

Abstract
Mucopolysaccharidosis type I-Hurler (MPS I-H) is a neurodegenerative lysosomal storage disorder (LSD) caused by inherited defects of the α-L-iduronidase (IDUA) gene. Current treatments are ineffective for treating central nervous system (CNS) manifestations because lysosomal enzymes do not effectively cross the blood-brain barrier (BBB). To enable BBB transport of the enzyme, we engineered a modified IDUA protein by adding a brain-targeting peptide from melanotransferrin. We demonstrated that fusion of melanotransferrin peptide (MTfp) at the N terminus of human IDUA (hIDUA) was enzymatically active and could efficiently cross the BBB in vitro. Then, liver-directed gene therapy using the adeno-associated virus 8 (AAV8) vector, which encoded the modified hIDUA cDNA driven by a liver-specific expression cassette was evaluated in an adult MPS I-H mouse model. The results showed that intravenous (i.v.) infusion of AAV8 resulted in sustained supraphysiological levels of IDUA activity and normalized glycosaminoglycan (GAG) accumulation in peripheral tissues. Addition of MTfp to the hIDUA N terminus allowed efficient BBB transcytosis and IDUA activity restoration in the brain, resulting in significant improvements in brain pathology and neurobehavioral deficits. Our results provide a novel strategy to develop minimally invasive therapies for treatment of MPS I-H and other neurodegenerative LSDs.
Keywords: MPS I, Hurler syndrome, CNS disease, MTfp, AAV, gene therapy
Graphical abstract
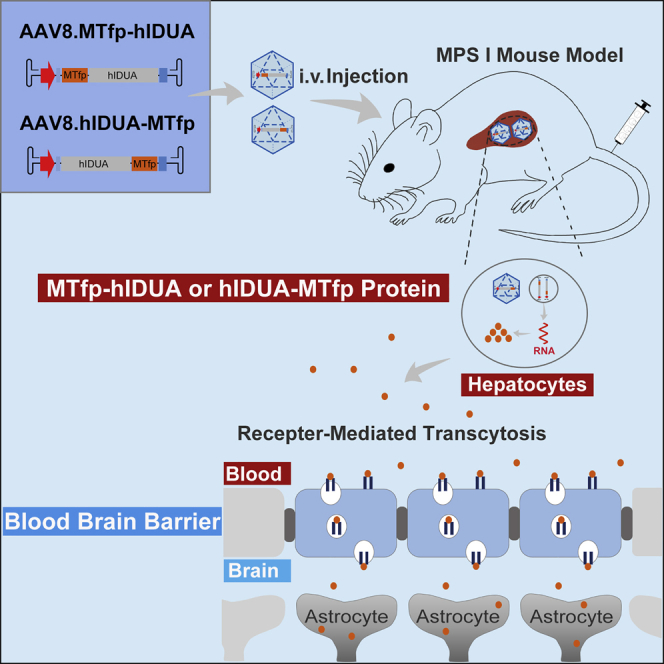
Jin et al. developed an AAV8 vector expressing human IDUA modified with melanotransferrin peptide and investigated its efficacy against central nervous system disease in MPS I-Hurler mice. They found that metabolic and cognitive deficits in treated mice were corrected, suggesting therapeutic potential of this strategy for MPS I-Hurler.
Introduction
Mucopolysaccharidosis type I (MPS I) is a rare autosomal recessive disease caused by deficiency of the α-l-iduronidase (IDUA) gene, which encodes a lysosomal enzyme required for degradation of glycosaminoglycans (GAGs). Excessive accumulation of GAGs results in widespread tissue pathology, mainly manifesting as hepatosplenomegaly, cardiac and pulmonary disease, corneal opacity, skeletal dysplasia, and degenerative neurological diseases.1 MPS I-Hurler (MPS I-H; MIM: 607014), the most severe phenotype, is characterized by severe developmental delay and cognitive impairment, so most individuals with MPS I-H usually die between 5 and 10 years of age when left untreated. The current standard treatment for individuals with MPS I-H is hematopoietic stem cell transplantation (HSCT). However, HSCT has 10%–15% mortality and severe morbidity, and this strategy does not significantly improve skeletal dysplasia and pre-existing cognitive decline.2 For individuals with MPS I with more attenuated phenotypes, MPS I-Hurler/Scheie (MPS I-HS; MIM: 607015) and MPS I-Scheie (MPS I-S; MIM: 607016), enzyme replacement therapy (ERT; laronidase) is the main treatment option. However, affected individuals need lifelong weekly injections of laronidase, and the treatment does not completely eliminate the clinical manifestations of the disease. Because of the inability of recombinant enzymes to cross the blood-brain barrier (BBB), there are no benefits of ERT for brain pathology and cognitive decline.3
For diseases such as MPS I that require lifelong systemic enzyme replacement, liver-directed gene transfer appears to be a potential therapeutic option. The liver has a high synthesis capacity and contains 10%–15% of the body’s total blood volume, making it ideal for use as a factory to secrete proteins into the circulation.4 Adeno-associated virus (AAV) vectors capable of safe and efficient hepatic targeting also provide conditions to maintain high levels of therapeutic enzyme concentrations in the bloodstream, driving greater enzyme uptake and improving efficacy in hard-to-treat tissues. For example, liver-directed AAV8 gene therapy significantly improves disease phenotypes in MPS I, MPS IVA, and Pompe disease models, including skeletal and cardiovascular damage, demonstrating the potential value of this treatment approach.5, 6, 7, 8 Nonetheless, because lysosomal enzymes do not effectively cross the BBB, this treatment approach is ineffective for treating central nervous system (CNS) manifestations. In addition, phase I/II gene therapy with an intracisternally administered AAV9 vector containing an IDUA expression cassette (RGX-111) is currently being tested as a strategy to directly target the CNS of individuals with MPS I (ClinicalTrials.gov: NCT03580083). However, this strategy may need to be combined with ERT to treat the somatic manifestations of the disease.9 It is necessary to find a novel and effective therapeutic strategy for simultaneous correction of somatic and CNS manifestations of MPS I.
The BBB prevents efficacious drugs from entering the CNS. By binding ligands for specific receptors on brain capillary endothelial cells of the BBB, active transport of therapeutic drugs to the brain was discovered.10 Studies show that drug delivery to the CNS via receptor-mediated transcytosis, a natural BBB-penetrating pathway, has significant advantages, manifested as minimal invasiveness and favorable efficacy. For example, weekly intravenous (i.v.) infusions of valanafusp alpha, an IDUA fusion protein that binds a monoclonal antibody against the human insulin receptor, stabilized CNS function and demonstrated long-term safety in phase I/II trials.11,12 To improve BBB delivery, many brain-targeting peptides have been identified. A study has reported persistent brain metabolic correction and normalization of cognitive behavioral defects in MPS I mice by hematopoietic stem cell-mediated expression of IDUA fused with receptor-binding peptides of apolipoprotein E.13 A highly conserved melanotransferrin peptide (MTfp) containing 12 amino acids has been screened. MTfp has been found to retain the ability of a soluble form of melanotransferrin to cross the BBB, distribute throughout the parenchyma, and enter endosomes and lysosomes within neurons, astrocytes, and microglia in the brain.14, 15, 16 For instance, MTfp-interleukin-1 receptor antagonist conjugates, administered peripherally, have been found to be able to cross the BBB and effectively improve neuropathic pain.17 In addition, MTfp effectively delivered NOX4-specific small interfering RNA (siRNA) to the brain, reducing ischemic stroke.18 These studies indicate that MTfp, as a brain-targeting peptide, has the potential to facilitate drug delivery to the brain.
In this study, we developed and tested a novel minimally invasive approach for simultaneous correction of the somatic and CNS manifestations of MPS I. This strategy makes use of liver-directed AAV8 gene transfer to evaluate the BBB-crossing ability of IDUA enzymes modified with MTfp after being secreted from the liver into the bloodstream and the therapeutic effect on disease phenotypes.
