

Highlights
An operando synthetic approach was exemplified to enhance catalyst stability for efficient reduction of CO2 to formate.
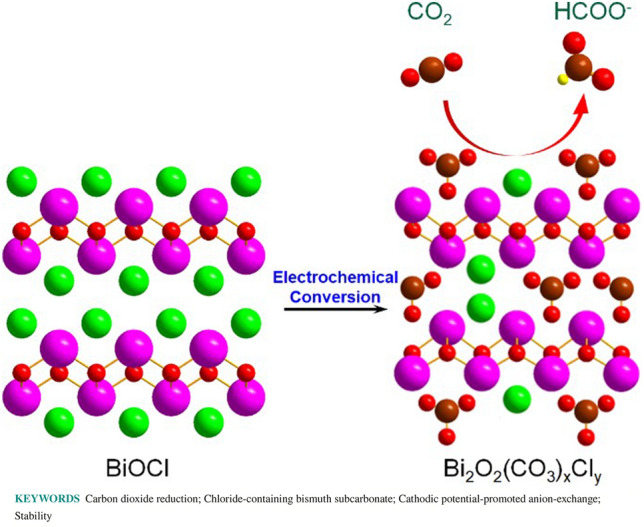
A highly stable Bi2O2(CO3)xCly electrocatalyst was synthesized by direct electrochemical conversion of BiOCl via a cathodic potential-promoted anion-exchange process under operando CO2RR conditions.
The surface Cl− in Bi2O2(CO3)xCly changes the p-orbital electron states to enhance the stability and alters the CO2RR pathway to markedly reduce the energy barrier.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40820-022-00862-0.
Keywords: Carbon dioxide reduction, Chloride-containing bismuth subcarbonate, Cathodic potential-promoted anion-exchange, Stability
Abstract
Bismuth-based materials (e.g., metallic, oxides and subcarbonate) are emerged as promising electrocatalysts for converting CO2 to formate. However, Bio-based electrocatalysts possess high overpotentials, while bismuth oxides and subcarbonate encounter stability issues. This work is designated to exemplify that the operando synthesis can be an effective means to enhance the stability of electrocatalysts under operando CO2RR conditions. A synthetic approach is developed to electrochemically convert BiOCl into Cl-containing subcarbonate (Bi2O2(CO3)xCly) under operando CO2RR conditions. The systematic operando spectroscopic studies depict that BiOCl is converted to Bi2O2(CO3)xCly via a cathodic potential-promoted anion-exchange process. The operando synthesized Bi2O2(CO3)xCly can tolerate − 1.0 V versus RHE, while for the wet-chemistry synthesized pure Bi2O2CO3, the formation of metallic Bio occurs at − 0.6 V versus RHE. At − 0.8 V versus RHE, Bi2O2(CO3)xCly can readily attain a FEHCOO- of 97.9%, much higher than that of the pure Bi2O2CO3 (81.3%). DFT calculations indicate that differing from the pure Bi2O2CO3-catalyzed CO2RR, where formate is formed via a *OCHO intermediate step that requires a high energy input energy of 2.69 eV to proceed, the formation of HCOO− over Bi2O2(CO3)xCly has proceeded via a *COOH intermediate step that only requires low energy input of 2.56 eV.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40820-022-00862-0.
Introduction
The renewable electricity-powered electrocatalytic carbon dioxide reduction reaction (CO2RR) to produce chemicals/fuels not only curbs greenhouse gas emissions but also reduces our reliance on the rapidly diminished petroleum resources [1]. In this regard, various C1 (e.g., carbon monoxide, formate, methane and methanol), C2 and C2+ (e.g., ethylene, ethanol, acetylene, acetate, acetaldehyde, oxalic acid and n-propanol) CO2RR products have been obtained [2, 3]. Among them, CO and HCOO−/HCOOH are the most energy-efficient CO2RR products as they can be formed by transferring two electrons to CO2. Comparing to CO, converting CO2 to HCOO−/HCOOH is more desirable because HCOO−/HCOOH are more valuable commodity chemicals [4, 5]. To date, the reported high-performance electrocatalysts for CO2 reduction to HCOO−/HCOOH are almost exclusively made of p-block metals-based materials such as In, Pb, Sn, Sb and Bi [6, 7].
Owning to their low toxicity and high selectivity toward HCOO−/HCOOH, Bi-based CO2RR electrocatalysts have attracted increasing attentions [8, 9]. Various Bi-based CO2RR electrocatalysts such as metallic Bio, oxides and subcarbonate (Table S1) have been employed to electrocatalytically convert CO2 to HCOO−/HCOOH. As shown in Table S1, in general, the metallic Bio-based ones perform better than other forms of bismuth-containing electrocatalysts. Nevertheless, the metallic Bio-based electrocatalysts usually require high overpotentials, consequently the high cathodic potentials, to achieve their optimal performances [10, 11], undesirable for energy efficiency. In addition, high cathodic potentials are favorable for the competing hydrogen evolution reaction (HER), which often leads to low Faradic efficiencies toward HCOO−/HCOOH (FEHCOO-/FEHCOOH) [12]. The bismuth oxides-based electrocatalysts were also reported (Table S1). Noticeably, such electrocatalysts often encounter stability issues because the bismuth oxides in these electrocatalysts can be easily converted to metallic Bio under CO2RR conditions [13]. For example, Deng et al. reported a Bi2O3 electrocatalyst with the optimal performance at − 0.9 V (vs RHE) to achieve a FEHCOO- of 91% with a partial HCOO− current density (JHCOO-) of ~ 8 mA cm−2 [14]. However, the as-synthesized Bi2O3 is found to be partially converted to metallic Bio under the CO2RR conditions at − 0.9 V vs RHE. In fact, the reported bismuth oxides electrocatalysts require cathodic potentials ≥ − 0.9 (vs RHE) to concurrently achieve FEHCOO- > 90% with JHCOO- ≥ 15 mA cm−2 [13, 15, 16]. Under such CO2RR conditions, the bismuth oxides in these electrocatalysts are either partially or completely converted to metallic Bio. Other than metallic Bio and bismuth oxides, Zhang’s group reported the use of ultrathin bismuth subcarbonate (Bi2O2CO3) nanosheets to catalyze CO2 reduction to HCOO− [17]. Their Bi2O2CO3 electrocatalyst exhibits a very low overpotential of 610 mV and can achieve a FEHCOO- of 85% with a JHCOO- of ~ 11 mA cm−2 at − 0.7 V (vs HRE), however, partial conversion of Bi2O2CO3 to the metallic Bio occurs within 30 min under − 0.65 V (vs RHE).
As reviewed above, under the required cathodic potentials to concurrently achieve high FEHCOO- and JHCOO-, the reported bismuth oxide and subcarbonate electrocatalysts are unavoidably reduced to metallic Bio, leading to the structural and compositional changes under operando CO2RR conditions. Critically, such operando structural transformation processes are progressive and potential-dependent, leading to great difficulties to confirm the actual active sites, hence the catalysis mechanisms. Parenthetically, the synthetic conditions of the reported bismuth oxide and subcarbonate electrocatalysts are vastly different to their electrocatalytic application conditions, which could be a cause of their structural transformation under the operando CO2RR conditions. If this is true, the severe operando stability issues might be effectively mitigated by employing identical synthesis and application conditions.
