

Abstract
Per- and polyfluoroalkyl substances (PFASs) are widely used anthropogenic chemicals. For environmental and toxicological analysis, it is important to understand the stability of PFASs, including novel per- and polyfluoroalkyl ether acids (PFEAs), in commonly used solvents. In this study, we investigated the effects of PFAS characteristics, solvent type, water-to-organic solvent ratio, and temperature on the stability of 21 PFASs including 18 PFEAs. None of the studied PFASs showed measurable degradation in deionized water, methanol, or isopropyl alcohol over 30 days; however, nine PFEAs degraded in the polar aprotic solvents acetonitrile, acetone, and dimethyl sulfoxide (DMSO). PFEA degradation followed first-order kinetics, and first-order rate constants increased with increasing temperature and with decreasing water-to-organic solvent ratio. Monoethers with a carboxylic acid functional group adjacent to a tertiary carbon (>CF-COOH) degraded more rapidly than multiethers, in which the carboxylic acid moiety was adjacent to repeating -CF2O- groups. In contrast, monoethers with a carboxylic acid moiety adjacent to a secondary carbon (-CF2-COOH), were stable in all tested solvents. Using high resolution mass spectrometry, we determined that PFEAs with a >CF-COOH group were stoichiometrically decarboxylated in aprotic solvents and formed products with a >CFH group; e.g., hexafluoropropylene oxide-dimer acid (HFPO-DA or GenX), HFPO-trimer acid, and HFPOtetramer acid were stoichiometrically converted to Fluoroethers E-1, E-2, and E-3, respectively. PFEA degradation results highlight the importance of solvent choice when preparing dosing solutions and performing extractions for environmental and toxicological assessments of PFEAs.
Keywords: PFAS, fluoroethers, GenX, acetonitrile, acetone, dimethyl sulfoxide, degradation, high-resolution mass spectrometry
Graphical Abstract
INTRODUCTION
Per- and polyfluoroalkyl substances (PFASs) are a class of anthropogenic chemicals produced and used since the 1940s.1 The unique stability and surface tension lowering properties of PFASs have led to a variety of applications, including their use in firefighting foams, metal plating, paper coatings, stain repellents, and fluoropolymer production.2–9 To date, almost 5,000 PFAS-related CAS numbers have been registered.10,11 Among the PFASs, perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) are the most widely studied. Concerns about their environmental persistence, bioaccumulation potential, toxicity, mobility, and widespread occurrence have led to the phase out of the production of PFOS, PFOA, their precursors, and other long-chain PFAAs starting in the early 2000s.11–14 In response, fluorochemical manufacturers have transitioned to producing fluorinated alternatives, including shorter-chain homologues [e.g. perfluorohexanoic acid (PFHxA)] of long-chain PFAAs and their precursors, as well as per- and polyfluoroalkyl ether acids (PFEAs).13 For example, the ammonium salt of hexafluoropropylene oxide-dimer acid (HFPO-DA) with the trade name “GenX” has replaced the ammonium salt of PFOA as a processing aid in fluoropolymer production since about 2010, and HFPO-DA has been detected in river water downstream of fluorochemical manufacturing sites.2,15–17 In North Carolina, HFPO-DA is also generated as a by-product during the production of fluoropolymer building blocks and has been discharged into the Cape Fear River (CFR).5,18 Sun et al. (2016)5 reported the average HFPO-DA concentration was 631 ng/L (range: 55–4,560 ng/L) in CFR water samples collected in 2013. Besides HFPO-DA, several other manufacturing by-products, likewise generated during the production of fluoropolymers and their building blocks, were discharged into the CFR. These manufacturing by-products can be broadly classified as (1) perfluoroalkyl monoether carboxylic acids (mono-ether PFECAs), (2) perfluoroalkyl multi-ether carboxylic acids (multi-ether PFECAs), and (3) polyfluoroalkyl ether acids (Table 1, Table S1, Figure S1).5,18–21 A recent study reported that among these manufacturing by-products, six PFEAs (i.e. Nafion byproduct 2, NVHOS, HydroEVE, PFO3OA, PFO4DA, and PFO5DoA) were commonly detected in blood serum of residents living in downstream communities that rely on the CFR as a source of drinking water.22
Table 1.
Examples of per- and polyfluoroalky ether acids (PFEAs)
| Compound | Formula | CAS # | Molecular structure |
|---|---|---|---|
| Perfluoroalkyl mono-ether carboxylic acids (mono-ether PFECas) | |||
|
| |||
| Perfluoro-2-methoxyacetic acid (PFMOAA) | C3HF5O3 | 674-13-5 |
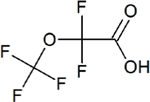
|
| Hexafluoropropylene oxide-dimer acid (HFPO-DA) = Perfluoro-2-propoxypropanoic acid (PFPrOPrA) | C6HF11O3 | 13252-13-6 |
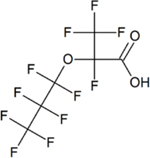
|
|
| |||
| Perfluoroalkyl multi-ether carboxylic acids (multi-ether PFECas) | |||
|
| |||
| Perfluoro(3,5-dioxahexanoic) acid (PFO2HxA) | C4HF7O4 | 39492-88-1 |
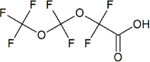
|
| Perfluoro(3,5,7,9-tetraoxadecanoic) acid (PFO4DA) | C6HF11O6 | 39492-90-5 |
|
| Hexafluoropropylene oxide-trimer acid (HFPO-TA) | C9HF17O4 | 13252-14-7 |
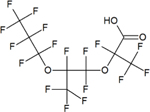
|
| Hexafluoropropylene oxide-tetramer acid (HFPO-TeA) | C12HF23O5 | 65294-16-8 |
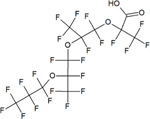
|
|
| |||
| Polyfluoroalkyl ether acids | |||
|
| |||
| Ethanesulfonic acid, 2-[1-[difluoro(1,2,2,2- tetrafluoroethoxy)methyl] −1,2,2,2- tetrafluoroethoxy] −1,1,2,2-tetrafluoro- (Nafion by-product 2) | C7H2F14SO5 | 749836-20-2 |
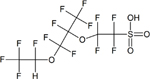
|
| 1,1,2,2-tetrafluoro-2-(1,2,2,2-tetrafluoro- ethoxy)ethane sulfonate (NVHOS) | C4H2F8SO4 | 801209-99-4 |
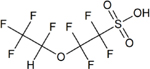
|
Homologues of HFPO-DA containing additional hexafluoropropylene units are also associated with fluorochemical production.2 For example, the trimer acid of HFPO, hexafluoropropylene oxide-trimer acid (HFPO-TA, Table 1), is used as a processing aid in fluoropolymer production and is an important building block in the synthesis of other fluorinated products.2,15 The presence of HFPO-TA was reported in the effluent of a fluoropolymer production plant in China and the concentration ranged from 5,200 to 68,500 ng/L, suggesting considerable amounts of this novel compound being produced in China.2,15 Also, the tetramer acid of hexafluoropropylene oxide (HFPO-TeA, Table 1) has been detected in a river near a fluoropolymer manufacturing facility, which was ascribed to its active use as a polymerization aid or its presence as an impurity therein.23,24
The discovery of novel PFASs requires development of compound-specific methods to conduct quantitative measurements of prevalence and assess the risk of toxicity. For these analyses, it is important to understand the stability of PFASs, including novel PFEAs, in organic solvents commonly used for stock standard solutions in analytical chemistry, during sample preparation (e.g., solvent extraction of soils), or for dosing solutions in toxicological studies. PFASs have long been known to be stable under a variety of conditions. For example, a previous study observed perfluoroalkyl carboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs) with chain length ≤10 to be stable (recovery = 79–110%) in water over a period of 28 days.25 Methanol or basic methanol is commonly used when preparing PFAS stock solutions for environmental analysis.26–29 However, PFOA and other PFCAs can form methyl esters in methanol if no base is added.30 In toxicological studies, dimethyl sulfoxide (DMSO) is often used as a carrier solvent due to its low toxicity and permeation across biological barriers.31–33 Recently, a few studies reported the apparent loss of HFPO-DA in DMSO in toxicological studies,33–36 and Liberatore et al.37 found that HFPO-DA degraded to an H-substituted ether derivative, Fluoroether E-1 (heptafluoropropyl 1,2,2,2-tetrafluoroethyl ether), in polar, aprotic solvents such as acetonitrile, acetone, and DMSO. However, the stability of other PFEAs in commonly used solvents that are relevant to environmental and toxicological analysis remains unknown.
