

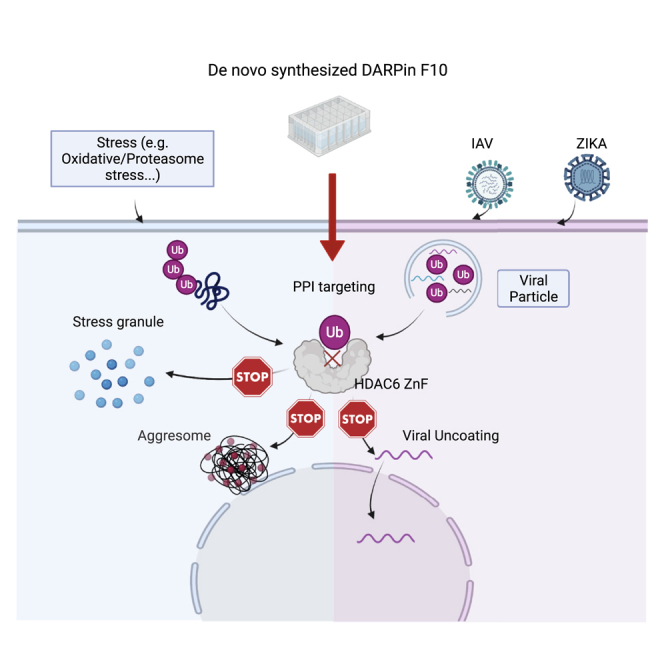
Summary
The deacetylase HDAC6 has tandem catalytic domains and a zinc finger domain (ZnF) binding ubiquitin (Ub). While the catalytic domain has an antiviral effect, the ZnF facilitates influenza A virus (IAV) infection and cellular stress responses. By recruiting Ub via the ZnF, HDAC6 promotes the formation of aggresomes and stress granules (SGs), dynamic structures associated with pathologies such as neurodegeneration. IAV subverts the aggresome/HDAC6 pathway to facilitate capsid uncoating during early infection. To target this pathway, we generate designed ankyrin repeat proteins (DARPins) binding the ZnF; one of these prevents interaction with Ub in vitro and in cells. Crystallographic analysis shows that it blocks the ZnF pocket where Ub engages. Conditional expression of this DARPin reversibly impairs infection by IAV and Zika virus; moreover, SGs and aggresomes are downregulated. These results validate the HDAC6 ZnF as an attractive target for drug discovery.
Keywords: HDAC6, influenza A virus, Zika virus, aggresome, stress granules, DARPin, ubiquitin, stress response, lysine deacetylases
Graphical abstract
Highlights
-
•
A small synthetic protein (DARPin) blocks interaction between HDAC6 and ubiquitin
-
•
This DARPin impairs infection by influenza and Zika virus at the uncoating step
-
•
Both viruses contain ubiquitin associated with their capsid
-
•
The DARPin also impacts the formation of aggresomes and stress granules
Wang et al. show that a designed ankyrin repeat protein (DARPin F10) targeting the HDAC6 zinc finger domain blocks its interaction with ubiquitin. F10 expression impairs influenza and Zika virus infection and aggresomes and stress granules formation. This highlights the importance of HDAC6-mediated ubiquitin recruitment for cellular stress response.
Introduction
RNA viruses like influenza A virus (IAV) and Zika virus (ZIKV) affect millions of people worldwide, and the current coronavirus disease 2019 (COVID-19) pandemic is a vivid reminder that viruses are a constant threat. Viruses have evolved mechanisms that intersect with cellular pathways and favor infection. One such pathway is ubiquitination, the process by which ubiquitin (Ub) is used to generate polymeric chains that can be conjugated to proteins. By modulating protein function (e.g., localization) or fate (e.g., degradation), ubiquitination impinges on cellular metabolism widely. Proteins ubiquitinated by K48-branched chains are targeted for degradation by the ubiquitin proteasome system (UPS) (Komander and Rape, 2012). Ub is critical in innate immunity, e.g., by modulating the activity of the RNA helicase retinoic acid-inducible gene I (RIG-I), a viral RNA sensor eliciting interferon production (Peisley et al., 2014).
Ub is also important for the cellular stress response and is part of membraneless organelles forming as defense mechanisms. When misfolded proteins fail to be degraded, they assemble with Ub chains in a perinuclear structure called aggresome (Johnston et al., 1998), which is degraded by autophagy; this is known as the aggresome processing pathway (APP). This process depends on protein transport along the microtubules, mediated by molecular motors, such as dynein, and on histone deacetylase 6 (HDAC6). Aggresomes have also been associated with viral replication (Wileman, 2006). Stress granules (SGs) are dynamic RNA-protein aggregates that build up following stress or viral infection (Nover et al., 1989). SGs contain RNAs and proteins including translational machinery components (Protter and Parker, 2016). UPS inhibition induces SG formation (Mazroui et al., 2007), and unconjugated Ub co-localizes with SGs (Markmiller et al., 2019). Like aggresomes, SGs have been associated with viral replication, neurodegeneration, and cancers (Anderson et al., 2015; Montero and Trujillo-Alonso, 2011; Reineke and Lloyd, 2013). SGs and aggresomes are viewed as temporary protective mechanisms against harmful cellular environments (Hao et al., 2013; Protter and Parker, 2016; Wheeler et al., 2016); their dynamics can be altered in pathological situations, as seen with the enrichment of mutated FUS or TDP-43 in SGs (Ash et al., 2014; Olzmann et al., 2008).
The lysine deacetylase HDAC6 is important for IAV infection and cellular granules formation. It has tandem catalytic domains (CDs) (Miyake et al., 2016) and an Ub-binding zinc finger domain (ZnF-UBP, hereafter ZnF) (Hook et al., 2002; Seigneurin-Berny et al., 2001). Major substrates of HDAC6 are tubulin (Hubbert et al., 2002; Zhang et al., 2003), HSP90 (Kovacs et al., 2005), cortactin (Zhang et al., 2007), or DDX3X (Saito et al., 2019). By regulating the acetylation of tubulin and cortactin, HDAC6 influences microtubules dynamics, cytoskeletal trafficking, and cellular motility (Boyault et al., 2007). HDAC6-specific CDs inhibitors have shown efficacy in cancer models (Brindisi et al., 2019; Cosenza and Pozzi, 2018; Mishima et al., 2015). In many cases the biological functions of HDAC6 depend, beyond the catalytic activity, on the ZnF that binds to the C-terminal -Gly-Gly of unanchored Ub chains (Boyault et al., 2006; Ouyang et al., 2012): formation of SGs and aggresomes requires an intact ZnF (Kawaguchi et al., 2003; Kwon et al., 2007; Legros et al., 2011). Due to distinct functions of the CDs and ZnF, HDAC6 has antiviral and pro-viral roles. Its CDs deacetylate RIG-I (Choi et al., 2016) and IAV RNA polymerase PA (Chen et al., 2019) to restrict viral genome amplification and induce the interferon response; in contrast, the ZnF facilitates IAV uncoating early in infection (Banerjee et al., 2014). Viruses often depend on the UPS (Isaacson and Ploegh, 2009) and intersect with Ub pathways during infection (Gustin et al., 2011); proteasome inhibitors block IAV entry and impact the replication of various viruses (Rudnicka and Yamauchi, 2016). For the flaviviruses Dengue and Zika, capsid or envelope protein ubiquitination is vital: Dengue uncoating requires a nondegradative ubiquitination step (Byk et al., 2016), and ZIKV E protein ubiquitination promotes virus attachment and entry into cells (Giraldo et al., 2020). A recent proteomics study found host proteins including Ub in viral particles of human immunodeficiency or vesicular stomatitis virus (Gale et al., 2019). We showed that IAV contains unanchored Ub (Banerjee et al., 2014) and that uncoating recapitulates key aspects of the APP, including HDAC6 and Ub recruitment (Arctibasova et al., in preparation). Moreover, the APP and the HDAC6-Ub interaction is critical for inflammasome activation (Magupalli et al., 2020). Hence, targeting the ZnF-Ub interaction is of great therapeutic interest.
Here we have generated designed ankyrin repeat proteins (DARPins) that recognize the ZnF of human HDAC6. We show that one DARPin binds tightly to the ZnF and blocks the Ub-binding pocket; owing to its high affinity, it displaces Ub from the ZnF in vitro and in cells. Conditional expression in cells leads to reduced infection by IAV and ZIKV, thereby establishing that this pathway can be used by enveloped RNA viruses beyond IAV. We found that Ub is associated with ZIKV particles, thus suggesting a conserved mechanism for uncoating. In these DARPin-expressing cells, aggresome and SG formation is impaired. Hence, manipulating the ZnF-Ub interaction can modulate the cellular response to stress and viral infection.
Results
Identification of DARPins selectively recognizing the HDAC6 ZnF
We set out to identify antibody-like molecules targeting the ZnF. The purified human HDAC6 ZnF was used as a bait for identification of nanobodies and DARPins (Binz et al., 2003, 2004; Plückthun, 2015) binding specifically (Figure 1A; see STAR Methods). Several nanobodies were identified, but their biological effect was weak (Figures S1A and S1B) and we concentrated on the DARPins (Binz et al., 2003, 2004; Plückthun, 2015). DARPins were selected by in vitro ribosome display (Brauchle et al., 2014; Dreier and Plückthun, 2012); following the high throughput screen, we identified by an HTFR assay (Table S1) 24 DARPins binding to the bait. We used them for binding assays with purified ZnF and Ub at equimolar ratios: all DARPins efficiently bound the ZnF (Figure S1C), but one, F10, prevented interaction with Ub (Figure 1B). Maximal interference was obtained when F10 was first incubated with the ZnF, but addition of F10 to a preformed ZnF-Ub complex was able to displace Ub (Figure S1D).
Figure 1.

DARPin F10 inhibits ZnF-Ub interaction in vitro and in cells
(A) Schematic of HDAC6, showing the catalytic domains (CD1, CD2) and the zinc finger domain (ZnF, amino acid 1,108–1,215) used to select binders. Generic nanobody and DARPin structures (PDB:1I3V and PDB:2QYJ) are shown. The HDAC6 sequence (aa 1,153–1,190) at top shows the Ub binding motifs (framed red) (Uniprot: Q9UBN7) (Ouyang et al., 2012).
(B) F10 blocks ZnF-Ub interaction in vitro. Purified His-tagged ZnF (aa 1,108–1,215), Flag A10 or F10 and mono-Ub were mixed; following incubation, DARPins and associated proteins were pulled down with anti-Flag beads. The precipitate was analyzed by immunoblotting, using antibodies against His, Ub, and Flag. PD, pull down; FT, flow through.
(C) In 293T cells, ZnF-Ub interaction is disrupted by F10, as monitored by a split-GFP assay. ZnF (aa 1,108–1,215) and Ub were fused to separate GFP fragments so that ZnF-Ub interaction is required to reconstitute a functional GFP molecule (scheme at top). GFP beta strands are GFP-(1–9), GFP-10, and GFP-11. A mutant ZnF (W1182A, ZnFm) was used as a control. An mRuby plasmid was included in all transfections (red signal). Scale bar: 1 mm.
(D) Immunoprecipitation of endogenous HDAC6 by F10. A GFP fusion protein to F10 or to control E3_5 was transiently expressed in A549 cells, and the DARPins were immunoprecipitated with GFP-trap beads. The immunoprecipitate (IP) was analyzed by immunoblotting, using antibodies against GFP or HDAC6.
(E) Mass spectrometry analysis to determine the F10 interactome. Immunoprecipitate (D above) was analyzed by mass spectrometry, and enriched proteins are annotated.