Results
Construction and validation of modified IDUA in vitro
To generate a modified IDUA that could cross the BBB and target the CNS via receptor-mediated transcytosis, we engineered human IDUA (hIDUA) by adding MTfp (DSSHAFTLDELR) to its N or C terminus using the (EAAAK)3 linker (referred to as MTfp-hIDUA or hIDUA-MTfp). To analyze the expression of the modified IDUA enzymes, we constructed codon-optimized MTfp-hIDUA or hIDUA-MTfp genes into an AAV backbone plasmid containing a liver-specific thyroxin-binding globulin (TBG) promoter, Kozak sequence, and bovine growth hormone (bGH) polyadenylation sequence, respectively (referred to as pAAV.TBG.MTfp-hIDUA or pAAV.TBG.hIDUA-MTfp; Figure 1A). A codon-optimized unmodified hIDUA gene was also constructed into the same AAV backbone plasmid and served as a control (referred to as pAAV.TBG.hIDUA; Figure 1A). These plasmids were transfected into human Huh7 and mouse H2.35 cells, respectively. Forty-eight hours after transfection, the cell lysates and the culture supernatants were collected for an IDUA enzyme activity assay and western blot analysis. As shown in Figures 1B and 1C, compared with unmodified hIDUA, MTfp addition at the N and C terminus did not compromise enzyme activity. MTfp N-terminal modification significantly increased IDUA enzyme activity. The pAAV.TBG.hIDUA-MTfp and pAAV.TBG.MTfp-hIDUA plasmids in Huh7 cells produced approximately 1.1- and 1.2-fold lysate IDUA activity and approximately 0.9- and 1.6-fold supernatant IDUA activity, respectively, relative to pAAV.TBG.hIDUA (Figure 1B). The pAAV.TBG.hIDUA-MTfp and pAAV.TBG.MTfp-hIDUA plasmids in H2.35 cells produced approximately the same level of lysate IDUA activity and approximately 0.9- and 1.5-fold supernatant IDUA activities, respectively, compared with pAAV.TBG.hIDUA (Figure 1C). Western blotting of cell lysates and culture supernatants showed that MTfp addition at the N and C terminus did not compromise IDUA expression, and precursors and mature forms of IDUA were detected in cell lysates after transfection with plasmids encoding unmodified hIDUA and modified hIDUA (Figure 1D). In addition, the effects of different flexible and rigid linkers between MTfp and hIDUA on enzyme activity were evaluated. The result showed that flexible linkers, including (GGS)3, XTEN, GSAGSAAGSGEF, and SIVAQLSRPDPA, compromise IDUA activity compared with the rigid (EAAAK)3 linker (Figure S1; Table S1).
Figure 1.

In vitro validation of engineered IDUA enzyme activity and transcytosis in the hCMEC/D3 BBB model
(A) Schematic of the encoding sequence of the plasmids expressing the different hIDUA proteins (unmodified, C-terminal modification, and N-terminal modification). (B) IDUA enzyme activity in Huh7 cell lysates and culture supernatants. The mock-transfected group (untreated) and pAAV.TBG.EGFP-transfected group served as controls. (C) IDUA enzyme activity in H2.35 cell lysates and culture supernatants. The mock-transfected group (untreated) and pAAV.TBG.EGFP-transfected group served as controls. (D) Western blot analysis. Culture supernatants (CSs) and cell lysates (CLs) were prepared from Huh7 cells transiently transfected for detection of IDUA protein. The mock-transfected group (untreated) and pAAV.TBG.EGFP-transfected group served as controls. (E) Schematic of the in vitro BBB permeability assay. (F) IDUA enzyme activity of basal lateral compartment medium collected 1, 2, and 4 h after culture was determined. Mock-transfected medium without IDUA was added to the apical compartment as the “untreated” group. (B, C, and F) n = 3 biological replicates each. Mean ± SD are shown. ns, nonsignificant; #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 compared with the pAAV.TBG.hIDUA group.
Next, the permeability of the modified IDUA was evaluated in a well-established in vitro human BBB model. This consists of hCMEC/D3 monolayer cells grown on collagen I-coated Transwell inserts separating an apical compartment from a basolateral compartment, representing the blood and brain sides of the capillary endothelium, respectively (Figure 1E). Equal amounts of enzyme were loaded into the apical compartment. After incubation at 37°C for 1, 2, and 4 h, basolateral IDUA enzyme activity was determined. As shown in Figure 1F, when unmodified hIDUA was added to the apical compartment, there was no change in basolateral IDUA activity within 4 h of incubation. The basolateral transfer amounts of hIDUA-MTfp were indistinguishable from those of unmodified hIDUA. However, addition of MTfp at the hIDUA N terminus increased apical-to-basolateral transfer by 2.4-fold after 4 h of incubation compared with unmodified hIDUA (Figure 1F). The result indicated that the fusion of MTfp at the N terminus of the hIDUA protein was enzymatically active and could efficiently cross the BBB in vitro.
High and stable circulating enzymatic activity in treated mice
To evaluate the therapeutic efficacy of the modified hIDUA enzymes in ameliorating the pathology of brain and peripheral tissues in the murine model of MPS I-H, we packaged AAV8 vectors encoding the modified hIDUA (MTfp-hIDUA or hIDUA-MTfp) or the unmodified hIDUA, all of which carry liver-specific TBG promoters (Figure 2A). The results of the AAV8 infection experiment showed that transduced Huh7 cells could efficiently secrete the modified hIDUA or unmodified hIDUA enzymes (Figure S2). Next, AAV8 vectors (3 × 1010 genome copies [GC]/mouse [low dose] or 3 × 1011 GC/mouse [high dose]) were injected intravenously into 4- to 6-week-old MPS I mice (Figure 2B). Dose-dependent increases in vector DNA copy numbers were observed in the liver 16 weeks after administration of AAV8 vectors (Figure S3). Serum samples were collected for measurement of IDUA activity throughout the 16-week study period. Untreated MPS I mice and wild-type mice served as untreated controls. All treated mice showed supraphysiological levels and a dose-dependent increase in IDUA enzyme activity in the serum for the duration of the study. 16 weeks after treatment, the mean serum activity (3 × 1010 GC/mouse dose) was 111 ± 32 nmol/h/mL in the AAV8.hIDUA-treated group, 35 ± 19 nmol/h/mL in the AAV8.hIDUA-MTfp-treated group, and 110 ± 32 nmol/h/mL in the AAV8.MTfp-hIDUA-treated group (Figure 2C). The mean serum IDUA activity in the low-dose-treated group was approximately 8-fold higher than the mean wild-type level (10.8 ± 0.7 nmol/h/mL). The mean serum IDUA activity in the high-dose-treated group was significantly higher (p < 0.05) than levels found in wild-type mice from week 3 through week 16. 16 weeks after treatment, the mean serum activity (3 × 1011 GC/mouse dose) was 8,904 ± 5,003 nmol/h/mL in the AAV8.hIDUA-treated group, 3,806 ± 1,081 nmol/h/mL in the AAV8.hIDUA-MTfp-treated group, and 11,488 ± 3,430 nmol/h/mL in the AAV8.MTfp-hIDUA-treated group. The mean serum IDUA activity in the high-dose-treated group was approximately 750-fold higher than the mean wild-type level (Figure 2C). The AAV8.MTfp-hIDUA-treated group showed the highest serum IDUA enzyme activity (>1,000-fold that of wild-type mice). A significant increase in serum enzyme activity from week 3 through week 16 indicated that liver-expressed enzymes were active and continuously secreted into the bloodstream.
Figure 2.

