the importance of complement as a driver of inflammation and tissue damage. Complement products and membrane attack complex are detected immunohistologically in necrotizing crescentic glomerulonephritis [76]. Wu et al. demonstrated that patients with both MPO-ANCA vasculitis and PR3-ANCA vasculitis have higher plasma levels of complement activation products C3a, C5a, and soluble C5b-9 than healthy controls [78]. In the same individuals, complement activation products tended to be lower in remission than during active disease, and patients achieving long-term remission off therapy had similar levels to healthy controls [78]. Xing et al. analyzed renal biopsies from 7 patients with anti-MPO necrotizing crescentic glomerulonephritis (NCGN) and observed soluble C5b-9, C3d, and the alternative pathway regulators factor B and factor P, but not in controls [76]. Furthermore, mannose-binding lectin and C4d were not detected in the biopsies of patients having anti-MPO ANCA vasculitis, suggesting a major role for the alternative pathway and less so for the classical and lectin pathways.
To further support this, Xiao et al. administered anti-MPO IgG to mice with knockout (KO) C5 required for all pathways of complement activation, C4 required for classical and lectin binding pathway complement activation, or factor B required for alternative pathway activation [79]. They found that C5 KO mice and C4 KO did not develop NCGN. They demonstrated that incubation of primed neutrophils with anti-MPO and anti-PR3 IgG led to significantly greater C3a generation than controls, suggesting that ANCA-induced neutrophil activation releases activators of the complement cascade [77]. Chen et al. demonstrated the importance of the alternative pathway in ANCA vasculitis by examining the role of complement factor H, an alternative pathway regulator that serves to prevent C3b-mediated amplification and facilitates the decay of C3-convertase [80]. They found that activation of neutrophils by ANCA and endothelial damage was prevented by factor H. They also found that factor H from patients with ANCA vasculitis was not as effective in inhibiting neutrophil activation and endothelial damage as factor H from healthy controls. Thus, aberrant factor H function may influence risk for developing ANCA vasculitis. Furthermore, endothelial damage due to ROS and proteolytic enzymes after neutrophil activation may lead to a loss of alternative pathway regulators normally found on the endothelial cell membrane such as decay-accelerating factor and membrane co-factor protein [81], predisposing to unchecked alternative pathway activation. These findings support the hypothesis that the alternative complement pathway plays prominently in causing vascular inflammation in ANCA vasculitis.
An important participant in the pathogenic role of complement in ANCA vasculitis is the complement anaphylatoxin C5a. ANCA-induced neutrophil activation generates C5a, which can bind to the C5a receptor (C5aR; CD88) on neutrophils, prime them, and create an activation and inflammatory loop [79, 82]. C5a has pro-inflammatory effects by increasing vascular permeability and expression of endothelial cell adhesion molecules, and as a chemoattractant recruiting more neutrophils [82]. Stimulation of neutrophils by C5a and ANCA results in respiratory burst and degranulation, activation of the coagulation cascade, generation of thrombin, and release of properdin and factor B, key activators of the alternative pathway [72, 76]. C5a also leads to the release of NETs, an important step in disease pathogenesis as detailed above, and C5a priming of neutrophils can release high mobility group box 1 (HMGB1), a nuclear protein found in chromatin, to the extracellular space. This may facilitate translocation of ANCA antigens, respiratory burst, degranulation, and NET formation from ANCA-activated neutrophils [83, 84]. Blockade of the C5aR prevents or reduces ANCA-induced glomerulonephritis in an experimental mouse model [24, 85]. A variant of this model with knocked out mouse C5aR and knocked in human C5aR demonstrated that an inhibitor of human C5aR reduced glomerulonephritis [86]. This led to clinical trials using avacopan that demonstrated efficacy in treating ANCA vasculitis [86, 87]. These studies and others highlight the central role of complement in the pathogenesis of ANCA vasculitis.
Tissue necrosis/damage in ANCA vasculitis — role of the monocyte
Inflammatory cell infiltration of the small blood vessel walls leading to vascular injury and tissue necrosis is the hallmark of ANCA vasculitis (Fig. 2). As discussed, primed neutrophils release MPO and PR3 at the cell surface and in the microenvironment of activated neutrophils that can bind to ANCA, which then initiates a cascade of events (cytoskeleton changes, vascular adhesion, ROS and proinflammatory cytokine release, complement activation, NET formation) that mediates acute inflammation and vascular damage. Factors that can prime neutrophils include infection-induced cytokines, complement proteins such as C5a, and proteins exposed on NETs among others [21, 88]. While neutrophils are integral to pathogenesis, other leukocytes such as monocytes also play an important role.
Fig. 2.
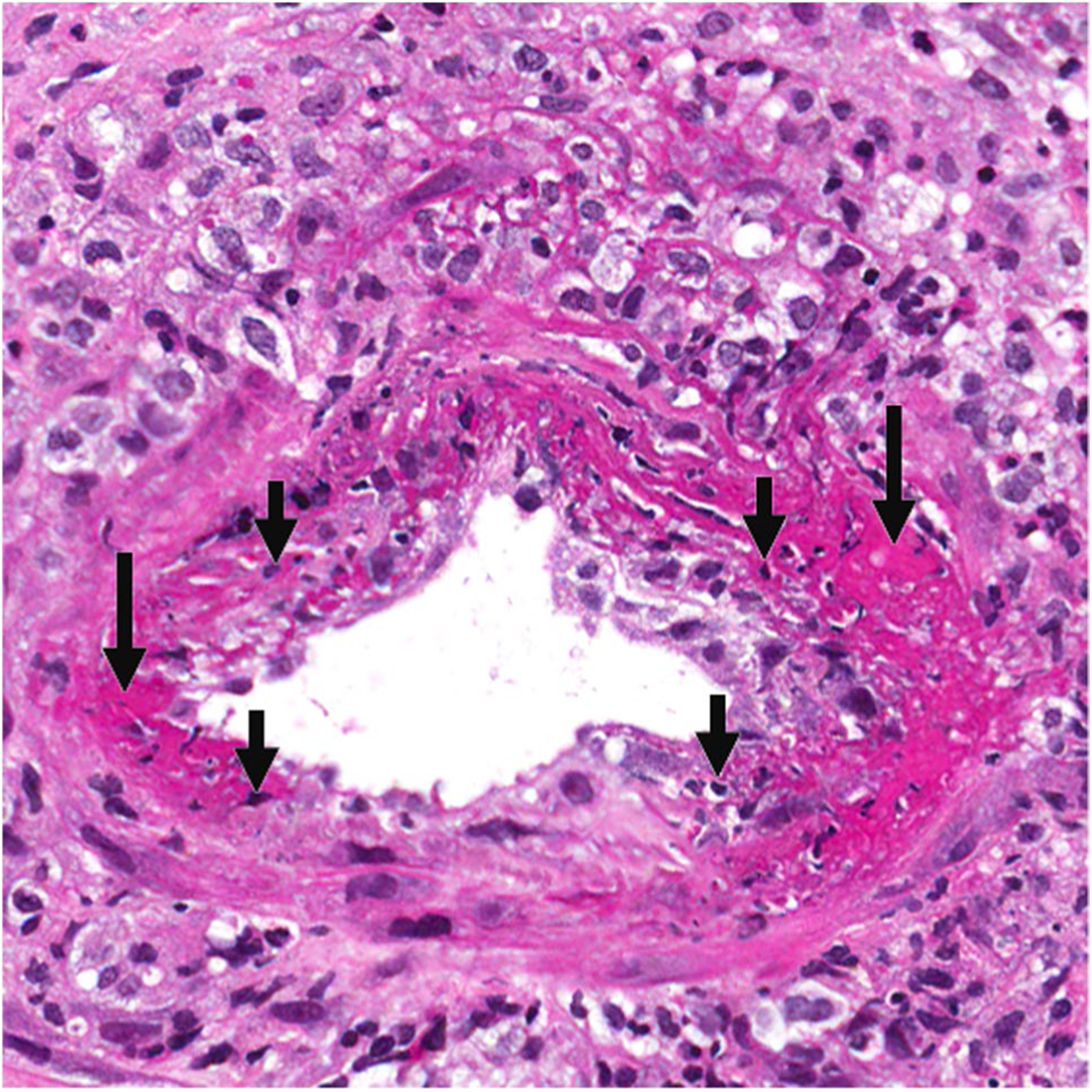
ANCA MPA arteritis. Arteritis in a kidney interlobular artery with segmental fibrinoid necrosis (long arrows), leukocytoclastic debris (short arrows), and transmural and perivascular mixed inflammatory infiltrate of neutrophils and mononuclear cells
ANCA vasculitis is a relapsing and remitting disease in which acute lesions during each flare are followed by resolution or tissue scarring [89] (Fig. 3). During the acute phase of a flare, neutrophils and monocytes are first recruited into tissues causing necrotizing vasculitis [90]. Within days, neutrophils undergo leukocytoclasis and disappear. If resolution of acute lesions does not resolve, neutrophils are replaced by mononuclear cells such as monocytes, macrophages, and T cells [89]. Most glomerular macrophages originate from peripheral blood monocyte precursors [91] that enter tissue by adhering to endothelial cells via vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) [92, 93]. In the glomeruli, monocytes differentiate into macrophages and express HLA class II antigens to present antigens to T cells and produce proinflammatory cytokines, such as TNF-α and IL-1 [91]. Increased intermediate (CD14 + CD16 +) monocytes were recently identified in the peripheral blood of patients with active ANCA vasculitis [94, 95]. These cells show an elevated expression of CD14, TLR 2/4, MHC II, and MPO, and PR3, suggesting increased activation and migration of these cells to inflamed tissue [94, 96]. CD68 + macrophages accumulate in the glomeruli of patients with ANCA vasculitis [97]. Macrophages amplify glomerular inflammation and, along with glomerular epithelial cells, form glomerular crescents in Bowmans space that are characteristic of severe GN in ANCA vasculitis [98–100]. Macrophages are heterogeneous and their functional phenotype is influenced by the cytokine milieu of their surrounding microenvironment [101]. M1-type macrophages release cytokines that damage tissue and M2-type macrophages release cytokines that promote the proliferation of cells for tissue repair [102]. Monocytes predominantly differentiate into M2-type macrophages that are involved in phagocytosis and cell-clearance but can lead to fibrosis [91]. In ANCA vasculitis, the phagocytic function of macrophages can be impaired by cytoplasmic proteases and apoptotic neutrophils [91], causing a cycle of macrophage inflammatory mediator release and subsequent neutrophil, T cell, and B cell activation [91].
Fig. 3.
