

Abstract
Histone deacetylases (HDACs) are clinically validated epigenetic drug targets for cancer treatment. HDACs inhibitors (HDACis) have been successfully applied against a series of cancers. First-generation inhibitors are mainly pan-HDACis that target multiple isoforms which might lead to serious side effects. At present, the next-generation HDACis are mainly focused on being class- or isoform-selective which can provide improved risk–benefit profiles compared to non-selective inhibitors. Because of the rapid development in next-generation HDACis, it is necessary to have an updated and state-of-the-art overview. Here, we summarize the strategies and achievements of the selective HDACis.
Histone deacetylases (HDACs) are clinically validated epigenetic drug targets for cancer treatment.
1. Introduction
Epigenetic regulations have been considered to be one of the important factors in tumor initiation and progression,1 among which acetylation is one of the most well studied modifications.2 The acetylation status is controlled by two counteracting enzyme families: the histone acetyltransferases (HATs) and the histone deacetylases (HDACs).3 Both of these enzymes exert their actions on the ε-amino group of Lys residues.4,5 Acetylation of the lysine side chains is linked to transcriptionally active genes, while hypoacetylated histones are associated with transcriptionally silent regions of the genomes.6–8 To date, 18 human HDACs were identified and classified into four classes according to their sequence homologies to yeast and domain organization: class I (HDACs 1, 2, 3 and 8), class II (HDACs 4, 5, 7, 9, 6 and 10), class III (SIRT1-7), and class IV containing a single isoform (HDAC11).9,10 As shown in Table 1. Class I, class II and class IV HDACs are Zn2+-dependent enzymes, whereas class III HDACs are structurally distinct and are characterized by their nicotinamide adenine dinucleotide (NAD+) dependency.11,12
Classification of histone deacetylases (HDACs) enzymesa.
| Class | Member | PDB code | Location | Expression |
|---|---|---|---|---|
| Class I (Zn2+-dependent) | HDAC1 | 4BKX 13 | Nucleus | Ubiquitous |
| HDAC2 | 4LXZ 14 | |||
| HDAC3 | 4A69 15 | |||
| HDAC8 | 1T64 16 | |||
| Class IIa (Zn2+-dependent) | HDAC4 | 2VQM 17 | Nucleus/cytoplasm | Specific |
| HDAC5 | — | |||
| HDAC7 | 3C10 18 | |||
| HDAC9 | — | |||
| Class IIb (Zn2+-dependent) | HDAC6 | 5EDU 19 | Mainly cytoplasm | Specific |
| HDAC10 | — | |||
| Class III (NAD+-dependent) | SIRT1 | 4ZZH 20 | Nucleus | Ubiquitous |
| SIRT2 | 1J8F 21 | Cytoplasm | ||
| SIRT3 | 3GLS 22 | Mitochondria | ||
| SIRT4 | — | Mitochondria | ||
| SIRT5 | 3RIG 23 | Mitochondria | ||
| SIRT6 | 3K35 24 | Nucleus | ||
| SIRT7 | 5IQZ 25 | Nucleus | ||
| Class IV (Zn2+-dependent) | HDAC11 | — | Nucleus/cytoplasm | Ubiquitous |
“–” This type Homo sapiens HDAC isoform don't display PDB code.
Class I HDACs include HDACs 1, 2, 3 and 8, which are primarily located in the nucleus and homologous to the yeast RPD3 protein,26 which are expressed ubiquitously in various human tissues and generally play an important role in cell proliferation, differentiation and cell cycle progression etc.27 A prominent feature of class I HDACs, with the exception of HDAC8, is that their functions are mediated as part of multiprotein complexes. These cofactors are necessary for their deacetylases activities and modulating the interactions of other cooperative HDACs with the promoters of genes.28 HDAC1 and HDAC2 interact with each other to form the catalytic core of several large complexes such as CoREST, NuRD and Sin3A complexes.29–31 It is essential to point out that HDAC1 and 2 are usually co-expressed and the homo and/or heterodimers are essential for their catalytic activities. HDAC3 is generally found in complex with HDAC4, 5 and 7 or the nuclear receptor corepressors (N-CoR) and SMRT.32,33 HDAC8, differing from the other class I HDACs, is mainly restricted in specific tissue and displays deacetylase activity as a single polypeptide (Fig. 1).34,35
Fig. 1. Representative class I HDAC complexes.28.
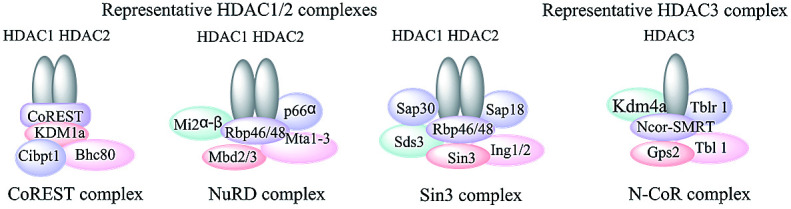
Class II HDACs (HDACs 4, 5, 6, 7, 9 and 10) are homologous to the yeast Hda1 protein, and have both histone and non-histone proteins targets.36 This class HDACs can be further classified in two subclasses based on the presence or absence of a double catalytic domain. Class IIa includes HDACs 4, 5, 7 and 9 and class IIb includes HDACs 6 and 10.37 Class IIa HDACs show poor deacetylase activities unless acting in concert with class I HDACs.38,39 For example, the catalytic domain of HDAC4 is interacted with HDAC3 within a larger NCoR–SMRT complex, and HDAC4 is essential for HDAC activity and transcriptional repression of target genes by the NcoR–SMRT–HDAC3 complex.40,41 HDAC6 is a unique histone deacetylase: it features two independent catalytic domains and which can deacetylate α-tubulin in vitro and in vivo.42,43 Through the deacetylation of tubulin, HDAC6 plays a central role in cytoskeleton regulation and cytoskeleton mediated processes, such as cell–cell interaction and cell migration.44
2. Histone deacetylase inhibitors (HDACis)
HDACs play a pivotal role in the regulation of multiple processes of life, from gene expressions to protein activities. Importantly, the high expression of HDACs were found in several types of cancers.45,46 For instance, transcriptional upregulation of HDAC1 is observed in colorectal and pancreatic cancers. In addition, HDAC3 in lung and colon cancers, HDAC8 in neuroblastoma were also found to be overexpressed.47–49 HDACs not only can remove the acetyl group from the histones but also deacetylate other non-histone proteins pertaining to the origin and development of tumors.50–53 In tumors, the biological functions of HDACs are mainly involved in the promotion of the proliferation, invasion, migration and angiogenesis of tumor cells.54,55 Therefore, HDACis are proposed to have great potentials in the therapies of human cancers.56
Most HDACis share the common pharmacophore models which consist of three parts: a cap part, a zinc binding group (ZBG), and a linker part connecting ZBG and cap part (Fig. 2).57,58 The cap group usually has an aromatic character or heteroaromatic hydrophobic moiety which mediates the interaction with the amino acids at the rim of the enzyme,59 and which is mainly responsible for the HDAC isoforms selectivities.60 ZBG acts as the chelating one for Zn2+ in the active site of HDACs, modification of the ZBG can introduce a change in potency of inhibitors.61
Fig. 2. The common pharmacophore model of HDACis. (A) Space-filling representation of TSA in the active-site pocket of HDLP. (B) Schematic representation of HDLP–TSA interactions (adapted with permission from 62). (C) Crystal structure of HDAC8 with SAHA. (D) Binding mode of SAHA with HDAC8 (this figure was cited from ref. 63).
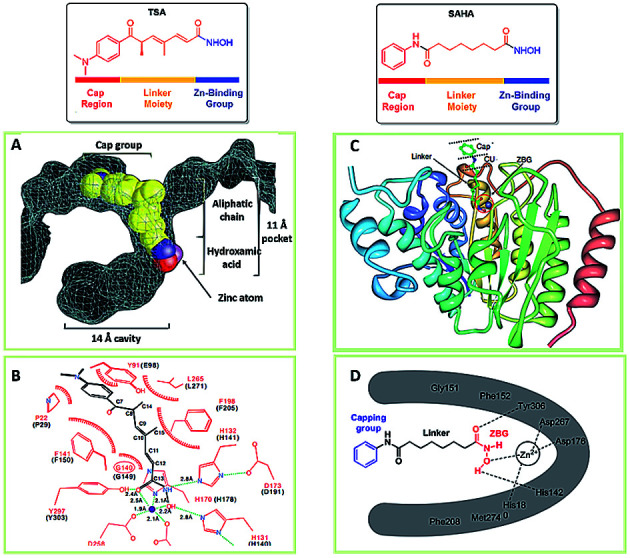
The first X-ray structure of histone deacetylase like protein (HDLP) with TSA was elucidated by Finnin et al. in 1999 (Fig. 2A and B).62 The structure revealed the presence of a tube-like, 11 Å internal channel for accommodation of the inhibitor. The hydroxamic acid is bound to the active site Zn2+ center, present at the bottom of the internal channel. Another 14 Å internal cavity, is adjacent to the active site. Crystal structure of HDAC8 with SAHA was shown in Fig. 2C, and the binding mode was similar to TSA with HDLP. Fig. 2D displayed the binding interaction of SAHA at active site of HDAC8, the hydroxamic acid group of SAHA makes the same contacts to the Zn2+ and active-site residues.63
Based on ZBG chemical structures, HDACis could be divided into five main classes: hydroxamates, cyclic peptides, benzamides, short-chain fatty acids, ketones and others.64,65 Among which, HDACis containing hydroxamates are the most investigated and potent.66 However, most hydroxamates are pan-HDACis, while the benzamides have increased class I selectivities. Until now, four HDACis, namely Vorinostat (1, SAHA), Belinostat (2, PXD101), Panobinostat (3, LBH589), Romidepsin (4, FK228) have been approved by the US FDA and one HDACi, namely Chidamide (5, CS005) was approved by the China FDA, for the treatments of hematologic malignancies and more than 20 inhibitors are now in clinical trials67–71 (Fig. 3).
Fig. 3. Examples of HDACis with different ZBG types.

