

Abstract
Red blood cells (RBCs) are essential for aerobic respiration through delivery of oxygen to distant tissues. However, RBCs are currently considered immunologically inert, and few, if any, secondary functions of RBCs have been identified. Here we showed that RBCs serve as critical immune sensors through surface expression of the nucleic acid-sensing toll-like receptor 9 (TLR9). Mammalian RBCs expressed TLR9 on their surface and bound CpG-containing DNA derived from bacteria, plasmodia, and mitochondria. RBC-bound mitochondrial DNA was increased during human and murine sepsis and pneumonia. In vivo, CpG-carrying RBCs drove accelerated erythrophagocytosis and innate immune activation characterized by increased interferon signaling. Erythroid-specific deletion of TLR9 abrogated erythrophagocytosis and decreased local and systemic cytokine production during CpG-induced inflammation and polymicrobial sepsis. Thus, detection and capture of nucleic acid by TLR9 expressing-RBCs regulated red cell clearance and inflammatory cytokine production, demonstrating that RBCs function as immune sentinels during pathologic states. Consistent with these findings, RBC-bound mitochondrial DNA was elevated in individuals with viral pneumonia and sepsis secondary to coronavirus disease 2019 (COVID-19) and associated with anemia and severity of disease. These findings uncover a previously unappreciated role of RBCs as critical players in inflammation distinct from their function in gas transport.
One Sentence Summary:
Red blood cells detect and bind cell-free nucleic acids, contributing to anemia and immune cell activation during acute inflammation.
INTRODUCTION
Red blood cells (RBCs) comprise the majority of circulating cells in mammals and are essential for respiration. Although non-gas exchanging functions of the red cell such as chemokine regulation, complement binding, and pathogen immobilization have been described, RBC immune function remains enigmatic (1–3). RBCs transit through all tissues and contact pathogen and self-derived inflammatory mediators in the circulation, positioning them as ideal messengers between distant organs. Indeed, we have recently demonstrated that RBCs bind and scavenge nucleic acids away from the lung during basal conditions at homeostasis (4), yet the role of RBC-nucleic acid-binding in the host immune response during inflammation is unknown.
Evolutionarily conserved nucleic acid-sensing toll-like receptors (TLRs) identify nucleic acids derived from self and pathogens and play a central role in inflammation by promoting inflammatory cytokine secretion, immune cell maturation, and proliferation (5–10). Elevated cell-free CpG-containing DNA is a hallmark of infection and sterile injury (7, 8, 11). Common to these inflammatory pathologies is acute anemia, a substantial cause of morbidity observed during sepsis and critical illness. Several studies have suggested a role for intracellular nucleic acids and TLR signaling in monocytes and macrophages in developing inflammatory anemia and cytopenia, yet whether RBCs themselves or cell-free DNA contribute to RBC clearance is unknown (12–14).
We recently discovered that RBCs express intracellular TLR9 and scavenge cell-free (CpG-containing) mitochondrial DNA (cf-mtDNA) under homeostatic conditions (4). These data suggested that TLR9-mediated sequestration of cf-mtDNA by RBCs represents a protective mechanism that clears toxic cell-free nucleic acids from the circulation in healthy hosts (4). However, how RBC-dependent CpG-binding contributes to the inflammatory response during infection remains unknown. Here we show that TLR9 is expressed on the RBC surface and that DNA binding by RBC TLR9 during inflammatory states leads to accelerated RBC clearance and systemic inflammation, thus linking RBC nucleic acid binding to innate immune activation and anemia during inflammatory states.
RESULTS
TLR9 is increased on the surface of RBCs during sepsis, and RBCs bind pathogen DNA.