Biochemical correction in peripheral tissues of treated mice
(A) Schematic of the AAV8.hIDUA, AAV8.hIDUA-MTfp, and AAV8.MTfp-hIDUA vectors. The AAV vectors contain the TBG promotor, a Kozak sequence, the signal peptide, the unmodified hIDUA or modified hIDUA (hIDUA-MTfp or MTfp-hIDUA) transgenes, and the bGH polyadenylation sequence flanked by AAV2 inverted terminal repeats (ITRs). (B) A summary of the in vivo experiments. (C) Serum IDUA activity from weeks 0–16 after injection was measured. Mean ± SEM are shown. IDUA activity in all high-dose-treated mice is significantly higher (p < 0.05) than levels found in wild-type mice from weeks 3–16. (D) IDUA activity in peripheral tissues was detected 16 weeks after injection. ####p < 0.0001 compared with the wild-type group. Mean ± SD are shown. (E) GAG levels in peripheral tissues were detected 16 weeks after injection. GAG levels in all groups are significantly lower (p < 0.001) than levels found in untreated MPS I mice. Mean ± SD are shown. (F) Urine GAGs were detected 16 weeks after injection. Mean ± SEM are shown. ####p < 0.0001 compared with the untreated MPS I group. (C–F) Wild-type mice, n = 7. Untreated MPS I mice, n = 7. 3 × 1011 GC/mouse dose group: AAV8.hIDUA-treated mice, n = 8; AAV8.hIDUA-MTfp-treated mice, n = 9; AAV8.MTfp-hIDUA-treated mice, n = 9. 3 × 1010 GC/mouse dose group, n = 7.
Biochemical correction in peripheral tissues of treated mice
To determine whether the modified IDUA enzyme secreted from the liver into the bloodstream was taken up by distal secondary tissues and was able to effectively clear the accumulated substrates, we quantified enzyme activity and GAG levels in a variety of tissues (heart, liver, spleen, lung, and kidney) 16 weeks after treatment. As shown in Figure 2D, liver enzyme activity was dose dependent and approximately 450-fold and 17-fold higher than the wild-type levels in the high-dose- and low-dose-treated groups, respectively. IDUA activity was significantly increased in the heart, spleen, lungs, and kidneys of all treated mice. The enzyme activity reached 34% (heart), 269% (spleen), 36% (lungs), and 52% (kidneys) of wild-type IDUA activity in the AAV8.MTfp-hIDUA low-dose group. The enzyme activity reached 193% (heart), 1931% (spleen), 216% (lungs), and 614% (kidneys) of wild-type IDUA activity in the AAV8.MTfp-hIDUA high-dose group (Figure 2D). Subsequently, we assessed whether cellular uptake of hIDUA enzymes expressed by the AAV8 vector was mediated by mannose-6-phosphate (M6P) receptors. Supernatants from transduced Huh7 cells were incubated with target cells for 24 h. When exogenous M6P was absent from hIDUA-enriched supernatants, intracellular IDUA activity was significantly increased, indicating that these enzymes were efficiently taken up by the cells. After adding 5 mM M6P, uptake of the hIDUA enzyme in target cells was significantly reduced. The result indicated that hIDUA enzymes expressed by AAV8 vectors were taken up by cells via M6P receptors (Figure S4). These results demonstrate that modified hIDUA produced by the liver contains the appropriate modifications necessary for uptake into secondary tissues via M6P receptor-mediated endocytosis.19
After determining that modified IDUA was secreted from the liver of treated mice in a form competent for uptake by distal, secondary tissues, we assessed whether the levels of modified IDUA uptake by secondary tissues could reduce IDUA natural substrate GAGs, whose accumulation is a hallmark of MPS I. The results showed that GAG levels in peripheral tissues and urine of all treated groups were significantly reduced and returned to normal 16 weeks after treatment (p < 0.001) (Figures 2E and 2F).
Biochemical correction in the brain tissues of treated mice expressing modified hIDUA
To evaluate the efficiency of BBB crossing and CNS uptake in treated mice, we measured IDUA activity and GAG storage in brain tissues (olfactory bulb, cortex, striatum, hippocampus, cerebellum, thalamus, and brain stem) 16 weeks after treatment. MPS I mice expressing unmodified hIDUA did not show any significant increase in IDUA activity in the brain. In contrast, there was a slight increase in enzyme activity in the brain of mice treated with AAV8.hIDUA-MTfp. Moreover, IDUA activity in the brain of AAV8.MTfp-hIDUA-treated mice showed a dose-dependent and significant increase, with IDUA activity reaching 16%–35% of that of wild-type mice in the low-dose group and 38%–59% of that of wild-type mice in the high-dose group (Figure 3A). IDUA mRNA expression analysis indicated no difference in the levels of IDUA mRNA expression in the brains of treated and untreated MPS I mice (Figure S5; Table S2). These data demonstrate the capability of the modified IDUA enzyme to cross the BBB and to be taken up by brain cells of treated mice upon secretion from the liver. This capability is specifically due to the presence of MTfp at the N terminus or C terminus of IDUA. Compared with modified hIDUA with MTfp at the C terminus, modified hIDUA with MTfp at the N terminus allows more efficient BBB transcytosis. We also assessed GAG levels in brain tissues (Figure 3B). The results showed that GAG levels of mice treated with AAV8.hIDUA were not decreased compared with those of untreated MPS I mice. In contrast, there was a slight decrease in GAG levels in the brains of mice treated with AAV8.hIDUA-MTfp. GAG levels were completely normalized (high-dose group) or close to normalized (low-dose group) in AAV8.MTfp-hIDUA-treated mice 16 weeks after treatment.
Figure 3.

Biochemical correction in brain tissues of treated mice expressing modified hIDUA
(A) IDUA activity in the brain was detected 16 weeks after treatment. (B) GAG levels in the brain were detected 16 weeks after treatment. (A and B) Wild-type mice, n = 7. Untreated MPS I mice, n = 7. 3 × 1011 GC/mouse dose group: AAV8.hIDUA-treated mice, n = 8; AAV8.hIDUA-MTfp-treated mice, n = 9; AAV8.MTfp-hIDUA-treated mice, n = 9. 3 × 1010 GC/mouse dose group, n = 7. Mean ± SEM are shown. #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 compared with the untreated MPS I group. No significant difference between the AAV8.hIDUA group and the untreated MPS I group was observed in any brain region.
Normalization of tissue pathology in treated mice
GAG accumulation leads to formation of characteristic vacuolation in macrophages or brain neurons in individuals with MPS I and the murine model, with emphasis on Purkinje cell vacuolation.20, 21, 22 Thus, we evaluated correction of the pathological phenotype of a subset of tissues (brain, heart, liver, spleen, lung, and kidney). The pathological findings showed a significant reduction in vacuolar cells in peripheral tissues (heart, liver, spleen, lung, and kidney) of all treated mice (Figures 4 and S6). Significant vacuolation of Purkinje and hippocampal pyramidal cells was observed in the brains of untreated MPS I mice (Figure 4). No improvement in vacuolation of Purkinje cells and hippocampal pyramidal cells in the brains of mice treated with AAV8.hIDUA was observed, which was consistent with the observed absence of IDUA activity in the brain (Figures 4 and S6). In contrast, mice expressing modified IDUA at the C terminus showed a slight improvement in vacuolation of Purkinje and hippocampal pyramidal cells, with only a few vacuolated Purkinje and hippocampal pyramidal neurons. Purkinje and hippocampal pyramidal cell vacuolation was completely corrected in the brains of mice expressing modified IDUA with MTfp at the N terminus. We also performed Alcian blue staining to evaluate GAG storage in tissues (brain, heart, liver, spleen, lung, and kidney). As expected, GAG accumulation was significantly higher in the tissues of untreated MPS I mice than in those of wild-type mice. In accordance with the reduced pathological vacuolization, a significant reduction in GAG storage was detected in heart, liver, spleen, lung, and kidney tissues of all treated mice (Figure S7). We observed a slight decrease in accumulated GAGs in the brains of mice expressing modified IDUA with MTfp at the C terminus. Moreover, GAG storage was completely reversed in the brains of mice expressing modified IDUA with MTfp at the N terminus, as shown in Figure S7. In addition, histochemical analysis also showed no signs of inflammation, such as lymphocyte or macrophage aggregation, in tissues of all treated mice. These data demonstrate that IDUA modified with MTfp at the N terminus produced from the liver efficiently cross the BBB at levels sufficient for normalization of GAGs in the brain.