To test whether F10 interfered with the ZnF-Ub interaction in cells, we used a split-GFP assay (Cabantous et al., 2013) (Figure 1C) in which reconstitution of a functional GFP molecule is mediated by ZnF-Ub interaction. Mutation of a ZnF residue (W1182A), which disrupts interaction with Ub (Hao et al., 2013), abolished the GFP signal. Expression of F10 suppressed the fluorescence signal, while expression of DARPin A10, which binds the ZnF without blocking Ub, did not interfere (Figures 1B and 1C).
When transiently expressed in A549 cells, F10 but not the control DARPin E3_5 (Binz et al., 2003) can be used to precipitate endogenous HDAC6 (Figure 1D). Mass spectrometric analysis of the precipitates identified five proteins besides HDAC6, none of which has a ZnF (Figure 1E). Thus, F10 can impair ZnF-Ub interaction in cells with high specificity. We also performed transcriptome analysis of A549 cells transiently expressing F10 or a control. The transfection process elicited some gene expression changes (Figures S2A and S2B), but none of these were DARPin specific; indeed, the changes induced by F10 or E3_5 showed no significant difference (Figure S2C).
DARPin F10 forms a stable complex with the ZnF
To understand how F10 inhibits HDAC6 ZnF-Ub binding, the ZnF-F10 complex structure (Figure S3A) was determined by crystallography. A 2.55-Å resolution structure was determined (Figure 2A) by molecular replacement using a ZnF-Ub C-terminal complex (PDB: 3GV4) as search model (see Table S2 for data collection and refinement statistics). An unbiased map was obtained by experimental phasing using the anomalous signal from the Zn2+ ions.
Figure 2.

2.55-Å crystal structure shows the detail of DARPin F10 inhibition on Ub recruitment
(A) F10 bound to HDAC6 ZnF. F10, ZnF (aa 1,108–1,215), and Zn2+ ions are colored purple, green, and gray. F10 α helices (α1 to α10) and β turns (L1 to L4) are numbered starting from the N-terminus (PDB: 7ATT).
(B) ZnF conformation alignment in two binding forms. Green shows the ZnF structure bound to F10, and cyan shows the ZnF bound to Ub (PDB: 3GV4). Right: zoomed-in view of the Ub binding “gatekeeper” amino acids, Arg1155 and Tyr1156. The shifting of side chains is indicated by dashed lines with the distance labeled.
(C) Zoomed-in views of the ZnF-Ub-binding pocket. The ZnF-F10 structure is superimposed onto the ZnF-Ub C-terminal tail structure (PDB: 3GV4). The Ub C-terminal tail is shown in surface representation (yellow): F10 L3 backbone clashes with the Ub tail. The region from His101 to Gly103 is indicated by the dashed line rectangle (right).
(D) Affinities of ZnF for F10 or free Ub by ITC assays. ZnF-F10: ΔS = 20.6 cal/mol/deg, N = 1.01. ZnF-Ub: ΔS = −12.8 cal/mol/deg, N = 1.46.
The asymmetric unit contains one copy of the ZnF-F10 complex, and the entire sequence, except for the first two N-terminal F10 amino acids (aa), is defined by electron density. The N-capping ankyrin repeat of F10 consists of two α helices followed by four complete repeats, together including α helices 1 to 10 (α1 to α10) and the β turns 1 to 4 (L1 to L4; Figure 2A). The complex interface was analyzed with QtPISA yielding a total buried interface surface area of 853.4 Å2. The extensive binding interface of F10, including L1-L4 and parts of α3, α5, and α7, encloses the tip of the protruding ZnF motif III (loops aa 1,133–1,142 and aa 1,153–1,160), and the four β turns of F10 insert like fingers into the canyon-like cleft between ZnF motif II (loops aa 1,184–1,187 and aa 1,112–1,116) and ZnF motif III of the ZnF (Figure S3B). ZnF-F10 interactions are governed by an intricate network of hydrophobic contacts and polar interactions between the two proteins (Figure S3C, electron densities shown in Figure S3D). They are in the central ankyrin repeats (Lys47 in α3, Asp67 in L2, Asp100 in L3, and Arg113 in α7) and ZnF motif III region, from Arg1155 to Asn1158, which we speculate to be critical for F10 binding specificity. Mutation of Lys47, Asp67, Asp100, and Arg113 to Alanine in F10 (F10KDDR) was sufficient to prevent complex formation, as shown by pull-down (Figure S4A and B) and isothermal titration calorimetry (ITC) assays (Figure S4C).
Aligning the F10-bound ZnF to the Ub-bound ZnF structure (3GV4) shows small differences between the conformations, with an overall root-mean-square deviation of 1.062 Å (Figure 2B). Limited backbone shift is observed; in contrast, the side chain of Arg1155 and Tyr1156, “gatekeepers” for Ub binding (Ouyang et al., 2012), are bent outward by 6 Å and 2.5 Å (Figure 2B right). Another ZnF key residue for Ub binding is W1182 (Hao et al., 2013; Ouyang et al., 2012); it makes hydrophobic contacts (distance >4 Å) with F10 (Met69), but its position was not altered (Figure S3E).
In the ZnF-Ub structure (3GV4), the negatively charged Ub C-terminus inserts into a positively charged pocket (ZnF binding pocket, ZBP; Figure S4D left), which is part of the characteristic ZnF cleft. When Ub is bound, the ZBP is filled by the Ub C-terminal -Leu-Arg-Gly-Gly peptide. Superposition of F10 onto the ZnF-Ub structure revealed that the Ub binding site is occupied by F10 residues (Figures 2C and S4E). Backbone atoms on L3 (His101 to Gly103; Figure 2C), and side chains of Lys102 (L3) and Met69 (L2) are directly clashing with Leu73 and Arg74 of Ub (Figure S4F). F10 binding to the positively charged ZnF-Ub-binding pocket is not significantly facilitated by charge complementarity as observed for Ub binding (Figure S4D right). Thus, complex formation and inhibition of Ub binding appear to rely on substantial shape complementarity and are driven by non-polar and polar interactions. Using ITC, we determined the in vitro binding affinities between the ZnF and mono-Ub or F10 (Figure 2D). This revealed that the ZnF has a 55-fold higher affinity for F10 (KD 95.05 ± 12.3 nM) than for Ub (KD 5.16 ± 0.47 μM), supporting a specific binding mode. Thus, by forming an interlocked interface comprising large parts of the ZnF, F10 binds stronger than Ub and competes for the Ub-binding pocket.
Targeting the HDAC6 ZnF impairs IAV infection during uncoating
To examine whether expression of F10 can impact HDAC6-related cellular processes, we generated A549 cell lines stably expressing F10, fused to an FKBPF36V degron (hereafter F10-FKBP cell line), as well as control cells expressing the FKBPF36V moiety (FKBP cells). In these cells, conditional degradation can be induced by dTAG-13 (hereafter dTAG), which engages FKBPF36V and Cereblon, leading to proteasomal degradation (Nabet et al., 2018) (Figure S5A). In F10-FKBP cells, dTAG addition led to complete degradation of the F10 fusion protein in ca. 6 h (Figure 3A). Pre-treatment with proteasome inhibitors, e.g., MG132, carfilzomib, and bortezomib, prevented degradation (Figure S5B). However, when MG132 was added after the dTAG treatment, F10 expression was not recovered for < 18 h (Figure S5C). F10 expression did not impact HDAC6 enzymatic activity, as evidenced by the fully deacetylated tubulin in these cells (Figure S5D). Next, we examined the F10-FKBP cell line transcriptome versus the parental A549 cells; importantly, the expression of genes involved in the interferon response e.g., DDX58 (encoding RIG-I), IRF3, IRF9, DDX60L and others, was not altered (Figure S5E).
Figure 3.

DARPin F10 interferes with IAV infection
(A) F10 can be efficiently degraded. Immunoblotting with lysates of F10-FKBP cells treated for the indicated times with dTAG (2 μM). The leftmost lane shows the parental A549 cells. The membrane was probed with antibodies against HDAC6, α-tubulin, and F10 (HA-F10-FKBPF36V, detected with anti-HA).
(B) F10 impairs IAV infection. A549 WT, F10-FKBP, or FKBP cells were infected with IAV at a MOI of 0.05 (n = 3), and culture supernatants were collected every 12 h until 72 h. Viral titer was quantified by plaque assay. Statistical analysis was done with one-way ANOVA test; p values show the significant (< 0.05) difference between cell lines expressing or not F10. Data are represented as mean ± SD.
(C) Treatment with dTAG restores IAV susceptibility. The indicated cell lines (−/+ dTAG pre-treatment) were infected with IAV at an MOI of 0.05 (n = 3). Culture supernatants were analyzed as in (B); p values show the significant (< 0.05) difference between A549 WT and F10-FKBP cells. Data are represented as mean ± SD.
(D) Effect of F10 on a single IAV life cycle. The indicated cell lines were infected with IAV at an MOI of 10 (n = 3), and culture supernatants were collected every 2 h up to 8 h. Viral titer was analyzed as in (C) above.
(E) IAV uncoating is impaired by F10. Left panels: confocal microscopy visualization of uncoating, staining for the capsid M1 protein (green). Parental A549 or F10-FKBP cells (−/+ dTAG pre-treatment) were used for IAV infection, and M1 localization was analyzed 3.5 hpi. Bafilomycin A1 treatment was used to block endosomal acidification. Total protein was stained to visualize the cell body (red); scale bar, 20 μm. Right panel, quantification of the image analysis in the different samples (n = 3); ca. 30 cells were selected per view (six to nine views for each condition), and M1 fluorescence intensity was analyzed. Statistical analysis as in (B); p values show the significant (< 0.05) difference against untreated A549 WT cells. Data are represented as mean ± SD.
Infection by IAV depends on the HDAC6/APP at the uncoating stage (Banerjee et al., 2014) (Arctibasova et al., in preparation). We therefore infected the different cell lines with IAV and analyzed the virus titer in the culture supernatant. Viral growth curve showed approximately a 10-fold reduction of virus titer in F10-FKBP cells compared with cells lacking the DARPin (Figure 3B). Remarkably, this difference was maintained through multiple replication cycles for up to 72 h post infection (hpi). F10 degradation by dTAG addition (Figure 3C top) prior to infection restored susceptibility to virus (Figure 3C bottom). We also examined the virus titer within one replication cycle (up to 8 h). Consistent with these results, a robustly reduced viral load was detected as early as 6 hpi in F10-FKBP cells (Figure 3D).
To visualize uncoating, we monitored the release of matrix protein M1 from viral particles and its cellular distribution. At 3.5 hpi the M1 signal (green) was detected as a weak, diffuse, dotted staining in untreated WT A549 cells (Figure 3E, top left panel). In bafilomycin-treated WT cells, in which endosomal acidification and transport are blocked, the M1 signal was strongly reduced, indicative of blocked uncoating. A similar pattern was observed in F10-FKBP cells, and dTAG addition restored the M1 signal; quantification confirmed that F10 inhibits IAV uncoating (Figure 3E, right panel).
To verify that the uncoating impairment was due to the interaction of F10 with the HDAC6 ZnF (and Ub recruitment blockade), we prepared A549 cell pools expressing either the WT F10 or the mutant F10KDDR (HA-F10 or HA-F10KDDR cells). Both pools express comparable DARPin levels; while the HA-F10 cells showed a strongly reduced infection as expected, the HA-F10KDDR cells showed a much weaker effect (Figures S5F and S5G).
