

Summary
Background
Schistosomiasis is an underestimated neglected tropical disease which affects over 236.6 million people worldwide. According to the CDC, the impact of this disease is second to only malaria as the most devastating parasitic infection. Affected individuals manifest chronic pathology due to egg granuloma formation, destroying the liver over time. The only FDA approved drug, praziquantel, does not protect individuals from reinfection, highlighting the need for a prophylactic vaccine. Schistosoma mansoni Cathepsin B (SmCB) is a parasitic gut peptidase necessary for helminth growth and maturation and confers protection as a vaccine target for intestinal schistosomiasis.
Methods
An SmCB expressing human adenovirus serotype 5 (AdSmCB) was constructed and delivered intramuscularly to female C57BL/6 mice in a heterologous prime and boost vaccine with recombinant protein. Vaccine induced immunity was described and subsequent protection from parasite infection was assessed by analysing parasite burden and liver pathology.
Findings
Substantially higher humoral and cell-mediated immune responses, consisting of IgG2c, Th1 effectors, and polyfunctional CD4+ T cells, were induced by the heterologous administration of AdSmCB when compared to the other regimens. Though immune responses favoured Th1 immunity, Th2 responses provided by SmCB protein boosts were maintained. This mixed Th1/Th2 immune response resulted in significant protection from S. mansoni infection comparable to other vaccine formulations which are in clinical trials. Schistosomiasis associated liver pathology was also prevented in a murine model.
Interpretation
Our study provides missing preclinical data supporting the use of adenoviral vectoring in vaccines for S. mansoni infection. Our vaccination method significantly reduces parasite burden and its associated liver pathology - both of which are critical considerations for this helminth vaccine.
Funding
This work was supported by the Canadian Institutes of Health Research, R. Howard Webster Foundation, and the Foundation of the McGill University Health Centre.
Keywords: Schistosoma mansoni, Parasite, Vaccine, Viral vector, Schistosomiasis, Heterologous, Prime and boost
Research in context.
Evidence before the study
There are currently no helminth vaccines approved by the FDA to date. Vaccines for schistosomiasis which are in clinical trials have demonstrated varying protective capacity reaching a maximum of 70% when tested in animals. These vaccines utilize a homologous vaccination strategy, consisting of 3 doses of adjuvanted recombinant protein, to confer protection that has yet to be validated in human trials. Adenovirus vectored vaccines are generally tested in high doses though cell-mediated responses can be delivered by lower doses.
Added value of the study
This low dose adenovirus vaccine is effective in mounting a robust immune response against a helminth antigen and conferring protection from schistosomiasis and its resulting pathology to the liver. The protection we demonstrated is higher than most in human clinical trials and provided by one less recombinant protein dose, without the need of an adjuvant.
Implications of all the available evidence
The development of a low-dose adenoviral vectored vaccine administered with less protein boosts which are unadjuvanted would greatly ease its production for the global population. This would increase vaccine availability while lowering both cost and potential adverse events. With the nearly one billion individuals at risk of schistosomiasis infection, the threat of resistance to chemotherapy, and lethal prognoses to affected individuals due to increased susceptibility to coinfections, the need for a preventative vaccine is urgent.
Alt-text: Unlabelled box
Introduction
Schistosomiasis is a neglected tropical disease affecting over 236.6 million individuals in over 70 countries worldwide.1 The impact of this disease has been estimated by the CDC to be the second most devastating parasitic infection behind malaria. Of the species that affect humans, the most widespread cause of schistosomiasis is Schistosoma mansoni, from which egg deposition in digestive tissues causes chronic disability and morbidity in endemic regions.
Current control strategies rely mainly on chemotherapy with praziquantel (PZQ). Although effective, PZQ does not protect from reinfection and drug resistance is a rising concern,2,3 justifying the development of vaccines for this parasite. S. mansoni Cathepsin B (SmCB) is a cysteine peptidase predominantly found in adult worms and migratory larvae and is involved in the digestion of host blood macromolecules for nutrient acquisition. We have previously described the protective efficacy of SmCB both as an adjuvanted protein4, 5, 6 and when expressed by a Salmonella vector.7,8
Adenovirus vectored vaccines have been developed for multiple infectious diseases and tested in both healthy9, 10, 11 and immunocompromised patients12, 13, 14 showing strong induction of cellular and humoral immune responses despite the presence of pre-existing immunity to the vector.12,15,16 A positive safety profile in immunocompromised patients becomes more important in areas where S. mansoni is endemic due to co-infections with other pathogens (e.g., HIV,17,18 hepatitis,19 malaria,20 tuberculosis,18,21 etc.). In response to the 2019 COVID-19 pandemic, several vaccines using adenovirus technology were engineered since they are cost effective, and production can scale up easily to meet the needs of a global disease.22
In this study we describe the construction and preclinical evaluation of a replication-incompetent recombinant human adenovirus serotype 5 (Ad) expressing SmCB (AdSmCB) when delivered in a heterologous prime-boost method with recombinant SmCB. The development of an efficacious anti-schistosome vaccine would aid in the elimination of this parasite, protecting nearly one billion individuals at risk of infection.23
Methods
Ethics statement
All animal procedures were performed in accordance with Institutional Animal Care and Use Guidelines approved by the Animal Care and Use Committee at McGill University (Animal Use Protocol 7625). Mouse housing, husbandry, and environmental enrichment can be found within McGill standard operating procedures (SOP) #502, #508, and #509. Animals were monitored for adverse events for three days post vaccination and weekly until the end of each experiment. Humane intervention points were monitored according to McGill SOP #410. All animals were humanely sacrificed at endpoint by anaesthesia with isoflurane before euthanasia by carbon dioxide asphyxiation followed by pneumothorax and blood collection by cardiac puncture.
Cell lines and reagents
Cell lines were obtained from commercial sources, passed quality control procedures, and were certified and validated by the manufacturer. SF-BMAd-R cells were validated for identity, as human derived.24 All reagents were validated by the manufacturer and/or has been cited previously in the literature. For most reagents RRID tags have been listed in text. Detailed information on others, and further validation of cell lines and reagents can also be found in in the Reagent Repository.
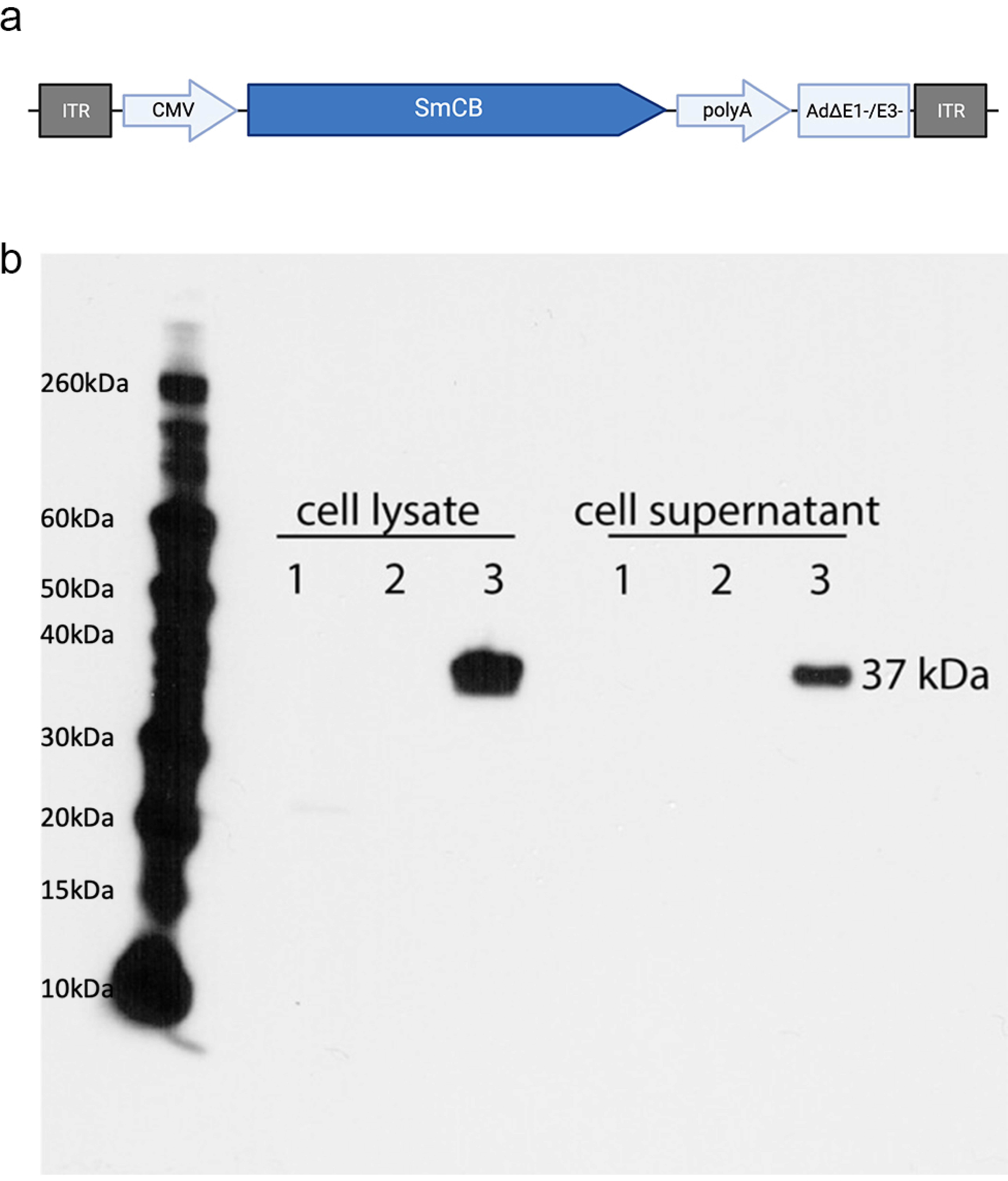
Generation of AdSmCB vector
The AdSmCB was developed following a similar protocol as described.25 Briefly, the SmCB gene cassette combined a Kozak sequence with the full length of SmCB (Genbank accession number M21309.1) followed by a proline-linked 6X histidine tag and the poly-A signal “AATAAAATATCTTTATTTTCATTACATCTGTGTGTT GGTTTTTTGTGTG” (GenScript, Piscataway, NJ, USA) (Supplemental Fig. 1). The full cassette was synthesized and codon optimized to mouse and human expression by Integrated DNA Technologies (Coralville, IA, USA) and cloned into the vector, pShuttle-CMV-Cuo.26 The plasmid containing our recombinant non-replicating human adenovirus serotype 5 (E1 and E3 genes removed (ΔE1-, ΔE3-); 1st generation) encoding the S. mansoni Cathepsin B gene was made through homologous recombination in AdEasier-1 cells (strain), a gift from Bert Vogelstein (Addgene plasmid #16399) (Addgene, Watertown, MA, USA).27 It was then linearized with PacI and transformed into HEK293A cells (RRID:CVCL_6910). Our recombinant adenovirus was then amplified using SF-BMAd-R cells,24 purified by ultracentrifugation on CsCl gradients as described previously,28 and titrated using the Adeno-X RapidTiter Kit (Clontech, Mountain View, CA, USA). A second human adenovirus serotype 5 (ΔE1-, ΔE3-; 1st generation), lacking a gene cassette, was used as a negative control.