3. Selective HDACis
The current HDACis are mostly belong to pan-inhibitors with several obvious side effects, such as fatigue, thrombo-cytopenia and gastrointestinal.72,73 Considering the distinct tissue distribution and cellular localization of individual HDACs, as well as the relationship between specific HDAC isoforms and different kinds of cancers, researchers hypothesized isoform-selective HDACis may possess better therapeutic index and fewer adverse effects.74–76 However, the therapeutic advantages of isoform-selective HDACis have not yet been proved clinically, and are still in course of studies.77,78
3.1. Class I selective HDACis
Crystal structures of the catalytic domain of all class I HDACs have been solved by X-ray diffraction. There is only one residue difference between HDAC 1 and 2, S263 in HDAC1 and A264 in HDAC2. In HDAC3, there is one insertion of phenylalanine (F199), which alters the orientation of tyrosine (Y198). The most noticeable difference in HDAC8 is loop L1, formed by seven amino acid residues (residues 30–36) (Fig. 4A).79 The flexible L1 loop is required for selective inhibitors binding to a HDAC8-specific pocket which is absent in other class I HDACs (Fig. 4B and C).80
Fig. 4. (A) Class I HDAC crystal structures (this figure was cited from ref. 79). (B) Surface representation of the pocket accommodating the linker and capping groups of the HDAC8-selective inhibitors. (C) Surface representation of the same region in hHDAC3 (this figure was cited from ref. 80).

3.1.1. Inhibitors of HDAC1-3
Many class I HDAC inhibitors contain 2-aminoanilide as ZBG, such as 12 (MS-275) and 13 (MGCD0103), which were selective for HDAC1-3.28 This type of inhibitors exploited a foot pocket close to the active site, which was only exist in HDAC1-3. Compound 12 and 13 displayed IC50 values in the nM range against HDAC1 and HDAC1&2, respectively.81,82 Compound 14 with bulky branched cap group displayed 10-fold and 20-fold potencies for HDAC1 compared to HDAC2 and HDAC3, respectively.83 Crystal structure of HDAC2 with 2-aminoanilide scaffold confirmed that the anilide scaffold could access the foot pocket next to the catalytic region. Then several 2-aminoanilides substituted in 5-position were designed for further occupying this foot pocket. Compound 15 displayed a 5-fold selectivity for HDAC1 over HDAC2.84 Compound 16 has been synthesized for the specific inhibition of HDAC1 and 2 versus HDACs 3–8 and compound 17 displayed high selectivity for HDAC1, with a 31-fold increase compared to HDAC2 (Fig. 5).85
Fig. 5. Structures of some benzamides selective inhibitors.

Based on a series of 504 triazole candidates, Suzuki et al. designed a series of HDAC3 selective inhibitors and found compound 18 showed potent HDAC3 inhibition with submicromolar IC50, and which didn't inhibit other HDAC isozymes even at 100 μM.86 Marson et al. described an oxazoline capping group with a N-(2-aminophenyl) benzamide unit, and compound 19 displayed good inhibitory activity against HDAC3, almost a 13-fold increase compared to HDAC1.87 Hsieh et al. developed a series of HDAC3 selective inhibitors, the target compounds 19A and 19B displayed high HDAC3 potency and selectivity. Especially, they exhibited efficacy in suppressing TNBC cells via the downregulation of β-catenin. Furthermore, compound 19B suppressed tumorigenesis in vivo (Fig. 6).88
Fig. 6. Structures of some benzamides selective inhibitors.

3.1.2. Inhibitors of HDAC8
HDAC8 is a HDAC isoform containing 377 amino acids residues with stand-alone deacetylase activity in vitro that can localize in either the nucleus or the cytoplasm.89 The L1 loop, formed by seven amino acids residues (Ser30, Leu31, Ala32, Lys33, Ile34, Pro35 and Lys36), is highly flexible, which is located toward the proximity of the active site and can undergo conformational changes for substrates binding. The Ser39 phosphorylation near the catalytic site is special for HDAC8,16 which will attribute to design specific HDAC8 inhibitors.
Compound 20 (PCI34051, Fig. 7.) was probably the most widely used HDAC8 specific inhibitor, reported by Balasubramanian et al. This hydroxamic acid possessed potent HDAC8 inhibition with an IC50 of 10 nM and was >200 fold selectivity over HDACs 1/2/3/6/10. The 4-methoxybenzyl fits into the sub-pocket of HDAC8 which may be a reason for the selectivity toward HDAC8 enzyme.90
Fig. 7. Chemical structure of compound 20 (PCI34051) and the docking pose of PCI34051 in the side pocket of HDAC8 (PDB ID 2VX5) (this figure was cited from ref. 90).

Zhang et al. reported a series of tetrahydroisoquinoline-based hydroxamic acid HDACis.91 The tertiary butyloxycarbonyl (Boc) group was located on the secondary amine atom and 4-methyloxy- phenyl group seemed beneficial to HDAC8 inhibitory activity. Molecular docking study displayed that the methoxyphenyl group could form π–π interaction with Tyrosine 100 (Y100). Zhang et al. further optimized the lead compound 21,92 compounds with Boc group were shown more potent activities than their corresponding deprotected analogues which indicated that the hydrophobicity of the cap group is very beneficial to the inhibitory activities. Based on the compound 21a, more potent compounds were synthesized. However, further cellular activities evaluations revealed that their antiproliferative activities were not as excellent as its HDACs inhibitory activities. Among these compounds, compound 22 showed the highest HDAC8 inhibitory activity (IC50 = 47 nM), about 2-fold selective over HeLa nuclear extract (HDAC1 and 2) and 4-fold selective over HDAC6 (Fig. 8).
Fig. 8. Structures of tetrahydroisoquinoline-based HDAC8 inhibitors.

Zhang et al. also designed and synthesized a series of tripeptidomimetics HDACis with spiro-ring containing sulfur atoms as a cap group, several compounds showed outstanding potencies against HDAC8.93 The results revealed that branched cap groups with aromatic ring could efficiently increase its inhibitory activities. In brief, a hydrophobic Boc-protected amino group was optimal for HDAC8 inhibitory activity compared with the Boc-cleaved counterpart. Compound 24a (IC50 = 35 nM) and compound 24b (IC50 = 21 nM) were 28- and 21-fold HDAC8 selective over HDAC1, respectively. In the following work, Zhang et al. developed a novel series of N-hydroxy-3-sulfamoylbenzamide-based HDAC8 selective inhibitors. Three compounds 25a–c showed effective HDAC8 inhibition with nanomolar IC50 values, and possessed selective anti-proliferative activities to two T-cell leukemia cell lines (Fig. 9).94
Fig. 9. Structures of novel HDAC8 inhibitors.

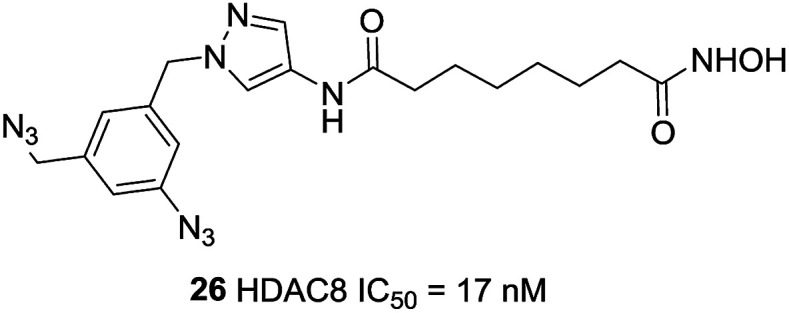
Neelarapu et al. described potent isoxazole- and pyrazole-based HDAC3 and HDAC8 diazide probes.95 The bulky substituent in the para position of the terminal phenyl ring of target compounds leads to its lower activities for HDAC8 and compounds with the azide group exhibited tolerable HDAC8 inhibitory activities. The most potent compound 26 was 8-fold more active toward HDAC8 than for HDAC3. Docking studies suggested that the pyrazole-based ligands are flexible enough to occupy the second binding site of HDAC8 (Fig. 10).
Fig. 10. Structures of pyrazole-based HDAC8 inhibitors.
Tang et al. developed an efficient synthetic method in 96-well plates for high throughput screening selective HDAC8 inhibitors.96 It could be found that compounds with comparatively bulky heteroaryl cap group as well as the aromatic linker displayed better HDAC8 inhibitory activities. Compound 29 possessed the best HDAC8 inhibition (IC50 = 23 nM) among the investigated ones (Fig. 11).
Fig. 11. Structures of heteroaryl-based HDAC8 inhibitors.