Figure 4.

Correction of histological abnormalities in treated mice
Shown is histological analysis of the brain, heart, liver, spleen, lungs, and kidneys 16 weeks after injection by H&E staining. Scale bars: 20 μm; hippocampus, 40 μm. Black arrows indicate vacuolation of Purkinje neurons and hippocampal pyramidal neurons or foamy macrophages in the heart, liver, spleen, lungs, and kidneys because of GAG accumulation. H&E staining of treated mice was obtained from high-dose groups. For low-dose groups, see Figure S6.
Reversal of bone abnormalities in treated mice
Bone abnormalities are often associated with lysosomal storage diseases, including MPS I.23 Therefore, we assessed bone development of mice 16 weeks after treatment. Untreated MPS I mice had a widened zygomatic arch, about 2.2-fold wider than wild-type mice, according to a microcomputed tomography (micro-CT) scan (Figures 5A and 5C). This abnormal appearance was consistent with the coarse facial features observed in individuals with MPS I.21 The zygomatic width abnormality was completely corrected (all high-dose-treated mice) or close to normalized (all low-dose treated mice) (Figures 5C and S8A). Similar results were obtained when assessing the width of the femur. The femur in untreated MPS I mice was found to be wider than in wild-type mice, with a 1.2-fold increase in femur width (Figures 5B and 5D). The width abnormality was also completely corrected in all treated mice (Figures 5D and S8B). However, there was no significant difference in femur length between 5-month-old wild-type and MPS I mice (Figures 5E and S8C).
Figure 5.

Reversal of bone abnormalities in treated mice
(A) Micro-CT shows a dilated zygomatic arch (white arrows) in a 5-month-old MPS I mouse, which is absent in all treated mice and wild-type mice. (B) Micro-CT shows a dilated femur (white arrows) in a 5-month-old MPS I mouse, which is absent in all treated mice and wild-type mice. (C) Quantification of zygomatic arch width. (D) Quantification of femur width. (E) Quantification of femur length. (C–E) Width and length were analyzed using the ImageJ program. Data represent the value relative to the mean of wild-type mice. Mean ± SD are shown. The data of treated mice were obtained from high-dose groups. For low-dose groups, see Figure S8. Wild-type mice, n = 7. Untreated MPS I mice, n = 7. 3 × 1011 GC/mouse dose group: AAV8.hIDUA-treated mice, n = 8; AAV8.hIDUA-MTfp-treated mice, n = 9; AAV8.MTfp-hIDUA-treated mice, n = 9. #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 compared with the untreated MPS I group.
Improvement of neurobehavioral deficits in treated mice expressing modified hIDUA 12 weeks after treatment
To determine whether the amelioration of CNS pathology in treated mice that were injected with AAV8 encoding the modified IDUA provided any cognitive benefit, we performed a delayed matching to place (DMP) dry maze test 12 weeks after injection based on a previous study.24 The DMP dry maze is a behavioral test that evaluates the spatial learning and memory abilities of mice by measuring the time it takes for the mice to find an escape route on a high platform.25 The results showed that there was no significant difference in the average speed of the mice on the platform in all groups (Figure S9). Over the course of 6 days of testing, the average escape latency of control wild-type mice decreased from 175 to 62 s (Figure 6A). In contrast, untreated MPS I mice showed a slow reduction in average escape latency from 160 to 117 s, with a significant difference from the wild-type mice, indicating cognitive deficits. There was no significant difference in mean escape latency between AAV8.hIDUA-treated and untreated MPS I groups (Figures 6A, 6B, S10A, and S10B). In contrast, treated mice expressing modified IDUA with MTfp at the C terminus had better learning and memory (Figures 6A, 6B, S10A, and S10B). The latency to escape of mice treated with high-dose AAV8.MTfp-hIDUA exhibited a significant improvement, with no significant difference from wild-type mice (Figures 6A and 6B). Mice treated with low-dose AAV8.MTfp-hIDUA also had greatly improved learning and memory, with average escape latency ranging from 167–81 s, which was not significantly different from wild-type mice on the last test day (Figures S10A and S10B). These data demonstrate that hIDUA modified with MTfp at the N terminus secreted from the liver can efficiently cross the BBB and confer significant cognitive benefits to MPS I mice and that low levels of enzymes in the brain may be sufficient for prevention of onset of cognitive deficits in this mouse model.
Figure 6.

Improvement of neurobehavioral deficits in treated mice expressing modified hIDUA 12 weeks after treatment
(A) Cognitive performance was assessed using the DMP dry maze 12 weeks after treatment. Data represent the time required to escape the platform over 6 days of testing. (B) Data represent the time required for individual mice from all high-dose groups to escape the platform on day 6 of testing. (A and B) Data are shown as the mean ± SEM. Wild-type mice, n = 7. Untreated MPS I mice, n = 7. 3 × 1011 GC/mouse dose group: AAV8.hIDUA-treated mice, n = 8; AAV8.hIDUA-MTfp-treated mice, n = 9; AAV8.MTfp-hIDUA-treated mice, n = 9. #Wild-type group, ∗AAV8.MTfp-hIDUA high dose group, #p < 0.05, ##p < 0.01, ∗∗p < 0.01 compared with the untreated MPS I group.
Discussion
In this study, a novel liver-directed AAV8 vector expressing modified hIDUA was developed and administered intravenously to the adult MPS I-H mouse model, resulting in a sustained and high level of IDUA enzyme activity in the blood circulation and improving hard-to-treat tissues like bone and brain. Dysostosis multiplex in MPS I is one of the most prevalent and incapacitating manifestations and less likely to be corrected by current therapies.2,26 Massive accumulation of GAGs in chondrocytes of individuals with MPS I and animal models result in skeletal development defects, typically including abnormal widening of the zygomatic arch and femur.23 In our study, the width of the zygomatic arch and femur in 5-month-old MPS I mice was 2.2- and 1.2-fold larger than in normal mice, respectively. The skeletal dysplasia of all treated mice could be significantly improved even when the AAV vector dose was as low as 3 × 1010 GC/mouse, demonstrating the effectiveness of this treatment (Figures 5 and S8). There were age-dependent differences in femur length between this MPS I mouse model and wild-type mice, as reported previously.21 In 5-week-old MPS I mice, the femur was 15% shorter than in wild-type mice. However, this decrease in femur length was less evident in 15-week-old MPS I mice and was no longer detectable in 35-week-old MPS I mice. The abnormal femoral length in the early stage may be due to abnormalities in the growth plate caused by GAG accumulation in MPS I mice.27 The osteoclast activity of the femur in this MPS I model was 4-fold higher than in wild-type femora.28 Our study showed that there was no significant difference in femur length between 5-month-old MPS I mice and wild-type mice, suggesting that the abnormal femur length of this MPS I mouse model might return to normal at 5 months of age.