In this contribution, we report an approach to electrochemically convert bismuth oxychloride (BiOCl) into chloride-containing bismuth subcarbonate (Bi2O2(CO3)xCly) under operando CO2RR conditions (at − 0.8 V vs RHE in CO2-saturated 0.5 M KHCO3 solution) and use it to exemplify that the operando synthesis can be an effective means to enhance the operando electrochemical stability of electrocatalysts. Systematic operando spectroscopic studies were conducted to depict the conversion mechanism and electrochemical stability. BiOCl is converted to Bi2O2(CO3)xCly via the cathodic potential-promoted anion-exchange process. The obtained Bi2O2(CO3)xCly can tolerate − 1.0 V versus RHE, while for the wet-chemistry synthesized pure tetragonal phased Bi2O2CO3, the formation of metallic Bio occurs at − 0.6 V versus RHE, signifying a markedly improved electrochemical stability. No notable structural change and performance decay are observed when Bi2O2(CO3)xCly is subjected to the stability test at − 0.8 V versus RHE over a 20 h period. At − 0.8 V versus RHE, Bi2O2(CO3)xCly can readily attain a FEHCOO- of 97.9%, much higher than that of Bi2O2CO3 (81.3%). The density functional theory (DFT) calculations indicate that differing from Bi2O2CO3-catalyzed CO2RR, where HCOOH is formed via a *OCHO intermediate step that requires a high energy input energy of 2.69 eV to proceed, the formation of HCOOH over Bi2O2(CO3)xCly has proceeded via a *COOH intermediate step that requires a notably reduced energy input of 2.56 eV.
Experimental and Calculation
Materials and Chemicals
Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O, 99%), potassium chloride (KCl, 99.5%), ethanol (C2H5OH, 99%) and ethylene glycol (C2H6O2, 99.8%) were purchased from Chem-Supply. Urea (CH4N2O), Nafion (5 wt%) was purchased from Sigma-Aldrich. Carbon paper (TGP-H-060) and Nafion 115 proton exchange membrane were purchased from Alfa Aesar. The carbon paper was ultrasonically treated in deionized water and ethanol, followed by emerging in the concentrated HNO3 at 100 °C for 12 h, thoroughly washed with the deionized water and ethanol and dried in air.
Synthesis of BiOCl-NSs
0.164 g of KCl and 0.868 g of Bi(NO3)3·5H2O were dissolved in 70 mL H2O and stirred for 1 h. The solution was transferred into a 100 mL Teflon-lined stainless-steel autoclave and kept at 120 °C for 24 h. The obtained BiOCl-NSs was adequately washed with deionized water and ethanol and dried at 60 °C for 6 h in vacuum oven.
Synthesis of Bi2O2(CO3)xCly
Twenty milligrams of the as-synthesized BiOCl-NSs was mixed with 80 μL Nafion solution (5 wt%) and dispersed 0.92 mL isopropanol under sonication for 40 min to form the ink. 100 μL ink was then cast onto the pre-treated carbon fiber paper substrate with an exposed area of 1 × 1 cm2 (2 mg cm−2 of BiOCl-NSs). The carbon fiber paper with loaded BiOCl-NSs was used as the working electrode and subjected to − 0.8 V (vs RHE) in CO2-saturated 0.5 M KHCO3 solution for 2 h to electrochemically transform BiOCl-NSs to Bi2O2(CO3)xCly.
Synthesis of Bi2O2CO3
For comparative purpose, pure Bi2O2CO3 was synthesized. Under constant stirring, 0.234 g of Bi(NO3)3·5H2O was dissolved into 10 mL H2O, followed by adding 1.502 g of CH4N2O and 10 mL of C2H5OH. The resultant solution was then placed in the oil bath under 90 °C for 4 h. The obtained pure Bi2O2CO3 was adequately washed with deionized water and ethanol and dried in a vacuum oven of 60 °C for 6 h.
Electrochemical Measurements
The electrochemical measurements were performed using a Nafion 115 proton exchange membrane separated two-compartment electrochemical cell consisting of a three-electrode system controlled by an electrochemical station (CHI 660E). For CO2RR, the Bi2O2(CO3)xCly working electrode (1 × 1 cm2) was fabricated by operando electrochemical transformation of the immobilized BiOCl-NSs on carbon fiber paper, while the Bi2O2CO3 working electrode was prepared by immobilizing 2 mg cm−2 of Bi2O2CO3 on carbon fiber paper (1 × 1 cm2). For all electrochemical measurements, an Ag/AgCl (3.5 M KCl) reference electrode, a Pt mesh counter electrode and CO2-saturated 0.5 M KHCO3 electrolyte (pH of 7.2) were employed. During CO2RR, the electrolyte in the cathode compartment was constantly stirred at a rate of 800 rpm and bubbled with CO2 at a flow rate of 5 mL min−1 controlled by a universal flow meter (Alicat Scientific, LK2). All reported potentials were converted to the reversible hydrogen electrode (RHE) in accordance with ERHE = EAg/AgCl + 0.059 × pH + 0.205. The gas chromatography (GC, RAMIN, GC2060) equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD) was used to qualitatively and quantitatively determine the gaseous products (e.g., H2 and CO or other gaseous hydrocarbons). The CO and H2 Faradaic efficiency were calculated as below:
| 1 |
| 2 |
where xCO and xH2 (vol%) are the volume fractions of CO and H2 in the exhaust gas, I (A) is the steady-state current, G = 5 mL min−1 is the CO2 flow rate, p = 1.013 × 105 Pa, T = 273.15 K, F = 96,485 C mol−1, R = 8.3145 J mol−1 K−1.
1H nuclear magnetic resonance (1H‐NMR) was used to qualitatively and quantitatively determine the liquid phase products, including HCOOH. After reaction, 0.5 mL electrolyte from the cathode compartment was mixed with 0.1 mL D2O containing 3-(trimethylsilyl)propanoic acid (TMSP) as the internal standard and subjected to NMR analysis. The Faradaic efficiency for formation of HCOO− was calculated as below:
| 3 |
Characterizations
The morphologies and structures of the samples were characterized by SEM (JEOL JSM-7100) and TEM (Tecnai F20, 200 kV). The STEM images were recorded using a probe corrected JEOL JEM-ARM200F instrument with at an acceleration voltage of 200 kV. AFM measurements were performed using a Bruker Dimension Icon system. XRD patterns were collected from a Bruker D8 diffractometer. The operando XRD patterns were recorded using a Bruker D8 diffractometer and a home-made three‐electrode electrochemical cell. Raman spectra were taken by a RENISHAW mVia Raman Microscope using a 532 nm excitation laser. The operando Raman studies were performed on a RENISHAW mVia Raman Microscope equipped with a microscopic lens immersed under the electrolyte to capture Raman signals and a home-made three‐electrode electrochemical cell consisting of a BiOCl-NSs or Bi2O2CO3 working electrode, an Ag/AgCl (3.5 M KCl) reference electrode and a Pt mesh counter electrode. The working electrodes were prepared by immobilizing BiOCl-NSs or Bi2O2CO3 on a commercial Si substrate (1 × 1 cm2). XPS spectra were recorded by Kratos Axis ULTRA using the C1 s at 284.8 eV as the internal standard. C and O K-edge XAS measurements were performed at the Soft X-ray spectroscopy beamline at Australian Synchrotron Facility, Australia's Nuclear Science and Technology Organisation (Clayton, Victoria, Australia). Bi L3-edge XAS measurements were performed at the 10-ID-B beamline of the Advanced Photon Source (APS), Argonne National Laboratory (ANL). Data reduction, processing and subsequent modeling were performed using the Demeter XAS software package [18]. Modeling of the EXAFS data of Bi2O2(CO3)xCly was performed using Bi–O, Bi–C and Bi–Bi backscattering paths from the crystal structure of Bi2O2CO3 [19], while the Bi–Cl contributions were generated from the optimized structure generated from the DFT calculations. All EXAFS fitting was performed using an S02 value of 0.868, which were obtained by modeling the EXAFS of a reference Bi foil (L3-edge at 13,419 eV). To minimize error in CN and NND values, Debye–Waller factors were optimized in initial rounds of EXAFS fitting and then held constant.