Infection by zika virus is inhibited by DARPin F10 at uncoating
ZIKV is a mosquito-borne, enveloped, single-stranded RNA virus; it has spread to the Americas (Campos et al., 2015) after outbreaks in Micronesia (Duffy et al., 2009) and French Polynesia (Cao-Lormeau et al., 2014). We set out to test if ZIKV is also sensitive to blockade of the HDAC6 ZnF. We used a low passage clinical isolate of Asian lineage ZIKV (Puerto Rico strain) to infect A549 WT and F10-FKBP cells, and the viral titer was determined. At 72 hpi the titer was strongly reduced in the F10-FKBP cells, in comparison to dTAG-treated cells or parental A549 cells (Figure 4A). To examine infection, we fixed cells at 72 or 16 hpi and stained for ZIKV envelope (E) protein and for DNA with DAPI to monitor cell number (Figures 4B and 4C). Based on the number of ZIKV E protein-positive cells, in WT A549 cells the infectivity ratio was about 20% at 72 h and > 10% at 16 h. In contrast, in F10-FKBP cells, this value was < 3% and largely recovered upon dTAG treatment.
Figure 4.

ZIKAV replication is inhibited by DARPin F10
(A) Reduction of ZIKV titer in F10-expressing cells. The indicated cell lines (−/+ dTAG pre-treatment) were infected with ZIKV at an MOI of 0.1 50% tissue culture infective dose (TCID50)/cell, and culture supernatant was analyzed at 72 h. Viral titers were determined by a TCID50 assay; the baseline titer obtained with A549 WT cells was set to 100%. Statistical analysis was done with one-way ANOVA test; p values refer to the significant (< 0.05) differences between samples (n = 3). Data are represented as mean ± SD.
(B) Reduced ZIKV infection in F10-FKBP cells. Left panels: microscopy visualization of ZIKV E protein expression. After ZIKV titer quantification in (A), the cells at 72 hpi were stained by DAPI (blue) and for ZIKV E protein (green). 10× objective; scale bar, 0.2 cm. Mock WT refers to non-infected A549 cells; all other samples were infected. Right graph: quantification of the ZIKV E protein-positive cells based on independent experiments (n = 3). Statistical analysis as in (A).
(C) ZIKV infection is halted by F10 at early time point. Analysis and quantification as in (B) but at 16 hpi. 20× objective.
(D) Ub is present in ZIKV particles. Purified ZIKV particles (African strain) were incubated with Proteinase K (ProK) or Triton X-100. Samples were analyzed by immunoblotting with antibodies detecting Ub and ZIKV E protein.
(E) Elevated ZIKV RNA early after infection in cells expressing F10. The indicated cells (−/+ dTAG pre-treatment) were infected with ZIKV at high MOI (= 10) and RNA was analyzed at 1 and 2 hpi. ZIKV E genes RNA level is shown as fold change after normalization to 18S and Actin RNA controls; WT A549 (1 h) was used as baseline for relative comparison with 2−ΔΔct method (n = 3). Statistical analysis was done with two-way ANOVA; p values (< 0.05) are shown. Data are represented as mean ± SD.
(F) No antiviral genes upregulation in ZIKV-infected F10-FKBP cells. The indicated cell lines (−/+ dTAG pre-treatment) were infected with ZIKV (MOI = 1), and total RNA was extracted at 24 hpi. Expression of interferon-related genes, DDX58 (RIG-I), ISG15 (ISG15), IFNL1 (IL-29 or IFN-λ), and IFNB1 (IFN-β1) was analyzed by qRT-PCR (n = 3). Actin and GAPDH served as double control. Mock (uninfected WT A549 cells) was used as baseline for relative comparison with 2−ΔΔct method. Statistical analysis as in (A); difference to Mock not shown.
We next examined whether Ub could be detected in ZIKV particles. For this we used ZIKV particles of an African lineage strain (Vielle et al., 2018) and tested Ub presence by immunoblotting, as we had done for IAV (Banerjee et al., 2014). As shown in Figure 4D, when the particles were either untreated or treated with only detergent, a high molecular weight Ub signal and the E capsid protein signal were detected. When the ZIKV particles were treated shortly with detergent together with Proteinase K to digest the capsid and release its contents, the high molecular weight Ub signal and the E protein signal disappeared, reflecting destruction of the viral capsid. Simultaneously low molecular weight Ub signals became visible. The presence of Ub was also evidenced in the ZIKV Puerto Rico strain (Figures S5H and S5I). Overall, this pattern is similar to what we observed with IAV (Banerjee et al., 2014) and is compatible with the presence of Ub in ZIKV particles.
Very early after infection the ZIKV RNA is protected by the capsid until it gets released in the cytoplasm, where it becomes rapidly degraded by nucleases (Gestuveo et al., 2021). Due to this, more ZIKV RNA can be detected at 1 than at 2 hpi; similar findings have been made for other flaviviruses (Pijlman et al., 2008). In this context, we reasoned that if ZIKV uncoating is blocked by F10, then higher ZIKV RNA levels should be seen in F10-FKBP cells than in WT cells or than in dTAG-treated F10-FKBP cells. We therefore infected cells at high multiplicity of infection (MOI) and measured ZIKV E RNA. As expected, in WT cells we found more ZIKV RNA at 1 than at 2 hpi; in contrast, in F10-FKBP cells the ZIKV RNA level was higher at either time point than in the WT cells. Furthermore, when F10 was degraded the ZIKV RNA level was as in WT cells (Figure 4E). These results demonstrate that for ZIKV, like for IAV, impinging on HDAC6-Ub interaction leads to a strong reduction in infectivity by affecting uncoating. We also examined the expression of interferon-related genes in ZIKV-infected cells: expression of DDX58 (encoding RIG-I), IFNB1 (type I IFN), IFNL1 (type III IFN), or other genes of this pathway was downregulated in F10-FKBP cells, indicating that the effect of F10 is not due to upregulation of antiviral pathways (Figure 4F).
Targeting the HDAC6 ZnF impairs cellular granules formation
After dTAG treatment, F10 re-expression takes more than 18 h (Figure S5C). Since an 18-h MG132 treatment is widely used to induce aggresome formation in human cells (Kawaguchi et al., 2003), this setup allowed to test the effect of F10 on this process. Aggresome was visualized by HDAC6 and Ub staining; in parental A549 cells a strong perinuclear staining was observed, which was disrupted in F10-FKBP cells (Figure 5A). When cells were first treated with dTAG, aggresome formation was restored (Figure 5A right panels). Quantification of Ub staining showed that aggresomes formed in < 60% of the F10-FKBP cells, compared to ca. 80% of the WT A549 or dTAG-treated F10-FKBP cells (Figure 5B). Higher magnification pictures show disruption of the aggresome structure by F10 (Figure S6C, left). When quantification was based on HDAC6, ca. 50% of WT and slightly above 20% of F10-FKBP cells were positive (Figures S6A and S6B). Scanning of the signal intensity across aggresome structures in the different cell lines shows how both signals are reduced in F10-FKBP cells (Figure S6C, right).
Figure 5.

DARPin F10 impairs cellular granules formation
(A) Aggresome formation was induced with MG132 in indicated cells (−/+ dTAG pre-treatment). Aggresomes were detected by staining for HDAC6 and Ub; DAPI was used to stain the nucleus. Scale bar, 20 μm.
(B) Quantification of aggresome formation by Ub signal (n = 3). The graph shows the percentage of aggresome-positive cells; each data point represents the percentage obtained in a randomly chosen micrograph containing ca. 50 cells. Statistical analysis was done by one-way ANOVA; p values refer to significant (< 0.05) differences between samples. Data are represented as mean ± SD.
(C) SGs were induced with NaAsO2 in indicated cells (−/+ dTAG pre-treatment); SGs were visualized and quantified by staining for G3BP1. Representative pictures are shown. Scale bar, 20 μm.
(D) Quantification of the number of SGs per cell area. Each data point represents the number of SGs per cell area in a sample containing one or a few (ca. 2 to 5) cells. Data were collected in five independent experiments (n = 5). Statistical analysis as in (B).
We next examined oxidative stress-induced SGs and monitored their presence by staining for G3BP1, a SG marker (Tourrière et al., 2003). F10-FKBP cells had approximately 30% fewer SGs than the parental A549 cells or dTAG-treated F10-FKBP cells (Figures 5C and 5D; a representative higher magnification view is shown in Figure S6D). However, size and roundness, two parameters related to SG maturation, were not altered by F10 expression (Figure S6E).
Discussion
HDAC6 is an established target for drug discovery: chemical inhibition of the CDs has shown antitumor activity and synergy with chemotherapy (Brindisi et al., 2019; Li et al., 2018). While drug discovery has been focused on HDAC6 CDs, our work highlights an alternative approach for interfering with HDAC6 ZnF-dependent pathways, such as the APP. Compared to the described small molecules binding the ZnF-Ub-binding pocket (Ferreira de Freitas et al., 2018; Harding et al., 2017), we used the entire Ub, not the C-terminal peptide, to show that its binding can be interfered with. Disruption of the ZnF-Ub interaction was achieved in vitro and in cells, where it leads to robust biological phenotypes. Indeed, displacing Ub from the HDAC6 ZnF has a strong impact on IAV infection, and the same effect was observed with ZIKV, another RNA virus. Like for IAV, we could evidence Ub in ZIKV particles, although final demonstration that it is unanchored awaits further testing. This suggests that the underlying mechanism may be similar for IAV and ZIKV and might also be used by other enveloped RNA viruses, such as Ebola virus and HIV, which contain Ub inside the virion (Gale et al., 2019). Ubiquitination of the ZIKV envelope protein was recently shown to be important for virus entry (Giraldo et al., 2020), indicating that Ub plays multiple roles in the life cycle of this virus.
Blocking the ZnF led to a reduced number of SGs following oxidative stress, but their size or shape was unaltered. In contrast, we found previously that impairment of HDAC6 catalytic activity results in smaller SGs. This impaired SG maturation is due to the lack of deacetylation and inefficient phase separation of the RNA helicase DDX3X, a critical SG component under some stress conditions (Saito et al., 2019). Thus, both functional domains of HDAC6 can impact distinct steps of SG formation. Aggresomes and SGs are regarded as cellular-protective inclusion bodies shielding cells from acute stress (Advani and Ivanov, 2020; Taylor et al., 2003). However, they have also been linked to neurodegenerative disease and cancers (Anderson et al., 2015; Mishima et al., 2015; Olzmann et al., 2008). Chronic stress associated with aging leads to the pathological accumulation of proteins like TDP-40, Tau, and Htt in SGs (Sweeney et al., 2017) and to their transformation from transient to persistent structures, accompanied by neuronal damages (Gao et al., 2019; Kawaguchi et al., 2003). Thus, downregulating SGs and aggresome formation by targeting the HDAC6 ZnF might synergize with therapies for neurodegeneration or cancer.
Viruses are often pathogenic and interfering with their infectivity is beneficial. Current treatments are based on targeting viral proteins like the IAV M2 ion channel and neuraminidase NA (Du et al., 2012; Hussain et al., 2017). Such antivirals easily lead to escape mutants as viral proteins are prone to mutations. Thus, targeting a cell-assisted viral uncoating mechanism by disrupting the Ub-HDAC6 interaction is an attractive alternative. Mice lacking HDAC6 are viable and do not show overt phenotypes under standard conditions other than elevated tubulin acetylation (Zhang et al., 2008). It is thus possible that transient impairment of the HDAC6 ZnF-Ub interaction might be tolerated. Hence, interfering selectively with the HDAC6 ZnF-Ub interaction represents a general therapeutic modality that could be combined with virus-specific approaches and might also be of interest in other pathological situations.