In this study, we determined the stability of 21 PFASs including 18 PFEAs in solvents including deionized water, methanol, acetonitrile (ACN), acetone, DMSO, and isopropyl alcohol (IPA). Specific objectives were to (1) investigate the effects of PFAS characteristics, solvent type, water-to-organic solvent ratio, and temperature on the stability of PFASs, (2) determine reaction rate constants associated with the degradation of reactive PFASs, and (3) identify degradation products for reactive PFASs using both liquid chromatography-high resolution mass spectrometry (LCHRMS) and gas chromatography-high resolution mass spectrometry (GC-HRMS).
MATERIALS AND METHODS
Materials
Twenty-one PFASs in four classes (i.e., 1 PFCA, 1 PFSA, 18 PFEAs and 1 fluorotelomer sulfonate) were studied (Table S1 and Figure S1). Analytical standards of PFASs were obtained from Wellington Laboratories (Guelph, ON, Canada), Fluoryx Labs (Carson City, NV), Cambridge Isotope Laboratories (Tewksbury, MA), The Chemours Company (Wilmington, DE), and SynQuest Laboratories (Alachua, FL) (see Table S1 for details). Isotopically labeled internal standards were purchased from Wellington Laboratories (Guelph, ON, Canada). Analytical standards of 1,2,2,2-tetrafluoroethyl trifluoromethyl ether (HFE 227), heptafluoropropyl 1,2,2,2tetrafluoroethyl ether (Fluoroether E-1), 2H-perfluoro-5-methyl-3,6-dioxanonane (Fluoroether E2), and 2H-perfluoro-5,8-dimethyl-3,6,9-trioxadodecane (Fluoroether E-3) were obtained from SynQuest Laboratories (Alachua, FL) (Table S2). All other chemicals and solvents were purchased from Fisher Scientific (Hampton, NH) and Sigma-Aldrich (St. Louis, MO).
PFAS stability test
Analytical standards of PFASs were either used as received in methanol or deionized water (Table S1), or they were diluted with methanol or deionized water to create PFAS stock solutions. An aliquot of a 50 ng/μL stock solution containing an individual PFAS or PFAS mixture (no differences were observed in experiments conducted with individual PFASs and PFAS mixtures) was added to polypropylene (PP) tubes and evaporated to dryness in a fume hood at room temperature (20.2°C) to eliminate effects of methanol or water on degradation experiments. The PFAS residue was subsequently dissolved in 10 mL of solvent [i.e. deionized water, methanol, acetonitrile (ACN), acetone, DMSO, or isopropyl alcohol (IPA)] at room temperature to yield a starting concentration of ~50 μg/L. For experiments involving various water-to-organic solvent ratios, solvents of different compositions (100% organic solvent, 90:10% (v/v) or 80:20% (v/v) organic solvent:water) were added. Then, PP tubes were vortexed and mixed to fully dissolve PFASs. Samples (100 μL) were taken from the PP tubes at times selected based on results of preliminary experiments to obtain sufficient data points with measurable concentrations and to determine degradation rate constants. Samples were diluted into 10 mL deionized water in PP tubes and stored at room temperature until analysis. To determine the effect of temperature on PFAS stability, experiments were conducted in constant temperature rooms at three temperatures [cold (3.4°C), room (20.2°C), and hot (32.4°C)]. Upon evaporation of methanol from the stock solution, PFAS residues were dissolved in solvents that had been previously brought to the temperature of interest. Temperature was recorded at least four times during the experiment, and the average temperature was reported. Experiments were not conducted at 3.4°C in DMSO because its melting point is 19°C.
Liquid chromatography-mass spectrometry (LC-MS) analysis
LC-MS analysis was performed using an Agilent 1290 Infinity II HPLC coupled to an Agilent 6545 quadrupole time-of-flight (QTOF) mass spectrometer. Details of the analytical method are described in the Supporting Information (Text S1). Authentic standards were used to determine all PFAS concentrations. When possible, quantification was conducted with an isotope dilution approach, in which the analyte response was normalized to that of an isotopically labeled analog.19,21 For other PFASs, the analyte response was normalized to that of an isotopically labeled PFAS with an LC retention time similar to that of the analyte (Table S1). Details of the PFAS quantitation and quality assurance/quality control (QA/QC) are provided in the Supporting Information (Texts S2–S3).
Degradation product identification
To identify and quantify the degradation products of HFPO-DA, HFPO-TA, and HFPO-TeA, an aliquot of a 5 mg/mL stock solution containing an individual HFPO acid homologue in methanol was added to solvent (ACN, acetone, or DMSO) in a 25-mL volumetric flask, yielding a starting concentration of ~5 mg/L. The volumetric flask was inverted twice to fully mix the added PFAS, and the first sample (t0, analyzed by LC-MS) was taken immediately after mixing. Then, an aliquot from the volumetric flask was transferred to a 20-mL headspace-free amber glass vial and stored at room temperature (20.2°C) for 5 days (d) to achieve complete degradation. After 5 d, samples were taken and analyzed using a headspace gas chromatography-mass spectrometry (GC-MS) method. Details of the headspace GC-MS method are provided in the Supporting Information (Text S4–S5).
