

Abstract
Wild‐type transthyretin amyloid cardiomyopathy (ATTRwt CM) is a more common disease than previously thought. Awareness of ATTRwt CM and its diagnosis has been challenged by its unspecific and widely distributed clinical manifestations and traditionally invasive diagnostic tools. Recent advances in echocardiography and cardiac magnetic resonance (CMR), non‐invasive diagnosis by bone scintigraphy, and the development of disease‐modifying treatments have resulted in an increased interest, reflected in multiple publications especially during the last decade. To get an overview of the scientific knowledge and gaps related to patient entry, suspicion, diagnosis, and systematic screening of ATTRwt CM, we developed a framework to systematically map the available evidence of (i) when to suspect ATTRwt CM in a patient, (ii) how to diagnose the disease, and (iii) which at‐risk populations to screen for ATTRwt CM. Articles published between 2010 and August 2021 containing part of or a full diagnostic pathway for ATTRwt CM were included. From these articles, data for patient entry, suspicion, diagnosis, and screening were extracted, as were key study design and results from the original studies referred to. A total of 50 articles met the inclusion criteria. Of these, five were position statements from academic societies, while one was a clinical guideline. Three articles discussed the importance of primary care providers in terms of patient entry, while the remaining articles had the cardiovascular setting as point of departure. The most frequently mentioned suspicion criteria were ventricular wall thickening (44/50), carpal tunnel syndrome (42/50), and late gadolinium enhancement on CMR (43/50). Diagnostic pathways varied slightly, but most included bone scintigraphy, exclusion of light‐chain amyloidosis, and the possibility of doing a biopsy. Systematic screening was mentioned in 16 articles, 10 of which suggested specific at‐risk populations for screening. The European Society of Cardiology recommends to screen patients with a wall thickness ≥12 mm and heart failure, aortic stenosis, or red flag symptoms, especially if they are >65 years. The underlying evidence was generally good for diagnosis, while significant gaps were identified for the relevance and mutual ranking of the different suspicion criteria and for systematic screening. Conclusively, patient entry was neglected in the reviewed literature. While multiple red flags were described, high‐quality prospective studies designed to evaluate their suitability as suspicion criteria were lacking. An upcoming task lies in defining and evaluating at‐risk populations for screening. All are steps needed to promote early detection and diagnosis of ATTRwt CM, a prerequisite for timely treatment.
Keywords: Amyloidosis, Cardiomyopathy, Transthyretin, ATTRwt CM, Suspicion, Diagnosis, Screening
Introduction
Once regarded a rare condition, wild‐type transthyretin amyloid cardiomyopathy (ATTRwt CM) is currently considered a more common cause of heart failure in the elderly. 1 The disease is characterized by progressive deposits of transthyretin (TTR) amyloid in the extracellular myocardial matrix, with development of an infiltrative cardiomyopathy that carries a poor prognosis. 2
Wild‐type transthyretin amyloid cardiomyopathy typically affects men over 60 years of age, though women can also be affected. Its genetic counterpart, variant ATTR (ATTRv) CM, is caused by mutations in the TTR gene, may appear at a younger age as compared with ATTRwt CM, and affects both genders equally. 3 This review will focus on ATTRwt CM.
The early stages of ATTRwt CM can manifest as heart failure with preserved ejection fraction (HFpEF) and may mimic hypertensive heart disease or hypertrophic cardiomyopathy, which increases the risk of misdiagnosis. 4 , 5 In more advanced stages, ATTRwt CM can present along other well‐known cardiac conditions like atrial fibrillation, atrioventricular conduction disorders, or aortic stenosis. 3
With the introduction of new effective treatment possibilities, 6 , 7 , 8 the relevance of identifying patients with ATTRwt CM has increased, as has the development and evaluation of non‐invasive tools to diagnose the disease. This enhanced interest has resulted in multiple publications on ATTRwt CM suspicion criteria, red flags, and diagnostic pathways over the last decade. As a natural next step in identifying and treating patients with ATTRwt CM, systematic screening of at‐risk subpopulations has also received some attention lately.
With this review, we seek to systematically describe the recent scientific literature dealing with suspicion, screening, and diagnosis of ATTRwt CM. This is performed with the purpose of analysing the scientific evidence underlying the available ATTRwt CM recommendations, thereby enabling a discussion of potential data gaps and needs to further substantiate optimal detection of patients affected by this detrimental disease.
Methods
To assess the literature on suspicion of and screening for ATTRwt as well as on diagnostic pathways for ATTRwt CM, we conducted a systematic literature review. Based on our clinical and professional research experience, we developed a new framework for analysing the resulting articles. This novel approach is described in the following section and illustrated in Figure 1 .
Figure 1.
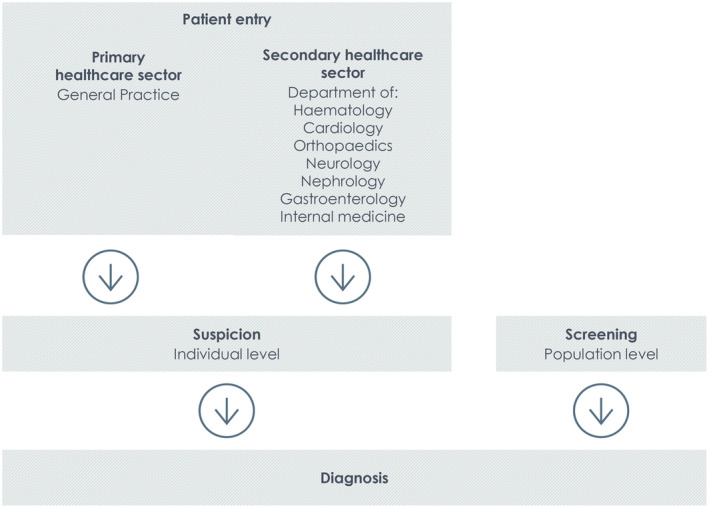
Analytical framework of the patient flow leading to a wild‐type transthyretin amyloid cardiomyopathy diagnosis.
Definitions and framework for the analyses
Patient entry is defined as the clinical setting in which suspicion of ATTRwt CM is first raised. As illustrated in Figure 1 , a patients' journey to an ATTRwt CM diagnosis can begin with a referral from the general practitioner or be initiated from different departments in the secondary healthcare sector. Patient entry is a relevant parameter to investigate as it often reflects the reason for suspicion in the first place and might provide important clues on the degree of ATTRwt CM awareness in the different parts of the healthcare system.
Suspicion of ATTRwt CM is defined as the physician's substantiated conjecture that an individual patient may have ATTRwt CM. Suspicion of ATTRwt CM is raised by a clinical evaluation initiated because of patient symptoms, abnormal imaging findings (which may be accidental) or cardiac biomarker levels, a relevant medical history, or a combination of these.
Screening for ATTRwt CM is defined as a systematic examination for ATTRwt CM in a population of patients defined as being at increased risk of ATTRwt CM. The screening entry criteria can be based on the presence of certain factors such as previous medical history, biomarkers, imaging findings, demographics, or a combination of these.
Diagnosis of ATTRwt CM is defined as the specific examinations recommended or required to confirm or reject a diagnosis of ATTRwt CM, including the order of these examinations and relevant thresholds or cut‐off levels.
Thus, while suspicion relates to the clinician's assessment of the individual patient based on clinical findings and the patient's symptoms and medical history, screening is a systematic assessment of a patient population who meet some predefined entry criteria. Figure 1 illustrates the analytical framework developed to analyse the articles included in this review.
Databases and search strategy
The two electronic databases Embase and MEDLINE were searched using the PubMed platform. The searches were conducted in September 2021 and covered the period from 2010 to August 2021. This systematic review was carried out following the principles of the PRISMA guidelines. 9
The search string was constructed of two clusters covering search terms relating to the ATTRwt CM population and diagnostic guideline terms, respectively (see Supporting Information, Table S1 , for the search strings tailored to the applied databases). References cited in recently published articles or other relevant articles were screened to confirm that the search captured all relevant articles.
Review process and eligibility criteria
Title and abstracts of all identified articles were reviewed for eligibility. Eligible articles were defined by a set of inclusion and exclusion criteria. Eligible articles were proceeded to full‐text review, and articles failing to meet the inclusion criteria were excluded. The review process was performed independently by two investigators. In the event of disagreement about the eligibility of an article, consensus was reached through discussion with a third investigator.
Inclusion criteria were as follows: articles describing a part of or a full diagnostic pathway, including but not limited to suspicion, screening, or diagnosis of ATTRwt CM. Articles were considered to report a diagnostic pathway if they graphically or visually presented an order of tests recommended to diagnose ATTRwt CM. Exclusion criteria were as follows: (i) articles not written in English and (ii) case reports or conference abstracts.
Data extraction
Extracted data included general information about the articles, information about suspicion and screening of ATTRwt CM (if addressed), information about clinical testing and diagnosing of ATTRwt CM, thresholds or cut‐off levels, and origin of the pathway (Table 1 ). Extracted suspicion criteria were not limited to symptoms or findings defined in the articles as suspicion criteria but included all symptoms or findings that could be interpreted as suspicion criteria, such as ‘red flags’, ‘clinical presentation’, ‘clinical clues’, ‘clinical features’, ‘common symptoms’, or ‘typical manifestations’.
Table 1.
Data extraction categories and variables
| Category | Variables |
|---|---|
| General information |
Publication year Country of research Journal Type of article (review, original research) Objective Methodology Main results Conclusion |
| Patient entry | Healthcare sector of initial contact |
| Suspicion |
Is suspicion addressed? (yes or no) Nature of suspicion criteria (e.g. specific symptoms, clinical findings, and medical history) Characteristics of suspicion criteria (e.g. their cut‐off values and evidence level) |
| Screening |
Is screening addressed? (yes or no) Patient populations suggested for screening Underlying evidence for suggested screening |
| Clinical testing and diagnosing |
Cardiac investigations and thresholds Diagnostic tools in suggested order Definite diagnose/rule out of ATTRwt Underlying evidence for suggested diagnostic elements |
| Origin of pathway | New pathway or modified from already published pathway |
To further evaluate the level of evidence underlying the suspicion, screening, and diagnosis of ATTRwt CM, relevant data from original prospective or retrospective studies referred to in the included articles were extracted. Only original studies including more than 20 patients with ATTRwt CM were included.
Data analysis
A descriptive analysis was performed to give a historical overview of the development of the published literature describing diagnostic pathways for ATTRwt CM. Pathways in the included articles were assessed in four subanalyses: (i) patients' entrance into the diagnostic pathways, (ii) suspicion of ATTRwt CM, (iii) systematic screening for ATTRwt CM, and (iv) establishment of the ATTRwt CM diagnosis. The analyses comprised a quantitative summary of extracted data and a qualitative assessment.
Results
From the two searches, 1706 articles were identified. Following removal of duplicates, 1318 articles were screened on titles and abstracts. Of these, 1225 articles were excluded, as they did not meet the inclusion criteria. A total of 93 articles were fully reviewed and assessed for eligibility. Finally, 48 articles describing a diagnostic pathway for ATTRwt were included in this systematic literature review. Two additional relevant articles 4 , 10 not captured in the primary literature search were included, resulting in a total of 50 articles (Figure 2 ).
Figure 2.
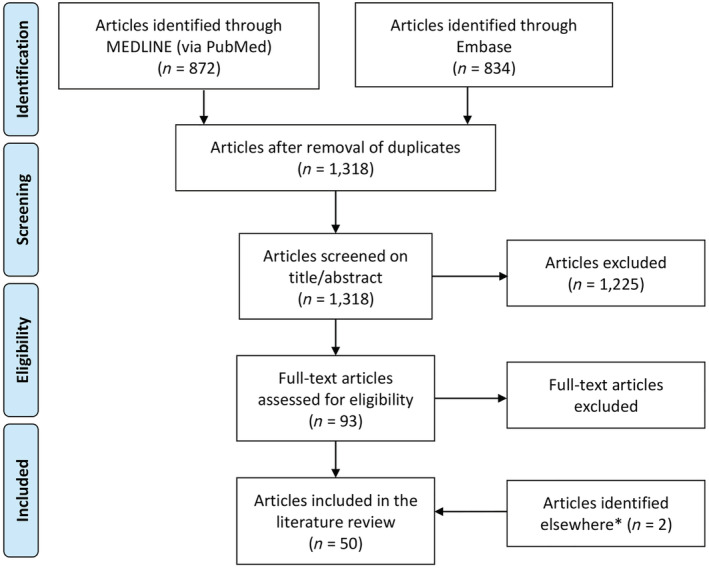
PRISMA chart. *Identified in connection with the analysis of the included articles.