Limitations of the study
Our previous work had shown the role of unanchored Ub chains, recruited by HDAC6, for IAV uncoating (Banerjee et al., 2014) (Arctibasova et al., in prep). Our DARPin disrupting the ZnF-Ub interaction allowed us to recapitulate the initial IAV observations and to extend them to ZIKV. With the tools available, we have not formally demonstrated that unanchored Ub chains are involved, but this is our favored working model; further work is required to rigorously establish this point. This will require the development of reagents allowing to unambiguously differentiate free from covalent forms of Ub. Finally, there is a formal possibility that the ZnF also has another, unidentified, function independent of Ub binding. Given our structural description of how F10 engages with the ZnF and the high affinity of the interaction, this seems unlikely.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human monoclonal HDAC6 antibody | Cell Signaling | Cat#7558; RRID: AB_10891804 |
| Monoclonal ANTI-FLAG® M2 antibody produced in mouse | Sigma-Aldrich | Cat#F3165; RRID: AB_259529 |
| Pan-Ubiquitin antibody(P4D1) | Santa Cruz | Cat#sc-8017; RRID: AB_2762364 |
| Pan-actin antibody | Sigma-Aldrich | Cat#SAB4502632; RRID: AB_10746710 |
| Anti-His tag antibody | Santa Cruz | Cat#sc-53073; RRID: AB_783791 |
| GFP-antibody | Cell Signaling | Cat#2037 |
| Monoclonal Anti-α-Tubulin antibody produced in mouse | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Monoclonal Anti-Acetylated Tubulin antibody produced in mouse | Sigma-Aldrich | Cat#T7451; RRID: AB_609894 |
| Goat anti-Mouse IgG (H+L), Superclonal™ Recombinant Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat#A28175; RRID: AB_2536161 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Invitrogen | Cat#A-11031; RRID: AB_144696 |
| Anti-HA tag antibody | Abcam | Cat#ab18181; RRID: AB_444303 |
| G3BP antibody - N-terminal region | Aviva Systems Biology | Cat#ARP37713_T100; RRID: AB_2107232 |
| Influenza A virus M1 antibody | ATCC | Cat#HB-64 |
| Ubiquitin antibody (clone: P4D1) | Cytoskeleton | Cat#AUB01; RRID: AB_2884970 |
| ZIKA virus E-protein antibody | ATCC | Cat#HB-112 |
| Bacterial and virus strains | ||
| E. coli strain BL21(DE3) | NEB | Cat#C2527H |
| E. coli strain DH5α | NEB | Cat#C2987H |
| Influenza A virus strain H3N2 X31A | Virapur | Cat#Lot B1707C |
| ZIKV, Asian lineage strain, Puerto Rico | Public Health England (PHE) | ZIKV PRVABC59 (GenBank KX377337) |
| ZIKV, African lineage strain | Public Health England (PHE) | MP1751 (Genbank DQ859059) |
| Chemicals, peptides, and recombinant proteins | ||
| MG132, Ready-Made solution | Sigma-Aldrich | Cat#M7449 |
| Sodium Arsenite Solution | Sigma-Aldrich | Cat#1.06277 |
| dTAG-13 | Novartis | N/A |
| Bafilomycin A1 | Sigma-Aldrich | Cat#SML1661 |
| Carfilzomib | Biovision | Cat#2385-5 |
| Bortezomib | Merck-Millipore | Cat#CAS 179324-69-7 |
| DARPin F10 | This work | N/A |
| Mono ubiquitin | BostonBioChem | Cat#U-100H |
| His-Avi-hsHDAC6(1108-1215) | This paper | N/A |
| His-hsHDAC6 (1108-1215) | This paper | N/A |
| hsHDAC6 (1108-1215) | This paper | N/A |
| 3C protease | This paper | FMI facility |
| Original DARPin proteins (A10 to H12) | This paper | Table S1 |
| Original Nanobody (including VHH733, ZnF_97, ZnF_166, ZnF_213, ZnF_214) | This paper | N/A |
| Critical commercial assays | ||
| Invitrogen™ NuPAGE™ 4 to 12%, Bis-Tris, 1.5 mm, Mini Protein Gel | Invitrogen | Cat#NP0335BOX |
| LDS sample buffer | Invitrogen | Cat#NP0007 |
| Ni-NTA agarose | Promega | Cat#30210 |
| Broadford Dye | Biorad | Cat#5000006 |
| FuGENE HD | Promega | Cat#E2311 |
| Q5® Site-Directed Mutagenesis Kit | New England BioLabs | Cat# E0554S |
| FastStart Universal SYBR Green Master (Rox) | Roche | Cat# 4913850001 |
| Reverse Transcription System | Promega | Cat#A3500 |
| Proteinase K | Merck | Cat# 3115887001 |
| DAPI | Invitrogen | Cat#D1306 |
| Opti-MEM | Gibco | Cat#31985062 |
| ProLong™ Gold Antifade Mountant | Invitrogen | Cat#P10144 |
| Trans-Blot Turbo Transfer System | Biorad | N/A |
| ECL™ Detection Reagents | GE healthcare | Cat#GERPN2209 |
| GFP_Trap M | Chromotek | Cat#Gtm-20 |
| Dynabeads Protein G | Invitrogen | Cat#10007D |
| Gibbson Assembly Master Mix | NEB | Cat#E2611 |
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| Lenti-X™ Concentrator | Takara | Cat#631231 |
| Nunc® Lab-Tek® Chamber Slide™ system | Millipore | Cat#C7182 |
| HiLoad 16/600 Superdex 200pg | Sigma-Aldrich | Cat#GE28-9893-35 |
| Superdex 200 increase 10/300 GL | Sigma-Aldrich | Cat#GE28-9909-44 |
| Deposited data | ||
| DARPin cell line RNA sequencing | This paper | ArrayExpress E-MTAB-11101 |
| RNA-seq of GFP-DARPin F10/CTR transfected & WT A549 cells | This paper | ArrayExpress E-MTAB-11496 |
| DARPin F10 interactome with AP-MS | This paper | ProteomeXchange PXD031799 |
| HDAC6 ZnF-DARPin F10 structure | This paper | PDB:7ATT |
| Original data | This paper | Mendeley https://doi.org/10.17632/s3xw35xczs.1 |
| HDAC6 ZnF-Ubiquitin C-terminal peptide RLRGG structure | Ouyang et al., 2012 | PDB: 3GV4 |
| Lama VHH domain unliganded structure | Spinelli et al., 2001 | PDB:1I3V |
| Crystal structure of a designed full consensus ankyrin | Merz et al., 2008 | PDB:2QYJ |
| Experimental models: Cell lines | ||
| HEK 293T | ATCC | Cat#ATCC® CRL-3216 |
| Vero | ATCC | Cat#CCL-81 |
| A549 | ATCC | Cat#ATCC® CCL-185 |
| MDCKII | ATCC | Cat#CCL-34 |
| Oligonucleotides | ||
| Oligonucleotide primers are listed in Table S4 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pOPINF-His-HDAC6(1108-1215) | This paper | N/A |
| Plasmid: pOPINF-His-Avi-HDAC6(1108-1215) | This paper | N/A |
| Plasmid: pLenti-Puro-Flag-HA-F10-FKBP12(F36V) | This paper | N/A |
| Plasmid: pLenti-Puro-Flag-HA-FKBP12(F36V) | This paper | N/A |
| Plasmid: pcDNA3.1-eGFP-DARPin E3_5/F10 | This paper | N/A |
| Plasmid: pcDNA3.1-GFP (1-9) | This paper | N/A |
| Plasmid: pcDNA3.1-GFP (10)-Ub | This paper | N/A |
| Plasmid: pcDNA3.1-GFP (11)-ZnF | This paper | N/A |
| Plasmid: pcDNA3.1-HA-F10/A10 | This paper | N/A |
| Plasmid: pcDNA3.1-GFP (11)-ZnF(W1182A) | This paper | N/A |
| Plasmid: pQiq_K_MRGS_His10-HA-3C-1766_F10 | This paper | N/A |
| Plasmid: pLVX-puro-(Nb1∼4) | This paper | N/A |
| Plasmid: Pol-Gag | This paper | N/A |
| Plasmid: VSV-G | This paper | N/A |
| Plasmid: pet21a-BirA | This paper | N/A |
| Plasmid: plenti-puro-PGK-POI (GFP cleaved) | Addgene | Cat#19070 |
| Software and algorithms | ||
| proteomicsDB | Schmidt et al., 2017 | https://doi.org/10.1093/nar/gkx1029 |
| Perseus | Max Planck Institute of Biochemistry | Version 1.5.2.6/ https://maxquant.net/perseus/ |
| ImageJ | Schneider et al., 2012 | Version 1.8.0_112/ https://imagej.nih.gov/ij/ |
| ImageJ/Stress Granule Counter | ImageJ Plugin | https://imagej.nih.gov/ij/plugins/stress_granule_counter/index.html |
| Graphpad-Prism | Graphpad Software | Version 8/ https://www.graphpad.com/scientific-software/prism/ |
| Phenix | Liebschner et al., 2019 | Version 1.18/ https://www.phenix-online.org/ |
| CCP4 | Winn et al., 2011 | Version 7.1/ https://www.ccp4.ac.uk/ |
| Pymol | Schrödinger, LLC | Version 1.2r3pre/ https://pymol.org/2/ |
| MicroCal ITC Origin Analysis software | Malvern Panalytical | Version 1.0/ https://www.malvernpanalytical.com/ |
| Rstudio | Rstudio | Version 1.3/ https://rstudio.com/ |
| Other | ||
| MicroCal VP-ITC | Malvern Panalytical | N/A |
| Spinning disk confocal scanning unit Yokogawa CSU W1 with Dual camera T2 | Yokogawa | N/A |
| Z1 ZEISS-Wide Field Microscope | ZEISS | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthias Patrick (Patrick.Matthias@fmi.ch).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement (MTA); published research reagents from the FMI are shared with the academic community under an MTA having terms and conditions corresponding to those of the UBMTA (Uniform Biological Material Transfer Agreement).
Experimental model and subject details
Cell lines
HEK 293T, Vero, MDCKII and A549 cells (ATCC) were maintained and passaged in DMEM medium containing 10% fetal bovine serum. Cells were maintained at 37°C with 5% CO2 before and after transfection.
Method details
Nanobody screens and preparation
The target protein was a 6xHis- and HALO-tagged human HDAC6 ZnF (aa 1106–1215, expressed from plasmid pHis6HaloTag-hHDAC6ZnF) which was prepared by expression in E. coli BL21(DE3) RIL+. As a control, 6xHis-HALO protein (expressed from plasmid pH6HTN His6HaloTag) was prepared in a similar manner. Cells were induced with 0.5 mM IPTG at 20°C for 20 h. E. coli BL21 (DE3) cells expressing 6xHis-HALO-tagged ZnF-UBP were pelleted, rapidly frozen in liquid nitrogen and stored at −80°C. The frozen cells were resuspended in ice-cold lysis buffer (20 mM Tris, pH7.5, 200 mM NaCl, 20 mM imidazole, 2 mM TCEP, 0.2% Tween 20) supplemented with Complete EDTA-free protease inhibitors (Roche) and 3 U/mL Benzonase (Sigma). After 30 min on ice the lysate was centrifuged at 30,000g for 30 min at 4°C. The clarified soluble lysate was incubated for 30 min at 4°C in batch mode with Ni-NTA IMAC agarose (Qiagen), and then transferred into a 10 mL Econo-Pac column (Bio-Rad) for washing with nickel wash buffer (20 mM Tris, pH 7.5, 200 mM NaCl, 20 mM imidazole, 2 mM TCEP). The target protein was eluted in nickel wash buffer containing 250 mM imidazole. The eluted protein was concentrated with Amicon ultra concentration device (30,000 MWCO) (Millipore) and separated using a DUO FLOW system (Bio-Rad) with a Sephacryl S-300 16/60 gel filtration column (GE Healthcare) equilibrated in 20 mM Tris, pH 7.5, 200 mM NaCl, 2 mM TCEP, 5% Glycerol and 0.02% NaN3. Protein fractions were analysed on a 4–12% Bis-Tris NuPAGE (Invitrogen) gels and pure fractions were pooled and concentrated to 15 mM for Nanobody production. Gels were stained with InstantBlue (Expedeon). Identification of nanobodies against the HDAC6 ZnF was done by Hybrigenics Services SAS. In brief, HALO-ZnF (or HALO as control) protein was biotinylated in vitro using HaloTag® PEG-Biotin Ligand (Promega: G8591 or G8592) following the manufacturer’s instruction and then used for three rounds of phage display with a naïve synthetic library based on a proprietary Lama scaffold. The Phage library was first incubated with the biotinylated His-HALO; the supernatant was then incubated with the biotinylated HALO-ZnF. Following selection, the positive hits were used to generate a yeast two-hybrid library, which was then screened against the human HDAC6 ZnF as bait (aa 1106–1215). Positive hits were isolated and validated by an intrabody assay. Following this, four different positive clones (Nb1 to 4) as well as a control clone were selected for further analysis.