Suzuki et al. designed a library of triazole-containing HDAC8 inhibitors using click chemistry-based Cu(i)-catalyzed-azide-alkyne cycloaddition (CuAAC).97 Compound 30, the most active HDAC8 selective inhibitor in this series, had a U-shaped conformation in the active site of HDAC8, especially, the phenylthiomethyl group binds to a hydrophobic pocket formed by Trp141, Ile34, and Pro35, which is unique to HDAC8. The triazole ring is also important to fix the orientation of the hydroxamate and the phenylthiomethyl group appropriately, which is mainly responsible for HDAC8 inhibitory activity and selectivity. In order to further optimizing the triazole linker motif, a new series of compounds were synthesized, in which the triazole ring of 30 (NCC149) was changed with phenyl or various five-membered aromatic rings.98 The results demonstrated that the five-membered ring is still necessary for fixing the orientation of hydroxamate and the hydrophobic pocket-binding phenylthiomethyl group. Compound 31, with reversed triazole, showed more potent HDAC8 inhibitory activity than 30 (Fig. 12).
Fig. 12. Structures of triazole-based HDAC8 inhibitors.

Ingham et al. discovered a highly potent HDAC8 inhibitor with IC50 = 0.8 nM.99 From the SAR study, it was observed that small hydrophobic groups at the R3 position, such as cyclopropane (32), were found to be beneficial and afforded greater inhibitions. At the same time, reduction of the alkyne resulted in the loss of inhibitory activity (Fig. 13).
Fig. 13. Structures of triazole-based HDAC8 inhibitors.

Huang et al. developed an ortho-aryl N-hydroxycinnamide HDAC8 inhibitors via structure-based drug design strategy (SBDD) and molecular modeling techniques.100 The results displayed that ortho-aryl substituted compounds showed higher potency than corresponding para substituted analogues, the ortho-oriented benzyl group may occupy the secondary hydrophobic surface pocket of HDAC8, thereby contributing to HDAC8 selectivity of compound 33. The molecular docking analysis showed that further hydrophobic incorporation into the ortho-phenyl moiety may make additional interactions to the active site of HDAC8, then more potent and more selective HDAC8 inhibitor (compound 34) was acquired, two to three-fold more potent than 33. In addition, compound 34 showed higher cytotoxicity against than 20 in three cancer cell lines (Fig. 14).
Fig. 14. Structures of ortho-aryl N-hydroxycinnamide-based HDAC8 inhibitors.

Taha et al. described the design and synthesis of a novel series of C1-substituted tetrahydroisoquinoline (TIQ)-based HDAC8 inhibitors.101 The TIQ-based compounds 35 and 36 displayed the highest potencies with IC50 values of 82 and 55 nM against HDAC8 and 330- and 135-fold selectivities over HDAC1, respectively. Docking studies showed that the C1-substituent pointed into the HDAC8 extended pocket (Fig. 15).
Fig. 15. Structures of tetrahydroisoquinoline-based HDAC8 inhibitors.

3.2. Class II selective HDACis
3.2.1. Inhibitors of HDAC6
The surface area of HDAC6 are different in shape and the channel of HDAC6 appears shallower and wider than other HDAC isoforms, which makes it possible for development of HDAC6 selective inhibitors.102 HDAC6, as the only HDAC containing two catalytic domains (Fig. 16A), is mainly expressed in the cytoplasm.103,104 The first catalytic domain (CD1) is enzymatically active. However, the second catalytic domain (CD2) is mainly responsible for the deacetylation of non-histones, such as α-tubulin, Hsp90 and cortactin,105 therefore HDAC6 plays a substantial role in chaperone activities and microtubule dynamics, which are responsible for cell mobility.106,107 Indeed, up-regulated expression of HDAC6 has been found closely linked to the invasive metastatic behavior of cancer cells.108 Therefore, HDAC6 represents a potential target for further investigation considering its therapeutic applications.
Fig. 16. (A) HDAC6 harbors a unique substrate-binding site (this figure was cited from ref. 104). (B) Superposition of broad-specificity HDACis (thin wheat stick figures) and the HDAC6 selective inhibitor HPOB (thin orange stick figure). (C) Close-up view of the Zn2+ binding site in the hCD2 complexes with broad-specificity HDACis. (D) Close-up view of the Zn2+ binding site in the hCD2-HPOB complex (this figure was cited from ref. 19).

Crystal structures of CD2 complexed with broad-specificity inhibitors and HDAC6-selective inhibitor HPOB revealed that the cap group of broad-specificity inhibitors clustered around a ‘hotspot’ on the L1 loop, and the hydroxamate hydroxy and carbonyl groups were interacted with H573 and Y745. However, only the hydroxy of hydroxamate group of HPOB coordinated to Zn2+(Fig. 16B–D).19 These data suggested that HDACis with bulky and short aromatic linker, big, rigid and hydrophobic cap are more efficient to achieve HDAC6 selectivity, which could preclude a closer approach of the hydroxamate group to Zn2+, and prevent the cap group of HDAC6 selective inhibitors from interacting with the L1 loop.109
Compound 37 (Tubacin), the first reported HDAC6 inhibitor, was developed by Schreiber and co-workers through cell-based screening of 7392 compounds.110 Tubacin was a “T” type structure with unique surface recognition region, including five aromatic rings and a dioxane scaffold. Studies have indicated that Tubacin showed different effects on HDAC1, HDAC6 and HDAC8, which were mainly derived from the surface difference between class I and class II HDACs.111 Unfortunately, its high lipophilicity made it more useful as a chemical tool other than a drug candidate. Compound 38 (ACY-1215) was the first HDAC6 selective inhibitors in clinical studies. Low doses of 38 combined with bortezomib or lenalidomide in anti-multiple myeloma can produce synergistic therapeutic effects.112 The structurally related drug ACY-241 was another selective HDAC6 inhibitor in phase I clinical development (Fig. 17).113
Fig. 17. Structures of novel HDAC6 inhibitors.

Kozikowski et al. prepared a series of biphenyl and phenylthiazole hydroxamic acid HDAC6 inhibitors.114 The phenylthiazole HDACi 40 was more potent than the biphenyl compound 39. Also, introduction of a Boc group led to an enhancement in the inhibitory activity toward HDAC6. However, the more HDAC6 selective analogues displayed reduced potencies against a panel of pancreatic cancer cell lines, which suggested that the anticancer properties of these compounds may not due to HDAC6 inhibition but with off-target effects. The same research group further developed a set of triazolylphenyl-based HDACi with modification at the cap region,115 the results showed that the substitution pattern of the central phenyl ring played a significant role in the selectivity for inhibition of HDAC6 over HDAC1. Using of nitrile oxide cycloaddition (NOC) chemistry, Kozikowski and co-workers reported potent HDACis which contained the aryl substituted isoxazole cap.116 Compound 43 displayed an impressive potency of 2 pM against HDAC6 together with a superb level of enzyme selectivity. The cap region interacted with only one side of the HDAC6 enzyme. The carbonyl group of the Boc group could interact with His499, which may be important in positioning the cap residue on the surface of protein (Fig. 18).
Fig. 18. Structures of heteroaryl-based HDAC6 inhibitors.

Based on molecular docking study and the feasibility analysis of synthesis, Yang et al. designed novel selective HDAC6 inhibitors using the quinazoline as the cap region.117 The SAR analysis indicated that the hydroxamic acid side chain on C-3 and introduction of a methoxy group to the C-4 position was necessary to keep the activity and selectivity on HDAC6. Compound 44, was the most potent selective inhibitor in these series of compounds for HDAC6 with an IC50 value of 17 nM and showed 25-fold and 200-fold selectivities relative to HDAC1 and HDAC8, respectively. This compound also displayed excellent low nanomolar antiproliferative effects against both the hematological and solid cancer cells (Fig. 19).
Fig. 19. Structures of quinazoline-based HDAC6 inhibitors.