Although TLR9 is an endosomal nucleic acid-sensing receptor, recent studies have identified the presence of TLR9 on the surface of intestinal epithelial cells, splenic dendritic cells, activated platelets, and a minor fraction of peripheral blood mononuclear cells (PBMCs) (15–18). We have previously detected intracellular TLR9 in RBCs but could not detect surface expression of TLR9 using permeabilization-dependent antibodies (4). However, when we used antibodies to a larger epitope in the TLR9 ectodomain, TLR9 was readily detected on intact non-permeabilized human and murine RBCs (Fig. 1A). We verified RBC TLR9 expression using confocal microscopy (Fig. 1B). We also asked whether RBCs from non-human primates express surface TLR9. Chimpanzee RBCs, like their human counterparts, express surface TLR9 (fig. S1). Thus, TLR9 expression is conserved in mammalian RBCs with the DNA-binding ectodomain on the RBC surface.
Figure 1. TLR9 is expressed on the surface of RBCs and RBCs bind pathogen DNA.
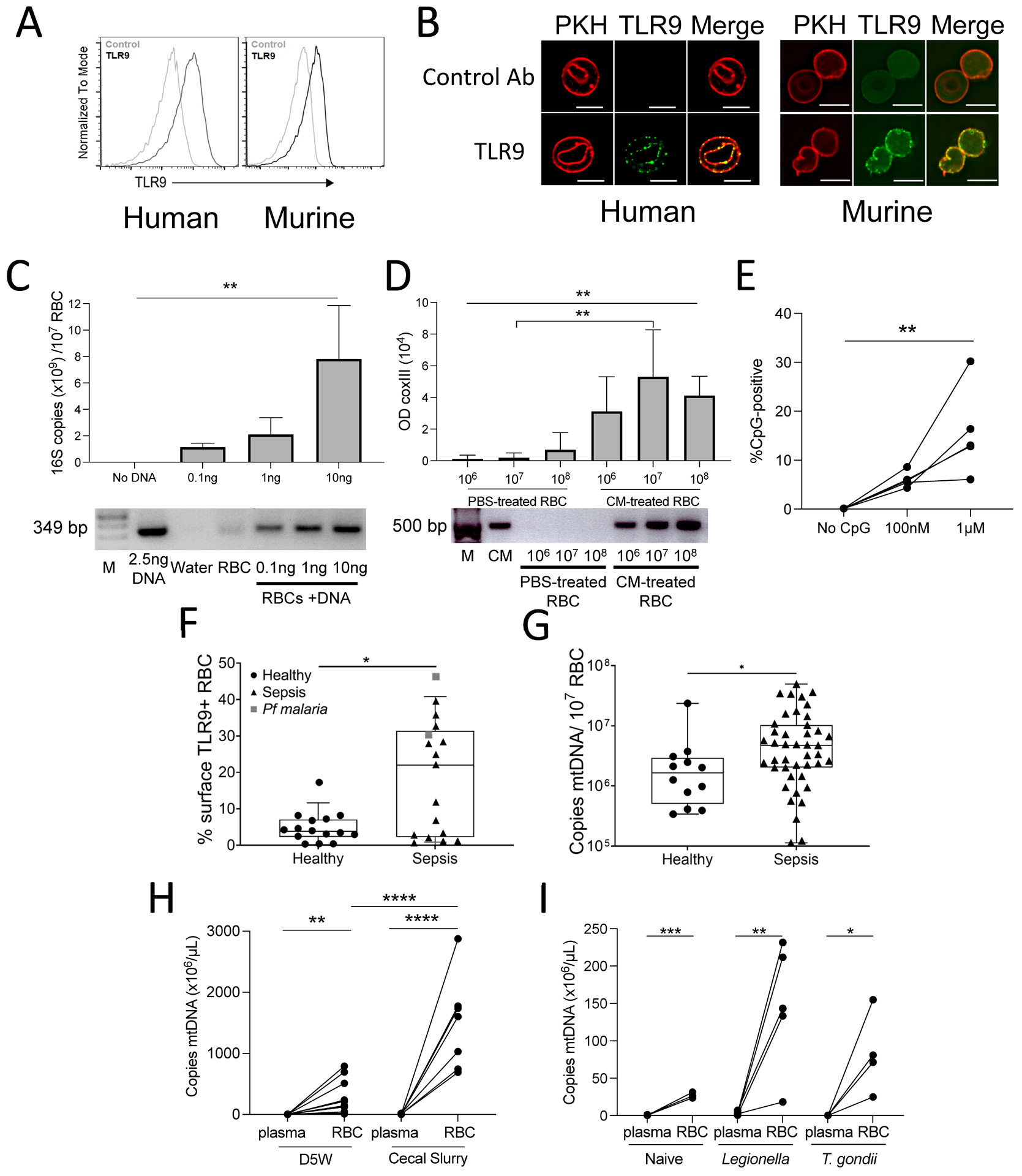
(A) Flow cytometry of TLR9 expression on non-permeabilized RBCs is shown. (B) Confocal images show TLR9 (green) and PKH 26 (red) in red blood cells, scale bar, 5μm. (C) RBCs from human donors were incubated with Legionella DNA and RBC-bound bacterial DNA was measured by qPCR for 16S (top panel). Data are representative of three independent studies and analyzed with one-way ANOVA with Dunnett’s correction for multiple comparisons. Amplicons from one representative study are shown (bottom panel). Lane 1:M=marker. (D) RBCs (106, 107, 108) were incubated with PBS or P. falciparum-positive culture medium (CM), and RBC-bound parasite DNA was quantified by P. falciparum mtDNA amplification (COX III). Optical density (OD) quantification of P. falciparum DNA bound to RBCs is shown (top panel). Data were analyzed by one-way ANOVA, with Sidak’s multiple comparisons test for PBS versus CM treated RBCs. RBCs from three donors were tested in three independent experiments. Amplicons from one representative study (bottom panel) are shown. Lane 