GFP-trap pull-down assay
C-terminally eGFP-tagged nanobodies were cloned into pLVX-puro lentiviral expression vectors. The lentiviral vector was co-transfected with Pol-Gag and VSV-G plasmids into HEK293T cells to produce lentivirus. Each eGFP-tagged nanobody was stably expressed in A549 cells after lentivirus infection, then eGFP-positive cells were sorted by FACS. A549 cells expressing each nanobody were harvested with ice-cold PBS from a 10 cm dish, spun down at 1,000g for 5 min. The pelleted samples were rapidly frozen at −80°C. The frozen pellet was treated with CSK (cytoskeleton) buffer (10 mM PIPES pH6.8, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 1 mM EGTA, 0.1%(v/v) Triton X-100) with 1× Complete EDTA free protease inhibitor cocktail (Roche#CO-RO) for 30 min on ice. The lysed cell extracts were subjected to low-speed centrifugation (3,000 rpm for 5 min) to separate the soluble cytoplasmic fraction. eGFP-tagged nanobodies were pulled-down with GFP-Trap agarose beads (Chromotek#gtm-20) equilibrated with CSK buffer containing 1% BSA. The soluble fractions were incubated with GFP-Trap beads for 30 min at 4°C, then spun down 1,000g for 2 min. The beads were washed with GFP-Trap Wash buffer (10 mM Tris, pH7.5, 200 mM NaCl, 0.5 mM EDTA, 5% Glycerol) twice, and finally washed with 10 mM Tris, pH7.5, 100 mM NaCl, 5% Glycerol buffer once. The bead samples were dissolved in Laemmli sample buffer supplemented with 10 mM DTT and boiled for 5 min at 95°C before loading on a 4%–12% Bis-Tris NuPAGE gels. Proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore#05317) by using the iBlot2 Dry Blot system (Thermo Fisher Scientific) following to instruction manual and detected with specific antibodies (anti-HDAC6 (D2E5, Cell Signaling Technology), 1:1,000, GFP (JL-8, Takara), 1:1,000, and α-Tubulin (Abcam ab4074) 1:1,000).
IAV infection assay (in nanobody expressing cells)
A549 cells stable expressing the HDAC6 ZnF nanobodies were infected with IAV X-31 (H3N2) strain. The cells were trypsinised and fixed in 4% FA at 5.5 h post infection. The cells were stained for FACS analysis in FACS buffer (PBS, 0.1% BSA) containing 0.1% Saponin. The primary antibody used was mAb HB-65 (anti-nucleoprotein, ATCC), 1:200, and secondary was goat anti-mouse IgG Alexa Fluor 647 (Invitrogen), 1:2500. Antibodies were incubated for 30 min at room temperature. The cells were washed by centrifugation at 2,500 rpm for 5 min, resuspended in 100 μL of FACS buffer and analysed using Novocyte Flow Cytometer (Aceabio). The fcs files were analysed using Flow Jo version 10.3.0.
Selection and screening of DARPins
To generate DARPin binders against HDAC6-ZnF, the biotinylated target protein (see below) was immobilized on MyOne T1 streptavidin-coated beads (Pierce). Ribosome display selections were performed essentially as described (Dreier and Plückthun, 2012). Selections were performed over four rounds with decreasing target concentration and increasing washing steps to enrich for binders with high affinities. After four rounds of selection, the enriched pool was cloned into a bacterial pQIq-based expression vector as fusion with an N-terminal MRGSH8-and C-terminal FLAG tag. After transformation into E. coli XL1-blue 380 single DARPin clones were expressed in 96 well format and lysed by addition of Cell lytic B reagent (Sigma), Lysozyme and Pierce nuclease. These bacterial crude extracts of single DARPin clones were subsequently used in a Homogeneous Time Resolved Fluorescence (HTRF)-based screen to identify potential binders. Binding of the FLAG-tagged DARPins to streptavidin-immobilized biotinylated HDAC6-ZnF was measured using FRET (donor: streptavidin-Tb cryptate, 610SATLB; acceptor: anti-FLAG-d2, 61FG2DLB; both Cisbio). Experiments were performed at room temperature in white 384-well Optiplate plates (PerkinElmer) using the Taglite assay buffer (Cisbio) at a final volume of 20 μL per well. FRET signals were recorded after an incubation time of 30 min using a Varioskan LUX Multimode Microplate (Thermo Scientific) with the following settings: Delay time: 60 μs, integration time: 200 μs, measurement time: 1′000 ms, dynamic range: automatic. HTRF ratios were obtained by dividing the acceptor signal (665 nm) by the donor signal (620 nm) and multiplying this value by 10′000 to derive the 665/620 ratio. The background signal was determined by using reagents in the absence of DARPins. From this result, potential binders were identified (Table S1)
Expression and purification of biotinylated His-Avi-HDAC6 ZnF (1108–1215), HDAC6 ZnF (1108–1215) and DARPin F10
E. coli BL21 (DE3), transfected by pOPINF-His-Avi-HDAC6 ZnF (in this case, bacteria was co-transfected with pet21a-BirA expressing plasmid, pOPINF-His-HDAC6 ZnF or DARPin F10 plasmid (pQiq_K_MRGS_His10-HA-3C-1766_F10), was cultured first in 50 mL LB medium overnight at 37°C, then 10 mL medium was transferred to 1 L 2xYT medium for continuous culturing in 2.5 L flask. When OD = 0.6 was reached, IPTG was added into the 1 L medium (final concentration 1 mM) and the temperature was reduced to 17°C (to induce ZnF protein biotinylation, D-biotin was added to 2xYT medium to a final concentration of 20 μM). Cultures were grown further for 18 h and bacteria were collected by centrifugation (4000 rpm, 15 min, 4°C). The pellet was frozen at −80°C. All constructs were purified as follows.
The bacterial pellet was lysed with an ultrasonic sonicator (15 cycles, one cycle = 20 s ON, 40 s OFF) to completely break the bacteria. The cell lysate was dissolved in Ni column loading buffer (buffer 1, containing 20 mM Tris pH = 7.5, 500 mM NaCl, 10 mM imidazole, supplied with protease inhibitor 1mM PMSF). The cell debris were separated from the protein by High-speed-centrifugation (17000rpm, 1 h, 4°C). After filtering (0.45 μM filter, Merck#SLHVM33RS) the supernatant, the protein solution was loaded onto a HisTrap column (#GE17-5248-01) using a peristaltic pump at a flow rate of 5 mL/min. The column was washed with 4–5 column volumes (CV) of buffer 1. To elute the target protein, we used a gradient elution by the AKTA system. The elution buffer (buffer 2, containing 20 mM Tris pH = 7.5, 500 mM NaCl, 250 mN imidazole) was used together with the buffer 1 to generate the gradient.
Eluted protein was digested by 3C protease (obtained from FMI protein facility) in dialysis buffer (20 mM Tris pH = 7.5, 500 mM NaCl) at 4°C. Digested protein was re-loaded onto a HisTrap column to remove the His tag. His-Avi-ZnF protein was not treated with 3C protease but loaded directly to the gel filtration step. The flow-through was collected and protein purity was determined by SDS-PAGE. Different fractions (with a purity >50%) from gradient elution were combined and applied onto an ion exchange column. Here, the protein solution was diluted 5 times with 20 mM Tris pH = 7.5 buffer and loaded onto a 5 mL pre-pack HiTrap Q HP(#GE29-0513-25) column. Protein was eluted over a 25 min period with a gradient generated by the AKTA system, by mixing buffer A (20 mM Tris pH = 7.5, 100 mM NaCl, 2 mM TCEP) and buffer B (20 mM Tris pH = 7.5, 1 M NacL, 2 mM TCEP). Eluted target protein was concentrated and loaded onto Gel filtration system, using either a Superdex® 200 Increase 10/300 GL (#GE28-9909-44) or HiLoad 16/600 Superdex 200pg (#GE28-9893-35) column. After Gel filtration, the separated target protein was flash-frozen with liquid nitrogen in Gel filtration buffer (20 mM Tris pH = 7.5, 100 mM NaCl, 1 mM TCEP). Protein concentration was determined by its absorbance at 280 nm.
Pull-down assay to identify inhibitory DARPin
10 μg Purified His-HDAC6 ZnF (1108–1215) was immobilized with 20 μL Ni-NTA agarose (Promega#30210) slurry at 4°C for 30 min. Subsequently, 20 μg DARPin protein and 10 μg Ub were added to the Ni-NTA-ZnF solution at same time (in Figure S1C, they were mixed as described in the figure legend). All proteins were diluted in Ni-NTA loading buffer (20 mM Tris pH = 7.5, 100 mM NaCl, 10 mM imidazole). The reaction volume was 500 μL, incubation was at 4°C on rotator for 30 min. After incubation, the beads were washed 3 times with washing buffer (20 mM Tris pH = 7.5, 150 mM NaCl). The supernatant was removed after spinning down the beads (500 g, 2 min), and 20 μL 1× LDS sample buffer (Invitrogen# NP0007) was added to each reaction. Following heating at 80°C for 10 min, all the samples were loaded onto NuPAGE 4%–12% gradient gels. The proteins were visualized with Instant Blue reagent (expedeon#ISB1L).
Crystallization, data collection, processing and refinement
ZnF-DARPin F10 complex crystal was obtained under condition 0.004 M Cobalt, 1.66 M Ammonium sulfate, 0.1 M MES pH = 6.45 (with additive screen: 30% 1,8-Diaminooctane) by sitting drop at 20°C. Drop size is 2 μL, and complex concentration is 43 mg/mL (determined by Bradford assay) in buffer (10 mM Tris pH = 7.5, 150 mM NaCl, 1 mM TCEP)
Diffraction data were collected at the Swiss Light Source (beamline ×06DA) with a Pilatus2M detector (Dectris) at a wavelength of 1 Å and a crystal cooled to 100 K. A total of 220° of data were collected with an oscillation of 0.1°/frame. Data were processed with autoPROC (Vonrhein et al., 2011) in combination with XDS (Karplus and Diederichs, 2012), AIMLESS (Evans and Murshudov, 2013), and other tools of the CCP4 suite (Winn et al., 2011) and converted to structure factor amplitudes with STARANISO (Tickle et al., 2018), applying a locally weighted I/δ = 1.2 resolution cutoff.