Western blot assays
Western blot analysis to determine protein expression of SmCB by AdSmCB was performed after infection of HEK293A cells. Briefly, cells were infected at a multiplicity of infection of 5 particles per cell and incubated for 48–72 h followed by the lysis of cells using Lysis Buffer (0.1M Tris, 10 μL EGTA, 50 μL Triton-100, 0.1M NaCl, 1mM EDTA, 25 μL 10% NaDeoxycholate, 1X protease inhibitor, in ddH2O). Cell supernatants and lysates were resolved on an SDS-PAGE gel under reducing conditions followed by transfer onto a nitrocellulose membrane. The membrane was subsequently blocked in phosphate buffered saline (PBS) with 0.05% Tween 20 (PBS-T) and (Fisher Scientific, Ottawa, ON, Canada) 5% milk (Smucker Foods of Canada Corp, Markham, ON, Canada) (PBS-TM). The membrane was then incubated with mouse monoclonal anti-polyHistidine (RRID:AB_258251) antibody diluted 1:5000 in PBS-TM overnight at 4°C. The membrane was then washed in PBS-T before incubation with horseradish peroxidase (HRP)-conjugated anti-mouse IgG (IgG-HRP) (Sigma Aldrich) diluted 1:20 000 in PBS-T for one hour at room temperature. After incubation the membrane was washed again and developed using SuperSignal West Pico Plus Chemiluminescent Substrate (ThermoFisher Scientific, Waltham, MA, USA).
S. mansoni Cathepsin B recombinant protein preparation
S. mansoni Cathepsin B was prepared and purified as previously described.6 Briefly, the PichiaPinkTM system (Thermo Fisher Scientific) was used, and recombinant yeast cells were cultured in a glycerol medium. After three days of growth, yeast cells were induced in a methanol medium to allow expression of recombinant protein. Recombinant protein was purified by Ni-NTA chromatography (Ni-NTA Superflow by QIAGEN, Venlo, Limburg, Netherlands), eluted, and dialysed into PBS. Recombinant SmCB was analysed by Western Blot using antibodies directed at the His-tag (RRID:AB_258251).
Animals and immunization protocol
Six- to eight-week-old female C57BL/6 mice were bred from mice purchased from Charles River Laboratories (RRID:IMSR_CRL:027) (Senneville, QC, Canada). Four groups of mice (n=8) were immunized for humoral and cell-mediated immunity assessment. Another four groups of mice (n=8) were immunized and subsequently infected for parasite burden assessment. Each mouse was immunized at weeks 0, 3, and 6 (Supplemental Fig. 2) by intramuscular injection in the thigh in a total volume of 50μL. Group 1 (PBS): mice were injected with PBS (Wisent Bioproducts, St. Bruno, QC, Canada). Group 2 (SmCB): mice were immunized with 20μg of recombinant SmCB three times. Group 3 (AdNeg:SmCB): mice were immunized with 105 infectious units (IU) of an empty adenovirus containing no gene cassette, followed by two boosts of 20μg SmCB. Group 4 (AdSmCB:SmCB): mice were immunized with 105 IU of AdSmCB, followed by two boosts of 20μg SmCB. A fifth group was included in the challenge study as a control for non-specific protection from the empty adenovirus vector. Group 5 (AdNeg): mice were immunized with 105 IU of an empty adenovirus containing no gene cassette, followed by two injections of PBS. Mice were bled from the saphenous vein at weeks 0, 3, and 6. Mice immunized for humoral and cell-mediated immunity assessment were euthanised three weeks after the final vaccination and blood and spleens were collected.
Schistosoma mansoni challenge
Biomphalaria glabrata snails infected with the Puerto Rican strain of S. mansoni were provided by NIAID Schistosomiasis Resource Center of the Biomedical Research Institute (Rockville, MD, USA). At week 9, S. mansoni cercariae were shed from snails and experimental groups immunized for the challenge study were blinded and challenged with 150 parasites via tail exposure for one hour. Seven weeks post infection, animals were euthanised to assess parasite burden. Images of mouse livers were taken during dissection using a Galaxy S10 cell phone camera (Samsung Group, Seoul, South Korea). Adult worms were perfused from the hepatic portal system and counted manually.4,6 Liver sections were suspended in 10% buffered formalin phosphate (Fisher Scientific) and processed for histology as described before.6,29 Remaining liver and intestines were weighed and digested overnight at 37°C in 4% potassium hydroxide. The following day, eggs present in these tissues were counted by microscopy and adjusted per gram of tissue. Burden reductions were calculated as previously described6,29:
Serum Total SmCB-specific IgG, IgG avidity, IgM, IgE, and IgA
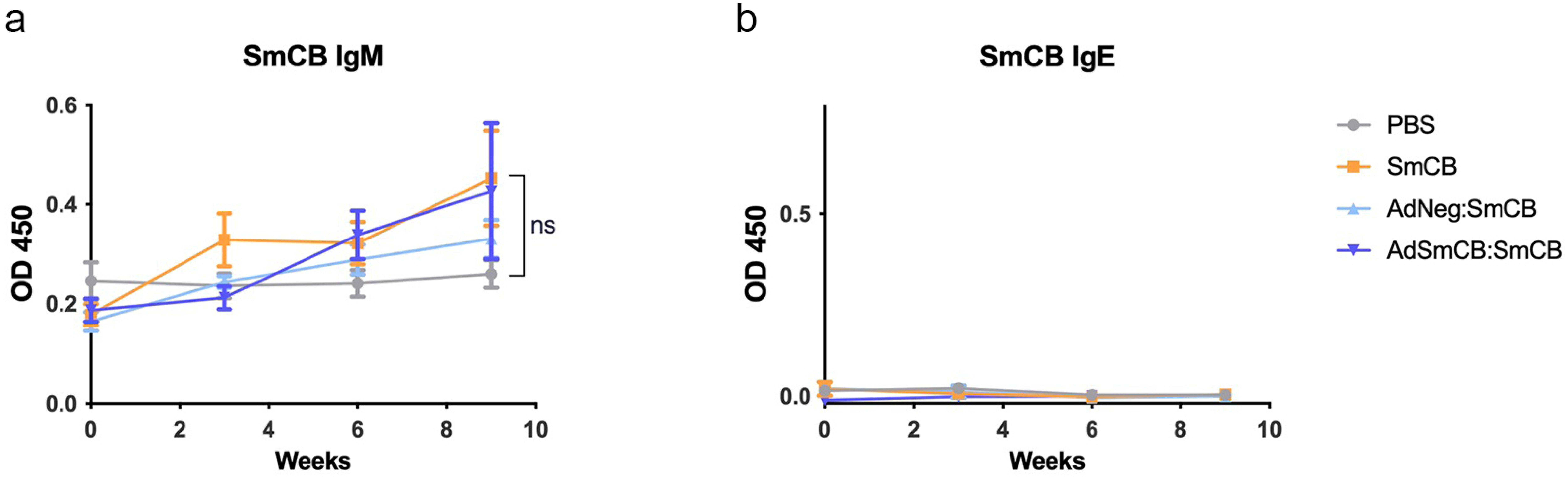
SmCB-specific serum IgG was assessed by ELISA as described elsewhere.29 Briefly, high binding 96-well plates (Greiner Bio-One, Frickenhausen, Germany) were coated with recombinant Cathepsin B (0.5 μg/mL) in 100 mM bicarbonate/carbonate buffer (pH 9.6) overnight at 4°C. Then plates were blocked with 2% bovine serum albumin (BSA; Sigma Aldrich) in PBS-T (blocking buffer) before serum samples were added in duplicate. When running serum for IgG, an additional set of serum samples were run in duplicate to determine IgG avidity. Plates were incubated for one hour at 37°C then washed with PBS (pH 7.4). IgG avidity assessment: 10M urea was added to one set of samples while blocking buffer was added to the other set and the standard curve. Plates were covered and incubated for 15 min at room temperature, washed four times, then blocked again with blocking buffer for one hour at 37°C. Next, plates were washed with PBS and anti-mouse IgG-HRP (Sigma Aldrich) was diluted 1:20 000 in blocking buffer and applied. For other immunoglobulins, the same protocol was followed without the additional avidity steps and the appropriate HRP-conjugated antibody was applied. HRP-conjugated anti-mouse IgM (RRID:AB_2794240, SouthernBiotech, Birmingham, AL, USA) or IgE (RRID:AB_2868311, Thermofisher) was diluted 1:6 000 in blocking buffer and applied. For IgA, HRP-conjugated anti-mouse IgA (Sigma Aldrich) was diluted 1:10 000 in blocking buffer and applied. Plates were washed a final time with PBS and 3,3’,5,5’-Tetramethyl benzidine (TMB) substrate (Sigma Aldrich) was added to each well. The reaction was stopped after 10 min using H2SO4 (0.5M; Fisher Scientific) and the optical density (OD) was measured at 450 nm with an EL800 microplate reader (BioTek Instruments Inc., Winooski, VT, USA). Concentrations of SmCB specific IgG and IgA were calculated by extrapolation from respective standard curves. IgG avidity indices were calculated by dividing the IgG titre in the urea condition by the IgG titre in the non-treated condition. IgM and IgE were reported as OD values.
Serum SmCB-specific IgG1, and IgG2c
SmCB-specific serum IgG1, and IgG2c were assessed by ELISA as described elsewhere.6,29 Briefly, Immunolon 2HB flat-bottom 96-well plates (Thermofisher) were coated with recombinant SmCB (0.5 μg/mL) in 100 mM bicarbonate/carbonate buffer (pH 9.6). Plates were washed with PBS-T and blocking buffer was applied for 90 min. A serial dilution of serum was applied to plates in duplicate and incubated for 2 h at 37°C. Plates were washed again with PBS-T, and goat anti-mouse IgG1-HRP (RRID:AB_2794426, SouthernBiotech) or goat anti-mouse IgG2c-HRP (RRID:AB_2794462, SouthernBiotech) was applied to plates for one hour at 37°C. After a final wash, TMB was added followed by H2SO4. Again, OD was measured as above. IgG1 and IgG2c endpoint titres were calculated as the reciprocal of the highest dilution which gave a reading above the cut-off. The endpoint titre cut-off was statistically established as described elsewhere30 using the sera of PBS immunized, unchallenged mice.
Cell-mediated immune responses
Three weeks after the last immunization, mice were euthanised, spleens were collected, and splenocytes were isolated as previously described.6 Splenocytes for multiplex ELISA assay were resuspended in RPMI-1640 supplemented with 10% foetal bovine serum, 1 mM penicillin/streptomycin, 10 mM HEPES, 1X MEM non-essential amino acids, 1 mM sodium pyruvate, 1 mM L-glutamine (Wisent Bioproducts), and 0.05 mM 2-mercaptoethanol (Sigma Aldrich) (fancy RPMI, fRPMI). Splenocytes for flow cytometry were resuspended in RPMI-1640 supplemented with 10% foetal bovine serum, 1 mM penicillin/streptomycin, and 10 mM HEPES (complete RPMI, cRPMI).