Lysosomal enzymes are usually unable to pass the BBB because of lack of the M6P receptor on the brain microvasculature in adult mice.29 To evaluate the therapeutic effect of modified IDUA enzymes in the brain, we selected adult MPS I mice with an entirely impenetrable BBB for gene transfer. In our current study, we did not detect any biochemical and pathological correction in the brains of MPS I mice expressing unmodified IDUA enzymes. In previous studies, however, repeated high-dose ERT has been found to facilitate BBB crossing of lysosomal enzymes and rescue cognitive deficits in disease model mice.30,31 A study also reported that a small but non-significant increase in IDUA activity in the brain may be associated with a partial rescue of the CNS phenotype in MPS I mice receiving i.v. injection of an AAV that encodes wild-type IDUA enzymes.32 The exact mechanism of high-dose ERT potentially crossing the BBB is unclear. In addition, studies indicate that a high serum dose of lysosomal enzymes alone may not be enough to result in BBB crossing, but the reason for the difference in the these findings is unknown.8,33 It has been found that coupling MTfp to the N terminus of the drug enabled the drug to efficiently cross the BBB.17 Some conjugates of brain-targeting peptides with the C terminus of lysosomal enzymes have also been reported to significantly cross the BBB.8,34 Therefore, we designed two modified hIDUA proteins by adding MTfp to the N or C terminus of hIDUA. Our findings revealed that therapeutic levels of IDUA enzyme activity were exclusively found in the brains of treated mice expressing hIDUA modified with MTfp, suggesting that MTfp modification enabled hIDUA to cross the BBB via receptor-mediated transcytosis. Interestingly, a significant increase in IDUA activity was only detected in the brains of treated mice expressing modified hIDUA with MTfp at the N terminus, implying that N-terminal MTfp modification allowed more effective BBB transcytosis than C-terminal MTfp modification, which was also consistent with the results of a transcytosis assay in the BBB model in vitro (Figures 1F and 3A). Because of the widespread lesion in the CNS of individuals with MPS I, an effective therapy strategy necessitates enzyme administration to all areas of the brain.11 In our study, an increase in IDUA enzyme activity was widely observed in different brain regions (olfactory bulb, cortex, striatum, hippocampus, cerebellum, thalamus, and brain stem), indicating that it was possible to deliver IDUA to all regions of the brain via the transvascular route across the BBB (Figure 3A).
The primary therapeutic goal of this study is to improve brain pathology in MPS I-H and stabilize cognitive decline. Cognitive decline in MPS I may include a two-step process: formation of intra-neuronal lysosomal inclusion bodies, followed by dystrophic neurites and cognitive decline caused by a secondary neuropathology.12 Studies found that cognitive deficits in IDUA knockout mice appear to generally occur at the age of 3–4 months or older and even at the age of 10 months.24,35, 36, 37, 38 In addition, it has also been found that 4-month-old IDUA knockout mice show significant spatial learning and memory deficits in the Morris water maze, but this difference decreases in 6- to 8-month-old IDUA knockout mice and wild-type mice.24 There are currently no behavioral studies of this MPS I-H mouse model, but abnormal lysosomal storage in the brain of this model was observed as early as 5 weeks of age, became more significant at 10 weeks of age, and was still increasing at a later time point.21 To test spatial learning and memory abilities, we used a modified Barnes maze (DMP dry maze).25 Significant spatial learning and memory deficits were observed in 4-month-old MPS I mice. The deficit was significantly improved in AAV8.MTfp-hIDUA-treated mice (Figure 6). The hippocampus has an important function in learning and memory processes.39 Spatial memory and navigation skills in hidden platform learning are thought to be primarily dependent on hippocampal place cells in the Morris water maze test.40 In the Morris water maze, gerbils with hippocampal pyramidal cell injury show deficits in spatial navigation.41 In our study, untreated MPS I mice showed severe hippocampal pyramidal cell vacuolation. However, vacuolization of hippocampus pyramidal cells in AAV8.MTfp-hIDUA-treated mice was corrected (Figure 4). In addition, restoration of IDUA activity and normalization of GAG storage were observed in the hippocampus in this treated group (Figure 3). We speculate that one of the most likely explanations for the improvement of spatial learning and memory deficits observed in treated mice may be restoration of IDUA levels in the hippocampus. However, this possibility is only based on behavioral data 12 weeks after treatment, and more long-term neurobehavioral evidence for this model is needed.
Although HSCT is the current gold standard for treatment of MPS I-H, its success is influenced by selection of affected individuals, the source of the transplant, the type of donor used, and especially the individual’s. Individuals with MPS I-H treated with HSCT after 2–2.5 years of age still have long-term cognitive-developmental deficits.2 A clinical study of intracisternal delivery of AAV9 is being performed to treat the CNS manifestations in individuals with MPS I (ClinicalTrials.gov: NCT03580083). Studies of intranasal or i.v. injection of AAV vectors that can cross the BBB for CNS manifestations of MPS I have been reported.37,42 Our study used liver-directed gene transfer based on i.v. delivery of AAV8 expressing modified IDUA proteins, which resulted in significant improvements in peripheral tissue and CNS manifestations of MPS I, indicating the potential safety and effectiveness of this therapeutic strategy. Because AAVs are non-integrating, most of the vector genome is lost during hepatocyte proliferation, which is especially important when the target population is children.4 However, evidence is emerging that the effects of AAV gene therapy, when targeted to the adult liver, can be maintained over long periods, although a small fraction of the vectors is lost.43, 44, 45 Our strategy may provide a safer and more appropriate treatment option for adults with MPS I-H, although more long-term data regarding therapeutic efficacy are required. More preclinical data on possible long-term toxic side effects of hIDUA enzyme overload in the blood and the immunogenic properties of modified hIDUA itself are also needed to promote future clinical applications based on liver-directed expression of modified hIDUA in individuals with MPS I.
Our study showed that liver-directed AAV8.MTfp-hIDUA gene therapy significantly restored IDUA enzyme activity in the brains of the MPS I-H mouse model, corrected brain pathology, and improved cognitive impairment 12 weeks after treatment. As a promising noninvasive strategy, this not only provides a feasible path for simultaneous reversal of lysosomal storage in peripheral tissues and the CNS of individuals with MPS I but also serves as a reference for minimally invasive treatment of other neurodegenerative LSDs.
Materials and methods
Plasmid construction
To construct the modified hIDUA constructs, MTfp (DSSHAFTLDELR) was fused to the N or C terminus of hIDUA using an (EAAAK)3 linker (supplemental information). The unmodified hIDUA and modified hIDUA genes (MTfp-hIDUA and hIDUA-MTfp) were codon optimized and synthesized by Genewiz (Suzhou, China). Then the codon-optimized genes and an enhanced green fluorescent protein (EGFP) gene were subcloned into an AAV backbone plasmid containing the full-length TBG promoter (two copies of enhancer elements of the α microglobulin/bikunin gene followed by a liver-specific TBG promoter), Kozak sequence, and bGH polyadenylation sequence flanked by AAV2 ITRs, yielding AAV packaging plasmids (pAAV.TBG.hIDUA, pAAV.TBG.hIDUA-MTfp, and pAAV.TBG.MTfp-hIDUA) and a pAAV.TBG.EGFP plasmid. All constructed plasmids were verified by sequencing.
Plasmid transfection
H2.35 (a mouse liver cell line) and Huh7 (a human hepatoma cell line) (ATCC, Manassas, VA) cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and cultured at 37°C with 5% CO2. The pAAV.TBG.hIDUA, pAAV.TBG.hIDUA-MTfp, pAAV.TBG.MTfp-hIDUA, and pAAV.TBG.EGFP plasmids were transfected into H2.35 or Huh7 cells using the TransIT-X2 Dynamic Delivery System (Mirus, Madison, WI) according to the manufacturer’s recommendations. 48 h after transfection, cell lysates and culture supernatants were harvested for an IDUA enzyme activity assay and western blot analysis, and culture supernatants were concentrated using a centrifuge filter (Millipore, Temecula, CA) prior to western blot analysis.
In vitro BBB permeability assay
hCMEC/D3 cells (Millipore), an immortalized human brain capillary endothelial cell line, were cultured in endothelial cell medium (ECM) (ScienCell, Carlsbad, CA). To establish the in vitro BBB model, hCMEC/D3 cells (passages 25–30) were seeded in type I collagen pre-coated Transwell filters (polyethylene terephthalate 24-well; pore size, 1 μm; Millipore) at a density of 1.2×105 cells/cm2 and cultured for 3–5 days as described previously.46 Then transfected culture supernatant containing equal amounts IDUA was added to each apical compartment and incubated at 37°C and 5% CO2. The media in the basolateral compartments were collected 1, 2, and 4 h after incubation for the IDUA enzyme activity assay. To determine the integrity of the in vitro BBB model, a sodium fluorescein permeability assay was performed in parallel with the transcytosis assay.