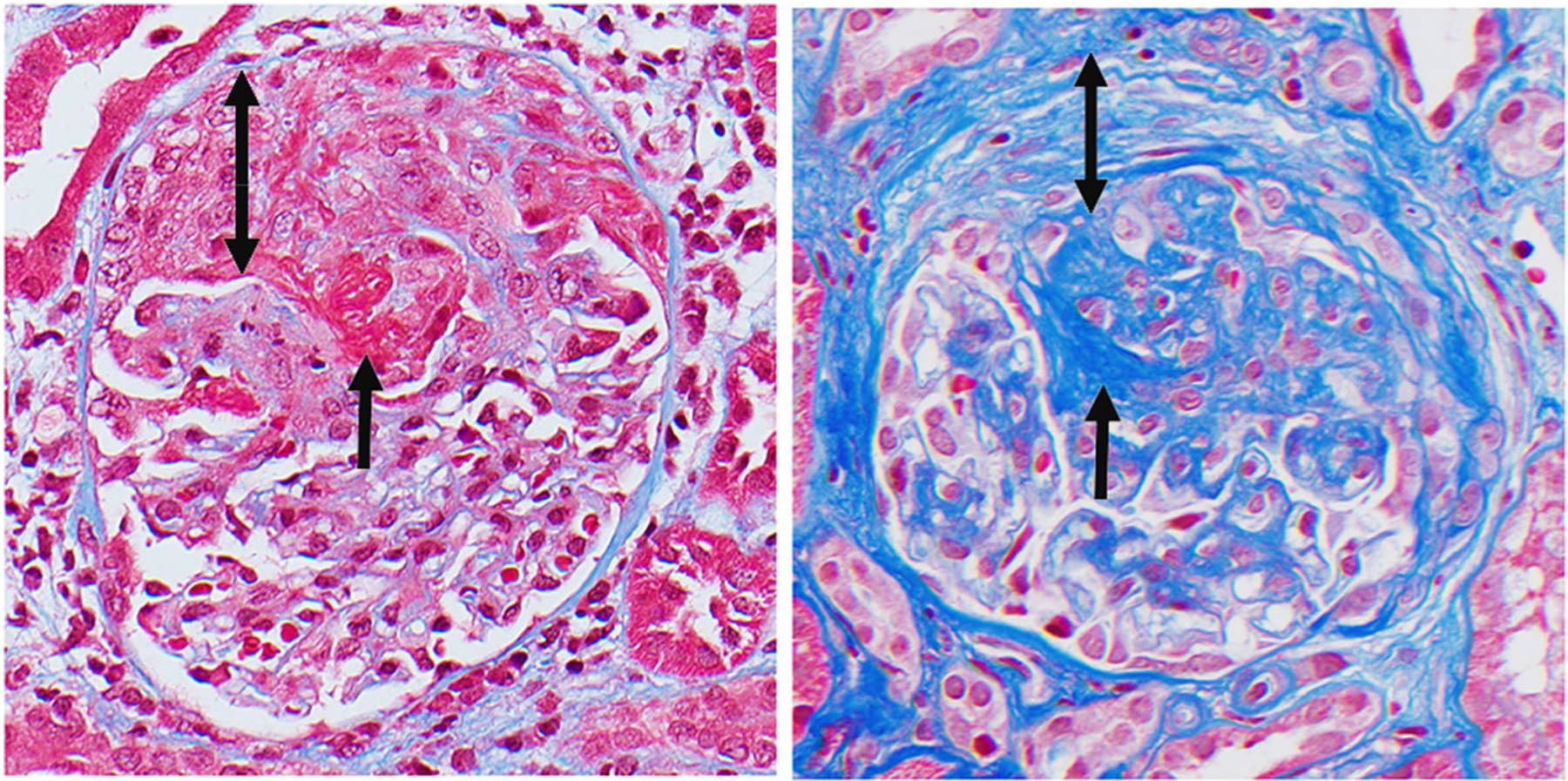
ANCA glomerulonephritis. Left panel: active glomerulonephritis with segmental fibrinoid necrosis (short arrow) and cellular crescent (double arrow). Right panel: sclerotic glomerulonephritis with segmental sclerosis (short error) and fibrotic crescent (double arrow)
ANCA vasculitis is a systemic disease that can affect multiple organs, especially the upper respiratory tract, lungs, and kidneys [103, 104]. Granulomatosis with polyangiitis (GPA) predominantly associated with PR3-ANCA [103] and eosinophilic granulomatosis with polyangiitis (EGPA) predominantly associated with MPO-ANCA, are characterized by necrotizing granulomatous inflammation that most often affects the respiratory tract. These granulomas, consisting of an organized collection of predominantly monocytes and macrophages forming a nodule, often develop along the respiratory tract of patients with ANCA vasculitis and are a key characteristic of GPA and EGPA [103, 104]. It is hypothesized that granulomatosis lesions form when a respiratory infection or other inflammatory condition primes neutrophils for activation by ANCA vasculitis in the airway mucosa [103]. Activated neutrophils lead to acute lesions resembling micro abscesses, and eventually mononuclear cells replace the neutrophils. Epithelioid macrophages then encapsulate or wall off the necrotic debris, forming the granuloma (Fig. 4). Over time, the necrotic debris may be replaced by collagen and lymphoid cells such as dendritic cells, memory T cells, B cells, and plasma cells. Depending on the timing of biopsy, a granuloma may reveal evidence of acute inflammatory damage or alternatively a healing phase to the process [103, 105]. However, chronic granulomas should not be considered clinically quiescent lesions as they may have immunogenic properties. Within granulomatous lesions, T cells may participate in the generation of ANCA and sustain autoimmune responses via interactions with dendritic cells and B cells [106]. This cellular activity within granulomas may provide a constant source of autoantibody formation and account for the greater relapse risks in patients with GPA [107]. In contrast to GPA and EGPA that predominantly affect the respiratory tract in addition to the kidneys, microscopic polyangiitis (MPA) does not have granulomatosis, more often has renal involvement, and its predominant ANCA type is MPO-ANCA [108, 109]. Approximately 80–100% of patients with MPA have renal manifestations [110, 111] that can range from asymptomatic hematuria to necrotizing crescentic GN causing end-stage kidney disease [110, 112]. Tissue necrosis and damage involve complex interactions among various immune cell types. Figure 5 summarizes the steps involved in the pathogenesis of vascular damage discussed so far. Given the substantial role that monocytes and macrophages play in the formation of necrotizing lesions and tissue injury resolution in GPA, EGPA, and MPA, they may present a promising target for therapy in ANCA vasculitis.
Fig. 4.
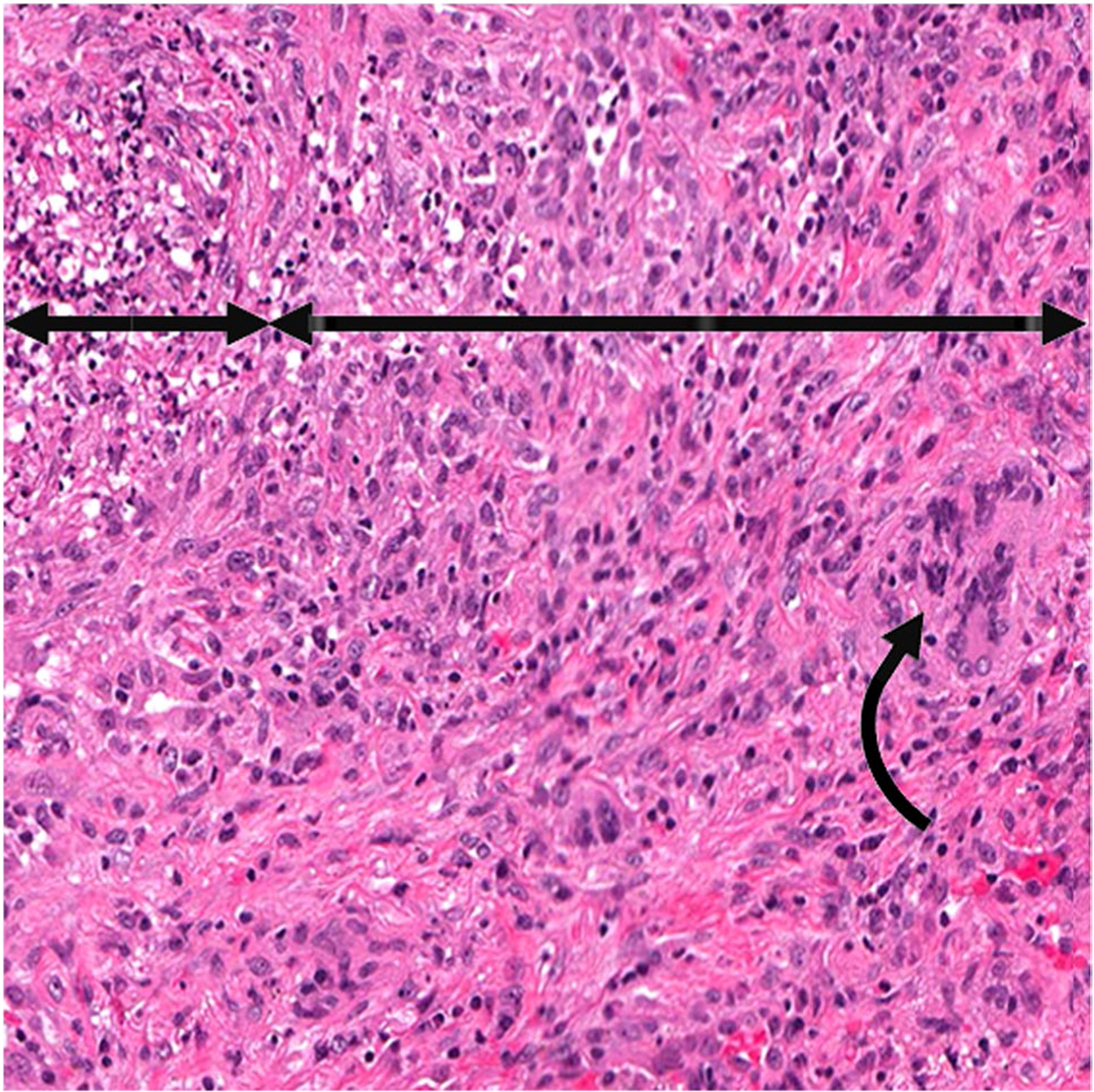
ANCA GPA granuloma. Pulmonary granulomatous inflammation with a central necrotic core of leukocytoclastic debris (short double arrow) surrounded by granulomatous inflammation with a predominance of monocytes and macrophages (long double arrow), and a few giant cells (curved arrow)
Fig. 5.