The ZnF-DARPin F10 complex formed crystals belonging to space group P3221 with one complex in the crystallographic asymmetric unit (ASU). The structure was determined using molecular replacement (MR) in PHASER (McCoy et al., 2007) with a search model derived from PDB entries 1MJ0 for DARPin F10 and 3GV4 for ZnF. A significant anomalous signal was detected in the data that could later be attributed to the coordinated zinc ions. This allowed experimental phasing using the single-wavelength anomalous dispersion (SAD) method in phenix.autosol (Terwilliger et al., 2009) yielding a map without model-bias. The initial model was improved by iterative cycles of building with COOT (Emsley et al., 2010) and refinement using phenix.rosetta_refine (DiMaio et al., 2013)and phenix.refine (Afonine et al., 2012). TLS groups were derived from analysis conducted with the TLSMD webserver (Painter and Merritt, 2006a, 2006b) and used for TLS refinement in phenix.refine. Data processing, phasing, and refinement statistics are in Table S2. Figures were made with PyMOL (Schrodinger, LCC).
Mutagenesis of DARPin F10 mutant (KDDR)
To generate DARPin F10 mutant (K47A, D67A, D100A, R113A, or F10KDDR), template plasmid pQiq_K_MRGS_His10-HA-3C-1766_F10 was used for mutagenesis by Q5 site-direct mutagenesis kit. Mutagenic primers are listed in key resources table. Mutated plasmid was transformed into E. coli BL21 strain for protein expression. Purification was performed as for the WT F10. Purified F10KDDR was dissolved in ITC buffer (10 mM Tris pH = 7.5, 100 mM NaCl, 2mM TCEP).
Isothermal titration calorimetry (ITC) for affinity determination
The experiment was performed on a MicroCal VP-ITC machine. Protein HDAC6 ZnF (1108–1215), DARPin F10 mutant (KDDR) and DARPin F10 were purified as described, and Ub was purchased from BostonBiochem(Cat# U-100H). All the proteins were dialyzed in ITC buffer (10 mM Tris pH = 7.5, 100 mM NaCl) for 3 h at 4°C before the experiment. We titrated 0.2 mM DARPin F10 protein (in the syringe) to 0.02 mM HDAC6 ZnF (1108–1215) protein in the cell (for ITC of Ub and ZnF, 0.25 mM Ub was titrated to the 0.01 mM ZnF, while for DARPin F10 mutant and ZnF, 0.1 mM mutant was titrated to 0.01 mM ZnF). The curve and statistics were done with the MicroCal ITC Origin Analysis software.
LC-MS analysis for DARPin F10 interacting protein
5 μg pcDNA3.1-GFP-DARPin F10 and pcDNA3.1-GFP-DARPin E3_5 were transfected into A549 cells; for each construct 3 × 10 cm dishes were used, each containing 2 × 10ˆ6 cells in 10 cm dishes. For each sample 15 μL Lipofectamine 3000 were used, and the cells were cultured for 2 days to allow for sufficient expression. After collecting the cells, each dish was lysed with 500 μL CoIP buffer (10 mM Tris pH = 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5%NP-40). The lysate was centrifuged at 14,000 rpm and 10 min, 4°C. Supernatant was transferred to a new tube, and the protein concentration was determined by Bradford Assay (Biorad#5000006). Same amount of protein lysate (1.0–1.5 μg) was incubated with 10 μL GFP_Trap M beads (Chromotek#gtm-20) on rotator at 4°C, overnight. In the following morning, the beads were washed with washing buffer (20 mM Tris pH = 7.5, 150 mM NaCl) 3 times.
To digest the protein on the beads, we used 10 μL Lys-C (0.2μg/μL in 50 mM Hepes pH 8.5) and 50 μL digestion buffer (3M GuaHCl, 20 mM EPPS pH 8.5, 10 mM CAA, 5 mM TCEP) to make master mix, and added 6 μL of this mixture to the beads from each 10 cm dish. After short spin down (<1000 g) and 4 h incubation at 37°C, we added 17 μL 50mM HEPES pH 8.5 as well as 1 μL 0.2 μg/μL trypsin to further digest the protein at 37°C overnight. Next morning, 1 more μL trypsin was added to the solution for 6 additional hours digestion. Then the sample was processed at the FMI Protein Analysis facility for mass spectrometry. The data was analysed by software Perseus (version 1.5.2.6). And protein was annotated with human (v2017-04) database.
Peptides of raw DARPin F10 data from the mass spectrometers were identified and quantified by MQ version 1.5.3.8. For data searches the search engine Andromeda was used. A costume DB with the DARPin_F10 and E3_5 sequence combined with the human sub-set of the UNIPROT DB and the contaminant library from MaxQuant was searched. Results were filtered with a FDR of 1%.
RNA seq analysis for DARPin F10 impact on cellular gene expression
A549 cells were transfected as described above section “LC-MS analysis”. Each plasmid was transfected into 3 10 cm dishes. After 2 days in culture, 1.5 million GFP positive cells were isolated from each dish by FACS (that is, 3 × 1.5 million cells for one transfected DARPin), and total RNA was extracted with the RNA extracting Kit (QIAGEN#74004). The samples were further processed at the FMI genomics Facility and sequenced on a Hi-Seq instrument; the results were analysed with R-Studio.
DARPin F10 and WT A549 cells (1.5 million cells) RNA were extracted by RNA extracting Kit (QIAGEN#74004) and analysed with FMI-Galaxy and R-studio.
Generation of A549 cells expressing a conditionally degradable DARPin F10 and F10KDDR cell lines
Plasmid Plenti-Puro-Flag-HA-DARPin F10-FKBPF36V and Plenti-Puro-Flag-HA-FKBPK36V (each 20 μg) were co-transfected with packaging plasmids (expressing tat, rev, gag, vsv-g, each at 1 μg) together with 75 μL FuGENE HD reagent (Promega#E2311) in 0.5 mL Opti-MEM medium (Sigma#31985062). After 25 min incubation at room temperature, the mix was added to 293T cells in 10 cm dish, seeded one day before with 1.6 million cells per dish. Cells were cultured at 37°C for 3 days; the medium was collected and filtered with 0.45 μm filter (Merck#SEIM003M00), and 1× LentiX concentrator (Takara#631231) was added (1/3 of the supernatant volume). The mixture was incubated at 4°C for 30 min and the lentivirus was precipitated by centrifugation at 1500 g, 45 min, 4°C. The pellet was re-suspended in 500 μL Opti-MEM (Gibco#31985062).
The re-suspended lentivirus pellet was added to the culture medium of A549 cells (10 cm dish, 0.6 million WT A549 cells seeded per dish one day before). Two days later the medium was changed to DMEM supplied with puromycin (final concentration 2 μg/mL). Puro-resistant cells were selected for 2 days and then single-cell sorted into 96 wells plates. After 1 month culturing, clones were expanded and analysed by western blot with HA antibody (Abcam#18181) to identify the cell lines expressing HA-DARPin F10-FKBPF36V or HA-FKBPF36V.
To generate HA-F10 and HA-F10KDDR cell lines, fragments encoding HA-F10 and HA-F10KDDR were cloned into lentivirus plasmid Plent-Puro-PGK-POI. Lentivirus preparation, infection of target cells and selection was done as above, but without selection of single cell clones. The cell lines expressing HA-F10 or HA-F10KDDR were used as pools for IAV infection in Figure S5.
Imaging and quantification of aggresomes and stress granules
For immunofluorescence, cells were seeded in 4 wells chamber slides (Millipore# C7182) to culture the cells. WT A549, DARPin F10-FKBPF36V and dTAG treated (2 μM, 6 h) F10 expressing cells were treated with 5 μM MG132 for 18 h to induce aggresome formation. Ubiquitin and HDAC6 were used as aggresome markers and stained by anti-Ub antibody (Cytoskeleton #AUB01, clone P4D1) or anti-HDAC6 antibody (CST#7558). The nucleus (DNA) was stained with DAPI (Sigma#D1306). Pictures were taken with 40× objective on a Zeiss Z1 widefield microscope (Ub stained aggresomes) and 60× objective on a Visitron spinning disk confocal microscope (HDAC6 stained aggresomes and zoomed aggresomes pictures in Figure S6); identical parameters (e. g. exposure time, binning, etc...) were used for all the conditions. Slide areas were identified where cell confluency was in the optimal range of ca. 50 to 70%; for each condition and biological replicate ca. 10 pictures were taken and analysed by ImageJ software. In some cases, pictures were selected manually, in other cases the first location was identified manually, and then further pictures were taken automatically at regular intervals around the initial point using the stage function of Visiview software. Aggresomes were identified by signal intensity and shape and quantitated by Particle analysis in ImageJ. For aggresome selection (for both HDAC6 and Ub markers), classification was done as follows: the image threshold was adjusted to 0.9 to 1.5%, then aggregates with a size >2 μm2 and a circularity between 0.4 and 1 were considered as aggresomes. For each biological replicate, ca. 400 to 800 cells were analysed for each condition (A549 WT, F10 and F10 + dTAG).
For stress granules, we used 1 mM Sodium Arsenite (30 min treatment) to induce SGs. G3BP1 was used as the marker for SGs and visualized by anti-G3BP1 antibody (Aviva Systems Biology#ARP37713_T100). Slide areas were identified where cell confluency was in the optimal range of ca. 50 to 70% and for each condition and biological replicate ca. 10 pictures were taken. For each picture groups of a few (ca. 2 to 5) cells were selected; in some cases, when cells were well separated, only the central cell was selected for analysis. Images were taken with 40× objective on a Zeiss Z1 widefield microscope. Quantification and analysis were done with ImageJ, installed with Stress Granule Counter Plugin (developed by Ann Sablina, Lomonosov Moscow State University, Russia). Settings for SGs selection were: threshold 3000, particle size between 1 and 10000 pixels, circularity >0.2 and number and smoothness 70. For each biological replicate, ca. 25 to 50 cells were analysed for each condition (A549 WT, F10 and F10 + dTAG). Plots and statistics were all done in software Graphpad Prism 8.
Split-GFP assay
Plasmid pcDNA3.1-GFP (1–9), pcDNA3.1-GFP(10)-ubiquitin, pcDNA3.1-GFP(11)-HDAC6 ZnF (1108–1215)/1182 mutant and pcDNA3.1-mRuby were co-transfected with FuGENE reagent (using the manufacturer’s protocol) (Promega#E2311) together to 0.5 million 293T cells in 6 well plate, with a molarity ratio of 1:1:1:1 (1 μg for pcDNA3.1-GFP(1–9), other plasmids were adjusted accordingly). After 2 days culturing at 37°C, the GFP signal was visualized by widefield microscopy (Zeiss Z1). mRuby expression served as a transfection control.
To investigate DARPin interference, non-fluorescent tagged DARPin F10 and DARPin A10 plasmid (1 μg for each) were transfected together with the plasmids mentioned above. Visualization procedures were the same.
IAV uncoating assay
A549 cells were seeded on glass slides and, after reaching ca. 70% confluence, incubated at 4°C with IAV ×31 (H3N2, MOI = 30 PFU/cell) for 1 h to synchronize infection. After that, the cells were incubated at 37°C for 3 h and fixed with paraformaldehyde 4% for 15min. For immunofluorescence, cells were permeabilized with 0.5% Triton X-100 in PBS for 5 min and incubated overnight in 1% BSA with M1-specific murine monoclonal antibody (ATCC#HB64, 1:4000). Cells were washed with PBS and incubated for 1 h with Alexa Fluor 488 goat anti-mouse (IgG) (H + L) (Thermo Fischer; 1:2000, 1% BSA) for 1 h at room temperature. Nuclei were stained for 5 min with DAPI (1:1000 in PBS). Glass slides were examined using spinning disk confocal scanning unit. Alex Fluor 647 NHS ester Tris was used to stain the total protein to visualize the cell body.