Western blot analysis
Western blot analyses were performed on cell lysates and culture supernatant. Total protein concentration was determined by bicinchoninic acid (BCA) assay (Thermo Scientific, Waltham, MA). IDUA protein was detected by a rabbit anti-IDUA antibody (1:4,000 dilution, 55158-1-AP, Proteintech). A mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (1:10,000 dilution, AC002, ABclonal) was used to detect GAPDH. Blots were imaged and analyzed using the iBright CL1000 imaging system (Thermo Scientific).
AAV8 vector production
The AAV8.hIDUA, AAV8.hIDUA-MTfp, and AAV8.MTfp-hIDUA vectors were obtained by packaging the pAAV.TBG.hIDUA, pAAV.TBG.hIDUA-MTfp, and pAAV.TBG.MTfp-hIDUA plasmids into AAV8, respectively (Figure 2A). All AAV8 vectors were produced by triple plasmid transfection of HEK293 cells as described previously.47 The genome titer (genome copies per milliliter) of the AAV8 vector was determined by quantitative PCR (qPCR) using the forward primer 5′-GCCAGCCATCTGTTGT-3′, reverse primer 5′-GGAGTGGCACCTTCCA-3′, and probe 5′-Fam-TCCCCCGTGCCTTCCTTGACC-Tamra-3’.48 All vectors used in this study passed the endotoxin assay using the QCL-1000 Chromogenic LAL Test Kit (Cambrex, East Rutherford, NJ).
AAV transduction in vitro
Huh7 cells were seeded into 96-well plates at a density of 5×104 cells/well in 200 μL DMEM containing 10% FBS and 100 U/mL penicillin/streptomycin. The cells were allowed to adhere for 24 h. After 24 h, cells were infected with a wild-type adenovirus (H5 serotype, ATCC) at a multiplicity of infection (MOI) of 30 GC/cell. Two hours after infection with the adenovirus, cells were transduced with AAV8 vectors at an MOI of 6 × 105 GC/cell in 100 μL FBS-free DMEM with 100 U/mL penicillin/streptomycin. After 4 h, the infection medium was replaced with 100 μL DMEM containing 2% FBS and 100 U/mL penicillin/streptomycin. Three days after AAV8 infection, the cell culture medium was collected for IDUA enzyme activity analysis.
Animal studies
The MPS I-H mouse model (Idua-W392X, stock number 017681) was purchased from Jackson Laboratory (Bar Harbor, ME). The background of the wild-type mice used in this study was C57BL/6J. All animal protocols were approved by the Institutional Animal Care and Concern Committee at Sichuan University, and animal care was performed in accordance with the committee’s guidelines. Mice were maintained on a chow diet at an animal facility at Sichuan University. Four-to six-week-old MPS I mice were enrolled in the study. The AAV8.hIDUA, AAV8.hIDUA-MTfp, and AAV8.MTfp-hIDUA vectors (3 × 1010 GC/mouse and 3 × 1011 GC/mouse) were diluted to 200 μL with PBS and then injected via the tail vein. Serum was collected by retro-orbital bleeding before vector dosing and every 1–2 weeks after dosing. Urine samples were collected by gently applying pressure to the bladder at the time of necropsy. 16 weeks after injection, age-matched untreated mice and treated mice were euthanized, and tissues were collected for various analyses.
IDUA enzyme activity assay
Tissue, serum, cell lysate, and culture supernatant samples were immediately frozen on dry ice and stored at −80°C until analysis. Serum and culture supernatants were used directly in IDUA enzyme activity assays. Tissue samples were homogenized in lysis buffer (0.9% NaCl, 0.2% Triton X-100 [pH 3.5]), freeze-thawed, and clarified by centrifugation. Protein concentrations of tissue and cell lysates were determined by BCA protein assay (Thermo Scientific). IDUA enzyme activity was determined with a fluorometric assay using the synthetic substrate 4-MU-iduronide (Glycosynth, Warrington, UK) as described previously.9 Units are given as nanomoles of 4-MU liberated per hour per milligram of protein or per milliliter of serum/culture supernatant.
GAG assay
Tissue and urine GAGs were determined using the Blyscan Glycosaminoglycan Assay Kit (Biocolor, Carrickfergus, UK) according to the manufacturer’s instructions.
AAV8 biodistribution
DNA was extracted from livers, and total vector genomes were quantified by TaqMan qPCR as described previously.49
H&E staining
Tissues were fixed in paraformaldehyde for 24 h, dehydrated through an ethanol series and xylene, and then embedded in paraffin. H&E staining was performed on 6-μm sections from paraffin-embedded tissues according to standard protocols.
GAG histochemistry
Tissue samples were prepared as H&E staining. Deparaffinized 6-μm sections were stained in 1% Alcian blue (Sigma-Aldrich, St. Louis, MO) for 15 min, rinsed in water for 2–3 min, and counterstained with Nuclear Fast Red (Sigma-Aldrich).
DMP dry maze assay
To detect whether this therapy provided any cognitive benefit to MPS I-H mice, we performed a DMP dry maze test 12 weeks after injection. The DMP dry maze assay was conducted using a modified Barnes maze apparatus and consisted of a circular platform (diameter, 122 cm; thickness, 1.2 cm) with 40 holes.50 An escape pipe was secured under one of the holes to allow the mice to escape the platform. The location of the escape hole changed every day. Visual cues were attached to each of the four walls for the mouse to use for spatial navigation. To begin the experiment, mice were placed on the edge of the platform some distance from the escape hole, and an opaque funnel covered the mouse. After a delay of approximately 30 s, a tone (2 kHz, 85 dB) was turned on, and the transfer box was immediately removed to expose the mice to bright light (1,200 lux). In response to these aversive conditions, the mice spontaneously sought out and burrowed into the escape hole. Mice were assessed during four trials per day on 4 consecutive days, with maximal escape time limited to 3 min. Data were collected and analyzed using the ANY-Maze program (SANS, Jiangsu, China).
Micro-CT
To detect whether liver-directed gene therapy could improve skeletal dysplasia in MPS I mice, the zygomata and femora of the mice were scanned using micro-CT (Quantum GX, PerkinElmer, Waltham, MA) 16 weeks after injection (approximately 5 months of age). Images were analyzed using the ImageJ program.
Statistics
GraphPad Prism 9 was used to perform all statistical tests. Values express mean ± SD or mean ± SEM. Statistical analysis was performed by Dunnett’s test, as indicated in the figure legends. In all tests, p < 0.05 was considered significant.
Acknowledgments
This work was supported by the Joint Funds of the National Natural Science Foundation of China (grant U19A2002), National Major Scientific and Technological Special Project for “Significant New Drugs Development” (2018ZX09733001-005-002), and the Science and Technology Major Project of Sichuan province (2017SZDZX0011).
Author contributions
Y.Y. and Y.W. conceived this study and designed the experiments. X.J. constructed the plasmid vectors. Q.Z. performed cell culture and transfection. J.X. produced AAV8 vector and endotoxin assays. X.J. and J.S. performed mouse studies. X.J., J.X., and L.S. performed the DMP dry maze assay. Y.L., X.Z., and X.J. performed IDUA and GAGs assays. K.S. and R.L. performed histopathology assays. X.J. wrote the manuscript. Y.Y., H.D., and Y.W. edited the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.04.010.