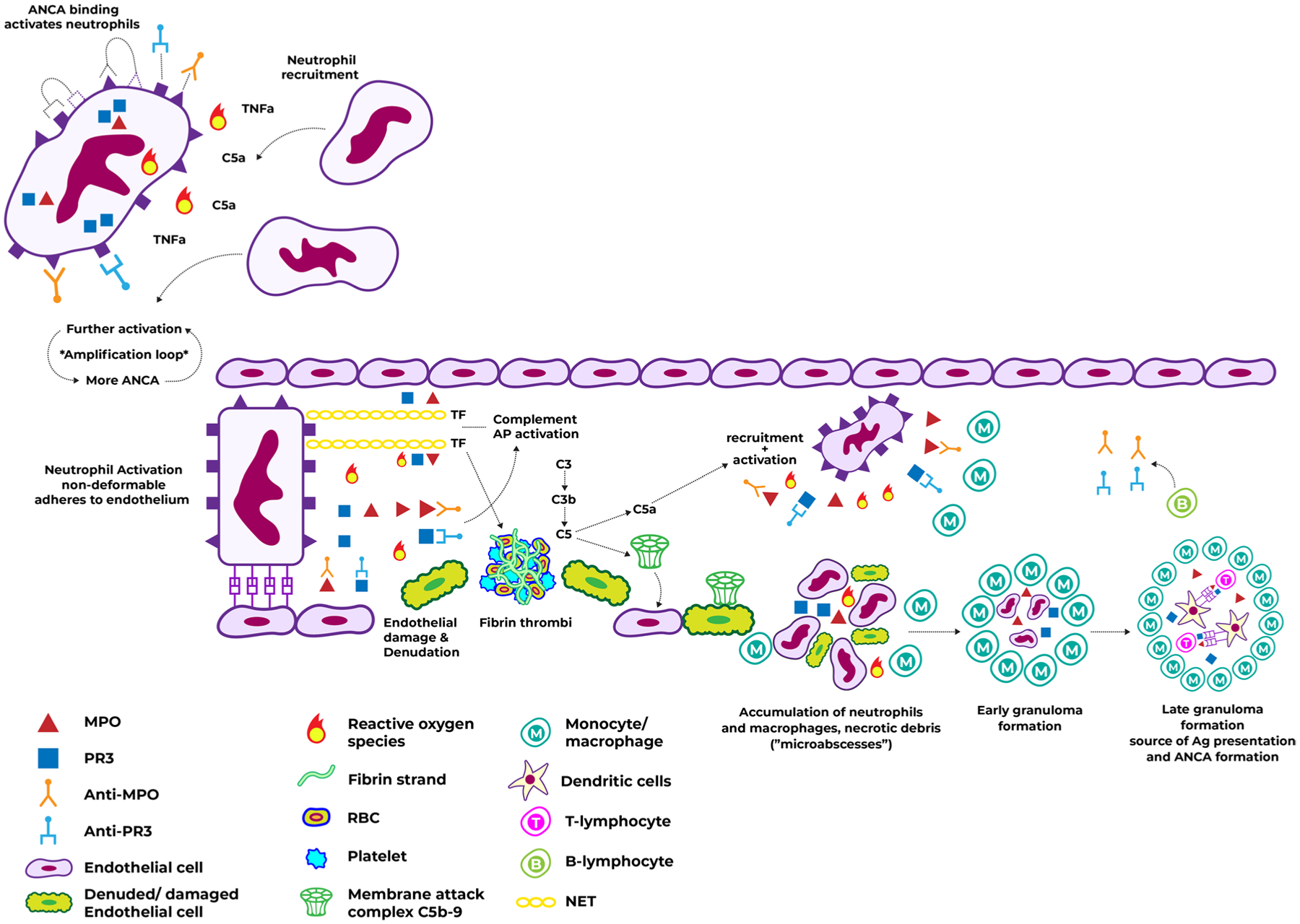
Pathogenesis of vascular damage in ANCA vasculitis. Caption: binding of ANCA to their antigens on the neutrophil surface leads to activation, degranulation, release of MPO/PR3 and neutrophil recruitment. This is enhanced in the context of inflammatory stimuli such as TNF-α and C5, and provides more ANCA antigens leading to a neutrophil activation amplification loop. Activation causes morphologic and surface protein expression changes which promote neutrophil adherence to the endothelium. The ensuing release of ROS and MPO/PR3 causes endothelial damage and denudation. Activation also leads to alternative complement pathway activation with subsequent MAC formation on the endothelium, and to NET release which may contain TF, thus promoting fibrin thrombi formation and massive recruitment with further activation of neutrophils and monocytes. The accumulation of inflammatory cells and necrotic debris lead to micro abscesses which may then become organized into early granulomas and then late granulomas. The late granulomas may contain ANCA antigens, and serve as a source of perpetual ANCA formation. Abbreviations: ANCA, anti-neutrophil cytoplasmic antibodies; MAC, membrane attack complex; MPO, myeloperoxidase; NET, neutrophil extracellular trap; PR3, proteinase 3; TF, tissue factor; TNF-α, tumor necrosis factor α
Biological and environmental factors influencing disease activity in ANCA vasculitis
ANCA vasculitis is a multifactorial disease in which numerous factors, such as genetics, infections, certain drugs, and environmental exposures, may contribute to disease initiation and relapse.
Genetic associations with ANCA vasculitis
GWAS have implicated genetic makeup in the onset and progression of ANCA vasculitis. Polymorphisms having strong associations with ANCA vasculitis have been found in HLA genes, as discussed in prior sections. In addition to polymorphisms in HLA genes, polymorphisms in the protein tyrosine phosphatase, non-receptor type 22 (PTPN22) gene have been associated with both MPA and GPA [113, 114]. PTPN22 encodes lymphoid tyrosine phosphatase and variations in PTPN22 can lead to dysregulated B cell receptor and T cell receptor signaling, causing autoimmunity. Additionally, cytotoxic T-lymphocyte antigen 4 (CTLA4) polymorphisms have been associated with GPA [115]. CTLA4 is expressed on CD4 + T cells and acts as a regulatory molecule of T cell activation; thus, CTLA4 polymorphisms may contribute to the dysregulation of regulatory T cell responses and contribute to sustaining autoimmunity. Furthermore, PRTN3, is of interest because it codes for PR3. Eight SNPs have been identified in the PRTN3 promotor region and exons [104, 116]. Polymorphisms in PRTN3 have also been associated with more severe disease and more frequent relapse in patients with PR3-ANCA vasculitis [117]. The proteolytic activity of PR3 is naturally inhibited by α1-antitrypsin, which is encoded by the SERPINA1 gene [118]. Polymorphisms of SERPINA1 may result in decreased function of α1-antitrypsin and increased damage to inflamed tissues in ANCA vasculitis [116]. Overall, additional studies to identify genetic associations in ANCA vasculitis will provide insight to new pathogenic mechanisms and potential therapeutic targets.
Infections
In addition to the role of genetics in ANCA vasculitis, infections can contribute to the onset of ANCA vasculitis and facilitate the autoimmune response. Pathogens are potential antigen sources that induce pro-inflammatory signals (such as stimulation of pathogen-associated molecular pattern receptors) and trigger innate and adaptive immune cell activation. The involvement of these factors may initiate a break in immune tolerance and the development of chronic or recurrent autoimmunity. For example, S. aureus has been associated with increased risk of relapse in patients with GPA. Approximately two thirds of patients with GPA are nasal carriers of S. aureus and have an increased risk for disease relapse [119]. S. aureus produces superantigens that are powerful, non-specific activators of T- and B-cell proliferation and can induce significant cytokine release [120, 121] and expansion of Th17 cells. These IL-17 producing CD4 + T cells are central in the development of autoimmunity [122, 123]. Th17 cells recruit inflammatory cells and generate a pro-inflammatory cytokine milieu, making them resistant to control by T-regulatory cells and presenting a break in tolerance [124]. Furthermore, bacterial antigens may initiate granuloma formation and contribute to autoantibody formation. The repertoire of immunoglobulin heavy chain genes found within granulomas of patients with GPA is suggestive of an antigen-driven process and bares similarities to heavy chain genes from B-cell with affinity to S. aureus superantigen [125]. A study examining toll-like receptor gene mutations in patients with ANCA vasculitis found mutations in TLR9 to be more closely associated with PR3-ANCA vasculitis than MPO-ANCA vasculitis [126]. Therefore, bacterial infections could play a major role in granuloma formation, creating a source of autoantibody formation, particularly PR3-ANCA. This could account for the greater relapse risk seen in patients with GPA, PR3-ANCA vasculitis, and respiratory tract involvement [107, 127]. Molecular mimicry is another theory through which S. aureus may contribute to autoimmunity where breaks in immune tolerance occur due to microbial antigens sharing sequence similarities to host proteins or autoantigens. Ooi et al. demonstrated that the peptide 6PGD391–410, part of a plasmid-encoded 6-phosphogluconate dehydrogenase found in some S. aureus strains, induced anti-MPO T- and B-cell autoimmunity in mice [128]. This implicates bacterial plasmid-derived antigens in the loss of tolerance to self-antigens and its potential role in the induction of autoimmunity in ANCA vasculitis.
Drugs associated with ANCA vasculitis disease onset and relapse
Drugs associated with ANCA vasculitis disease induction and relapse include hydralazine, a common vasodilator used to treat hypertension. One study correlated hydralazine administration and the onset of ANCA vasculitis with pulmonary-renal syndrome in a patient [129]. Hydralazine may induce vasculitis because it decreases DNA methyltransferase expression and inhibits extracellular signal-regulated kinase, thus inducing autoimmunity by dysregulating the suppression of MPO and PR3 antigen expression [130]. Hydralazine is also hypothesized to be metabolized by MPO to form reactive intermediates that lead to the formation of anti-MPO antibodies [131]. Other drugs associated with ANCA vasculitis include penicillamine, levamisole-adulterated cocaine, propylthiouracil, minocycline, allopurinol, and diphenylhydantoin [103, 132].
Environmental exposures
Environmental triggers have been associated with the onset of ANCA vasculitis, including silica exposure [133, 134]. Silica can activate the NLRP3 inflammasome complex and generate IL-1β and other proinflammatory mediators that propagate autoimmune responses in ANCA vasculitis [135]. In addition, occupational exposures to inhaled pesticides, dust, fumes, and hydrocarbons have been associated with ANCA vasculitis [136, 137].
Overall, an interplay of multiple factors such as genetics, infectious triggers, certain drugs, and environmental exposures can contribute to the onset, progression, and relapse of ANCA vasculitis. Polymorphisms in innate and adaptive immune cell genes contribute to the disruption of immune tolerance. A better understanding of signaling pathways of these genes may lead to targeting pathways in ANCA vasculitis. In addition, studies investigating host genetic and environmental interactions may lead to a better understanding of interactions in ANCA vasculitis and may elucidate avenues for disease prevention and treatment.
Conclusion and future directions
The initial treatment of ANCA vasculitis focused on decreasing inflammation and suppressing the immune system in a non-specific manner. For decades, standard treatment has been glucocorticosteroids and cyclophosphamide. Over the last decade, a better understanding of the pathogenesis and of the main drivers of tissue damage in ANCA vasculitis led to randomized controlled trials examining more targeted therapies. Table 1 summarizes major clinical trials in ANCA vasculitis, the main mechanisms of action of the agents, and conclusions. Our understanding of the pathogenesis of ANCA vasculitis continues to improve, with novel pathways and cell types implicated in the pathogenesis. We are starting to gain a better understanding of the differences which underlie MPO- and PR3-ANCA vasculitis and how, even though they share a similar nomenclature, they likely represent distinct disease processes (Table 2). This will lead to the discovery of additional targeted therapies in ANCA vasculitis.
