

Graphical abstract

Keywords: Thymidine analogs, Antimicrobial activities, SARS-CoV-2 main protease, Molecular docking, Molecular dynamics, PASS and POM pharmacophore sites identification
Abstract
Nucleoside precursors and nucleoside analogs occupy an important place in the treatment of viral respiratory pathologies, especially during the current COVID-19 pandemic. From this perspective, the present study has been designed to explore and evaluate the synthesis and spectral characterisation of 5́-O-(lauroyl) thymidine analogs 2–6 with different aliphatic and aromatic groups through comprehensive in vitro antimicrobial screening, cytotoxicity assessment, physicochemical aspects, molecular docking and molecular dynamics analysis, along with pharmacokinetic prediction. A unimolar one-step lauroylation of thymidine under controlled conditions furnished the 5́-O-(lauroyl) thymidine and indicated the selectivity at C-5́ position and the development of thymidine based potential antimicrobial analogs, which were further converted into four newer 3́-O-(acyl)-5́-O-(lauroyl) thymidine analogs in reasonably good yields. The chemical structures of the newly synthesised analogs were ascertained by analysing their physicochemical, elemental, and spectroscopic data. In vitro antimicrobial tests against five bacteria and two fungi, along with the prediction of activity spectra for substances (PASS), indicated promising antibacterial functionality for these thymidine analogs compared to antifungal activity. In support of this observation, molecular docking experiments have been performed against the main protease of SARS-CoV-2, and significant binding affinities and non-bonding interactions were observed against the main protease (6LU7, 6Y84 and 7BQY), considering hydroxychloroquine (HCQ) as standard. Moreover, the 100 ns molecular dynamics simulation process was performed to monitor the behaviour of the complex structure formed by the main protease under in silico physiological conditions to examine its stability over time, and this revealed a stable conformation and binding pattern in a stimulating environment of thymidine analogs. Cytotoxicity determination confirmed that compounds were found less toxic. Pharmacokinetic predictions were investigated to evaluate their absorption, distribution, metabolism and toxic properties, and the combination of pharmacokinetic and drug-likeness predictions has shown promising results in silico. The POM analysis shows the presence of an antiviral (O1δ-, O2δ-) pharmacophore site. Overall, the current study should be of great help in the development of thymidine-based, novel, multiple drug-resistant antimicrobial and COVID-19 drugs.
1. Introduction
Nucleoside analogs have, in recent times, been used as the backbone for many drugs in the treatment of infectious diseases caused by HIV, hepatitis B or C viruses, and the herpes viruses [1]. Nucleoside analogs are the subunits of DNA and RNA, and structurally they consist of a sugar moiety linked to a nitrogen base through an N-β-glycosidic bond. Interestingly, biological activity, as well as the chemical and physical properties of nucleoside analog, can be tuned remarkably by the simple alteration of the sugar moiety of a nucleoside with different substituents and heteroatoms, among others [2]. The nucleoside analogs usually show antiviral activity by inhibiting viral replication through the blockage of cellular division; impairment of DNA/RNA synthesis or inhibition of cellular or viral enzyme activity. The nucleoside analogs, Telbivudine (hepatitis B inhibitor), Entecavir (HIV/ AIDS and Hepatitis B inhibitor), Clevudine (Hepatitis B inhibitor), Zalcitabine (reverse-transcriptase inhibitor), Taribavirin (prodrug of Ribavirin), Stavudine (HIV/AIDS inhibitors), Lamivudine (first-generation nucleoside reverse transcriptase inhibitor), Cordycepin (RNA synthesis inhibitor), and Cordycepin Triphosphate (polyadenylation inhibitors, antineoplastic, antioxidant, and anti-inflammatory agent), have been used in the treatment of many viral diseases. Moreover, nucleoside analogs have immense clinical importance as medicinal agents because of their antiviral and anticancer activities [3]; they have been the “drug of choice” for the treatment of various viral diseases, such as ebola [11], dengue, and Zika [4]. Additionally, 2ʹ-deoxynucleosides such as doxuridne, trifluridine, doxudine, vidarabine, and brivudine, have been used to treat herpes virus infections [5], [6]. The modifications in the sugar moiety, namely ribofuranose or deoxyribofuranose of nucleosides, include changes in the sugar substituents, replacement of the oxygen with another atom, and the addition of heteroatoms in the sugar ring, ring size variations, and replacement with acyclic moiety [7], [8], [9], [10], [11], [12]. These alterations can produce remarkable variations in biological activity and the degree of selective toxicity, according to their respective chemical and physical properties [13], [14], [15], [16], [17], [18], [19], [20]. The modified compounds have exhibited broad-spectrum biological activity. Thymidine (1) derivatives such as telbivudine are antiviral drugs used in HBV treatment [21]. Another nucleoside, cytidine, is found in offal meat and pyrimidine-rich foods such as beer, tomatoes, broccoli and oats. Cytidine is one of the RNA components that translate instructions from DNA to proteins [22]. Some recent research revealed that thymidine analogs, as well as nucleobases, are a pharmacologically diverse family, which includes cytotoxic compounds, antimicrobial agents and immune-suppressive molecules [23], [24], [25], [26]. However, one study indicates that selective acylation of thymidine (1) causes complexities due to the appearance of one primary OH group, one secondary OH group and an –NH group. Hence, various methods like direct acylation [8], the dibutyltin oxide method [27], [28], coupling techniques, and the enzymatic method have been reported. Among these methods, the direct method generally furnished 5́-O-substituted analogs [27], [28], whereas the dibutyltin oxide method gave 3́-O-substituted analogs [8], [27], [28]. However, most of the time, the direct method produced higher yields at a lower cost. The novel COVID-19 virus spread around the world from Wuhan, China in December 2019. The World Health Organization (WHO) declared the disease a public pandemic emergency on March 11th 2020, as the cause of Severe Acquired Respiratory Syndrome (SARS-CoV-2). As a result, the development of multiple drug-resistant solutions for pathogenic organisms and coronavirus disease 2019 (COVID-19) has become a current global concern for researchers. Most of the proposed COVID-19 drugs, such as remdesivir and favipiravir, are based on a nucleoside structure, and modifications of the hydroxyl (–OH) group of nucleosides, including thymidine, have revealed some potent SARS-CoV-2 candidates [29], [30], [31] and antimicrobial agents in the current study. However, there is an urgent need for the development of new chemotherapeutic agents with a novel mode of action to combat these multiple drug-resistant organisms, including SARS CoV-2. One of our recent works based on PASS prediction, In vitro antimicrobial, physicochemical and molecular docking studies revealed that thymidine derivatives are potential inhibitors of SARS CoV (1Q2W) [32].
Keeping in mind these features mentioned above, as well as the subsequent aim of finding novel drugs [32], we have made some advancements in our present study. Different acylating agents are inserted in the 5́ and 3́ thymidine structures to synthesize novel thymidine analogs. The in vitro antimicrobial evaluation of several thymidine-based analogs 2–6 with different aliphatic and aromatic chains against eight pathogens, molecular docking against the coronavirus (SARS CoV-2) main protease (6LU7, 6Y84 and 7BQY), along with the prediction of activity spectra for substances (PASS), are reported on herein. To confirm the stability of the docked complexes, molecular dynamics was performed for 100 ns. Cytotoxicity was determined to explore the safety profile synthesize thymidine analogs. Then we analyze the series of analogs 2–6 to identify their pharmacophore sites. Furthermore, steps have been taken to optimize the synthesized thymidine analogs to investigate their physicochemical behavior based on the DFT approach with pharmacokinetic and drug-likeness properties.
2. Materials and methods
2.1. Material and equipment
The solvents used were of analytical grade and were purified using standard procedures. All reagents used are commercially available from Sigma-Aldrich (Germany) and were used as received unless otherwise specified. Melting points (mp) were determined using an electrothermal melting point apparatus. Evaporations were performed under diminished pressure on a Büchi rotary evaporator. Infrared spectral analyses were recorded using a Fourier-transform infrared (FTIR) spectrophotometer (IR Prestige-21, Shimadzu, Japan) at the Department of Chemistry, University of Chittagong. A Brucker advance DPX 400 MHz using tetramethylsilane as the internal standard was employed for recording the proton nuclear magnetic resonance (1H NMR) spectra at WMSRC, JU, Bangladesh. Mass spectra of the synthesised compounds were obtained by liquid chromatography-electrospray ionisation tandem mass spectrometry in positive ionization mode. Thin-layer chromatography (TLC) was performed on Kieselgel GF254 (Germany), and the chromatogram was visualised by spraying the plates with 1% H2SO4, followed by heating the plates at 150–200 °C until coloration appeared. Column chromatography was performed using silica gel G60. To identify the drug interactions with receptor proteins, molecular docking is the most suitable tool. The blind docking method employs a search throughout the whole surface of the protein molecule for binding sites. The following software was used in the present study:
-
(i)
Gaussian 09,
-
(ii)
AutoDock 4.2.6,
-
(iii)
Swiss-Pdb 4.1.0,
-
(iv)
Python 3.8.2,
-
(v)
Discovery Studio 4.1,
-
(vi)
PyMOL 2.3,
-
(vii)
(https://www.pharmaexpert.ru/passonline/) to predict PASS properties
-
(viii)
admetSAR server (https://lmmd.ecust.edu.cn/admetsar2/about) and SwissADME free web tools (https://www.swissadme.ch) were employed to calculate the pharmacokinetic properties, and
-
(ix)
YASARA for molecular dynamics.
2.2. Synthesis
2.2.1. 5́-O-(Lauroyl)thymidine (2)
A solution of thymidine (1) (200 mg, 0.82 mmol) in dry pyridine (3 ml) was cooled to −5°C, whereupon lauroyl chloride (0.205 ml, 1.1 M eq.) was added to it. The mixture was stirred at this temperature for 6 h and then stirred overnight at room temperature. T.L.C. examination (methanol-chloroform, 1:4) indicated full conversion of the starting material into a single product (Rf = 0.55). The solution was poured into ice water with constant stirring. It was then extracted with chloroform (3 × 10 ml). The combined chloroform layer was washed successively with dilute hydrochloric acid (10%), saturated aqueous sodium hydrogen carbonate (NaHCO3) solution and distilled water. The organic layer was dried with anhydrous magnesium sulphate (MgSO4) and filtered, and the filtrate was concentrated under reduced pressure to leave a syrup. Purification using silica gel column chromatography (with methanol-chloroform 1:4 as eluant) afforded the title compound (2) (130 mg, 62.86%) as a white crystalline solid which was used in the next stage. Recrystallisation from chloroform-n-hexane provided the lauroyl derivatives (2) as needles (mp. 65–66 °C).
Yield %62.86, Mp: 65–66 °C. FTIR: ν max 1680, 1708 (–CO), 3408 ∼ 3502 (br) (–OH) cm−1.1H NMR (400 MHz, CDCl3): δH 8.71 (1H, s, –NH), 7.30 (1H, d, J = 2.3 Hz, H-6), 6.29 (1H, t, J = 6.6 Hz, H-1́), 4.42 (1H, dd, J = 12.0 and 4.5 Hz, H-5́a), 4.31 (1H, dd, J = 12.1 and 4.5 Hz, H-5́b), 4.22 (1H, m, H-3́), 3.61 (1H, ddd, J = 3.6, 4.6 and 4.2 Hz, H-4́), 3.35 (1H, s, 3́–OH), 3.08 (1H, ddd, J = 13.5, 6.5 and 4.0 Hz, H-2́a), 2.38 {2H, m, CH3(CH2)9CH 2CO-}, 2.17 (1H, ddd, J = 13.5, 6.5 and 6.7 Hz, H-2́b), 1.95 (3H, d, J = 1.3 Hz, 5-CH3), 1.66 {2H, m, CH3(CH2)8CH 2CH2CO-}, 1.28 {16H, m, CH3(CH 2)8CH2CH2CO-}, 0.90 {3H, m, CH 3(CH2)10CO-}. 13C NMR (100 MHz, CDCl3): δC 172.29 {CH3(CH2)10 CO-}, 163.30 (C-1), 150.10 (C-4), 134.45 (C-3), 123.75(C-2), 87.80 (C-1ˊ), 77.20 (C-4ˊ), 76.25 (C-3ˊ), 67.25 (C-5ˊ), 51.50 (C-2ˊ), 34.38, 31.90, 29.59, 29.32, 29.24 (×2), 25.01, 22.65, 21.72, 20.09 {CH3(CH2)10CO-}, 16.10 (C-5), 13.51 {CH3(CH2)10CO-}. LC-MS [M + 1]+ 425.54. Calcd. for C21H36O5N2CO: C = 62.18%, H = 8.47%; found: C = 62.19%, H = 8.48%.
General procedure for the preparation of lauroyl derivatives (3–6).
2.2.2. 3́-O-(4-t-Butylbenzoyl)-5́-O-(lauroyl)thymidine (3)
A suspension of lauroyl derivative (2) (130 mg, 0.30 mmol) in dry pyridine (3 ml) was cooled to −5°C whereupon 4-t-butyl benzoyl chloride (129 mg, 2.2 M eq.) was added to it. The reaction mixture was stirred for 6 h at this temperature and then stirred overnight at room temperature. The progress of the reaction was monitored using T.L.C. (methanol-chloroform, 1:8), which showed the complete conversion of reactant into a single product (Rf = 0.51). The excess reagent was destroyed by the addition of a few pieces of ice, and the reaction mixture was processed as described earlier. The resulting syrup was purified using column chromatography to afford compound (3) (111 mg, 61.99%) as a crystalline solid mp. 58–59 °C.
Yield %61.99, Mp: 58–59 °C. FTIR: ν max 1704 (–CO) cm−1. 1H NMR (400 MHz, CDCl3): δH 9.04 (1H, s, –NH), 8.04 (2H, m, Ar-H), 7.49 (2H, m, Ar-H), 7.33 (1H, d, J = 1.8 Hz, H-6), 6.31 (1H, m, H-1́), 4.44 (1H, m, H-3́), 4.36 (1H, dd, J = 12.0 and 4.5 Hz, H-5́a), 4.24 (1H, dd, J = 12.2 and 3.6 Hz, H-5́b), 3.21 (1H, m, H-4́), 3.01 (1H, ddd, J = 13.6, 6.6 and 4.6 Hz, H-2́a), 2.93 (1H, ddd, J = 12.5, 6.1 and 6.2 Hz, H-2́b), 2.39 {2H, m, CH3(CH2)9CH 2CO-}, 1.97 (3H, d, J = 1.3 Hz, 5-CH3), 1.66 {2H, m, CH3(CH2)8CH 2CH2CO-}, 1.37, 1.29, 1.22 {9H, 3 s, (CH 3) 3C–}, 1.28 {16H, m, CH3(CH 2)8CH2CH2CO-}, 0.91 {3H, m, CH 3(CH2)10CO-}. 13C NMR (100 MHz, CDCl3): δC 174.40 {(CH3)3CC6H4 CO-}, 172.45 {CH3(CH2)10 CO-}, 169.44 (C-1), 155.45 (C-4), 139.49 (C-3), 132.62, 132.67, 130.94, 129.95, 127.50, 126.52 {(CH3)3CC 6H4CO-}, 115.21 (C-2), 89.82 (C-1ˊ), 87.54 (C-4ˊ), 73.89 (C-3ˊ), 64.21 (C-5ˊ), 40.92 (C-2ˊ), 35.69 {(CH3)3 CC6H4CO-}, 34.39, 31.44, 29.92, 29.87, 29.32 (×2), 25.22, 22.12, 21.77, 20.11 {CH3(CH2)10CO-}, 16.17 (C-5), 13.68, 13.66, 13.48 {(CH3)3CC6H4CO-}, 13.41 {CH3(CH2)10CO-}. LC-MS [M + 1]+ 585.75. Calcd. for C32H48O6N2CO: C = 67.72%, H = 8.20%; found: C = 67.74%, H = 8.22%.
Similar reaction and purification methods were employed to synthesize a myristoyl derivative (4) (102 mg, 71.80%, as needles, mp. 68–69 °C), palmitoyl derivative (5) (100 mg, 66.03%, as needles, mp. 79–80 °C), and bromobenzoyl derivative (6) (98.5 mg, 54.15%, as needles, mp. 74–75 °C.
5́-O-Lauroyl-3́-O-(myristoyl)thymidine (4).
A suspension of 5́-O-lauroylthymidine (2) (95 mg, 0.22 mmol) in dry pyridine (3 ml) was cooled to −5°C whereupon myristoyl chloride (0.129 ml, 2.2 M eq.) was added to it. The mixture was stirred at this temperature for 6 h and then stirred overnight at room temperature. The progress of the reaction was monitored by T.L.C. (methanol-chloroform, 1:8) which exhibited complete conversion of the starting material into a single product (Rf = 0.52). The excess reagent was destroyed by the addition of a few pieces of ice and the reaction mixture was processed as described earlier. Work-up as usual and purification by passage through a silica gel column chromatography with methanol-chloroform (1:8) furnished the title compound (4) (102 mg, 71.80%) as a crystalline solid mp. 68–69 °C.
Yield %71.80, Mp: 68–69 °C. FTIR: ν max 1701 (–CO) cm−1.1H NMR (400 MHz, CDCl3): δH 9.0 (1H, s, –NH), 7.29 (1H, d, J = 1.3 Hz, H-6), 6.25 (1H, t, J = 6.5 Hz, H-1́), 4.81 (1H, m, H-3́), 4.49 (1H, dd, J = 12.0 and 4.5 Hz, H-5́a), 4.33 (1H, dd, J = 12.0 and 3.5 Hz, H-5́b), 4.0 (1H, ddd, J = 3.5, 4.5 and 3.9 Hz, H-4́), 2.39 (1H, ddd, J = 13.5, 6.5 and 4.0 Hz, H-2́a), 2.36 {2H, m, CH3(CH2)9CH 2CO-}, 2.32 {2H, m, CH3(CH2)11CH 2CO-}, 2.24 (1H, ddd, J = 13.5, 6.5 and 6.7 Hz, H-2́b), 1.95 (3H, d, J = 1.3 Hz, 5-CH3), 1.76 {2H, m, CH3(CH2)10CH 2CH2CO-}, 1.65 {20H, m, CH3(CH 2)10CH2CH2CO-}, 1.64 {2H, m, CH3(CH2)8CH 2CH2CO-}, 1.28 {16H, m, CH3(CH 2)8CH2CH2CO-}, 0.90 {3H, m, CH 3(CH2)10CO-}, 0.88 {3H, t, J = 6.8 Hz, CH 3(CH2)12CO-}. 13C NMR (100 MHz, CDCl3): δC 172.69 {CH3(CH2)10 CO-}, 172.48 {CH3(CH2)12 CO-}, 169.28 (C-1), 155.33 (C-4), 138.40 (C-3), 114.21 (C-2), 89.34 (C-1ˊ), 88.74 (C-4ˊ), 74.11 (C-3ˊ), 65.21 (C-5ˊ), 40.67 (C-2ˊ), 34.44, 31.67, 29.43, 29.87, 29.22, 29.18, 25.01, 22.60, 21.34, 20.11 {CH3(CH2)10CO-}, 34.21, 34.02, 31.92, 29.59 (×2), 29.37, 29.24, 24.96, 24.92, 22.65, 21.72, 20.09 {CH3(CH2)12CO-},16.12 (C-5), 14.51 {CH3(CH2)10CO-}, 14.08 {CH3(CH2)12CO-}. LC-MS [M + 1]+ 635.90. Calcd. For C35H62O6N2CO: C = 68.04%, H = 9.76%; found: C = 68.03%, H = 9.77%.
2.2.3. 5′-O-Lauroyl-3′-O-(palmitoyl)thymidine (5)
A solution of lauroyl derivative (2) (97 mg, 0.22 mmol) in dry pyridine (3 ml) was cooled to −5°C whereupon palmitoyl chloride (0.15 ml, 2.2 M eq.) was added to it. The mixture was stirred at this temperature for 6 h and then stirred overnight at room temperature. T.L.C. (methanol-chloroform, 1:8) examination indicated complete conversion of the starting material into a faster-moving product (Rf = 0.53). Chromatographic purification by silica gel column chromatography with methanol-chloroform (1:8) as eluant furnished the palmitoyl derivative (5) (100 mg, 66.03%) as a white crystalline solid. Recrystallization from chloroform-n-hexane gave the palmitoyl derivatives (5) as needles, (mp. 79–80 °C).