DFT Calculations
All computation studies were performed using density functional theory (DFT) implemented in the Vienna Ab-initio Simulation Package (VASP) code in this study [20, 21]. For the effects of electron–electron exchange and correlation, the Perdew–Burke–Ernzerhof (PBE) functional at the generalized gradient approximation (GGA) level was employed [22]. The projected augmented wave (PAW) potentials were used throughout for ion–electron interactions [23], with the 5d106s26p3, 2s22p2, 2s22p4, 3s23p5 and 1s1 treated as valence electrons of Bi, C, O, Cl and H, respectively. The plane-wave cutoff of 520 eV was set for all the computations. The (1 × 2) clean {001} faceted Bi2O2CO3 was modeled by a 14-atomic layer slab separated by a vacuum layer of 20 Å in this study. When geometries of all structures were optimized, top seven layers of the surfaces including adsorbate were relaxed, while the bottom seven layers were fixed. The gamma-centered Monkhorst–Pack k-point meshes with a reciprocal space resolution of 2π × 0.04 Å−1 were utilized for structural optimization. For the calculations on CO2 and formic acid molecules, a (20 × 20 × 20) Å3 unit cell and a Γ-only k-point grid were used. All atoms were allowed to relax until the Hellmann–Feynman forces were smaller than 0.01 eV Å−1, and the convergence criterion for the electronic self-consistent loop was set to 10–5 eV. The adsorption energy of each adsorbate [ΔE (eV/n)] was calculated as follows:
| 4 |
where Ead, Esurf and Ead/surf are the energies of an adsorbate, the clean {001} facet and the surface with adsorbates, respectively. And n is the number of adsorbates on the surface.
Based on computational hydrogen electrode (CHE) model [24, 25], each electrochemical reaction step can be regarded as a simultaneous transfer of the proton–electron pair as a function of the applied potential. The reaction mechanism of CO2 reduction should consist of the following elementary reactions:
| 5 |
| 6 |
or
| 7 |
| 8 |
where * means the corresponding surface and adsorbed states. The free energy for all intermediate states and non-adsorbed gas-phase molecule is calculated as:
| 9 |
where the Eelec is the electronic energy obtained from DFT calculation; EZPE is the zero-point vibrational energy estimated by harmonic approximation; ∫CpdT is the enthalpic correction and TS is the entropy. Here, reported values of EZPE, ∫CpdT and TS are adopted [24]. The solvation effect has been considered for *COOH by stabilizing 0.25 eV [24].
Results and Discussion
Synthesis and Characterization of Bi2O2(CO3)xCly
In this work, Bi2O2(CO3)xCly was synthesized by direct electrochemical conversion of the pre-synthesized BiOCl under operando CO2RR conditions (Fig. 1a). The BiOCl nanosheets (BiOCl-NSs) were firstly synthesized as the precursor via a one-pot hydrothermal method [26]. The X-ray diffraction (XRD) pattern of the as-synthesized BiOCl-NSs (Fig. S1) can be indexed to the tetragonal BiOCl (PDF No. 06-0249). The Raman spectrum (Fig. S2) displays two strong peaks centered at 143 and 199 cm−1, assignable to (external) and (internal) Bi–Cl vibration modes, respectively, while the weak peak at 400 cm−1 can be attributed to mode [27]. The atomic force microscopy (AFM) and field-emission scanning electron microscopy (FE-SEM) images (Fig. S3a, b) disclose that the obtained BiOCl-NSs are octagonal shaped with sizes between 600 and 800 nm and thicknesses of ~ 150 nm. The high-resolution inverse fast Fourier transformation transmission electron microscopy (IFFT-TEM) image perpendicular to the nanosheet plane (Fig. S3c) reveals lattice spacings of 2.75 and 2.75 Å with an interplanar angle of 90°, corresponding to the (110) and (1͞10) facets of BiOCl. The selected area electron diffraction (SAED) pattern (Fig. S3d) coincides to the diffraction pattern of the single crystal BiOCl (k±l0, k = l = n) from [001] zone axis. The aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and the corresponding energy-dispersive X-ray spectroscopy (EDX) elemental mapping images confirm the homogeneous distribution of Bi, O and Cl throughout the entire BiOCl-NSs (Fig. S3e).
Fig. 1.

a Schematic illustrating electrochemical conversion of BiOCl to Bi2O2(CO3)xCly (Bi: pink, O: red, C: brown, Cl: green). b XRD pattern, c Raman spectrum, d FE-SEM images, e TEM image and SAED pattern, f HRTEM image, g IFFT-HRTEM image and h HAADF-STEM image and corresponding EDX element mapping images of Bi2O2(CO3)xCly resulted from the electrochemical treatment of BiOCl-NSs under − 0.8 V versus RHE in CO2-saturated 0.5 M KHCO3 solution for 2 h. (Color figure online)
The as-synthesized BiOCl-NSs were then immobilized onto a conductive carbon paper substrate (1.0 × 1.0 cm2) with a loading density of 2.0 mg cm−2 (Fig. S4) and subject to − 0.8 V (vs RHE) for 2 h in CO2-saturated 0.5 M KHCO3 solution to electrochemically convert the loaded BiOCl-NSs into Bi2O2(CO3)xCly. The XRD pattern (Fig. 1b) of the resultant Bi2O2(CO3)xCly can be assigned to the tetragonal phased Bi2O2CO3 (PDF No. 41–1488). The Raman spectrum (Fig. 1c) displays two strong peaks at 163 and 1068 cm−1, attributing to the external vibration of Bi2O2CO3 crystal and the ν1 mode of the intercalated CO32− between the (BiO)22+ planes [28, 29]. Raman peak at 182 cm−1 could be assigned to the A1g mode of the intercalated Cl− in the interlayer [29, 30]. The FE-SEM and AFM images (Figs. 1d and S5) unveil that Bi2O2(CO3)xCly possesses a sheeted structure with lateral sizes of 600–800 nm and thicknesses of 130–140 nm. The TEM image (Fig. 1e) shows that Bi2O2(CO3)xCly is formed by multiple thin-layer structures with “doughnuts-like” shape, resulting from the substitution of chloride by carbonate. The SAED pattern normal to the nanosheets (inset of Fig. 1e) manifests the reflections of Bi2O2CO3 (k00 and 0l0, k = l = n) with [001] zone axis. The high-resolution TEM image (HRTEM, Fig. 1f) displays a lattice spacing of 0.273 nm, corresponding to Bi2O2CO3 (110) plane, which is also confirmed by the high-resolution IFFT-HRTEM image (Fig. 1g). The HAADF-STEM image and the corresponding EDX elemental mapping (Fig. 1h) unveil the homogenously distributed Bi, O, C and Cl. The EDX estimated Bi/Cl atomic ratio in Bi2O2(CO3)xCly is 17.7:1 (Fig. S6), significantly higher than that of BiOCl (1.2:1), confirming the presence of Cl in Bi2O2(CO3)xCly.
The X-ray photoelectron spectroscopy (XPS) analysis was then carried out. The high-resolution XPS Bi 4f spectra (Fig. 2a) confirm the presence of Bi3+ and Bi–O bonds (160.0 and 165.3 eV) [31] in BiOCl. The Bi3+ peaks of Bi2O2(CO3)xCly show a negative shift of 0.26 eV, consistent with that of reported Bi2O2CO3 [32]. Figure 2b shows the high-resolution XPS O 1s spectra of BiOCl and Bi2O2(CO3)xCly. The former could be deconvoluted into the binding energy peaks assignable to the Bi–O lattice O (530.8 eV), the surface adsorbed hydroxyl (~ 531.9 eV) and O species in Nafion (536.3, 533.3 and 531.9 eV) [33, 34], while the deconvoluted binding energy peaks at 530.2 and 531.0 eV from the later are ascribed to the Bi–O lattice O and C=O, respectively [35, 36]. The lattice O peaks in Bi2O2(CO3)xCly shifted to lower energies due to the substitution of chloride by carbonate. The two binding energy peaks at 199.2 and 200.8 eV assignable to Cl 2p3/2 and Cl 2p1/2 can be deconvoluted from the high-resolution XPS Cl 2p spectra of both BiOCl and Bi2O2(CO3)xCly (Fig. 2c), indicating the presence of the lattice Cl− [37]. Notably, a Bi/Cl atomic ratio of 61.5:1 is determined from the XPS Cl 2p spectrum of Bi2O2(CO3)xCly (Fig. S7), confirming the presence of chemically bonded Cl on the surface of Bi2O2(CO3)xCly.