Cytokine production by multiplex ELISA
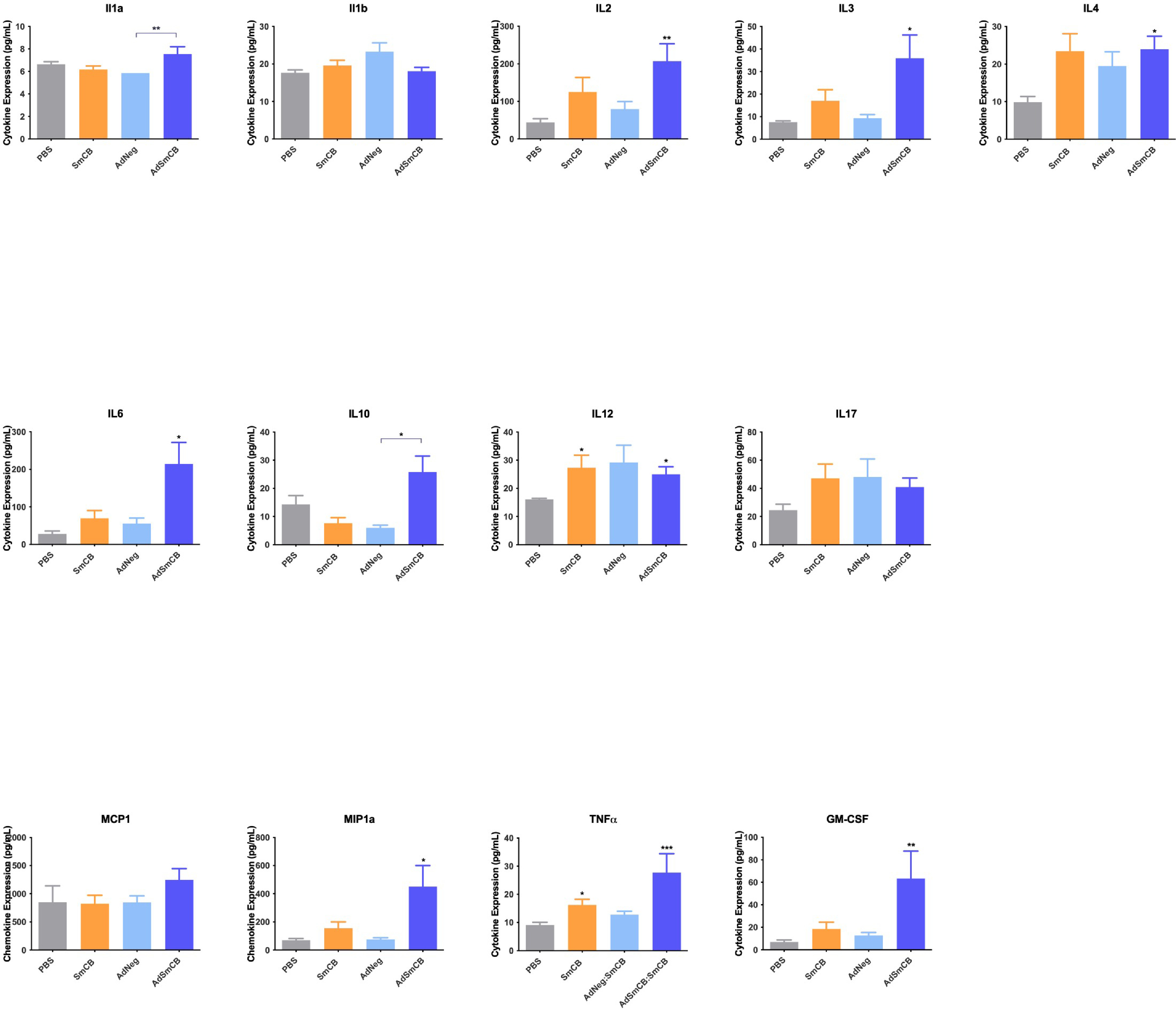
Splenocytes were incubated at 106 cells in 200 μL with SmCB in fRPMI (2.5 μg/mL recombinant protein). After 72 h at 37°C + 5% CO2, plates were centrifuged, and supernatant was collected and stored at -80°C until analysis. Cell supernatants were assessed for the presence of 16 cytokines and chemokines (IL1α, IL1β, IL2, IL3, IL4, IL5, IL6, IL10, IL12p70, IL17, IFNγ, TNFα, MCP-1 (CCL2), MIP-1α (CCL3), GM-CSF (CSF2), and RANTES (CCL5)) using Q-plex Mouse Cytokine – Screen (16-plex) multiplex ELISA following the manufacturer's guidelines (Quansys Biosciences, Logan, UT, USA). Samples were run in singlet.
T cell-mediated cytokine secretion by flow cytometry
Splenocytes were seeded into 96-well U-bottom plates (BD Falcon) at 106 cells in 200 uL/well. Duplicate cultures were stimulated with or without SmCB in cRPMI (2.5 μg/mL) for 18 h at 37°C + 5% CO2. For the last 6 h of incubation, protein transport inhibitor was prepared according to the manufacturer's guidelines (RRID:AB_2869014, BD Science, San Jose, CA, USA) and added to all samples. Cells stimulated with phorbol 12-myristate 13-acetate (Thermofisher) and ionomycin (Thermofisher) were processed as positive controls. Plates were then processed for flow cytometry as described elsewhere.31 To minimize spectral overlapping: single stain, fluorescence minus one, and unstained controls were also included. All staining and fixation steps took place at 4°C protected from light. Briefly, splenocytes were washed twice with cold PBS, and stained with 50 μL/well fixable viability dye eFluor 780 (Thermofisher) diluted at 1:300 for 20 min. Cells were washed twice using PBS with 1% BSA (PBS-BSA), and then blocked with Fc block (RRID:AB_394656, BD Science) diluted 1:50, for 15 min at 4°C protected from light. All surface stains were diluted 1:50 in PBS-BSA and 50 μL/well of extracellular cocktail was applied for 30 min. The following antibodies made up the extracellular cocktail: CD3-FITC (RRID:464883, Thermofisher), CD4-V500 (RRID:AB_1937327, BD Bioscience) and CD8-PerCP-Cy5 (RRID:AB_394081, BD Science). Cells were then washed as before and fixed with 1X fixation buffer (RRID:AB_2869005, BD Science) overnight. The next day, plates were washed twice with 1X permeabilization buffer (perm buffer) (RRID:AB_2869011, BD Science) and stained with an intracellular cocktail of antibodies diluted 1:50 in perm buffer applied as 50 μL/well for 30 min. The intracellular cocktail was made up of: IL-2-Pe-Cy5 (RRID:AB_2123674, Biolegend, San Diego, CA, USA), IFNγ-PE (RRID:AB_395376, BD Science), and TNFα-efluor450 (RRID:AB_1548825, Thermofisher). After staining, cells were washed once with perm buffer, once with PBS-BSA, and resuspended in PBS-BSA and acquired on a BD LSRFortessa X-20 (BD Science). Flow data were analysed using Flowjo software (version 10.0.8r1) (Treestar, Ashland, OR, USA) and SPICE software (version 6.1).32 Our gating strategy is shown in Supplemental Fig. 3.
Histology, egg granuloma assessment, and fibrotic area measurements
Liver sections in 10% buffered formalin phosphate were processed for histopathology and stained using haematoxylin and eosin to assess granuloma size and egg morphology and Masson's trichrome to measure fibrotic area. Granuloma sizes were measured using Zen Blue software (version 2.5.75.0; Zeiss) as previously reported.6,29,33, 34, 35, 36 Briefly, while working at 400X magnification, the pointer was used to trace the perimeter of 24-32 granulomas in an exudative-productive stage with a clearly visible egg per experimental group, which the software converted into an area. Hepatic eggs were classified as abnormal if their internal structure was lost or the perimeter of the egg was crenelated. Abnormal eggs were counted and reported as a percent of the total eggs counted per field of vision. Eighteen to 32 different fields of vision were assessed per experimental group over two independent experiments. Slides stained with Masson's trichrome were imaged using the Aperio AT Turbo digital whole slide scanning system (Leica Biosystems, Concord, ON, Canada) at 20X magnification. Twenty-five to 37 single egg formed granulomas per group were delimited and the area of visible blue was measured using QuPath 0.3.0.37
Statistical analysis
Experimental units are defined as individual animals. Sample size determination: Sample sizes (n=7) were calculated using G*Power (3.1.9.3)38 based on the means and standard deviation of preliminary data to achieve at least 90% power and allow for a five percent type I error. One mouse was added (n=8) to each group to compensate for a possible attrition rate of 10%. To minimise potential confounders, mice were matched for age, sex, and body weight. Randomisation: Mice were randomised into experimental groups before the start of each study. Blinding: For challenge experiments, staff performing infections and sample harvesting were blinded to groups, and unblinded after data analysis. Inclusion/Exclusion: No animals were excluded. For the assessment of granuloma size and fibrotic area around single eggs, outliers were calculated using the ROUT method (Q=1) and if present, they were excluded.
Statistical analysis was performed using GraphPad Prism 9 software (La Jolla, CA, USA). Data were assessed for normality using Shapiro-Wilk tests. Non-parametric data were analysed by Kruskal-Wallis tests with Dunn's multiple comparisons. When appropriate, one-way and two-way ANOVAs were employed with Tukey's multiple comparisons. P values <0.05 were considered significant.
Role of funders
Funding agencies did not have a role in the study design, data collection, data analyses, interpretation, or writing of this manuscript.
Results
Vaccination with AdSmCB:SmCB results in robust humoral responses
Humoral responses were determined throughout the immunization schedule. No mice had detectable SmCB specific IgG at baseline, and the PBS control remained negative throughout the study. Mice receiving SmCB developed IgG antibody titres by week 3, whereas mice receiving recombinant Ad as a primary immunization showed detectable IgG titres only after immunization with a protein boost. However, by week 6 IgG titres between SmCB and AdSmCB:SmCB groups were no longer significantly different and at the end of the immunization period (week 9) AdSmCB:SmCB produced significantly higher titres than the AdNeg:SmCB group (p=0.0261, Kruskal-Wallis test) (Figure 1a). We also sought to determine antigen-specific IgG avidity (Figure 1b) and IgG subtypes at the time of infection. All vaccinated animals produced highly avid IgG antibodies. Although there was no significant difference in IgG avidity between groups SmCB and AdSmCB:SmCB, mice which received the AdSmCB prime showed significantly greater avidity compared to the group which was primed with the empty Ad vector followed by 2x SmCB protein (AdNeg:SmCB) (p=0.0371, one-way ANOVA). There were no statistical differences between the amount of IgG1 produced by any of the experimental groups (Figure 1c), however AdSmCB:SmCB significantly increased the production of SmCB specific IgG2c (3.66e5 ± 1.39e5) when compared to both the SmCB (9.64e3 ± 7.78e3) (p=0.0017) and AdNeg:SmCB groups (3.05e3 ± 9.04e2) (p=0.0037, Kruskal-Wallis test) (Figure 1d). Finally, when compared to the SmCB group, the ratio of IgG1 to IgG2c was significantly reduced in mice first given a priming immunization of recombinant adenovirus (p=0.0046, Kruskal-Wallis test) (Figure 1e). Throughout the immunization schedule, all vaccinated animals saw a trend of increasing antigen-specific IgM, however this trend was not significant. Additionally, no animals developed antigen-specific IgE or IgA in response to vaccination (Supplemental Figure 4).