Characteristics of the included articles
Among the 50 articles containing a diagnostic pathway for ATTRwt CM, 36 articles were review studies. 4 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 The 14 remaining articles were consensus recommendation papers, 2 , 5 , 45 , 46 , 47 , 48 , 49 , 50 , 51 cohort studies, 52 , 53 , 54 , 55 , 56 or guidelines. 57 The majority of the identified articles were not country or regional specific.
Eighteen articles focused specifically on ATTRwt CM or on ATTRwt CM plus ATTRv CM (in the following referred to as ATTR‐CM), 4 , 5 , 10 , 11 , 13 , 17 , 19 , 21 , 25 , 32 , 35 , 37 , 39 , 40 , 45 , 49 , 52 , 53 while the rest covered CA in general, including ATTR‐CM.
Historical development
We found an overall increase in the rate of published articles suggesting a diagnostic pathway for ATTRwt CM from 2010 to August 2021, particularly since 2019 when also articles reproducing an already published diagnostic pathway started to appear (Figure 3 ).
Figure 3.
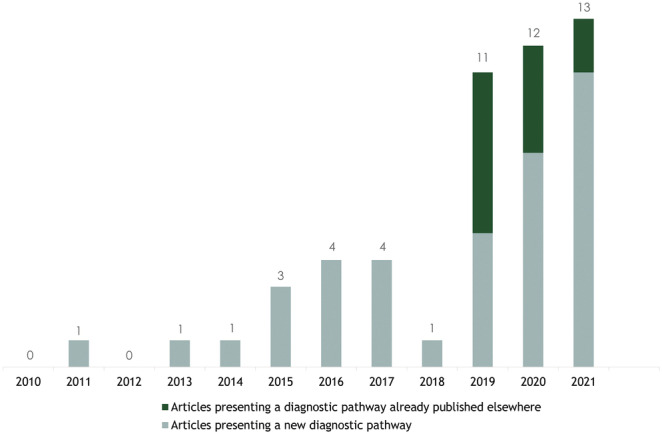
Number of articles published between 2010 and November 2020 presenting a diagnostic pathway for wild‐type transthyretin amyloid cardiomyopathy. Please note that the numbers of articles add up to 51 because the article by Dorbala et al. is divided into two publications. 47 , 48
Analyses of the diagnostic pathways
Patients' entrance into the diagnostic pathways
Of the 50 identified articles for ATTRwt CM, three explicitly mentioned the importance of the primary care providers. 5 , 28 , 50 The remaining 47 articles did not report the initial contact with the healthcare system. The speciality areas of the journals publishing the included articles can be considered an indicator of the clinical societies focusing on ATTRwt CM patients. Notably, 43 of 50 articles were published in a journal within cardiology or haematology, 2 , 4 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 40 , 41 , 42 , 43 , 45 , 46 , 47 , 48 , 49 , 51 , 52 , 53 , 54 , 55 , 56 , 57 while the remaining seven articles were published in papers of general or internal medicine 5 , 29 , 30 , 31 , 39 , 50 or oncology. 44 No articles were published in journals within neurology or orthopaedics.
Suspicion of wild‐type transthyretin amyloid cardiomyopathy
All included articles, except three, 52 , 54 , 55 described an initial suspicion of ATTRwt CM based on clinical presentation and red flags. Table 2 lists the typical suspicion criteria as described in the identified articles, divided into cardiac symptoms/diagnoses, extracardiac manifestations, cardiac biomarkers, and findings based on electrocardiography (ECG), echocardiography (ECHO), and cardiac magnetic resonance (CMR). Most of the suspicion criteria did not show any historical trend in that they appeared throughout the investigated period. Exceptions were, however, the apical sparing global longitudinal strain pattern on ECHO, which was not described in the included articles until 2015, 11 , 29 , 34 and aortic stenosis and lumbar spinal stenosis, which were not included as suspicion criteria until 2016 32 and 2017, 13 respectively.
Table 2.
Frequency of the most prevalent suspicion criteria/red flags mentioned in the identified articles
| Clinical symptoms and medical history | |
|---|---|
| Symptoms of heart failure | 34 |
| HFpEF | 30 |
| Carpal tunnel syndrome | 42 |
| Lumbar spinal stenosis | 29 |
| Neuropathy (peripheral or autonomic) | 35 |
| ECG findings | |
|---|---|
| Discordance between QRS voltage on ECG and wall thickness on ECHO | 32 |
| Atrial fibrillation | 24 |
| Low QRS voltage | 30 |
| Conduction abnormality (AV block, left and right bundle block) | 25 |
| Pseudo‐infarction pattern | 27 |
| ECHO findings | |
|---|---|
| Ventricular wall thickening | 44 |
| Apical sparing global longitudinal strain pattern | 36 |
| Small pericardial effusion | 28 |
| Reduced global longitudinal strain | 28 |
| Biatrial enlargement/dilatation | 27 |
| Granular sparkling appearance of myocardium | 25 |
| Aortic stenosis | 23 |
| CMR findings | |
|---|---|
| Late gadolinium enhancement | 43 |
| Increased extracellular volume | 36 |
| Elevated native T1 mapping sequences | 33 |
| Diffuse subendocardial or transmural late gadolinium enhancement | 30 |
| Cardiac biomarkers | |
|---|---|
| Abnormal troponin levels | 26 |
| Abnormal NT‐proBNP level | 21 |
AV, atrioventricular; CMR, cardiac magnetic resonance imaging; ECG, electrocardiography; ECHO, echocardiography; HFpEF, heart failure with preserved ejection fraction; NT‐proBNP, N‐terminal pro‐brain natriuretic peptide.
Evaluation of the evidence for selected suspicion criteria
The most prevalent suspicion criteria for each of the categories listed in Table 2 were evaluated in terms of underlying evidence, except for ‘Symptoms of heart failure’ due to its diffuse expression and its intrinsic linkage to ATTRwt CM. Original studies referred to and relevant results are listed in Tables 3 and 4 and outlined in short in the succeeding text.
Table 3.
Original studies referred to in relation to suspicion criteria in the form of symptoms and diagnoses
| Suspicion criteria | Study type | |||
|---|---|---|---|---|
| Prospective | Retrospective | Autopsy | Registry | |
| HFpEF |
ATTRwt CM in 13% of patients admitted with HFpEF and vwt ≥ 12 mm 1 ATTRwt CM in 18% of patients >65 years with HFpEF 58 ATTRwt CM in 6% of patients with HFpEF as evaluated by myocardial biopsy analysis 59 |
Broad description of ejection fraction in patients with ATTRwt CM 60 | 5% of patients with HFpEF had ATTRwt CM as the primary reason for HFpEF 61 | |
| Carpal tunnel syndrome |
TTR amyloid in tenosynovial tissue from patients undergoing CTS surgery was found in 34% (no check for amyloid in the heart) 62 TTR amyloid in tenosynovial tissue from patients undergoing CTS surgery was found in 10.2% (cardiac involvement confirmed in one patient) 63 Confirmed ATTRwt CM was reported in 0.85% of patients undergoing CTS surgery 64 |
CTS prevalence of 20–60% reported in patients with ATTRwt CM 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 | Risk of ATTRwt CM 12× increased in patients undergoing CTS surgery 75 | |
| Lumbar spinal stenosis |
TTR amyloid found in 33% of LSS surgery samples, no check for cardiac symptoms 76 TTR amyloid found in 45% of LSS surgery samples, no cardiac symptoms found 77 |
14% of patients with ATTRwt CM had a history of LSS 67 | ||
| Neuropathy |
Of ATTRwt CM patients, 12% had neuropathic pain, 12% had numbness, and 13% had tingling CM 65 , 78 12% of patients with ATTRwt MS had autonomic neuropathy manifesting as orthostasis 70 |
|||
| Atrial fibrillation | Atrial fibrillation was reported in 27–67% patients with ATTRwt CM 3 , 60 , 66 , 70 , 79 , 80 | |||
| Aortic stenosis |
ATTRwt CM was reported in 6% of patients undergoing SAVR 81 ATTRwt CM was reported in 12–16% in patients undergoing TAVR 82 , 83 , 84 ATTRwt CM was reported in 12% of patients undergoing SAVR or TAVR 85 |
|||
ATTRwt CM, wild‐type transthyretin amyloid cardiomyopathy; CTS, carpal tunnel syndrome; HFpEF, heart failure with preserved ejection fraction; LSS, lumbar spinal stenosis; SAVR, surgical aortic valve replacement; TAVR, transcatheter aortic valve replacement; TTR, transthyretin.
Table 4.
Original studies referred to in relation to suspicion criteria in the form of imaging findings and cardiac biomarkers
| Imaging technique/biomarker | Study type | ||
|---|---|---|---|
| Prospective | Descriptive | Comparative | |
| ECG | Applicability of ECG and ECHO in diagnosing CA, not specifically ATTRwt CM 86 | Description of ATTRwt CM in specific 3 |
Comparison of ECG in different types of CA 79 , 87 , 88 , 89 , 90 Comparison between CA and other diseases 91 |
| Echocardiography | Description of ATTRwt CM in specific 3 |
Comparison of ECHO in different types of CA 88 , 92 , 93 , 94 , 95 Comparison between CA and other diseases or controls 96 , 97 , 98 , 99 , 100 |
|
| CMR |
Comparison of CMR between CA and other cardiac diseases 101 , 102 , 103 , 104 , 105 Comparison of CMR between different types of CA 106 , 107 , 108 , 109 |
||
| Cardiac biomarkers | Comparison of high‐sensitivity cardiac troponin T levels between CA and other causes of cardiac hypertrophy 110 | ||
ATTRwt CM, wild‐type transthyretin amyloid cardiomyopathy; CA, cardiac amyloidosis; CMR, cardiac magnetic resonance; ECG, electrocardiography; ECHO, echocardiography.
Heart failure with preserved ejection fraction
Thirty articles 2 , 3 , 5 , 11 , 12 , 16 , 18 , 19 , 20 , 21 , 22 , 23 , 25 , 26 , 27 , 32 , 33 , 34 , 36 , 37 , 40 , 41 , 42 , 44 , 45 , 46 , 47 , 48 , 49 , 56 , 57 mentioned HFpEF as a suspicion criterion for wild‐type ATTRwt CM, or at least for CA. Prospective studies referred to reported between 6% and 18% of patients with HFpEF to have ATTRwt. 1 , 58 , 59 Some articles 10 , 13 , 32 , 33 , 34 , 47 , 48 raised the awareness that while HFpEF can be a sign of ATTRwt CM, studies have reported 30–50% of patients with ATTRwt CM to have heart failure with reduced ejection fraction (HFrEF). 60 , 111
Carpal tunnel syndrome
All, except eight 15 , 26 , 31 , 52 , 53 , 54 , 55 articles, mentioned carpal tunnel syndrome (CTS) as a common and very early finding in patients with ATTRwt. Three prospective studies investigated the frequency of amyloid and/or ATTRwt CM in patients undergoing surgery for CTS. 62 , 63 , 64 One registry study showed that while patients undergoing CTS surgery have a low risk of being diagnosed with amyloidosis (0.10% incidence within 10 years of surgery), the risk was still 12 times higher than for people without CTS. 75
Lumbar spinal stenosis
Sixteen articles 2 , 4 , 5 , 13 , 14 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 36 , 37 , 38 , 39 , 40 , 42 , 43 , 44 , 45 , 46 , 49 , 50 , 51 , 56 , 57 mentioned lumbar spinal stenosis (LSS) as a disease often associated to ATTRwt CM. Original studies referred to included two relatively small prospective studies investigating for amyloid in ligamentum flavum samples from patients undergoing LSS surgery and reporting the presence of TTR amyloid in, respectively, 33% and 45% of the samples. 76 , 77
Neuropathy
Autonomic and peripheral neuropathy was included as a typical symptom in 35 articles. 2 , 4 , 5 , 10 , 11 , 14 , 15 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 27 , 28 , 29 , 30 , 31 , 33 , 34 , 37 , 39 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 56 , 57 Most articles, however, primarily discussed neuropathy in relation to ATTRv CM and, accordingly, only referred to ATTRv CM data. No original prospective studies were referred to that investigated the prevalence of ATTRwt CM in patients with neuropathy.
Atrial fibrillation
Twenty‐four articles mentioned atrial fibrillation as a typical feature of ATTRwt CM. 2 , 5 , 10 , 11 , 13 , 14 , 18 , 19 , 20 , 22 , 24 , 25 , 26 , 29 , 30 , 32 , 33 , 34 , 36 , 42 , 43 , 46 , 47 , 48 , 56 Original studies referred were all retrospective and documented a high occurrence (27–67%) of atrial fibrillation in ATTRwt CM patients. No studies were designed to investigate the prevalence of ATTRwt CM in patients with atrial fibrillation.