Supplemental information
References
- 1.Parker E.I., Xing M., Moreno-De-Luca A., Harmouche E., Terk M.R. Radiological and clinical characterization of the lysosomal storage disorders: non-lipid disorders. Br. J. Radiol. 2014;87:20130467. doi: 10.1259/bjr.20130467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor M., Khan S., Stapleton M., Wang J., Chen J., Wynn R., Yabe H., Chinen Y., Boelens J.J., Mason R.W., et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: past, present, and future. Biol. Blood Marrow Transpl. 2019;25:e226–e246. doi: 10.1016/j.bbmt.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scarpa M., Orchard P.J., Schulz A., Dickson P.I., Haskins M.E., Escolar M.L., Giugliani R. Treatment of brain disease in the mucopolysaccharidoses. Mol. Genet. Metab. 2017;122:25–34. doi: 10.1016/j.ymgme.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Maestro S., Weber N.D., Zabaleta N., Aldabe R., Gonzalez-Aseguinolaza G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021;3:100300. doi: 10.1016/j.jhepr.2021.100300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinderer C., Bell P., Gurda B.L., Wang Q., Louboutin J.P., Zhu Y., Bagel J., O'Donnell P., Sikora T., Ruane T., et al. Liver-directed gene therapy corrects cardiovascular lesions in feline mucopolysaccharidosis type I. Proc. Natl. Acad. Sci. U S A. 2014;111:14894–14899. doi: 10.1073/pnas.1413645111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawamoto K., Karumuthil-Melethil S., Khan S., Stapleton M., Bruder J.T., Danos O., Tomatsu S. Liver-targeted AAV8 gene therapy ameliorates skeletal and cardiovascular pathology in a mucopolysaccharidosis IVA murine model. Mol. Ther. Methods Clin. Dev. 2020;18:50–61. doi: 10.1016/j.omtm.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costa-Verdera H., Collaud F., Riling C.R., Sellier P., Nordin J.M.L., Preston G.M., Cagin U., Fabregue J., Barral S., Moya-Nilges M., et al. Hepatic expression of GAA results in enhanced enzyme bioavailability in mice and non-human primates. Nat. Commun. 2021;12:6393. doi: 10.1038/s41467-021-26744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sorrentino N.C., D'Orsi L., Sambri I., Nusco E., Monaco C., Spampanato C., Polishchuk E., Saccone P., De Leonibus E., Ballabio A., Fraldi A. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol. Med. 2013;5:675–690. doi: 10.1002/emmm.201202083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinderer C., Bell P., Gurda B.L., Wang Q., Louboutin J.P., Zhu Y., Bagel J., O'Donnell P., Sikora T., Ruane T., et al. Intrathecal gene therapy corrects CNS pathology in a feline model of mucopolysaccharidosis I. Mol. Ther. 2014;22:2018–2027. doi: 10.1038/mt.2014.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banks W.A. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016;15:275–292. doi: 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- 11.Boado R.J., Hui E.K.W., Lu J.Z., Zhou Q.H., Pardridge W.M. Reversal of lysosomal storage in brain of adult MPS-I mice with intravenous Trojan horse-iduronidase fusion protein. Mol. Pharm. 2011;8:1342–1350. doi: 10.1021/mp200136x. [DOI] [PubMed] [Google Scholar]
- 12.Giugliani R., Giugliani L., de Oliveira Poswar F., Donis K.C., Corte A.D., Schmidt M., Boado R.J., Nestrasil I., Nguyen C., Chen S., Pardridge W.M. Neurocognitive and somatic stabilization in pediatric patients with severe Mucopolysaccharidosis Type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): an open label phase 1-2 trial. Orphanet J. Rare Dis. 2018;13:110. doi: 10.1186/s13023-018-0849-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Amouri S.S., Dai M., Han J.F., Brady R.O., Pan D. Normalization and improvement of CNS deficits in mice with Hurler syndrome after long-term peripheral delivery of BBB-targeted iduronidase. Mol. Ther. 2014;22:2028–2037. doi: 10.1038/mt.2014.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karkan D., Pfeifer C., Vitalis T.Z., Arthur G., Ujiie M., Chen Q., Tsai S., Koliatis G., Gabathuler R., Jefferies W.A. A unique carrier for delivery of therapeutic compounds beyond the blood-brain barrier. PLoS One. 2008;3:e2469. doi: 10.1371/journal.pone.0002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nounou M.I., Adkins C.E., Rubinchik E., Terrell-Hall T.B., Afroz M., Vitalis T., Gabathuler R., Tian M.M., Lockman P.R. Anti-cancer antibody trastuzumab-melanotransferrin conjugate (BT2111) for the treatment of metastatic HER2+ breast cancer tumors in the brain: an in-vivo study. Pharm. Res. 2016;33:2930–2942. doi: 10.1007/s11095-016-2015-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh C.S.B., Eyford B.A., Abraham T., Munro L., Choi K.B., Okon M., Vitalis T.Z., Gabathuler R., Lu C.J., Pfeifer C.G., et al. Discovery of a highly conserved peptide in the iron transporter melanotransferrin that traverses an intact blood brain barrier and localized in neural cells. Front. Neurosci. 2021;15:596976. doi: 10.3389/fnins.2021.596976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thom G., Tian M.M., Hatcher J.P., Rodrigo N., Burrell M., Gurrell I., Vitalis T.Z., Abraham T., Jefferies W.A., Webster C.I., Gabathuler R. A peptide derived from melanotransferrin delivers a protein-based interleukin 1 receptor antagonist across the BBB and ameliorates neuropathic pain in a preclinical model. J. Cereb. Blood Flow Metab. 2019;39:2074–2088. doi: 10.1177/0271678x18772998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eyford B.A., Singh C.S.B., Abraham T., Munro L., Choi K.B., Hill T., Hildebrandt R., Welch I., Vitalis T.Z., Gabathuler R., et al. A nanomule peptide carrier delivers siRNA across the intact blood-brain barrier to attenuate ischemic stroke. Front. Mol. Biosci. 2021;8:611367. doi: 10.3389/fmolb.2021.611367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kornfeld S., Mellman I. The biogenesis of lysosomes. Annu. Rev. Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- 20.Jones M.Z., Alroy J., Boyer P.J., Cavanagh K.T., Johnson K., Gage D., Vorro J., Render J.A., Common R.S., Leedle R.A., et al. Caprine mucopolysaccharidosis-IIID: clinical, biochemical, morphological and immunohistochemical characteristics. J. Neuropathol. Exp. Neurol. 1998;57:148–157. doi: 10.1097/00005072-199802000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Wang D., Shukla C., Liu X., Schoeb T.R., Clarke L.A., Bedwell D.M., Keeling K.M. Characterization of an MPS I-H knock-in mouse that carries a nonsense mutation analogous to the human IDUA-W402X mutation. Mol. Genet. Metab. 2010;99:62–71. doi: 10.1016/j.ymgme.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrer I., Cusí V., Pineda M., Galofré E., Vila J. Focal dendritic swellings in Purkinje cells in mucopolysaccharidoses types I, II and III. A Golgi and ultrastructural study. Neuropathol. Appl. Neurobiol. 1988;14:315–323. doi: 10.1111/j.1365-2990.1988.tb00891.x. [DOI] [PubMed] [Google Scholar]
- 23.Hampe C.S., Eisengart J.B., Lund T.C., Orchard P.J., Swietlicka M., Wesley J., McIvor R.S. Mucopolysaccharidosis type I: a review of the natural history and molecular pathology. Cells. 2020;9:1838. doi: 10.3390/cells9081838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan D., Sciascia A., 2nd, Vorhees C.V., Williams M.T. Progression of multiple behavioral deficits with various ages of onset in a murine model of Hurler syndrome. Brain Res. 2008;1188:241–253. doi: 10.1016/j.brainres.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng X., Krukowski K., Jopson T., Rosi S. Delayed-matching-to-place task in a dry maze to measure spatial working memory in mice. Bio Protoc. 2017;7:e2389. doi: 10.21769/BioProtoc.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawamoto K., Stapleton M., Alméciga-Díaz C.J., Espejo-Mojica A.J., Losada J.C., Suarez D.A., Tomatsu S. Therapeutic options for mucopolysaccharidoses: current and emerging treatments. Drugs. 2019;79:1103–1134. doi: 10.1007/s40265-019-01147-4. [DOI] [PubMed] [Google Scholar]