Yield %66.03, Mp: 79–80 °C. FTIR data: ν max 1700 (–CO) cm−1.1H NMR (400 MHz, CDCl3) data: δH 9.0 (1H, s, –NH), 7.30 (1H, d, J = 1.6 Hz, H-6), 6.36 (1H, t, J = 6.4 Hz, H-1́), 4.74 (1H, m, H-3́), 4.40 (1H, dd, J = 12.2 and 4.6 Hz, H-5́a), 4.37 (1H, dd, J = 12.1 and 3.6 Hz, H-5́b), 4.10 (1H, ddd, J = 3.5, 4.5 and 3.9 Hz, H-4́), 2.46 (1H, ddd, J = 13.5, 6.5 and 4.0 Hz, H-2́a), 2.37 {2H, m, CH3(CH2)9CH 2CO-}, 2.34 {2H, m, CH3(CH2)13CH 2CO-}, 2.25 (1H, ddd, J = 13.5, 6.5 and 6.7 Hz, H-2́b), 1.96 (3H, d, J = 1.3 Hz, 5-CH3), 1.75 {2H, m, CH3(CH 2)12CH 2CH2CO-}, 1.65 {2H, m, CH3(CH2)8CH 2CH2CO-}, 1.29 {26H, m, CH3(CH 2)13CH2CO-}, 1.28 {16H, m, CH3(CH 2)8CH2CH2CO-}, 0.90 {3H, m, CH 3(CH2)14CO-}, 0.89 {3H,m,CH 3(CH2)10CO-}. 13C NMR (100 MHz, CDCl3): δC 172.33 {CH3(CH2)10 CO-}, 172.21 {CH3(CH2)14 CO-}, 168.98 (C-1), 158.10 (C-4), 139.67 (C-3), 114.33 (C-2), 89.43 (C-1ˊ), 87.11 (C-4ˊ), 74.22 (C-3ˊ), 64.22 (C-5ˊ), 40.44 (C-2ˊ), 34.65, 31.54, 29.76, 29.21, 29.12, 25.76, 25.11, 22.63, 21.70, 20.05 {CH3(CH2)10CO-}, 34.31, 34.36, 34.12, 31.95, 29.52, 29.15, 29.11, 25.11, 24.77, 24.63, 22.60, 22.55, 21.33, 20.01 {CH3(CH2)14CO-}, 16.23 (C-5), 14.02 {CH3(CH2)12CO-}, 13.53 {CH3(CH2)10CO-}. LC-MS [M + 1]+ 663.95. Calcd. for C37H66O6N2CO: C = 66.78%, H = 9.95% found: C = 66.79%, H = 9.97%.
2.2.4. 3́-O-(4-Bromobenzoyl)-5́-O-(lauroyl)thymidine (6)
A solution of 2 (127 mg, 0.29 mmol) in dry pyridine (3 ml) was cooled to −5°C whereupon 4-bromobenzoyl chloride (0.108 mg, 2.2 M eq.) was added to it. The mixture was stirred at this temperature for 6 h and then stirred overnight at room temperature. T.L.C. (methanol-chloroform, 1:8) displayed complete conversion of reactant into one product (Rf = 0.52). The usual work-up procedure, followed by silica gel column chromatographic purification (with methanol-chloroform, 1:8, as eluant), afforded 4-bromobenzoyl derivative (6) (98.5 mg, 54.15%) as a semi-solid.
Yield %54.15, Mp: 74–75 °C. FTIR data: ν max 1700 (–CO) cm−1.1H NMR (400 MHz, CDCl3) data: δH 9. 0 (1H, s, –NH), 7.96 (2H, m, Ar-H), 7.63 (2H, m, Ar-H), 7.31 (1H, d, J = 1.7 Hz, H-6), 6.29 (1H, t, J = 6.6 Hz, H-1́), 5.34 (1H, m, H-3́), 5.09 (1H, dd, J = 12.1 and 4.7 Hz, H-5́a), 4.45 (1H, dd, J = 11.9 and 3.8 Hz, H-5́b), 4.42(1H, ddd, J = 3.7, 4.8 and 4.2 Hz, H-4́), 2.42 (1H, m, H-2́a), 2.37 {2H, m, CH3(CH2)9CH 2CO-}, 2.34 (1H, m, H-2́b), 1.95 (3H, d, J = 1.6 Hz, 5-CH3), 1.65 {2H, m, CH3(CH2)8CH 2CH2CO-}, 1.28 {16H, m, CH3(CH 2)8CH2CH2CO-}, 0.89 {3H, m, CH 3(CH2)10CO-}. 13C NMR (100 MHz, CDCl3): δC 172.21 {CH3(CH2)10 CO-}, 170.02 (C-1), 168.38 (4-Br.C6H4.CO–), 155.22 (C-4), 139.65 (C-3), 131.91, 131.58, 131.13, 129.41 (×2), 128.92 (4-Br.C6H4.CO–), 114.34 (C-2), 89.22 (C-1ˊ), 87.67 (C-4ˊ), 73.32 (C-3ˊ), 64.11 (C-5ˊ), 41.21 (C-2ˊ), 34.58, 31.48, 29.21, 29.12, 29.08, 29.01, 25.21, 22.56, 21.28, 20.11 {CH3(CH2)10CO-}, 16.32 (C-5), 13.26 {CH3(CH2)10CO-}. LC-MS [M + 1]+ 607.90. Calcd. for BrC28H39O6N2CO: C = 57.34%, H = 6.43%; found: C = 57.36%, H = 6.45%.
2.3. In vitro antimicrobial activity test
2.3.1. Test organisms
The tested microorganisms (bacteria and fungi) were collected from the Microbiology Laboratory, Department of Microbiology, University of Chittagong, Chattogram, Bangladesh. As shown in Table S1, five human pathogenic bacteria and two plant pathogenic fungi were used for the antimicrobial evaluation.
2.4. Disc diffusion test to check antibacterial susceptibility
Antibacterial susceptibility testing of the compounds was performed using standard disk diffusion assay according to CLSI protocol [33], [34]. The inoculum was prepared by picking the test bacteria from plates with a sterile inoculating loop and suspending it in sterile normal saline (0.85%). The density of the suspension to be inoculated was determined through a comparison with the McFarland 0.5 standard. After that, the test bacteria were swabbed uniformly over the Mueller-Hinton agar (MHA) plates, and filter paper discs (6 mm, Hi-media) incorporated into the test compounds were placed in the agar medium. After that, the plates were incubated for 18 h at 37 °C and the diameters of zones of inhibition around the discs were measured in millimeters (mm). The experiments were repeated three times.
2.5. Determination of MIC and MBC using the micro-broth dilution method
Microdilution methods are the most appropriate ones for the determination of MIC and MBC values since they offer the possibility of estimating the concentration of the antimicrobial agent being tested in the broth medium, and they may be used to quantitatively measure the in vitro antimicrobial activity against bacteria. A two-fold dilution of the test compound, 0.125, 0.25, 0.5, 1, 2, 4, 8, 16, 32, 64 and 128 µg/mL) was prepared by dispensing approximately 100 μL of each 2x compound solution in column 1 of a 96-well microtiter plate containing 100 μL of MHB broth, and from columns 2 to 10, compound solutions were serially diluted. Each well, except the sterility control well (blank), was inoculated with a 5 μL test strain inoculum of 104 to 105 CFU/mL prepared after dilution of a standardised suspension adjusted to a 0.5 McFarland scale. The microplate was loosely wrapped with clingfilm wrap to prevent bacterial dehydration. Each plate included a growth control in column 11 (without compound) and sterility control (only media) in column 12. Then, the inoculated 96-well microtitre plates were incubated at 37 °C for 24 h. To each well, 10 µL of 2, 3 and 5-triphenyl tetrazolium chloride 0.5% (w/v) solution was added. The cultures were then incubated at 37 °C for 24 hrs. The appearance of a red color indicated the growth of bacteria, and MIC and MBC were interpreted visually.
2.6. Screening of mycelial growth
The poisoned food technique [6], [12] was employed to screen the antifungal activity in which potato dextrose agar (PDA) was used as the culture medium. The test compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1% (w/v), and 0.1 ml of the solution (containing 1 mg of the respective test compound) was transferred to a sterile petri dish using a sterilised pipette. After that, 20 ml of the medium was poured into the Petri dish and allowed to solidify. The inoculation was then performed in the centre of each petri dish with a 5 mm mycelium block of each fungus. The blocks were placed at the centre of each petri dish in an inverted position to maximise the contact between the mycelium and the culture medium. The inoculation plates were incubated at 27 ± 2 °C, and the experiment was conducted in triplicate. A control sample (i.e., PDA without test chemicals) was also maintained under the same conditions. After 5 days of incubation, the diameter of the fungal radial mycelial growth was measured. An average of three measurements was considered for the radial mycelia growth of the fungus in mm.
2.7. Structure-activity relationship (SAR)
Structure–activity relationship (SAR) analysis was employed to predict antimicrobial activity from the molecular structure of the pharmaceutical target. This well-known technology is often used in the drug designing process to guide the acquisition or synthesis of new compounds with desirable properties. In the present study, the SAR research was analyzed according to the Hunt [34] and Kim [35] membrane permeation concept.
2.8. PASS and bioactivity prediction
The online web application PASS (https://www.pharmaexpert.ru/passonline/) has been employed to calculate the antimicrobial activity spectrum of the selected thymidine analogs [36]. Firstly, the thymidine analog structures were drawn, and then changed into their smiles formats by using the renowned SwissADME free online application (https://www.swissadme.ch) to determine the antimicrobial spectrum using the PASS web tool. This server can surmise over 4000 types of antimicrobial functions, together with drug and non-drug activity, which helps to suggest the best potential objects with 90% validity. PASS outcomes are revealed by Pa (probability for active molecule) and Pi (probability for inactive molecule). Having potentialities, the Pa and Pi scores vary in the range of 0.00 to 1.00, and usually, Pa + Pi ≠ 1, as these potentialities are predicted freely. The biological actions with Pa > Pi are only thought of as probable for a selected drug molecule. PASS calculation outcomes were explained and used flexibly, viz. (i) when Pa is greater than 0.7, the probability to identify the activity is analytically high, (ii) if 0.5 < Pa < 0.7, the probability to identify the activity is analytically low, again, the molecule is perhaps not very similar to well conversant pharmaceutically used drugs and (iii) if Pa < 0.5, the potentiality to identify the activity analytically is less. In the present study, the Molinspiration online server (https://www.molinspiration.com/cgi-bin/properties) was utilised to analyse the drug-like properties of lead compounds. The Molinspiration cheminformatics engine allows the fast prediction of biological activity and virtual screening of large collections of molecules, and the selection of molecules with the highest probability, to show biological activity. The screening is based on the identification of fragments or substructure features typical of the active molecules.
2.9. Details of geometry optimization
In computational chemistry, quantum mechanical methods are widely used to calculate thermal, molecular orbital and molecular electrostatic properties [37]. Geometry optimisation and further modification of all synthesised analogs were carried out using the Gaussian 09 program [38]. Density functional theory (DFT) with Beck’s (B) [39] three-parameter hybrid model, Lee, Yang and Parr’s (LYP) [40] correlation functional under a 3-21G basis set, was employed to optimize and predict their thermal and molecular orbital properties. Dipole moment, enthalpy, free energy, and electrical energy were calculated for all the analogs. Frontier molecular orbital features, HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) were counted at the same level of theory. For each of the thymidine analogs, the HOMO-LUMO energy gap, hardness (η), and softness (S) were calculated from the energies of the frontier HOMO and LUMO as reported, considering Parr and Pearson’s interpretation of DFT and Koopman’s theorem [40] on the correlation of chemical potential (µ), electronegativity (χ) and electrophilicity (ω) with HOMO and LUMO energy (ε). The following equations were used to calculate global chemical reactivity by analyzing molecular orbital features.
2.10. Protein selection and molecular docking
The crystal 3D structure of SARS-CoV-2 main protease proteins (pdb: 6LU7, 6Y84 and 7BQY) were recuperated in the pdb format from the protein data bank [41]. PyMol (version 1.3) software packages were employed to remove all heteroatoms and water molecules [42]. Energy minimization of the protein was performed by using a Swiss-PdbViewer (version 4.1.0) [43]. Furthermore, a molecular docking study against the SARS-CoV-2 main protease proteins 6LU7, 6Y84 and 7BQY (Fig. S1) was conducted on the optimized drugs. Finally, the PyRx application (version 0.8) was used to carry out molecular docking simulation [44], envisaging the target protein as a macromolecule and the thymidine analogs as a ligand. The protein and ligands were input by converting the pdb format to pdbqt, and the AutoDock Tools of the MGL software package were used to perform this job. In AutoDockVina, the size of the grid box was maintained at (53.3467, 65.9465, and 57.6051 Å for 6LU7), (36.0681, 63.8808, and 61.9745 Å for 6Y84) and (49.8375, 64.9838, and 59.1841 Å for 7BQY) along the X, Y, and Z axes. After docking, both the structures of macromolecule and ligand were saved in pdbqt format, and Accelrys Discovery Studio (version 4.1) was employed to explore the results of docking and to predict the non-bonding interactions among cytidine derivatives and amino acid chains of the receptor protein [45]. PDBsum online server was also used to check the validation of the main protease receptor with a Lig-plot and Ramachandran plot (Fig. S2), which revealed that >95.0% of residues were in the allowed region and no residues were missed.
2.11. Molecular dynamics simulation
The molecular dynamics simulation study was conducted in YASARA dynamics [46] with the aid of the AMBER14 force field [47]. The docked complexes were initially cleaned and optimised, and hydrogen bond networking was oriented. A cubic simulation cell was created whereby the TIP3P solvation model was used under periodic boundary conditions [48]. The simulation cell was extended 20 Å for each case from the docked complexes. The physiological condition of the simulation cell was set at 310 K, pH 7.4 and 0.9% NaCl. The initial energy minimisation was conducted using the steepest gradient algorithms through simulated annealing methods (5000 cycles) [49]. The long-range electrostatic interactions were calculated by the Particle Mesh Ewald (PME) using a cut off radius of 8.0 Å [50]. The time step of the simulation cell was set at 2.0 fs. The simulation’s trajectories were saved after every 100 ps. By following the constant pressure and Berendsen thermostat, the simulation was extended for 100 ns. The simulation’s trajectories were used to calculate the root means square deviation (RMSD), root mean square fluctuation (RMSF), solvent accessible surface area (SASA), and the radius of gyration (Rg), and hydrogen bond [51], [52], [53], [54], [55].
Therefore, the YASARA trajectories were utilised in the calculation of the binding free energy from MM-PBSA methods, where positive energy indicates better energy [56]. The equation used for the calculations for the binding free energy calculations was:
| Binding Energy = EpotRecept + EsolvRecept + EpotLigand + EsolvLigand − EpotComplex − EsolvComplex |
2.12. Cytotoxic activity evaluation
To determine the toxicity of the thymidine analogs the brine shrimp lethality assay (BSLA) technique was performed that stated by [57]. The tested thymidine analogs were dissolved in DMSO and prepared at four different concentrations of 20, 40, 80 and 160 µL by adding NaCl solution to each vial up to 5 ml volume. The final concentrations of the sample in the vials were 4, 8, 16 and 32 µL in vials labelled as A, B, C and D, respectively. The experiment was performed three times for each concentration and 10 brine shrimps nauplii were added to each vial. A control test was performed in a vial containing 10 nauplii in 5 ml seawater. Then the vials were kept for incubation at room temperature for 24 hr and 48 hr. After incubation, the vials were observed using a magnifying glass and the number of survivors in each vial was enumerated and noted. From the data, the average percentage of mortality of nauplii was calculated for each concentration. No deaths were found for controls.
2.13. Pharmacokinetic and drug-likeness prediction
The prediction of ADMET properties in drug development is important to research that helps to prevent drug failure during the clinical stages. For this reason, the best-identified esters were evaluated using pkCSM [58] for their in silico pharmacokinetic parameters to avoid the failure of the esters during clinical trials and to improve their chances of reaching the stage of potential candidate drugs. Its absorption in the human intestine; percolation of the blood–brain barrier and the central nervous system, and its metabolism indicate the chemical biotransformation of a drug by the body, total clearance of drugs and the toxicity levels of the molecules. The drug-likeliness of a molecule was expressed using Lipinski’s rule of five parameters (molecular weight < 500 Da, no more than 5 hydrogen bond donors, no more than 10 hydrogen bond acceptors and logP should not be greater than 5). Lipinski’s rule of five properties was obtained from the SwissADME server (https://www.swissadme.ch/index.php).
2.14. Statistical analysis
For each parameter investigated, experimental results were presented as mean ± standard error for three replicates. Two-tailed Student t-tests were used as appropriate for statistical analysis. Only ρ values that were<0.05 were considered to be statistically significant.
3. Results and discussion
3.1. Characterization
The main objective of this research work (Fig. 1 ) reported in this paper was to carry out selective lauroylation (Scheme 1 ) of thymidine (1) with lauroyl chloride using the direct acylation method. The resulting lauroylation product was transformed into several analogs employing various aliphatic and aromatic agents (Table 1 ). These thymidine analogs were used in an attempt to perform an antimicrobial assessment, along with the prediction of PASS and bioactivity spectra, to realize the mode of their antimicrobial behavior. As a result of screening synthesized thymidine analogs for potential antimicrobial activity, it was observed that most of the analogs have potent bactericidal and fungicidal in vitro activity against tested bacterial and fungal pathogens. Furthermore, synthesized analogs were analyzed for their physicochemical properties using DFT-geometry optimization. The observed results were then rationalized by molecular docking and molecular dynamics simulation, along with the prediction of in silico pharmacokinetic and drug-likeness properties.
Fig. 1.