Aortic stenosis
Aortic stenosis was mentioned in 23 articles, 2 , 3 , 10 , 20 , 21 , 22 , 24 , 32 , 35 , 37 , 38 , 39 , 40 , 42 , 43 , 44 , 46 , 47 , 48 , 49 , 50 , 51 , 56 , 57 21 of which were published after 2019. The increased attention of aortic stenosis in patients with ATTRwt CM is based on the original studies referred to, which are all prospective and have been published in 2016 or later. 81 , 82 , 83 , 84 , 85 Two of the studies suggested that patients with concomitant ATTRwt CM and aortic stenosis had a worse prognosis. 81 , 84
Electrocardiography
The most typical ECG findings mentioned were atrial fibrillation (discussed earlier), conduction abnormalities, pseudo‐infarction pattern, low QRS voltage, and a discordance between QRS voltage on ECG and ventricular wall thickness on ECHO. Original studies referred to described ECG patterns in patients diagnosed with ATTRwt CM or compared ECG characteristics in the different types of CA or in CA and other cardiac diseases. None of the studies were designed to investigate the prevalence of ATTRwt CM in patients with specific ECG features. Notably, although a criterion like low voltage QRS was mentioned often, it was also characterized as a low sensitive criterion for ATTRwt CM and more typical of amyloid light‐chain (AL) amyloidosis. Articles referred to in this context were retrospective. 3 , 87 , 88
Echocardiography
Ventricular wall thickening was mentioned in all studies discussing ECHO findings. Other typical ECHO findings are listed in Table 2 . Original studies referred to described ECHO patterns in patients diagnosed with ATTRwt CM or compared characteristics in the different types of CA or in CA and other cardiac diseases. Conclusions from these studies were that ECHO, and especially apical sparing global longitudinal strain pattern, is indeed useful for raising the suspicion of CA. 96 , 97 , 98 , 99 There were no prospective studies evaluating the prevalence of ATTRwt CM in patients with specific ECHO features, the closest being a study investigating the prevalence of ATTRv CM in patients with a ventricular wall thickness ≥15 mm. 112
Cardiac magnetic resonance
The most typical CMR findings suggestive of CA are listed in Table 2 . Original studies referred to were comparative and investigated the use of CMR in distinguishing between CA and other cardiac diseases or between the different types of CA. Native myocardial T1 mapping and extracellular volume appear to be good indicators of ATTRwt CM. 101 , 102 , 103 One study found that a native T1 < 1,036 ms was associated with 98% negative predictive value for CA whereas a native T1 > 1,164 ms was associated with 98% positive predictive value for CA and argued that the application of these thresholds can limit the use of gadolinium contrast to those with intermediate T1 levels. 101 No prospective studies were referred to that evaluated the prevalence of ATTRwt CM in patients with specific CMR features.
Cardiac biomarkers—troponin and N‐terminal pro‐brain natriuretic peptide
Cardiac biomarkers in the form of troponin or N‐terminal pro‐brain natriuretic peptide (NT‐proBNP) were mentioned as a marker of suspicion in 13 papers. 4 , 5 , 11 , 12 , 13 , 21 , 22 , 24 , 32 , 33 , 45 , 46 , 49 Generally, troponin and NT‐proBNP are considered non‐specific markers of myocardial injury or heart failure, and only one article was referred to which investigated the value of high‐sensitivity cardiac troponin T levels in discriminating between CA and other causes of cardiac hypertrophy. 110 Rather, their role as prognostic markers for patients already diagnosed with ATTR‐CM was widely discussed, based primarily on three original studies. 60 , 113 , 114
Systematic screening for wild‐type transthyretin amyloid cardiomyopathy
Sixteen articles used wording regarding systematic screening for ATTRwt CM. 4 , 5 , 11 , 18 , 21 , 22 , 23 , 25 , 32 , 33 , 43 , 44 , 45 , 49 , 50 , 51 Most of these were published in 2019 or later. 4 , 5 , 18 , 21 , 23 , 33 , 43 , 44 , 49 , 50 , 51 Discussed themes included the worsening of disease outcomes associated with delayed diagnosis, the importance of systematic screening in enabling early disease identification and thereby maximizing the benefit of therapy, and the existing knowledge gaps that complicate the implementation of systematic screening. Ten of the articles suggested specific at‐risk patient populations who could be relevant for ATTRwt CM screening, such as elderly patients with neuropathy, patients with aortic stenosis, patients with established or suspected non‐cardiac amyloidosis, patients with monoclonal gammopathy, and older adults with heart failure and a history or symptoms of CTS, LSS, or spontaneous rupture of the distal biceps tendon. 4 , 5 , 11 , 18 , 21 , 23 , 25 , 33 , 50 , 51 Witteles et al. (2019) discussed in detail how to best screen for ATTRwt CM in everyday practice and specifically suggested, based on data, that men >65 years and women >70 years with heart failure or one or more specific red flags and a wall thickness ≥14 mm should be screened for ATTRwt CM. Likewise, in the position statement from the European Society of Cardiology (ESC), it was recommended that individuals with a wall thickness ≥12 mm and heart failure, aortic stenosis, or red flag symptoms should be screened for ATTRwt CM, especially if they are older than 65 years. 51 The red flags mentioned included many of the suspicion criteria listed in Table 2 , and the evidence underlying the specific suggestions overlapped substantially with the original studies described earlier. 4 , 51 Thus, while many articles discussed the need for systematic screening of certain populations, no prospective screening studies of at‐risk populations were referred to.
Establishment of the wild‐type transthyretin amyloid cardiomyopathy diagnosis
Essential diagnostic tools to reach a conclusive ATTRwt CM diagnosis include serological and in some cases bone marrow examinations to exclude AL amyloidosis, bone scintigraphy, myocardial biopsy, and genetic test. In alignment with this, one or more of these tools are included in all the diagnostic algorithms reviewed. Whether all types of tests are needed typically depends on the results of the other tests. Figure 4 provides an overview of the most prevalent test result combinations that according to the reviewed literature are required to reach a conclusive ATTRwt CM diagnosis.
Figure 4.
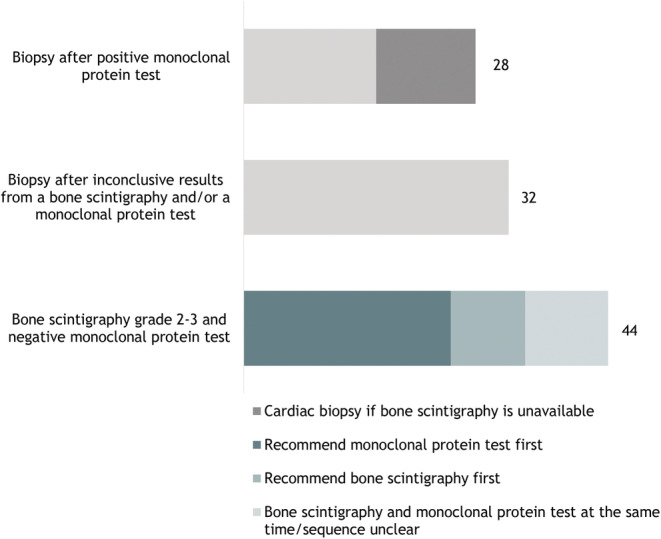
Overview of the frequency of diagnostic tool combinations suggested in the 50 articles included for review. The category ‘Biopsy after inconclusive results from a bone scintigraphy and/or monoclonal protein test’ consists of diagnostic pathways suggesting one or more of the following to diagnose ATTRwt CM: (i) bone scintigraphy grade 2–3, positive monoclonal protein test, and biopsy; (ii) bone scintigraphy grade 0/1, positive monoclonal protein test, and biopsy; and (iii) bone scintigraphy grade 0/1, negative monoclonal protein test, and biopsy.
While most published algorithms feature both a bone scintigraphy and tests to exclude AL amyloidosis, the order of these tests differs. Thus, 25 articles suggest to exclude AL amyloidosis before carrying out a bone scintigraphy, 5 , 10 , 12 , 14 , 16 , 17 , 18 , 19 , 21 , 22 , 23 , 24 , 25 , 29 , 32 , 35 , 36 , 37 , 41 , 42 , 44 , 45 , 46 , 49 , 56 while nine articles suggest the reverse order. 13 , 15 , 20 , 33 , 38 , 47 , 48 , 52 , 53 , 55 Whether a biopsy is also needed depends on the outcome of the other tests. Also, whether this should be an endomyocardial biopsy (EMB) or an extracardiac biopsy is discussed in many articles.
The history, diagnostic role, and underlying evidence for each of the tools are outlined in the succeeding text.
Exclusion of amyloid light‐chain amyloidosis
The relevance of excluding AL amyloidosis when diagnosing ATTRwt CM was reflected in the suggested diagnostic algorithms throughout the studied period, and all but three articles 26 , 31 , 54 included it as a part of the diagnostic pathway.
Especially, the 18 articles focusing on ATTR‐CM in specific 4 , 5 , 10 , 11 , 13 , 17 , 19 , 21 , 25 , 32 , 35 , 37 , 39 , 40 , 45 , 49 , 52 , 53 (rather than CA in general) underlined the importance of excluding AL amyloidosis and agreed that the required tests are immunofixation electrophoresis (IFE) of serum and urine and the serum free light‐chain (FLC) assay. Still, only few 25 , 39 , 44 , 45 , 51 , 53 referred to original studies providing evidence for using these tests. Original studies referred to include an article in which the serum reference intervals for free κ light chains, free λ light chains, and the κ/λ ratio were defined. Especially the latter is used in relation to exclusion of AL amyloidosis and is calculated to be 0.26–1.65 as based on measurement of 282 serum samples. 115 Also, two articles were referred to which investigated new mass spectrometry (MS)‐based methodologies that may eliminate the need for FLC assays in the future. 116 , 117 Finally, in connection with discussing the tests required to exclude AL amyloidosis, awareness of monoclonal gammopathy of undetermined significance (MGUS) was often mentioned, because patients with MGUS present with abnormal values in the tests used to exclude AL amyloidosis. The original study primarily referred to in this connection reported an MGUS prevalence of 39% in patients with ATTR‐CM. 118
Bone scintigraphy
The earliest articles did not include the use of bone scintigraphy in their diagnostic algorithm. 28 , 30 However, since 2014, the method has gained ground and was included in all reviewed articles beginning with Hutt et al. (2014). 52
The evidence underlying the use of bone scintigraphy came from multiple studies that have evaluated the applicability of different technetium (99mTc)‐labelled radiotracers in diagnosing ATTR‐CM. The studied radiotracers referred to in the included articles are 99mTc‐3,3‐diphosphono‐1,2‐propanodicarboxylic acid (99mTc‐DPD), 99mTc‐pyrophosphate (99mTc‐PYP), and 99mTc‐hydroxymethylene diphosphonate (99mTc‐HMDP). Retrospective studies were referred to which all reported that 99mTc‐DPD had a high sensitivity for cardiac amyloid in patients with ATTR‐CM, 119 , 120 , 121 , 122 as had 99mTc‐PYP 123 , 124 , 125 and 99mTc‐HMDP. 126 , 127 Importantly, prospective studies reached similar results, 52 , 53 , 54 , 128 and in line with the individual studies, a meta‐analysis from 2018 129 referred to concluded that bone scintigraphy using 99mTc‐labelled radiotracers provides very high diagnostic accuracy in the assessment of ATTR‐CM. Of note, clinical studies suggesting that bone scintigraphy can detect cardiac amyloid at a very early stage were also referred to. 126 , 130 , 131
There was a general consensus among the included articles to use the visual scoring system developed by Perugini et al. (2005) 119 and ranging from 0 (absent cardiac uptake and normal bone uptake) to 3 (strong cardiac uptake with mild/absent bone uptake) to evaluate the results of the bone scintigraphy, with a score ≥2 suggesting an ATTR‐CM diagnosis. By use of the Perugini scoring system, the largest prospective study referred to reported a sensitivity of bone scintigraphy for cardiac TTR amyloid of >99%. 53 Meanwhile, the specificity was somewhat lower (86%), which could be explained by the cardiac uptake of 99mTc‐labelled radiotracers in some patients with AL amyloidosis. This underlines the importance of combining bone scintigraphy with tests to exclude AL amyloidosis, and by doing this, Gillmore et al. (2016) reached a specificity and positive predictive value for ATTR‐CM of 100%. 53
Biopsies
All but four 26 , 29 , 52 , 54 articles included a biopsy as a part of their diagnostic pathway. As illustrated in Figure 3 , 20 of these articles argued that a biopsy is needed in case of inconclusive results from the bone scintigraphy and/or abnormal results from the tests conducted to exclude AL amyloidosis. 2 , 4 , 12 , 13 , 15 , 16 , 17 , 18 , 20 , 21 , 22 , 23 , 25 , 27 , 33 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 43 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 53 , 55 , 56 Also, 28 articles 2 , 4 , 5 , 10 , 12 , 14 , 16 , 17 , 18 , 19 , 21 , 22 , 23 , 24 , 25 , 28 , 30 , 32 , 34 , 38 , 39 , 40 , 41 , 45 , 46 , 49 , 51 , 57 suggested that one way to diagnose ATTR‐CM is with a biopsy after a positive monoclonal protein test (Figure 3 ). Biopsy types mentioned included fat tissue, bone marrow, involved organ, and in some cases an endomyocardial biopsy if less‐invasive biopsies are negative, but suspicion remain. Twelve of these 28 articles only suggested conducting a cardiac biopsy if bone scintigraphy was unavailable. 2 , 5 , 10 , 14 , 19 , 21 , 24 , 36 , 44 , 45 , 46 , 49
The evidence underlying the choice of tissue for biopsy included original studies investigating the suitability of endomyocardial biopsies 132 , 133 , 134 , 135 as well as extracardiac biopsies for the detection of amyloid in patients with ATTR‐CM. 136 , 137 , 138 , 139 , 140 , 141 These studies reported amyloid detection rates of ~100% in cardiac tissue, 132 , 133 14–45% in fat tissue, 138 , 139 , 140 30% in bone marrow, 139 and 44% in gastrointestinal tract and subcutaneous tissue combined 141 in patients with ATTR‐CM.