On the basis of the strategy for creating multifunctional drugs, Duan et al. reported a novel series of phenylsulfonyl-furoxan-based hydroxamates with HDAC inhibitory and NO donating activities.118 The most potent hybrid, 45, displayed potent HDAC6 activity with IC50 of 7.4 nM, however, it exhibited pan-HDAC inhibition in a Western blot assay, which was likely due to class I HDACs inhibition caused by NO release at the cellular level. Further studies revealed that this compound induced a much stronger apoptotic effect and G1 phase arrest in HeLa cells as well as exhibited greater oral antitumor potency than SAHA in vivo (Fig. 20).
Fig. 20. Structures of novel HDAC6 inhibitors.

Based on celecoxib, Yang et al. designed and synthesized a series of HDACis bearing phenylpyrazol moiety.119 SAR analysis indicated that locating the linker group at 1′ of pyrazole gave the most selectivity, the most selective HDAC6 inhibitor 46, was with IC50 value of 20 nM. Docking studies revealed that compound 46 could occupy the hydrophobic pocket of HDAC6 with a π–π stack between phenyl ring and Phe679, which could account for the selectivity of 46 against HDAC6.
Yang et al. described a pharmacophore combining the pacritinib and vorinostat with JAK2 and HDAC bispecific inhibitors.120 Compound 47, with single-digit nanomolar potencies against JAK2 and HDAC6, showed broad antiproliferative potencies across a range of solid and hematological cell lines. Based on Ruxolitinib and Vorinostat, this group developed a new series of pyrazole substituted pyrrolopyrimidine based hydroxamates with HDAC and JAK inhibitory activities.121 Finally, compound 48 with the optimal length of six carbons and methyl group, displayed single digit nanomolar potency against HDAC6. On this basis, Yao et al. explored alternative attachment points for the hydroxamate bearing chain and the pyrazole substituent.122 When the pyrazole was substituted with homoallyl, compound 49, had a sub-nanomolar IC50 against HDAC6 of 0.25 nM and selectivity over HDAC1 with more than 300-fold potency. Binding models displayed that the homoallyl pyrazole substituent occupied in a shallow hydrophobic pocket around Thr678 and Met682 which could explain the observed strong inhibition of HDAC6 (Fig. 21).
Fig. 21. Structures of pyrrolopyrimidine based HDAC6 inhibitors.

Butler et al. found that the active channel of HDAC6 is wider and shallower than HDAC1 (17.5 Å vs. 12.5 Å), thus replacement of the traditional long methylene linker with bulkier and shorter aromatic moieties may help to enhance the selectivity for HDAC6.123 Then a series of carbazole based hydroxamic acids with alkylaryl linker were synthesized. However, as the carbazoles are too lipophilic, a tertiary amine was introduced to the tricyclic ring. The tetrahydro-γ-carboline analogue, compound 50 (Tubastatin A), with an IC50 of 15 nM against HDAC6 and over 1000-fold selectivity against HDAC1. In order to investigate a second generation of these compounds, a series of substituted β- and γ-carbolines based hydroxamic acids were designed,124 the subsequent SAR analysis found that substitutions to the 6-, 7-, 8-, and 9-positions of the cap group were not beneficial to increasing the selectivities of these compounds, while introducing aromatic functionalities at the 2-position were beneficial. Compound 51, with a 4-pyridyl group, showed subnanomolar potency against HDAC6 as well as approximately 5000-fold selectivity over HDAC1. By using a bicyclic cap as the structural scaffold, Shen et al. developed a set of benzimidazole based inhibitors that showed comparable potency and selectivity against HDAC6.125 Especially, compound 52, with a fluorine atom at the 3-position of the aryl linker, led to the increased lipophilicity, which may improve binding energies correlating with improved HDAC6 inhibition. Modeling studies revealed that benzimidazole could stabilize π–π stacking with Phe680 and the methyl group fitted into a small sub-pocket that aided in cap region of HDAC6. At the same time, this compound possessed improved pharmacokinetic properties compared to Tubastatin A. Lee et al. developed a series of 5-aroylindolyl substituted hydroxamic acids HDAC6 inhibitors for Alzheimer's disease. The most potent compound 53, with an IC50 value of 3.92 nM, was able to reduce tau phosphorylation and aggregation. The docking studies showed that the cap region of compound bound to the specific pocket of HDAC6 and the anisole moiety could form van der Waals interactions with residues N494, D496, and W497 (Fig. 22).
Fig. 22. Structures of carboline based HDAC6 inhibitors.

A series of sulfur analogues of 50 were prepared by Vreese et al.126 It was found that a sulfone unit could form hydrogen bonds between the oxygen atoms and the backbone nitrogen of residues Asp567 and Gly619, and sulfur oxidation seemed beneficial for bioactivities. Sulfones compounds 54 and 55, had IC50 values of 1.9 and 3.7 nM, respectively, as well as a 5789-fold and a 3243-fold selectivities against HDAC1. In further modification, compound 56, with a five-membered thiaheterocyclic ring, was also a potent HDAC6 inhibitor. Meanwhile, five membered sulfones compounds display appropriate preliminary ADME/Tox profile (Fig. 23).127
Fig. 23. Structures of carboline based HDAC6 inhibitors.

Based on the findings of Butler et al., Leonhardt et al. designed a set of tetracyclic tetrahydro-β-carboline derivatives.128 The additional piperazine-2,5-dione structure was proven to be more potent than the imidazolidine-2,4-dione structure. Lead compound 57, with IC50 value of 3.73 nM, was a stronger HDAC6 inhibitor than 50 as well as had a selective index of >200 against HDAC1, 2, 4, 8 and 10. By combination of the fluorophore of 4 MS and 50, Zhang et al. designed HDAC6 selective inhibitors 58a and 58b as small-molecular fluorescent probes.12958a displayed satisfying HDAC6 selectivity with the selective index of over 140, 700 and 210 against HDAC1/2/3 and could label MG132-induced aggresome because of its high HDAC6 selectivity. Encouraged by these positive results, Ho et al. synthesized a series of fluorescent HDAC6 selective inhibitors with substituents on the naphthalimide skeleton. Compound 59 with the methoxy group at position 4 of naphthalimide exhibited the greatest HDAC6 inhibitory activity (IC50 = 0.1 nM) and HDAC6/1 selectivity (1080-fold). Compound 59 could also increase α-tubulin acetylation in a dose-dependent manner and showed the most effective activity against six cancer cell lines (Fig. 24).102
Fig. 24. Structures of carboline based HDAC6 inhibitors.

Through virtual screening, Schlimme et al. identified compound 60 as a HDAC6 inhibitor with an IC50 of 300 nM.130 In order to further improving the potency and selectivity of 60, this group reported the preparation of thiazole-, oxazole-, and oxadiazole-containing biarylhydroxamic acids.131 In general, the thiazole- and oxadiazole-containing derivatives were much less potent and selective than the oxazole containing compounds, and the para-position substitutions were better for selectivities. The 4-bromophenyl substituted oxazole hydroxamate 61 was the most potent analogues between these series of compounds, with IC50 value of 59 nM. Docking studies revealed that the oxazole interacted with Phe566 and Phe520, which could explain the high selectivity of para-substituted phenyl-oxazoles based compounds (Fig. 25).
Fig. 25. Structures of thiazole and oxazole based HDAC6 inhibitors.

Blackburn et al. screened several hundred hydroxamic acids for inhibitory activity of HDAC6 and identified 4-(aminomethyl)-N-hydroxybenzamide as potent inhibitors of HDAC6 with improved selectivity.132 Heterocyclic analogues such as compound 62 (tetrahydroisoquinolines) showed enhanced HDAC6 selectivity, and had an IC50 of 36 nM against HDAC6. In addition, compound 62 showed negligible inhibition against matrix metalloproteases such as MMPs 2, 9, and 4 (>100 μM). In further modifications, this group designed three additional series based on 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazine, 4,5,6,7-tetrahydropyrazolopyrazine and 5,6,7,8-tetrahydro-imidazo[1,2-a]pyrazines.133 And 63 was the most potent HDAC6 inhibitors in these series of compounds, with IC50 value of 33 nM, almost 100-fold selectivity versus HDAC8. Homology modeling revealed that fused ring spacer was more effectively occupied the channel of HDAC6, and the hydrophobic capping group interacted with the protein surface (Fig. 26).
Fig. 26. Structures of novel HDAC6 inhibitors.

Based on the homology model, Bergman et al. found that the entrance to the binding site was wider and shallower for HDAC6 than HDAC1, then a series of compounds with the introduction of certain branched-elements to the linker were synthesized.134 Structure–activity relationship studies exhibited that compounds bearing a branched linker group resulted in the increased potency and selectivity for HDAC6. The n-butyl-containing compound 64 displayed nanomolar inhibitory potency against HDAC6 and indicated excellent selectivity over HDAC1 (600-fold). The same group further reported SAR studies in a series of analogues based on the scaffold of compound 64.135 The new compound 65, bearing added indazole ring, exhibited improved potency against HDAC6 (IC50 = 1.6 nM) and retained selectivity (450-fold) over HDAC1. Docking simulations revealed that one of the nitrogen atoms in the indazole ring engage in hydrogen bonding interactions with the carbonyl group of Ala641, which may be the reason for the improving HDAC6 activity and selectivity. The in vivo studies demonstrated that this compound possessed improving capability to inhibit tumor growth in melanoma models through regulating inflammatory and immune responses (Fig. 27).
Fig. 27. Structures of branched linker based HDAC6 inhibitors.