Flow diagram of the whole study.
Scheme 1.

Reagents and conditions: (a) dry Py, 0 °C, 6 h; DMAP, (b) R1-Cl = several acyl halides, 0 °C to rt, stirrer for 6 h. (3–6).
Table 1.
Structure of the synthesized thymidine analogs 2–6.
| Drugs | Chemical Structure | R1 | R2 |
|---|---|---|---|
| 2 | 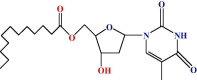 |
CH3(CH2)10CO- | H |
| 3 |  |
CH3(CH2)10CO- | (CH3)3CC6H4CO- |
| 4 | 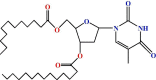 |
CH3(CH2)10CO- | CH3(CH2)12CO- |
| 5 | 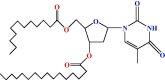 |
CH3(CH2)10CO- | CH3(CH2)14CO- |
| 6 | 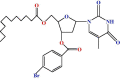 |
CH3(CH2)10CO- | 4-Br.C6H4CO- |
The initial goal was to react thymidine (1) with a unimolecular amount of lauroyl chloride in anhydrous pyridine at a low temperature. Purification using silica gel chromatography furnished the pivaloyl derivative (2) in 62.86% as needles (mp. 65–66 °C). The FTIR spectrum (Fig. S3) showed carbonyl stretching bands at 1680, 1708 cm−1 and hydroxyl stretching at 3408 ∼ 3502 (br) cm−1. The 1H NMR spectrum (Fig. S4) of 2 exhibited the following peaks: two two-proton multiplets at δ 2.38 {CH3(CH2)9CH 2CO-}and δ 1.66 {CH3(CH2)8CH 2CH2CO-}, a sixteen-proton multiplet at δ 1.28 {CH3(CH 2)8CH2CH2CO-} and a three-proton multiplet at δ 0.90 {CH 3(CH2)10CO-}, thereby suggesting the introduction of one lauroyl group in the compound, 2. There was a downfield shift of the H-5/ protons to δ 4.42 (dd, J = 12.0 and 4.5 Hz, H-5́a), and δ 4.31 (dd, J = 12.1 and 4.5 Hz, H-5́b) from their usual value (∼4.0) [32], and these δ values showed the attachment of one lauroyl group at position 5/. The rest of the 1H NMR spectrum conformed with the structure according to it. Molecular ion peak at m/z [M + 1]+ 425.54 corresponding to molecular formula, C22H36O5N2CO. The formation of compound 2 may be explained by assuming that the lauroyl group is attached to the most reactive and less sterically hindered primary hydroxyl group of the ribose moiety at position 5/, thereby forming the 5/-O-(lauroyl) thymidine (2) as the sole product.
3.2. 2d-NMR
The COSY spectrum of compound (2) showed that the starting point could well be the signal from Ar-NH proton which is the most downfield and therefore readily assigned. Thus the signal from Ar-NH at the bottom left of the diagonal has a cross-peak labelled as Ar-NH, H-5́b connecting it to the signal from H-5́b. Thus, the Ar-NH proton around δ 8.71 is coupled to the hydrogen whose signal appears around δ 4.31 (i.e. H-5́b proton). Similarly, the signal from H-5́b is further connected by a cross-peak to the signal from 2H, CH3(CH2)9 CH2CO- to show the coupling between H-5́b and 2H, CH3(CH2)9 CH2CO-.
The downfield shift of H-6, H-1́, H-4́, H-2́, H-3́ and H-5́a as compared to precursor compound (2) (Table 2 ) demonstrated the attachment of lauroyl groups at C-5́ positions of thymidine. Assignments of the signals by analyzing its COSY, HSQC, and HMBC spectral experiments (Fig. 2 ) along with the 13C NMR spectrum confirmed the structure as 5́-O-(lauroyl)thymidine (2).
Table 2.
1H NMR and 13C NMR shift values of compound (2). 1H and 13C assignments were obtained from HSQC and HMBC experiments performed on Bruker DPX-400 spectrometer (CDCl3, 400 MHz).
| Position | δH (ppm) (J Hz) | (HSQC) δC (ppm) |
HMBC |
|---|---|---|---|
| Ar-NH | 8.71 (s) | 134.45 | H: 6, |
| H-6 | 7.30 (d, J = 2.3) | 87.14 | H: 1, Ar-CO |
| H-1́ | 6.29 (t, J = 6.6) | 145.09 | H: 6, 2 |
| H-5́a | 4.42 (dd, J = 12.0 and 4.5) | 105.13 | H: 1, 3́ |
| H-5́b | 4.31 dd, J = 12.1 and 4.5) | 103.23 | H: 3́, 4́ |
| H-3́ | 4.22 (m) | 76.25 | H: 3́, 4́ |
| H-4́ | 3.61 (ddd, J = 3.6, 4.6 and 4.2) | 77.20 | H: 4́, CO |
| 3́–OH | 3.35 (s) | 89.10 | H: 4, 5́a, 5́b |
| H-2́a | 3.08 (ddd, J = 13.5, 6.5 and 4.0) | 51.50 | H: 3́,5́a, 5́b |
| H-2́b | 2.17 (ddd, J = 13.5, 6.5 and 6.7) | ||
| 5-CH3 | 1.95 (d, J = 1.6) | ||
| 5́–CH3(CH2)10CO- | – | 172.29 | H: 5́a, 5́b |
Fig. 2.