Figure 1.

Humoral response to vaccination. (a) SmCB-specific IgG titres measured by ELISA. Immunizations are denoted by arrows. (b–e) Further analysis of the IgG antibody response at time of challenge (week 9). (b) Avidity of antigen-specific IgG reported as the avidity index. SmCB binding (c) IgG1 and (d) IgG2c measured by endpoint titre ELISA. (e) IgG immune skewing represented by the ratio of IgG1/IgG2c. All data are presented as the mean ± SEM of two independent experiments. (n=8). *p<0.05, **p<0.01, ***p<0.001 ((a,d-e) analysed using Kruskal-Wallis test (b-c) analysed using one-way ANOVA).
AdSmCB:SmCB enhances cytokine and chemokine expression
To determine the immune landscape of lymphocyte responses created by vaccination, we ran a multiplex ELISA on the supernatants of stimulated splenocytes. For many of the cytokines and chemokines tested, the AdSmCB:SmCB group generated elevated levels of molecular signals as shown in the radar plot (Figure 2a). Each vaccine formulation can be seen to produce a unique cytokine and chemokine signature. Notably, AdSmCB:SmCB maintains the significant expression of IL5 (p=0.0100) also seen in the SmCB group (p=0.0308, Kruskal-Wallis test) (Figure 2b), while enhancing expression of IFNγ from both PBS (p=0.0009) and AdNeg:SmCB groups (p=0.0152, Kruskal-Wallis test) (Figure 2c), and RANTES (CCL5) compared to SmCB alone (p=0.0100, Kruskal-Wallis test) (Figure 2d), among others (Supplemental Figure 5).
Figure 2.

Cell-mediated memory responses to SmCB. (a) Mean levels of cytokines and chemokines from restimulated splenocytes shown in the radar plot. Data are calculated as the fold change above the PBS control along the axis in log scale. Bar graphs depicting expression levels of cytokines (b) IL5, (c) IFNγ, and chemokine (d) RANTES (CCL5). All data are presented as the mean ± SEM of two independent experiments. (n=8). *p<0.05, **p<0.01, ***p<0.001 (Kruskal-Wallis test).
AdSmCB:SmCB increases IFNγ+ T cell frequency and promotes CD4+ T cell polyfunctionality
IFNγ is a key contributor of protection in Schistosoma radiation attenuated vaccine models, so we were interested in its increased expression in mice vaccinated with our vectored vaccine. Since AdSmCB:SmCB immunized animals also displayed elevated levels of RANTES, a T-cell associated chemokine, we used flow cytometry to determine if T cells could be responsible for IFNγ production. Indeed, when splenic T cells were stimulated ex vivo with SmCB, we observed an increased frequency of CD4+ T cells (Figure 3a) expressing IFNγ in mice vaccinated with AdSmCB:SmCB (0.141 ± 4.27e-2%) compared to PBS (0.013 ± 8.40e-3%) (p=0.0032), SmCB (0.015 ± 8.86e-3%) (p=0.0040), and AdNeg:SmCB (0.026 ± 8.00e-3%) (p=0.0105, two-way ANOVA). The percent of CD4+ T cells expressing IL2 was also increased in the recombinant adenovirus group (0.098 ± 5.85e-2%) when compared to the PBS control (0.001 ± 7.89e-4%) (p=0.0429, two-way ANOVA). Using Boolean and SPICE analyses, we then assessed the polyfunctional profiles of our experimental groups. Figure 3b shows the distribution of CD4+ T cell populations expressing one, two, and three cytokines. We saw that our recombinant adenovirus elicited a larger repertoire of polymorphic CD4+ T cells than the recombinant protein and empty viral vector groups, with an emergence in triple positive cells (IFNγ+TNFα+IL2+), as well as IFNγ+TNFα+, and IL2+TNFα+ cells. Since these pie charts are not to scale, we included a heat map which graphically represents the frequencies of cells in each polymorphic category. Each category depicts a different cytokine expression profile which has been established in the legend to the right of the heat map. By the increased intensity of red observed in our recombinant adenovirus group, we saw that AdSmCB:SmCB has a larger proportion of each CD4+ T cell type (categories 1-6) except for those expressing only TNFα (category 7) which was higher in the SmCB vaccinated mice. While there was a marked increase in cells expressing IFNγ alone (category 4) and IL2 alone (category 6), the increased percentage of CD4+ T cells expressing more than one cytokine can be easily visualized (categories 1-3, and 5) within the heat map. When looking at the proportion of AdSmCB:SmCB CD8+ T cells expressing IFNγ (0.158 ± 6.68e-2%) we again see a striking increase when compared to all other groups: PBS (0.021 ± 1.29e-2%) (p=0.0026), SmCB (0.041 ± 1.41e-2%) (p=0.0137), and AdNeg:SmCB (0.014 ± 1.10e-2%) (p=0.0014, two-way ANOVA) (Figure 3c). We saw similar trends of increased TNFα expression from both CD4+ and CD8+ T cells in groups SmCB and AdSmCB:SmCB; however, these were not significant. Although Boolean analysis and pie chart depictions of each vaccine resulted in a unique CD8+ T cell signature (Figure 3d), the differences between groups were far less drastic than in the case of the CD4+ T cells, as seen in the corresponding heat map. In summary, our polymorphic T cell analysis nicely corroborated the striking expression of IFNγ and IL2 witnessed in our AdSmCB:SmCB vaccinated animals.
Figure 3.

Responding T cell signature. Frequencies of (a) CD4+ and (c) CD8+ T cells expressing IFNγ, IL2, and TNFα shown as % of the parent population. Polyfunctional signatures of both (b) CD4+ and (d) CD8+ T cells shown in representative pie charts. Heat maps were included, for each subset of T cell, to describe the relative amounts of each polyfunctional profile. Both heat maps show the percentage of CD4+ or CD8+ T cell in each category on a continuum from 0% (blue) to increasing % (red). Numbered and colour coded categories within each pie chart/heat map represent various T cell profiles of cytokine expression and are explained in the included legends. All data are presented as the mean ± SEM of net values (stimulated cells – unstimulated cells) from two independent experiments. (n=8). *p<0.05, **p<0.01 (two-way ANOVA).
AdSmCB:SmCB significantly reduces parasite burden
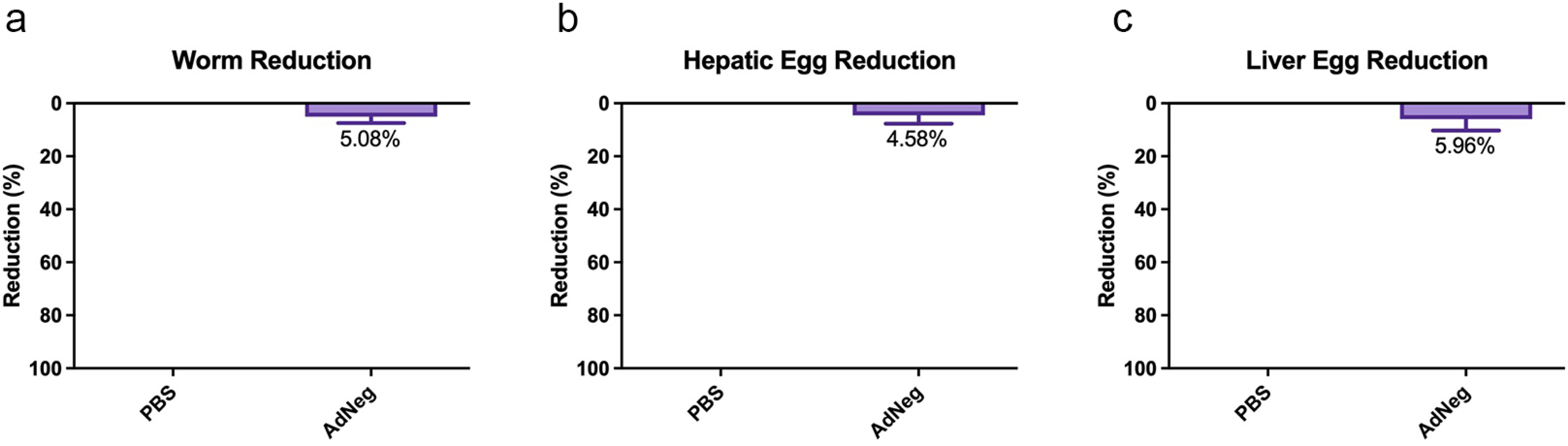
To determine the protective efficacy of our Ad vaccine, immunized animals were infected with S. mansoni and assessed for adult worms, hepatic eggs, and intestinal eggs. A fifth group of mice, vaccinated with an empty adenovirus vector without protein boosts, was included to control for any non-specific protective capacity of the vector itself. The average amount of adult worms collected from control mice was 37 ± 7 worms over two independent experiments. Relative reduction was calculated against the PBS control group within the same experiment to reduce batch discrepancy between infections. The AdNeg vector group was unable to significantly reduce adult worm burden from the PBS control (Supplemental Figure 6). However, when this empty vector was boosted twice with recombinant protein, protection increased to 24.2 ± 8.1% (p=0.0380, one-way ANOVA) (Figure 4a). Worm burden was further reduced in animals vaccinated with 3 doses of recombinant protein, and those initially primed with our recombinant Ad by 42.8 ± 4.2% (p=0.0001) and 71.7 ± 7.8% (p<0.0001, one-way ANOVA), respectively. The main cause of pathology in schistosomiasis is egg deposition by adult worms. Therefore, egg burden reductions in both livers (Figure 4b) and intestines (Figure 4c) were also calculated. Hepatic eggs averaged 14 096 ± 3 953 eggs per gram and intestinal eggs averaged 15 327 ± 4 705 eggs per gram of tissue in the PBS control. Similar to worm reduction, AdNeg alone was unable to confer any significant protection from egg deposition. When boosted twice with recombinant protein, mice initially immunized with the AdNeg vector had liver and intestinal egg reductions of 22.3 ± 7.1% (p=0.0245) and 22.4 ± 7.4% (p=0.0798, one-way ANOVA), respectively. Animals immunized with recombinant protein alone reduced liver and intestinal eggs by 42.9 ± 4.8% (p<0.0001) and 41.6 ± 5.4% (p=0.0004), respectively, whereas animals immunized with AdSmCB:SmCB were protected from liver and intestinal eggs by 68.6 ± 5.8% (p<0.0001) and 75.7 ± 8.7% (p<0.0001, one-way ANOVA), respectively.