1: marker, data are presented as mean ± SD (E) CpG-binding to RBCs from healthy donors is shown (n=5), analyzed by Kruskal-Wallis test, with Dunn’s correction for multiple comparisons. (F) Surface TLR9 expression on RBCs was measured in samples from 15 healthy donors and 19 patients with sepsis; data were analyzed by a Mann Whitney U-test. (G) mtDNA on RBCs is shown for samples isolated from healthy donors (n=12) and patients with sepsis (n=23); data were analyzed by a Mann-Whitney U-test. Boxes display medians with interquartile ranges, each dot represents a healthy donor or patient for G and F. (H) mtDNA (mtCo1) in the plasma and on RBCs was measured 6 hours following injection of mice with cecal slurry (CS) or D5W; data were analyzed by an unpaired t-test for RBCs versus plasma mtCo1 within treatment group and between treatment groups; n=7 to 11 mice per group, three independent studies. (I) mtCo1 was measured on RBC and plasma of L. pneumophila- or T. gondii-infected mice; significance calculated by an unpaired t-test. For all panels, *P<0.05, **P<0.01, ***P<0.005, and ****P<0.001 denote significant findings.
Immunostimulatory unmethylated CpG motifs are a feature of microbial DNA; we therefore asked whether RBCs could detect pathogen DNA. The ability of RBCs from healthy human donors to bind bacterial DNA or malarial mtDNA was tested by incubating RBCs with genomic DNA from Legionella pneumophila or media from Plasmodium falciparum erythrocyte culture. Following incubation with bacterial DNA or P. falciparum DNA, RBCs were isolated, and polymerase chain reaction (PCR) for the 16s ribosomal RNA gene (bacterial DNA) or coxIII (malarial mtDNA) was performed. We found a dose-dependent increase in amplifiable microbial DNA on RBCs following incubation with bacterial or malarial DNA (Fig. 1C and D). We confirmed human RBCs’ ability to bind malarial DNA by examining the binding of synthetic CpG based on sequences found in the P. falciparum genome to RBCs (Fig. 1E) (19). Collectively, these data demonstrate that human RBCs can bind pathogen DNA.