Table 1.
Trials of current therapies for ANCA vasculitis
| Major trials and indication | Patients | Study groups | Findings |
|---|---|---|---|
| Avacopan | |||
| C5 receptor blocker | |||
| ADVOCATE [87] Induction |
New or relapsed GPA or MPA with ANCA positivity Total N = 331 |
Avacopan 30 mg bid vs Prednisone taper (on background CYC/RTX induction) | Avacopan non-inferior for remission, more sustained remissions at 52 weeks, less steroid-related adverse events |
| Azathioprine | |||
| Nucleotide analogue anti-metabolite, non-specific cytotoxicity affecting all leukocytes | |||
| CYCAZAREM [138] Maintenance |
Remission of GPA, MPA or renal-limited vasculitis with oral CYC Total N = 144 |
AZA 2 mg/kg vs PO CYC 1.5 mg/kg up to month 12 | No difference in rates of relapse nor of serious adverse events |
| Belimumab | |||
| Monoclonal antibody blocking B-lymphocyte stimulator protein leading to decreased B-lymphocyte survival | |||
| BREVAS [139] Maintenance |
Remission of after CYC or RTX Total N = 105 |
Belimumab vs Placebo add-on to AZA-prednisone maintenance | Similar rates of relapses, treatment failure, and worsening BVAS |
| Cyclophosphamide | |||
| Alkylating agent; non-specific cytotoxicity affecting all leukocytes | |||
| CYCLOPS [140] Induction |
New diagnosis GPA or MPA with renal involvement and positive ANCA serology (or confirmatory biopsy) Total N = 149 |
CYC IV vs PO | IV non-inferior to oral for remission, less cumulative dose with IV, but more relapses |
| CORTAGE [141] Induction |
> 65 years old with new diagnosis GPA, MPA, EGPA, or PAN (not HBV-related) Total N = 104 |
IV CYC 500 mg×6 every 2–3 weeks (with 9 months GCS) vs IV CYC 500 mg/m2 every 2–3 weeks until remission (with 26 months GCS) | Fixed dose IV CYC had less adverse events, similar remission rates but possibly more relapses |
| Etanercept | |||
| Recombinant TNF receptor linked to human IgG which will bind TNF blocking its effect | |||
| WGET [142] Induction |
New or relapsing GPA | Etanercept twice per week vs Placebo (add-on to CYC or MTX induction) | Similar rates of remission and relapses |
| IVIg | |||
| Binding to and neutralization of ANCA | |||
| IVIg study [143] Induction |
Treated ANCA vasculitis with persisting activity, with intention to escalate therapy Total N = 34 |
1 course IVIg (2 mg/kg) vs Placebo | Significantly more treatment response with IVIg |
| Leflunomide | |||
| Immunomodulatory effect via inhibition of de novo pyrimidine ribonucleotides preventing proliferation of activated lymphocytes [144] | |||
| Methotrexate vs leflunomide [145] Maintenance |
Remission of GPA after oral CYC Total N = 54 |
Leflunomide vs weekly PO MTX for 2 years | Less relapses with leflunomide (study ended early) but more adverse events |
| Methotrexate | |||
| Folate antimetabolite, non-specific cytotoxicity affecting all leukocytes | |||
| NORAM [146]) | New diagnosis GPA or MPA Total N = 100 |
Weekly PO MTX vs daily PO CYC | MTX non-inferior for remissions overall, but less effective in patients with severe disease. More relapses with MTX |
| WEGENT [147] Maintenance |
Remission of GPA or MPA after IV CYC Total N = 126 |
Weekly PO MTX vs AZA up to month 12 | No difference in rates of relapses nor of serious adverse events |
| Mycophenolate | |||
| Guanosine nucleotide depleting anti-metabolite preferentially affecting lymphocytes (B and T) | |||
| MYCYC [148] Induction |
New diagnosis GPA or MPA with positive ANCA or biopsy-proven disease Total N = 140 |
MMF vs IV CYC 15 mg/kg every 2–3 weeks | MMF non-inferior to CYC but more relapses with MMF (for PR3-ANCA) |
| IMPROVE [149] Maintenance |
Remission of GPA or MPA after PO or IV CYC Total N = 156 |
MMF vs AZA up to month 12 | AZA more effective in preventing relapse; similar adverse events |
| Plasmapheresis | |||
| Removal of ANCA | |||
| MEPEX [150] Induction |
New diagnosis ANCA vasculitis with renal biopsy and creatinine > 5.8 mg/dL Total N = 137 |
PLEX × 7 vs Methylprednisolone 1 g × 3 | Less ESKD at 3- and 12-months with PLEX |
| PEXIVAS [151] Induction |
New diagnosis or relapsed severe ANCA vasculitis (eGFR < 50 ml/min or pulmonary hemorrhage) Total N = 704 |
PLEX × 7 sessions vs no PLEX Low-dose prednisone taper vs standard-dose prednisone taper |
No difference in rates of remission, ESKD or death with PLEX vs no PLEX, and with low-dose vs standard-dose prednisone |
| Rituximab | |||
| CD20 monoclonal antibody causing targeted B-lymphocyte depletion | |||
| MAINRITS AN 1 [152] Maintenance |
Remission of GPA, MPA or renal-limited vasculitis after IV CYC Total N = 115 |
RTX day 0, day 14, 6 month, 12 month, 18 month vs AZA × 24 months | Less major relapses with RTX |
| MAINRITSAN2 [153] Maintenance |
Remission of GPA or MPA after CYC, RTX, or MTX Total N = 162 |
RTX fixed-dose (day 0, day 14, 6 months, 12 months, 18 months) vs Bcell tailored (day 0 then upon repopulation) | Similar relapse rates but less RTX requirement in Bcell-tailored |
| MAINRITSAN3 [154] Maintenance |
Remission of GPA or MPA at end of MAINRITSAN2 Total N = 97 |
RTX continuation every 6 months × 4 vs placebo | Less relapses with RTX continuation |
| RITUXVAS [155] Induction |
New diagnosis ANCA vasculitis with positive ANCA and renal involvement N = 44 |
RTX 375 mg/m2 IV weekly × 4 + IV CYC 15 mg/kg × 2 vs IV CYC 15 mg/kg every 2–3 weeks | Similar remission rates and adverse events with RTX |
| RAVE [156] Induction |
New or relapsing GPA or MPA with positive ANCA Total N = 197 |
RTX 375 mg/m2 weekly × 4 vs PO CYC | RTX non-inferior to CYC; RTX more effective in relapsing disease; similar adverse events |
| Sulfamethoxazole-trimethoprime | |||
| Antibiotic which inhibits bacterial folic acid synthesis with coverage against upper respiratory tract bacteria | |||
| Dutch co-trimoxazole study (157)) Maintenance |
Remission of GPA after CYC Total N = 81 |
SMX-TMP vs placebo for 2 years | Less relapses and less infections with SMX-TMP; more side effects |
Abbreviations: ANCA, anti-neutrophil cytoplasmic antibody; AZA, azathioprine; BVAS, Birmingham vasculitis activity score; CYC, cyclophosphamide; ESKD, end-stage kidney disease; GCS, glucocorticosteroids; GPA, granulomatosis with polyangiitis; IVIg, intravenous immunoglobulin; MMF, mycophenolate mofetil; MPA, microscopic polyangiitis; MTX, methotrexate; PAN, polyarteritis nodosa; PLEX, plasma exchange; RTX, rituximab; SMX-TMP, sulfamethoxazole-trimethoprime
Table 2.
Hypotheses of potential mechanisms for clinical differences in the different forms of ANCA vasculitis
| Process | Pathophysiologic difference between MPO and PR3 ANCA | Clinical correlate |
|---|---|---|
| Complementary PR3 | Demonstration of antibodies against a peptide derived from anti-sense (complementary) PR3 in PR3-ANCA patients | More frequent relapses in PR3-ANCA, possibly due to molecular mimicry from exposure to microbial antigens |
| Activation of neutrophils by ANCA | PR3-ANCA antibodies show a greater potency for neutrophil activation, degranulation, and ROS release | PR3-ANCA vasculitis more often has active lesions on kidney biopsy such as cellular crescents and fibrinoid necrosis, compared to MPO-ANCA vasculitis |
| Endothelial cell internalization of ANCA antigens | Internalization of MPO leads to ROS formation which may then predispose to fibrogenesis, whereas internalization of PR3 leads to endothelial cell apoptosis Small airway epithelial cells internalize MPO but not PR3 |
MPO-ANCA more commonly seen with interstitial lung disease associated with ANCA vasculitis |
| Cytotoxic T-lymphocyte antigen 4 | Polymorphisms in CTLA4, which regulates T-cell activation, have been associated with GPA, which could lead to aberrant regulatory responses and sustained auto-immunity | More frequent relapses seen with GPA compared to MPA |
| Bacterial antigens | Possibility of infection driving pathogenesis in PR3-ANCA and GPA since mutations in TLR9 are more often associated with PR3- than with MPO-ANCA vasculitis. Also, Ig heavy chains in granulomas of patients with GPA share affinities to heavy chains against S. aureus superantigens | Granulomatous disease in PR3-ANCA More frequent relapses seen in GPA and PR3-ANCA |
| Drug-induced vasculitis | Hydralazine may be metabolized by MPO, forming intermediary substances triggering the formation of anti-MPO antibodies | High anti-MPO antibodies commonly seen in hydralazine-induced vasculitis |
Abbreviations: ANCA, anti-neutrophil cytoplasmic antibody; CTLA4, cytotoxic T-lymphocyte antigen 4; GPA, granulomatosis with polyangiitis; Ig, immunoglobulin; MPA, microscopic polyangiitis; MPO, myeloperoxidase; PR3, proteinase 3; TLR, toll-like receptor
Increased understanding of the immunogenicity of MPO and PR3 combined with the specific targeting of the adaptive immune system has provided the foundation for future antigen-specific immunotherapies. Utilization of the MPO peptide to induce tolerance in a mouse model of MPO-induced GN and ameliorate established disease is incredibly informative to begin the generation of antigen-specific therapies [158]. Generation of chimeric autoantibody receptor (CAAR) T cells analogous to those being tested in pemphigus vulgaris may now be possible [159]. As we learn more about autoantigen-targeted therapies, the field is poised to produce therapies tailored to directly address the autoimmune response thereby decreasing overall immunosuppression and risks for infection.