Figure 4.

Parasite burden reduction. Reduction from the PBS control of (a) adult worms, (b) hepatic eggs, and (c) intestinal eggs at week 16. All data are presented as the mean ± SEM of two independent experiments. (n=8). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (one-way ANOVA).
Liver pathology is markedly reduced in vaccinated animals
During animal dissection, images were taken of whole livers. Visual analysis showed an increased number of granulomas (white formations) and hepatomegaly in infected PBS mice. Despite the presence of granuloma formation, livers in all vaccinated animals showed reduced pathology post infection, which was marked in groups SmCB and AdSmCB:SmCB (Figure 5a). Microscopic examination of liver tissue stained by haematoxylin and eosin or Masson's trichrome was used to assess granuloma formation (Figure 5b) and egg-induced liver fibrosis (Figure 5c), respectively. Granulomas in control mice were large and well formed with an average size of 69 982 ± 7 636 μm2 harbouring intact eggs with normal appearances. When compared to both the PBS control and the AdNeg:SmCB groups, recombinant protein (SmCB) and recombinant adenovirus (AdSmCB:SmCB) groups were able to reduce granuloma sizes to 38 902 ± 2 954 μm2 (p=0.0095, p=0.0003) and 37 796 ± 4 189 μm2 (p=0.0089, p=0.0004, Kruskal-Wallis test), respectively (Figure 5d). Larger and more developed granulomas were found to have collagen deposition within them. Only animals vaccinated with AdSmCB:SmCB displayed a reduction in egg-induced fibrotic areas compared to control mice, from 114 815 ± 13 575 μm2 to 64 891 ± 7 146 μm2 (p=0.0159, Kruskal-Wallis test) (Figure 5e). Through microscopic visualization, we also determined the amount of liver eggs which were abnormal in structure. Although the number of abnormal eggs increased in all mice which received recombinant protein, only the AdSmCB:SmCB group showed a significantly increased proportion (Supplemental Figure 7), reaching 32 ± 4% compared to the 14 ± 4% of the PBS control (p=0.0136) and the 12 ± 4% (p=0.0135, Kruskal-Wallis test) of the SmCB group.
Figure 5.

Pathological outcomes. (a) Whole livers imaged post-infection showing visual pathology. Liver portions were subsequently prepared for histology and stained using haematoxylin and eosin or Masson's trichrome. Representative images show qualitative (b) granuloma sizes (outlined in orange) and (c) egg-induced fibrosis (collagen stained in blue) for each group. Scale bars represent 100 μm. (n=8). Quantitative (d) granuloma sizes (n=24-32) and (e) fibrotic areas (n=25-37) delimited and measured using ZenBlue and QuPath software, respectively. All data are presented as the mean ± SEM of two independent experiments. *p<0.05, **p<0.01, ***p<0.001 (Kruskal-Wallis test).
Discussion
Schistosomiasis continues to be a major public health problem despite ongoing control efforts. The emergence of drug resistant strains and high reinfection rates after drug therapy highlight the need for additional anti-schistosome tools.39, 40, 41 The development of an effective vaccine against Schistosoma is of global importance. Although many preclinical efforts are in the pipeline, none yet have been approved for human use. Current Schistosoma vaccine strategies include recombinant protein and DNA-based vaccines; however, recent work has demonstrated protection via pathogen-vectored vaccines, for example Salmonella YS1646.7,8 With the development, proposed safety, and wide distribution of adenoviral vectored vaccines during the SARS-CoV-2 pandemic, we decided to develop our own vaccine using this technology. Only a single other adenoviral vectored vaccine has been tested in models of schistosomiasis.42,43 In our work, we targeted the most widespread species causing human infection, S. mansoni. Our group has previously demonstrated the protective efficacy of recombinant SmCB4, 5, 6 which acts primarily through Th2 mediated immunity.44 Therefore, in our present study, we focused on increasing this protection through the use of viral vectoring and heterologous prime-boosting.
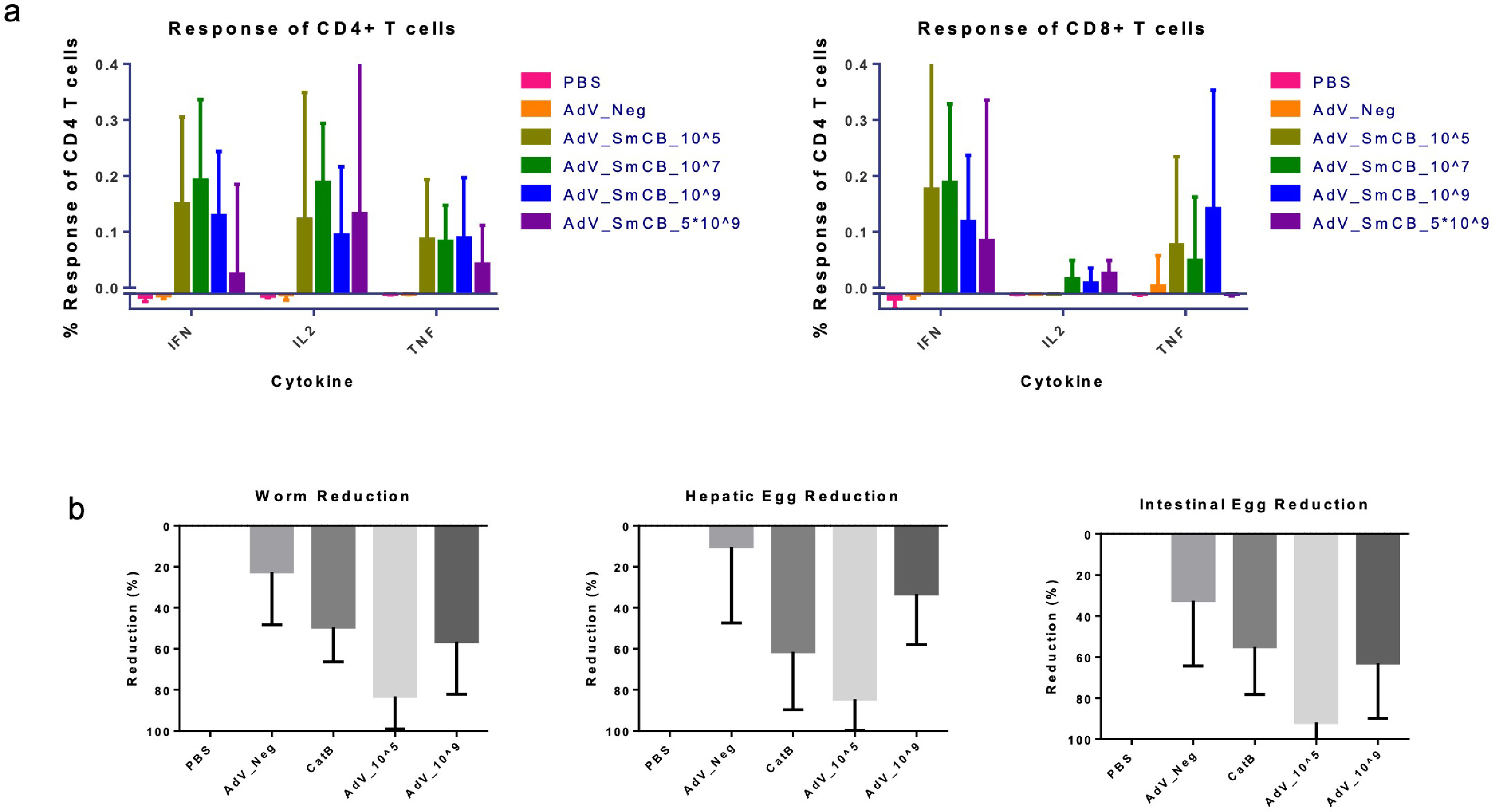
Based on preliminary dose response studies, we found that contrary to the common delivery of high doses of viral vector in the literature (>10^7 IU), our AdSmCB elicited similar T cell cytokine expression and higher protective capacity at very low doses (10^5 IU) (Supplemental Figure 8), which may be preferable to prevent vaccine related adverse events. Our vaccine strategy, using a recombinant viral vector prime followed by protein boosts, offered protection from S. mansoni infection, well surpassing the WHO 40% threshold indicating significance, and practically reaching the 75% threshold proposed at a National Institute of Allergy and Infectious Diseases schistosomiasis vaccine meeting.45 Parasite burden reduction seems to be dependent on priming with a SmCB-expressing adenovirus followed by recombinant protein boosts as protection was lower in mice which received homologous immunizations of SmCB protein alone and abrogated in animals which received either an empty adenovirus vector alone or the empty vector boosted by SmCB.
Humoral responses and antibodies targeting secreted proteins, such as the abundantly expressed SmCB, have been suggested to play a key role in cure from schistosomiasis.46 Intramuscular immunization with our recombinant adenovirus vectored vaccine resulted in trends of increased antigen-specific IgM and significant expansions of highly avid antigen-specific IgG. The role of IgM in schistosomiasis is not well defined. Although some groups have shown putative effects of IgM hindering protection mediated through other antibody isotypes47,48 it has also been shown to recognize Schistosoma epitopes, kill larvae in vitro, and provide passive protection in vivo.49 We hypothesize that pentameric IgM may play a helping role in our vaccine by broadly sequestering the peptidase activity of SmCB contributing to parasite starvation, prior to specialized antibody isotype switching. More solidly, the protective effects of IgG have been demonstrated numerous times.50, 51, 52 Although antibody production was delayed in contrast to mice which received recombinant protein alone, mice primed with AdSmCB exhibited comparable levels of antigen-specific IgG by the time of infection. Antibodies produced by both AdSmCB:SmCB and SmCB vaccinated mice displayed high avidity, likely due to boosting immunizations of antigen, and given the Th1 skewing nature of adenoviral vectors, we were not surprised to see a dramatic increase in antigen-specific IgG2c antibodies. Despite overall humoral responses skewing towards Th1 immunity, our AdSmCB:SmCB vaccine maintained analogous levels of antigen-specific IgG1 when compared with the recombinant protein alone group. IgG1 has been explicitly correlated with protection from schistosomiasis in animal models53,54; protection by IgG2 antibodies has also been described.55,56 Additionally, we assessed antigen-specific IgA and IgE, which did not seem to be elicited by our vaccines. Although total IgE has shown protection from many parasitic worms,48,57, 58, 59 the lack of vaccine-induced antigen-specific IgE is a promising feature of AdSmCB:SmCB to avoid allergy-type hypersensitivity reactions which have been detrimental to helminth vaccine safety.60
Splenocyte memory responses to antigen revealed an increased level of cytokine and chemokine expression from our recombinant adenovirus vaccine superior to both protein alone and AdNeg:SmCB. Splenocytes from mice vaccinated with recombinant protein alone showed significant production of IL5, a key mediator of eosinophil activation and differentiation. Several early studies have demonstrated schistosomula killing dependent on eosinophils.61, 62, 63 SmCB is expressed as early as the schistosomula stage and its suppression by RNA interference resulted in growth retardation.64 We saw that our recombinant adenovirus vaccine also increased IL5, which may mean targeting lung-stage larvae before their maturity into egg-laying adult worms.