To identify the degradation products of PMPA, PEPA, and multi-ether PFECAs (PFO2HxA, PFO3OA, PFO4DA, and PFO5DoA), an aliquot of a 0.1 mg/mL stock solution containing an individual multi-ether PFECA was added to solvent (ACN, acetone, or DMSO) in a 2-mL glass vial, yielding a starting concentration of ~1 mg/L. Samples were stored at room temperature (20.2°C) for 5 d for PMPA and PEPA, and ~60 d for four multi-ether PFECAs to achieve complete or significant degradation. Subsequently, samples were analyzed by GC-HRMS as described in the Supporting Information (Text S5).
RESULTS AND DISCUSSION
Effect of solvent type on PFAS stability
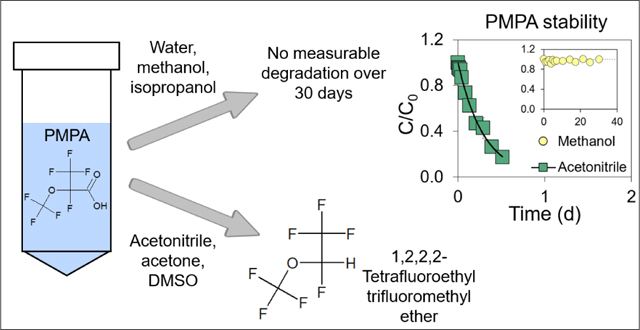
To assess the effect of solvent type on the stability of 21 PFASs (i.e., 1 PFCA, 1 PFSA, 18 PFEAs and 1 fluorotelomer sulfonate as shown in Table S1 and Figure S1), batch experiments were conducted in deionized water and five organic solvents [methanol, acetonitrile (ACN), acetone, DMSO, and isopropyl alcohol (IPA)] at room temperature (20.2°C). None of the studied PFASs showed measurable degradation in deionized water after ~30 d (Table S3). Percent recoveries for PFHxA and PFHxS in deionized water after ~30 d were 93 and 91%, respectively, in agreement with a previous study for PFCAs and PFSAs with chain length ≤ 10 (recovery = 79–110% in water in 28 d).25 In methanol and IPA, PFHxA and PFHxS were also stable, with recoveries of 98–100% in methanol and 99% in IPA after ~30 d (Tables S4–S5). The latter results are also consistent with a previous study indicating no measurable degradation of PFCAs and PFSAs with chain length ≤ 8 in solvents composed of water and methanol.9 Furthermore, neither 6:2 FtS nor the 18 studied PFEAs showed measurable degradation in methanol and IPA after ~30 d (Tables S4–S5).
In the polar aprotic solvents ACN, acetone, and DMSO, all of the branched mono-ether PFECAs (PMPA, PEPA, HFPO-DA, HFPO-TA, HFPO-TeA) and all of the multi-ether PFECAs (PFO2HxA, PFO3OA, PFO4DA, PFO5DoA) degraded (Figure 1, Figure S3, Tables S6–S8). Branched mono-ether PFECAs degraded most rapidly in acetone, followed by ACN and DMSO (Figure 1 and Figure S3). For example, after 1 h (0.04 d), percent recoveries of HFPO-DA were 25, 74, and 88% in acetone, ACN, and DMSO, respectively (Figure 1a). Our observations for HFPO-DA are consistent with a previous study of HFPO-DA degradation in polar aprotic solvents, with half-lives on the order of hours.37 Another study used computational modeling and found decarboxylation of trichloroacetic acid was likewise solvent-dependent, being most favored in polar aprotic solvents such as DMSO.38 The degradation of multi-ether PFECAs happened most rapidly in acetone, but degradation rates were slower than those of branched mono-ether PFECAs. For example, after 21 d, PFO4DA recoveries were 22, 77, and 77% in acetone, ACN, and DMSO, respectively (Figure 1b).
Figure 1.

Stability of (a) HFPO-DA and (b) PFO4DA in water, methanol, isopropyl alcohol (IPA), dimethyl sulfoxide (DMSO), acetonitrile (ACN), and acetone at room temperature (20.2°C). The inset in panel (a) shows the stability of HFPO-DA in water, methanol, and IPA. Curves describe results of first-order kinetic models, and the corresponding rate constants are given in Table 2. To facilitate comparability, all analyte concentrations were normalized to the initial concentration (Ct/C0). Error bars represent standard deviations of duplicate measurements. See Figure S1 for compound structures.
Effect of PFAS characteristics on PFAS stability
PFHxA and PFHxS did not display measurable degradation in the studied solvents over ~30 d at room temperature (20.2°C) (Tables S3–S8). The same was also observed for 6:2 fluorotelomer sulfonate (6:2 FtS) and all studied polyfluoroalkyl ether acids including Nafion by-product 2, NVHOS, HydroEVE, ADONA, and F-53B. For example, percent recoveries of polyfluoroalkyl ether acids were 92–104% in deionized water, 96–103% in methanol, 96–106% in ACN, 95–105% in acetone, 93–104% in DMSO, and 102–108% in IPA in ~30 d (Tables S3–S8).
Linear mono-ether PFECAs including PFMOAA, PFMOPrA, and PFMOBA, in which the carboxylic acid moiety was adjacent to a secondary carbon (-CF2-COOH), also did not exhibit measurable degradation in any of the studied solvents over ~30 d (Tables S3–S8). In contrast, branched mono-ether PFECAs containing a carboxylic acid functional group adjacent to a tertiary carbon (>CF-COOH), including PMPA (branched isomer of PFMOPrA), PEPA (branched isomer of PFMOBA), and HFPO-DA, degraded rapidly in ACN, acetone, and DMSO (Figure 2, Figures S4–S5, Tables S6–S8). For instance, in ACN, percent recoveries of PMPA, PEPA, and HFPO-DA declined to 47, 35 and 26%, respectively, after ~0.2 d (Figure 2a).
Figure 2.

Stability of perfluoroalkyl ether carboxylic acids (PFECAs): (a) mono-ether PFECAs, (b) HFPO acid homologues, and (c) multi-ether PFECAs in acetonitrile (ACN) at room temperature (20.2°C). Curves describe results of first-order kinetic models, and the corresponding rate constants are given in Table 2. HFPO-DA is plotted in both panels (a) and (b) for comparison. To facilitate comparability, all analyte concentrations were normalized to the initial concentration (Ct/C0). Error bars represent standard deviations of duplicate measurements. See Figure S1 for compound structures.