As regards the method used for analysing the type of amyloid present in the biopsy samples, there was a general consensus that MS is superior to immunohistochemistry (IHC). Indeed, the superiority of MS was mentioned throughout the study period, beginning with Kapoor et al. (2011). 30 Studies referred to in this connection included a study documenting the risk of false positive immunostaining for TTR amyloid in patients with AL amyloidosis, 142 and studies documenting a high diagnostic accuracy of laser dissection MS. 143 , 144 , 145 One study reported that IHC had a diagnostic accuracy of 76% while using laser dissection MS increased this to 96%, 143 and a recent study based on 700 biopsies also concluded that laser dissection MS is superior to IHC for identifying the specific type of amyloid. 144 The study mostly referred to documented a 100% specificity and sensitivity in a training set of 50 cases of amyloidosis and 98% in a validation set composed of 41 cases of CA. 145
Genetic testing
All but three 11 , 15 , 52 of the included articles mentioned the need for genetic testing in their diagnostic algorithm. Most state that distinguishing between ATTRv CM and ATTRwt CM is important in terms of treatment options, screening of family members, and improved assessment of prognosis. No original articles were referred to in relation to the importance of genetic testing.
Consensus recommendation papers and guidelines
Six of the identified consensus recommendation papers were from national and international academic societies. 2 , 45 , 46 , 47 , 48 , 51 , 57 The Canadian Cardiovascular Society/Canadian Heart Failure Society (CCS/CHFS) published a joint position statement providing recommendations that support the early recognition and optimal diagnostic approach for patients with CA, 46 the American Heart Association (AHA) published a scientific statement intended to guide clinical practice and cover current diagnostic strategies in ATTR‐CM, 45 and a collaboration of multiple American and European societies within imaging techniques and cardiovascular disease (ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI) published recommendations for multimodality imaging in CA. 47 , 48 Most recently, both the ESC and the German Cardiac Society (DGK) published position statements with the purpose of assisting cardiologists and other physicians in recognizing, diagnosing, and treating patients with CA. 2 , 51 One guideline was also identified, namely, a guideline from the Japanese Circulation Society (JCS) on diagnosis and treatment of CA. 57
In accordance with the majority of review articles identified, these position papers conclude that modern cardiac imaging techniques, including bone scintigraphy, allow accurate non‐invasive diagnosis of ATTR‐CM without the need for confirmatory cardiac biopsies. 2 , 45 , 46 , 47 , 48 , 51 CCS/CHFS only suggests conducting an EMB if bone scintigraphy is unavailable, 46 while AHA also suggests conducting a cardiac biopsy in case of a negative monoclonal protein test and a negative bone scintigraphy if suspicion of ATTR‐CM remains high. 45 DGK and ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI argue that a cardiac biopsy remains necessary in the context of equivocal imaging or the coexistence of a monoclonal gammopathy. 2 , 47 , 48 ESC clearly distinguishes between invasive and non‐invasive diagnostic possibilities and supports both. 51
Discussion
Historical development
The number of articles suggesting a diagnostic pathway for ATTR‐CM has increased substantially over the studied period (Figure 3 ). This reflects the augmented interest in the disease spurred by the development of new effective treatments for ATTR‐CM. 6 , 7 , 8 The use of bone scintigraphy in enabling a non‐invasive diagnosis of ATTR‐CM is among the most important clinical advancements within the suspicion, screening, and diagnosis of ATTRwt CM since 2010. 52 , 53 , 55 Also, this period of time has offered the development of more sensitive imaging analyses like native myocardial T1 mapping and extracellular volume in CMR 101 , 102 , 103 and apical sparing global longitudinal strain pattern in ECHO, 96 , 97 , 98 which together with bone scintigraphy has improved the possibilities of suspecting ATTRwt CM significantly.
Patient entrance
Patient entrance received very little attention in the reviewed literature. Patients with extracardiac symptoms like CTS, LSS, or neuropathy typically enter the healthcare system via orthopaedics or neurology, an entry that could enable an early ATTRwt CM diagnosis if processed properly. Still, this was not in focus in the identified literature. Combined with the fact that none of the included articles were published in journals within neurology or orthopaedics, this could raise the concern that patients debuting with extracardiac symptoms are not identified as early as they could be. That said, recent papers published in orthopaedics and surgical journals 146 , 147 , 148 , 149 suggest that focus on ATTRwt CM is increasing outside of cardiology as well.
Suspicion
While multiple red flags exist for ATTRwt CM, none of them are specific for the disease. Most findings and symptoms overlap substantially with those of AL amyloidosis and other cardiac disease. The red flags might, however, differ in their suitability of detecting ATTRwt CM at an early stage. While CTS and LSS belong in the category of medical history and can be seen years before cardiac involvement, native T1 mapping on CMR was described as enabling early detection of ATTR‐CM as was bone scintigraphy. Also, HFpEF progresses into HFrEF as disease advances, while low voltage may primarily be found in late‐stage disease.
The suitability of specific ECHO and CMR findings in raising a suspicion of ATTRwt CM seems indisputable. These imaging tools have been widely investigated for their ability to diagnose ATTRwt CM, and while they do not possess this quality due to too much overlap with AL amyloidosis, they are indeed able to distinguish between CA and other causes of thick hearts. In many cases, an ECHO will be sufficient to raise the suspicion of ATTRwt CM. However, the ability of CMR to distinguish between CA, hypertensive heart disease, and hypertrophic cardiomyopathy, which are all characterized by increased wall thickness on an ECHO, makes a CMR useful in some cases.
The diversity and lack of specificity of ATTRwt CM symptoms indeed complicates the development of a structured approach to how to detect ATTRwt CM patients in the first place and renders the initial suspicion of the disease the weak link in ATTRwt CM patients' journey to diagnosis. Indeed, a recent British study demonstrated substantial delays in establishing the ATTR‐CM diagnosis following presentation with cardiac symptoms by documenting a delay of more than 4 years in more than 40% of the patients. 150 The reviewed articles are characterized by listing numerous typical symptoms, but only few translate these lists into a more guided approach of how to define at‐risk patients. An exception worth mentioning is the combination of increased wall thickness and one or more red flags in people of advanced age, as suggested by Witteles et al. (2019) and in the ESC consensus statement (2021). 4 , 51
In general, the suggested red flags are not specific and have not been evaluated prospectively in terms of their sensitivity, specificity, and positive predictive value. Rather, data are based on observations, experience, and consensus among experts.
Screening
To enable further progression towards solid recommendations and workable guidelines for when to suspect ATTRwt CM, large prospective studies investigating the suitability of specific red flag combinations in identifying the right patients are needed. This would also be the natural step towards implementing systematic screening for the disease. To the best of our knowledge, none of such studies are available at present. While the screening discussion is currently at a premature stage, newer articles highlighted the need to raise awareness for ATTRwt CM and to identify straightforward and cost‐effective tools and algorithms for large‐scale screening of populations at risk. This possibility is expected to unfold in coming years as a natural consequence of ATTR‐CM treatments becoming increasingly available. Indeed, a search on clinicaltrials.gov for ongoing screening studies identified trials with titles like Global prevalence of ATTR‐CM in participants with HFpEF (NCT04424914), Screening for amyloidosis before aortic valve elective replacement (NCT04869631), and Prevalence of Amyloidosis in Patients With Lumbar Spinal Stenosis or Carpal Tunnel Syndrome (NCT03966105).
Diagnosis
There was a high degree of consensus among the identified articles, including the recommendation papers from academic societies, on how to diagnose ATTR‐CM once the suspicion has been raised. Bone scintigraphy and tests to exclude AL amyloidosis are central and inevitable tools, as is the need for a cardiac biopsy in inconclusive cases.
Multiple recommendations were, however, provided for (i) whether to conduct the bone scintigraphy or the tests to exclude AL amyloidosis first and (ii) when to make a biopsy and which biopsy to choose. As for (i), the rationale behind starting with the tests to exclude AL amyloidosis is that the bone scintigraphy might be unnecessary in patients, for whom the AL amyloidosis test results point towards an AL amyloidosis diagnosis. Conversely, tests to exclude AL amyloidosis are always needed, regardless of the results of the bone scintigraphy, because a bone scintigraphy can never be used to rule out AL amyloidosis. As for (ii), the time point and type of biopsy likely reflect that the complete clinical picture should always be considered in inconclusive cases as should the pros and cons of biopsy types. Hence, the high diagnostic value as well as the risk (though minor) of performing an invasive EMB should be compared with both the advantage of avoiding an EMB by performing a less‐invasive extracardiac biopsy (of low diagnostic value), and the disadvantage of having to perform both an extracardiac biopsy and an EMB in case of a negative extracardiac biopsy.
The underlying evidence referred to for strengths and possible pitfalls of the individual diagnostic tools seemed strong for bone scintigraphy and biopsies, but somewhat limited for tests to exclude AL amyloidosis. The reason for this is unknown but may relate to a limited interest in these somewhat simple laboratory tests among cardiologists. The need for a correct interpretation of the FLC assay results should, however, not be overlooked, especially as the reference range referred to deviate in patients with renal problems, 151 which can constitute a significant part of patients with suspected ATTRwt CM.
Conclusions
Wild‐type transthyretin amyloid cardiomyopathy is an underdiagnosed disease that fortunately has gained interest, primarily due to an improvement in the diagnostic tools and the discovery and approval of disease‐modifying medical treatment. The comparison of diagnostic pathways described in 50 different articles shows that diagnostic pathways are similar, albeit with minor differences, and should ensure a fairly stringent diagnostic process across the world.
Meanwhile, this systematic review has identified some significant knowledge gaps. Firstly, patient entry receives little if no attention in the literature. More focus on initial patient contact and increased awareness among general practitioners and relevant non‐cardiological specialities could shorten the way to diagnosis and minimize the risk of misdiagnosis and improper treatment. Secondly, a major challenge lies in raising the initial suspicion of the disease. While multiple red flags are described, there is a general lack of high‐quality prospective studies designed to evaluate the sensitivity and specificity of specific red flags or red flag combinations, not to mention their mutual ranking. Thirdly, a task lies in defining relevant at‐risk screening populations. This should be performed by prospectively evaluating the value of systematic screening in predefined populations. This would lead the way to detection of ATTRwt CM at an early stage, a prerequisite for timely and relevant treatment.
Conflict of interest
K.B. was a paid consultant to Pfizer in connection with the development of this manuscript and reports financial relationships outside of the submitted work with Pfizer Denmark. F.G., M.M., and S.H.P. did not receive any funding from Pfizer to conduct this study and have paid for their own work in this project, albeit they report financial relationships outside of the submitted work with Pfizer Denmark. A.B.‐B., A.M.S., and T.P. are employees at Pfizer Denmark.
Funding
This study was sponsored by Pfizer.
Supporting information
Table S1. Search string used for Embase and MEDLINE.