The HMBC correlations of compound 2; CO with H-5´a,b and H-6 protons.
The lauroyl derivative (2) was then allowed to react with a unimolecular amount of 4-t-butyl benzoyl chloride in dry C6H5N at freezing temperature. A usual work-up procedure provided the 4-t-butyl benzoyl derivative (3) in good yields. In its 1H NMR spectrum, two two-proton multiplets at δ 8.04 (Ar-H) and δ 7.49 (Ar-H) and three singlets at δ 1.37, δ 1.29 and δ 1.22 {9H, (CH 3)3C–} corresponded to the presence of one 4-t-butyl benzoyl group in compound (3). Further support for the structure accorded to compound (2) was obtained through the preparation of its myristoyl derivative (4), and palmitoyl derivative (5). Thus, the treatment of compound (2) with myristoyl chloride and palmitoyl chloride, in dry pyridine, followed by conventional work-up and chromatographic purification provided the myristoyl derivative (4) and palmitoyl derivative (5) with good yields. It was possible to propose a structure for compounds 4 and 5 by analysing their complete spectroscopic and elemental data.
Finally, 4-bromobenzoyl chloride was used to derivatise compound 2 using the direct acylation method. After the usual workup and purification procedure, the 4-bromobenzoyl derivative (6) was obtained. The formation of a monosubstitution product was revealed by its 1H NMR spectrum, which showed one two-proton multiplet at δ 7.96 (as m, Ar-H), and one two-proton multiplet at δ 7.63 (as m, Ar-H), corresponding to the aromatic ring protons of one 4-bromobenzoyl group in the molecule. The downfield shift of C-3/ to δ 5.34 (as m) from the usual value in the precursor compound 2 and the resonances of other protons in their anticipated positions, showed the presence of the 4-bromobenzoyl group at position 3/. Based on a complete analysis of the FTIR, 1H NMR, mass spectra, and other properties, the structure of this compound was assigned as 3́-O-(4-bromobenzoyl)-5́-O-(lauroyl)thymidine (6).
3.3. Antibacterial potentiality
The use of nucleoside analogs can be considered a novel option as they are expected to act at the genomic level, thereby interfering with the transcription or replication processes required for microbial survival. As there are no alternative pathways in the pathogens for these basic metabolic processes, the nucleoside analogs, by inhibiting these basic pathways, are effective antimicrobial agents [59]. The synthesized thymidine analogs (2–6) were tested for their in vitro antimicrobial activity against a series of strains of Gram-positive bacteria (Bacillus subtilis and Bacillus cereus), and Gram-negative bacteria (Escherichia coli, Salmonella typhi and Pseudomonas aeruginosa). The methods included the disk diffusion method and broth microdilution method for MIC and MBC determination, in accordance with [33]. The results of the preliminary antibacterial activities are shown in Table 3 . Analog 3 exhibited the highest zone of inhibition against Bacillus cereus (15.25 mm) and Bacillus subtilis (13.25 mm), and analog 4 showed a notable zone of inhibition against Bacillus subtilis (12.75 mm) and Bacillus cereus (12.75 mm). Furthermore, analog 2 also exhibited a promising zone of inhibition against both Gram-positive bacteria Bacillus subtilis (12.75 mm) and Bacillus cereus (10.75 mm).
Table 3.
A zone of inhibition was observed against Gram-positive and Gram-negative bacteria by analogs 2–6.
| Diameter of inhibition zone (mm) |
|||||
|---|---|---|---|---|---|
| Drugs | B. subtilis (+ve) | S. aureus (+ve) |
E. coli (-ve) |
S. typhi (-ve) |
P. aeruginosa (-ve) |
| 1 | NI | NI | NI | NI | NI |
| 2 | *12.75 ± 0.1 | 10.75 ± 0.3* | *13.00 ± 0.1 | *14.50 ± 0.3 | *12.75 ± 0.1 |
| 3 | 13.25 ± 0.2 | 15.25 ± 0.3* | NI | 9.75 ± 0.2 | *11.75 ± 0.4 |
| 4 | 12.75 ± 0.1 | 12.75 ± 0.4* | *12.5 ± 0.2 | NI | NI |
| 5 | 14.5 ± 0.1* | 9.00 ± 0.3 | 9.00 ± 0.2 | *15.25 ± 0.2 | 8.50 ± 0.2 |
| 6 | NI | 9.50 ± 0.1 | NI | 9.5 ± 0.1 | NI |
| Azithromycin | 18.5 ± 0.3** | 17.75 ± 0.3** | **17.25 ± 0.1 | **18 ± 0.2 | **18.5 ± 0.3 |
The data are presented as mean ± SD and the values are represented for triplicate experiments. Statistically significant inhibition (p < 0.05) is marked with an asterisk (*) for test compounds and a double asterisk (**) for the reference antibiotic azithromycin. NI = No inhibition.
Interestingly, analog 5 displayed a fluctuating zone of inhibition, where a 14.50 mm zone was found for Bacillus subtilis and a 9.00 mm inhibition zone was observed against Bacillus cereus. Analog 6 exhibited a moderate zone of inhibition against only one Gram-positive bacteria Bacillus cereus (9.50 mm). Based on the above observation, the Gram-positive antibacterial activity of the thymidine analogs can be ordered as 3˃4˃2˃5˃6˃ (Fig. S5).
Furthermore, based on the data presented, analogs 2 and 5 exhibited a zone of inhibition against all the three Gram-negative bacteria: Escherichia coli, Salmonella typhi and Pseudomonas aeruginosa, and analog 3 showed inhibition against only two bacteria: Salmonella typhi and Pseudomonas aeruginosa. Moreover, analog 4, showed inhibition against Escherichia coli, and analog 6 showed inhibition against Salmonella typhi. These derivatives provided moderate to high activity against all three Gram-negative bacteria: Escherichia coli, Salmonella typhi and Pseudomonas aeruginosa. Based on this activity, the compounds can be ordered as 2˃5˃3˃4˃6. Based on the results for zones of inhibition, the compounds showed better activity against Gram-positive bacteria than against Gram-negative bacteria (Fig. S6).
Moreover, to determine the antibacterial effects against the pathogenic bacteria (Fig. S7), the minimal inhibitory concentration (MIC) was measured, along with the minimal bactericidal concentration (MBC) values of the most active thymidine analogs. The results are listed in Table S2 and Fig. 3, Fig. 4 . The best antibacterial effects against the tested strains were found for thymidine analogs 2 and 3, which showed MIC values in the range of 0.125–8.0 µg/mL. Analog 2 was active against all of the test bacteria, and the best activity for this compound was recorded against B subtilis (0.125 µg/mL). Interestingly, analog 3 recorded good activity against most of the test pathogens in impressively low concentrations, and the best activity was recorded against S. typhi (0.5 µg/mL). The lowest value of MBC was found for both analogs 2 and 3 (8.00 µg/mL) against B. subtilis and S. typhi. Besides, the highest MBC value (16.00 µg/mL) was obtained for these derivatives against E. coli, B. subtilis and P. aeruginosa. The MBC values for these compounds killing the other tested organism, lie between these ranges (8.00–16.00 µg/mL).
Fig. 3.

MIC values of some compounds against tested organisms.
Fig. 4.

MBC values of some compounds against tested organisms.
3.4. Antifungal susceptibility
Most of the methyl thymidine (1) analogs were found to have outstanding inhibition to the mycelial growth of both A. niger and A. flavus (Table 4 ). Among the tested analogs, 2 inhibited 82.76 ± 1.0% against the A. flavus and 60.16 ± 1.2% against the A. niger in their antifungal potential assessment. Remarkable mycelial growth prevention also built up for analog 3 against the A. niger (65.25 ± 1.1%) and A. flavus (72.95 ± 1.0%) in their mycelial growth test. Moreover, promising mycelial growth prevention also built up for analog 5 against the A. niger (57.62 ± 1.1%) and A. flavus (81.55 ± 1.0%) in the mycelial growth test. It is reaveled that analogs 2, 3 and 5 were very much effective against A. niger and A. flavus, and their zone of inhibition was higher than standard antibiotic nystatin (Figs. S8 and S9). Furthermore, analog 6 exhibited high inhibition against A. flavus, while no inhibition was observed against A. niger. The only synthesized analog 4 was inactive against both the pathogens; likewise, the parent molecule thymidine 1. It was found that the acylation of thymidine improves antimicrobial activity. The observed results reveal that the insertion of different acyl moieties, including 4-tert-butylbenzoyl, lauroyl, palmitoyl and 4-bromobenzoyl groups, significantly enhanced the antimicrobial activity of thymidine analogs.
Table 4.
Antifungal activities of the synthesised analogs in (%) of inhibition.
| Drugs | % inhibition of fungal mycelial growth in mm (20 μg/μL) |
|
|---|---|---|
| Aspergillus niger | Aspergillus. flavus | |
| 1 | NI | NI |
| 2 | *60.16 ± 1.2 | *82.76 ± 1.0 |
| 3 | *65.25 ± 1.1 | *72.95 ± 1.0 |
| 4 | NI | NI |
| 5 | 57.62 ± 1.1 | *81.55 ± 1.0 |
| 6 | NI | *79.92 ± 1.1* |
| Nystatin | **65.4 ± 1.0 | **64.1 ± 1.0 |
The data are presented as mean ± SD and the values are represented for triplicate experiments. Statistically significant inhibition (p < 0.05) is marked with an asterisk (*) for test compounds and a double asterisk (**) for the reference antibiotic azithromycin. NI = No inhibition.
3.5. SAR study
The development of antimicrobial agents is an area of great urgency and activity due to the emergence of multidrug resistance in common pathogens, and the appearance of new infections. The majority of antimicrobial agents are diverse five- and six-membered heterocyclic molecules that play a crucial role in the metabolism of all living cells [59]. Moreover, a great deal of interest has been directed toward condensed ring systems due to their various types of physiological activities and the success in utilizing them as privileged medicinal scaffolds. Considering that nucleosides play an essential role in most of the fundamental cellular metabolic functions, it is not surprising that nucleoside analogs are capable of targeting a variety of enzymes, such as those involved in bacterial peptidoglycan biosynthesis, fungal chitin biosynthesis, and those involved in protein synthesis. SAR analysis is important in understanding the mechanisms of antibacterial activity for thymidine derivatives (Fig. 5 ). The structure–activity relationship (SAR) of thymidine analogs can be established from the results of the antimicrobial activities reported in Table 3, Table 4. The thymidine itself showed no activity against pathogenic bacteria, so a change in the thymidine skeleton greatly affected the antibacterial activity. It is possible to see that there is a tendency for decreasing influence in the following order: fused butylbenzoyl > lauroyl > palmitoyl for Gram-positive bacteria and lauroyl > palmitoyl > butylbenzoyl for Gram-negative bacteria respectively (Fig. 6 ).
Fig. 5.

Structure-activity relationship study of the synthesized thymidine analogs.
Fig. 6.