Other HFPO acid homologues [i.e. GenX (ammonium salt of HFPO-DA), HFPO-TA, and HFPOTeA] also degraded rapidly in ACN, acetone, and DMSO (Figure 2b, Figures S4–S5). For example, percent recoveries of HFPO-DA, GenX, HFPO-TA, and HFPO-TeA in ACN were 26, 28, 53 and 46%, respectively, after ~0.2 d. Liberatore et al.37 proposed that HFPO-DA degraded in aprotic solvents via a solvent-mediated proton transfer mechanism that was facilitated by the H-bond accepting nature of the polar aprotic solvents ACN, acetone, and DMSO. In our study, rates of HFPO-DA and GenX decomposition were practically identical (Figure 2b), suggesting that protonation of the polar, aprotic solvents was possible regardless of whether the proton was associated with the -COOH group of HFPO-DA or with the NH4+ cation of GenX (see Figure S1 for HFPO-DA and GenX structures).
Multi-ether PFECAs, in which the carboxylic acid was adjacent to repeating -CF2O- groups (i.e. PFO2HxA, PFO3OA, PFO4DA, and PFO5DoA), degraded at a rate that was ~2 orders of magnitude slower than that of branched mono-ether PFECAs (Figure 2c). Furthermore, there was no correlation between degradation rate and chain length for multi-ether PFECAs. For example, after ~30 d, percent recoveries of PFO2HxA, PFO3OA, PFO4DA, and PFO5DoA dropped to 67, 66, 60, and 67%, respectively, in ACN (Figure 2c).
Kinetics and reaction order
To determine the reaction order for the degradation of branched mono-ether PFECAs (i.e. PMPA, PEPA, HFPO-DA, and GenX) and multi-ether PFECAs (i.e. PFO2HxA, PFO3OA, PFO4DA, PFO5DoA, HFPO-TA, and HFPO-TeA) in ACN, acetone, and DMSO, we performed kinetic experiments with HFPO-DA at different initial concentrations (10, 50, and 100 μg/L) in ACN, acetone, and DMSO (Figure S6), and experiments at initial concentrations of 10 and 50 μg/L were conducted in duplicate. Degradation rates at the tested initial concentrations were similar and did not exhibit consistent changes with initial concentration (Figure S6). Furthermore, degradation rates were linear when log-transformed concentrations were plotted as a function of time. Thus, the degradation of branched mono-ether PFECAs and multi-ether PFECAs in ACN, acetone, and DMSO can be expressed by first-order kinetics (Text S6). First-order rate constants (k) for the degradation of mono- and multi-ether PFECAs in ACN, acetone, and DMSO are summarized in Table 2 and Table S9 along with 95% confidence intervals. For example, k values describing the degradation of PMPA were 3.03, 19.2, and 2.00 d−1 in ACN, acetone, and DMSO, respectively (Table 2). These k values translate into PMPA half-lives that range from 52 min in acetone to 8.3 hours in DMSO. For branched mono-ether PFECAs, k values increased with increasing chain length; e.g. k values for HFPO-DA were approximately twice those of PMPA in acetone and DMSO. For the HFPO acid homologues GenX, HFPO-TA, and HFPO-TeA, k values were in the same order of magnitude as those for HFPO-DA. For example, in acetone, the first-order rate constants were 47.9, 27.8, and 32.1 d−1 for GenX, HFPO-TA, and HFPO-TeA, respectively (compared with k = 45.4 d−1 for HFPO-DA). Thus, half-lives of the tested HFPO acid homologues range in acetone from about 21 min for GenX to 36 min for HFPO-TA. For multi-ether PFECAs, first-order rate constants were several orders of magnitude lower than those for branched monoether PFECAs, i.e. 0.0128–0.0170 d−1 in ACN, 0.0578–0.106 d−1 in acetone, and 0.0139–0.0195 d−1 in DMSO (Table 2). Corresponding half-lives of multi-ether PFECAs were 41–54 d in ACN, 6.512 d in acetone, and 36–50 d in DMSO. No correlations were found between k values and the chain length of multi-ether PFECAs (Table 2).
Table 2.
First-order rate constants (k) describing the degradation of PFECAs in 100% acetonitrile (ACN), acetone, and dimethyl sulfoxide (DMSO) at room temperature (20.2°C)
| Compound | ACN | Acetone | DMSO | |||
|---|---|---|---|---|---|---|
| k (d−1) | R2 | k (d−1) | R2 | k (d−1) | R2 | |
| PFMOAA | No measurable degradation in ~30 d | No measurable degradation in ~30 d | No measurable degradation in ~30 d | |||
| PFMOPrA | No measurable degradation in ~30 d | No measurable degradation in ~30 d | No measurable degradation in ~30 d | |||
| PMPA | 3.03 a,b 0.906 (2.74, 3.31) | 19.2 b 0.909 (17.1, 21.2) | 2.00 b 0.826 (1.73, 2.27) | |||
| PFMOBA | No measurable degradation in ~30 d | No measurable degradation in ~30 d | No measurable degradation in ~30 d | |||
| PEPA | 4.75 b (4.41, 5.10) | 0.945 | 27.0 b (24.7, 29.3) | 0.943 | 2.63 b (2.28, 2.97) | 0.838 |
| HFPO-DA (PFPrOPrA) | 4.96 b (4.39, 5.52) | 0.882 | 45.4 b (43.5, 47.3) | 0.988 | 3.92 b (3.34, 4.51) | 0.819 |
| GenX | 5.86 (5.50, 6.22) | 0.997 | 47.9 (42.3, 53.5) | 0.993 | 4.64 (4.18, 5.11) | 0.992 |
| PFO2HxA | 0.0136 (0.0122, 0.0151) | 0.972 | 0.0578 (0.0547, 0.0608) | 0.993 | 0.0168 (0.0153, 0.0183) | 0.980 |
| PFO3OA | 0.0128 (0.0114, 0.0142) | 0.970 | 0.0743 (0.0680, 0.0806) | 0.982 | 0.0195 (0.0176, 0.0213) | 0.978 |
| PFO4DA | 0.0170 (0.0152, 0.0188) | 0.973 | 0.106 (0.0946, 0.116) | 0.979 | 0.0160 (0.0143, 0.0178) | 0.971 |
| PFO5DoA | 0.0129 (0.0116, 0.0143) | 0.972 | 0.0732 (0.0691, 0.0773) | 0.994 | 0.0139 (0.0124, 0.0154) | 0.970 |
| HFPO-TA | 3.39 (3.05, 3.73) | 0.990 | 27.8 (23.4, 32.2) | 0.987 | 3.52 (3.17, 3.88) | 0.992 |
| HFPO-TeA | 3.71 (3.21, 4.22) | 0.998 | 32.1 (22.0, 42.1) | 0.931 | 3.68 (2.91, 4.45) | 0.987 |
Experimental results were analyzed using the regression data analysis tool in Microsoft Excel and the average kinetics constant was reported. Values in parentheses represent the lower and upper limit of 95% confidence interval.
Experiment was conducted in replicates starting at different initial concentrations (10, 50 and 100 μg/L).