The TLR9 ligand, CpG-containing mitochondrial DNA, is elevated in the circulation during sepsis, a deadly syndrome defined by the dysregulated host response to infection (11, 20, 21). We therefore examined TLR9 expression on RBCs from critically ill patients with sepsis. RBCs were prospectively collected from patients enrolled in a cohort designed to study sepsis (Molecular Epidemiology of SepSis in the Intensive Care Unit (ICU), MESSI cohort) at the University of Pennsylvania. Flow cytometry for TLR9 was performed on intact non-permeabilized RBCs obtained on ICU presentation. We found that surface TLR9 was increased on RBCs from patients with sepsis compared with RBCs from healthy donors (Fig. 1F). We next performed quantitative PCR (qPCR) to determine the mtDNA content of RBCs obtained from healthy volunteers and critically ill patients with sepsis (4, 11). Consistent with our in vitro findings of CpG-DNA acquisition by RBCs, mtDNA is elevated on RBCs during human sepsis (Fig. 1G). Thus, both RBC-expressed TLR9 and its ligand, mtDNA, are elevated during human sepsis.
We next asked whether RBCs bind CpG-containing host mitochondrial DNA during infection. We measured mitochondrial DNA (coxI) in the plasma and on RBCs in a murine model of sepsis and murine models of bacterial pneumonia and systemic parasite infection (L. pneumophila and Toxoplasma gondii). mtDNA was elevated on RBCs when compared with plasma in all animal models of infection (Fig. 1H and I). These data demonstrate that CpG-containing mtDNA is sequestered on RBCs during pneumonia, parasitic infection, and polymicrobial infection (4).
DNA binding results in altered RBC structure and function
During sterile inflammation and infection, cell-free CpG-containing mtDNA is elevated in the plasma (9, 11, 22). Because altered RBC morphology is a common feature of sepsis, critical illness, and the recently reported multi-system inflammatory syndrome in children (MIS-C), we examined the effect of excess cell-free CpG on RBC morphology and function (23–28). Extensive alterations of RBC morphology following high dose CpG treatment (100 nM) were visualized by electron microscopy (Fig. 2A and B). RBC shape changes were associated with marked differences in the distribution of the cytoskeletal proteins spectrin and actin following CpG binding (Fig. 2C). We next examined the distribution of the RBC membrane protein Band 3 following CpG DNA treatment. As a control, we used GpC DNA, which binds TLR9 without causing TLR9 activation or conformational changes (29). Treatment with CpG DNA, but not GpC DNA, led to alterations in Band 3 distribution (Fig. 2D and E). Taken together, these data indicate that CpG DNA binding alters RBC morphology in a TLR9-dependent manner. We next examined the morphology of RBCs following CpG incubation using imaging flow cytometry. This analysis identified a population of RBCs with aberrant morphology following incubation with CpG. The automated feature finder was utilized to discriminate RBC subpopulations based on morphology, and mean pixel and intensity features were identified to best differentiate smooth from altered cells (Fig. 2F); the altered cells were all TLR9 positive (Fig. 2G). In the presence of low concentrations of extracellular CpG DNA, RBCs remained morphologically unaltered. However, the addition of increasing amounts of CpG resulted in malformed RBCs (Fig. 2H and I). Because we observed increased surface TLR9 accessibility in patients with sepsis, we examined RBC surface TLR9 expression following treatment with diverse stimuli. Lipopolysaccharide (LPS), tumor necrosis factor (TNF-α), and phorbol 12-myristate 13-acetate (PMA) did not alter surface TLR9 detection, whereas the lipid disrupting agent (hydrogen peroxide) increased surface TLR9 abundance (fig. S2).
Figure 2. DNA binding results in altered RBC structure and function.