Molecular orbital distribution plots of HOMO-LUMO of thymidine analogs (2–6) at DFT/B3LYP/3-21G.
For most of the bacteria tested, fused butyl benzoyl moieties were more active than the palmitoyl, but myristoyl and bromobenzoyl containing derivatives were weaker inhibitors than lauroyl analogs. In the present study, higher concentrations of synthesised compounds were required to inhibit the growth of Gram-negative bacteria (MIC values in the 2 to 8 mg/L) in comparison to Gram-positive. This type of different behaviour is probably due to the different structures of the cell wall between Gram-positive and Gram-negative bacteria. An explanation for this could be the fact that the peptidoglycan of Gram-negative bacteria is surrounded by an outer membrane which restricts diffusion through its lipopolysaccharide (LPS) covering. The LPS layer plays an essential role in providing selective permeability [60], [61] and serves as an efficient barrier against the rapid penetration of various compounds. Moreover, the periplasmic space of Gram-negative bacteria contains hydrolytic enzymes, which can inactivate foreign molecules introduced from the environment. On the other hand, Gram-positive bacteria lack the outer membrane but are characterised by a thick, hydrophilic, porous structure that makes them more permeable. Therefore, Gram-positive bacteria are expected to be more sensitive to synthesized derivatives compared to Gram-negative bacteria. Moreover, the introduction of the lauroyl and palmitoyl groups gradually increased the hydrophobicity of the thymidine derivatives. The hydrophobicity of the compound is essential for bioactivity, as it may affect the toxicity or alter the integrity of the membrane because it is directly related to membrane permeation [10]. Hunt [34] and Judge [62] suggest that the potency of aliphatic alcohols is directly related to their lipid solubility due to the hydrophobic interactions between the alkyl chains of the alcohol and the lipid membrane regions. Hydrophobic interaction might occur between acyl chains of thymidine accumulated in the lipid-like nature of bacteria membranes. As a consequence of their hydrophobic interaction, bacteria lose their membrane permeability, ultimately causing the death of the bacteria.
3.6. Assessment of antimicrobial activities (PASS) and bioactivity
The antimicrobial spectrum was also predicted by applying the web server PASS to all the thymidine analogs 2–6. The PASS results are expressed as Pa and Pi, and are presented in Table 5 . It can be seen in Table 5 that the thymidine analogs 2–6 showed 0.30 < Pa < 0.41 for antibacterial, 0.27 < Pa < 0.32 for antifungal, 0.51 < Pa < 0.72 for antiviral and 0.69 < Pa < 0.83 for anti-carcinogenic. These results reveal that these molecules were more efficient against bacterial and virus pathogens in comparison with fungal pathogens. The attachment of additional aliphatic acyl chains (C12 to C16) increased antibacterial activity (Pa = 0.415) of thymidine (1, Pa = 0.332), whereas the insertion of Br- and -C(CH3)3 substituted aromatic groups decreased the activity somewhat. The same scenario was observed for antifungal and anti-carcinogenic activity, where acyl chain analogs revealed improved values compared to the benzoyl analogs. However, analog 2, which contain the lauroyl group, exhibited the highest antifungal and anti-carcinogenic activities. An attempt was also made to predict the antiviral parameters of these analogs.
Table 5.
Prediction of antimicrobial activity of the thymidine analogs using the PASS web tool.
| Biological activity | ||||||||
|---|---|---|---|---|---|---|---|---|
| Drugs | Antiviral |
Antibacterial |
Antifungal |
Anti-carcinogenic |
||||
| Pa | Pa | Pa | Pi | Pa | Pi | Pa | Pi | |
| 1 | 0.806 | 0.432 | 0.332 | 0.112 | 0.240 | 0.112 | 0.806 | 0.005 |
| 2 | 0.724 | 0.415 | 0.415 | 0.074 | 0.319 | 0.074 | 0.830 | 0.004 |
| 3 | 0.723 | 0.304 | 0.304 | 0.092 | 0.277 | 0.092 | 0.795 | 0.005 |
| 4 | 0.515 | 0.410 | 0.410 | 0.073 | 0.322 | 0.073 | 0.800 | 0.005 |
| 5 | 0.515 | 0.410 | 0.410 | 0.073 | 0.322 | 0.073 | 0.800 | 0.005 |
| 6 | 0.610 | 0.348 | 0.348 | 0.089 | 0.282 | 0.089 | 0.697 | 0.004 |
Therefore, PASS determination exhibited 0.51 < Pa < 0.72 for antiviral, which shows that the thymidine analogs have more potential as antiviral agents than other antimicrobial parameters.
3.7. Thermodynamic analysis
A simple alteration of the chemical structure significantly influences structural properties, including thermal and molecular orbital properties. The spontaneity of a reaction and the stability of a product can be calculated from the free energy and enthalpy values [63]. Highly negative values are more likely to gain thermal stability. In drug design, hydrogen bond formation and nonbonded interactions are also influenced by the dipole moment. Free energy (G) is a significant criterion to represent the interaction of binding partners, where negative values are favorable for spontaneous binding and interaction. In the current study, the thymidine analogs possessed greater negative values for E, H and G than the parent thymidine, thus indicating that the attachment of an acyl group could improve interaction and the binding of these molecules with different microbial enzymes. The highest free energy is (-4316.309 Hartree) observed for thymidine analog (6), which has also shown the highest enthalpy (-4316.195 Hartree) and highest electronic energy (-4316.194 Hartree). The dipole moment value of a molecule is very significant to describe its electronic properties, where a high dipole moment value of a molecule causes more intermolecular interactions. A high dipole moment value reveals a more polar nature [64]. As shown in Table 6 , two thymidine analogs (4 and 6) have an improved dipole moment, which enhances the polar nature of a molecule, and promotes binding affinity, hydrogen bonding, and nonbonding interaction with the receptor protein. The highest dipole moment (5.911 Debye) was observed for analog (6), whereas (3) showed the lowest value (3.229 Debye). The scores for all parameters gradually increased with the number of carbon atoms in the substituents (2–6). The bromobenzoyl analog had better scores for all parameters, as evidenced by analog (6) (4-Br.Bz), which had the highest free energy of the therapeutics under investigation and showed markedly improved dipole moment. Overall, it is reasonable to propose that the modification of hydroxyl (–OH) groups of thymidine significantly increase its thermodynamic properties.
Table 6.
Molecular formula, molecular weight, electronic energy (E), enthalpy (H), Gibb’s free energy (G) in Hartree and dipole moment (p, Debye) of thymidine analogs.
| Drugs | MF | MW | E | H | G | p |
|---|---|---|---|---|---|---|
| 1 | C10H14N2O5 | 242.23 | −870.052 | −870.051 | −870.111 | 5.839 |
| 2 | C22H36O5N2CO | 424.54 | −1412.551 | −1412.550 | −1412.652 | 4.677 |
| 3 | C32H48O6N2CO | 584.75 | −1911.288 | −1911.243 | −1911.373 | 3.229 |
| 4 | C35H62O6N2CO | 634.90 | −2033.183 | −2033.182 | −2033.327 | 5.872 |
| 5 | C37H66O6N2CO | 662.95 | −2111.334 | −2111.333 | −2111.485 | 5.836 |
| 6 | C28H39O6BrN2CO | 607.54 | −4316.195 | −4316.194 | −4316.309 | 5.911 |
3.8. Frontier molecular orbitals (FMO)
The frontier molecular orbitals are the principal orbitals in a molecule, and they are considered to study chemical reactivity and kinetic stability. These frontier molecular orbitals are known as the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The electronic absorption relates to the transition from the ground to the first excited state, and is mainly represented by one electron excitation from HOMO to LUMO [65].
The extended energy gap of a molecule relates to high chemical stability and low chemical reactivity. A low energy gap is related to low chemical stability and high chemical reactivity because of the ease of the transition of electrons. As a result, the removal of electrons from ground state HOMO to excited state LUMO requires more energy.
The HOMO and LUMO energies, HOMO-LUMO gap (), hardness (η), softness (S), chemical potential (µ) and electronegativity (χ) and electrophilicity (ω) index of all analogs are presented in Table S3. Table S3 and Figs. 6, S10 and S11 show the thymidine analog (3) had a slightly higher energy gap value (5.575 eV), and the thymidine analog (6) had a slightly lower energy gap value (5.122 eV) than that of the other analogs. The thymidine analog (3) had chemical hardness and softness values of 2.788 eV and 0.359 eV respectively, where the hardness value was highest from among all the analogs. On the other hand, the thymidine analog (6) had the lowest chemical hardness (2.561 eV) as well as the highest chemical softness (0.391 eV). Analog (6) had a Br- functional group in the aromatic ring, which is the reason behind this high chemical reactivity.
3.9. Molecular electrostatic potential map analysis
In computer-aided drug design, atomic charges are employed to investigate the connectivity between the structure and biological activity of a drug. The molecular electrostatic potential (MEP) is used globally as a reactivity map displaying the most suitable region for the electrophilic and nucleophilic attack of charged point-like reagents on organic molecules [66]. It helps to illustrate the biological recognition process and hydrogen bonding interaction [67].
An MEP counter map is a simple way of predicting how different geometry could interact. The MEP of the title analog is obtained based on the B3LYP with the basis set at 3-21G for an optimised result, as shown in Fig. 7 . The importance of MEP lies in the fact that it simultaneously shows the molecular size and shape, as well as positive, negative and neutral electrostatic potential regions in terms of colour grading, which is very useful in the research of molecular structure, along with the physicochemical properties relationship [68]. Molecular electrostatic potential (MEP) was calculated to forecast the reactive sites for the electrophilic and nucleophilic attack of the optimized structure of thymidine (1) and analogs (2–6). The different values of electrostatic potential are represented by different colours, with potential increases in the order red < orange < yellow < green < blue. The red colour displays the maximum negative area, which shows favorable sites for electrophilic attack; the blue colour indicates the maximum positive area favorable for the nucleophilic attack, and the green colour represents zero potential areas (Figs. 8 and 9 ).
Fig. 7.

Map of the molecular electrostatic potential of thymidine (1) and analogs (2–6).
Fig. 8.

Nonbonding interactions of thymidine (a); analog 2, (b); analog 4 and (c, d); analog 6 with the active site of SARS-CoV-2 generated by Discovery Studio.
Fig. 9.

A: Docking pose (space-filling model) and 2D interaction map of analogs 2 and 3 with the main protease. B: Superimposed views of ligands 2 and 3 after rigid docking. C: Docking pose (space-filling model) and 2D interaction map of analogs 3 and 6 with the main protease.
3.10. Molecular docking simulation
Molecular docking is a type of bioinformatics modeling which deals with the interaction between two or more molecules to give a stable adduct. The key aim of molecular docking is to predict the potential binding geometries of a putative ligand of a known three-dimensional structure with a target protein. In this investigation, a series of thymidine analogs was studied in silico to highlight their possible binding energy and interaction modes with the active site of SARS-CoV-2 main protease (Tables 7, 8 and S4), using the AutoDock Vina software. The multiple sequence alignment of the closest homologs of the main protease 6LU7, 6Y84 and 7BQY structure is displayed in Fig. S12. According to the outcomes obtained from docking screening, three analogs (2, 3 and 6) with the strongest binding energies were selected to describe the binding mode of the potential thymidine analogs. As shown in Table 7 , the aromatic analogs displayed better binding scores than the aliphatic analogs. The interactions between the inhibitor and bordering residues of the SARS-CoV-2 main protease are illustrated in the 2D schematics, which were attained by importing the docking results into the Discovery Studio Visualizer. Fig. 8 and S13 showed the amino acids that participated in the pattern of interactions between the ligand and enzyme with an important contribution to the total energy of the interaction. Most of these interactions included hydrophobic contacts, Van der Waals interactions, hydrogen bonds, electrostatic, carbonyl, and one specific atom-aromatic ring, as well as providing an insight into understanding molecular recognition. Fig. 9 and S14 depicts the docked conformation of the most active molecules (2, 3 and 6) based on docking studies. The results show that thymidine analogs (3 and 6) are the most promising ligand (-7.9, −6.3, −7.2, −8.6, −6.6 and −7.5 kcal/mol), which bound with SARSCoV-2 main protease via many hydrophobic bonding and hydrogen interactions. The binding sites were mainly located in a hydrophobic cleft bordered by the amino acid residues Cys145, His41, Phe294, Met49, Met165, Val104, Pro293, Thr199 and Thr25. There are six hydrogen bond contacts with four different amino acids: Asp153, Cys145, Thr26, Ser46, His164, Leu287 and Gln110 respectively. The thymidine analogs (3 and 6) had an additional benzene ring in the thymidine, providing a high density of electrons in the molecule, indicated by the highest binding score. These results show that modification of –OH group along with an aromatic ring molecule increased the binding affinity, while the addition of hetero groups like –C(CH3)3, -Br caused some fluctuations in binding affinities; however, modification with halogenated aromatic rings increased the binding affinity. The docked pose clearly shows that the drug molecules bind within the active site of the SARS-CoV-2 main protease macromolecular structure.
Table 7.
Binding Energy of the thymidine analogs against main protease.
| Drugs | 6LU7 (kcal/mol) | 6Y84 (kcal/moL) | 7BQY (kcal/moL) |
|---|---|---|---|
| 1 | −6.5 | −5.6 | −6.5 |
| 2 | −7.6 | −6.2 | 6.9 |
| 3 | −7.9 | −6.3 | −7.2 |
| 4 | −4.0 | −5.3 | −5.3 |
| 5 | −5.7 | −5.5 | −4.8 |
| 6 | −8.6 | −6.6 | −7.5 |
| HCQ | −6.7 | −6.8 | −6.6 |
The parent molecule, thymidine (1), exhibited interactions with the key residues of the main protease Cys145 and HIS41 through hydrogen and hydrophobic bonding within a closer bond distance (2.7547 Å). Additionally, Met165, Glu166 and Ser144 interactions were found; whereas Glu166 revealed a shorter distance (2.7547 Å) because of the unique interaction of branched alkyl chain with cytosine base. Aliphatic chain substituted analogs (2 and 4–5) revealed a lower binding score with the main protease, which indicated the burying of the ligand in the receptor cavity. In spite of having lower binding affinity, they also interacted with the catalytic binding of the main protease, such as Cys145, His41, Arg22, His164, Phe294, Cys44, Asn23, Glu166, Met49, Arg298, Thr111, Asp153 and Gly143. These analogs exhibited diverse nonbonding interactions, such as pi-alkyl, pi-sigma, amide pi-stacked, and pi-donor hydrogen bonds with the active site of the main protease.
These outcomes clearly show that, due to having high electron density, aromatic substituents can easily increase the binding ability as well as the antimicrobial ability of the thymidine analogs. Along with Phe294, all the analogs displayed the maximum π-π interactions with the His41, denoting tight binding with the active site. Reports suggest that Phe294 is thought to be the principal component of the PPS, APS and PPT responsible for the accessibility of small molecules at the active site. Binding energy and binding mode were exalted in some of the analogs (2, 3, and 6) because of significant hydrogen bonding. It was observed that the alterations of the –OH group in thymidine increased the π–π interactions with the amino acid chain at the binding site, whereas their polarity improvement resulted in the formation of hydrogen bond interactions. The maximum numbers of H-bonds were observed for analog (2), with Cys145, Gly143, and Glu166 residues.
It has already been reported that ten commercial medicines may possibly form H-bonds with key residues of 2019-nCoV main protease [69]. H-bonds execute a vital function in shaping the specificity of ligand binding with the receptor; drug design in chemical and biological processes, and molecular recognition and biological activity. The H-bond surface and hydrophobic surfaces of analogs (2 and 5) with the main protease are therefore represented in Fig. 10 . It was accomplished from the docking study of all thymidine analogs with the SARS-CoV-2 main protease that the molecules are generally surrounded by the above-mentioned residues, like the standard drug, this suggests that this molecule may prevent the viral replication of SARS-CoV-2.
Fig. 10.