The search continues for the drug that provides optimal treatment of ANCA vasculitis in a targeted manner with minimal adverse effects. As discussed, ANCA vasculitis pathogenesis is a multi-step process with multiple potential triggers, implicating multiple different cell types. Combination therapies will likely remain essential for induction of remission of active ANCA vasculitis. Current therapies offer high rates of remission and the focus for induction therapy is to find less toxic means of inducing remission. However, the main challenge in management of ANCA vasculitis is our limited understanding and ability to prevent the high rate of long-term relapses despite newer therapies. As our understanding of the pathogenesis continues to improve, there is hope in eventually preventing autoantigen formation, tolerance induction, and maintenance of remission with disease-specific treatments.
Acknowledgements
Special thanks to Jean Brown who provided word editing and inserted the reference citations, and to Joshua Terrell who helped create the illustrative Figures 1 and 5.
Footnotes
Conflict of interest The authors declare no competing interests.
References
- 1.Little MA, Smyth CL, Yadav R, Ambrose L, Cook HT, Nourshargh S, Pusey CD (2005) Antineutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood 106:2050–2058 [DOI] [PubMed] [Google Scholar]
- 2.Xiao H, Heeringa P, Liu Z, Hu P, Zhao M, Aratani Y, Falk RJ (2002) Induction of necrotizing and crescentic glomerulonephritis (NCGN) and small-vessel vasculitis (SVV) by adoptive transfer of anti-myeloperoxidase (anti-MPO) lymphocytes into recombinase activating gene-2 deficient (RAG-2 −/−) mice. Cleve Clin J Med 69:13 [Google Scholar]
- 3.Schlieben DJ, Korbet SM, Kimura RE, Schwartz MM, Lewis EJ (2005) Pulmonary-renal syndrome in a newborn with placental transmission of ANCAs. Am J Kidney Dis 45:758–761 [DOI] [PubMed] [Google Scholar]
- 4.Cui Z, Zhao MH, Segelmark M, Hellmark T (2010) Natural autoantibodies to myeloperoxidase, proteinase 3, and the glomerular basement membrane are present in normal individuals. Kidney Int 78:590–597 [DOI] [PubMed] [Google Scholar]
- 5.Roth AJ, Ooi JD, Hess JJ, van Timmeren MM, Berg EA, Poulton CE, McGregor J, Burkart M, Hogan SL, Hu Y, Winnik W, Nachman PH, Stegeman CA, Niles J, Heeringa P, Kitching AR, Holdsworth S, Jennette JC, Preston GA, Falk RJ (2013) Epitope specificity determines pathogenicity and detectability in ANCA-associated vasculitis. J Clin Invest 123:1773–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falk RJ, Jennette JC (1988) Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med 318:1651–7 [DOI] [PubMed] [Google Scholar]
- 7.Goldschmeding R, van der Schoot CE, ten Bokkel HD, Hack CE, van den Ende ME, Kallenberg CG, dem Borne AE (1989) Wegener’s granulomatosis autoantibodies identify a novel diisopropylfluorophosphate-binding protein in the lysosomes of normal human neutrophils. J Clin Invest 84:1577–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aratani Y, Koyama H, Nyui S, Suzuki K, Kura F, Maeda N (1999) Severe impairment in early host defense against Candida albicans in mice deficient in myeloperoxidase. Infect Immun 67:1828–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kessenbrock K, Fröhlich L, Sixt M, Lämmermann T, Pfister H, Bateman A, Belaaouaj A, Ring J, Ollert M, Fässler R, Jenne DE (2008) Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest 118:2438–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joosten LA, Netea MG, Fantuzzi G, Koenders MI, Helsen MM, Sparrer H, Pham CT, van der Meer JW, Dinarello CA, van den Berg WB (2009) Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum 60:3651–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, Nester CM, Pendergraft WF III, Magnuson TR, Jennette JC, Falk RJ (2010) Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest 120:3209–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Rossum AP, Rarok AA, Huitema MG, Fassina G, Limburg PC, Kallenberg CG (2004) Constitutive membrane expression of proteinase 3 (PR3) and neutrophil activation by anti-PR3 antibodies. J Leukoc Biol 76:1162–1170 [DOI] [PubMed] [Google Scholar]
- 13.Halbwachs-Mecarelli L, Bessou G, Lesavre P, Lopez S, Witko-Sarsat V (1995) Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett 374:29–33 [DOI] [PubMed] [Google Scholar]
- 14.Rarok AA, Stegeman CA, Limburg PC, Kallenberg CG (2002) Neutrophil membrane expression of proteinase 3 (PR3) is related to relapse in PR3-ANCA-associated vasculitis. J Am Soc Nephrol 13:2232–2238 [DOI] [PubMed] [Google Scholar]
- 15.Schreiber A, Busjahn A, Luft FC, Kettritz R (2003) Membrane expression of proteinase 3 is genetically determined. J Am Soc Nephrol 14:68–75 [DOI] [PubMed] [Google Scholar]
- 16.Yang JJ, Tuttle RH, Hogan SL, Taylor JG, Phillips BD, Falk RJ, Jennette JC (2000) Target antigens for anti-neutrophil cytoplasmic autoantibodies (ANCA) are on the surface of primed and apoptotic but not unstimulated neutrophils. Clin Exp Immunol 121:165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witko-Sarsat V, Lesavre P, Lopez S, Bessou G, Hieblot C, Prum B, Noel LH, Guillevin L, Ravaud P, Sermet-Gaudelus I, Timsit J, Grunfeld JP, Halbwachs-Mecarelli L (1999) A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J Am Soc Nephrol 10:1224–1233 [DOI] [PubMed] [Google Scholar]
- 18.Jones BE, Yang J, Muthigi A, Hogan SL, Hu Y, Starmer J, Henderson CD, Poulton CJ, Brant EJ, Pendergraft WF 3rd, Jennette JC, Falk RJ, Ciavatta DJ (2016) Gene-specific DNA methylation changes predict remission in patients with ANCA-associated vasculitis. J Am Soc Nephrol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miralda I, Uriarte SM, McLeish KR (2017) Multiple phenotypic changes define neutrophil priming. Front Cell Infect Microbiol 7:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyabayashi M, Yasui K (1996) Regulation of neutrophil O2- production by neutrophil-endothelial cell interaction via CD11b: its modulation by tumor necrosis factor-alpha (TNF-alpha) and lipopolysaccharide (LPS). Int J Hematol 65:49–59 [DOI] [PubMed] [Google Scholar]
- 21.Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL (1994) Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol 95:244–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falk RJ, Terrell RS, Charles LA, Jennette JC (1990) Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA 87:4115–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Z, Richard C, Menard HA (2000) De novo synthesis of proteinase 3 by cytokine primed circulating human polymorphonuclear neutrophils and mononuclear cells. J Rheumatol 27:2406–2411 [PubMed] [Google Scholar]
- 24.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R (2009) C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol 20:289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martins A, Ximenes V, Fonseca L (2013) Serum myeloperoxidase level is increased in heavy smokers. Open J Clin Diagnostics 5–8 [Google Scholar]
- 26.Watts RA, Mahr A, Mohammad AJ, Gatenby P, Basu N, Flores-Suarez LF (2015) Classification, epidemiology and clinical subgrouping of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Nephrol Dial Transplant 30(Suppl 1):i14–22 [DOI] [PubMed] [Google Scholar]
- 27.Pendergraft WF, Preston GA, Shah RR, Tropsha A, Carter CW, Jennette JC, Falk RJ (2004) Autoimmunity is triggered by cPR-3(105–201), a protein complementary to human autoantigen proteinase-3. Nat Med 10:72–79 [DOI] [PubMed] [Google Scholar]
- 28.Gilligan HM, Bredy B, Brady HR, Hebert MJ, Slayter HS, Xu Y, Rauch J, Shia MA, Koh JS, Levine JS (1996) Antineutrophil cytoplasmic autoantibodies interact with primary granule constituents on the surface of apoptotic neutrophils in the absence of neutrophil priming. J Exp Med 184:2231–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pieterse E, van der Vlag J (2014) Breaking immunological tolerance in systemic lupus erythematosus. Front Immunol 5:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, Grone HJ, Brinkmann V, Jenne DE (2009) Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 15:623–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, Zal T, Mellman I, Schröder JM, Liu YJ, Gilliet M (2007) Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449:564–569 [DOI] [PubMed] [Google Scholar]
- 32.(2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, Baslund B, Brenchley P, Bruchfeld A, Chaudhry AN, Cohen Tervaert JW, Deloukas P, Feighery C, Gross WL, Guillevin L, Gunnarsson I, Harper L, Hruskova Z, Little MA, Martorana D, Neumann T, Ohlsson S, Padmanabhan S, Pusey CD, Salama AD, Sanders JS, Savage CO, Segelmark M, Stegeman CA, Tesar V, Vaglio A, Wieczorek S, Wilde B, Zwerina J, Rees AJ, Clayton DG, Smith KG (2012) Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med 367:214–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merkel PA, Xie G, Monach PA, Ji X, Ciavatta DJ, Byun J, Pinder BD, Zhao A, Zhang J, Tadesse Y, Qian D, Weirauch M, Nair R, Tsoi A, Pagnoux C, Carette S, Chung S, Cuthbertson D, Davis JC Jr, Dellaripa PF, Forbess L, Gewurz-Singer O, Hoffman GS, Khalidi N, Koening C, Langford CA, Mahr AD, McAlear C, Moreland L, Seo EP, Specks U, Spiera RF, Sreih A, St Clair EW, Stone JH, Ytterberg SR, Elder JT, Qu J, Ochi T, Hirano N, Edberg JC, Falk RJ, Amos CI, Siminovitch KA (2017) Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Rheumatol 69:1054–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Free ME, Stember KG, Hess JJ, McInnis EA, Lardinois O, Hogan SL, Hu Y, Mendoza C, Le AK, Guseman AJ, Pilkinton MA, Bortone DS, Cowens K, Sidney J, Karosiene E, Peters B, James E, Kwok WW, Vincent BG, Mallal SA, Jennette JC, Ciavatta DJ, Falk RJ (2020) Restricted myeloperoxidase epitopes drive the adaptive immune response in MPO-ANCA vasculitis. J Autoimmun 106:102306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen DP, McInnis EA, Wu EY, Stember KG, Hogan SL, Hu Y, Henderson C, Blazek L, Mallal S, Karosiene E, Peters B, Sidney J, James E, Kwok WW, Jennette JC, Ciavatta DJ, Falk RJ, Free ME (2021) Maintenance of remission is influenced by HLA-DPB1*04:01 and interaction with PR3225–239. J Am Soc Nephrol (In Revision) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erdbrugger U, Hellmark T, Bunch DO, Alcorta DA, Jennette JC, Falk RJ, Nachman PH (2006) Mapping of myeloperoxidase epitopes recognized by MPO-ANCA using human-mouse MPO chimers. Kidney Int 69:1799–1805 [DOI] [PubMed] [Google Scholar]