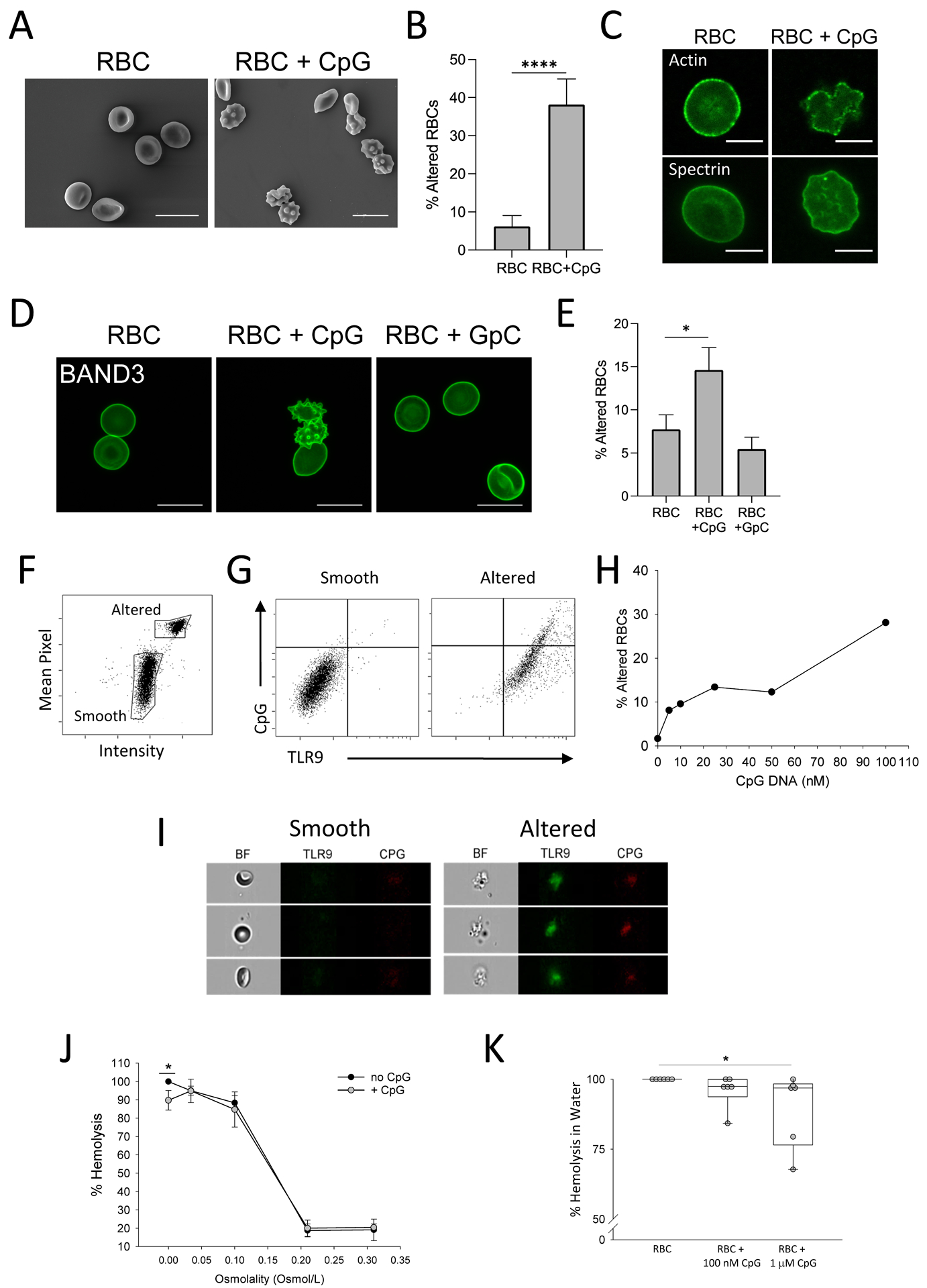
(A) Scanning electron microscopy of human RBCs following CpG treatment. Scale bar, 10μm. (B) The quantification of RBC alterations as observed by electron microscopy is shown. Alteration is defined by loss of biconcave disk shape and formation of echinocytes. Cells from five separate fields were counted and averaged; an unpaired t-test was used to calculate significance. (C) Confocal images show RBC cytoskeletal proteins actin and spectrin following CpG treatment. Scale bar, 5μm. (D) Confocal images show Band 3 expression following CpG and control GpC treatment. Naïve RBCs are also shown. Scale bar, 10μm. (E) Quantification of RBC alterations by Band 3 staining is shown. RBCs from four individual donors were tested. A Kruskal-Wallis test with Dunn’s post-hoc analysis was used to calculate significance. (F to I) Imaging flow cytometry analysis on CpG-treated human RBCs is shown. (F) Imaging flow cytometry reveals smooth and altered RBC populations as defined by Mean Pixel and Intensity parameters. (G) Smooth and altered RBC populations were analyzed for CpG binding and TLR9 expression. (H) The percent of altered RBCs with increasing doses of CpG is shown. (I) Images of smooth and altered RBCs are shown. (J) Osmotic fragility is shown for of healthy human RBCs pre-treated with PBS or 1 μM CpG; data were analyzed by a Mann-Whitney U test. (K) Hemolysis in water of RBCs pre-treated with PBS, 100 nM CpG, or 1 μM CpG is shown. RBCs from six independent donors were tested and data were analyzed by Kruskal-Wallis test with Dunn’s post-hoc analysis. Boxes display medians with interquartile ranges, each dot represents a healthy donor. For all panels, *P<0.05, **P<0.01, ***P<0.005, and ****P<0.001 denote significant findings.