(a): Hydrogen bond surface of SARS-CoV-2 main protease with analog 2 (b); the hydrophobic bond surface of SARS-CoV-2 main protease with analog 5.
The distance of the ligands, along with the change in the accessible area of the two important catalytic residues (Cys145 and His41) within the active site of the protease, is shown in Table 8 . It is noted that the analyzed thymidine analogs bind within the active site of the main protease of SARS-CoV-2, like the standard drug hydroxychloroquine (HCQ), which is inevitable to prevent the protein mutarotation of the virus by minimizing the viral replication. The binding affinities calculated varied in the range of (-4.0 to −8.6 kcal/mol), suggesting that the synthesized molecules can spontaneously interact within the binding site of the SARS-CoV-2 main protease. Although the blind docking studies revealed that all the molecules can act as potential agents for the establishment of COVID-19 drugs, from the estimated free energy of binding values, it may be inferred that the analogs (2, 3 and 6) with the highest negative minimum binding energy value amongst all the studied analogs could be the best possible SARS-CoV-2 inhibitors.
Table 8.
Nonbonding interaction data of thymidine analogs against main protease.
| 6LU7 | ||||
|---|---|---|---|---|
| Drugs | Bond category | Residues in contact | Interaction type | Distance (Å) |
| 1 | H | Ser144 | CH | 2.2804 |
| H | Ser144 | CH | 2.8882 | |
| H | Glu166 | CH | 2.7547 | |
| H | Cys145 | CH | 2.9402 | |
| H | Glu166 | PDH | 2.8758 | |
| Hydrophobic | Met165 | A | 5.2418 | |
| 2 | H | Asp197 | CH | 2.4976 |
| H | Tyr237 | CH | 2.2034 | |
| H | Cys145 | CH | 2.5146 | |
| H | Gly143 | CH | 2.0837 | |
| H | Thr199 | CH | 2.8164 | |
| H | Asn23 | CH | 2.5491 | |
| Hydrophobic | His41 | A | 5.4329 | |
| Hydrophobic | Leu287 | A | 4.0128 | |
| 3 | H | Cys145 | CH | 2.9281 |
| H | Lys137 | CH | 2.9048 | |
| H | Gly143 | CH | 2.3611 | |
| H | Thr199 | CH | 1.8735 | |
| Hydrophobic | Leu287 | PS | 3.8043 | |
| Hydrophobic | His41 | A | 4.6492 | |
| 4 | H | Arg222 | CH | 2.1832 |
| Hydrophobic | Leu71 | A | 4.5601 | |
| Hydrophobic | Arg222 | A | 3.8938 | |
| Hydrophobic | Trp218 | PA | 5.3572 | |
| 5 | H | Ile249 | CH | 2.7595 |
| H | Gln110 | CH | 2.5110 | |
| H | Phe294 | CH | 2.6424 | |
| Hydrophobic | Met49 | PS | 3.4474 | |
| Hydrophobic | Glu166 | A | 5.3002 | |
| Hydrophobic | Ile200 | A | 5.2995 | |
| Hydrophobic | Val202 | A | 5.0707 | |
| Hydrophobic | Ile249 | A | 4.0245 | |
| Hydrophobic | Pro293 | A | 5.1850 | |
| Hydrophobic | Phe294 | PA | 4.5719 | |
| 6 | H | Cys145 | CH | 2.3894 |
| H | Gln110 | CH | 2.4455 | |
| H | Asp153 | C | 3.5234 | |
| Hydrophobic | Phe294 | PPS | 3.8467 | |
| Hydrophobic | His41 | A | 4.6591 | |
| Hydrophobic | Pro293 | A | 4.1825 | |
| Hydrophobic | Val104 | A | 3.8944 | |
| Hydrophobic | Val104 | PA | 4.9265 | |
| HCQ | H | Gly143 | CH | 2.6960 |
| H | Ser144 | CH | 2.2254 | |
| H | Ser144 | CH | 2.4119 | |
| H | Cys145 | CH | 2.4609 | |
| H | Phe140 | C | 3.7263 | |
| Hydrophobic | Met49 | A | 4.7454 | |
| Hydrophobic | Met165 | A | 5.4899 | |
| Hydrophobic | His41 | PA | 4.7413 | |
CH = Conventional Hydrogen Bond; C = Carbon Hydrogen Bond; A = Alkyl; PA = Pi-Alkyl; PS = Pi-sigma; APS = Amide Pi-stacked; PDH = Pi-Donor Hydrogen Bond; PPS = Pi-Pi Stacked; PPT = Pi-Pi T-shaped.
3.11. Molecular dynamics
The molecular dynamics simulation study was conducted to understand the structural stability of the docked complexes. Therefore, the root means square deviations of C-alpha atoms from the simulation trajectories were explored to examine flexibility across the simulation periods. Fig. 11 (a-f) indicates that analogs 2, 3, and 4 had the upper trend from the very beginning, which indicates the flexible nature of the docked complexes. Therefore, the complexes reached a steady-state after 35 ns times and maintained a lower degree of deviations in RMSD. The complexes had an RMSD below 2.5 Å for the entire atomistic simulation times, which defines the structural integrity of the complexes (Fig. 11a). Also, the solvent-accessible surface area of the complexes was examined to understand the changes in the protein volume, where a higher SASA correlates with the expansion of the surface area, and a lower SASA defines the truncated nature of the protein.
Fig. 11.

The molecular dynamics simulation trajectories from 100-ns simulation time, here (a) root mean square deviation of the C-alpha atoms; (b) solvent accessible surface area of the docked complex to understand the change in protein area; (c) Radius of the gyration of the complexes (d) hydrogen bond of the complexes (e) root mean square fluctuation and (f) free energy of the complexes.
Fig. 11b indicates that all complexes extended their surface volumes upon ligand binding during the initial phases, but complexes 2 and 3 had relatively stable profiles and maintained a similar trend. Moreover, analog 6 exhibited lower SASA than the other complexes, which defines the condensed nature of the complexes. The radius of gyrations of the complexes defines their labile nature, where a higher Rg is related to greater mobility, and a lower Rg indicates the stable nature of the complexes. Fig. 11c indicates that all complexes had a similar Rg profile; also, lower degrees of the deviations were observed for the three simulated complexes. This trend in the radius of gyration profiling defines the strict and stable nature of the complexes. The hydrogen bond plays an important role in defining the stable nature of the complexes, where Fig. 11d indicates that the three complexes lowered fluctuations and maintained integrity in hydrogen bond profiling. The root means square fluctuations were also analyzed to understand the flexibility across the amino acid residues. Fig. 11e demonstrates that almost every residue had a lower RMSF than 2.5 Å, which indicates the stable nature of the three complexes. Furthermore, the binding free energy of the complexes was explored from the MM-PBSA methods, where a positive score defines more favorable bindings according to the algorithms of YASARA dynamics (Fig. 11f). The average MM-PBSA scores of analogs 2, 3 and 4 were 25.47, 45.488 and 3.054 KJ/mol respectively, which indicates the favorable bindings of the three complexes.
3.12. Cytotoxic activity of the thymidine analogs
The cytotoxic activity of the synthesized thymidine analogs (2–6) in the brine shrimp lethality bioassay method [57] is displayed in Fig. 12 , which exhibited the percentage of mortality of shrimps at 24 h and 48 h. A long alkyl chain increased the cytotoxicity of the compounds and the introduction of a phenyl ring was suggested to increase hydrophobicity and cytotoxicity [70]. From the observed data, it was found that thymidine analog 2 (5́-O-lauroylthymidine) showed the lowest level of toxicity i.e. 36.25% death. Analogs 4 {5́-O-lauroyl-3́-O-(myristoyl)thymidine} and 5 {5′-O-lauroyl-3′-O-(palmitoyl)thymidine} showed highest levels of toxicity (i.e. 52.25%-53.25% death) indicating its higher mortality.
Fig. 12.

Cytotoxic activity of thymidine analogs (2–6).
The rest of the thymidine analogs 3́-O-(4-t-butylbenzoyl)-5́-O-(lauroyl)thymidine (3) and 3́-O-(4-bromobenzoyl)-5́-O-(lauroyl)thymidine (6) were found less toxic for brine shrimp (i.e. 41. 25% and 38.75% death). It was evident from the observation, that the benzoyl analogs had lower cytotoxic activity than the alkyl chain analogs. Moreover, the cytotoxic activity of the alkyl chain derivatives increases with the concentration.
3.13. The pharmacokinetic profile and drug-likeness analysis
In an attempt to predict pharmacokinetic properties such as absorption, distribution, metabolism, excretion (ADME) and toxicity of the compounds, the pkCSM ADMET descriptors algorithm protocol was used. Drug absorption depends on factors such as membrane permeability [indicated by the cell line of colon cancer (Caco-2)], intestinal absorption, skin permeability thresholds, and substrate or inhibitor of P-glycoprotein. A value of intestinal absorbance below 30% suggests poor absorbance. According to the results shown in Table 9 , all the analogs showed excellent absorption as the absorption values are more than 30. Skin permeability is an important consideration for improving drug efficacy, and it is particularly of interest in the development of transdermal drug delivery. A molecule will barely penetrate the skin if log Kp is more than −2.5 cm/h. From Table 9, it can be seen that the skin permeability (Kp) of the thymidine analogs is −2.732 to −2.829 cm/h (<-2.5). Therefore, it can be predicted that all analogs have good skin penetrability. For the pkCSM predictive model, high Caco-2 permeability is translated into predicted log Papp values greater than 0.90 cm/s. As Table 9 shows, the value of the Caco-2 permeability (log Papp) of the thymidine analogs ranged from 0.47 to 0.78 cm/s, log Papp < 0.9 cm/s, so it is predicted that these have low Caco-2 permeability.
Table 9.
Prediction of in silico absorption of thymidine analogs.
| Drugs | Water solubility (log mol/L) | Caco-2 permeability | Intestinal absorption | Skin permeability |
|---|---|---|---|---|
| 1 | −2.77 | −0.123 | 60.686 | −3.029 |
| 2 | −4.536 | 0.479 | 65.54 | −2.829 |
| 3 | −6.143 | 0.788 | 81.92 | −2.736 |
| 4 | −4.976 | 0.511 | 88.511 | −2.732 |
| 5 | −4.573 | 0.487 | 89.962 | −2.734 |
| 6 | −5.331 | 0.734 | 80.473 | −2.737 |
For the discovery of oral administrative drugs, solubility is one of the major descriptors. High water solubility is useful for delivering active ingredients in sufficient quantities in small volumes of such pharmaceutical dosage. The values for water solubility are given in log (mol/l) (Insoluble ≤ -10 < poorly soluble < -6 < Moderately < -4 < soluble < -2 < very soluble < 0 ≤ highly soluble). From the results shown in Table 9, it can be observed that the analogs tested are soluble.
Distribution volume (Vd) is a pharmacokinetic parameter that reflects the tendency of an individual substance to either linger in the plasma or redistribute to another tissue compartment. According to Pires et al. [58], VDss is considered low if it is below 0.71 L/kg (log VDss < -0.15), and high if it is above 2.81 L/kg (log VDss greater than 0.45. It can be seen in Table 10 that the value of thymidine analogs VDss ranged from −0.551 to 0.433, with only one analog having a VDss value of < -0.15 (analog 3). The blood–brain partitioning and brain distribution are critical properties for drugs targeting the central nervous system. The analogs tested a logBB < -1 considered poorly distributed to the brain. From Table 10, it can be seen that the logPS (the central nervous system (CNS) permeability) value of thymidine analog range from –3.74 to −3.25, logPS < -3 so it can be predicted that analogs (2–6) are unable to penetrate the CNS. The model provided by pkCSM pharmacokinetics predicts the total clearance log(CLtot) of a given compound in log(mL/min/kg). The larger the CLtot value of the compound, the faster the excretion processes. It can be seen in Table 10 that the log CLtot value of thymidine analogs ranges from 0.02 to 1.65 ml/min/kg, and from those values, the rate of excretion of the analog can be predicted. Metabolism is predicted based on the CYP models for substrate or inhibition (CYP2D6, CYP1A2, CYP2C19 and CYP3A4). Table S5 shows that all the thymidine analogs do not affect or inhibit all the enzymes, except CYP3A4 for analog 6, so it can be predicted that analogs (2–5) in the body tend to be metabolized by the P450 enzyme. Bioactivity radar Charts of the thymidine analogs (Fig. S15) reveal the promising pharmacokinetic profiles of all candidates. The toxicity results of the thymidine analogs are described in Table S6, and their high LD50 values (2.23 to 2.47) suggest that the analogs are lethal only at very high doses. A negative result in the AMES test suggests that an analog is not mutagenic. The results also suggest that all analogs tested may not inhibit the hERG channel and may not have skin sensitization. Molecular parameters like the membrane permeability of lead molecules depend on some fundamental features of molecules, such as partition coefficient (logP), molecular weight (MW), and several hydrogen bond acceptors/donors, which is associated with Lipinski’s ‘rule of five’ [71]. Table S7 showed that analogs (3–6) violate the rule of five in two cases, indicating the good bioavailability of thymidine analogs, whereas analog (2) followed the rules of Lipinski. Low molecular weight containing derivatives are easily absorbed, diffused and transported, as compared to high molecular weight derivatives. As molecular weight increases past a certain limit, the bulkiness of the molecules also increases comparably. TPSA (Topological Polar Surface Area) is a very important physicochemical property of molecules that provides information on the polarity of analogs. This parameter was screened to analyse drug transport properties. Polar surface area is the sum of all polar atoms, mainly oxygen and nitrogen, and includes attached hydrogen.
Table 10.
Prediction in silico of distribution and execration of thymidine analogs.
| Drugs | Distribution |
Execration |
|||
|---|---|---|---|---|---|
| Vdss | BBB permeability | CNS permeability | Total Clearance | Renal OCT2 substrate | |
| 1 | −0.295 | −0.982 | −3.649 | 0.729 | No |
| 2 | −0.439 | −1.210 | −3.884 | 1.665 | No |
| 3 | −0.061 | −1.235 | −3.421 | 0.688 | No |
| 4 | 0.433 | −1.589 | −3.279 | 1.512 | No |
| 5 | −0.551 | −1.632 | −3.256 | 1.475 | No |
| 6 | 0.182 | −1.350 | −3.741 | 0.022 | No |
3.14. POM analysis: Identification of the pharmacophore sites
POM Theory (Petra/Osiris/Molinspiration) that is invented by the group of Taibi Ben Hadda in collaboration with the American NCI and TAACF led us to real success in pharmacology and drug design fields [72]. By using the POM (Petra/Osiris/Molinspiration) Theory, it becomes now easier, to identify and optimize most of the antibacterial [73], [74], [75], antifungal [76], [77], [78], antiviral [79], [80], [81], antiparasitic [82], [83], and antitumor [85] pharmacophore sites, one by one, on the basis of their different physicochemical parameters and their different electronic charge repartition of corresponding heteroatoms. This young POM Theory was extended, with success, to other various and different bio targets [86]. Here we treat the series of compounds 2–6 to identify their pharmacophore sites. So the identification of the type of pharmacophore sites of these compounds was derived from the physical and chemical properties of the tested compounds by using Petra, Osiris and Molinspiration (POM) platform. Results of pharmacokinetic properties and bioactivity score analysis are shown in (Table 11 ).
Table 11.
Prediction of Molecular properties and bioactivity scores.
| Drugs | Molecular Properties | Bioactivity Scores |
|---|---|---|
| 2 | miLogP 4.23 TPSA 110.63 MW 424.54 nOHNH 2 nviolations 0 volume 411.72 |
GPCR ligand 0.22 Ion channel modulator −0.22 Kinase inhibitor −0.10 Nuclear receptor ligand −0.54 Protease inhibitor −0.19 Enzyme inhibitor 0.58 |
| 3 | miLogP 8.31 TPSA 116.71 MW 584.75 nOHNH 1 nviolations 2 volume 569.26 |
GPCR ligand 0.04 Ion channel modulator −0.54 Kinase inhibitor −0.27 Nuclear receptor ligand −0.55 Protease inhibitor −0.21 Enzyme inhibitor 0.15 |
| 4 | miLogP 9.36 TPSA 116.71 MW 634.90 nOHNH 1 nviolations 2 volume 649.85 |
GPCR ligand −0.09 Ion channel modulator −0.87 Kinase inhibitor −0.47 Nuclear receptor ligand −0.81 Protease inhibitor −0.20 Enzyme inhibitor −0.07 |
| 5 | miLogP 9.59 TPSA 116.71 MW 662.95 nOHNH 1 nviolations 2 volume 683.46 |
GPCR ligand −0.25 Ion channel modulator −1.13 Kinase inhibitor −0.69 Nuclear receptor ligand −1.03 Protease inhibitor −0.29 Enzyme inhibitor −0.28 |
| 6 | miLogP 7.11 TPSA 116.71 MW 542.67 nOHNH 1 nviolations 2 volume 519.64 |
GPCR ligand 0.06 Ion channel modulator −0.40 Kinase inhibitor −0.17 Nuclear receptor ligand −0.52 Protease inhibitor −0.23 Enzyme inhibitor 0.28 |
Most compounds of series 2–6 present attractive bioactivity scores. Their 3D structure presentation presents an interesting combination of two principal antifungal/antiviral Oδ-,O’δ-) pharmacophore sites (Fig. 13 ).
Fig. 13.