Effect of water-to-organic solvent ratio on PFAS stability
To assess the impact of water-to-organic solvent ratio on PFAS stability, we conducted experiments with branched mono-ether PFECAs (PMPA, PEPA, and HFPO-DA) and multi-ether PFECAs (PFO2HxA, PFO3OA, PFO4DA, and PFO5DoA) in ACN, acetone, and DMSO with different compositions (100% organic solvent, 90:10% (v/v), and 80:20% (v/v) organic solvent:water). Increasing water-to-organic solvent ratio led to a decrease in the degradation rate of branched mono-ether PFECAs in ACN, acetone, and DMSO (Figure 3, Figures S7–S8, Table S10). For instance, in 100% ACN, k for HFPO-DA was 4.96 d−1; in 90% ACN+10% water, it was 0.0380 d−1; and in 80% ACN+20% water, HFPO-DA degradation was negligible after ~30 d (Figure 3a, Table S10). Thus, the half-life of HFPO-DA increased from 3.4 h in 100% ACN to 18 d in 90:10% (v/v) ACN:water. Results obtained for HFPO-DA, PEPA, and PMPA with 80% ACN+20% water suggest that ACN can be used as a solvent for sample preparation (e.g. extraction of food samples, preparation of blood serum samples) as long as the water content of the sample exceeds 20%. However, degradation of HFPO-DA, PEPA, and PMPA would remain a concern for acetone and DMSO even when the water content is 20%.
Figure 3.

Stability of HFPO-DA in (a) acetonitrile (ACN), (b) acetone, and (c) dimethyl sulfoxide (DMSO) with different water-to-organic solvent ratios at room temperature (20.2°C). Curves describe results of first-order kinetic models, and the corresponding rate constants are given in Table 2 and Table S10. The inset in each panel highlights data for HFPO-DA in 100% organic solvent. To facilitate comparability, all analyte concentrations were normalized to the initial concentration (Ct/C0). Error bars represent standard deviations of duplicate measurements. See Figure S1 for compound structures.
Results of degradation experiments conducted with multi-ether PFECAs (PFO2HxA, PFO3OA, PFO4DA, and PFO5DoA) in ACN, acetone, and DMSO at different water-to-organic solvent ratios are depicted in Figure S9. After ~30 d, multi-ether PFECA recoveries ranged from 60–67% in 100% ACN, 0–19% in 100% acetone, and 52–63% in 100% DMSO. However, adding 10 or 20% water to solvents led to no measurable degradation of multi-ether PFECAs after ~30 d (Figure S9). Overall, increasing the water percentage in ACN, acetone and DMSO led to substantial decreases in PFECA degradation rates. Liberatore et al.37 suggested that protic solvents, such as water, stabilize the carboxylic acid moiety of PFECAs via hydrogen-bonding, and observing increased PFECA stability with increasing water content in ACN, acetone, and DMSO is consistent with the suggested stabilization mechanism.
Effect of temperature on PFAS stability
Experiments were conducted to evaluate the effect of temperature [cold (3.4°C), room (20.2°C), and hot (32.4°C)] on the stability of branched mono-ether PFECA (i.e. PMPA, PEPA, and HFPODA) in ACN, acetone, and DMSO. Calculated first-order rate constants are shown in Table S11. A positive correlation between first-order rate constants and temperature (from 3.4 to 32.4°C) was observed for PMPA, PEPA, and HFPO-DA in ACN, acetone, and DMSO. For instance, first-order rate constants describing the degradation of HFPO-DA in ACN were 0.383, 4.96, and 31.8 d−1 at 3.4, 20.2, and 32.4°C, respectively (Table S11). In DMSO, experiments were not conducted at 3.4°C because the melting point of DMSO is 19°C. Compared with HFPO-DA, similar patterns were observed for PMPA and PEPA; i.e., k values describing the degradation rates of PMPA and PEPA in ACN, acetone, and DMSO increased with increasing temperature (Table S11).
The temperature-dependence of first-order rate constants describing the degradation of branched mono-ether PFECAs in ACN, acetone, and DMSO was described by the Arrhenius equation (Text S7). Arrhenius plots for PMPA, PEPA, and HFPO-DA in ACN, acetone, and DMSO are depicted in Figure 4 and Figures S10–S11, and the corresponding activation energies and pre-exponential factors are shown in Table S12. The activation energies for PMPA, PEPA, and HFPO-DA degradation in ACN were similar at 108.8, 108.5, and 106.7 kJ/mol, respectively, suggesting chain length had no effect for branched mono-ether PFECAs (Figure 4a, Table S12). For HFPO-DA, activation energies were 106.7, 112.0, and 147.1 kJ/mol in ACN, acetone, and DMSO, respectively, suggesting solvent effects were small (Figure 4b, Table S12).
Figure 4.

(a) Arrhenius plots describing the temperature-dependence of first-order rate constants of HFPO-DA, PEPA, and PMPA in acetonitrile (ACN) at three temperatures [cold (3.4°C), room (20.2°C), and hot (32.4°C)]; and (b) the Arrhenius plot of the first-order rate constants of HFPODA in ACN, acetone, and dimethyl sulfoxide (DMSO). The first-order rate constants at different temperatures are given in Table S11. Temperature was recorded at least four times during the test and average temperature was reported. Experiment was not conducted at 3.4°C in DMSO due to the high melting point of DMSO (19°C). See Figure S1 for compound structures.
Degradation product identification
We observed the degradation of branched mono-ether PFECAs (PMPA, PEPA, and HFPO-DA), HFPO acid homologues (HFPO-TA, and HFPO-TeA), and multi-ether PFECAs (PFO2HxA, PFO3OA, PFO4DA, and PFO5DoA) in polar, aprotic solvents (i.e. ACN, acetone, and DMSO). To identify degradation products, reactive PFASs were contacted with polar aprotic solvents for 5 days, after which time samples were analyzed by GC-Orbitrap HRMS (Figure 5, Figures S12–S16). When PMPA degraded in acetone, the GC-Orbitrap total ion chromatogram (TIC) for the sample collected after 5 days exhibited a new chromatographic peak with a retention time of 1.73 min, consistent with the retention time for 1,2,2,2-tetrafluoroethyl trifluoromethyl ether (HFE 227) (Figure 5). Also, the most abundant ion in the mass spectrum (m/z = 166.99269) for the PMPA degradation product in acetone matched that for the HFE 227 standard (m/z = 166.99259), which was identified as the [M-F]+ ion of HFE 227 (Figure 5). Other prominent ions in the mass spectrum for the PMPA degradation product in acetone included m/z = 68.99472 and 101.00096. Annotation of the spectra with predicted structures based on exact mass and anticipated components reveals the likely chemical identity of these fragments as CF3 and C2HF4; and each of these formulas is a subset of the molecular precursor (HFE 227, C3HOF7). Retention times and abundance ratios were consistent between the observed PMPA degradation product and the HFE 227 standard, thus demonstrating that PMPA transformed to HFE 227 in acetone. Similarly, in ACN and DMSO, TIC and mass spectra show PMPA transformed to HFE 227 (Figure S15). The GC-Orbitrap TIC and mass spectrum of the PEPA degradation product in acetone are shown in Figure S16. We hypothesized PEPA transformed in aprotic solvents via decarboxylation, and the proposed molecular structure of the degradation product is depicted in Figure S16.