Acknowledgements
We thank Incentive Denmark ApS for their invaluable advice and support in the initial literature search and data analysis.
Bay, K. , Gustafsson, F. , Maiborg, M. , Bagger‐Bahnsen, A. , Strand, A. M. , Pilgaard, T. , and Poulsen, S. H. (2022) Suspicion, screening, and diagnosis of wild‐type transthyretin amyloid cardiomyopathy: a systematic literature review. ESC Heart Failure, 9: 1524–1541. 10.1002/ehf2.13884.
References
- 1. González‐López E, Gallego‐Delgado M, Guzzo‐Merello G, de Haro‐del Moral FJ, Cobo‐Marcos M, Robles C, Bornstein B, Salas C, Lara‐Pezzi E, Alonso‐Pulpon L, Garcia‐Pavia P. Wild‐type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36: 2585–2594. [DOI] [PubMed] [Google Scholar]
- 2. Yilmaz A, Bauersachs J, Bengel F, Büchel R, Kindermann I, Klingel K, Knebel F, Meder B, Morbach C, Nagel E, Schulze‐Bahr E, aus dem Siepen F, Frey N. Diagnosis and treatment of cardiac amyloidosis: position statement of the German Cardiac Society (DGK). Clin Res Cardiol 2021; 110: 479–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. González‐López E, Gagliardi C, Dominguez F, Quarta CC, de Haro‐del Moral FJ, Milandri A, Salas C, Cinelli M, Cobo‐Marcos M, Lorenzini M, Lara‐Pezzi E, Foffi S, Alonso‐Pulpon L, Rapezzi C, Garcia‐Pavia P. Clinical characteristics of wild‐type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J 2017; 38: 1895–1904. [DOI] [PubMed] [Google Scholar]
- 4. Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, Gospodinova M, Obici L, Rapezzi C, Garcia‐Pavia P. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail 2019; 7: 709–716. [DOI] [PubMed] [Google Scholar]
- 5. Gertz M, Adams D, Ando Y, Beirão JM, Bokhari S, Coelho T, Comenzo RL, Damy T, Dorbala S, Drachman BM, Fontana M, Gillmore JD, Grogan M, Hawkins PN, Lousada I, Kristen AV, Ruberg FL, Suhr OB, Maurer MS, Nativi‐Nicolau J, Quarta CC, Rapezzi C, Witteles R, Merlini G. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract 2020; 21: 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007–1016. [DOI] [PubMed] [Google Scholar]
- 7. Adams D, Gonzalez‐Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Planté‐Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH III, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 11–21. [DOI] [PubMed] [Google Scholar]
- 8. Benson MD, Waddington‐Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Planté‐Bordeneuve V, Barroso FA, Merlini G, Obici L, Scheinberg M, Brannagan TH III, Litchy WJ, Whelan C, Drachman BM, Adams D, Heitner SB, Conceição I, Schmidt HH, Vita G, Campistol JM, Gamez J, Gorevic PD, Gane E, Shah AM, Solomon SD, Monia BP, Hughes SG, Kwoh TJ, McEvoy BW, Jung SW, Baker BF, Ackermann EJ, Gertz MA, Coelho T. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. Int J Surg 2010; 8: 336–341. [DOI] [PubMed] [Google Scholar]
- 10. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state‐of‐the‐art review. J Am Coll Cardiol 2019; 73: 2872–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, Berk JL, Plante‐Bordeneuve V, Schmidt HHJ, Merlini G. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol 2015; 66: 2451–2466. [DOI] [PubMed] [Google Scholar]
- 12. Zhao L, Fang Q. Recent advances in the noninvasive strategies of cardiac amyloidosis. Heart Fail Rev 2016; 21: 703–721. [DOI] [PubMed] [Google Scholar]
- 13. González‐López E, López‐Sainz Á, Garcia‐Pavia P. Diagnosis and treatment of transthyretin cardiac amyloidosis. Progress and hope. Rev Esp Cardiol (Engl Ed) 2017; 70: 991–1004. [DOI] [PubMed] [Google Scholar]
- 14. Nativi‐Nicolau J, Maurer MS. Amyloidosis cardiomyopathy: update in the diagnosis and treatment of the most common types. Curr Opin Cardiol 2018; 33: 571–579. [DOI] [PubMed] [Google Scholar]
- 15. Pelletier‐Galarneau M, Abikhzer G, Giraldeau G, Harel F. Molecular imaging of cardiac amyloidosis. Curr Cardiol Rep 2019; 21: 12. [DOI] [PubMed] [Google Scholar]
- 16. Chacko L, Martone R, Cappelli F, Fontana M. Cardiac amyloidosis: updates in imaging. Curr Cardiol Rep 2019; 21: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Di Giovanni B, Gustafson D, Delgado DH. Amyloid transthyretin cardiac amyloidosis: diagnosis and management. Expert Rev Cardiovasc Ther 2019; 17: 673–681. [DOI] [PubMed] [Google Scholar]
- 18. Ihne S, Morbach C, Obici L, Palladini G, Störk S. Amyloidosis in heart failure. Curr Heart Fail Rep 2019; 16: 285–303. [DOI] [PubMed] [Google Scholar]
- 19. Griffin JM, Maurer MS. Transthyretin cardiac amyloidosis: a treatable form of heart failure with a preserved ejection fraction. Trends Cardiovasc Med 2021; 31: 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Çavuşoğlu Y, Özpelit E, Çelik A, İkitimur B, Kayıkçıoğlu M, Tokgözoğlu L, Tüfekçioğlu O, Yılmaz MB. Cardiac amyloidosis: recent advances in the diagnosis and therapy. Turk Kardiyol Dern Ars 2019; 47: 1–34. [DOI] [PubMed] [Google Scholar]
- 21. Hafeez AS, Bavry AA. Diagnosis of transthyretin amyloid cardiomyopathy. Cardiol Ther 2020; 9: 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Papathanasiou M, Carpinteiro A, Rischpler C, Hagenacker T, Rassaf T, Luedike P. Diagnosing cardiac amyloidosis in every‐day practice: a practical guide for the cardiologist. Int J Cardiol Heart Vasc 2020; 28: 100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Varga C, Dorbala S, Lousada I, Polydefkis MJ, Wechalekar A, Maurer MS, Comenzo RL. The diagnostic challenges of cardiac amyloidosis: a practical approach to the two main types. Blood Rev 2021; 45: 100720. [DOI] [PubMed] [Google Scholar]
- 24. Wolfson AM, Shah KS, Patel JK. Amyloid and the heart. Curr Cardiol Rep 2019; 21: 164. [DOI] [PubMed] [Google Scholar]
- 25. Hanna M, Ruberg FL, Maurer MS, Dispenzieri A, Dorbala S, Falk RH, Hoffman J, Jaber W, Soman P, Witteles RM, Grogan M. Cardiac scintigraphy with technetium‐99m‐labeled bone‐seeking tracers for suspected amyloidosis: JACC review topic of the week. J Am Coll Cardiol 2020; 75: 2851–2862. [DOI] [PubMed] [Google Scholar]
- 26. Lo Presti S, Mihos CG, Yucel E, Horvath SA, Santana O. A focused review on the pathophysiology, diagnosis, and management of cardiac amyloidosis. Rev Cardiovasc Med 2017; 18: 123–133. [DOI] [PubMed] [Google Scholar]
- 27. Jurcuţ R, Onciul S, Adam R, Stan C, Coriu D, Rapezzi C, Popescu BA. Multimodality imaging in cardiac amyloidosis: a primer for cardiologists. Eur Heart J Cardiovasc Imaging 2020; 21: 833–844. [DOI] [PubMed] [Google Scholar]
- 28. Esplin BL, Gertz MA. Current trends in diagnosis and management of cardiac amyloidosis. Curr Probl Cardiol 2013; 38: 53–96. [DOI] [PubMed] [Google Scholar]
- 29. Patel KS, Hawkins PN. Cardiac amyloidosis: where are we today? J Intern Med 2015; 278: 126–144. [DOI] [PubMed] [Google Scholar]
- 30. Kapoor P, Thenappan T, Singh E, Kumar S, Greipp PR. Cardiac amyloidosis: a practical approach to diagnosis and management. Am J Med 2011; 124: 1006–1015. [DOI] [PubMed] [Google Scholar]
- 31. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet 2016; 387: 2641–2654. [DOI] [PubMed] [Google Scholar]
- 32. Narotsky DL, Castano A, Weinsaft JW, Bokhari S, Maurer MS. Wild‐type transthyretin cardiac amyloidosis: novel insights from advanced imaging. Can J Cardiol 2016; 32: 1166.e1–1166.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vergaro G, Aimo A, Barison A, Genovesi D, Buda G, Passino C, Emdin M. Keys to early diagnosis of cardiac amyloidosis: red flags from clinical, laboratory and imaging findings. Eur J Prev Cardiol 2020; 27: 1806–1815. [DOI] [PubMed] [Google Scholar]
- 34. Kourelis TV, Gertz MA. Improving strategies for the diagnosis of cardiac amyloidosis. Expert Rev Cardiovasc Ther 2015; 13: 945–961. [DOI] [PubMed] [Google Scholar]
- 35. Addison D, Slivnick JA, Campbell CM, Vallakati A, Jneid H, Schelbert E. Recent advances and current dilemmas in the diagnosis and management of transthyretin cardiac amyloidosis. J Am Heart Assoc 2021; 10: e019840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ash S, Shorer E, Ramgobin D, Vo M, Gibbons J, Golamari R, Jain R, Jain R. Cardiac amyloidosis—a review of current literature for the practicing physician. Clin Cardiol 2021; 44: 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bistola V, Parissis J, Foukarakis E, Valsamaki PN, Anastasakis A, Koutsis G, Efthimiadis G, Kastritis E. Practical recommendations for the diagnosis and management of transthyretin cardiac amyloidosis. Heart Fail Rev 2021; 26: 861–879. [DOI] [PubMed] [Google Scholar]
- 38. Cappelli F, Perfetto F, Martone R, Di Mario C. Cardiac amyloidosis in patients undergoing TAVR: why we need to think about it. Cardiovasc Revasc Med 2021; 22: 109–114. [DOI] [PubMed] [Google Scholar]
- 39. Garcia‐Pavia P, Domínguez F, Gonzalez‐Lopez E. Transthyretin amyloid cardiomyopathy. Med Clin (Barc) 2021; 156: 126–134. [DOI] [PubMed] [Google Scholar]
- 40. Inomata T, Tahara N, Nakamura K, Endo J, Ueda M, Ishii T, Kitano Y, Koyama J. Diagnosis of wild‐type transthyretin amyloid cardiomyopathy in Japan: red‐flag symptom clusters and diagnostic algorithm. ESC Heart Fail 2021; 8: 2647–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jamal F, Rosenzweig M. Amyloidosis with cardiac involvement: identification, characterization, and management. Curr Hematol Malig Rep 2021; 16: 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Joury A, Gupta T, Krim SR. Cardiac amyloidosis: presentations, diagnostic work‐up and collaborative approach for comprehensive clinical management. Curr Probl Cardiol 2021; 46: 100910. [DOI] [PubMed] [Google Scholar]
- 43. Sabbour H, Hasan KY, al Badarin F, Alibazoglu H, Rivard AL, Romany I, Perlini S. From clinical clues to final diagnosis: the return of detective work to clinical medicine in cardiac amyloidosis. Front Cardiovasc Med 2021; 8: 644508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stern LK, Kittleson MM. Updates in cardiac amyloidosis diagnosis and treatment. Curr Oncol Rep 2021; 23: 47. [DOI] [PubMed] [Google Scholar]
- 45. Kittleson MM, Maurer MS, Ambardekar AV, Bullock‐Palmer RP, Chang PP, Eisen HJ, Nair AP, Nativi‐Nicolau J, Ruberg FL, American Heart Association Heart Failure and Transplantation Committee of the Council on Clinical Cardiology . Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation 2020; 142: e7–e22. [DOI] [PubMed] [Google Scholar]
- 46. Fine NM, Davis MK, Anderson K, Delgado DH, Giraldeau G, Kitchlu A, Massie R, Narayan J, Swiggum E, Venner CP, Ducharme A, Galant NJ, Hahn C, Howlett JG, Mielniczuk L, Parent MC, Reece D, Royal V, Toma M, Virani SA, Zieroth S. Canadian Cardiovascular Society/Canadian Heart Failure Society joint position statement on the evaluation and management of patients with cardiac amyloidosis. Can J Cardiol 2020; 36: 322–334. [DOI] [PubMed] [Google Scholar]