A hallmark of schistosomiasis protection, which was brought to light during the evaluation of radiation attenuated schistosome vaccines, is IFNγ.65,66 This cytokine was only increased in those mice which received our adenovirus vectored vaccine. Since the production of RANTES (CCL5) was also increased, even compared to the SmCB group, we were curious to determine if T cells could be responsible for IFNγ expression. We found that not only was the frequency of AdSmCB:SmCB T cells expressing IFNγ elevated compared to all other groups, but CD4+ T cells expressing IL2 was also increased. Of note, when we assessed memory responses from restimulated T cells, we found varying functionalities between vaccine groups. While CD8+ T cell phenotypes were similar between groups we saw a marked increase in the polyfunctionality of CD4+ T cells when mice were vaccinated with our recombinant adenovirus. Although the role of polyfunctional T cells in schistosomiasis protection remains elusive, their contributions have been described in models of yellow fever67,68 and influenza as functionally superior cells which exhibit increased degranulation and expression of CD40L and Th1 cytokines: IFNγ, IL2, and TNFα.69,70 Data from both murine and human studies corroborate polyfunctional cell protection from influenza lethality and disease severity respectively.71,72 Interestingly, these cells have also been identified as key players in immunity conferred by vectored vaccines. A smallpox vaccine elicited polyfunctional T cells specific to vaccinia virus which extended to vaccine expressed HIV gene products.73 Further, triple positive CD4+ T cells (IFNγ+IL2+TNFα+), a subset of cells which were increased only in our AdSmCB:SmCB group, delivered protection in a parasite infection model where an adenovirus vectored vaccine was tested against Leishmania major.74
Schistosomiasis pathology is caused by the induction of Th2 responses by the release of soluble egg antigens from eggs trapped in host tissue. As SmCB expression is continued into the adult worm life cycle stage, we hypothesize that steady pressure on a worms’ ability to acquire nutrients will lead to a reduction in its fitness and, in turn, that of the eggs it produces. In support, when we evaluated visual fields of egg clusters under a microscope, there was a significantly larger proportion of eggs which were crenelated with a loss of internal structures in mice vaccinated with AdSmCB:SmCB. Through vaccination we were able to prevent many manifestations of liver pathology, including granuloma size and fibrotic area, normally caused by S. mansoni infection. We also witnessed visual protective effects on gross livers in those mice which were vaccinated with SmCB alone or our recombinant adenovirus prior to challenge.
Though encouraging, there are limitations to our current study. The use of adenovirus based vaccines has been criticised due to neutralising antibodies to the vector and the induction of vaccine related adverse events.75 Although some research has shown antigen-specific immune responses despite pre-existing anti-adenovirus immunity, these responses may be futile if adverse events are inherent of adenoviral vectors. To circumvent these issues, we are exploring the expression of our target antigen from other viral vectors. A second limitation is the use of the mouse model for testing S. mansoni vaccine efficacy. It has been proposed that, due to physiological features of the murine pulmonary system, vaccine efficacy in mouse models may be over-exaggerated (caused by non-specific, vaccine-induced systemic T cell activation and cytokine levels being maximal at the time schistosomes passage through the lungs).76 Despite mice being the most feasible animal model for screening schistosomiasis vaccines, future studies will be needed to determine if protection can be replicated in other animals (e.g., non-human primates) and when parasite challenge is delayed. Finally, this vaccine was tested in a prophylactic capacity without drug intervention, which is not fully reflective of endemic areas where many cases go undiagnosed and individuals are likely already infected. Future directions include testing our adenovirus vectored vaccine in therapeutic models and in reinfection models after chemotherapy.
Protective correlates of immunity for helminthic infections are widely debated, thus we broadly assessed immune responses (including immunoglobulins, cytokines, and chemokines). However, the careful balancing of targeted Th1 and Th2 responses has been proposed.77 Due to the complex nature of parasitic infections and their inherent modulation of the host immune system, we expect that a multipronged immune response would be necessary for cure. Our data suggest that the use of adenovirus as a vector alters the natural Th2 skewing of the immune system to SmCB, facilitating enhanced cell-mediated immunity without hindering protection offered by the humoral response. We believe our heterologous strategy could be improved by adjuvanting protein boosts to further augment immune responses thereby increasing protection.
In summary, our findings describe a viral vectored vaccine which prophylactically protects from schistosomiasis, at levels comparable to others in pre-clinical work and those currently in clinical trials, through a platform which has been widely used in humans and can be easily up scaled for global production. Our adenovirus vectored vaccine elicits strong humoral immunity and cellular effectors, balancing Th2 and Th1 arms of immunity to target SmCB-expressing larvae and adult worms. More importantly, parasite burden reduction by our vaccine led to a prevention of pathology caused by S. mansoni egg deposition, which is crucial to alleviating chronic morbidities and may significantly aid regions where coinfections make liver pathologies lethal.
Contributors
Experimental design was conducted by DJP and MN in collaboration with RDW and RG. Adenoviral constructs were designed and engineered by DJP with assistance from SME and CG. Experiments were conducted by DJP. ASH and SSL assisted with animal sacrifice, sample collection, and sample processing. The manuscript was prepared by DJP and MN, with revisions by RDW and RG. The data presented here were verified by DJP and ASH. All authors have read and approved the final version of this manuscript.
Data sharing statement
The data supporting the conclusions of this study are available within the article and its Supplementary Data files. Raw data files will be made available by the authors upon request.
Declaration of Competing Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
We thank Annie Beauchamp for her assistance with animal work, and Lydia Labrie, Francesca Battelli, and Jonathan Starr for their assistance with sample collection and processing. We would also like to thank Dr. Margaret Mentink-Kane and Kenia V. Benitez from the Biomedical Research Institute (Rockville, MD) for supplying us with infected Biomphalaria snails, as well as the Immunophenotyping and Histopathology cores at the Research Institute of the McGill University Health Centre (Montreal, QC). The National Reference Centre for Parasitology is supported by Public Health Agency of Canada/National Microbiology Laboratory, the Foundation of the Montreal General Hospital, the Foundation of the McGill University Health Centre, the Research Institute of the McGill University Health Centre, and the R. Howard Webster Foundation. This work was supported by the Canadian Institutes of Health Research, R. Howard Webster Foundation, and the Foundation of the McGill University Health Centre.
Footnotes
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.ebiom.2022.104036.
Appendix. Supplementary materials
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.World Health Organization . World Health Organization; 2022. Schistosomiasis.https://www.who.int/news-room/fact-sheets/detail/schistosomiasis [Internet]. 2022 [cited 2022 Mar 26]. Available from: [Google Scholar]
- 2.McManus D.P., Bergquist R., Cai P., Ranasinghe S., Tebeje B.M., You H. Schistosomiasis-from immunopathology to vaccines. Semin Immunopathol. 2020;42(3):355–371. doi: 10.1007/s00281-020-00789-x. https://link.springer.com/article/10.1007/s00281-020-00789-x Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vale N., Gouveia M.J., Rinaldi G., Brindley P.J., Gärtner F., Da Costa J.M.C. Praziquantel for schistosomiasis: Single-drug metabolism revisited, mode of action, and resistance. Antimicrob Agents Chemother. 2017;61(5) doi: 10.1128/AAC.02582-16. https://journals-asm-org.proxy3.library.mcgill.ca/doi/full/10.1128/AAC.02582-16 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ricciardi A., Visitsunthorn K., Dalton J.P., Ndao M. A vaccine consisting of Schistosoma mansoni cathepsin B formulated in Montanide ISA 720 VG induces high level protection against murine schistosomiasis. BMC Infect Dis. 2016;16(1):112. doi: 10.1186/s12879-016-1444-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ricciardi A., Dalton J.P., Ndao M. Evaluation of the immune response and protective efficacy of Schistosoma mansoni Cathepsin B in mice using CpG dinucleotides as adjuvant. Vaccine. 2015;33(2):346–353. doi: 10.1016/j.vaccine.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 6.Perera D.J., Hassan A.S., Jia Y., et al. Adjuvanted schistosoma mansoni-cathepsin b with sulfated lactosyl archaeol archaeosomes or AddaVaxTM provides protection in a pre-clinical schistosomiasis model. Front Immunol. 2020;0:2990. doi: 10.3389/fimmu.2020.605288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hassan A.S., Zelt N.H., Perera D.J., Ndao M., Ward B.J. Vaccination against the digestive enzyme Cathepsin B using a YS1646 Salmonella enterica Typhimurium vector provides almost complete protection against Schistosoma mansoni challenge in a mouse model. PLoS Negl Trop Dis. 2019;13(12) doi: 10.1371/journal.pntd.0007490. https://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0007490 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hassan A.S., Perera D.J., Ward B.J., Ndao M. Therapeutic activity of a Salmonella-vectored Schistosoma mansoni vaccine in a mouse model of chronic infection. Vaccine. 2021;39(39):5580–5588. doi: 10.1016/j.vaccine.2021.08.031. [DOI] [PubMed] [Google Scholar]
- 9.Zhu F.C., Guan X.H., Li Y.H., et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2020;396(10249):479–488. doi: 10.1016/S0140-6736(20)31605-6. http://www.thelancet.com/article/S0140673620316056/fulltext Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baden L.R., Stieh D.J., Sarnecki M., et al. Safety and immunogenicity of two heterologous HIV vaccine regimens in healthy, HIV-uninfected adults (TRAVERSE): a randomised, parallel-group, placebo-controlled, double-blind, phase 1/2a study. Lancet HIV. 2020;7(10):e688–e698. doi: 10.1016/S2352-3018(20)30229-0. http://www.thelancet.com/article/S2352301820302290/fulltext Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cicconi P., Jones C., Sarkar E., et al. First-in-human randomized study to assess the safety and immunogenicity of an investigational respiratory syncytial virus (RSV) vaccine based on chimpanzee-adenovirus-155 viral vector–expressing RSV fusion, nucleocapsid, and antitermination viral proteins in healthy adults. Clin Infect Dis. 2020;70(10):2073–2081. doi: 10.1093/cid/ciz653. https://academic.oup.com/cid/article/70/10/2073/5537685 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balint J.P., Gabitzsch E.S., Rice A., et al. Extended evaluation of a phase 1/2 trial on dosing, safety, immunogenicity, and overall survival after immunizations with an advanced-generation Ad5 [E1-, E2b-]-CEA(6D) vaccine in late-stage colorectal cancer. Cancer Immunol Immunother. 2015;64(8):977–987. doi: 10.1007/s00262-015-1706-4. https://link.springer.com/article/10.1007/s00262-015-1706-4 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morse M.A., Chaudhry A., Gabitzsch E.S., et al. Novel adenoviral vector induces T-cell responses despite anti-adenoviral neutralizing antibodies in colorectal cancer patients. Cancer Immunol Immunother. 2013;62(8):1293–1301. doi: 10.1007/s00262-013-1400-3. https://link.springer.com/article/10.1007/s00262-013-1400-3 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.University Hospitals of North Midlands NHS Trust. Covid-19 Vaccine Response in Immunocompromised Haematology Patients (COVAC-IC). 2021 Mar 18 [last updated 2022 Mar 4]. In: ClinicalTrials.gov [Internet]. Bethesda (MD): U.S. National Library of Medicine. Available from: https://clinicaltrials.gov/ct2/show/NCT04805216?term=BNT162b2&cond=immunocompromised&draw=2 Identifier: NCT04805216.