- 38.Fujii A, Tomizawa K, Arimura Y, Nagasawa T, Ohashi YY, Hiyama T, Mizuno S, Suzuki K (2000) Epitope analysis of myeloperoxidase (MPO) specific anti-neutrophil cytoplasmic autoantibodies (ANCA) in MPO-ANCA-associated glomerulonephritis. Clin Nephrol 53:242–252 [PubMed] [Google Scholar]
- 39.Bruner BF, Vista ES, Wynn DM, James JA (2011) Epitope specificity of myeloperoxidase antibodies: identification of candidate human immunodominant epitopes. Clin Exp Immunol 164:330–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams JM, Ben-Smith A, Hewins P, Dove SK, Hughes P, McEwan R, Wakelam MJ, Savage CO (2003) Activation of the G(i) heterotrimeric G protein by ANCA IgG F(ab′)2 fragments is necessary but not sufficient to stimulate the recruitment of those downstream mediators used by intact ANCA IgG. J Am Soc Nephrol 14:661–669 [DOI] [PubMed] [Google Scholar]
- 41.Hewins P, Williams JM, Wakelam MJ, Savage CO (2004) Activation of Syk in neutrophils by antineutrophil cytoplasm antibodies occurs via Fcgamma receptors and CD18. J Am Soc Nephrol 15:796–808 [DOI] [PubMed] [Google Scholar]
- 42.Rarok AA, Limburg PC, Kallenberg CG (2003) Neutrophil-activating potential of antineutrophil cytoplasm autoantibodies. J Leukoc Biol 74:3–15 [DOI] [PubMed] [Google Scholar]
- 43.Ohlsson S, Holm L, Hansson C, Ohlsson SM, Gunnarsson L, Pettersson Å, Skattum L (2019) Neutrophils from ANCA-associated vasculitis patients show an increased capacity to activate the complement system via the alternative pathway after ANCA stimulation. PLoS One 14:e0218272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franssen CF, Huitema MG, Muller Kobold AC, Oost-Kort WW, Limburg PC, Tiebosch A, Stegeman CA, Kallenberg CG, Tervaert JW (1999) In vitro neutrophil activation by antibodies to proteinase 3 and myeloperoxidase from patients with crescentic glomerulonephritis. J Am Soc Nephrol 10:1506–1515 [DOI] [PubMed] [Google Scholar]
- 45.Charles LA, Caldas ML, Falk RJ, Terrell RS, Jennette JC (1991) Antibodies against granule proteins activate neutrophils in vitro. J Leukoc Biol 50:539–546 [DOI] [PubMed] [Google Scholar]
- 46.Franssen CF, Gans RO, Arends B, Hageluken C, ter Wee PM, Gerlag PG, Hoorntje SJ (1995) Differences between anti-myeloperoxidase and anti-proteinase 3-associated renal disease. Kidney Int 47:193–199 [DOI] [PubMed] [Google Scholar]
- 47.Franssen C, Gans R, Kallenberg C, Hageluken C, Hoorntje S (1998) Disease spectrum of patients with antineutrophil cytoplasmic autoantibodies of defined specificity: distinct differences between patients with anti-proteinase 3 and anti-myeloperoxidase autoantibodies. J Intern Med 244:209–216 [DOI] [PubMed] [Google Scholar]
- 48.Yang JJ, Jennette JC, Falk RJ (1994) Immune complex glomerulonephritis is induced in rats immunized with heterologous myeloperoxidase. Clin Exp Immunol 97:466–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brons RH, de Jong MC, de Boer NK, Stegeman CA, Kallenberg CG, Tervaert JW (2001) Detection of immune deposits in skin lesions of patients with Wegener’s granulomatosis. Ann Rheum Dis 60:1097–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brouwer E, Huitema MG, Klok PA, de Weerd H, Tervaert JW, Weening JJ, Kallenberg CG (1993) Antimyeloperoxidase-associated proliferative glomerulonephritis: an animal model. J Exp Med 177:905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cream JJ, Bryceson AD, Ryder G (1971) Disappearance of immunoglobulin and complement from the Arthus reaction and its relevance to studies of vasculitis in man. Br J Dermatol 84:106–109 [DOI] [PubMed] [Google Scholar]
- 52.Neal CR, Arkill KP, Bell JS, Betteridge KB, Bates DO, Win-love CP, Salmon AHJ, Harper SJ (2018) Novel hemodynamic structures in the human glomerulus. Am J Physiol Renal Physiol 315:F1370–F1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tse WY, Nash GB, Hewins P, Savage CO, Adu D (2005) ANCA-induced neutrophil F-actin polymerization: implications for microvascular inflammation. Kidney Int 67:130–139 [DOI] [PubMed] [Google Scholar]
- 54.Yen A, Braverman IM (1976) Ultrastructure of the human dermal microcirculation: the horizontal plexus of the papillary dermis. J Invest Dermatol 66:131–142 [DOI] [PubMed] [Google Scholar]
- 55.Horsfield K (1978) Morphometry of the small pulmonary arteries in man. Circ Res 42:593–597 [DOI] [PubMed] [Google Scholar]
- 56.Ojima Y, Sawada K, Fujii H, Shirai T, Saito A, Kagaya S, Aoki S, Takeuchi Y, Ishii T, Nagasawa T (2018) Anti-neutrophil cytoplasmic antibody-associated vasculitis (AAV) restricted to the limbs. Intern Med 57:1301–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun WY, Pitson SM, Bonder CS (2010) Tumor necrosis factor-induced neutrophil adhesion occurs via sphingosine kinase-1-dependent activation of endothelial {alpha}5{beta}1 integrin. Am J Pathol 177:436–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu N, Westra J, Rutgers A, Doornbos-Van der Meer B, Huitema MG, Stegeman CA, Abdulahad WH, Satchell SC, Mathieson PW, Heeringa P, Kallenberg CG (2011) Decreased CXCR1 and CXCR2 expression on neutrophils in anti-neutrophil cytoplasmic autoantibody-associated vasculitides potentially increases neutrophil adhesion and impairs migration. Arthritis Res Ther 13:R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radford DJ, Savage CO, Nash GB (2000) Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheum 43:1337–1345 [DOI] [PubMed] [Google Scholar]
- 60.Varani J, Fligiel SE, Till GO, Kunkel RG, Ryan US, Ward PA (1985) Pulmonary endothelial cell killing by human neutrophils. Possible involvement of hydroxyl radical. Lab Invest 53:656–663 [PubMed] [Google Scholar]
- 61.Westlin WF, Gimbrone MA Jr (1993) Neutrophil-mediated damage to human vascular endothelium. Role of cytokine activation. Am J Pathol 142:117–128 [PMC free article] [PubMed] [Google Scholar]
- 62.Yang JJ, Preston GA, Pendergraft WF, Segelmark M, Heeringa P, Hogan SL, Jennette JC, Falk RJ (2001) Internalization of proteinase 3 is concomitant with endothelial cell apoptosis and internalization of myeloperoxidase with generation of intracellular oxidants. Am J Pathol 158:581–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pendergraft WF III, Rudolph EH, Falk RJ, Jahn JE, Grimmler M, Hengst L, Jennette JC, Preston GA (2004) Proteinase 3 sidesteps caspases and cleaves p21(Waf1/Cip1/Sdi1) to induce endothelial cell apoptosis. Kidney Int 65:75–84 [DOI] [PubMed] [Google Scholar]
- 64.Yang JJ, Kettritz R, Falk RJ, Jennette JC, Gaido ML (1996) Apoptosis of endothelial cells induced by the neutrophil serine proteases proteinase 3 and elastase. Am J Pathol 149:1617–1626 [PMC free article] [PubMed] [Google Scholar]
- 65.Savage CO, Gaskin G, Pusey CD, Pearson JD (1993) Anti-neutrophil cytoplasm antibodies can recognize vascular endothelial cell-bound anti-neutrophil cytoplasm antibody-associated autoantigens. Exp Nephrol 1:190–195 [PubMed] [Google Scholar]
- 66.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535 [DOI] [PubMed] [Google Scholar]
- 67.Söderberg D, Kurz T, Motamedi A, Hellmark T, Eriksson P, Segelmark M (2015) Increased levels of neutrophil extracellular trap remnants in the circulation of patients with small vessel vasculitis, but an inverse correlation to anti-neutrophil cytoplasmic antibodies during remission. Rheumatology (Oxford) 54:2085–2094 [DOI] [PubMed] [Google Scholar]
- 68.Abreu-Velez AM, Smith JG Jr, Howard MS (2009) Presence of neutrophil extracellular traps and antineutrophil cytoplasmic antibodies associated with vasculitides. N Am J Med Sci 1:309–313 [PMC free article] [PubMed] [Google Scholar]
- 69.Imamoto T, Nakazawa D, Shida H, Suzuki A, Otsuka N, Tomaru U, Ishizu A (2014) Possible linkage between microscopic polyangiitis and thrombosis via neutrophil extracellular traps. Clin Exp Rheumatol 32:149–150 [PubMed] [Google Scholar]
- 70.Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, Piconese S, Parenza M, Guiducci C, Vitali C, Colombo MP (2012) Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 120:3007–3018 [DOI] [PubMed] [Google Scholar]
- 71.Wang H, Wang C, Zhao MH, Chen M (2015) Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol 181:518–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang YM, Wang H, Wang C, Chen M, Zhao MH (2015) Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol 67:2780–2790 [DOI] [PubMed] [Google Scholar]
- 73.Kambas K, Chrysanthopoulou A, Vassilopoulos D, Apostolidou E, Skendros P, Girod A, Arelaki S, Froudarakis M, Nakopoulou L, Giatromanolaki A, Sidiropoulos P, Koffa M, Boumpas DT, Ritis K, Mitroulis I (2014) Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis 73:1854–1863 [DOI] [PubMed] [Google Scholar]
- 74.Stassen PM, Derks RP, Kallenberg CG, Stegeman CA (2008) Venous thromboembolism in ANCA-associated vasculitis–incidence and risk factors. Rheumatology (Oxford) 47:530–534 [DOI] [PubMed] [Google Scholar]
- 75.Noris M, Remuzzi G (2013) Overview of complement activation and regulation. Semin Nephrol 33:479–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xing GQ, Chen M, Liu G, Heeringa P, Zhang JJ, Zheng X, J E, Kallenberg CG, Zhao MH (2008) Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol [DOI] [PubMed] [Google Scholar]