- 47. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans AWJM, Hanna MA, Hazenberg BPC, Kristen AV, Kwong RY, Maurer MS, Merlini G, Miller EJ, Moon JC, Murthy VL, Quarta CC, Rapezzi C, Ruberg FL, Shah SJ, Slart RHJA, Verberne HJ, Bourque JM. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2—evidence base and standardized methods of imaging. J Nucl Cardiol 2019; 26: 2065–2123. [DOI] [PubMed] [Google Scholar]
- 48. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans AWJM, Hanna MA, Hazenberg BPC, Kristen AV, Kwong RY, Maurer MS, Merlini G, Miller EJ, Moon JC, Murthy VL, Quarta CC, Rapezzi C, Ruberg FL, Shah SJ, Slart RHJA, Verberne HJ, Bourque JM. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI EXPERT consensus recommendations for multimodality imaging in cardiac amyloidosis: part 2 of 2—diagnostic criteria and appropriate utilization. J Nucl Cardiol 2020; 27: 659–673. [DOI] [PubMed] [Google Scholar]
- 49. Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, Grogan M, Kristen AV, Lousada I, Nativi‐Nicolau J, Cristina Quarta C, Rapezzi C, Ruberg FL, Witteles R, Merlini G. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 2019; 12: e006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bonderman D, Pölzl G, Ablasser K, Agis H, Aschauer S, Auer‐Grumbach M, Binder C, Dörler J, Duca F, Ebner C, Hacker M, Kain R, Kammerlander A, Koschutnik M, Kroiss AS, Mayr A, Nitsche C, Rainer PP, Reiter‐Malmqvist S, Schneider M, Schwarz R, Verheyen N, Weber T, Zaruba MM, Badr Eslam R, Hülsmann M, Mascherbauer J. Diagnosis and treatment of cardiac amyloidosis: an interdisciplinary consensus statement. Wien Klin Wochenschr 2020; 132: 742–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Garcia‐Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, Burazor I, Caforio ALP, Damy T, Eriksson U, Fontana M, Gillmore JD, Gonzalez‐Lopez E, Grogan M, Heymans S, Imazio M, Kindermann I, Kristen AV, Maurer MS, Merlini G, Pantazis A, Pankuweit S, Rigopoulos AG, Linhart A. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2021; 42: 1554–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hutt DF, Quigley AM, Page J, Hall ML, Burniston M, Gopaul D, Lane T, Whelan CJ, Lachmann HJ, Gillmore JD, Hawkins PN, Wechalekar AD. Utility and limitations of 3,3‐diphosphono‐1,2‐propanodicarboxylic acid scintigraphy in systemic amyloidosis. Eur Heart J Cardiovasc Imaging 2014; 15: 1289–1298. [DOI] [PubMed] [Google Scholar]
- 53. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, Lachmann HJ, Bokhari S, Castano A, Dorbala S, Johnson GB, Glaudemans AWJM, Rezk T, Fontana M, Palladini G, Milani P, Guidalotti PL, Flatman K, Lane T, Vonberg FW, Whelan CJ, Moon JC, Ruberg FL, Miller EJ, Hutt DF, Hazenberg BP, Rapezzi C, Hawkins PN. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133: 2404–2412. [DOI] [PubMed] [Google Scholar]
- 54. Galat A, van der Gucht A, Guellich A, Bodez D, Cottereau AS, Guendouz S, Hittinger L, Dubois‐Randé JL, Plante‐Bordeneuve V, Itti E, Meignan M, Damy T, Rosso J. Early phase 99Tc‐HMDP scintigraphy for the diagnosis and typing of cardiac amyloidosis. JACC Cardiovasc Imaging 2017; 10: 601–603. [DOI] [PubMed] [Google Scholar]
- 55. Moore PT, Burrage MK, Mackenzie E, Law WP, Korczyk D, Mollee P. The utility of 99mTc‐DPD scintigraphy in the diagnosis of cardiac amyloidosis: an Australian experience. Heart Lung Circ 2017; 26: 1183–1190. [DOI] [PubMed] [Google Scholar]
- 56. Zhang KW, Zhang R, Deych E, Stockerl‐Goldstein KE, Gorcsan J 3rd, Lenihan DJ. A multi‐modal diagnostic model improves detection of cardiac amyloidosis among patients with diagnostic confirmation by cardiac biopsy. Am Heart J 2021; 232: 137–145. [DOI] [PubMed] [Google Scholar]
- 57. Kitaoka H, Izumi C, Izumiya Y, Inomata T, Ueda M, Kubo T, Koyama J, Sano M, Sekijima Y, Tahara N, Tsukada N, Tsujita K, Tsutsui H, Tomita T, Amano M, Endo J, Okada A, Oda S, Takashio S, Baba Y, Misumi Y, Yazaki M, Anzai T, Ando Y, Isobe M, Kimura T, Fukuda K, Japanese Circulation Society Joint Working Group . JCS 2020 guideline on diagnosis and treatment of cardiac amyloidosis. Circ J 2020; 84: 1610–1671. [DOI] [PubMed] [Google Scholar]
- 58. Bennani Smires Y, Victor G, Ribes D, Berry M, Cognet T, Méjean S, Huart A, Roussel M, Petermann A, Roncalli J, Carrié D, Rousseau H, Berry I, Chauveau D, Galinier M, Lairez O. Pilot study for left ventricular imaging phenotype of patients over 65 years old with heart failure and preserved ejection fraction: the high prevalence of amyloid cardiomyopathy. Int J Cardiovasc Imaging 2016; 32: 1403–1413. [DOI] [PubMed] [Google Scholar]
- 59. Hahn VS, Yanek LR, Vaishnav J, Ying W, Vaidya D, Lee YZJ, Riley SJ, Subramanya V, Brown EE, Hopkins CD, Ononogbu S, Perzel Mandell K, Halushka MK, Steenbergen C Jr, Rosenberg AZ, Tedford RJ, Judge DP, Shah SJ, Russell SD, Kass DA, Sharma K. Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. JACC Heart Fail 2020; 8: 712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, Klarich KW, Miller WL, Maleszewski JJ, Dispenzieri A. Natural history of wild‐type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016; 68: 1014–1020. [DOI] [PubMed] [Google Scholar]
- 61. Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, Roger VL, Gertz MA, Dispenzieri A, Zeldenrust SR, Redfield MM. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail 2014; 2: 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sekijima Y, Uchiyama S, Tojo K, Sano K, Shimizu Y, Imaeda T, Hoshii Y, Kato H, Ikeda SI. High prevalence of wild‐type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: a common cause of carpal tunnel syndrome in the elderly. Hum Pathol 2011; 42: 1785–1791. [DOI] [PubMed] [Google Scholar]
- 63. Sperry BW, Reyes BA, Ikram A, Donnelly JP, Phelan D, Jaber WA, Shapiro D, Evans PJ, Maschke S, Kilpatrick SE, Tan CD, Rodriguez ER, Monteiro C, Tang WHW, Kelly JW, Seitz WH Jr, Hanna M. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol 2018; 72: 2040–2050. [DOI] [PubMed] [Google Scholar]
- 64. Zegri‐Reiriz I, de Haro‐del Moral FJ, Dominguez F, Salas C, de la Cuadra P, Plaza A, Krsnik I, Gonzalez‐Lopez E, Garcia‐Pavia P. Prevalence of cardiac amyloidosis in patients with carpal tunnel syndrome. J Cardiovasc Transl Res 2019; 12: 507–513. [DOI] [PubMed] [Google Scholar]
- 65. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, Steidley DE, Ventura H, Murali S, Silver MA, Jacoby D, Fedson S, Hummel SL, Kristen AV, Damy T, Planté‐Bordeneuve V, Coelho T, Mundayat R, Suhr OB, Waddington Cruz M, Rapezzi C, THAOS Investigators . Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (transthyretin amyloid outcome survey). J Am Coll Cardiol 2016; 68: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, Wechalekar A, Gibbs SD, Venner CP, Wassef N, McCarthy C, Gilbertson JA, Rowczenio D, Hawkins PN, Gillmore JD, Lachmann HJ. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc 2013; 2: e000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. aus dem Siepen F, Hein S, Prestel S, Baumgärtner C, Schönland S, Hegenbart U, Röcken C, Katus HA, Kristen AV. Carpal tunnel syndrome and spinal canal stenosis: harbingers of transthyretin amyloid cardiomyopathy? Clin Res Cardiol 2019; 108: 1324–1330. [DOI] [PubMed] [Google Scholar]
- 68. Nakagawa M, Sekijima Y, Yazaki M, Tojo K, Yoshinaga T, Doden T, Koyama J, Yanagisawa S, Ikeda SI. Carpal tunnel syndrome: a common initial symptom of systemic wild‐type ATTR (ATTRwt) amyloidosis. Amyloid 2016; 23: 58–63. [DOI] [PubMed] [Google Scholar]
- 69. Papoutsidakis N, Miller EJ, Rodonski A, Jacoby D. Time course of common clinical manifestations in patients with transthyretin cardiac amyloidosis: delay from symptom onset to diagnosis. J Card Fail 2018; 24: 131–133. [DOI] [PubMed] [Google Scholar]
- 70. Connors LH, Sam F, Skinner M, Salinaro F, Sun F, Ruberg FL, Berk JL, Seldin DC. Heart failure resulting from age‐related cardiac amyloid disease associated with wild‐type transthyretin: a prospective, observational cohort study. Circulation 2016; 133: 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cornwell GG 3rd, Murdoch WL, Kyle RA, Westermark P, Pitkanen P. Frequency and distribution of senile cardiovascular amyloid: a clinicopathologic correlation. Am J Med 1983; 75: 618–623. [DOI] [PubMed] [Google Scholar]
- 72. Milandri A, Farioli A, Gagliardi C, Longhi S, Salvi F, Curti S, Foffi S, Caponetti AG, Lorenzini M, Ferlini A, Rimessi P, Mattioli S, Violante FS, Rapezzi C. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail 2020; 22: 507–515. [DOI] [PubMed] [Google Scholar]
- 73. Sekijima Y, Yazaki M, Ueda M, Koike H, Yamada M, Ando Y. First nationwide survey on systemic wild‐type ATTR amyloidosis in Japan. Amyloid 2018; 25: 8–10. [DOI] [PubMed] [Google Scholar]
- 74. Yamada T, Takashio S, Arima Y, Nishi M, Morioka M, Hirakawa K, Hanatani S, Fujisue K, Yamanaga K, Kanazawa H, Sueta D, Araki S, Usuku H, Nakamura T, Suzuki S, Yamamoto E, Ueda M, Kaikita K, Tsujita K. Clinical characteristics and natural history of wild‐type transthyretin amyloid cardiomyopathy in Japan. ESC Heart Fail 2020; 7: 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fosbøl EL, Rørth R, Leicht BP, Schou M, Maurer MS, Kristensen SL, Kober L, Gustafsson F. Association of carpal tunnel syndrome with amyloidosis, heart failure, and adverse cardiovascular outcomes. J Am Coll Cardiol 2019; 74: 15–23. [DOI] [PubMed] [Google Scholar]
- 76. Westermark P, Westermark GT, Suhr OB, Berg S. Transthyretin‐derived amyloidosis: probably a common cause of lumbar spinal stenosis. Ups J Med Sci 2014; 119: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yanagisawa A, Ueda M, Sueyoshi T, Okada T, Fujimoto T, Ogi Y, Kitagawa K, Tasaki M, Misumi Y, Oshima T, Jono H, Obayashi K, Hirakawa K, Uchida H, Westermark P, Ando Y, Mizuta H. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol 2015; 28: 201–207. [DOI] [PubMed] [Google Scholar]
- 78. Coelho T, Maurer MS, Suhr OB. THAOS—the Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild‐type transthyretin amyloidosis. Curr Med Res Opin 2013; 29: 63–76. [DOI] [PubMed] [Google Scholar]
- 79. Longhi S, Quarta CC, Milandri A, Lorenzini M, Gagliardi C, Manuzzi L, Bacchi‐Reggiani ML, Leone O, Ferlini A, Russo A, Gallelli I, Rapezzi C. Atrial fibrillation in amyloidotic cardiomyopathy: prevalence, incidence, risk factors and prognostic role. Amyloid 2015; 22: 147–155. [DOI] [PubMed] [Google Scholar]