- 15.Osada T., Yang X.Y., Hartman Z.C., et al. Optimization of vaccine responses with an E1, E2b and E3-deleted Ad5 vector circumvents pre-existing anti-vector immunity. Cancer Gene Ther. 2009;16(9):673–682. doi: 10.1038/cgt.2009.17. https://www.nature.com/articles/cgt200917 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smaill F., Jeyanathan M., Smieja M., et al. A human type 5 adenovirus-based tuberculosis vaccine induces robust T cell responses in humans despite preexisting anti-adenovirus immunity. Sci Transl Med. 2013;5(205) doi: 10.1126/scitranslmed.3006843. https://www.science.org/doi/abs/10.1126/scitranslmed.3006843 Available from: [DOI] [PubMed] [Google Scholar]
- 17.Furch B.D., Koethe J.R., Kayamba V., Heimburger D.C., Kelly P. Interactions of Schistosoma and HIV in Sub-Saharan Africa: a systematic review. Am J Trop Med Hyg. 2020;102(4):711–718. doi: 10.4269/ajtmh.19-0494. https://www.ajtmh.org/view/journals/tpmd/102/4/article-p711.xml Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLaughlin T.A., Nizam A., Hayara F.O., et al. Schistosoma mansoni infection is associated with a higher probability of tuberculosis disease in HIV-infected adults in Kenya. J Acquir Immune Defic Syndr. 2021;86(2):157–163. doi: 10.1097/QAI.0000000000002536. https://journals-lww-com.proxy3.library.mcgill.ca/jaids/Fulltext/2021/02010/Schistosoma_mansoni_Infection_Is_Associated_With_a.3.aspx Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abruzzi A., Friedx B., Alikhan S.B. Coinfection of schistosoma species with hepatitis B or hepatitis C viruses. Adv Parasitol. 2016;91:111–231. doi: 10.1016/bs.apar.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Getie S., Wondimeneh Y., Getnet G., et al. Prevalence and clinical correlates of Schistosoma mansoni co-infection among malaria infected patients, Northwest Ethiopia. BMC Res Notes. 2015;8(1):1–6. doi: 10.1186/s13104-015-1468-2. https://bmcresnotes-biomedcentral-com.proxy3.library.mcgill.ca/articles/10.1186/s13104-015-1468-2 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLaughlin T.A., Khayumbi J., Ongalo J., et al. CD4 T cells in mycobacterium tuberculosis and Schistosoma mansoni co-infected individuals maintain functional TH1 responses. Front Immunol. 2020;11:127. doi: 10.3389/fimmu.2020.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mendonça S.A., Lorincz R., Boucher P., Curiel D.T. Adenoviral vector vaccine platforms in the SARS-CoV-2 pandemic. NPJ Vaccines. 2021;6(1):1–14. doi: 10.1038/s41541-021-00356-x. https://www.nature.com/articles/s41541-021-00356-x Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lago E.M., Silva M.P., Queiroz T.G., et al. Phenotypic screening of nonsteroidal anti-inflammatory drugs identified mefenamic acid as a drug for the treatment of schistosomiasis. eBioMedicine. 2019;43:370–379. doi: 10.1016/j.ebiom.2019.04.029. http://www.thelancet.com/article/S2352396419302683/fulltext Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilbert R., Guilbault C., Gagnon D., et al. Establishment and validation of new complementing cells for production of E1-deleted adenovirus vectors in serum-free suspension culture. J Virol Methods. 2014;208:177–188. doi: 10.1016/j.jviromet.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 25.Haq K., Jia Y., Elahi S.M. Evaluation of recombinant adenovirus vectors and adjuvanted protein as a heterologous prime-boost strategy using HER2 as a model antigen. Vaccine. 2019;37(47):7029–7040. doi: 10.1016/j.vaccine.2019.08.079. Nov 8. [DOI] [PubMed] [Google Scholar]
- 26.Mullick A., Xu Y., Warren R., et al. The cumate gene-switch: a system for regulated expression in mammalian cells. BMC Biotechnol. 2006;6(1):1–18. doi: 10.1186/1472-6750-6-43. https://bmcbiotechnol.biomedcentral.com/articles/10.1186/1472-6750-6-43 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He T.C., Zhou S., da Costa L.T., Yu J., Kinzler K.W., Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci. 1998;95(5):2509–2514. doi: 10.1073/pnas.95.5.2509. https://www.pnas.org/content/95/5/2509 [Internet]Mar 3 [cited 2021 Sep 17]Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oualikene W, Lamoureux L, Weber JM, Massie B. Protease-Deleted Adenovirus Vectors and Complementing Cell Lines: Potential Applications of Single-Round Replication Mutants for Vaccination and Gene Therapy. Human Gene Therapy. 2004;11(9):1341–1353. 10.1089/10430340050032438. [DOI] [PubMed]
- 29.Hassan A.S., Zelt N.H., Perera D.J., Ndao M., Ward B.J. Vaccination against the digestive enzyme Cathepsin B using a YS1646 Salmonella enterica Typhimurium vector provides almost complete protection against Schistosoma mansoni challenge in a mouse model. PLoS Negl Trop Dis. 2019;13(12) doi: 10.1371/journal.pntd.0007490. http://www.ncbi.nlm.nih.gov/pubmed/31790394 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frey A., Di Canzio J., Zurakowski D. A statistically defined endpoint titer determination method for immunoassays. J Immunol Methods. 1998;221(1–2):35–41. doi: 10.1016/s0022-1759(98)00170-7. [DOI] [PubMed] [Google Scholar]
- 31.Hodgins B., Pillet S., Landry N., Ward B.J. A plant-derived VLP influenza vaccine elicits a balanced immune response even in very old mice with co-morbidities. PLoS One. 2019;14(1):e0210009. doi: 10.1371/journal.pone.0210009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roederer M., Nozzi J.L., Nason M.C. SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytom Part A. 2011;79A(2):167–174. doi: 10.1002/cyto.a.21015. https://onlinelibrary.wiley.com/doi/full/10.1002/cyto.a.21015 [Internet]Feb 1 [cited 2021 Dec 9]Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cronan M.R., Matty M.A., Rosenberg A.F., et al. An explant technique for high-resolution imaging and manipulation of mycobacterial granulomas. Nat Methods. 2018;15(12):1098–1107. doi: 10.1038/s41592-018-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagen J., Young N.D., Every A.L., et al. Omega-1 knockdown in Schistosoma mansoni eggs by lentivirus transduction reduces granuloma size in vivo. Nat Commun. 2014;5(1):1–9. doi: 10.1038/ncomms6375. www.nature.com/naturecommunications Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ebenezer J.A., Christensen J.M., Oliver B.G. Periostin as a marker of mucosal remodelling in chronic rhinosinusitis. Rhinol J. 2017;55(3):234–241. doi: 10.4193/Rhino16.215. [DOI] [PubMed] [Google Scholar]
- 36.Tang C., Pan Q., Xie Y., Xiong Y., Zhang R., Huang J. Effect of cytotoxic T-lymphocyte antigen-4 on the efficacy of the fatty acid-binding protein vaccine against schistosoma japonicum. Front Immunol. 2019;10(May):1022. doi: 10.3389/fimmu.2019.01022. https://www.frontiersin.org/article/10.3389/fimmu.2019.01022/full Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bankhead P., Loughrey M.B., Fernández J.A., et al. QuPath: Open source software for digital pathology image analysis. Sci Rep. 2017;7(1):1–7. doi: 10.1038/s41598-017-17204-5. https://www.nature.com/articles/s41598-017-17204-5 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Faul F., Erdfelder E., Lang A.G., Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39(2):175–191. doi: 10.3758/bf03193146. https://pubmed.ncbi.nlm.nih.gov/17695343/ Available from: [DOI] [PubMed] [Google Scholar]
- 39.Melman S.D., Steinauer M.L., Cunningham C., et al. Reduced susceptibility to praziquantel among naturally occurring Kenyan isolates of Schistosoma mansoni. PLoS Negl Trop Dis. 2009;3(8):e504. doi: 10.1371/journal.pntd.0000504. https://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0000504 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zacharia A., Mushi V., Makene T. A systematic review and meta-analysis on the rate of human schistosomiasis reinfection. PLoS One. 2020;15(12) doi: 10.1371/journal.pone.0243224. Available from:/pmc/articles/PMC7714137/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woldegerima E., Bayih A.G., Tegegne Y., Aemero M., Zeleke A.J. Prevalence and reinfection rates of schistosoma mansoni and praziquantel efficacy against the parasite among primary school children in Sanja Town, Northwest Ethiopia. J Parasitol Res. 2019 doi: 10.1155/2019/3697216. Available from: /pmc/articles/PMC6507171/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai Y., Wang X., Zhao S., et al. Construction and evaluation of replication-defective recombinant optimized triosephosphate isomerase adenoviral vaccination in Schistosoma japonicum challenged mice. Vaccine. 2014;32(7):771–778. doi: 10.1016/j.vaccine.2013.12.059. [DOI] [PubMed] [Google Scholar]
- 43.Dai Y., Wang X., Tang J., et al. Enhancement of protective efficacy through adenoviral vectored vaccine priming and protein boosting strategy encoding triosephosphate isomerase (SjTPI) against schistosoma japonicum in Mice. PLoS One. 2015;10(3) doi: 10.1371/journal.pone.0120792. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0120792 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El Ridi R., Tallima H., Selim S., et al. Cysteine peptidases as schistosomiasis vaccines with inbuilt adjuvanticity. PLoS One. 2014;9(1) doi: 10.1371/journal.pone.0085401. https://pubmed-ncbi-nlm-nih-gov.proxy3.library.mcgill.ca/24465551/ Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mo A.X., Colley D.G. Workshop report: schistosomiasis vaccine clinical development and product characteristics. Vaccine. 2016;34(8):995–1001. doi: 10.1016/j.vaccine.2015.12.032. [DOI] [PubMed] [Google Scholar]
- 46.Amaral M.S., Santos D.W., Pereira A.S.A., et al. Rhesus macaques self-curing from a schistosome infection can display complete immunity to challenge. Nat Commun. 2021;12(1):1–17. doi: 10.1038/s41467-021-26497-0. https://www-nature-com.proxy3.library.mcgill.ca/articles/s41467-021-26497-0 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yi X.Y., Omer-Ali P., Kelly C., Simpson A.J., Smithers S.R. IgM antibodies recognizing carbohydrate epitopes shared between schistosomula and miracidia of Schistosoma mansoni that block in vitro killing. J Immunol. 1986;137(12):3946–3954. [PubMed] [Google Scholar]