Lee et al. found 66 (HPOB) was a HDAC6 selective inhibitor, with an IC50 value of 52 nM against HDAC6, almost 50-fold selectivity over HDAC1. 66 enhanced Etoposide, Doxorubicin, or SAHA-induced transformed cell death, which indicated that 66 had advantage in combination therapy to enhance the potency of antitumor drugs.136 In further study, Lee et al. described a new selective HDAC6 inhibitor 67 (HPB), with an IC50 value of 31 nM, almost 15 to 400-fold more potent than other HDACs.13767 selectively induced accumulation of acetylated α-tubulin and other substrates of HDAC6. 67 inhibited cell growth but did not influence cell death of normal and transformed cells. However, HPB enhanced transformed cell death caused by the anticancer drugs Paclitaxel or SAHA (Fig. 28).
Fig. 28. Structures of branched linker based HDAC6 inhibitors.

Diedrich et al. designed a series of selective HDAC6 inhibitors utilizing peptoid-based branched cap groups.138 The most potent HDAC6 inhibitor 68 (IC50 = 1.59 nM) exhibited selectivity index of 46 289 against HDAC4 and 126 against HDAC2. Especially, compound 68 displayed remarkable chemo-sensitizing properties and completely resensitized Cal27 CisR cell line towards cisplatin. Porter et al. reported that 69 (Bavarostat) exhibited more than 16 000-fold selectivity in comparison with HDAC1-3.139 Docking studies revealed that the hybridization of the linker benzylic nitrogen and the steric bulky capping group affected the affinity of hydroxamate-Zn2+ coordination which contributed to HDAC6 selectivity (Fig. 29).
Fig. 29. Structures of peptoid-based HDAC6 inhibitors.

Bracher et al. reported the preparation of phenothiazines based benzhydroxamic acid HDAC6 inhibitors.140 They confirmed the beneficial effect of the benzylic linker on potency and selectivity for HDAC6 in these series of inhibitors. The lead compound 70 showed impressive HDAC6 inhibition (IC50 = 22 nM) and a selectivity factor (SF) of 231 over HDAC1. The selective inhibition of HDAC6 by analogues of 70 (71 and 72) was also validated in cell culture with western blotting of tubulin versus histone acetylation. Docking studies of compound 70 to HDAC6 displayed that the tricyclic ring system can interact with the rim of the binding tunnel. Song et al. utilized structure-based virtual screening and provided the novel HDAC6 inhibitor 73. This compound inhibited HDAC6 with an IC50 value of 56 nM and displayed excellent selectivity for HDAC6 over other HDAC isoforms. At the same time, compound 73 could increase the acetylation of a-tubulin but not histone H3, which also confirmed the selectivity for HDAC6 of this inhibitor (Fig. 30).141
Fig. 30. Structures of novel HDAC6 inhibitors.