- 80. Martinez‐Naharro A, Gonzalez‐Lopez E, Corovic A, Mirelis JG, Baksi AJ, Moon JC, Garcia‐Pavia P, Gillmore JD, Hawkins PN, Fontana M. High prevalence of intracardiac thrombi in cardiac amyloidosis. J Am Coll Cardiol 2019; 73: 1733–1734. [DOI] [PubMed] [Google Scholar]
- 81. Treibel TA, Fontana M, Gilbertson JA, Castelletti S, White SK, Scully PR, Roberts N, Hutt DF, Rowczenio DM, Whelan CJ, Ashworth MA, Gillmore JD, Hawkins PN, Moon JC. Occult transthyretin cardiac amyloid in severe calcific aortic stenosis: prevalence and prognosis in patients undergoing surgical aortic valve replacement. Circ Cardiovasc Imaging 2016; 9. [DOI] [PubMed] [Google Scholar]
- 82. Scully PR, Treibel TA, Fontana M, Lloyd G, Mullen M, Pugliese F, Hartman N, Hawkins PN, Menezes LJ, Moon JC. Prevalence of cardiac amyloidosis in patients referred for transcatheter aortic valve replacement. J Am Coll Cardiol 2018; 71: 463–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Castaño A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, Rubin J, Chiuzan C, Nazif T, Vahl T, George I, Kodali S, Leon MB, Hahn R, Bokhari S, Maurer MS. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017; 38: 2879–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nitsche C, Scully PR, Patel KP, Kammerlander AA, Koschutnik M, Dona C, Wollenweber T, Ahmed N, Thornton GD, Kelion AD, Sabharwal N, Newton JD, Ozkor M, Kennon S, Mullen M, Lloyd G, Fontana M, Hawkins PN, Pugliese F, Menezes LJ, Moon JC, Mascherbauer J, Treibel TA. Prevalence and outcomes of concomitant aortic stenosis and cardiac amyloidosis. J Am Coll Cardiol 2021; 77: 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Longhi S, Lorenzini M, Gagliardi C, Milandri A, Marzocchi A, Marrozzini C, Saia F, Ortolani P, Biagini E, Guidalotti PL, Leone O, Rapezzi C. Coexistence of degenerative aortic stenosis and wild‐type transthyretin‐related cardiac amyloidosis. JACC Cardiovasc Imaging 2016; 9: 325–327. [DOI] [PubMed] [Google Scholar]
- 86. Rahman JE, Helou EF, Gelzer‐Bell R, Thompson RE, Kuo C, Rodriguez ER, Hare JM, Baughman KL, Kasper EK. Noninvasive diagnosis of biopsy‐proven cardiac amyloidosis. J Am Coll Cardiol 2004; 43: 410–415. [DOI] [PubMed] [Google Scholar]
- 87. Damy T, Maurer MS, Rapezzi C, Planté‐Bordeneuve V, Karayal ON, Mundayat R, Suhr OB, Kristen AV. Clinical, ECG and echocardiographic clues to the diagnosis of TTR‐related cardiomyopathy. Open Heart 2016; 3: e000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Quarta CC, Solomon SD, Uraizee I, Kruger J, Longhi S, Ferlito M, Gagliardi C, Milandri A, Rapezzi C, Falk RH. Left ventricular structure and function in transthyretin‐related versus light‐chain cardiac amyloidosis. Circulation 2014; 129: 1840–1849. [DOI] [PubMed] [Google Scholar]
- 89. Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol 2014; 114: 1089–1093. [DOI] [PubMed] [Google Scholar]
- 90. Cappelli F, Vignini E, Martone R, Perlini S, Mussinelli R, Sabena A, Morini S, Gabriele M, Taborchi G, Bartolini S, Lossi A, Nardi G, Marchionni N, di Mario C, Olivotto I, Perfetto F. Baseline ECG features and arrhythmic profile in transthyretin versus light chain cardiac amyloidosis. Circ Heart Fail 2020; 13: e006619. [DOI] [PubMed] [Google Scholar]
- 91. Sperry BW, Vranian MN, Hachamovitch R, Joshi H, McCarthy M, Ikram A, Hanna M. Are classic predictors of voltage valid in cardiac amyloidosis? A contemporary analysis of electrocardiographic findings. Int J Cardiol 2016; 214: 477–481. [DOI] [PubMed] [Google Scholar]
- 92. Givens RC, Russo C, Green P, Maurer MS. Comparison of cardiac amyloidosis due to wild‐type and V122I transthyretin in older adults referred to an academic medical center. Aging Health 2013; 9: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nochioka K, Quarta CC, Claggett B, Roca GQ, Rapezzi C, Falk RH, Solomon SD. Left atrial structure and function in cardiac amyloidosis. Eur Heart J Cardiovasc Imaging 2017; 18: 1128–1137. [DOI] [PubMed] [Google Scholar]
- 94. Cappelli F, Baldasseroni S, Bergesio F, Perlini S, Salinaro F, Padeletti L, Attanà P, Paoletti Perini A, Moggi Pignone A, Grifoni E, Fabbri A, Marchionni N, Gensini GF, Perfetto F. Echocardiographic and biohumoral characteristics in patients with AL and TTR amyloidosis at diagnosis. Clin Cardiol 2015; 38: 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Moñivas Palomero V, Durante‐Lopez A, Sanabria MT, Cubero JS, González‐Mirelis J, Lopez‐Ibor JV, Navarro Rico SM, Krsnik I, Dominguez F, Mingo AM, Hernandez‐Perez FJ, Cavero G, Santos SM. Role of right ventricular strain measured by two‐dimensional echocardiography in the diagnosis of cardiac amyloidosis. J Am Soc Echocardiogr 2019; 32: 845–53.e1. [DOI] [PubMed] [Google Scholar]
- 96. Phelan D, Collier P, Thavendiranathan P, Popović ZB, Hanna M, Plana JC, Marwick TH, Thomas JD. Relative apical sparing of longitudinal strain using two‐dimensional speckle‐tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012; 98: 1442–1448. [DOI] [PubMed] [Google Scholar]
- 97. Phelan D, Thavendiranathan P, Popovic Z, Collier P, Griffin B, Thomas JD, Marwick TH. Application of a parametric display of two‐dimensional speckle‐tracking longitudinal strain to improve the etiologic diagnosis of mild to moderate left ventricular hypertrophy. J Am Soc Echocardiogr 2014; 27: 888–895. [DOI] [PubMed] [Google Scholar]
- 98. Pagourelias ED, Mirea O, Duchenne J, van Cleemput J, Delforge M, Bogaert J, Kuznetsova T, Voigt JU. Echo parameters for differential diagnosis in cardiac amyloidosis: a head‐to‐head comparison of deformation and nondeformation parameters. Circ Cardiovasc Imaging 2017; 10: e005588. [DOI] [PubMed] [Google Scholar]
- 99. Lee GY, Kim HK, Choi JO, Chang SA, Oh JK, Jeon ES, Sohn DW. Visual assessment of relative apical sparing pattern is more useful than quantitative assessment for diagnosing cardiac amyloidosis in borderline or mildly increased left ventricular wall thickness. Circ J 2015; 79: 1575–1584. [DOI] [PubMed] [Google Scholar]
- 100. Boldrini M, Cappelli F, Chacko L, Restrepo‐Cordoba MA, Lopez‐Sainz A, Giannoni A, Aimo A, Baggiano A, Martinez‐Naharro A, Whelan C, Quarta C, Passino C, Castiglione V, Chubuchnyi V, Spini V, Taddei C, Vergaro G, Petrie A, Ruiz‐Guerrero L, Moñivas V, Mingo‐Santos S, Mirelis JG, Dominguez F, Gonzalez‐Lopez E, Perlini S, Pontone G, Gillmore J, Hawkins PN, Garcia‐Pavia P, Emdin M, Fontana M. Multiparametric echocardiography scores for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 2020; 13: 909–920. [DOI] [PubMed] [Google Scholar]
- 101. Baggiano A, Boldrini M, Martinez‐Naharro A, Kotecha T, Petrie A, Rezk T, Gritti M, Quarta C, Knight DS, Wechalekar AD, Lachmann HJ, Perlini S, Pontone G, Moon JC, Kellman P, Gillmore JD, Hawkins PN, Fontana M. Noncontrast magnetic resonance for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 2020; 13: 69–80. [DOI] [PubMed] [Google Scholar]
- 102. Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, Pica S, Castelletti S, Piechnik SK, Robson MD, Gilbertson JA, Rowczenio D, Hutt DF, Lachmann HJ, Wechalekar AD, Whelan CJ, Gillmore JD, Hawkins PN, Moon JC. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging 2014; 7: 157–165. [DOI] [PubMed] [Google Scholar]
- 103. Martinez‐Naharro A, Kotecha T, Norrington K, Boldrini M, Rezk T, Quarta C, Treibel TA, Whelan CJ, Knight DS, Kellman P, Ruberg FL, Gillmore JD, Moon JC, Hawkins PN, Fontana M. Native T1 and extracellular volume in transthyretin amyloidosis. JACC Cardiovasc Imaging 2019; 12: 810–819. [DOI] [PubMed] [Google Scholar]
- 104. Martinez‐Naharro A, Treibel TA, Abdel‐Gadir A, Bulluck H, Zumbo G, Knight DS, Kotecha T, Francis R, Hutt DF, Rezk T, Rosmini S, Quarta CC, Whelan CJ, Kellman P, Gillmore JD, Moon JC, Hawkins PN, Fontana M. Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 2017; 70: 466–477. [DOI] [PubMed] [Google Scholar]
- 105. White JA, Kim HW, Shah D, Fine N, Kim KY, Wendell DC, Al‐Jaroudi W, Parker M, Patel M, Gwadry‐Sridhar F, Judd RM, Kim RJ. CMR imaging with rapid visual T1 assessment predicts mortality in patients suspected of cardiac amyloidosis. JACC Cardiovasc Imaging 2014; 7: 143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Dungu JN, Valencia O, Pinney JH, Gibbs SD, Rowczenio D, Gilbertson JA, Lachmann HJ, Wechalekar A, Gillmore JD, Whelan CJ, Hawkins PN, Anderson LJ. CMR‐based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging 2014; 7: 133–142. [DOI] [PubMed] [Google Scholar]
- 107. Fontana M, Pica S, Reant P, Abdel‐Gadir A, Treibel TA, Banypersad SM, Maestrini V, Barcella W, Rosmini S, Bulluck H, Sayed RH, Patel K, Mamhood S, Bucciarelli‐Ducci C, Whelan CJ, Herrey AS, Lachmann HJ, Wechalekar AD, Manisty CH, Schelbert EB, Kellman P, Gillmore JD, Hawkins PN, Moon JC. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2015; 132: 1570–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fontana M, Banypersad SM, Treibel TA, Abdel‐Gadir A, Maestrini V, Lane T, Gilbertson JA, Hutt DF, Lachmann HJ, Whelan CJ, Wechalekar AD, Herrey AS, Gillmore JD, Hawkins PN, Moon JC. Differential myocyte responses in patients with cardiac transthyretin amyloidosis and light‐chain amyloidosis: a cardiac MR imaging study. Radiology 2015; 277: 388–397. [DOI] [PubMed] [Google Scholar]
- 109. Kristen AV, Aus dem Siepen F, Scherer K, Kammerer R, Andre F, Buss SJ, Bauer R, Lehrke S, Voss A, Giannitsis E, Katus HA, Steen H. Comparison of different types of cardiac amyloidosis by cardiac magnetic resonance imaging. Amyloid 2015; 22: 132–141. [DOI] [PubMed] [Google Scholar]
- 110. Takashio S, Yamamuro M, Izumiya Y, Hirakawa K, Marume K, Yamamoto M, Ueda M, Yamashita T, Ishibashi‐Ueda H, Yasuda S, Ogawa H, Ando Y, Anzai T, Tsujita K. Diagnostic utility of cardiac troponin t level in patients with cardiac amyloidosis. ESC Heart Fail 2018; 5: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E, Coccolo F, Cooke RMT, Bacchi‐Reggiani L, Sangiorgi D, Ferlini A, Cavo M, Zamagni E, Fonte ML, Palladini G, Salinaro F, Musca F, Obici L, Branzi A, Perlini S. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 2009; 120: 1203–1212. [DOI] [PubMed] [Google Scholar]
- 112. Damy T, Costes B, Hagège AA, Donal E, Eicher JC, Slama M, Guellich A, Rappeneau S, Gueffet JP, Logeart D, Planté‐Bordeneuve V, Bouvaist H, Huttin O, Mulak G, Dubois‐Randé JL, Goossens M, Canoui‐Poitrine F, Buxbaum JN. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J 2016; 37: 1826–1834. [DOI] [PubMed] [Google Scholar]