- 48.Capron A., Dessaint J.P. Immunologic aspects of schistosomiasis. Annu Rev Med. 1992;43:209–227. doi: 10.1146/annurev.me.43.020192.001233. www.annualreviews.org Available from: [DOI] [PubMed] [Google Scholar]
- 49.Jwo J., LoVerde P.T. The ability of fractionated sera from animals vaccinated with irradiated cercariae of Schistosoma mansoni to transfer immunity to mice. J Parasitol. 1989;75(2):252–260. https://pubmed-ncbi-nlm-nih-gov.proxy3.library.mcgill.ca/2466975/ Available from: [PubMed] [Google Scholar]
- 50.Mangold B.L., Dean D.A. Passive transfer with serum and IgG antibodies of irradiated cercaria-induced resistance against Schistosoma mansoni in mice. J Immunol. 1986;136(7):2644–2648. [PubMed] [Google Scholar]
- 51.Wilson R.A., Langermans J.A.M., van Dam G.J., et al. Elimination of schistosoma mansoni adult worms by rhesus macaques: basis for a therapeutic vaccine? PLoS Negl Trop Dis. 2008;2(9):e290. doi: 10.1371/journal.pntd.0000290. https://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0000290 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang W., Le L., Ahmad G., et al. Fifteen years of Sm-p80-based vaccine trials in nonhuman primates: antibodies from vaccinated baboons confer protection in vivo and in vitro from schistosoma mansoni and identification of putative correlative markers of protection. Front Immunol. 2020;11:1246. doi: 10.3389/fimmu.2020.01246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Molehin A.J., Sennoune S.R., Zhang W., et al. Cross species prophylactic efficacy of Sm-p80-based vaccine and intracellular localization of Sm-p80/Sm-p80 ortholog proteins during development in Schistosoma mansoni, Schistosoma japonicum and Schistosoma haematobium. Parasitol Res. 2017;116(11):3175. doi: 10.1007/s00436-017-5634-4. Available from: /pmc/articles/PMC5660642/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delgado V., Mclaren D.J. Evidence for enhancement of IgGl subclass expression in mice polyvaccinated with radiation-attenuated cercariae of Schistosoma mansoni and the role of this isotype in serum-transferred immunity. Parasite Immunol. 1990;12(1):15–32. doi: 10.1111/j.1365-3024.1990.tb00933.x. https://onlinelibrary-wiley-com.proxy3.library.mcgill.ca/doi/full/10.1111/j.1365-3024.1990.tb00933.x Available from: [DOI] [PubMed] [Google Scholar]
- 55.Hewitson J.P., Hamblin P.A., Mountford A.P. Immunity induced by the radiation-attenuated schistosome vaccine. Parasite Immunol. 2005;27(7–8):271–280. doi: 10.1111/j.1365-3024.2005.00764.x. https://onlinelibrary-wiley-com.proxy3.library.mcgill.ca/doi/full/10.1111/j.1365-3024.2005.00764.x Available from: [DOI] [PubMed] [Google Scholar]
- 56.Lam H.Y.P., Yang T.H., Liang T.R., Cheng P.C., Chang K.C., Peng S.Y. Heat-killed Propionibacterium acnes augment the protective effect of 28-kDa glutathione S-transferases antigen against Schistosoma mansoni infection. Acta Trop. 2021;222 doi: 10.1016/j.actatropica.2021.106033. [DOI] [PubMed] [Google Scholar]
- 57.Rihet P., Demeure C.E., Bourgois A., Prata A., Dessein A.J. Evidence for an association between human resistance to Schistosoma mansoni and high anti-larval IgE levels. Eur J Immunol. 1991;21(11):2679–2686. doi: 10.1002/eji.1830211106. https://onlinelibrary.wiley.com/doi/full/10.1002/eji.1830211106 Available from: [DOI] [PubMed] [Google Scholar]
- 58.Jiz M., Friedman J.F., Leenstra T., et al. Immunoglobulin E (IgE) responses to paramyosin predict resistance to reinfection with Schistosoma japonicum and are attenuated by IgG4. Infect Immun. 2009;77(5):2051–2058. doi: 10.1128/IAI.00012-09. https://journals.asm.org/doi/abs/10.1128/IAI.00012-09 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu L.C., Zarrin A.A. The production and regulation of IgE by the immune system. Nat Rev Immunol. 2014;14(4):247–259. doi: 10.1038/nri3632. https://www-nature-com.proxy3.library.mcgill.ca/articles/nri3632 2014 144 [Internet]Mar 14 [cited 2022 Jan 13]Available from: [DOI] [PubMed] [Google Scholar]
- 60.Diemert D.J., Pinto A.G., Freire J., et al. Generalized urticaria induced by the Na-ASP-2 hookworm vaccine: Implications for the development of vaccines against helminths. J Allergy Clin Immunol. 2012;130(1):169–176. doi: 10.1016/j.jaci.2012.04.027. [DOI] [PubMed] [Google Scholar]
- 61.Caulfield J.P., Lenzi H.L., Elsas P., Dessein A.J. Ultrastructure of the attack of eosinophils stimulated by blood mononuclear cell products on schistosomula of Schistosoma mansoni. Am J Pathol. 1985;120(3):380. Available from: /pmc/articles/PMC1887990/?report=abstract. [PMC free article] [PubMed] [Google Scholar]
- 62.Jong E.C., Chi E.Y., Klebanoff S.J. Human neutrophil-mediated killing of schistosomula of Schistosoma Mansoni: augmentation by schistosomal binding of eosinophil peroxidase. Am J Trop Med Hyg. 1984;33(1):104–115. doi: 10.4269/ajtmh.1984.33.104. https://www.ajtmh.org/view/journals/tpmd/33/1/article-p104.xml Available from: [DOI] [PubMed] [Google Scholar]
- 63.Dessein A., Samuelson J.C., Butterworth A.E., et al. Immune evasion by Schistosoma mansoni: loss of susceptibility to antibody or complement-dependent eosinophil attack by schistosomula cultured in medium free of macromolecules. Parasitology. 1981;82(3):357–374. doi: 10.1017/s0031182000066890. https://www-cambridge-org.proxy3.library.mcgill.ca/core/journals/parasitology/article/immune-evasion-by-schistosoma-mansoni-loss-of-susceptibility-to-antibody-or-complementdependent-eosinophil-attack-by-schistosomula-cultured-in-medium-free-of-macromolecules/A2851C4C857923FDA4386E2C172EA551 Available from: [DOI] [PubMed] [Google Scholar]
- 64.Tchoubrieva E.B., Ong P.C., Pike R.N., Brindley P.J., Kalinna B.H. Vector-based RNA interference of cathepsin B1 in Schistosoma mansoni. Cell Mol Life Sci. 2010;67(21):3739–3748. doi: 10.1007/s00018-010-0345-3. https://link-springer-com.proxy3.library.mcgill.ca/article/10.1007/s00018-010-0345-3 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilson R.A. Interferon gamma is a key cytokine in lung phase immunity to schistosomes but what is its precise role? Braz J Med Biol Res. 1998;31(1):157–161. doi: 10.1590/s0100-879x1998000100022. http://www.scielo.br/j/bjmbr/a/h8SxVK4bTqrjQ6qyP4QrGFL/?lang=en Available from: [DOI] [PubMed] [Google Scholar]
- 66.Jankovic D., Wynn T.A., Kullberg M.C., et al. Optimal vaccination against Schistosoma mansoni requires the induction of both B cell- and IFN-gamma-dependent effector mechanisms. J Immunol. 1999;162(1):345–351. https://pubmed-ncbi-nlm-nih-gov.proxy3.library.mcgill.ca/9886405/ Available from: [PubMed] [Google Scholar]
- 67.Gaucher D., Therrien R., Kettaf N., et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J Exp Med. 2008;205(13):3119–3131. doi: 10.1084/jem.20082292. www.jem.org/cgi/doi/10.1084/jem.20082292 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Akondy R.S., Monson N.D., Miller J.D., et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. 2009;183(12):7919–7930. doi: 10.4049/jimmunol.0803903. https://www.jimmunol.org/content/183/12/7919 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kannanganat S., Ibegbu C., Chennareddi L., Robinson H.L., Amara R.R. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. J Virol. 2007;81(16):8468–8476. doi: 10.1128/JVI.00228-07. https://journals-asm-org.proxy3.library.mcgill.ca/doi/abs/10.1128/JVI.00228-07 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.L'Huillier A.G., Ferreira V.H., Hirzel C., et al. T-cell responses following natural influenza infection or vaccination in solid organ transplant recipients. Sci Rep. 2020;10(1):1–9. doi: 10.1038/s41598-020-67172-6. https://www-nature-com.proxy3.library.mcgill.ca/articles/s41598-020-67172-6 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Savic M., Dembinski J.L., Laake I., et al. Distinct T and NK cell populations may serve as immune correlates of protection against symptomatic pandemic influenza A(H1N1) virus infection during pregnancy. PLoS One. 2017;12(11) doi: 10.1371/journal.pone.0188055. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0188055 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown D.M., Lee S., Garcia-Hernandez M de la L., Swain S.L. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J Virol. 2012;86(12):6792–6803. doi: 10.1128/JVI.07172-11. https://journals-asm-org.proxy3.library.mcgill.ca/doi/abs/10.1128/JVI.07172-11 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Precopio M.L., Betts M.R., Parrino J., et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8+ T cell responses. J Exp Med. 2007;204(6):1405–1416. doi: 10.1084/jem.20062363. www.jem.org/cgi/doi/10.1084/jem.20062363 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Darrah P.A., Patel D.T., De Luca P.M., et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13(7):843–850. doi: 10.1038/nm1592. https://www.nature.com/articles/nm1592 Available from: [DOI] [PubMed] [Google Scholar]
- 75.Kelton J.G., Arnold D.M., Nazy I. Lessons from vaccine-induced immune thrombotic thrombocytopenia. Nat Rev Immunol. 2021;21(12):753–755. doi: 10.1038/s41577-021-00642-8. https://www-nature-com.proxy3.library.mcgill.ca/articles/s41577-021-00642-8 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilson R.A., Li X.H., Castro-Borges W. Do schistosome vaccine trials in mice have an intrinsic flaw that generates spurious protection data? Parasites Vectors. 2016;9(1):1–16. doi: 10.1186/s13071-016-1369-9. https://parasitesandvectors-biomedcentral-com.proxy3.library.mcgill.ca/articles/10.1186/s13071-016-1369-9 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perera D.J., Ndao M. Promising technologies in the field of helminth vaccines. Front Immunol. 2021;12:3220. doi: 10.3389/fimmu.2021.711650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.