4. Conclusions
The development of the selective HDACis have made great progresses through modifying the cap region and linker, however, the design of selective HDACis remains a great challenge. The high sequence homology between catalytic sites as well as the tendency of HDACs in vivo to become functionally active multiprotein aggregates limited the development of isotype specific HDACis, since inhibitory activity of single HDAC could be different from which in protein complexes. It is noting that the development of X-ray crystallography, molecular modeling studies and computer-aided drug design may provide further contributions to the design of novel selective HDACis.
HDACis are more active in hematological cancers than solid tumors, yet the reason is not clear. In order to overcome this issue, the possible strategy is to design multifunctional or multitarget HDACis synthesized by combining pharmacophores of typical HDACis and other inhibitors. A huge number of multi-targets inhibitors based on HDAC inhibitory activity have now been developed and showed remarkable anti-tumor activity. Numerous multi-target inhibitors have been demonstrated the advantages in enhancing anti-tumor efficacy and reducing side effects. However, the design and synthesis of multi-target compounds still require more exploration due to the multifactorial mechanistic nature of cancer. Therefore, the research of multi-target inhibitors is also another important direction for next generation HDACis.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
We acknowledge the financial support from National Natural Science Foundation of China (No. 81673304 and 81703360), Open Research Project Foundation of Shanghai Key Laboratory of Regulatory Biology (2018LBR03), the Natural Science Foundation of Shandong Province (No. ZR2016HB43), the Young Taishan Scholars Program (tsqn20161037), and the Shandong Talents Team Cultivation Plan of University Preponderant Discipline (No. 10027).
References
- Abdelfatah E. Kerner Z. Nanda N. Ahuja N. Ther. Adv. Gastroenterol. 2016;9:560–579. doi: 10.1177/1756283X16644247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M. Montgomery R. L. Olson E. N. Nat. Rev. Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaskos C. Tomos I. Garmpis N. Karakatsani A. Dimitroulis D. Garmpi A. Spartalis E. Kampolis C. F. Tsagkari E. Loukeri A. A. Margonis G.-A. Spartalis M. Andreatos N. Schizas D. Kokkineli S. Antoniou E. A. Nonni A. Tsourouflis G. Markatos K. Kontzoglou K. Kostakis A. Tomos P. Anticancer Res. 2018;38:37–43. doi: 10.21873/anticanres.12189. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Fang H. Jiao J. Xu W. Curr. Med. Chem. 2008;15:2840–2849. doi: 10.2174/092986708786242796. [DOI] [PubMed] [Google Scholar]
- Li Z. Zhu W.-G. Int. J. Biol. Sci. 2014;10:757–770. doi: 10.7150/ijbs.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A. J. Li H. Patel D. J. Allis C. D. Nat. Rev. Mol. Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganai S. A. Ramadoss M. Mahadevan V. Curr. Neuropharmacol. 2016;14:55–71. doi: 10.2174/1570159X13666151021111609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallinari P. Di Marco S. Jones P. Pallaoro M. Steinkuhler C. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- de Ruijter A. J. M. van Gennip A. H. Caron H. N. Kemp S. van Kuilenburg A. B. P. Biochem. J. 2003;370:737–749. doi: 10.1042/bj20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoretti I. V. Lee Y.-M. Goodson H. V. J. Mol. Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Khan O. La Thangue N. B. Immunol. Cell Biol. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- Haberland M. Johnson A. Mokalled M. H. Montgomery R. L. Olson E. N. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7751–7755. doi: 10.1073/pnas.0903139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard C. J. Watson P. J. Celardo I. Gordiyenko Y. Cowley S. M. Robinson C. V. Fairall L. Schwabe J. W. R. Mol Cell. 2013;51:57–67. doi: 10.1016/j.molcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer B. E. L. Mintzer R. Fong R. Mukund S. Tam C. Zilberleyb I. Flicke B. Ritscher A. Fedorowicz G. Vallero R. Ortwine D. F. Gunzner J. Modrusan Z. Neumann L. Koth C. M. Lupardus P. J. Kaminker J. S. Heise C. E. Steiner P. J. Biol. Chem. 2013;288:26926–26943. doi: 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P. J. Fairall L. Santos G. M. Schwabe J. W. R. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somoza J. R. Skene R. J. Katz B. A. Mol C. Ho J. D. Jennings A. J. Luong C. Arvai A. Buggy J. J. Chi E. Tang J. Sang B.-C. Verner E. Wynands R. Leahy E. M. Dougan D. R. Snell G. Navre M. Knuth M. W. Swanson R. V. McRee D. E. Tari L. W. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Bottomley M. J. Lo Surdo P. Di Giovine P. Cirillo A. Scarpelli R. Ferrigno F. Jones P. Neddermann P. De Francesco R. Steinkuhler C. Gallinari P. Carfi A. J. Biol. Chem. 2008;283:26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz A. Min J. Allali-Hassani A. Schapira M. Shuen M. Loppnau P. Mazitschek R. Kwiatkowski N. P. Lewis T. A. Maglathin R. L. McLean T. H. Bochkarev A. Plotnikov A. N. Vedadi M. Arrowsmith C. H. J. Biol. Chem. 2008;283:11355–11363. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai Y. Christianson D. W. Nat. Chem. Biol. 2016;12:741–747. doi: 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H. Case A. W. Riera T. V. Considine T. Lee J. E. Hamuro Y. Zhao H. Jiang Y. Sweitzer S. M. Pietrak B. Schwartz B. Blum C. A. Disch J. S. Caldwell R. Szczepankiewicz B. Oalmann C. Ng P. Y. White B. H. Casaubon R. Narayan R. Koppetsch K. Bourbonais F. Wu B. Wang J. Qian D. Jiang F. Mao C. Wang M. Hu E. Wu J. C. Perni R. B. Vlasuk G. P. Ellis J. L. Nat. Commun. 2015;6:7645. doi: 10.1038/ncomms8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnin M. S. Donigian J. R. Pavletich N. P. Nat. Struct. Biol. 2001;8:621–625. doi: 10.1038/89668. [DOI] [PubMed] [Google Scholar]
- Jin L. Wei W. Jiang Y. Peng H. Cai J. Mao C. Dai H. Choy W. Bemis J. E. Jirousek M. R. Milne J. C. Westphal C. H. Perni R. B. J. Biol. Chem. 2009;284:24394–24405. doi: 10.1074/jbc.M109.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J. Zhou Y. Su X. Yu J. J. Khan S. Jiang H. Kim J. Woo J. Kim J. H. Choi B. H. He B. Chen W. Zhang S. Cerione R. A. Auwerx J. Hao Q. Lin H. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan P. W. Feldman J. L. Devries M. K. Dong A. Edwards A. M. Denu J. M. J. Biol. Chem. 2011;286:14575–14587. doi: 10.1074/jbc.M111.218990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyanka A. Solanki V. Parkesh R. Thakur K. G. Proteins: Struct., Funct., Bioinf. 2016;84:1558–1563. doi: 10.1002/prot.25085. [DOI] [PubMed] [Google Scholar]
- Yang X.-J. Seto E. Nat. Rev. Mol. Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo C. B. Jones P. A. Nat. Rev. Drug Discovery. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- Thaler F. Mercurio C. ChemMedChem. 2014;9:523–536. doi: 10.1002/cmdc.201300413. [DOI] [PubMed] [Google Scholar]
- Denslow S. A. Wade P. A. Oncogene. 2007;26:5433–5438. doi: 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- You A. Tong J. K. Grozinger C. M. Schreiber S. L. Proc. Natl. Acad. Sci. U. S. A. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazin M. J. Kadonaga J. T. Cell. 1997;89:325–328. doi: 10.1016/S0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- Wen Y. D. Perissi V. Staszewski L. M. Yang W. M. Krones A. Glass C. K. Rosenfeld M. G. Seto E. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7202–7207. doi: 10.1073/pnas.97.13.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagianni P. Wong J. Oncogene. 2007;26:5439–5449. doi: 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- Buggy J. J. Sideris M. L. Mak P. Lorimer D. D. McIntosh B. Clark J. M. Biochem. J. 2000;350(1):199–205. doi: 10.1042/bj3500199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. J. Seto E. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- Yang X.-J. Gregoire S. Mol. Cell. Biol. 2005;25:2873–2884. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryder B. E. Sodji Q. H. Oyelere A. K. Future Med. Chem. 2012;4:505–524. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganai S. A. Curr. Drug Targets. 2018;19:593–601. doi: 10.2174/1389450117666160527143257. [DOI] [PubMed] [Google Scholar]
- Fischle W. Dequiedt F. Fillion M. Hendzel M. J. Voelter W. Verdin E. J. Biol. Chem. 2001;276:35826–35835. doi: 10.1074/jbc.M104935200. [DOI] [PubMed] [Google Scholar]
- Fischle W. Dequiedt F. Hendzel M. J. Guenther M. G. Lazar M. A. Voelter W. Verdin E. Molecular cell. 2002;9:45–57. doi: 10.1016/S1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- Stahl M. Gore S. D. Vey N. Prebet T. Expert Opin. Invest. Drugs. 2016;25:307–317. doi: 10.1517/13543784.2016.1146251. [DOI] [PubMed] [Google Scholar]