- 113. Kristen AV, Maurer MS, Rapezzi C, Mundayat R, Suhr OB, Damy T, THAOS investigators . Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis—report from the Transthyretin Amyloidosis Outcome Survey (THAOS). PLoS ONE 2017; 12: e0173086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez‐Naharro A, Quarta CC, Rezk T, Whelan CJ, Gonzalez‐Lopez E, Lane T, Gilbertson JA, Rowczenio D, Petrie A, Hawkins PN. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018; 39: 2799–2806. [DOI] [PubMed] [Google Scholar]
- 115. Katzmann JA, Clark RJ, Abraham RS, Bryant S, Lymp JF, Bradwell AR, Kyle RA. Serum reference intervals and diagnostic ranges for free κ and free λ immunoglobulin light chains: relative sensitivity for detection of monoclonal light chains. Clin Chem 2002; 48: 1437–1444. [PubMed] [Google Scholar]
- 116. Barnidge DR, Dispenzieri A, Merlini G, Katzmann JA, Murray DL. Monitoring free light chains in serum using mass spectrometry. Clin Chem Lab Med 2016; 54: 1073–1083. [DOI] [PubMed] [Google Scholar]
- 117. Muchtar E, Gertz MA, Kyle RA, Lacy MQ, Dingli D, Leung N, Buadi FK, Hayman SR, Kapoor P, Hwa YL, Fonder A, Hobbs M, Gonsalves W, Kourelis TV, Warsame R, Russell S, Lust JA, Lin Y, Go RS, Zeldenrust S, Rajkumar SV, Kumar SK, Dispenzieri A. A modern primer on light chain amyloidosis in 592 patients with mass spectrometry‐verified typing. Mayo Clin Proc 2019; 94: 472–483. [DOI] [PubMed] [Google Scholar]
- 118. Phull P, Sanchorawala V, Connors LH, Doros G, Ruberg FL, Berk JL, Sarosiek S. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid 2018; 25: 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, Leone O, Farsad M, Ciliberti P, Bacchi‐Reggiani L, Fallani F, Branzi A, Rapezzi C. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc‐3,3‐diphosphono‐1,2‐propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 2005; 46: 1076–1084. [DOI] [PubMed] [Google Scholar]
- 120. Rapezzi C, Quarta CC, Guidalotti PL, Longhi S, Pettinato C, Leone O, Ferlini A, Salvi F, Gallo P, Gagliardi C, Branzi A. Usefulness and limitations of 99mTc‐3,3‐diphosphono‐1,2‐propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. Eur J Nucl Med Mol Imaging 2011; 38: 470–478. [DOI] [PubMed] [Google Scholar]
- 121. Quarta CC, Guidalotti PL, Longhi S, Pettinato C, Leone O, Ferlini A, Biagini E, Grigioni F, Bacchi‐Reggiani ML, Lorenzini M, Milandri A, Branzi A, Rapezzi C. Defining the diagnosis in echocardiographically suspected senile systemic amyloidosis. JACC Cardiovasc Imaging 2012; 5: 755–758. [DOI] [PubMed] [Google Scholar]
- 122. Pilebro B, Suhr OB, Naslund U, Westermark P, Lindqvist P, Sundstrom T. 99mTc‐DPD uptake reflects amyloid fibril composition in hereditary transthyretin amyloidosis. Ups J Med Sci 2016; 121: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. 99mTc‐pyrophosphate scintigraphy for differentiating light‐chain cardiac amyloidosis from the transthyretin‐related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013; 6: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, Pozniakoff T, Ruberg FL, Miller EJ, Berk JL, Dispenzieri A, Grogan M, Johnson G, Bokhari S, Maurer MS. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol 2016; 1: 880–889. [DOI] [PubMed] [Google Scholar]
- 125. Wizenberg TA, Muz J, Sohn YH, Samlowski W, Weissler AM. Value of positive myocardial technetium‐99m‐pyrophosphate scintigraphy in the noninvasive diagnosis of cardiac amyloidosis. Am Heart J 1982; 103: 468–473. [DOI] [PubMed] [Google Scholar]
- 126. Glaudemans AW, van Rheenen RW, van den Berg MP, Noordzij W, Koole M, Blokzijl H, Dierckx RAJO, Slart RHJA, Hazenberg BPC. Bone scintigraphy with 99mtechnetium‐hydroxymethylene diphosphonate allows early diagnosis of cardiac involvement in patients with transthyretin‐derived systemic amyloidosis. Amyloid 2014; 21: 35–44. [DOI] [PubMed] [Google Scholar]
- 127. Cappelli F, Gallini C, di Mario C, Costanzo EN, Vaggelli L, Tutino F, Ciaccio A, Bartolini S, Angelotti P, Frusconi S, Farsetti S, Vergaro G, Giorgetti A, Marzullo P, Genovesi D, Emdin M, Perfetto F. Accuracy of 99mTc‐hydroxymethylene diphosphonate scintigraphy for diagnosis of transthyretin cardiac amyloidosis. J Nucl Cardiol 2019; 26: 497–504. [DOI] [PubMed] [Google Scholar]
- 128. Masri A, Bukhari S, Ahmad S, Nieves R, Eisele YS, Follansbee W, Brownell A, Wong TC, Schelbert E, Soman P. Efficient 1‐hour technetium‐99 m pyrophosphate imaging protocol for the diagnosis of transthyretin cardiac amyloidosis. Circ Cardiovasc Imaging 2020; 13: e010249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Treglia G, Glaudemans A, Bertagna F, Hazenberg BPC, Erba PA, Giubbini R, Ceriani L, Prior JO, Giovanella L, Slart RHJA. Diagnostic accuracy of bone scintigraphy in the assessment of cardiac transthyretin‐related amyloidosis: a bivariate meta‐analysis. Eur J Nucl Med Mol Imaging 2018; 45: 1945–1955. [DOI] [PubMed] [Google Scholar]
- 130. Haq M, Pawar S, Berk JL, Miller EJ, Ruberg FL. Can 99mTc‐pyrophosphate aid in early detection of cardiac involvement in asymptomatic variant TTR amyloidosis? JACC Cardiovasc Imaging 2017; 10: 713–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Longhi S, Guidalotti PL, Quarta CC, Gagliardi C, Milandri A, Lorenzini M, Potena L, Leone O, Bartolomei I, Pastorelli F, Salvi F, Rapezzi C. Identification of TTR‐related subclinical amyloidosis with 99mTc‐DPD scintigraphy. JACC Cardiovasc Imaging 2014; 7: 531–532. [DOI] [PubMed] [Google Scholar]
- 132. Kristen AV, Dengler TJ, Katus HA. Suspected cardiac amyloidosis: endomyocardial biopsy remains the diagnostic gold‐standard. Am J Hematol 2007; 82: 328. [DOI] [PubMed] [Google Scholar]
- 133. Pellikka PA, Holmes DR Jr, Edwards WD, Nishimura RA, Tajik AJ, Kyle RA. Endomyocardial biopsy in 30 patients with primary amyloidosis and suspected cardiac involvement. Arch Intern Med 1988; 148: 662–666. [PubMed] [Google Scholar]
- 134. Sławek S, Araszkiewicz A, Gaczkowska A, Koszarska J, Celiński D, Grygier M, Lesiak M, Grajek S. Endomyocardial biopsy via the femoral access—still safe and valuable diagnostic tool. BMC Cardiovasc Disord 2016; 16: 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yilmaz A, Kindermann I, Kindermann M, Mahfoud F, Ukena C, Athanasiadis A, Hill S, Mahrholdt H, Voehringer M, Schieber M, Klingel K, Kandolf R, Böhm M, Sechtem U. Comparative evaluation of left and right ventricular endomyocardial biopsy: differences in complication rate and diagnostic performance. Circulation 2010; 122: 900–909. [DOI] [PubMed] [Google Scholar]
- 136. Ansari‐Lari MA, Ali SZ. Fine‐needle aspiration of abdominal fat pad for amyloid detection: a clinically useful test? Diagn Cytopathol 2004; 30: 178–181. [DOI] [PubMed] [Google Scholar]
- 137. Sjölander D, Bijzet J, Hazenberg BP, Nilsson KP, Hammarström P. Sensitive and rapid assessment of amyloid by oligothiophene fluorescence in subcutaneous fat tissue. Amyloid 2015; 22: 19–25. [DOI] [PubMed] [Google Scholar]
- 138. Quarta CC, Gonzalez‐Lopez E, Gilbertson JA, Botcher N, Rowczenio D, Petrie A, Rezk T, Youngstein T, Mahmood S, Sachchithanantham S, Lachmann HJ, Fontana M, Whelan CJ, Wechalekar AD, Hawkins PN, Gillmore JD. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J 2017; 38: 1905–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Fine NM, Arruda‐Olson AM, Dispenzieri A, Zeldenrust SR, Gertz MA, Kyle RA, Swiecicki PL, Scott CG, Grogan M. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol 2014; 113: 1723–1727. [DOI] [PubMed] [Google Scholar]
- 140. Fernández de Larrea C, Verga L, Morbini P, Klersy C, Lavatelli F, Foli A, Obici L, Milani P, Capello GL, Paulli M, Palladini G, Merlini G. A practical approach to the diagnosis of systemic amyloidoses. Blood 2015; 125: 2239–2244. [DOI] [PubMed] [Google Scholar]
- 141. Ueda M, Horibata Y, Shono M, Misumi Y, Oshima T, Su Y, Tasaki M, Shinriki S, Kawahara S, Jono H, Obayashi K, Ogawa H, Ando Y. Clinicopathological features of senile systemic amyloidosis: an ante‐ and post‐mortem study. Mod Pathol 2011; 24: 1533–1544. [DOI] [PubMed] [Google Scholar]
- 142. Satoskar AA, Efebera Y, Hasan A, Brodsky S, Nadasdy G, Dogan A, Nadasdy T. Strong transthyretin immunostaining: potential pitfall in cardiac amyloid typing. Am J Surg Pathol 2011; 35: 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gilbertson JA, Theis JD, Vrana JA, Lachmann H, Wechalekar A, Whelan C, Hawkins PN, Dogan A, Gillmore JD. A comparison of immunohistochemistry and mass spectrometry for determining the amyloid fibril protein from formalin‐fixed biopsy tissue. J Clin Pathol 2015; 68: 314–317. [DOI] [PubMed] [Google Scholar]
- 144. Rezk T, Gilbertson JA, Mangione PP, Rowczenio D, Rendell NB, Canetti D, Lachmann HJ, Wechalekar AD, Bass P, Hawkins PN, Bellotti V, Taylor GW, Gillmore JD. The complementary role of histology and proteomics for diagnosis and typing of systemic amyloidosis. J Pathol Clin Res 2019; 5: 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR 3rd, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry‐based proteomic analysis in clinical biopsy specimens. Blood 2009; 114: 4957–4959. [DOI] [PubMed] [Google Scholar]
- 146. Donnelly JP, Hanna M, Sperry BW, Seitz WH Jr. Carpal tunnel syndrome: a potential early, red‐flag sign of amyloidosis. J Hand Surg Am 2019; 44: 868–876. [DOI] [PubMed] [Google Scholar]
- 147. Hara Y, Tajiri Y, Kawano K, Hoshikawa S, Kita Y. The tenosynovitis of fingers associated with transthyretin amyloidosis. J Hand Surg Asian Pac 2020; 25: 340–344. [DOI] [PubMed] [Google Scholar]
- 148. Sood RF, Kamenko S, McCreary E, Sather BK, Schmitt M, Peterson SL, Lipira AB. Diagnosing systemic amyloidosis presenting as carpal tunnel syndrome: a risk nomogram to guide biopsy at time of carpal tunnel release. J Bone Joint Surg Am 2021; 103: 1284–1294. [DOI] [PubMed] [Google Scholar]
- 149. Triguero A, González‐Costello J, López‐Marne S, Llop A, Pané M, Yun S. Hand surgeons and amyloidosis specialists warning: transthyretin‐associated amyloidosis with bifid median nerve as a cause of bilateral carpal tunnel syndrome. A case report and literature review. Eur J Orthop Surg Traumatol 2021. [DOI] [PubMed] [Google Scholar]
- 150. Lane T, Fontana M, Martinez‐Naharro A, Quarta CC, Whelan CJ, Petrie A, Rowczenio DM, Gilbertson JA, Hutt DF, Rezk T, Strehina SG, Caringal‐Galima J, Manwani R, Sharpley FA, Wechalekar AD, Lachmann HJ, Mahmood S, Sachchithanantham S, Drage EPS, Jenner HD, McDonald R, Bertolli O, Calleja A, Hawkins PN, Gillmore JD. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation 2019; 140: 16–26. [DOI] [PubMed] [Google Scholar]
- 151. Hutchison CA, Harding S, Hewins P, Mead GP, Townsend J, Bradwell AR, Cockwell P. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol 2008; 3: 1684–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Search string used for Embase and MEDLINE.