Figure 5.

(A) GC-Orbitrap total ion chromatogram (TIC) comparisons of 1,2,2,2-Tetrafluoroethyl trifluoromethyl ether standard (top) and degradation product of PMPA in acetone in 5 days in black color and acetone blank in gray color (bottom); (B) mass spectra of 1,2,2,2-Tetrafluoroethyl trifluoromethyl ether standard (top) and degradation product of PMPA in acetone in 5 days (bottom); (C) mass spectra list of 1,2,2,2-Tetrafluoroethyl trifluoromethyl ether standard (top) and degradation product of PMPA in acetone in 5 days (bottom).
We observed the formation of Fluoroethers E-1, E-2, and E-3 from the degradation of HFPO-DA, HFPO-TA, and HFPO-TeA, respectively, in polar, aprotic solvents including ACN, acetone, and DMSO (TICs and mass spectra are shown in Figures S12–S14). For example, the TIC exhibits chromatographic peaks with a retention time of 1.90 min for the degradation product of HFPODA in acetone, consistent with the retention time for Fluoroether E-1 (Figure S12). The most abundant feature for HFPO-DA degradation product in acetone is 266.98624, which was identified as the [M-F]+ ion of Fluoroether E-1. Similarly, TICs and mass spectra show HFPO-TA and HFPO-TeA transformed to Fluoroethers E-2 and E-3, respectively, in ACN, acetone, and DMSO (Figures S13–S14). A GC-triple quadrupole MS was used to quantify concentrations of Fluoroethers E-1, E-2, and E-3 resulting from the degradation of HFPO-DA, HFPO-TA, and HFPO-TeA, respectively, in aprotic solvents. The rapid disappearance of HFPO-DA was accompanied by the formation of Fluoroether E-1, with molar yields of 129% ± 14% in ACN, 120% ± 26% in acetone, and 92% ± 3% in DMSO in 5 d (Table S13). Similarly, the degradation of HFPO-TA and HFPO-TeA was accompanied by the stoichiometric formation of Fluoroether E-2 and E-3, respectively. For example, molar yields of Fluoroether E-2 from the degradation of HFPO-TA were 90% ± 13% in ACN, 109% ± 16% in acetone, and 84% ± 16% in DMSO (Table S13).
To gain insights into the degradation mechanism, we conducted experiments with (1) HFPO-DA, (2) HFPO-TA, (3) HFPO-TeA, and (4) a mixture of Fluoroethers E-1, E-2, and E-3 (control experiment) in fully deuterated acetone (detailed in Text S4), and the abundance of [M-F]+ and [M-F+1]+ ions for Fluoroethers E-1, E-2, and E-3 was determined (Table S14). Compared to the control experiments conducted with Fluoroethers E-1, E-2, and E-3, for which percentages of the heavy isotopologue [M-F+1]+ were consistent with the natural abundance (1.109%) of 13C (Table S14), percentages of the heavy isotopologues were substantially higher when Fluoroethers E-1, E2, and E-3 were produced from the degradation of HFPO-DA, HFPO-TA, and HFPO-TeA in deuterated acetone (Table S14). As proposed by Liberatore et al. (2020),37 the degradation of HFPO-DA likely involves the formation of a carbanion intermediate, and this carbanion has nucleophilic properties.39 Formation of the carbanion may be a result of proton transfer from the carboxylic acid group of HFPO-DA to the ketone oxygen of acetone and subsequent CO2 elimination from deprotonated HFPO-DA,37 or it may result from an addition/elimination reaction analogous to the haloform reaction (Figure S17).40 The latter may involve the nucleophilic addition of the aprotic polar solvent (e.g. acetone) and/or hydroxide (if trace levels of water were present, which is likely, e.g. from humidity in ambient air), followed by the elimination of a carbocation in the case of acetone or water and CO2 in the case of hydroxide (Figure S17). Finally, the carbanion abstracts H+ or D+ from the protonated solvent,37 the carbocation, and/or water (Figure S17). If hydroxide served as a nucleophile, both light and heavy hydroxide (OH-, OD-) may have been present as a result of H/D interchanges41 between trace levels of H2O and deuterated acetone, such that both light and heavy water could have served as a source of H+/D+ for the formation of Fluoroether E-1. Alternatively, formation of heavy Fluoroether E-1 suggests that the carbanion not only abstracts H associated with the oxygen of protonated acetone37 or the carbocation (Figure S17), but also deuterium from deuterated methyl groups followed by rearrangement to form a -CD2H group (Figure S17). Similar reactions can explain the formation of Fluoroethers E-2 and E-3 from HFPO-TA and HFPO-TeA, respectively. Based on averages of triplicate experiments, the prevalence of heavy isotopologues increased with increasing chain length of the degradation products (Table S14). Because of higher variability in the data, only the increase from Fluoroether E-1 to Fluoroether E-3 was statistically significant and was likely due to isotopologues containing 13C.
For multi-ether PFECAs (PFO2HxA, PFO3OA, PFO4DA, and PFO5DoA), we were unable to identify degradation products in aprotic solvents. We analyzed total ion chromatograms and extracted ion chromatograms for fragments anticipated for the possible breakdown structures and typical to fluorinated compounds (i.e. m/z=68.99466, CF3+) obtained on high-resolution and lowresolution GC-MS and the LC-HRMS instruments. However, no byproducts were detected, possibly because of low responses and/or incompatibility with analytical methods as described in detail in Text S5.
Supplementary Material
SYNOPSIS.
Nine of 18 evaluated per- and polyfluoroalkyl ether acids degraded in three organic solvents commonly used in environmental and toxicological studies.
Implications.
We assessed the stability and reactivity of 21 PFASs including 18 novel PFEAs in six solvents commonly used in toxicology and environmental analytical chemistry (deionized water, methanol, IPA, ACN, acetone, and DMSO). All selected PFASs were stable in deionized water, methanol, and IPA at room temperature over a period 30 days; however, nine of the selected PFEAs degraded in the polar aprotic solvents ACN, acetone, and DMSO. PFEA half-lives in pure solvents ranged from 0.35 hours for GenX in acetone to 54 d for PFO3OA in ACN. Results of this study highlight that solvent selection is important when preparing (1) PFEA dosing solutions for toxicological studies, (2) stock standard solutions for calibrating analytical instrumentation for quantitative analysis of PFEAs, and (3) environmental or biological samples by solvent extraction, liquid-liquid extraction, or solid-phase extraction approaches. PFEA degradability in aprotic solvents can be mitigated by the presence of water and by low temperatures. Further research should focus on identifying the mechanism(s) involved in PFEA degradation to support the development of models that can predict (1) the stability of a wide range of PFASs in solvents of interest and (2) the formation of degradation products. Furthermore, future research could explore whether solvent-mediated processes can be used for PFAS destruction.