- Grozinger C. M. Hassig C. A. Schreiber S. L. Proc. Natl. Acad. Sci. U. S. A. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliszczak M. Trousil S. Aberg O. Perumal M. Nguyen Q. D. Aboagye E. O. Br. J. Cancer. 2013;108:342–350. doi: 10.1038/bjc.2012.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T. Zhang C. Hassan S. Liu X. Song F. Chen K. Zhang W. Yang J. J. Hematol. Oncol. 2018;11:111. doi: 10.1186/s13045-018-0654-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose P. Dai Y. Grant S. Pharmacol. Ther. 2014;143:323–336. doi: 10.1016/j.pharmthera.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabarska A. Dmoszynska-Graniczka M. Nowosadzka E. Stepulak A. Postepy Hig. Med. Dosw. 2013;67:722–735. doi: 10.5604/17322693.1061381. [DOI] [PubMed] [Google Scholar]
- Weichert W. Roske A. Gekeler V. Beckers T. Stephan C. Jung K. Fritzsche F. R. Niesporek S. Denkert C. Dietel M. Kristiansen G. Br. J. Cancer. 2008;98:604–610. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichert W. Roske A. Gekeler V. Beckers T. Ebert M. P. A. Pross M. Dietel M. Denkert C. Rocken C. Lancet Oncol. 2008;9:139–148. doi: 10.1016/S1470-2045(08)70004-4. [DOI] [PubMed] [Google Scholar]
- Wilson A. J. Byun D.-S. Popova N. Murray L. B. L'Italien K. Sowa Y. Arango D. Velcich A. Augenlicht L. H. Mariadason J. M. J. Biol. Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- Giannini G. Cabri W. Fattorusso C. Rodriquez M. Future Med. Chem. 2012;4:1439–1460. doi: 10.4155/fmc.12.80. [DOI] [PubMed] [Google Scholar]
- Choudhary C. Kumar C. Gnad F. Nielsen M. L. Rehman M. Walther T. C. Olsen J. V. Mann M. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Marks P. Rifkind R. A. Richon V. M. Breslow R. Miller T. Kelly W. K. Nat. Rev. Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- Glozak M. A. Sengupta N. Zhang X. Seto E. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- West A. C. Johnstone R. W. J. Clin. Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick R. D. Swendeman S. L. Coffey D. C. Rifkind R. A. Marks P. A. Richon V. M. La Quaglia M. P. Cancer Res. 1999;59:4392–4399. [PubMed] [Google Scholar]
- West A. C. Johnstone R. W. J. Clin. Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafa V. Miceli M. Altucci L. Nebbioso A. Expert Opin. Ther. Pat. 2013;23:1–17. doi: 10.1517/13543776.2013.736493. [DOI] [PubMed] [Google Scholar]
- Laubach J. P. San-Miguel J. F. Hungria V. Hou J. Moreau P. Lonial S. Lee J. H. Einsele H. Alsina M. Richardson P. G. Expert Rev. Hematol. 2017;10:229–237. doi: 10.1080/17474086.2017.1280388. [DOI] [PubMed] [Google Scholar]
- Juliandi B. Abematsu M. Sanosaka T. Tsujimura K. Smith A. Nakashima K. Neurosci. Res. 2012;72:23–31. doi: 10.1016/j.neures.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Zhan P. Itoh Y. Suzuki T. Liu X. J. Med. Chem. 2015;58:7611–7633. doi: 10.1021/acs.jmedchem.5b00229. [DOI] [PubMed] [Google Scholar]
- Micelli C. Rastelli G. Drug Discovery Today. 2015;20:718–735. doi: 10.1016/j.drudis.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Finnin M. S. Donigian J. R. Cohen A. Richon V. M. Rifkind R. A. Marks P. A. Breslow R. Pavletich N. P. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- Sangwan R. Rajan R. Mandal P. K. Eur. J. Med. Chem. 2018;158:620–706. doi: 10.1016/j.ejmech.2018.08.073. [DOI] [PubMed] [Google Scholar]
- Wagner F. F. yi U. M. W. Lewis M. C. Holson E. B. Neurotherapeutics. 2013;10:589–604. doi: 10.1007/s13311-013-0226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellacci L. Perinelli D. R. Maggi F. Grifantini M. Petrelli R. Curr. Med. Chem. 2018;16 doi: 10.2174/0929867325666181016163110. [DOI] [PubMed] [Google Scholar]
- Eckschlager T. Plch J. Stiborova M. Hrabeta J. Int. J. Mol. Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann B. S. Johnson J. R. Cohen M. H. Justice R. Pazdur R. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- Grant C. Rahman F. Piekarz R. Peer C. Frye R. Robey R. W. Gardner E. R. Figg W. D. Bates S. E. Expert Rev. Anticancer Ther. 2011;11:1622. doi: 10.1586/era.11.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien W. Lee D. H. Zheng Y. Wuensche P. Alvarez R. Wen D. L. Aribi A. M. Thean S. M. Doan N. B. Said J. W. Koeffler H. P. Mol. Carcinog. 2014;53:722–735. doi: 10.1002/mc.22024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraj D. Green M. M. Gasparetto C. Future Oncol. 2017;13:477–488. doi: 10.2217/fon-2016-0329. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H. Bountra C. Fish P. V. Lee K. Schapira M. Nat. Rev. Drug Discovery. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Falkenberg K. J. Johnstone R. W. Nat. Rev. Drug Discovery. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Liao D. Drug Discovery Today: Technol. 2015;18:24–28. doi: 10.1016/j.ddtec.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P. Weykamp M. Dennahy I. S. Williams A. M. Bhatti U. F. Liu B. Nikolian V. C. Li Y. Alam H. B. J. Trauma Acute Care Surg. 2018;84:795–801. doi: 10.1097/TA.0000000000001824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micelli C. Rastelli G. Drug Discovery Today. 2015;20:718–735. doi: 10.1016/j.drudis.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Zhang L. Han Y. Jiang Q. Wang C. Chen X. Li X. Xu F. Jiang Y. Wang Q. Xu W. Med. Res. Rev. 2015;35:63–84. doi: 10.1002/med.21320. [DOI] [PubMed] [Google Scholar]
- Thaler F. Mercurio C. ChemMedChem. 2014;9:523–526. doi: 10.1002/cmdc.201300413. [DOI] [PubMed] [Google Scholar]
- Taha T. Y. Aboukhatwa S. M. Knopp R. C. Ikegaki N. Abdelkarim H. Neerasa J. Lu Y. Neelarapu R. Hanigan T. W. Thatcher G. R. J. Petukhov P. A. ACS Med. Chem. Lett. 2017;8:824–829. doi: 10.1021/acsmedchemlett.7b00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maolanon A. R. Madsen A. S. Olsen C. A. Cell Chem. Biol. 2016;23:759–768. doi: 10.1016/j.chembiol.2016.06.011. [DOI] [PubMed] [Google Scholar]
- Marek M. Shaik T. B. Heimburg T. Chakrabarti A. Lancelot J. Ramos-Morales E. Da Veiga C. Kalinin D. Melesina J. Robaa D. Schmidtkunz K. Suzuki T. Holl R. Ennifar E. Pierce R. J. Jung M. Sippl W. Romier C. J. Med. Chem. 2018;61:10000–10016. doi: 10.1021/acs.jmedchem.8b01087. [DOI] [PubMed] [Google Scholar]
- Beckers T. Burkhardt C. Wieland H. Gimmnich P. Ciossek T. Maier T. Sanders K. Int. J. Cancer. 2007;121:1138–1148. doi: 10.1002/ijc.22751. [DOI] [PubMed] [Google Scholar]
- Khan N. Jeffers M. Kumar S. Hackett C. Boldog F. Khramtsov N. Qian X. Mills E. Berghs S. C. Carey N. Finn P. W. Collins L. S. Tumber A. Ritchie J. W. Jensen P. B. Lichenstein H. S. Sehested M. Biochem. J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Siliphaivanh P. Harrington P. Witter D. J. Otte K. Tempest P. Kattar S. Kral A. M. Fleming J. C. Deshmukh S. V. Harsch A. Secrist P. J. Miller T. A. Bioorg. Med. Chem. Lett. 2007;17:4619–4624. doi: 10.1016/j.bmcl.2007.05.080. [DOI] [PubMed] [Google Scholar]
- Witter D. J. Harrington P. Wilson K. J. Chenard M. Fleming J. C. Haines B. Kral A. M. Secrist J. P. Miller T. A. Bioorg. Med. Chem. Lett. 2008;18:726–731. doi: 10.1016/j.bmcl.2007.11.047. [DOI] [PubMed] [Google Scholar]
- Moradei O. M. Mallais T. C. Frechette S. Paquin I. Tessier P. E. Leit S. M. Fournel M. Bonfils C. Trachy-Bourget M.-C. Liu J. Yan T. P. Lu A.-H. Rahil J. Wang J. Lefebvre S. Li Z. Vaisburg A. F. Besterman J. M. J. Med. Chem. 2007;50:5543–5546. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- Suzuki T. Kasuya Y. Itoh Y. Ota Y. Zhan P. Asamitsu K. Nakagawa H. Okamoto T. Miyata N. PLoS One. 2013;8:e68669. doi: 10.1371/journal.pone.0068669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson C. M. Matthews C. J. Atkinson S. J. Lamadema N. Thomas N. S. J. Med. Chem. 2015;58:6803–6818. doi: 10.1021/acs.jmedchem.5b00545. [DOI] [PubMed] [Google Scholar]
- Hsieh H.-Y. Chuang H.-C. Shen F.-H. Detroja K. Hsin L.-W. Chen C.-S. Eur. J. Med. Chem. 2017;140:42–51. doi: 10.1016/j.ejmech.2017.08.069. [DOI] [PubMed] [Google Scholar]
- Van den Wyngaert I. de Vries W. Kremer A. Neefs J. Verhasselt P. Luyten W. H. Kass S. U. FEBS Lett. 2000;478:77–83. doi: 10.1016/S0014-5793(00)01813-5. [DOI] [PubMed] [Google Scholar]
- Chakrabarti A. Melesina J. Kolbinger F. R. Oehme I. Senger J. Witt O. Sippl W. Jung M. Future Med. Chem. 2016;8:1609–1634. doi: 10.4155/fmc-2016-0117. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Feng J. Liu C. Zhang L. Jiao J. Fang H. Su L. Zhang X. Zhang J. Li M. Wang B. Xu W. Bioorg. Med. Chem. 2010;18:1761–1772. doi: 10.1016/j.bmc.2010.01.060. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Feng J. Jia Y. Wang X. Zhang L. Liu C. Fang H. Xu W. J. Med. Chem. 2011;54:2823–2838. doi: 10.1021/jm101605z. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Feng J. Jia Y. Xu Y. Liu C. Fang H. Xu W. Eur. J. Med. Chem. 2011;46:5387–5397. doi: 10.1016/j.ejmech.2011.08.045. [DOI] [PubMed] [Google Scholar]
- Zhao C. Zang J. Ding Q. g. Inks E. S. Xu W. Chou C. J. Zhang Y. Eur. J. Med. Chem. 2018;150:282–291. doi: 10.1016/j.ejmech.2018.03.002. [DOI] [PubMed] [Google Scholar]