- 27.Oestreich A.K., Garcia M.R., Yao X., Pfeiffer F.M., Nobakhti S., Shefelbine S.J., Wang Y., Brodeur A.C., Phillips C.L. Characterization of the MPS I-H knock-in mouse reveals increased femoral biomechanical integrity with compromised material strength and altered bone geometry. Mol. Genet. Metab. Rep. 2015;5:3–11. doi: 10.1016/j.ymgmr.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunn G., Dai Y., Du M., Belakhov V., Kandasamy J., Schoeb T.R., Baasov T., Bedwell D.M., Keeling K.M. Long-term nonsense suppression therapy moderates MPS I-H disease progression. Mol. Genet. Metab. 2014;111:374–381. doi: 10.1016/j.ymgme.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urayama A., Grubb J.H., Sly W.S., Banks W.A. Mannose 6-phosphate receptor-mediated transport of sulfamidase across the blood-brain barrier in the newborn mouse. Mol. Ther. 2008;16:1261–1266. doi: 10.1038/mt.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blanz J., Stroobants S., Lüllmann-Rauch R., Morelle W., Lüdemann M., D'Hooge R., Reuterwall H., Michalski J.C., Fogh J., Andersson C., Saftig P. Reversal of peripheral and central neural storage and ataxia after recombinant enzyme replacement therapy in alpha-mannosidosis mice. Hum. Mol. Genet. 2008;17:3437–3445. doi: 10.1093/hmg/ddn237. [DOI] [PubMed] [Google Scholar]
- 31.Ou L., Herzog T., Koniar B.L., Gunther R., Whitley C.B. High-dose enzyme replacement therapy in murine Hurler syndrome. Mol. Genet. Metab. 2014;111:116–122. doi: 10.1016/j.ymgme.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ou L., DeKelver R.C., Rohde M., Tom S., Radeke R., St Martin S.J., Santiago Y., Sproul S., Przybilla M.J., Koniar B.L., et al. ZFN-mediated in vivo genome editing corrects murine Hurler syndrome. Mol. Ther. 2019;27:178–187. doi: 10.1016/j.ymthe.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rozaklis T., Beard H., Hassiotis S., Garcia A.R., Tonini M., Luck A., Pan J., Lamsa J.C., Hopwood J.J., Hemsley K.M. Impact of high-dose, chemically modified sulfamidase on pathology in a murine model of MPS IIIA. Exp. Neurol. 2011;230:123–130. doi: 10.1016/j.expneurol.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 34.Wang D., El-Amouri S.S., Dai M., Kuan C.Y., Hui D.Y., Brady R.O., Pan D. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc. Natl. Acad. Sci. U S A. 2013;110:2999–3004. doi: 10.1073/pnas.1222742110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ou L., Przybilla M.J., Koniar B., Whitley C.B. RTB lectin-mediated delivery of lysosomal α-l-iduronidase mitigates disease manifestations systemically including the central nervous system. Mol. Genet. Metab. 2018;123:105–111. doi: 10.1016/j.ymgme.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf D.A., Lenander A.W., Nan Z., Belur L.R., Whitley C.B., Gupta P., Low W.C., McIvor R.S. Direct gene transfer to the CNS prevents emergence of neurologic disease in a murine model of mucopolysaccharidosis type I. Neurobiol. Dis. 2011;43:123–133. doi: 10.1016/j.nbd.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belur L.R., Temme A., Podetz-Pedersen K.M., Riedl M., Vulchanova L., Robinson N., Hanson L.R., Kozarsky K.F., Orchard P.J., Frey W.H., 2nd, et al. Intranasal adeno-associated virus mediated gene delivery and expression of human iduronidase in the central nervous system: a noninvasive and effective approach for prevention of neurologic disease in mucopolysaccharidosis type I. Hum. Gene Ther. 2017;28:576–587. doi: 10.1089/hum.2017.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belur L.R., Romero M., Lee J., Podetz-Pedersen K.M., Nan Z., Riedl M.S., Vulchanova L., Kitto K.F., Fairbanks C.A., Kozarsky K.F., et al. Comparative effectiveness of intracerebroventricular, intrathecal, and intranasal routes of AAV9 vector administration for genetic therapy of neurologic disease in murine mucopolysaccharidosis type I. Front. Mol. Neurosci. 2021;14:618360. doi: 10.3389/fnmol.2021.618360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang S.H., Morris R.G. Hippocampal-neocortical interactions in memory formation, consolidation, and reconsolidation. Annu. Rev. Psychol. 2010;61:49–79. doi: 10.1146/annurev.psych.093008.100523. c41-44. [DOI] [PubMed] [Google Scholar]
- 40.Poucet B., Save E., Lenck-Santini P.P. Sensory and memory properties of hippocampal place cells. Rev. Neurosci. 2000;11:95–111. doi: 10.1515/revneuro.2000.11.2-3.95. [DOI] [PubMed] [Google Scholar]
- 41.Onifer S.M., Low W.C. Chapter 40 Spatial memory deficit resulting from ischemia-induced damage to the hippocampus is ameliorated by intra-hippocampal transplants of fetal hippocampal neurons. Prog. Brain Res. 1990;82:359–366. doi: 10.1016/s0079-6123(08)62623-0. [DOI] [PubMed] [Google Scholar]
- 42.Belur L.R., Podetz-Pedersen K.M., Tran T.A., Mesick J.A., Singh N.M., Riedl M., Vulchanova L., Kozarsky K.F., McIvor R.S. Intravenous delivery for treatment of mucopolysaccharidosis type I: a comparison of AAV serotypes 9 and rh10. Mol. Genet. Metab. Rep. 2020;24:100604. doi: 10.1016/j.ymgmr.2020.100604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nathwani A.C., Reiss U., Tuddenham E., Chowdary P., McIntosh J., Riddell A., Pie J., Mahlangu J.N., Recht M., Shen Y.-M., et al. Adeno-associated mediated gene transfer for hemophilia B:8 Year follow up and impact of removing “empty viral particles” on safety and efficacy of gene transfer. Blood. 2018;132:491. doi: 10.1182/blood-2018-99-118334. [DOI] [Google Scholar]
- 44.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.George L.A., Monahan P.E., Eyster M.E., Sullivan S.K., Ragni M.V., Croteau S.E., Rasko J.E.J., Recht M., Samelson-Jones B.J., MacDougall A., et al. Multiyear factor VIII expression after AAV gene transfer for hemophilia A. N. Engl. J. Med. 2021;385:1961–1973. doi: 10.1056/NEJMoa2104205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helms H.C., Abbott N.J., Burek M., Cecchelli R., Couraud P.O., Deli M.A., Förster C., Galla H.J., Romero I.A., Shusta E.V., et al. In vitro models of the blood-brain barrier: an overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016;36:862–890. doi: 10.1177/0271678x16630991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Q., Su J., Liu Y., Jin X., Zhong X., Mo L., Wang Q., Deng H., Yang Y. In vivo PCSK9 gene editing using an all-in-one self-cleavage AAV-CRISPR system. Mol. Ther. Methods Clin. Dev. 2021;20:652–659. doi: 10.1016/j.omtm.2021.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lock M., Alvira M.R., Chen S.J., Wilson J.M. Absolute determination of single-stranded and self-complementary adeno-associated viral vector genome titers by droplet digital PCR. Hum. Gene Ther. Methods. 2014;25:115–125. doi: 10.1089/hgtb.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Y., Wang L., Bell P., McMenamin D., He Z., White J., Yu H., Xu C., Morizono H., Musunuru K., et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barnes C.A. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.