ACKNOWLEDGMENTS
The authors would like to acknowledge Dr. Seth Newton for helping with GC-HRMS analysis, and Drs. Kevin O’Shea and Danielle Westerman for providing insights into the degradation mechanisms. This research was supported in part by the United States Environmental Protection Agency (R839482), the National Institute for Environmental Health Sciences Superfund Research Program (P42ES027706), the North Carolina Policy Collaborative through an appropriation from the North Carolina General Assembly, and North Carolina State University. This document has been reviewed by the U.S. Environmental Protection Agency, Office of Research and Development, and approved for publication. The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
Supporting information includes tables, figures, descriptions of LC-MS and GC-MS methods and associated data analysis.
REFERENCES
- (1).Ahrens L; Benskin JP; Cousins IT; Crimi M; Higgins CP Themed Issues on Per- And Polyfluoroalkyl Substances. Environ. Sci. Water Res. Technol. 2019, 5 (11), 1808–1813. [DOI] [PubMed] [Google Scholar]
- (2).Pan Y; Zhang H; Cui Q; Sheng N; Yeung LWY; Guo Y; Sun Y; Dai J. First Report on the Occurrence and Bioaccumulation of Hexafluoropropylene Oxide Trimer Acid: An Emerging Concern. Environ. Sci. Technol. 2017, 51 (17), 9553–9560. [DOI] [PubMed] [Google Scholar]
- (3).Dombrowski PM; Kakarla P; Caldicott W; Chin Y; Sadeghi V; Bogdan D; Barajas-Rodriguez F; Chiang SYD Technology Review and Evaluation of Different Chemical Oxidation Conditions on Treatability of PFAS. Remediation 2018, 28 (2), 135–150. [Google Scholar]
- (4).Hatton J; Holton C; DiGuiseppi B. Occurrence and Behavior of Per- and Polyfluoroalkyl Substances from Aqueous Film-Forming Foam in Groundwater Systems. Remediation 2018, 28 (2), 89–99. [Google Scholar]
- (5).Sun M; Arevalo E; Strynar M; Lindstrom A; Richardson M; Kearns B; Pickett A; Smith C; Knappe DRU Legacy and Emerging Perfluoroalkyl Substances Are Important Drinking Water Contaminants in the Cape Fear River Watershed of North Carolina. Environ. Sci. Technol. Lett 2016, 3 (12), 415–419. [Google Scholar]
- (6).Buck RC; Franklin J; Berger U; Conder JM; Cousins IT; Voogt P. De; Jensen AA; Kannan K; Mabury SA; van Leeuwen S. J. P Perfluoroalkyl and Polyfluoroalkyl Substances in the Environment: Terminology, Classification, and Origins. Integr. Environ. Assess. Manag. 2011, 7 (4), 513–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lindstrom AB; Strynar MJ; Libelo EL Polyfluorinated Compounds: Past, Present, and Future. Environ. Sci. Technol. 2011, 45 (19), 7954–7961. [DOI] [PubMed] [Google Scholar]
- (8).Wang Z; Dewitt JC; Higgins CP; Cousins IT A Never-Ending Story of Per- and Polyfluoroalkyl Substances (PFASs)? Environ. Sci. Technol. 2017, 51 (5), 2508–2518. [DOI] [PubMed] [Google Scholar]
- (9).Point AD; Holsen TM; Fernando S; Hopke PK; Crimmins BS Towards the Development of a Standardized Method for Extraction and Analysis of PFAS in Biological Tissues. Environ. Sci. Water Res. Technol. 2019, 5 (11), 1876–1886. [Google Scholar]
- (10).OECD. 033–066-C609–51.Pdf. Ser. Risk Manag. 2018, No. 39. (39), 1–24. [Google Scholar]
- (11).Koch A; Aro R; Wang T; Yeung LWY Towards a Comprehensive Analytical Workflow for the Chemical Characterisation of Organofluorine in Consumer Products and Environmental Samples. TrAC - Trends Anal. Chem 2020, 123, 115423. [Google Scholar]
- (12).Scheringer M; Trier X; Cousins IT; de Voogt P; Fletcher T; Wang Z; Webster TF Helsingør Statement on Poly- and Perfluorinated Alkyl Substances (PFASs). Chemosphere 2014, 114, 337–339. [DOI] [PubMed] [Google Scholar]
- (13).Wang Z; Cousins IT; Scheringer M; Hungerbuehler K. Hazard Assessment of Fluorinated Alternatives to Long-Chain Perfluoroalkyl Acids (PFAAs) and Their Precursors: Status Quo, Ongoing Challenges and Possible Solutions. Environ. Int 2015, 75, 172–179. [DOI] [PubMed] [Google Scholar]
- (14).Barzen-Hanson KA; Field JA Discovery and Implications of C2and C3perfluoroalkyl Sulfonates in Aqueous Film-Forming Foams and Groundwater. Environ. Sci. Technol. Lett 2015, 2 (4), 95–99. [Google Scholar]
- (15).Pan Y; Zhang H; Cui Q; Sheng N; Yeung LWY; Sun Y; Guo Y; Dai J. Worldwide Distribution of Novel Perfluoroether Carboxylic and Sulfonic Acids in Surface Water. Environ. Sci. Technol. 2018, 52 (14), 7621–7629. [DOI] [PubMed] [Google Scholar]
- (16).Heydebreck F; Tang J; Xie Z; Ebinghaus R. Alternative and Legacy Perfluoroalkyl Substances: Differences between European and Chinese River/Estuary Systems. Environ. Sci. Technol. 2015, 49 (14), 8386–8395. [DOI] [PubMed] [Google Scholar]
- (17).Gebbink WA; Van Asseldonk L; Van Leeuwen SPJ Presence of Emerging Per- and Polyfluoroalkyl Substances (PFASs) in River and Drinking Water near a Fluorochemical Production Plant in the Netherlands. Environ. Sci. Technol. 2017, 51 (19), 11057–11065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Strynar M; Dagnino S; McMahen R; Liang S; Lindstrom A; Andersen E; McMillan L; Thurman M; Ferrer I; Ball C. Identification of Novel Perfluoroalkyl Ether Carboxylic Acids (PFECAs) and Sulfonic Acids (PFESAs) in Natural Waters Using Accurate Mass Time-of-Flight Mass Spectrometry (TOFMS). Environ. Sci. Technol. 2015, 49 (19), 11622–11630. [DOI] [PubMed] [Google Scholar]
- (19).Hopkins ZR; Sun M; DeWitt JC; Knappe DRU Recently Detected Drinking Water Contaminants: GenX and Other Per- and Polyfluoroalkyl Ether Acids. J. Am. Water Works Assoc. 2018, 110 (7), 13–28. [Google Scholar]