- 77.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC (2007) Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol 170:52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu EY, McInnis EA, Boyer-Suavet S, Mendoza CE, Aybar LT, Kennedy KB, Poulton CJ, Henderson CD, Hu Y, Hogan SL, Hu P, Xiao H, Nachman PH, Jennette JC, Falk RJ, Bunch DO (2019) Measuring circulating complement activation products in myeloperoxidase- and proteinase 3-antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol 71:1894–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Charles Jennette J, Xiao H, Hu P (2013) Complement in ANCA-associated vasculitis. Semin Nephrol 33:557–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen SF, Wang FM, Li ZY, Yu F, Chen M, Zhao MH (2018) Complement factor H inhibits anti-neutrophil cytoplasmic autoantibody-induced neutrophil activation by interacting with neutrophils. Front Immunol 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kerr H, Richards A (2012) Complement-mediated injury and protection of endothelium: lessons from atypical haemolytic uraemic syndrome. Immunobiology 217:195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen M, Daha MR, Kallenberg CG (2010) The complement system in systemic autoimmune disease. J Autoimmun 34:J276–J286 [DOI] [PubMed] [Google Scholar]
- 83.Ma YH, Ma TT, Wang C, Wang H, Chang DY, Chen M, Zhao MH (2016) High-mobility group box 1 potentiates antineutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res Ther 18:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang C, Wang H, Hao J, Chang DY, Zhao MH, Chen M (2015) Involvement of high mobility group box 1 in the activation of C5a-primed neutrophils induced by ANCA. Clin Immunol 159:47–57 [DOI] [PubMed] [Google Scholar]
- 85.Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, Seitz LC, Penfold ME, Gan L, Hu P, Lu B, Gerard NP, Gerard C, Schall TJ, Jaen JC, Falk RJ, Jennette JC (2014) C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol 25:225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, Burst V, Grundmann F, Jadoul M, Szombati I, Tesar V, Segelmark M, Potarca A, Schall TJ, Bekker P (2017) Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol 28:2756–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jayne DRW, Merkel PA, Schall TJ, Bekker P (2021) Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med 384:599–609 [DOI] [PubMed] [Google Scholar]
- 88.Jennette JC, Falk RJ (1998) Pathogenesis of the vascular and glomerular damage in ANCA-positive vasculitis. Nephrol Dial Transplant 13(Suppl 1):16–20 [DOI] [PubMed] [Google Scholar]
- 89.Xiao H, Hu P, Falk RJ, Jennette JC (2016) Overview of the pathogenesis of ANCA-associated vasculitis. Kidney Dis (Basel) 1:205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jennette JC, Falk RJ (2007) The role of pathology in the diagnosis of systemic vasculitis. Clin Exp Rheumatol 25:S52–S56 [PubMed] [Google Scholar]
- 91.Vegting Y, Vogt L, Anders HJ, de Winther MPJ, Bemelman FJ, Hilhorst ML (2021) Monocytes and macrophages in ANCA-associated vasculitis. Autoimmun Rev 20:102911. [DOI] [PubMed] [Google Scholar]
- 92.Arrizabalaga P, Sole M, Iglesias C, Escaramis G, Ascaso C (2006) Renal expression of ICAM-1 and VCAM-1 in ANCA-associated glomerulonephritis-are there differences among sero-logic subgroups? Clin Nephrol 65:79–86 [DOI] [PubMed] [Google Scholar]
- 93.Wuthrich RP (1992) Intercellular adhesion molecules and vascular cell adhesion molecule-1 and the kidney. J Am Soc Nephrol 3:1201–1211 [DOI] [PubMed] [Google Scholar]
- 94.O’Brien EC, Abdulahad WH, Rutgers A, Huitema MG, O’Reilly VP, Coughlan AM, Harrington M, Heeringa P, Little MA, Hickey FB (2015) Intermediate monocytes in ANCA vasculitis: increased surface expression of ANCA autoantigens and IL-1beta secretion in response to anti-MPO antibodies. Sci Rep 5:11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Matsumoto K, Suzuki K, Yoshimoto K, Seki N, Tsujimoto H, Chiba K, Takeuchi T (2020) Longitudinal immune cell monitoring identified CD14(++) CD16(+) intermediate monocyte as a marker of relapse in patients with ANCA-associated vasculitis. Arthritis Res Ther 22:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tarzi RM, Liu J, Schneiter S, Hill NR, Page TH, Cook HT, Pusey CD, Woollard KJ (2015) CD14 expression is increased on monocytes in patients with anti-neutrophil cytoplasm antibody (ANCA)-associated vasculitis and correlates with the expression of ANCA autoantigens. Clin Exp Immunol 181:65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aasarød K, Bostad L, Hammerstrøm J, Jørstad S, Iversen BM (2001) Wegener’s granulomatosis: inflammatory cells and markers of repair and fibrosis in renal biopsies–a clinicopathological study. Scand J Urol Nephrol 35:401–410 [DOI] [PubMed] [Google Scholar]
- 98.Rastaldi MP, Ferrario F, Crippa A, Dell’antonio G, Casartelli D, Grillo C, D’Amico G (2000) Glomerular monocyte-macrophage features in ANCA-positive renal vasculitis and cryoglobulinemic nephritis. J Am Soc Nephrol 11:2036–2043 [DOI] [PubMed] [Google Scholar]
- 99.Cattell V (1994) Macrophages in acute glomerular inflammation. Kidney Int 45:945–952 [DOI] [PubMed] [Google Scholar]
- 100.Anguiano L, Kain R, Anders HJ (2020) The glomerular crescent: triggers, evolution, resolution, and implications for therapy. Curr Opin Nephrol Hypertens 29:302–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.O’Laughlin S, Braverman M, Smith-Jefferies M, Buckley P (1992) Macrophages (histiocytes) in various reactive and inflammatory conditions express different antigenic phenotypes. Hum Pathol 23:1410–1418 [DOI] [PubMed] [Google Scholar]
- 102.Wang N, Liang H, Zen K (2014) Molecular mechanisms that influence the macrophage m1–m2 polarization balance. Front Immunol 5:614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alba MA, Jennette JC, Falk RJ (2018) Pathogenesis of ANCA-associated pulmonary vasculitis. Semin Respir Crit Care Med 39:413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Flint J, Morgan MD, Savage CO (2010) Pathogenesis of ANCA-associated vasculitis. Rheum Dis Clin North Am 36:463–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jennette JC, Falk RJ (2014) Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol 10:463–473 [DOI] [PubMed] [Google Scholar]
- 106.Hilhorst M, Shirai T, Berry G, Goronzy JJ, Weyand CM (2014) T cell-macrophage interactions and granuloma formation in vasculitis. Front Immunol 5:432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pagnoux C, Hogan SL, Chin H, Jennette JC, Falk RJ, Guillevin L, Nachman PH (2008) Predictors of treatment resistance and relapse in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis: Comparison of two independent cohorts. Arthritis Rheum 58:2908–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chung SA, Seo P (2010) Microscopic polyangiitis. Rheum Dis Clin North Am 36:545–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lionaki S, Blyth ER, Hogan SL, Hu Y, Senior BA, Jennette CE, Nachman PH, Jennette JC, Falk RJ (2012) Classification of antineutrophil cytoplasmic autoantibody vasculitides: the role of antineutrophil cytoplasmic autoantibody specificity for myeloperoxidase or proteinase 3 in disease recognition and prognosis. Arthritis Rheum 64:3452–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM (1985) Microscopic polyarteritis: presentation, pathology and prognosis. Q J Med 56:467–483 [PubMed] [Google Scholar]
- 111.Guillevin L, Durand-Gasselin B, Cevallos R, Gayraud M, Lhote F, Callard P, Amouroux J, Casassus P, Jarrousse B (1999) Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 42:421–430 [DOI] [PubMed] [Google Scholar]
- 112.Lhote F, Cohen P, Guillevin L (1998) Polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome. Lupus 7:238–258 [DOI] [PubMed] [Google Scholar]
- 113.Jagiello P, Aries P, Arning L, Wagenleiter SE, Csernok E, Hellmich B, Gross WL, Epplen JT (2005) The PTPN22 620W allele is a risk factor for Wegener’s granulomatosis. Arthritis Rheum 52:4039–4043 [DOI] [PubMed] [Google Scholar]
- 114.Gregersen PK, Lee HS, Batliwalla F, Begovich AB (2006) PTPN22: Setting thresholds for autoimmunity. Semin Immunol 18:214–223 [DOI] [PubMed] [Google Scholar]
- 115.Chung SA, Xie G, Roshandel D, Sherva R, Edberg JC, Kravitz M, Dellaripa PF, Hoffman GS, Mahr AD, Seo P, Specks U, Spiera RF, St Clair EW, Stone JH, Plenge RM, Siminovitch KA, Merkel PA, Monach PA (2012) Meta-analysis of genetic polymorphisms in granulomatosis with polyangiitis (Wegener’s) reveals shared susceptibility loci with rheumatoid arthritis. Arthritis Rheum 64:3463–3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li W, Huang H, Cai M, Yuan T, Sheng Y (2021) Antineutrophil cytoplasmic antibody-associated vasculitis update: Genetic pathogenesis. Front Immunol 12:624848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Weidner S, Geuss S, Hafezi-Rachti S, Wonka A, Rupprecht HD (2004) ANCA-associated vasculitis with renal involvement: an outcome analysis. Nephrol Dial Transplant 19:1403–1411 [DOI] [PubMed] [Google Scholar]
- 118.Campbell EJ, Campbell MA, Owen CA (2000) Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibition. J Immunol 165:3366–3374 [DOI] [PubMed] [Google Scholar]
- 119.Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, De Jong PE, Kallenberg CG (1994) Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis [see comments]. Ann Intern Med 120:12–17 [DOI] [PubMed] [Google Scholar]
- 120.Proft T, Fraser JD (2003) Bacterial superantigens. Clin Exp Immunol 133:299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zouali M (2007) Exploitation of host signaling pathways by B cell superantigens–potential strategies for developing targeted therapies in systemic autoimmunity. Ann N Y Acad Sci 1095:342–354 [DOI] [PubMed] [Google Scholar]
- 122.Li H, Nooh MM, Kotb M, Re F (2008) Commercial peptidoglycan preparations are contaminated with superantigen-like activity that stimulates IL-17 production. J Leukoc Biol 83:409–418 [DOI] [PubMed] [Google Scholar]
- 123.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G (2007) Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8:639–646 [DOI] [PubMed] [Google Scholar]
- 124.Oukka M (2008) Th17 cells in immunity and autoimmunity. Ann Rheum Dis 67(Suppl 3):iii26–9 [DOI] [PubMed] [Google Scholar]
- 125.Voswinkel J, Krämer J, Müller A, Herlyn K, Lamprecht P, Gross WL, Gause A (2004) B lymphocytes infiltrating Wegener’s granuloma: the immunoglobulin VH gene repertoire from granulomatous tissues displays an antigen-driven maturation and suggests a microbial trigger. Arthritis Res Ther 6:74 [Google Scholar]