3D Structure of compounds 3–6 and Identification of their two potential combined antiviral/antifungal (Oδ-,O’δ-) pharmacophore sites.
4. Conclusion
In this research, a series of thymidine analogs were analysed in vitro and in silico for their antimicrobial, thermodynamic, molecular docking, molecular dynamics and drug-likeness properties. Most of the thymidine analogs have a HOMO-LUMO gap lower than thymidine, resulting in modified analogs being more reactive than the parent drug. Insertion of various aliphatic and aromatic groups into the thymidine structure can significantly improve their biological activity mode. The study revealed that 4-t-butyl and bromo substituted benzoyl analogs 3 and 6 had more potential against bacterial organisms with better pharmacokinetic and biological spectra. These observations were rationalised by conducting molecular docking, which revealed promising antiviral efficacy for thymidine analogs. Thymidine analogs (2, 3 and 6) showed promising binding interactions and binding energy with SARS-CoV-2 main protease which revealed analogs (2, 3 and 6) showed an in silico potent ability to fight SARS-CoV-2. The molecular electrostatic potential study also showed the most negative and positive surface area of the investigated ligand, and thus, anticipated the most suitable areas for hydrogen bonding sites. This result greatly supports molecular dynamic studies up to 100 ns, keeping in mind protein, which confirms the binding stability of the docked complex in the trajectory analysis. This also means that the protein–ligand complex is highly stable in biological systems. In addition, these analogs were analysed for their cytotoxicity and pharmacokinetic properties, which revealed that a combination of toxicity determination, in silico ADMET prediction, and drug-likeness has promising results because most of the designed molecules showed improved kinetic parameters, and maintained all drug-likeness rules as well as provided an interesting result in terms of biological activity. As this study has been carried out using synthetic, antimicrobial and in silico computational methods, these results require further wet-lab experiments to be carried out under in vivo as well as in vitro conditions for these analogs to be potential drug candidates to treat SARS-CoV-2. This hypothesis is also predicted by POM Theory which shows the presence of an antiviral (O1δ-, O2δ-) pharmacophore site.
Author Contributions.
MAH and NSM synthetic experimental analysis; MAG and MB validation of article and improve the manuscript; FAA, TBH and HL performed the POM study; FA antimicrobial; SM and MAS performed the computational, dynamics and interpretation of the results; SMAK conceptualization, methodology, article writing, and supervision. All authors reviewed and approved the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
The authors are very much thankful to the Ministry of Education, Government of Bangladesh [Ref: 37.20.0000.004.33.006.2020-3782] for providing the financial support. The authors are also indebted to the Director, Wazed Miah Science Research Centre, JU, and Dhaka, Bangladesh for the recording of spectra.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2022.105850.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.De Clercq E., Li G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/cmr.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahmoud S., Hasabelnaby S., Hammad S.F., Sakr T.M. Antiviral nucleoside and nucleotide analogs a review. J. Adv. Pharm. Res. 2018;2:73–88. doi: 10.21608/aprh.2018.5829. [DOI] [Google Scholar]
- 3.Guinan M., Benckendorff C., Smith M., Miller G.J. Recent advances in the chemical synthesis and evaluation of anticancer nucleoside analogues. Molecules. 2020;25:2050–2075. doi: 10.3390/molecules25092050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eyer L., Nencka R., Huvarová I., Palus M., Alves M.J., Gould E.A., De Clercq E., Ruzek D. Nucleoside inhibitors of Zika virus. J. Infect. Dis. 2016;214:707–711. doi: 10.1093/infdis/jiw226. [DOI] [PubMed] [Google Scholar]
- 5.Hamuy R., Berman B. Topical antiviral agents for herpes simplex virus infections. Drugs Today. 1998;34:1013–1025. doi: 10.1358/dot.1998.34.12.487486. [DOI] [PubMed] [Google Scholar]
- 6.X. Rabasseda, Brivudine: A herpes virostatic with rapid antiviral activity and once-daily dosing. Drugs Today, 39(2003), 359–371, doi.org./10.1358/dot.2003.39.5.740221. [DOI] [PubMed]
- 7.S. M. A. Kawsar, M. Islam, S. Jesmin, M. A. Manchur, I. Hasan, S. Rajia, Evaluation of the antimicrobial activity and cytotoxic effect of some uridine derivatives. Int. J. Biosci., 12 (2018) 211–219, 10.12692/ijb/12.6.211-219.
- 8.Kawsar S.M.A., Hamida A.A., Sheikh A.U., Hossain M.K., Shagir A.C., Sanaullah A.F.M., Manchur M.A., Imtiaj H., Ogawa Y., Fujii Y., Koide Y., Ozeki Y. Chemically modified uridine molecules incorporating acyl residues to enhance antibacterial and cytotoxic activities. Int. J. Org. Chem. 2015;5:232–245. doi: 10.4236/ijoc.2015.54023. [DOI] [Google Scholar]
- 9.Shagir A.C., Bhuiyan M.M.R., Ozeki Y., Kawsar S.M.A. Simple and rapid synthesis of some nucleoside derivatives: structural and spectral characterization. Curr. Chem. Lett. 2016;5:83–92. doi: 10.5267/j.ccl.2015.12.001. [DOI] [Google Scholar]
- 10.Maowa J., Alam A., Rana K.M., Dey S., Hosen A., Fujii Y., Hasan I., Ozeki Y., Kawsar S.M.A. Synthesis, characterization, synergistic antimicrobial properties and molecular docking of sugar modified uridine derivatives. Ovidius Univ. Ann. Chem. 2021;32:6–21. doi: 10.2478/auoc-2021-0002. [DOI] [Google Scholar]
- 11.Bulbul M.Z.H., Chowdhury T.S., Misbah M.M.H., Ferdous J., Dey S., Hasan I., Fujii Y., Ozeki Y., Kawsar S.M.A. Synthesis of new series of pyrimidine nucleoside derivatives bearing the acyl moieties as potential antimicrobial agents. Pharmacia. 2021;68:23–34. doi: 10.3897/pharmacia.68.e56543. [DOI] [Google Scholar]
- 12.Arifuzzaman M., Islam M.M., Rahman M.M., Mohammad A.R., Kawsar S.M.A. An efficient approach to the synthesis of thymidine derivatives containing various acyl groups: characterization and antibacterial activities. ACTA Pharm. Sci. 2018;56:7–22. doi: 10.23893/1307-2080.APS.05622. [DOI] [Google Scholar]
- 13.Mirajul M.I., Arifuzzaman M., Monjur M.R., Atiar M.R., Kawsar S.M.A. Novel methyl 4,6-O-benzylidene-α-D-glucopyranoside derivatives: synthesis, structural characterization and evaluation of antibacterial activities. Hacettepe J. Biol. Chem. 2019;47:153–164. doi: 10.15671/hjbc.622038. [DOI] [Google Scholar]
- 14.Kawsar S.M.A., Hasan T., Chowdhury S.A., Islam M.M., Hossain M.K., Mansur M.A. Synthesis, spectroscopic characterization and in vitro antibacterial screening of some D-glucose derivatives. Int. J. Pure App Chem. 2013;8:125–135. [Google Scholar]
- 15.Misbah M.M.H., Ferdous J., Bulbul M.Z.H., Chowdhury T.S., Dey S., Hasan I., Kawsar S.M.A. Evaluation of MIC, MBC, MFC and anticancer activities of acylated methyl β-D-galactopyranoside esters. Int. J. Biosci. 2020;16:299–309. doi: 10.12692/ijb/16.4.299-309. [DOI] [Google Scholar]
- 16.A. Alam, M. A. Hosen, M. Islam, J. Ferdous, Y. Fujii, Y. Ozeki. S. M. A. Kawsar, Synthesis, Antibacterial and cytotoxicity assessment of modified uridine molecules. Curr. Adv. Chem. Biochem., 6(2021) 114-129, doi.org/10.9734/bpi/cacb/v6/8670D.
- 17.Kawsar S.M.A., Kumar A. Computational investigation of methyl α-D-glucopyranoside derivatives as inhibitor against bacteria, fungi and COVID-19 (SARS-2) J. Chil. Chem. Soci. 2021;66:5206–5214. [Google Scholar]
- 18.Kawsar S.M.A., Faruk M.O., Rahman M.S., Fujii Y., Ozeki Y. Regioselective synthesis, characterization, and antimicrobial activities of some new monosaccharide derivatives. Scientia Pharm. 2014;82:1–20. doi: 10.3797/scipharm.1308-03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Y. Farhana, M. R. Amin, M. A. Hosen, M. Z. H. Bulbul, S. Dey, S. M. A. Kawsar Monosaccharide derivatives: Synthesis, antimicrobial, PASS, antiviral, and molecular docking studies against sars-cov-2 mpro inhibitors. J. Cellul. Chem. Technol., 55(2021), 477-499, doi.org/10.17516/1998-2836-0226.
- 20.Kawsar S.M.A., Hosen M.A., Fujii Y., Ozeki Y. Thermochemical, DFT, molecular docking and pharmacokinetic studies of methyl β-D-galactopyranoside esters. J. Comput. Chem. Mol. Model. 2020;4:452–462. doi: 10.25177/JCCMM.4.4.RA.10663. [DOI] [Google Scholar]
- 21.L. L. Ching, G. Edward, F. L. Yun, C.W. Hsu, M. D. S. Thongsawat, Telbivudine versus Lamivudine in patients with Chronic Hepatitis B. N. Engl. J. Med., 357(2007), 2576–2588, doi.org./10.1056/NEJMoa066422. [DOI] [PubMed]
- 22.Rana K.M., Ferdous J., Hosen A., Kawsar S.M.A. Ribose Moieties Acylation and Characterization of Some Cytidine Analogs. J. Siberian Fed. Univ. Chem. 2020;13:465–478. doi: 10.17516/1998-2836-0199. [DOI] [Google Scholar]
- 23.Alam A., Hosen M.A., Hosen A., Fujii Y., Ozeki Y., Kawsar S.M.A. Synthesis, characterization, and molecular docking against a receptor protein FimH of Escherichia coli (4XO8) of Thymidine Derivatives. J. Mex. Chem. Soc. 2021;65:256–276. doi: 10.29356/jmcs.v65i2.1464. [DOI] [Google Scholar]
- 24.Devi S.R., Jesmin S., Rahman M., Manchur M.A., Fujii Y., Kanaly R.A., Ozeki Y., Kawsar S.M.A. Microbial efficacy and two step synthesis of uridine derivatives with spectral characterization. Acta Pharm. Sci. 2019;57:47–68. doi: 10.23893/1307-2080.APS.05704. [DOI] [Google Scholar]
- 25.M. A. Hosen, A. Alam, M. Islam, Y. Fujii, Y. Ozeki, S. M. A. Kawsar, Geometrical optimization, PASS prediction, molecular docking, and in silico ADMET studies of thymidine derivatives against FimH adhesin of Escherichia coli. Bulg. Chem. Commun. 53(2021), 327-342, doi.org./10.34049/bcc.53.3.5375.
- 26.Kawsar S.M.A., Hosen M.A. An optimization and pharmacokinetic studies of some thymidine derivatives. Turkish Comp. Theo. Chem. 2020;4:59–66. doi: 10.33435/tcandtc.718807. [DOI] [Google Scholar]
- 27.Kabir A.K.M.S., Matin M.M., Kawsar S.M.A., Anwar M.N. Antibacterial activities of some selectively acylated uridine derivatives. Chittagong Univ. J. Sci. 1998;22:37–41. [Google Scholar]
- 28.A. Alam, K. M. Rana, M. A. Hosen, S. Dey, B. Bezbaruah, S. M. A. Kawsar, Modified thymidine derivatives as potential inhibitors of SARS-CoV: PASS, in vitro antimicrobial, physicochemical and molecular docking studies. Phys. Chem. Res., 10(2022), 391-409, doi.org./10.22036/PCR.2022.317494.1996.
- 29.Kawsar S.M.A., Hosen M.A., Alam A., Islam M., Ferdous J., Fujii Y., Ozeki Y. Thymidine Derivatives as Inhibitors against Novel Coronavirus (SARS-CoV-2) Main Protease: Theoretical and Computational Investigations. Adv. Chem. Res. 2021;69:89–129. [Google Scholar]
- 30.Maowa J., Hosen M.A., Alam A., Rana K.M., Fujii Y., Ozeki Y., Kawsar S.M.A. Pharmacokinetics and Molecular Docking Studies of Uridine Derivatives as SARS- COV-2 Mpro Inhibitors. Phys. Chem. Res. 2021;9:385–412. doi: 10.22036/pcr.2021.264541.1869. [DOI] [Google Scholar]
- 31.Bulbul M.Z.H., Hosen M.A., Ferdous J., Misbah M.M.H., Kawsar S.M.A. Thermochemical, DFT study, physicochemical, molecular docking and ADMET predictions of some modified uridine derivatives. Int. J. New Chem. 2021;8:88–110. doi: 10.22034/ijnc.2020.131337.1124. [DOI] [Google Scholar]
- 32.Y. Farhana, M.R Amin, A. Hosen, S. M. A. Kawsar, Bromobenzoylation of methyl α-D-mannopyranoside: synthesis and spectral characterization. J. Sib. Fed. Univ. Chem., 14(2021), 171-183, doi.org./10.17516/1998-2836-0226.
- 33.Clinical Laboratory Standards Institute (CLSI). (2012). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard-Ninth Edition. Wayne, PA: Clinical and Laboratory Standards Institute. CLSI Documents M07-A9.