Fig. 2.

High-resolution XPS spectra of a Bi 4f, b O 1s and c Cl 1s obtained from the as-synthesized BiOCl-NSs and Bi2O2(CO3)xCly. d O K-edge spectra, e C K-edge spectra and f Bi L3-edge spectra of BiOCl, Bi2O2CO3, Bi2O2(CO3)xCly and referenced samples. g Bi L3-edge k2-weighted FT-EXAFS spectra of Bi2O2(CO3)xCly and Bi2O2CO3 in R space. h Fitting analysis of Bi2O2(CO3)xCly using Bi–O, Bi–C and Bi–Cl paths. i Proposed geometric configuration of Bi2O2(CO3)xCly
The X-ray absorption spectroscopy (XAS) measurements were then conducted to probe the electronic structure and local atomic environments. The O K-edge near-edge X-ray absorption fine structure (NEXAFS) spectra of BiOCl, Bi2O2(CO3)xCly and reference samples are shown in Fig. 2d. The observed binding energy peaks at 532.0 and 537.0 eV from Bi2O2(CO3)xCly are assignable to the hybridization of O 2p with Bi 6 s orbitals [38, 39], while the binding energy peak at 534 eV corresponds to the π* C=O transition, indicating the presence of lattice carbonyl oxygen species [40]. The displayed binding energy peaks at 539.4 and 543.2 eV in the spectrum of Bi2O2(CO3)xCly are ascribed to the non-equivalent σ* C–O bonds in the carboxylic group originated from the adsorbed carbonate [41]. Based on the C K-edge NEXAFS spectra of Bi2O2(CO3)xCly and reference samples (Fig. 2e), the binding energy peak at 289.5 eV can be attributed to the σ* states of C–O [41], while the peaks at 297.2 and 299.8 eV are assignable to the σ* C = O resonances associated with the presence of carbonate species [42]. It is to note that the O K-edge and C K-edge NEXAFS spectra obtained from Bi2O2CO3 and Bi2O2(CO3)xCly exhibit very similar characteristics, implying that the crystal structure of Bi2O2CO3 in Bi2O2(CO3)xCly is not noticeably altered by the presence of Cl−. According to the Bi L3-edge X-ray absorption near-edge structure (XANES) spectra (Fig. 2f), the same valence states of Bi3+ exist in BiOCl, Bi2O2CO3 and Bi2O2(CO3)xCly. Figure 2g shows the k2-weighted Fourier transformed Bi L3-edge extended X-ray absorption fine structure (k2-weighted FT-EXAFS) spectra of Bi2O2CO3 and Bi2O2(CO3)xCly. The peaks at 1.74, 2.33 and 3.58 Å assignable to the Bi–O and Bi–Bi bonds in the (BiO)22+ ab plane are observed from both Bi2O2CO3 and Bi2O2(CO3)xCly, indicating an identical (BiO)22+ ab plane in both Bi2O2CO3 and Bi2O2(CO3)xCly. The spectrum of Bi2O2CO3 shows a peak at 2.93 Å, corresponding to the interactions of Bi in (BiO)22+ ab plane with the intercalated CO32− between the (BiO)22+ ab plane layers (the interlayer). Notably, for Bi2O2(CO3)xCly, the peak is shifted to 3.01 Å, implying the differences in the interactions of Bi in (BiO)22+ ab plane with the interlayer anions due to the presence of the intercalated Cl− in the interlayer of Bi2O2(CO3)xCly. The fittings of the Bi L3-edge k2-weighted FT-EXAFS spectra of Bi2O2(CO3)xCly and Bi2O2CO3 in R space and k space were then performed (Figs. 2h, S8 and Table S2) [43]. It unveils that the spectrum of Bi2O2(CO3)xCly fits well with Bi2O2CO3 and Bi–Cl path, confirming the presence of the intercalated Cl− in the interlayer. The coordination numbers (CNs) of Bi–O in the first coordination sphere of Bi2O2(CO3)xCly and Bi2O2CO3 are 2.34 at 2.22 Å and 2.27 at 2.24 Å, respectively, further confirming an almost unchanged (BiO)22+ ab plane coordination environment. For Bi2O2CO3, a Bi–C CN of 1.75 at 3.36 Å represents the interactions between the Bi atoms in the (BiO)22+ ab plane and the intercalated CO32− in the interlayer. For Bi2O2(CO3)xCly, the measured Bi–C CN of 1.51 at 3.38 Å indicates a reduction in the Bi–C coordination. Notably, the fitting of the EXAFS spectrum of Bi2O2(CO3)xCly using Bi–Cl backscattering path from the optimized structure corresponds to a Bi–Cl CN of 0.33, which is closely approximated to the dropped Bi–C CN, unambiguously confirming the presence of the intercalated Cl− between (BiO)22+ ab plane layers in Bi2O2(CO3)xCly. A likely Bi2O2(CO3)xCly structure is shown in Fig. 2i. The above results confirm that under − 0.8 V (vs RHE) cathodic potential in CO2-saturated 0.5 M KHCO3 solution for 2 h, the BiOCl-NSs are electrochemically
Operando Converting BiOCl into Bi2O2(CO3)xCly
It is known that both of the tetragonal BiOCl and Bi2O2CO3 crystals (Fig. S9) belong to the Sillén crystal family, featuring a matlockite-type positively charged (BiO)22+ ab plane layer structure stacking between the negatively charged bichloride and “standing-on-end” CO32− anions slabs, respectively. Comparing to the d spacing of {001} faceted BiOCl along the c axis (7.83 Å), the d spacing of {002} faceted Bi2O2CO3 is markedly reduced to 6.84 Å. Therefore, under apt cathodic potentials, due to the layer structure similarity, and the apparently decreased d spacing of {002} faceted Bi2O2CO3, the transformation of BiOCl to Bi2O2CO3 could occur via the intercalative substitution of the interlayer Cl− with CO32− through a glide of the neighboring (BiO)22+ ab planes along [100] and [010] directions with the translational distances of ½ a and ½ b, respectively [17]. To depict the structural evolution processes under the operando CO2RR conditions, the BiOCl-NSs immobilized on the carbon fiber paper were subjected to different cathodic potentials (EApp) in CO2-saturated 0.5 M KHCO3 solution, and the XRD patterns were operando recorded. The XRD patterns recorded under the open circuit potential (OCP) and EApp ≤ − 0.2 V (Fig. 3a) are almost identical to that of the as-synthesized BiOCl (Fig. S1). The initial conversion of BiOCl to Bi2O2(CO3)xCly occurs at EApp = − 0.3 V as indicated by the observed diffraction peak at 56.9° corresponding to {123} faceted Bi2O2CO3. When EApp is increased from − 0.3 to − 0.7 V, although the recorded XRD patterns are still dominated by the diffraction patterns of BiOCl, the progress of converting BiOCl to Bi2O2(CO3)xCly is evidenced by the progressively increased intensities of Bi2O2CO3 diffraction peaks and the accompanied decrease in the intensities of BiOCl diffraction peaks. With EApp = − 0.8 V, all recorded diffraction peaks belong to Bi2O2CO3 (PDF No. 41-1488), signifying the complete conversion of BiOCl to Bi2O2(CO3)xCly. The structural evolution and the time required to completely convert BiOCl to Bi2O2(CO3)xCly under EApp = − 0.8 V were subsequently investigated (Fig. S10). As can be seen, BiOCl is fully covered to Bi2O2(CO3)xCly within 60 min under EApp = − 0.8 V. As disclosed in Fig. 3a, Bi2O2(CO3)xCly remains as the sole product when − 0.8 V ≤ EApp ≤ − 1.0 V. With EApp = − 1.1 V, the bismuth in Bi2O2(CO3)xCly is partially reduced to the metallic phased Bio as evidenced by the appearance of the diffraction peaks assignable to rhombohedral phased Bio (PDF No. 05–0519). With EApp = − 1.2 V, all of the recorded diffraction peaks belong to the rhombohedral phased Bio, confirming the ultimate conversion of Bi2O2(CO3)xCly to the metallic Bio phase. As shown in Fig. S11, the BiOCl-derived Bio at EApp = − 1.2 V is formed by the aggregated Bio NSs with the exposed {001} facets. Notably, the required cathodic potential to convert Bi2O2(CO3)xCly to Bio is more negative than those reported potentials to reduce Bi2O2CO3 to Bio [44], {Lv, 2017 #1765}inferring a superior electrochemical stability of Bi2O2(CO3)xCly over Bi2O2CO3, which might be attributed to the presence of Cl− in Bi2O2(CO3)xCly. To confirm this, the pure tetragonal phased Bi2O2CO3 nanosheets (Figs. S12–S14) were synthesized by a wet-chemistry method [43] and subjected to different cathodic potentials in CO2-saturated 0.5 M KHCO3 solution. Figure 3b shows the operando recorded XRD patterns. The formation Bio occurs at EApp = − 0.6 V, while the Bi2O2CO3 is fully converted to the rhombohedral phased metallic Bio (PDF No. 05-0519) at EApp = − 0.8 V, confirming that the presence of Cl− in Bi2O2(CO3)xCly is responsible for the improved electrochemical stability. It is noteworthy that compared to the characteristic diffraction peaks of Bi2O2CO3, all of the recorded characteristic diffraction peaks from Bi2O2(CO3)xCly are shifted slightly toward lower angles (Fig. S15), indicating an expended d spacing in Bi2O2(CO3)xCly due to the presence of Cl− in the interlayer. The above operando XRD studies unveil that the electrochemical conversion of BiOCl to Bi2O2(CO3)xCly is realized by the cathodic potential-promoted anion-exchange in the interlayer between the (BiO)22+ ab planes.