The mean fluorescence green intensity (MFI) was quantified using ImageJ. One representative out of three independent experiments is shown. For all panels, error bars represent standard deviation of the pictures analysed (approximately 40 cells per picture and more than 200 cells in total) and statistical significance was determined by one-way ANOVA (ns (non-significant) = p > 0.05).
IAV growth curve
A549 cells were infected in 6 well/plates with Influenza virus (Virapur, H3N2, purified Influenza A/×31, #B1707C) at 37°C in infection medium (DMEM 0.2% BSA, 2 mM L-glutamine and 1 μg/mL of TPCK-treated trypsin). For single cycle infection assays, cells and viruses were pre-incubated at 4°C for 1 h and the cells were washed with PBS before incubation at 37°C for the indicated time points. Viruses from the harvested supernatants were quantified using plaque assay in MDCKII cells with 2% agar overlay.
A Zika virus stock from a low passage clinical isolate of Asian lineage (Vielle et al., 2018) passaged on Vero cells and purified by ultracentrifugation was used to infect A549 cells in 24 well/plates at 37°C with in DMEM 10% FCS. Virus titers collected from the culture supernatant were determined on Vero cells (ATCC#CCL-81) and expressed as 50% tissue culture infective dose per ml (TCID50/ml) using the Reed and Muench method.
Detection of ubiquitin inside ZIKV particles
To detect the ubiquitin inside the Zika virus particles, we used Trition X-100 0.1% for opening the virus envelope and Proteinase K to destroy the virus capsid. Unpurified African Zika virus strain, 3 × 107 TCID50/ml, and Puerto Rico strain 1 × 107 TCID50/ml were incubated on ice for 1 h with a lysis buffer containing Triton X-100 0.1% in a proportion of virus solution and buffer of 1:1. Proteinase K with varying concentrations was added to samples and incubated at room temperature for exactly 2 min. LDS sample buffer (5×) and PMSF (2 mM) were added to each sample and immediately heated at 99°C for 10 min to stop the Proteinase K reaction. 13 μL mixed sample was loaded to each well of 12-well Bis-tris NuPAGE Gel (4%–12%) Ubiquitin and Zika protein E were analysed by SDS-PAGE.
Investigation of ZIKV uncoating by real-time PCR
A549 cells were incubated with Zika virus (Puerto Rico strain, MOI of 10) in media containing cycloheximide (1 μg/μL, Sigma-Aldrich) on ice for 30 min for synchronizing the virus infection. The inoculum was replaced with fresh media containing cycloheximide 1 μg/μL and incubated at 37°C before total RNA was isolated at 1 and 2 hpi accordingly to the kit manufacturer (Macherey-Nagel™ NucleoSpin™ RNA). cDNA was synthesized using the Omniscript RT Kit from QIAGEN used for RT-qPCR.
Real-time PCR of interferon genes in ZIKV infection
A549 WT cells, F10-FKBP cells (treated with dTAG 1 μM, 12 h prior to infection) were infected by Zika virus (strain Puerto Rico) with MOI = 1. Mock group (A549 WT cells) was non-infected. At 24 h after transfection, cells were washed with PBS twice to remove detached cells, and remaining cells were lysed by lysis buffer (provided in RNA extraction kit) in plate, and RNA was extracted by following the manufacturer’s instructions. 1 μg RNA was used to reverse transcription (in 20 μL reaction). cDNA was further diluted (10×) and applied to real-time PCR. Gene IFNB1, IFNL1, DDX58 and ISG15 were chosen to analyse the antiviral response after ZIKV infection. Among them, IFN-α was undetectable. 2ˆ-ΔΔct method was used for comparing the upregulation of each gene as compared to the Mock group.
Immunofluorescence for zika virus
For detection of Zika virus protein on infected cells at 16 and 72hpi, the A549 cells were washed with PBS, fixed with 4% PFA and incubated with anti-E protein Flavivirus group antibody 4G2 in PBS supplemented with 1% BSA for 1h at 37°C. Cells were washed with PBS and incubated for 1 h with Alexa Fluor 488 goat anti-mouse (IgG) (H + L) (Thermo Fischer; 1:2000, 1% BSA) for 1 h at room temperature. Nuclei were stained for 5 min with DAPI (Thermo Fischer, 1:1000 in PBS). Picture was taken by 10× (72 hpi) and 20× (16hpi) objectives from confocal microscope.
Quantification and statistical analysis
Details of specific statistical tests and experimental design for immunofluorescent experiments are given in the relevant figure legends. All Virus growth curve experiments were performed in triplicate for technical replication. Each experiment was independently repeated 2 to 3 times to consolidate the conclusions. Statistical analysis was performed with GraphPad Prism 8; Method details have been described in Figure legends; p values are indicated in the figures. All data are represented as mean ± SD.
Acknowledgments
We thank L. Gelman and S. Bourke for help with microscopy, D. Hess, J. Seebacher, and V. Iesmantavicius for mass spectrometry, and H. Gut for help with X-ray data collection. We thank H. Rice and A. Herman (FACS facility, Bristol University) and H. Kohler (FMI FACS facility). We thank C. Cao and G. Matthias for excellent support, and T. Reinberg, S. Furler, J. Marinho for help with DARPin screening, P. Nigg and M. Saito for help with cloning, S. Iketani and J. Tormo for nanobody experiments, and G. Langousis and J. Sanchez for comments. Financial support through the SystemsX.ch MRD project VirX 2014/264, evaluated by the Swiss National Science Foundation, and by the European Research Council—ERC Synergy grant number 856581 CHUbVi is gratefully acknowledged. This work was supported by the Novartis Research Foundation.
Author contributions
L.W. and P.M. designed the project; L.W. performed biochemical and microscopy experiments and interpreted the data with supervision by P.M.; E.A.M. and L.W. performed IAV experiment and analysis; E.A.M., B.I.O.E., A.F., L.W., and M.P.A. performed ZIKV experiments and analysis; L.W. and G.K performed X-ray data processing and structure refinement; J.V.S., B.D., and A.P. performed experimental design and analysis for DARPin selection; Y.Y. and Y.M. performed nanobody-related experiments; L.W and P.M wrote the manuscript, and all authors contributed to the final version.
Declaration of interests
Part of the results presented herein have been used in the patent application EP-A-20213494.6.
Published: April 26, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110736.
Supplemental information
Data and code availability
-
•
RNA sequencing data have been deposited at ArrayExpress and are publicly available as of the date of publication. Mass Spectrometry data (Search results are in Table S3) have been deposited at the ProteomeXchange Consortium via the PRIDE partner repository and are publicly available as of the date of publication. Structural data have been deposited with the Protein DataBank and are publicly available as of the date of publication. All the accession numbers are listed in the key resources table.
Original data have been deposited at Mendeley and are publicly available as of the date of publication. DOIs are listed in the key resources table.
Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Advani V., Ivanov P. Stress granule subtypes: an emerging link to neurodegeneration. Cell Mol. Life Sci. 2020;77:4827–4845. doi: 10.1007/s00018-020-03565-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P., Kedersha N., Ivanov P. Stress granules, P-bodies and cancer. Biochim. Biophys. Acta. 2015;1849:861–870. doi: 10.1016/j.bbagrm.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash P., Vanderweyde T., Youmans K., Apicco D., Wolozin B. Pathological stress granules in Alzheimer's disease. Brain Res. 2014;1584:52–58. doi: 10.1016/j.brainres.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee I., Miyake Y., Nobs S.P., Schneider C., Horvath P., Kopf M., Matthias P., Helenius A., Yamauchi Y. Influenza A virus uses the aggresome processing machinery for host cell entry. Science. 2014;346:473–477. doi: 10.1126/science.1257037. [DOI] [PubMed] [Google Scholar]
- Binz H.K., Amstutz P., Kohl A., Stumpp M.T., Briand C., Forrer P., Grutter M.G., Pluckthun A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 2004;22:575–582. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- Binz H.K., Stumpp M.T., Forrer P., Amstutz P., Plückthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003;332:489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- Boyault C., Gilquin B., Zhang Y., Rybin V., Garman E., Meyer-Klaucke W., Matthias P., Muller C.W., Khochbin S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25:3357–3366. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault C., Sadoul K., Pabion M., Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene. 2007;26:5468–5476. doi: 10.1038/sj.onc.1210614. [DOI] [PubMed] [Google Scholar]
- Brauchle M., Hansen S., Caussinus E., Lenard A., Ochoa-Espinosa A., Scholz O., Sprecher S.G., Plückthun A., Affolter M. Protein interference applications in cellular and developmental biology using DARPins that recognize GFP and mCherry. Biol. Open. 2014;3:1252–1261. doi: 10.1242/bio.201410041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindisi M., Saraswati A.P., Brogi S., Gemma S., Butini S., Campiani G. Old but gold: tracking the new guise of histone deacetylase 6 (HDAC6) enzyme as a biomarker and therapeutic target in rare diseases. J. Med. Chem. 2019;63:23–29. doi: 10.1021/acs.jmedchem.9b00924. [DOI] [PubMed] [Google Scholar]
- Byk L.A., Iglesias N.G., De Maio F.A., Gebhard L.G., Rossi M., Gamarnik A.V. Dengue virus genome uncoating requires ubiquitination. mBio. 2016;7 doi: 10.1128/mBio.00804-16. e00804–00816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous S., Nguyen H., Pedelacq J., Koraïchi F., Chaudhary A., Ganguly K., Lockard M., Favre G., Terwilliger T., Waldo G. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci. Rep. 2013;3:2854. doi: 10.1038/srep02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos G.S., Bandeira A.C., Sardi S.I. Zika virus outbreak, bahia, Brazil. Emerging Infect. Dis. 2015;21:1885–1886. doi: 10.3201/eid2110.150847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao-Lormeau V.M., Roche C., Teissier A., Robin E., Berry A.L., Mallet H.P., Sall A.A., Musso D. Zika virus, French polynesia, South pacific, 2013. Emerging Infect. Dis. 2014;20:1085–1086. doi: 10.3201/eid2006.140138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Qian Y., Chen X., Ruan Z., Ye Y., Chen H., Babiuk L.A., Jung Y.-S., Dai J. HDAC6 restricts influenza A virus by deacetylation of the RNA polymerase PA subunit. J. Virol. 2019;93 doi: 10.1128/JVI.01896-18. e01896–01818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.J., Lee H.C., Kim J.H., Park S.Y., Kim T.H., Lee W.K., Jang D.J., Yoon J.E., Choi Y.I., Kim S., et al. HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J. 2016;35:429–442. doi: 10.15252/embj.201592586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosenza M., Pozzi S. The therapeutic strategy of HDAC6 inhibitors in lymphoproliferative disease. Int. J. Mol. Sci. 2018;19:2337. doi: 10.3390/ijms19082337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio F., Echols N., Headd J.J., Terwilliger T.C., Adams P.D., Baker D. Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat. Methods. 2013;10:1102–1104. doi: 10.1038/nmeth.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier B., Plückthun A. Rapid selection of high-affinity binders using ribosome display. Methods Mol. Biol. (Clifton, NJ) 2012;805:261–286. doi: 10.1007/978-1-61779-379-0_15. [DOI] [PubMed] [Google Scholar]
- Du J., Cross T.A., Zhou H.X. Recent progress in structure-based anti-influenza drug design. Drug Discov. Today. 2012;17:1111–1120. doi: 10.1016/j.drudis.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy M.R., Chen T.H., Hancock W.T., Powers A.M., Kool J.L., Lanciotti R.S., Pretrick M., Marfel M., Holzbauer S., Dubray C., et al. Zika virus outbreak on yap island, federated states of micronesia. New Engl. J. Med. 2009;360:2536–2543. doi: 10.1056/NEJMoa0805715. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P.R., Murshudov G.N. How good are my data and what is the resolution? Acta Crystallogr. Ds Biol. Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira de Freitas R., Harding R.J., Franzoni I., Ravichandran M., Mann M.K., Ouyang H., Lautens M., Santhakumar V., Arrowsmith C.H., Schapira M. Identification and structure-activity relationship of HDAC6 zinc-finger ubiquitin binding domain inhibitors. J. Med. Chem. 2018;61:4517–4527. doi: 10.1021/acs.jmedchem.8b00258. [DOI] [PubMed] [Google Scholar]