- 34.Hunt W.A. The effects of aliphatic alcohols on the biophysical and biochemical correlates of membrane function. Adv. Exp. Med. Biol. 1975;56:195–210. doi: 10.1007/978-1-4684-7529-6_9. [DOI] [PubMed] [Google Scholar]
- 35.Kim Y.M., Farrah S., Baney R.H. Structure–antimicrobial activity relationship for silanols, a new class of disinfectants, compared with alcohols and phenols. Int. J. Antimicrob. Agents. 2007;29:217–222. doi: 10.1016/j.ijantimicag.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 36.S. Kumaresan, V. Senthilkumar, A. Stephen, B. S. Balakumar, GC-MS analysis and PASS-assisted prediction of biological activity spectra of extract of Phomopsis sp. Isolated from Andrographis paniculata. World J. Pharm. Res., 4(2015), 1035-1053.
- 37.Seeberger P.H., Werz D.B. Synthesis and medical applications of oligosaccharides. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- 38.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, M. Mennucci, G.A. Petersson et al. Gaussian 09. Gaussian Inc, Wallingford, CT, 2009. doi.org/gaussian.com/g09citation/.
- 39.Becke A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 40.Lee C., Yang W., Parr R.G. Development of the colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 41.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Delano W.L. De-Lano Scientific; San Carlos, CA, USA: 2002. The PyMOL molecular graphics system. [Google Scholar]
- 43.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 44.C. H. Williams, C. C. (Eds.). Hong, Chemical biology: methods and protocols, Springer, New York, USA, (2015), .
- 45.Version ADS 4.0. Accelrys, San Diego, USA, 2017.
- 46.A. Adji, N. J. Niode, V. V. Memah, J. Posangi, G. J. P. Wahongan, Y. Ophinni, R. Idroes, S. Mahmud, T. Bin. Emran, F. Nainu, T. E.Tallei, H. Harapan, Designing an epitope vaccine against Dermatophagoides pteronyssinus: An in silico study. Acta Trop., 222(2021),106028, doi.org/10.1016/j.actatropica.2021.106028. [DOI] [PubMed]
- 47.Dash R., Ali M.C., Dash N., Azad M.A.K., Hosen S.M.Z., Hannan M.A., Moon I.S. Structural and dynamic characterizations highlight the deleterious role of SULT1A1 R213H polymorphism in substrate binding. Int. J. Mol. Sci. 2019;20:6256. doi: 10.3390/ijms20246256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dutta M., Tareq A.M., Rakib A., Mahmud S., Sami S.A., Mallick J., Islam M.N., Majumder M., Uddin M.Z., Alsubaie A., Almalki A.S.A., Khandaker M.U., Bradley D.A., Rana M.S., Bin T. Emran, phytochemicals from Leucas zeylanica targeting main protease of SARS-CoV-2: chemical profiles, molecular docking, and molecular dynamics simulations. Biology (Basel) 2021;10:789. doi: 10.3390/biology10080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 50.Harrach M.F., Drossel B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014;140 doi: 10.1063/1.4872239. [DOI] [PubMed] [Google Scholar]
- 51.Krieger E., Vriend G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015;36:996–1007. doi: 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Land H., Humble M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. Methods Mol. Biol. 2018;1685:43–67. doi: 10.1007/978-1-4939-7366-8_4. [DOI] [PubMed] [Google Scholar]
- 53.S. Mahmud, S. Biswas, G. K. Paul, M. A. Mita, S. Afrose, M. R. Hasan, M. S. S. Shimu, M. A. R. Uddin, M. S. Uddin, S. Zaman, K. M. K. Kibria, M. A. Khan, T. Bin. Emran, M. A. Saleh, Antiviral peptides against the main protease of SARS-CoV-2: A molecular docking and dynamics study. Arab. J. Chem., 14(2021), 103315, doi.org/10.1016/j.arabjc.2021.103315. [DOI] [PMC free article] [PubMed]
- 54.S. Mahmud, S. Biswas, G. K. Paul, M. A. Mita, M. M. Promi, Afrose, M. R. Hasan, S. Zaman, M. S. Uddin, K. Dhama, T. Bin. Emran, M. A. Saleh, J. Simal-Gandara, Plant-based phytochemical screening by targeting main protease of sars-cov-2 to design effective potent inhibitors. Biology (Basel)., 10(2021), 589, doi.org/10.3390/biology10070589. [DOI] [PMC free article] [PubMed]
- 55.S. Mahmud, M. A. Mita, S. Biswas, G. K. Paul, M. M. Promi, Afrose, M. R. Hasan, M. S. S. Shimu, S. Zaman, S. Uddin, T. E. Tallei, T. Bin. Emran, M. A. Saleh, Molecular docking and dynamics study to explore phytochemical ligand molecules against the main protease of SARS-CoV-2 from extensive phytochemical datasets. Expert Rev. Clin. Pharmacol., 14(2021), 1305-1315, doi.org/10.1080/17512433.2021.1959318. [DOI] [PubMed]
- 56.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 57.Pires D.E.V., Blundell T.L., Ascher B.D. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Isono K. Nucleoside antibiotics: structure, biological activity, and biosynthesis. J. Antibiot. 1988;41:1711–1739. doi: 10.7164/antibiotics.41.1711. [DOI] [PubMed] [Google Scholar]
- 59.Saini M.S., Kumar A., Dwivedi J., Singh R., Review A. Biological Significances of Heterocyclic Compounds. Int. J. Pharm. Sci. Res. 2013;4:66–77. [Google Scholar]
- 60.Li W.R., Xie X.B., Shi Q.S., Zeng H.Y., Ou-Yang Y.S., Chen Y.B. Antibacterial activity and mechanism of silver nanoparticles on Escherichia coli. Appl. Microbiol. Biotechnol. 2010;85:1115–1122. doi: 10.1007/s00253-009-2159-5. [DOI] [PubMed] [Google Scholar]
- 61.V. Judge, B. Narasimhan, M. Ahuja, D. Sriram, P. Yogeeswari, E. D. Clercq, C. Pannecouque, J. Balzarini, Synthesis, antimycobacterial, antiviral, antimicrobial activity and QSAR studies of N(2)-acyl isonicotinic acid hydrazide derivatives. Med.l Chem., 9(2013), 53–76, doi.org/10.2174/157340613804488404. [DOI] [PubMed]
- 62.Cohen N., Benson S.W. Estimation of heats of formation of organic compounds by additivity methods. Chem. Rev. 1993;93:2419–2438. doi: 10.1021/cr00023a005. [DOI] [Google Scholar]
- 63.Lien E.J., Guo Z.R., Li R.L., Su C.T. Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. J. Pharm. Sci. 1982;71:641–655. doi: 10.1002/jps.2600710611. [DOI] [PubMed] [Google Scholar]
- 64.S. Saravanan, V. Balachandran, Quantum chemical studies, natural bond orbital analysis and thermodynamic function of 2,5-dichlorophenylisocyanate. Spectrochim, Acta A Mol. Biomol. Spectrosc., 120(2014), 351-364, doi.org/10.1016/j.saa.2013.10.042. [DOI] [PubMed]
- 65.Amin M.L. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights. 2013;7:27–34. doi: 10.4137/DTI.S12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Politzer P., Murray J.S. Molecular electrostatic potentials and chemical reactivity. Rev. Comput. Chem. 1991;2:273–312. doi: 10.1002/wcms.1326. [DOI] [Google Scholar]
- 67.P. Politzer, D. G. Truhlar, (Eds.), Chemical applications of atomic and molecular electrostatic potentials. Reactivity, Structure, Scattering, and Energetics of Organic, Inorganic, and Biological Systems. Springer, USA, (1981), www.springer.com/gp/book/9780306406577.
- 68.X. Liu, X. J. Wang, Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genom., 7(2020), 119–121, doi.org./10.1016/j.jgg.2020.02.001. [DOI] [PMC free article] [PubMed]
- 69.C. A. Lipinski, F. Lombardo, B. W. Dominy, P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development. Adv. Drug Deliv. Rev., 46(2001), 3-25, doi.org./10.1016/s0169-409x(00)00129-0. [DOI] [PubMed]
- 70.Note: https://www.maroc.ma/fr/actualites/research-excellence-awards-une-vingtaine-de-travaux-de-recherche-et-dinnovation-primes.
- 71.M. Rbaa, A. Hichar, P. Dohare, E. H. Anouar, Y. Lakhrissi, B. Lakhrissi, M. Berredjem, F. Almalki, V. Rastija, M. Rajabi, T. Ben Hadda, A. Zarrou, Synthesis, Characterization, Biocomputational Modeling and Antibacterial Study of Novel Pyran Based on 8-Hydroxyquinoline. Arab. J. Sci. Eng., 46(2021), 5533–5542. doi.org/10.1007/s13369-020-05089-y.
- 72.A. R. Bhat, R. S. Dongre, F. A. Almalki, M. Berredjem, M. Aissaoui, R. Touzani, T. Ben Hadda, M. S. Akhter, Synthesis, Biological Activity and POM/DFT/Docking Analyses of Annulated Pyrano[2,3-d]pyrimidine Derivatives: Identification of Antibacterial and Antitumor Pharmacophore sites. Bioorg. Chem., 106(2021), 104480, doi.org/10.1016/j.bioorg.2020.104480. [DOI] [PubMed]
- 73.I. Grib, M. Berredjem, K. O. Rachedi, S. E. Djouad, S. Bouacida, R. Bahadi, T. S. Ouk, M. Kadri, T. Ben Hadda, B. Belhania, Novel N-sulfonylphthalimides: Efficient synthesis, X-ray characterization, spectral investigations, POM analyses, DFT computations and antibacterial activity. J. Mol. Struct., 1217(2020), 128423, doi.org/10.1016/j.molstruc.2020.128423.
- 74.J. Sheikh, T. Ben Hadda, Antibacterial, antifungal and antioxidant activity of some new water-soluble b-diketones. Med. Chem. Res., 22(2013), 964-975, doi.org./10.1007/s00044-012-0089-8.
- 75.Y. N. Mabkhot, M. Arfan, H. Zgou, Z. K. Genc, M. Genc, A. Rauf, S. Bawazeer, T. Ben Hadda, How to Improve Antifungal Bioactivity: POM and DFT Study of some Chiral Amides Derivatives of Diacetyl-L-tartaric Acid and Amines. Res. Chem. Intermed., 42(2016), 8055–8068, doi.org./10.1007/s11164-016-2578-8.
- 76.Y. N. Mabkhot, A. Alatibi, N. El-sayed, N. Kheder, A. Wadood, A. Rauf, S. Bawazeer, S. Al-Showiman, T. Ben Hadda, Experimental-Computational Evaluation of Antimicrobial Activity of Some Novel Armed Thiophene Derivatives. Molecules, 21(2016), 222, doi.org./10.3390/molecules21020222. [DOI] [PMC free article] [PubMed]
- 77.T. Ben Hadda, M. A. Ali, V. Masand, S. Gharby, T. Fergoug and I. Warad. Tautomeric Origin of Dual Effects of N1-nicotinoyl-3-(4’-hydroxy-3’-methyl phenyl)-5-[(sub)phenyl]-2-pyrazolines on Bacterial and Viral Strains: POM Analyses as New Efficient Bioinformatics' Platform to Predict and Optimize Bioactivity of Drugs. Med. Chem. Res., 22(2013), 1438–1449, doi.org./10.1007/s00044-012-0143-6.
- 78.K. T. Rachedi, T. -S. Ouk, R. Bahadi, A. Bouzina, S. -E. Djouad, K. Bechlem, R. Zerrouki, T. Ben Hadda, F. A. Almalki, M. Berredjem , Synthesis, DFT and POM analyses of cytotoxicity activity of α-amidophosphonates derivatives: Identification of potential antiviral O,O-pharmacophore site. J. Mol. Struct., 1197(2019), 196-203, doi.org/10.1016/j.molstruc.2019.07.053.
- 79.E. R. Esharkawy, F. A. Almalki, T. Ben Hadda, In vitro Potential Antiviral SARS-CoV-19- Activity of Natural Product Thy-mohydroquinone and Dithymoquinone from Nigella sativia. Bioorg. Chem., 120(2022), 105587, doi.org/10.1016/j.bioorg.2021.105587. [DOI] [PMC free article] [PubMed]
- 80.M. Berredjem, A. Bouzina, R. Bahadi, S. Bouacida, V. Rastija, S. - E. Djouad, T. O. Sothea, F. A. Almalki, T. Ben Hadda, M. Aissaoui, Antitumor activity, X-Ray crystallography, in silico study of some-sulfamido-phosphonates.Identification of pharmacophore sites. J. Mol. Struct., 1250(2022), 131886, https://doi.org/10.1016/j.molstruc.2021.131886.
- 81.A. Rauf, S. Bawazeer, M. Raza, E. El-Sharkawy, M. H. Rahman, M. A. EI-Esawi, G. Uddin, B. S. Siddiqui, A. A. Khalil, J. Molnar, Á. Csonka, D. Szabó, H. Khan, M. S. Mubarak,T. Ben Hadda, M. Kamaruddin, Reversal of multidrug resistance and antitumor promoting activity of 3-oxo-6β-hydroxy- β-amyrin isolated from Pistacia integerrima. Biocell, 45(2021), 139-147, doi.org./10.32604/biocell.2021.013277.
- 82.M. J. Waring, T. Ben Hadda, A. T. Kotchevar, A. Ramdani, R. Touzani, S. Elkadiri, A. Hakkou, M. Bouakka and T. Ellis, 2,3-Bifunctionalized Quinoxalines: Synthesis, DNA Interactions and Evaluation of Anticancer, Antituberculosis and Antifungal Activity. Molecules, 7(2002), 641-656, doi.org./10.3390/70800641.
- 83.Y. N. Mabkhot, A. Barakat, S. Yousuf, M. I. Choudhary, W. Frey, T. Ben Hadda, M. S. Mubarak, Substituted thieno[2,3-b]thiophenes and related congeners: Synthesis, <beta>-glucuronidase inhibition activity, crystal structure, and POM analyses. Bioorg. Med. Chem., 22(2014), 6715–6725, doi.org./10.1016/j.bmc.2014.08.014. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.