- (20).McCord J; Strynar M. Identification of Per- and Polyfluoroalkyl Substances in the Cape Fear River by High Resolution Mass Spectrometry and Nontargeted Screening. Environ. Sci. Technol. 2019, 53, 4717–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang C; Hopkins ZR; McCord J; Strynar MJ; Knappe DRU Fate of Per- And Polyfluoroalkyl Ether Acids in the Total Oxidizable Precursor Assay and Implications for the Analysis of Impacted Water. Environ. Sci. Technol. Lett 2019, 6 (11), 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kotlarz N; McCord J; Collier D; Lea C; Strynar M; Lindstron A; Wilkie A; Islam J; Matney K; Tarte P; et al. Measurement of Novel, Drinking Water-Associated PFAS in Blood from Adults and Children in Wilmington, North Carolina. Env. Heal. Perspect 2020, 128 (7), 077005 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Song X; Vestergren R; Shi Y; Huang J; Cai Y. Emissions, Transport, and Fate of Emerging Per- and Polyfluoroalkyl Substances from One of the Major Fluoropolymer Manufacturing Facilities in China. Environ. Sci. Technol. 2018, 52 (17), 9694–9703. [DOI] [PubMed] [Google Scholar]
- (24).Bao Y; Cagnetta G; Huang J; Yu G. Degradation of Hexafluoropropylene Oxide Oligomer Acids as PFOA Alternatives in Simulated Nanofiltration Concentrate: Effect of Molecular Structure. Chem. Eng. J. 2020, 382, 122866. [Google Scholar]
- (25).Berger U; Kaiser MA; Kärrman A; Barber JL; Van Leeuwen SPJ Recent Developments in Trace Analysis of Poly- and Perfluoroalkyl Substances. Anal. Bioanal. Chem. 2011, 400 (6), 1625–1635. [DOI] [PubMed] [Google Scholar]
- (26).Xiao F; Hanson RA; Golovko SA; Golovko MY; Arnold WA PFOA and PFOS Are Generated from Zwitterionic and Cationic Precursor Compounds During Water Disinfection with Chlorine or Ozone. Environ. Sci. Technol. Lett 2018, 5, 382–388. [Google Scholar]
- (27).Boone JS; Guan B; Vigo C; Boone T; Byrne C; Ferrario J. A Method for the Analysis of Perfluorinated Compounds in Environmental and Drinking Waters and the Determination of Their Lowest Concentration Minimal Reporting Levels. J. Chromatogr. A 2014, 1345, 68–77. [DOI] [PubMed] [Google Scholar]
- (28).Xiao F; Golovko SA; Golovko MY Identification of Novel Non-Ionic, Cationic, Zwitterionic, and Anionic Polyfluoroalkyl Substances Using UPLC–TOF–MS E HighResolution Parent Ion Search. Anal. Chim. Acta 2017, 988, 41–49. [DOI] [PubMed] [Google Scholar]
- (29).Woudneh MB; Chandramouli B; Hamilton C; Grace R. Effect of Sample Storage on the Quantitative Determination of 29 PFAS: Observation of Analyte Interconversions during Storage. Environ. Sci. Technol. 2019, 53 (21), 12576–12585. [DOI] [PubMed] [Google Scholar]
- (30).Hanari N; Itoh N; Ishikawa K; Yarita T; Numata M. Variation in Concentration of Perfluorooctanoic Acid in Methanol Solutions during Storage. Chemosphere 2014, 94, 116–120. [DOI] [PubMed] [Google Scholar]
- (31).Sant KE; Venezia OL; Sinno PP; Timme-Laragy AR Perfluorobutanesulfonic Acid Disrupts Pancreatic Organogenesis and Regulation of Lipid Metabolism in the Zebrafish, Danio Rerio. Toxicol. Sci 2019, 167 (1), 157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dasgupta S; Reddam A; Liu Z; Liu J; Volz DC High-Content Screening in Zebrafish Identifies Perfluorooctanesulfonamide as a Potent Developmental Toxicant. Environ. Pollut. 2020, 256, 113550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gaballah S; Swank A; Sobus JR; Hines E; Strynar M; Tal T; Howey XM; Schmid J; Catron T; Mccord J. Evaluation of Developmental Toxicity, Developmental Neurotoxicity, and Tissue Dose in Zebrafish Exposed to GenX and Other PFAS. Environ. Health Perspect. 2020, 128 (4), 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sheng N; Cui R; Wang J; Guo Y; Wang J; Dai J. Cytotoxicity of Novel Fluorinated Alternatives to Long-Chain Perfluoroalkyl Substances to Human Liver Cell Line and Their Binding Capacity to Human Liver Fatty Acid Binding Protein. Arch. Toxicol. 2018, 92 (1), 359–369. [DOI] [PubMed] [Google Scholar]
- (35).Blake BE; Cope HA; Hall SM; Keys RD; Mahler BW; McCord J; Scott B; Stapleton HM; Strynar MJ; Elmore SA; et al. Evaluation of Maternal, Embryo, and Placental Effects in CD-1 Mice Following Gestational Exposure to Perfluorooctanoic Acid (PFOA) or Hexafluoropropylene Oxide Dimer Acid (HFPO-DA or GenX). Environ. Health Perspect. 2020, 128 (2), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Conley JM; Lambright CS; Evans N; Strynar MJ; McCord J; McIntyre BS; Travlos GS; Cardon MC; Medlock-Kakaley E; Hartig PC; et al. Adverse Maternal, Fetal, and Postnatal Effects of Hexafluoropropylene Oxide Dimer Acid (GenX) from Oral Gestational Exposure in Sprague-Dawley Rats. Environ. Health Perspect. 2019, 127 (3), 37008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liberatore HK; Jackson SR; Strynar MJ; Mccord JP Solvent Suitability for HFPO-DA ( “ GenX ” Parent Acid) in Toxicological Studies. Environ. Sci. Technol. Lett 2020, 7, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Da Silva GCQ; Cardozo TM; Amarante GW; Abreu CRA; Horta BAC Solvent Effects on the Decarboxylation of Trichloroacetic Acid: Insights from: Ab Initio Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2018, 20 (34), 21988–21998. [DOI] [PubMed] [Google Scholar]
- (39).Harada KH; Fujii Y. Comment on “Solvent Suitability for HFPO-DA (‘GenX’ Parent Acid) in Toxicological Studies.” Environ. Sci. Technol. Lett. 2021, 8 (3), 285–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Smith MB March’s Advanced Organic Chemistry – Reactions, Mechanisms, and Structures, 8th Ed.; 2020; pp 757–758. [Google Scholar]
- (41).Kostyukevich Y; Acter T; Zherebker A; Ahmed A; Kim S; Nikolaev E. Hydrogen/Deuterium Exchange in Mass Spectrometry. Mass Spectrom. Rev. 2018, 37 (6), 811–853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.