Fig. 3.

a, b Operando XRD patterns of the as-synthesized BiOCl-NSs and Bi2O2CO3 recorded from CO2-saturated 0.5 M KHCO3 solution under different cathodic potentials. c, d Operando Raman spectra of the as-synthesized BiOCl-NSs and Bi2O2CO3 recorded from CO2-saturated 0.5 M KHCO3 solution under different cathodic potentials
To further elaborate the electrochemical conversion pathway, the operando potential-dependent Raman spectra of BiOCl immobilized on the carbon fiber paper were recorded under different cathodic potentials in CO2-saturated 0.5 M KHCO3 solution (Fig. 3c). Under the OCP and EApp ≤ − 0.3 V conditions, the recorded spectra are almost identical to that of the as-synthesized BiOCl-NSs (Fig. S2). With EApp = − 0.4 V, the characteristic peaks of BiOCl at 143 () and 199 cm−1 () are markedly reduced and disappeared, respectively, which are accompanied by the appearance of a new peak at 182 cm−1 that might be assigned to the A1g mode of the intercalated Cl− in the interlayer, although this could be complicated by the reduced crystal symmetry due to the disorder or free rotation of CO32− in the interlayer [29, 30]. These observed changes in the Raman spectrum signify the initial conversion of BiOCl to Bi2O2(CO3)xCly at EApp = − 0.4 V, consistent with the operando XRD observation shown in Fig. 3a. At EApp = − 0.8 V, two new peaks at 163 and 1068 cm−1 attributed to the external vibration of Bi2O2CO3 crystal and the ν1 mode of CO32− anions between the (BiO)22+ planes appear, signifying the complete conversion of BiOCl to Bi2O2(CO3)xCly. Within − 0.8 V ≤ EApp ≤ − 1.0 V, the peak at 182 cm−1 (A1g mode) is rapidly decreased, while the peaks at 163 and 1068 cm−1 are evolved and intensified, which are likely due to the changes in the local atomic symmetry rather than the crystal cell parameters because of the unchanged operando XRD patterns within the same potential range (Fig. 3a). It is known that the high-intensity Raman bands between 150 and 200 cm−1 normally correspond to the out-of-plane lattice vibrations of the Bi atoms perpendicular to the (BiO)22+ layers [45]. Because the phonon frequencies are sensitive to the dopant-induced asymmetry and the interlayer thickness, therefore, the blueshifted phonons from 182 to 163 cm−1 could be resulted from the suppressed vibrations of the (BiO)22+ layers along c direction due to the replaced Cl− by CO32− at relatively high cathodic potentials. The characteristic Raman peaks of Bi2O2CO3 at 163 and 1068 cm−1 are dramatically decreased under EApp = − 1.1 V and vanished at EApp = − 1.2 V due to the formation of metallic Bio, consistent with the operando XRD observations. For comparative purpose, the operando potential-dependent Raman spectra of the pure tetragonal phased Bi2O2CO3 nanosheets were obtained (Fig. 3d). When EApp ≤ − 0.5 V, the peak at 182 cm−1 associating with the A1g mode of the intercalated Cl− in the CO32− slab is absent, while the characteristic Raman peaks of Bi2O2CO3 at 163 and 1068 cm−1 are apparent, however, rapidly extinct when EApp ≥− 0.6 V due to the formation of metallic Bio, consistent with the operando XRD observations shown in Fig. 3b. This further confirms that compared to the chloride-free Bi2O2CO3, the reduction of Bi2O2(CO3)xCly to metallic Bio requires a much higher cathodic potential due to presence of the intercalated Cl− in the CO32− slab, signifying a noticeably improved electrochemical stability. These operando Roman studies further suggest that the conversion of BiOCl to Bi2O2(CO3)xCly is achieved by the cathodic potential-promoted anion-exchange in the interlayer.