- Neelarapu R. Holzle D. L. Velaparthi S. Bai H. Brunsteiner M. Blond S. Y. Petukhov P. A. J. Med. Chem. 2011;54:4350–4364. doi: 10.1021/jm2001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W. Luo T. Greenberg E. F. Bradner J. E. Schreiber S. L. Bioorg. Med. Chem. Lett. 2011;21:2601–2605. doi: 10.1016/j.bmcl.2011.01.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T. Ota Y. Ri M. Bando M. Gotoh A. Itoh Y. Tsumoto H. Tatum P. R. Mizukami T. Nakagawa H. Iida S. Ueda R. Shirahige K. Miyata N. J. Med. Chem. 2012;55:9562–9575. doi: 10.1021/jm300837y. [DOI] [PubMed] [Google Scholar]
- Suzuki T. Muto N. Bando M. Itoh Y. Masaki A. Ri M. Ota Y. Nakagawa H. Iida S. Shirahige K. Miyata N. ChemMedChem. 2014;9:657–664. doi: 10.1002/cmdc.201300414. [DOI] [PubMed] [Google Scholar]
- Ingham O. J. Paranal R. M. Smith W. B. Escobar R. A. Yueh H. Snyder T. Porco Jr J. A. Bradner J. E. Beeler A. B. ACS Med. Chem. Lett. 2016;7:929–932. doi: 10.1021/acsmedchemlett.6b00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.-J. Wang Y.-C. Chao S.-W. Yang C.-Y. Chen L.-C. Lin M.-H. Hou W.-C. Chen M.-Y. Lee T.-L. Yang P. Chang C.-I. ChemMedChem. 2012;7:1815–1824. doi: 10.1002/cmdc.201200300. [DOI] [PubMed] [Google Scholar]
- Taha T. Y. Aboukhatwa S. M. Knopp R. C. Ikegaki N. Abdelkarim H. Neerasa J. Lu Y. Neelarapu R. Hanigan T. W. Thatcher G. R. J. Petukhov P. A. ACS Med. Chem. Lett. 2017;8:824–829. doi: 10.1021/acsmedchemlett.7b00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y.-H. Wang K.-J. Hung P.-Y. Cheng Y.-S. Liu J.-R. Fung S.-T. Liang P.-H. Chern J.-W. Yu C.-W. Org. Biomol. Chem. 2018;16:7820–7832. doi: 10.1039/C8OB00966J. [DOI] [PubMed] [Google Scholar]
- Asthana J. Kapoor S. Mohan R. Panda D. J. Biol. Chem. 2013;288:22516–22526. doi: 10.1074/jbc.M113.489328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Li L. Min J. Nat. Chem. Biol. 2016;12:660–661. doi: 10.1038/nchembio.2158. [DOI] [PubMed] [Google Scholar]
- Liu Y. Li L. Min J. Nat. Chem. Biol. 2016;12:885. doi: 10.1038/nchembio1016-885. [DOI] [PubMed] [Google Scholar]
- Luxton G. W. G. Gundersen G. G. Dev. Cell. 2007;13:161–162. doi: 10.1016/j.devcel.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Aldana-Masangkay G. I. Sakamoto K. M. J. Biomed. Biotechnol. 2011;2011:875824. doi: 10.1155/2011/875824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. X. Wan R. Z. Liu Z. P. Eur. J. Med. Chem. 2018;143:1406–1418. doi: 10.1016/j.ejmech.2017.10.040. [DOI] [PubMed] [Google Scholar]
- Liang T. Fang H. Curr. Top. Med. Chem. 2018;18:2429–2447. doi: 10.2174/1568026619666181129141822. [DOI] [PubMed] [Google Scholar]
- Haggarty S. J. Koeller K. M. Wong J. C. Butcher R. A. Schreiber S. L. Chem. Biol. 2003;10:383–396. doi: 10.1016/S1074-5521(03)00095-4. [DOI] [PubMed] [Google Scholar]
- Estiu G. Greenberg E. Harrison C. B. Kwiatkowski N. P. Mazitschek R. Bradner J. E. Wiest O. J. Med. Chem. 2008;51:2898–2906. doi: 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- Santo L. Hideshima T. Kung A. L. Tseng J.-C. Tamang D. Yang M. Jarpe M. van Duzer J. H. Mazitschek R. Ogier W. C. Cirstea D. Rodig S. Eda H. Scullen T. Canavese M. Bradner J. Anderson K. C. Jones S. S. Raje N. Blood. 2012;119:2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P. Almeciga-Pinto I. Jarpe M. van Duzer J. H. Mazitschek R. Yang M. Jones S. S. Quayle S. N. Oncotarget. 2017;8:2694–2707. doi: 10.18632/oncotarget.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozikowski A. P. Chen Y. Gaysin A. M. Savoy D. N. Billadeau D. D. Kim K. H. ChemMedChem. 2008;3:487–501. doi: 10.1002/cmdc.200700314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Lopez-Sanchez M. Savoy D. N. Billadeau D. D. Dow G. S. Kozikowski A. P. J. Med. Chem. 2008;51:3437–3448. doi: 10.1021/jm701606b. [DOI] [PubMed] [Google Scholar]
- Kozikowski A. P. Tapadar S. Luchini D. N. Kim K. H. Billadeau D. D. J. Med. Chem. 2008;51:4370–4373. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Wang T. Wang F. Niu T. Liu Z. Chen X. Long C. Tang M. Cao D. Wang X. Xiang W. Yi Y. Ma L. You J. Chen L. J. Med. Chem. 2016;59:1455–1470. doi: 10.1021/acs.jmedchem.5b01342. [DOI] [PubMed] [Google Scholar]
- Duan W. Li J. Inks E. S. Chou C. J. Jia Y. Chu X. Li X. Xu W. Zhang Y. J. Med. Chem. 2015;58:4325–4338. doi: 10.1021/acs.jmedchem.5b00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. Cheng G. Xu Q. Luan S. Wang S. Liu D. Zhao L. Bioorg. Med. Chem. 2018;26:1418–1425. doi: 10.1016/j.bmc.2017.08.029. [DOI] [PubMed] [Google Scholar]
- Yang E. G. Mustafa N. Tan E. C. Poulsen A. Ramanujulu P. M. Chng W. J. Yen J. J. Y. Dymock B. W. J. Med. Chem. 2016;59:8233–8262. doi: 10.1021/acs.jmedchem.6b00157. [DOI] [PubMed] [Google Scholar]
- Yao L. Mustafa N. Tan E. C. Poulsen A. Singh P. Minh-Dao D.-T. Lee J. X. T. Ramanujulu P. M. Chng W. J. Yen J. J. Y. Ohlson S. Dymock B. W. J. Med. Chem. 2017;60:8336–8357. doi: 10.1021/acs.jmedchem.7b00678. [DOI] [PubMed] [Google Scholar]
- Yao L. Ramanujulu P. M. Poulsen A. Ohlson S. Dymock B. W. Bioorg. Med. Chem. Lett. 2018;28:2636–2640. doi: 10.1016/j.bmcl.2018.06.037. [DOI] [PubMed] [Google Scholar]
- Butler K. V. Kalin J. Brochier C. Vistoli G. Langley B. Kozikowski A. P. J. Am. Chem. Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin J. H. Butler K. V. Akimova T. Hancock W. W. Kozikowski A. P. J. Med. Chem. 2012;55:639–651. doi: 10.1021/jm200773h. [DOI] [PubMed] [Google Scholar]
- Shen S. Benoy V. Bergman J. K. Kalin J. H. Frojuello M. Vistoli G. Haeck W. Van Den Bosch L. Kozikowski A. P. ACS Chem. Neurosci. 2016;7:240–258. doi: 10.1021/acschemneuro.5b00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vreese R. Verhaeghe T. Desmet T. D'Hooghe M. Chem. Commun. 2013;49:3775–3777. doi: 10.1039/C3CC41422A. [DOI] [PubMed] [Google Scholar]
- De Vreese R. Depetter Y. Verhaeghe T. Desmet T. Benoy V. Haeck W. Van Den Bosch L. D'Hooghe M. Org. Biomol. Chem. 2016;14:2537–2549. doi: 10.1039/C5OB02625C. [DOI] [PubMed] [Google Scholar]
- Leonhardt M. Sellmer A. Kraemer O. H. Dove S. Elz S. Kraus B. Beyer M. Mahboobi S. Eur. J. Med. Chem. 2018;152:329–357. doi: 10.1016/j.ejmech.2018.04.046. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Yan J. Yao T.-P. Eur. J. Med. Chem. 2017;141:596–602. doi: 10.1016/j.ejmech.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlimme S. Hauser A.-T. Carafa V. Heinke R. Kannan S. Stolfa D. A. Cellamare S. Carotti A. Altucci L. Jung M. Sippl W. ChemMedChem. 2011;6:1193–1198. doi: 10.1002/cmdc.201100007. [DOI] [PubMed] [Google Scholar]
- Senger J. Melesina J. Marek M. Romier C. Oehme I. Witt O. Sippl W. Jung M. J. Med. Chem. 2016;59:1545–1555. doi: 10.1021/acs.jmedchem.5b01493. [DOI] [PubMed] [Google Scholar]
- Blackburn C. Barrett C. Chin J. Garcia K. Gigstad K. Gould A. Gutierrez J. Harrison S. Hoar K. Lynch C. Rowland R. S. Tsu C. Ringeling J. Xu H. J. Med. Chem. 2013;56:7201–7211. doi: 10.1021/jm400385r. [DOI] [PubMed] [Google Scholar]
- Blackburn C. Barrett C. Brunson M. Chin J. England D. Garcia K. Gigstad K. Gould A. Gutierrez J. Hoar K. Rowland R. S. Tsu C. Ringeling J. Wager K. Xu H. Bioorg. Med. Chem. Lett. 2014;24:5450–5454. doi: 10.1016/j.bmcl.2014.10.022. [DOI] [PubMed] [Google Scholar]
- Bergman J. A. Woan K. Perez-Villarroel P. Villagra A. Sotomayor E. M. Kozikowski A. P. J. Med. Chem. 2012;55:9891–9899. doi: 10.1021/jm301098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares M. T. Shen S. Knox T. Hadley M. Kutil Z. Barinka C. Villagra A. Kozikowski A. P. ACS Med. Chem. Lett. 2017;8:1031–1036. doi: 10.1021/acsmedchemlett.7b00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-H. Mahendran A. Yao Y. Ngo L. Venta-Perez G. Choy M. L. Kim N. Ham W.-S. Breslow R. Marks P. A. Proc. Natl. Acad. Sci. U. S. A. 2013;110:15704–15709. doi: 10.1073/pnas.1313893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-H. Yao Y. Mahendran A. Lang N. Venta-Perez G. Choy M. L. Breslow R. Marks P. A. Proc. Natl. Acad. Sci. U. S. A. 2015;112:E5899. doi: 10.1073/pnas.1519546112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedrich D. Hamacher A. Gertzen C. G. W. Avelar L. A. A. Reiss G. J. Kurz T. Gohlke H. Kassack M. U. Hansen F. K. Chem. Commun. 2016;52:3219–3222. doi: 10.1039/C5CC10301K. [DOI] [PubMed] [Google Scholar]
- Porter N. J. Osko J. D. Diedrich D. Kurz T. Hooker J. M. Hansen F. K. Christianson D. W. J. Med. Chem. 2018;61:8054–8060. doi: 10.1021/acs.jmedchem.8b01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voegerl K. Ong N. Senger J. Herp D. Schmidtkunz K. Marek M. Mueller M. Bartel K. Shaik T. B. Porter N. J. Robaa D. Christianson D. W. Romier C. Sippl W. Jung M. Bracher F. J. Med. Chem. 2019;62:1138–1166. doi: 10.1021/acs.jmedchem.8b01090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y. Lim J. Seo Y. H. Eur. J. Med. Chem. 2019;164:263–272. doi: 10.1016/j.ejmech.2018.12.056. [DOI] [PubMed] [Google Scholar]