We next examined the effect of excess cell-free CpG on RBC function by measuring osmotic fragility of human RBCs and viability. The addition of CpG resulted in reduced osmotic fragility (Fig. 2J and K). Collectively, these data suggest that surface accessibility of the TLR9 ectodomain is tunable by CpG exposure and that CpG binding to RBCs alters RBC structure and function.
CpG binding by RBCs leads to loss of CD47 detection
RBC survival is determined by multiple factors, including membrane integrity, phosphatidylserine (PS) externalization, and CD47 expression. RBC viability was assessed by loss of membrane integrity using the dye calcein-AM. CpG did not lead to a loss of RBC membrane integrity (fig. S3A and B). Because PS externalization serves as a phagocytosis signal and CD47 expression serves as a self-preservation signal, we examined both PS externalization and CD47 expression following CpG treatment of RBCs. Although we did not observe an increase in PS positive cells (fig. S3C and D), we observed loss of CD47 as measured by binding of the antibody CC2C6, which detects the antiphagocytic epitope of CD47 (Fig. 3A and B). We observed that cells that were CC2C6 negative following the addition of CpG DNA had bound higher amounts of DNA than those that were CC2C6 positive (fig. S4A and B), suggesting that CpG-acquisition by RBCs led to a loss of CD47 detection. These findings were confirmed with imaging flow cytometry, which revealed that most CD47 Dim cells were altered (fig. S4C to F). We next asked whether loss of CD47 detection was dependent on RBC-TLR9. CD47 masking by CpG was attenuated in the presence of monoclonal TLR9 antibody, (fig. S4G).
Figure 3. CpG binding by RBCs leads loss of CD47 detection.