CO2RR Performance
All electrochemical measurements were performed using a three-electrode electrochemical system with Bi2O2(CO3)xCly or Bi2O2CO3 working electrode in CO2- or Ar-saturated 0.5 M KHCO3 solution. When EApp > − 0.5 V (vs EHE), the linear sweep voltammetry (LSV) responses of Bi2O2(CO3)xCly in CO2-saturated solution display higher cathodic current densities than that obtained from the Ar-saturated solution (Fig. S16), indicating a superior electrocatalytic activity of Bi2O2(CO3)xCly toward CO2RR. The potentiostatic experiments were then performed under different cathodic potentials to examine the electrocatalytic CO2RR activity and selectivity. Figure 4a shows the chronoamperometric curves of Bi2O2(CO3)xCly from the CO2-saturated 0.5 M KHCO3 solution. The reaction products in gaseous and aqueous phases were qualitatively identified and quantitatively determined by the gas chromatography (GC) and nuclear magnetic resonance (NMR). For all cases investigated, H2 is identified as the sole product in the gaseous phase, while the formate is found to be the sole product in the aqueous phase. The NMR determined formate concentrations (Figs. S17 and S18) corresponding to the chronoamperometric curves shown in Fig. 4a were used to calculate the corresponding FEHCOO- and JHCOO- values. Figure 4b shows the plot of JHCOO- against EApp. For both Bi2O2(CO3)xCly and Bi2O2CO3, an increase in EApp leads to an increase in JHCOO-. For a given EApp, the observed JHCOO- from Bi2O2(CO3)xCly is higher than that observed from Bi2O2CO3, implying a superior CO2RR activity of Bi2O2(CO3)xCly over Bi2O2CO3. At EApp = − 0.8 V, the JHCOO- attained by Bi2O2(CO3)xCly is 18.4 mA cm−2, higher than that of Bi2O2CO3 (14.2 mA cm−2). Figure 4c shows the plot of FEHCOO- (derived from Fig. 4a) against EApp. For Bi2O2(CO3)xCly, an increase in EApp from − 0.4 to − 0.6 V leads to a rapidly increased FEHCOO- from 79.2 to 96.2%, and further increasing EApp to − 0.8 V leads to an increased FEHCOO- to 97.9%. FEHCOO- remains almost unchanged when EApp is further increased to − 1.0 V, while the corresponding JHCOO- is increased to 40.5 mA cm−2 (Fig. 4b). Based on the operando XRD and Raman observations (Fig. 3a, c), the formation of the metallic phased Bio will not occur with EApp ≤ − 1.0 V, therefore, the observed changes in FEHCOO- from the potential range of − 0.4 V ≤ EApp ≤ − 1.0 V reflect the influence of potential on CO2RR selectivity of Bi2O2(CO3)xCly. Although Bi2O2(CO3)xCly is partially converted to metallic Bi–NSs within − 1.0 V ≤ EApp ≤ − 1.2 V (Fig. 3a, c), the high FEHCOO- can still be attained due to the electrocatalytic activity of Bio toward CO2RR under high cathodic potentials [17]. When EApp > − 1.2 V, the observed decrease in FEHCOO- is due to the intensified competition from HER [7]. Interestingly, for pure Bi2O2CO3, a rapidly increased FEHCOO- from 65.5 to 77.5% is observed when EApp is increased from − 0.4 to − 0.6 V and reached a maxima FEHCOO- of 83.0% at EApp = − 0.7 V, where Bi2O2CO3 is partially converted to the metallic Bio. With − 0.8 V ≤ EApp ≤ − 1.2 V, Bi2O2CO3 is fully converted to the metallic Bio and the slightly decreased FEHCOO- reflects the influence of potential on CO2RR selectivity of metallic Bio rather than that of Bi2O2CO3. When EApp > − 1.2 V, FEHCOO- is rapidly decreased due to the intensified competition from HER.
Fig. 4.

a Chronoamperometric curves of Bi2O2(CO3)xCly recorded from CO2-saturated 0.5 M KHCO3 solution under different cathodic potentials. b, c Plots of HCOOH partial current density and Faradic efficiency against catholic potential for Bi2O2(CO3)xCly- and Bi2O2CO3-catalyzed CO2RR. d Chronoamperometric curves and FEHCOOH of Bi2O2(CO3)xCly at − 0.8 V versus RHE. e Free energy diagrams of Bi2O2(CO3)xCly- and Bi2O2CO3-catalyzed CO2 reduction to HCOOH. f PDOSs plots of Bi2O2(CO3)xCly- and Bi2O2CO3-catalyzed CO2 reduction to HCOOH
The chronoamperometric stability of Bi2O2(CO3)xCly (Fig. 4d) was evaluated over a 20 h period in CO2-saturated 0.5 M KHCO3 solution at EApp = − 0.8 V vs RHE. While FEHCOO- of ~ 95% is well retained, a 12.1% increase in the cathodic current density is observed, indicating an increased JHCOO-. Interestingly, when the used electrolyte is replaced by the fresh one, an almost identical chronoamperometric curve is obtainable with ~ 12% increase in the cathodic current density at 20 h, indicating an excellent long-term stability of Bi2O2(CO3)xCly [46]. The excellent electrocatalytic stability of Bi2O2(CO3)xCly can be attributed to its excellent structural stability as evidenced by the almost unchanged XRD pattern (Fig. S19), Raman spectra (Fig. S20), as well as SEM and TEM images (Fig. S21) of Bi2O2(CO3)xCly after the chronoamperometric stability test. The chronoamperometric stability of Bi2O2CO3 was also evaluated at EApp = − 0.8 V vs RHE (Fig. S22). Over a 20 h testing period, FEHCOO- is decreased from 86.2 to 80.0%, while the cathodic current density is increased from 12.5 to 14.2 mA cm−2. However, after the chronoamperometric stability test, the pure tetragonal phased Bi2O2CO3 is fully converted to the metallic phased Bio (Figs. S23–S26). The stability test result confirms that Bi2O2(CO3)xCly fabricated under operando CO2RR conditions possesses excellent stability.
DFT Calculations
It is known that electrocatalytic CO2RR to HCOOH has normally proceeded via a proton-coupled electron transfer (PCET) step to form *COOH or *OCHO intermediates and followed by another PCET step to generate HCOOH [47]. It is also known that the CO2RR pathway depends strongly on the adsorption energy of the intermediates [48]. DFT calculations were therefore carried out to determine the preferential intermediates of the pure tetragonal phased Bi2O2CO3- and Bi2O2(CO3)xCly-catalyzed CO2 reduction to HCOOH. Our DFT calculations unveil that *OCHO intermediate can preferentially adsorb on the {001} faceted Cl-free Bi2O2CO3 surface [49] with an adsorption free energy (ΔG*OCHO) of − 2.67 eV (Fig. S27a), while no stable structure of *COOH intermediate adsorbed on the {001} faceted Cl-free Bi2O2CO3 can be obtained. These results imply that the pure tetragonal phased Bi2O2CO3-catalyzed CO2 reduction to HCOOH has proceeded via a *OCHO intermediate pathway. In contrast, our initial DFT calculations are failed to obtain a stable structure of *OCHO intermediate adsorbed on Bi2O2(CO3)xCly surface. Nonetheless, further DFT calculations unveil that the *COOH intermediate is apt to absorb to the Bi2O2(CO3)xCly surface with a ΔG*COOH of − 1.25 eV (Fig. S27b), inferring that the Bi2O2(CO3)xCly-catalyzed CO2 reduction to HCOOH has proceeded via a *COOH intermediate pathway. Figure 4e illustrates the free energy diagrams of Bi2O2CO3- and Bi2O2(CO3)xCly-catalyzed CO2 reduction to HCOOH. During the 1st PCET step, the formation of *OCHO on Bi2O2CO3 surface and *COOH on Bi2O2(CO3)xCly surface is exothermic. During the 2nd PCET step, the formation of *HCOOH on Bi2O2(CO3)xCly is exothermic, while on Bi2O2CO3 is endothermic. The desorption of *HCOOH from Bi2O2CO3 and Bi2O2(CO3)xCly to form HCOOH are energetically uphill. However, the desorption of *HCOOH from Bi2O2(CO3)xCly requires 2.56 eV to proceed, which is 0.13 eV lower than that of Bi2O2CO3 (2.69 eV), indicating a better kinetic activity of Bi2O2(CO3)xCly over the Cl-free Bi2O2CO3. The poor kinetic activity of Bi2O2CO3 could be resulted from the excessively high adsorption energy of *OCHO impeded active sites turnover. Figure 4f shows the projected density of states (PDOS) of the p bands of Bi sites in Bi2O2(CO3)xCly and the {001} faceted Bi2O2CO3. As can be seen, the PDOS of the {001} faceted Bi2O2CO3 near Fermi level is dominated by the p-orbital electron states with much higher electronic densities than that of Bi2O2(CO3)xCly, indicating a higher reactivity for *OCHO intermediate adsorption, hence an impeded active sites regeneration [50, 51]. In addition, the high PDOS density of the {001} faceted Bi2O2CO3 near Fermi level indicates high densities of the unoccupied p-orbital states of Bi in the {001} faceted Bi2O2CO3 with an estimated lowest unoccupied molecular orbital (LUMO) energy of 0.4 eV above Fermi level, corresponding to a LUMO potential of − 0.4 eV. In strong contrast, for Bi2O2(CO3)xCly, the p-orbital electron states can only be observed at 2.3 eV above the Fermi level with low densities, indicating low unoccupied p-orbital states of Bi in Bi2O2(CO3)xCly, corresponding to a LUMO potential of − 2.3 eV. It is known that for semiconductor electrodes, the reduction reaction takes place via the injection of electrons into LUMO. Therefore, the LUMO potential corresponds to the minimum required cathodic potential for electron injection. As illustrated in Fig. S28, compared to Bi2O2CO3, the higher LUMO potential of Bi2O2(CO3)xCly infers that a higher cathodic potential is required to convert Bi3+ in Bi2O2(CO3)xCly to metallic Bio, which explains the superior electrochemical stability of Bi2O2(CO3)xCly over Bi2O2CO3 under CO2RR conditions.