- 126.Husmann CA, Holle JU, Moosig F, Mueller S, Wilde B, Cohen Tervaert JW, Harper L, Assmann G, Gross WL, Epplen JT, Wieczorek S (2014) Genetics of toll like receptor 9 in ANCA associated vasculitides. Ann Rheum Dis 73:890–896 [DOI] [PubMed] [Google Scholar]
- 127.Walsh M, Flossmann O, Berden A, Westman K, Hoglund P, Stegeman C, Jayne D (2012) Risk factors for relapse of anti-neutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 64:542–548 [DOI] [PubMed] [Google Scholar]
- 128.Ooi JD, Jiang JH, Eggenhuizen PJ, Chua LL, van Timmeren M, Loh KL, O’Sullivan KM, Gan PY, Zhong Y, Tsyganov K, Shochet LR, Ryan J, Stegeman CA, Fugger L, Reid HH, Rossjohn J, Heeringa P, Holdsworth SR, Peleg AY, Kitching AR (2019) A plasmid-encoded peptide from Staphylococcus aureus induces anti-myeloperoxidase nephritogenic autoimmunity. Nat Commun 10:3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Al-Abdouh A, Siyal AM, Seid H, Bekele A, Garcia P (2020) Hydralazine-induced antineutrophil cytoplasmic antibody-associated vasculitis with pulmonary-renal syndrome: a case report. J Med Case Rep 14:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Deng C, Lu Q, Zhang Z, Rao T, Attwood J, Yung R, Richardson B (2003) Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum 48:746–756 [DOI] [PubMed] [Google Scholar]
- 131.Magro CM, Momtahen S, Harp J (2017) The distinctive histo-pathology of hydralazine-associated ANCA positive vasculitis: in vivo demonstration of NETosis. Eur J Dermatol 27:91–92 [DOI] [PubMed] [Google Scholar]
- 132.Dhillon SS, Singh D, Doe N, Qadri AM, Ricciardi S, Schwarz MI (1999) Diffuse alveolar hemorrhage and pulmonary capillaritis due to propylthiouracil. Chest 116:1485–1488 [DOI] [PubMed] [Google Scholar]
- 133.Hogan SL, Satterly KK, Dooley MA, Nachman PH, Jennette JC, Falk RJ (2001) Silica exposure in anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and lupus nephritis. J Am Soc Nephrol 12:134–142 [DOI] [PubMed] [Google Scholar]
- 134.Gomez-Puerta JA, Gedmintas L, Costenbader KH (2013) The association between silica exposure and development of ANCA-associated vasculitis: systematic review and meta-analysis. Auto-immun Rev 12:1129–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gómez DM, Urcuqui-Inchima S, Hernandez JC (2017) Silica nanoparticles induce NLRP3 inflammasome activation in human primary immune cells. Innate Immun 23:697–708 [DOI] [PubMed] [Google Scholar]
- 136.Pai P, Bone JM, Bell GM (1998) Hydrocarbon exposure and glomerulonephritis due to systemic vasculitis. Nephrol Dial Transplant 13:1321–1323 [DOI] [PubMed] [Google Scholar]
- 137.Duna GF, Cotch MF, Galperin C, Hoffman DB, Hoffman GS (1998) Wegener’s granulomatosis: role of environmental exposures. Clin Exp Rheumatol 16:669–674 [PubMed] [Google Scholar]
- 138.Jayne D, Rasmussen N, Andrassy K, Bacon P, Cohen Tervaert JW, Dadoniene J, Ekstrand A, Gaskin G, Gregorini G, de Groot K, Gross W, Hagen EC, Mirapeix E, Pettersson E, Siegert C, Sinico A, Tesar V, Westman K, Pusey C (2003) A randomized trial of maintenance therapy for vasculitis associated with anti-neutrophil cytoplasmic autoantibodies. N.Engl. J Med 349:36–44 [DOI] [PubMed] [Google Scholar]
- 139.Jayne D, Blockmans D, Luqmani R, Moiseev S, Ji B, Green Y, Hall L, Roth D, Henderson RB, Merkel PA, Collaborators BS (2019) Efficacy and safety of belimumab and azathioprine for maintenance of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled study. Arthritis Rheumatol 71:952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Harper L, Morgan MD, Walsh M, Hoglund P, Westman K, Flossmann O, Tesar V, Vanhille P, de GK, Luqmani R, Flores-Suarez LF, Watts R, Pusey C, Bruchfeld A, Rasmussen N, Blockmans D, Savage CO, Jayne D (2012) Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis 71:955–60 [DOI] [PubMed] [Google Scholar]
- 141.Pagnoux C, Quemeneur T, Ninet J, Diot E, Kyndt X, de Wazieres B, Reny JL, Puechal X, le Berruyer PY, Lidove O, Vanhille P, Godmer P, Fain O, Blockmans D, Bienvenu B, Rollot F, Ait el Ghaz-Poignant S, Mahr A, Cohen P, Mouthon L, Perrodeau E, Ravaud P, Guillevin L (2015) Treatment of systemic necrotizing vasculitides in patients aged sixty-five years or older: results of a multicenter, open-label, randomized controlled trial of corticosteroid and cyclophosphamide-based induction therapy. Arthritis Rheumatol 67:1117–1127 [DOI] [PubMed] [Google Scholar]
- 142.Group WR (2005) Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med 352:351–361 [DOI] [PubMed] [Google Scholar]
- 143.Jayne DR, Chapel H, Adu D, Misbah S, O’donoghue D, Scott D, Lockwood CM (2000) Intravenous immunoglobulin for ANCA-associated systemic vasculitis with persistent disease activity. QJM 93:433–9 [DOI] [PubMed] [Google Scholar]
- 144.Fox RI, Herrmann ML, Frangou CG, Wahl GM, Morris RE, Strand V, Kirschbaum BJ (1999) Mechanism of action for leflunomide in rheumatoid arthritis. Clin Immunol 93:198–208 [DOI] [PubMed] [Google Scholar]
- 145.Metzler C, Miehle N, Manger K, Iking-Konert C, de GK, Hellmich B, Gross WL, Reinhold-Keller E (2007) Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener’s granulomatosis. Rheumatology (Oxford) 46:1087–91 [DOI] [PubMed] [Google Scholar]
- 146.de Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Gregorini G, Gross WL, Luqmani R, Jayne DR (2005) Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 52:2461–2469 [DOI] [PubMed] [Google Scholar]
- 147.Pagnoux C, Mahr A, Hamidou MA, Boffa JJ, Ruivard M, Ducroix JP, Kyndt X, Lifermann F, Papo T, Lambert M, Le NJ, Khellaf M, Merrien D, Puechal X, Vinzio S, Cohen P, Mouthon L, Cordier JF, Guillevin L (2008) Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. 359:2790–803 [DOI] [PubMed] [Google Scholar]
- 148.Jones RB, Hiemstra TF, Ballarin J, Blockmans DE, Brogan P, Bruchfeld A, Cid MC, Dahlsveen K, de Zoysa J, Espigol-Frigole G, Lanyon P, Peh CA, Tesar V, Vaglio A, Walsh M, Walsh D, Walters G, Harper L, Jayne D, European Vasculitis Study G (2019) Mycophenolate mofetil versus cyclophosphamide for remission induction in ANCA-associated vasculitis: a randomised, non-inferiority trial. Ann Rheum Dis 78:399–405 [DOI] [PubMed] [Google Scholar]
- 149.Hiemstra TF, Walsh M, Mahr A, Savage CO, de GK, Harper L, Hauser T, Neumann I, Tesar V, Wissing KM, Pagnoux C, Schmitt W, Jayne DR (2010) Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA 304:2381–8 [DOI] [PubMed] [Google Scholar]
- 150.Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, Guillevin L, Mirapeix E, Savage CO, Sinico RA, Stegeman CA, Westman KW, Van Der Woude FJ, dLvW RA, Pusey CD. (2007) Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 18:2180–2188 [DOI] [PubMed] [Google Scholar]
- 151.Walsh M, Merkel PA, Peh CA, Szpirt WM, Puechal X, Fujimoto S, Hawley CM, Khalidi N, Flossmann O, Wald R, Girard LP, Levin A, Gregorini G, Harper L, Clark WF, Pagnoux C, Specks U, Smyth L, Tesar V, Ito-Ihara T, de Zoysa JR, Szczeklik W, Flores-Suarez LF, Carette S, Guillevin L, Pusey CD, Casian AL, Brezina B, Mazzetti A, McAlear CA, Broadhurst E, Reidlinger D, Mehta S, Ives N, Jayne DRW, Investigators P (2020) Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med 382:622–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Guillevin L, Pagnoux C, Karras A, Khouatra C, Aumaitre O, Cohen P, Maurier F, Decaux O, Ninet J, Gobert P, Quemeneur T, Blanchard-Delaunay C, Godmer P, Puechal X, Carron PL, Hatron PY, Limal N, Hamidou M, Ducret M, Daugas E, Papo T, Bonnotte B, Mahr A, Ravaud P, Mouthon L (2014) Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med 371:1771–1780 [DOI] [PubMed] [Google Scholar]
- 153.Charles P, Terrier B, Perrodeau E, Cohen P, Faguer S, Huart A, Hamidou M, Agard C, Bonnotte B, Samson M, Karras A, Jourde-Chiche N, Lifermann F, Gobert P, Hanrotel-Saliou C, Godmer P, Martin-Silva N, Pugnet G, Matignon M, Aumaitre O, Viallard JF, Maurier F, Meaux-Ruault N, Riviere S, Sibilia J, Puechal X, Ravaud P, Mouthon L, Guillevin L, French Vasculitis Study G (2018) Comparison of individually tailored versus fixed-schedule rituximab regimen to maintain ANCA-associated vasculitis remission: results of a multicentre, randomised controlled, phase III trial (MAINRITSAN2). Ann Rheum Dis 77:1143–9 [DOI] [PubMed] [Google Scholar]
- 154.Charles P, Perrodeau E, Samson M, Bonnotte B, Neel A, Agard C, Huart A, Karras A, Lifermann F, Godmer P, Cohen P, Hanrotel-Saliou C, Martin-Silva N, Pugnet G, Maurier F, Sibilia J, Carron PL, Gobert P, Meaux-Ruault N, Le Gallou T, Vinzio S, Viallard JF, Hachulla E, Vinter C, Puechal X, Terrier B, Ravaud P, Mouthon L, Guillevin L (2020) Long-term rituximab use to maintain remission of antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 173:179–187 [DOI] [PubMed] [Google Scholar]
- 155.Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, Savage CO, Segelmark M, Tesar V, van PP, Walsh D, Walsh M, Westman K, Jayne DR (2010) Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 363:211–20 [DOI] [PubMed] [Google Scholar]
- 156.Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, Kallenberg CG, St Clair EW, Turkiewicz A, Tchao NK, Webber L, Ding L, Sejismundo LP, Mieras K, Weitzenkamp D, Ikle D, Seyfert-Margolis V, Mueller M, Brunetta P, Allen NB, Fervenza FC, Geetha D, Keogh KA, Kissin EY, Monach PA, Peikert T, Stegeman C, Ytterberg SR, Specks U (2010) Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 363:221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stegeman CA, Tervaert JW, De Jong PE, Kallenberg CG (1996) Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N.Engl. J Med 335:16–20 [DOI] [PubMed] [Google Scholar]
- 158.Gan PY, Tan DS, Ooi JD, Alikhan MA, Kitching AR, Holdsworth SR (2016) Myeloperoxidase peptide-based nasal tolerance in experimental ANCA-associated GN. J Am Soc Nephrol 27:385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, Milone MC, Payne AS (2016) Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353:179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]