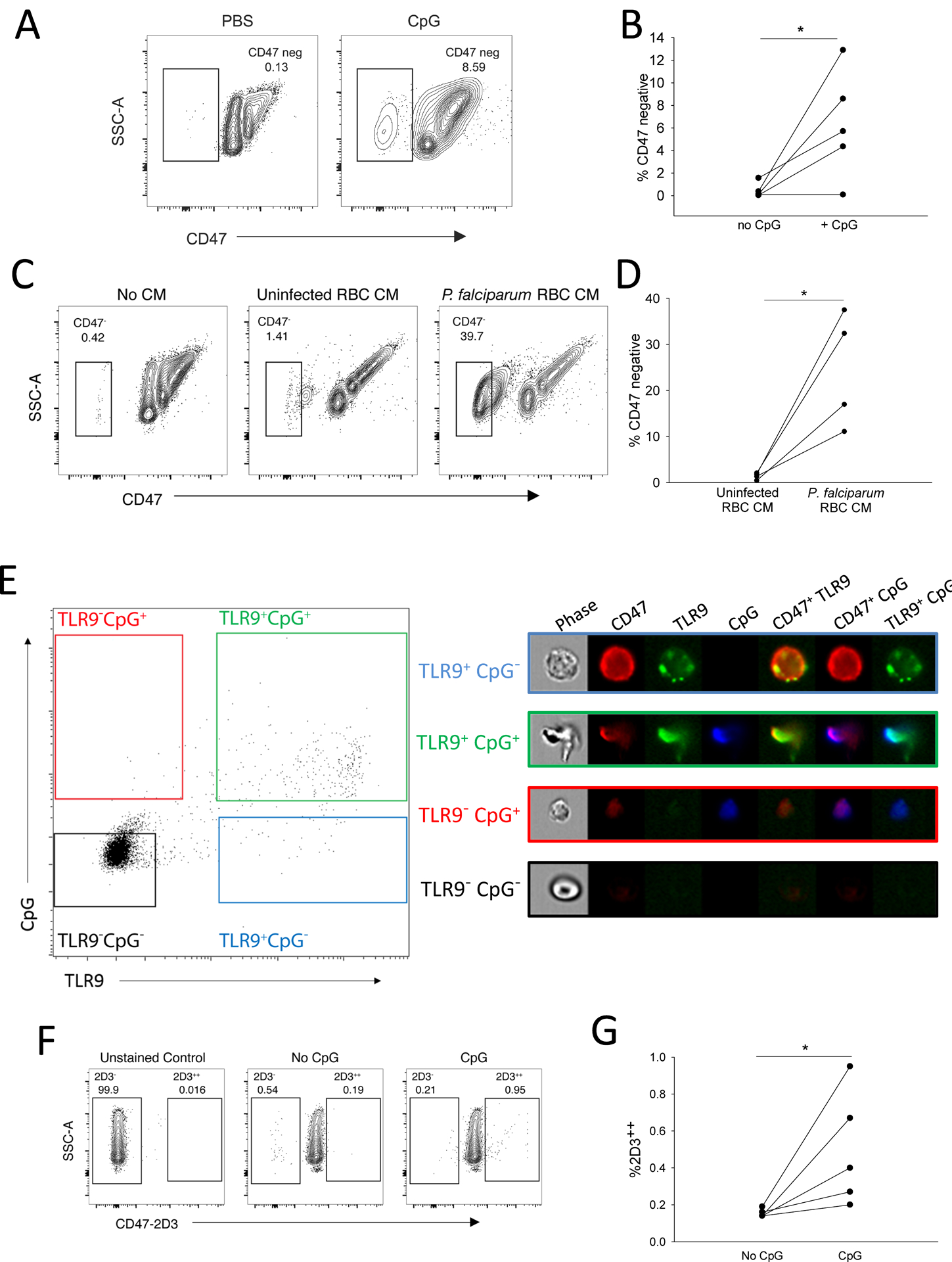
(A and B) Representative results (A) and quantification from five donors (B) are shown for CD47 detection (with antibody clone CC2C6) on human RBCs following incubation with 10μg/mL CpG. Data were analyzed by a paired t-test. SSC-A, side scatter area. (C and D) Representative results (C) and quantification from four donors (D) are shown for CD47 detection in human RBCs treated with conditioned medium (CM) derived from P. falciparum-infected RBC culture. Data were analyzed by a paired t-test. (E) Imaging flow cytometry is shown for CpG-treated human RBCs probed for CD47 and TLR9. (F) Induction of a conformational change in CD47 by CpG was observed by binding of anti-CD47 (clone 2D3), a damaged-associated conformational epitope. One representative flow cytometry analysis is shown. (G) Quantification of 2D3 binding is shown for five individual donors. Significance between CpG-treated and un-treated RBCs were calculated using t test, *P=0.038 by a t-test, P=0.06 by a paired t-test, tests were performed on the same data set. For all panels, *P<0.05, **P<0.01, ***P<0.005, and ****P<0.001 denote significant findings.