Conclusions
In summary, we reported an approach to electrochemically convert bismuth oxychloride (BiOCl) into chloride-containing bismuth subcarbonate (Bi2O2(CO3)xCly) under operando CO2RR conditions. We demonstrated that the operando synthesis is an effective strategy to enhance the electrochemical stability of bismuth-based electrocatalysts. The exemplified approach in this work could be widely applicable to enhance the electrochemical stabilities of other electrocatalysts for other reactions.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was financially supported by Australian Research Council Discovery Project (DP200100965). Bi L3-edge XAS measurements were performed at the 10-ID-B beamline of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Operations at 10-ID-B are further supported by members of the Materials Research Collaborative Access Team. C and O K-edge measurements were performed at the SXR beamline of the Australian Synchrotron, part of the Australian Nuclear Science and Technology Organisation.
Funding
Open access funding provided by Shanghai Jiao Tong University.
Contributor Information
Porun Liu, Email: p.liu@griffith.edu.au.
Huijun Zhao, Email: h.zhao@griffith.edu.au.
References
- 1.Luna PD, Hahn C, Higgins D, Jaffer SA, Jaramillo TF, et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science. 2019;364(6438):3506. doi: 10.1126/science.aav3506. [DOI] [PubMed] [Google Scholar]
- 2.Li J, Abbas SU, Wang H, Zhang Z, Hu W. Recent advances in interface engineering for electrocatalytic CO2 reduction reaction. Nano-Micro Lett. 2021;13:216. doi: 10.1007/s40820-021-00738-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu ZZ, Zhang XL, Niu ZZ, Gao FY, Yang PP, et al. Identification of Cu(100)/Cu(111) interfaces as superior active sites for CO dimerization during CO2 electroreduction. J. Am. Chem. Soc. 2022;144(1):259–269. doi: 10.1021/jacs.1c09508. [DOI] [PubMed] [Google Scholar]
- 4.Zheng X, Luna PD, Arquer FPG, Zhang B, Becknell N, et al. Sulfur-modulated tin sites enable highly selective electrochemical reduction of CO2 to formate. Joule. 2017;1:794–805. doi: 10.1016/j.joule.2017.09.014. [DOI] [Google Scholar]
- 5.Lu X, Wu Y, Yuan X, Wang H. An integrated CO2 electrolyzer and formate fuel cell enabled by a reversibly restructuring Pb-Pd bimetallic catalyst. Angew. Chem. Int. Ed. 2019;58(12):4031–4035. doi: 10.1002/anie.201814257. [DOI] [PubMed] [Google Scholar]
- 6.Zhao S, Li S, Guo T, Zhang S, Wang J, et al. Advances in Sn-based catalysts for electrochemical CO2 reduction. Nano-Micro Lett. 2019;11:62. doi: 10.1007/s40820-019-0293-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu D, Feng R, Xu C, Sui PF, Zhang J, et al. Regulating the electron localization of metallic bismuth for boosting CO2 electroreduction. Nano-Micro Lett. 2021;14:38. doi: 10.1007/s40820-021-00772-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong Q, Ding P, Xu M, Zhu X, Wang M, et al. Structural defects on converted bismuth oxide nanotubes enable highly active electrocatalysis of carbon dioxide reduction. Nat. Commun. 2019;10:2807. doi: 10.1038/s41467-019-10819-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia C, Zhu P, Jiang Q, Pan Y, Liang W, et al. Continuous production of pure liquid fuel solutions via electrocatalytic CO2 reduction using solid-electrolyte devices. Nat. Energy. 2019;4:776–785. doi: 10.1038/s41560-019-0451-x. [DOI] [Google Scholar]
- 10.Yang F, Elnabawy AO, Schimmenti R, Song P, Wang J, et al. Bismuthene for highly efficient carbon dioxide electroreduction reaction. Nat. Commun. 2020;11:1088. doi: 10.1038/s41467-020-14914-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng P, Yang F, Wang Z, Chen S, Zhou Y, et al. Metal-organic frameworks-derived carbon nanorods encapsulated bismuth oxides for rapid and selective CO2 electroreduction to formate. Angew. Chem. Int. Ed. 2020;59(27):10807–10813. doi: 10.1002/anie.202000657. [DOI] [PubMed] [Google Scholar]
- 12.Zhu DD, Liu JL, Qiao SZ. Recent advances in inorganic heterogeneous electrocatalysts for reduction of carbon dioxide. Adv. Mater. 2016;28(18):3423–3452. doi: 10.1002/adma.201504766. [DOI] [PubMed] [Google Scholar]
- 13.Lee CW, Hong JS, Yang KD, Jin K, Lee JH, et al. Selective electrochemical production of formate from carbon dioxide with bismuth-based catalysts in an aqueous electrolyte. ACS Catal. 2018;8(2):931–937. doi: 10.1021/acscatal.7b03242. [DOI] [Google Scholar]
- 14.Deng P, Wang H, Qi R, Zhu J, Chen S, et al. Bismuth oxides with enhanced bismuth-oxygen structure for efficient electrochemical reduction of carbon dioxide to formate. ACS Catal. 2019;10(1):743–750. doi: 10.1021/acscatal.9b04043. [DOI] [Google Scholar]
- 15.Tran-Phu T, Daiyan R, Fusco Z, Ma Z, Amal R, et al. Nanostructured β-Bi2O3 fractals on carbon fibers for highly selective CO2 electroreduction to formate. Adv. Funct. Mater. 2019;30(3):1906478. doi: 10.1002/adfm.201906478. [DOI] [Google Scholar]
- 16.Liu S, Lu XF, Xiao J, Wang X, Lou XWD. Bi2O3 nanosheets grown on multi-channel carbon matrix catalyze efficient CO2 electroreduction to HCOOH. Angew. Chem. Int. Ed. 2019;58(39):13828–13833. doi: 10.1002/anie.201907674. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Zhang X, Ling Y, Li F, Bond AM, et al. Controllable synthesis of few-layer bismuth subcarbonate by electrochemical exfoliation for enhanced CO2 reduction performance. Angew. Chem. Int. Ed. 2018;57(40):13283–13287. doi: 10.1002/anie.201807466. [DOI] [PubMed] [Google Scholar]
- 18.Ravel B, Newville M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005;12:537–541. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- 19.Greaves C, Blower SK. Structural relationships between Bi2O2CO3 and β-Bi2O3. Mater. Res. Bull. 1988;23(7):1001–1008. doi: 10.1016/0025-5408(88)90055-4. [DOI] [Google Scholar]
- 20.Kresse G, Hafner J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B. 1993;48(17):13115–13118. doi: 10.1103/PhysRevB.48.13115. [DOI] [PubMed] [Google Scholar]
- 21.Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996;6(1):15–50. doi: 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- 22.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996;77(18):3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 23.Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999;59(3):1758–1775. doi: 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- 24.Peterson AA, Abild-Pedersen F, Studt F, Rossmeisl J, Nørskov JK. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010;3(9):1311–1315. doi: 10.1039/c0ee00071j. [DOI] [Google Scholar]