- Gale T.V., Horton T.M., Hoffmann A.R., Branco L.M., Garry R.F. Host proteins identified in extracellular viral particles as targets for broad-spectrum antiviral inhibitors. J. Proteome Res. 2019;18:7–17. doi: 10.1021/acs.jproteome.8b00204. [DOI] [PubMed] [Google Scholar]
- Gao X., Jiang L., Gong Y., Chen X., Ying M., Zhu H., He Q., Yang B., Cao J. Stress granule: a promising target for cancer treatment. Br. J. Pharmacol. 2019;176:4421–4433. doi: 10.1111/bph.14790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestuveo R.J., Royle J., Donald C.L., Lamont D.J., Hutchinson E.C., Merits A., Kohl A., Varjak M. Analysis of Zika virus capsid-Aedes aegypti mosquito interactome reveals pro-viral host factors critical for establishing infection. Nat. Commun. 2021;12:2766. doi: 10.1038/s41467-021-22966-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldo M.I., Xia H., Aguilera-Aguirre L., Hage A., van Tol S., Shan C., Xie X., Sturdevant G.L., Robertson S.J., McNally K.L., et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature. 2020;585:414–419. doi: 10.1038/s41586-020-2457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin J.K., Moses A.V., Fruh K., Douglas J.L. Viral takeover of the host ubiquitin system. Front Microbiol. 2011;2:161. doi: 10.3389/fmicb.2011.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao R., Nanduri P., Rao Y., Panichelli R.S., Ito A., Yoshida M., Yao T.P. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell. 2013;51:819–828. doi: 10.1016/j.molcel.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding R., Ferreira de Freitas R., Collins P., Franzoni I., Ravichandran M., Ouyang H., Juarez-Ornelas K., Lautens M., Schapira M., von Delft F., et al. Small molecule antagonists of the interaction between the histone deacetylase 6 zinc-finger domain and ubiquitin. J. Med. Chem. 2017;60:9090–9096. doi: 10.1021/acs.jmedchem.7b00933. [DOI] [PubMed] [Google Scholar]
- Hook S.S., Orian A., Cowley S.M., Eisenman R.N. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. U S A. 2002;99:13425–13430. doi: 10.1073/pnas.172511699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X., Yao T. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Hussain M., Galvin H., Haw T., Nutsford A., Husain M. Drug resistance in influenza A virus: the epidemiology and management. Infect. Drug Resist. 2017;10:121–134. doi: 10.2147/IDR.S105473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson M.K., Ploegh H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe. 2009;5:559–570. doi: 10.1016/j.chom.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J.A., Ward C.L., Kopito R.R. Aggresomes: a cellular response to misfolded proteins. J. Cel. Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus P.A., Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y., Kovacs J., McLauri A., Vance J., Ito A., Yao T. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Komander D., Rape M. The ubiquitin code. Annu. Rev. Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Kovacs J.J., Murphy P.J., Gaillard S., Zhao X., Wu J.T., Nicchitta C.V., Yoshida M., Toft D.O., Pratt W.B., Yao T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Kwon S., Zhang Y., Matthias P. The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev. 2007;21:3381–3394. doi: 10.1101/gad.461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros S., Boxus M., Gatot J.S., Van Lint C., Kruys V., Kettmann R., Twizere J.C., Dequiedt F. The HTLV-1 Tax protein inhibits formation of stress granules by interacting with histone deacetylase 6. Oncogene. 2011;30:4050–4062. doi: 10.1038/onc.2011.120. [DOI] [PubMed] [Google Scholar]
- Li T., Zhang C., Hassan S., Liu X., Song F., Chen K., Zhang W., Yang J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018;11:111. doi: 10.1186/s13045-018-0654-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D., Afonine P.V., Baker M.L., Bunkóczi G., Chen V.B., Croll T.I., Hintze B., Hung L.W., Jain S., McCoy A.J., et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019;75:861–877. doi: 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magupalli V., Negro R., Tian Y., Hauenstein A., Di Caprio G., Skillern W., Deng Q., Orning P., Alam H., Maliga Z., et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. 2020;369:eaas8995. doi: 10.1126/science.aas8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S., Fulzele A., Higgins R., Leonard M., Yeo G.W., Bennett E.J. Active protein neddylation or ubiquitylation is dispensable for stress granule dynamics. Cell Rep. 2019;27:1356–1363.e3. doi: 10.1016/j.celrep.2019.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazroui R., Di Marco S., Kaufman R.J., Gallouzi I.E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cel. 2007;18:2603–2618. doi: 10.1091/mbc.E06-12-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz T., Wetzel S.K., Firbank S., Plückthun A., Grütter M.G., Mittl P.R. Stabilizing ionic interactions in a full-consensus ankyrin repeat protein. J. Mol. Biol. 2008;376:232–240. doi: 10.1016/j.jmb.2007.11.047. [DOI] [PubMed] [Google Scholar]
- Mishima Y., Santo L., Eda H., Cirstea D., Nemani N., Yee A.J., O'Donnell E., Selig M.K., Quayle S.N., Arastu-Kapur S., et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 2015;169:423–434. doi: 10.1111/bjh.13315. [DOI] [PubMed] [Google Scholar]
- Miyake Y., Keusch J.J., Wang L., Saito M., Hess D., Wang X., Melancon B.J., Helquist P., Gut H., Matthias P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016;12:748–754. doi: 10.1038/nchembio.2140. [DOI] [PubMed] [Google Scholar]
- Montero H., Trujillo-Alonso V. Stress granules in the viral replication cycle. Viruses. 2011;3:2328–2338. doi: 10.3390/v3112328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A., et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nover L., Scharf K.D., Neumann D. Cytoplasmic heat shock granules are formed from precursor particles and are associated with a specific set of mRNAs. Mol. Cell Biol. 1989;9:1298–1308. doi: 10.1128/mcb.9.3.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann J., Li L., Chin L. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr. Med. Chem. 2008;15:47–60. doi: 10.2174/092986708783330692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang H., Ali Y.O., Ravichandran M., Dong A., Qiu W., MacKenzie F., Dhe-Paganon S., Arrowsmith C.H., Zhai R.G. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 2012;287:2317–2327. doi: 10.1074/jbc.M111.273730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter J., Merritt E.A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Painter J., Merritt E.A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Cryst. 2006;39:109–111. [Google Scholar]
- Peisley A., Wu B., Xu H., Chen Z.J., Hur S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature. 2014;509:110–114. doi: 10.1038/nature13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijlman G.P., Funk A., Kondratieva N., Leung J., Torres S., van der Aa L., Liu W.J., Palmenberg A.C., Shi P.Y., Hall R.A., et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe. 2008;4:579–591. doi: 10.1016/j.chom.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Plückthun A. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015;55:489–511. doi: 10.1146/annurev-pharmtox-010611-134654. [DOI] [PubMed] [Google Scholar]
- Protter D.S.W., Parker R. Principles and properties of stress granules. Trends Cell Biol. 2016;26:668–679. doi: 10.1016/j.tcb.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke L.C., Lloyd R.E. Diversion of stress granules and P-bodies during viral infection. Virology. 2013;436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicka A., Yamauchi Y. Ubiquitin in influenza virus entry and innate immunity. Viruses. 2016;8:293. doi: 10.3390/v8100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M., Hess D., Eglinger J., Fritsch A.W., Kreysing M., Weinert B.T., Choudhary C., Matthias P. Acetylation of intrinsically disordered regions regulates phase separation. Nat. Chem. Biol. 2019;15:51–61. doi: 10.1038/s41589-018-0180-7. [DOI] [PubMed] [Google Scholar]
- Schmidt T., Samaras P., Frejno M., Gessulat S., Barnert M., Kienegger H., Krcmar H., Schlegl J., Ehrlich H.-C., Aiche S., et al. ProteomicsDB. Nucleic Acids Res. 2017;46:D1271–D1281. doi: 10.1093/nar/gkx1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneurin-Berny D., Verdel A., Curtet S., Lemercier C., Garin J., Rousseaux S., Khochbin S. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol. Cell Biol. 2001;21:8035–8044. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli S., Tegoni M., Frenken L., van Vliet C., Cambillau C. Lateral recognition of a dye hapten by a llama VHH domain. J. Mol. Biol. 2001;311:123–129. doi: 10.1006/jmbi.2001.4856. [DOI] [PubMed] [Google Scholar]
- Sweeney P., Park H., Baumann M., Dunlop J., Frydman J., Kopito R., McCampbell A., Leblanc G., Venkateswaran A., Nurmi A., et al. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener. 2017;6:6. doi: 10.1186/s40035-017-0077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor P., Tanaka F., Robitschek J., Sandoval C.M., Taye A., Markovic-Plese S., Fischbeck K. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003;12:749–757. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C., Adams P.D., Read R.J., McCoy A.J., Moriarty N.W., Grosse-Kunstleve R.W., Afonine P.V., Zwart P.H., Hung L.W. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 2009;65:582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickle I.J., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G. Global Phasing Ltd; 2018. STARANISO.http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi [Google Scholar]
- Tourrière H., Chebli K., Zekri L., Courselaud B., Blanchard J.M., Bertrand E., Tazi J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cel. Biol. 2003;160:823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Vielle N.J., Zumkehr B., García-Nicolás O., Blank F., Stojanov M., Musso D., Baud D., Summerfield A., Alves M.P. Silent infection of human dendritic cells by African and Asian strains of Zika virus. Sci. Rep. 2018;8:5440. doi: 10.1038/s41598-018-23734-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler J.R., Matheny T., Jain S., Abrisch R., Parker R. Distinct stages in stress granule assembly and disassembly. Elife. 2016;5:e18413. doi: 10.7554/eLife.18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wileman T. Aggresomes and autophagy generate sites for virus replication. Science. 2006;312:875–878. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Yuan Z., Zhang Y., Yong S., Salas-Burgos A., Koomen J., Olashaw N., Parsons J.T., Yang X.J., Dent S.R., et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Kwon S., Yamaguchi T., Cubizolles F., Rousseaux S., Kneissel M., Cao C., Li N., Cheng H.L., Chua K., et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Li N., Caron C., Matthias G., Hess D., Khochbin S., Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA sequencing data have been deposited at ArrayExpress and are publicly available as of the date of publication. Mass Spectrometry data (Search results are in Table S3) have been deposited at the ProteomeXchange Consortium via the PRIDE partner repository and are publicly available as of the date of publication. Structural data have been deposited with the Protein DataBank and are publicly available as of the date of publication. All the accession numbers are listed in the key resources table.
Original data have been deposited at Mendeley and are publicly available as of the date of publication. DOIs are listed in the key resources table.
Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.