- 25.Peterson AA, Nørskov JK. Activity descriptors for CO2 electroreduction to methane on transition-metal catalysts. J. Phys. Chem. Lett. 2012;3(2):251–258. doi: 10.1002/aenm.201903083. [DOI] [Google Scholar]
- 26.Tong X, Yang Z, Feng J, Li Y, Zhang H. BiOCl/UiO-66 composite with enhanced performance for photo-assisted degradation of dye from water. Appl. Organomet. Chem. 2018;32(2):e4049. doi: 10.1002/aoc.4049. [DOI] [Google Scholar]
- 27.Lu J, Zhou W, Zhang X, Xiang G. Electronic structures and lattice dynamics of layered BiOCl single crystals. J. Phys. Chem. Lett. 2020;11(3):1038–1044. doi: 10.1021/acs.jpclett.9b03575. [DOI] [PubMed] [Google Scholar]
- 28.Zhao W, Wang Y, Wang A, Qian J, Zhu W, et al. Novel Bi2O2CO3/polypyrrole/g-C3N4 nanocomposites with efficient photocatalytic and nonlinear optical properties. RSC Adv. 2017;7(13):7658–7670. doi: 10.1039/c6ra28346b. [DOI] [Google Scholar]
- 29.Tobon-Zapata GE, Etcheverry SB, Baran EJ. Vibrational spectrum of bismuth subcarbonate. J. Mater. Sci. Lett. 1997;16:656–657. doi: 10.1023/A:1018527602604. [DOI] [Google Scholar]
- 30.Grice JD. A solution to the crystal structures of bismutite and beyerite. Can. Mineral. 2002;40(2):693–698. doi: 10.2113/gscanmin.40.2.693. [DOI] [Google Scholar]
- 31.Miao Z, Wang Q, Zhang Y, Meng L, Wang X. In situ construction of S-scheme AgBr/BiOBr heterojunction with surface oxygen vacancy for boosting photocatalytic CO2 reduction with H2O. Appl. Catal. B Environ. 2022;301:120802. doi: 10.1016/j.apcatb.2021.120802. [DOI] [Google Scholar]
- 32.Huang H, Xiao K, Yu S, Dong F, Zhang T, et al. Iodide surface decoration: a facile and efficacious approach to modulating the band energy level of semiconductors for high-performance visible-light photocatalysis. Chem. Commun. 2016;52(2):354–357. doi: 10.1039/c5cc08239k. [DOI] [PubMed] [Google Scholar]
- 33.Wei S, Zhong H, Wang H, Song Y, Jia C, et al. Oxygen vacancy enhanced visible light photocatalytic selective oxidation of benzylamine over ultrathin Pd/BiOCl nanosheets. Appl. Catal. B Environ. 2022;305:121032. doi: 10.1016/j.apcatb.2021.121032. [DOI] [Google Scholar]
- 34.Friedman AK, Shi W, Losovyj Y, Siedle AR, Baker LA. Mapping microscale chemical heterogeneity in nafion membranes with X-ray photoelectron spectroscopy. J. Electrochem. Soc. 2018;165(11):H733–H741. doi: 10.1149/2.0771811jes. [DOI] [Google Scholar]
- 35.Cen W, Xiong T, Tang C, Yuan S, Dong F. Effects of morphology and crystallinity on the photocatalytic activity of (BiO)2CO3 nano/microstructures. Ind. Eng. Chem. Res. 2014;53(39):15002–15011. doi: 10.1021/ie502670n. [DOI] [Google Scholar]
- 36.Kar P, Maji TK, Nandi R, Lemmens P, Pal SK. In-situ hydrothermal synthesis of Bi-Bi2O2CO3 heterojunction photocatalyst with enhanced visible light photocatalytic activity. Nano-Micro Lett. 2017;9:18. doi: 10.1007/s40820-016-0118-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji W, Niu J, Zhang W, Li X, Yan W, et al. An electroactive ion exchange hybrid film with collaboratively-driven ability for electrochemically-mediated selective extraction of chloride ions. Chem. Eng. J. 2022;427:130807. doi: 10.1016/j.cej.2021.130807. [DOI] [Google Scholar]
- 38.Rajeevan NE, Kumar R, Shukla DK, Thakur P, Brookes NB, et al. Bi-substitution-induced magnetic moment distribution in spinel BixCo2-xMnO4 multiferroic. J. Phys. Condens. Matter. 2009;21(40):406006. doi: 10.1088/0953-8984/21/40/406006. [DOI] [PubMed] [Google Scholar]
- 39.Shukla DK, Kumar R, Mollah S, Choudhary RJ, Thakurm P, et al. Swift heavy ion irradiation induced magnetism in magnetically frustrated BiMn2O5 thin films. Phys. Rev. B. 2010;82(17):174432. doi: 10.1103/PhysRevB.82.174432. [DOI] [Google Scholar]
- 40.Qiao R, Chuang YD, Yan S, Yang W. Soft X-ray irradiation effects of Li2O2, Li2CO3 and Li2O revealed by absorption spectroscopy. PLoS ONE. 2012;7(11):e49182. doi: 10.1371/journal.pone.0049182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Han J, Zhu Y, Zhou R, Jaye C, et al. Probing the dependence of electron transfer on size and coverage in carbon nanotube-quantum dot heterostructures. J. Phy. Chem. C. 2015;119(47):26327–26338. doi: 10.1021/acs.jpcc.5b08681. [DOI] [Google Scholar]
- 42.Ye Y, Kawase A, Song MK, Feng B, Liu YS, et al. X-ray absorption spectroscopy characterization of a Li/S cell. Nanomaterials. 2016;6(1):14. doi: 10.3390/nano6010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu PF, Zu MY, Zheng LR, Yang HG. Bismuth oxyiodide microflower-derived catalysts for efficient CO2 electroreduction in a wide negative potential region. Chem. Commun. 2019;55(82):12392–12395. doi: 10.1039/c9cc05089b. [DOI] [PubMed] [Google Scholar]
- 44.Lv W, Bei J, Zhang R, Wang W, Kong F, et al. Bi2O2CO3 nanosheets as electrocatalysts for selective reduction of CO2 to formate at low overpotential. ACS Omega. 2017;2(6):2561–2567. doi: 10.1021/acsomega.7b00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng T, Tan C, Zhang S, Tu T, Peng H, et al. Raman spectra and strain effects in bismuth oxychalcogenides. J. Phys. Chem. C. 2018;122(34):19970–19980. doi: 10.1021/acs.jpcc.8b05475. [DOI] [Google Scholar]
- 46.Zhang M, Wei W, Zhou S, Ma DD, Cao A, et al. Engineering conductive network of atomically thin bismuthene with rich defects enables CO2 reduction to formate with industry-compatible current densities and stability. Energy Environ. Sci. 2021;14:4998–5008. doi: 10.1039/d1ee01495a. [DOI] [Google Scholar]
- 47.Han N, Wang Y, Yang H, Deng J, Wu J, et al. Ultrathin bismuth nanosheets from in situ topotactic transformation for selective electrocatalytic CO2 reduction to formate. Nat. Commun. 2018;9:1320. doi: 10.1038/s41467-018-03712-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen T, Liu T, Ding T, Pang B, Wang L, et al. Surface oxygen injection in tin disulfide nanosheets for efficient CO2 electroreduction to formate and syngas. Nano-Micro Lett. 2021;13:189. doi: 10.1007/s40820-021-00703-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao C, Ma DD, Gu JF, Xie X, Zeng G, et al. Metal-organic layers leading to atomically thin bismuthene for efficient carbon dioxide electroreduction to liquid fuel. Angew. Chem. Int. Ed. 2020;132(35):15124–15130. doi: 10.1002/anie.202005577. [DOI] [PubMed] [Google Scholar]
- 50.Xing Y, Kong X, Guo X, Liu Y, Li Q, et al. Bi@Sn core-shell structure with compressive strain boosts the electroreduction of CO2 into formic acid. Adv. Sci. 2020;7(22):1902989. doi: 10.1002/advs.201902989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Norskov JK, Bligaard T, Rossmeisl J, Christensen CH. Towards the computational design of solid catalysts. Nat. Chem. 2009;1:37–46. doi: 10.1038/nchem.121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.