Because naïve RBCs bind malarial DNA, we asked if malarial DNA would induce loss of the CD47 antiphagocytic epitope in uninfected erythrocytes from healthy human donors. CpG sequences, based on sequences found in the P. falciparum genome, led to a loss of CD47 detection on RBCs obtained from healthy donors (fig. S5A and B) (19). Incubation of naïve human RBCs with P. falciparum culture supernatants also led to robust loss of the antiphagocytic epitope (Fig. 3C and D).
CD47 associates with the Band 3 complex, a macro-complex of proteins in the RBC membrane (30, 31). We therefore asked whether CD47 was also in complex with TLR9. We found that these two proteins co-immunoprecipitated (fig. S5C). We confirmed this physical interaction using confocal microscopy, which revealed co-localization of TLR9 and CD47 in the RBC membrane (fig. S5D). Because we had observed morphological alterations in the RBC membrane following CpG binding, we performed imaging flow cytometry to better characterize the distribution of surface TLR9 and CD47 in intact RBCs following CpG DNA addition. We incubated RBCs with 100nM CpG and found that CpG DNA led to alterations in RBC structure and CD47 and TLR9 redistribution (Fig. 3E). TLR9-positive, CpG-negative cells demonstrated punctate surface TLR9 staining and uniform surface CD47 distribution, whereas TLR9-positive, CpG-positive cells showed alteration of the membrane and clustering of TLR9 and CD47. These findings suggest that a conformational change of CD47 occurs following CpG-binding by RBCs.
We next asked whether conformational changes of CD47 were associated with the altered localization observed after CpG binding. Conformational changes in CD47 can be detected by an increase in binding of the anti-CD47 antibody 2D3 (32). This antibody detects an epitope on CD47 that has undergone conformational changes and is present on “damaged”, experimentally aged, and sickled RBCs (32, 33). Incubation of RBCs with CpG for 2 hours led to increased detection of this CD47 epitope using the 2D3 antibody (Fig. 3F and G).
DNA-carrying RBCs undergo accelerated erythrophagocytosis and initiate innate immune responses
Because CD47 is a marker of self, and loss of CD47 leads to accelerated erythrophagocytosis by red pulp F4/80-positive splenic macrophages (RPM) (34), we asked whether CpG binding by RBCs would result in accelerated clearance of RBCs by RPM. Green fluorescent protein (GFP)-expressing RBCs were treated with phosphate-buffered saline (PBS) or CpG DNA for 2 hours before being washed and infused into mice. Analysis of spleens one hour following infusion revealed that F4/80hi macrophages ingested GFP RBCs (fig. S6A). Mice that received CpG-treated RBCs demonstrated a higher percentage of erythrophagocytic macrophages than mice that received PBS-treated RBCs (Fig. 4A to C). Spleen weights of animals receiving CpG-treated RBCs were also elevated at 20 hours post-infusion compared with animals that received PBS-treated RBCs, consistent with increased erythrophagocytosis and splenic congestion (Fig. 4D). Collectively, these findings demonstrate that exposure of RBCs to high concentrations of CpG DNA leads to accelerated erythrophagocytosis in vivo.
Figure 4. DNA-carrying RBCs undergo accelerated erythrophagocytosis and initiate innate immune responses in naïve mice.
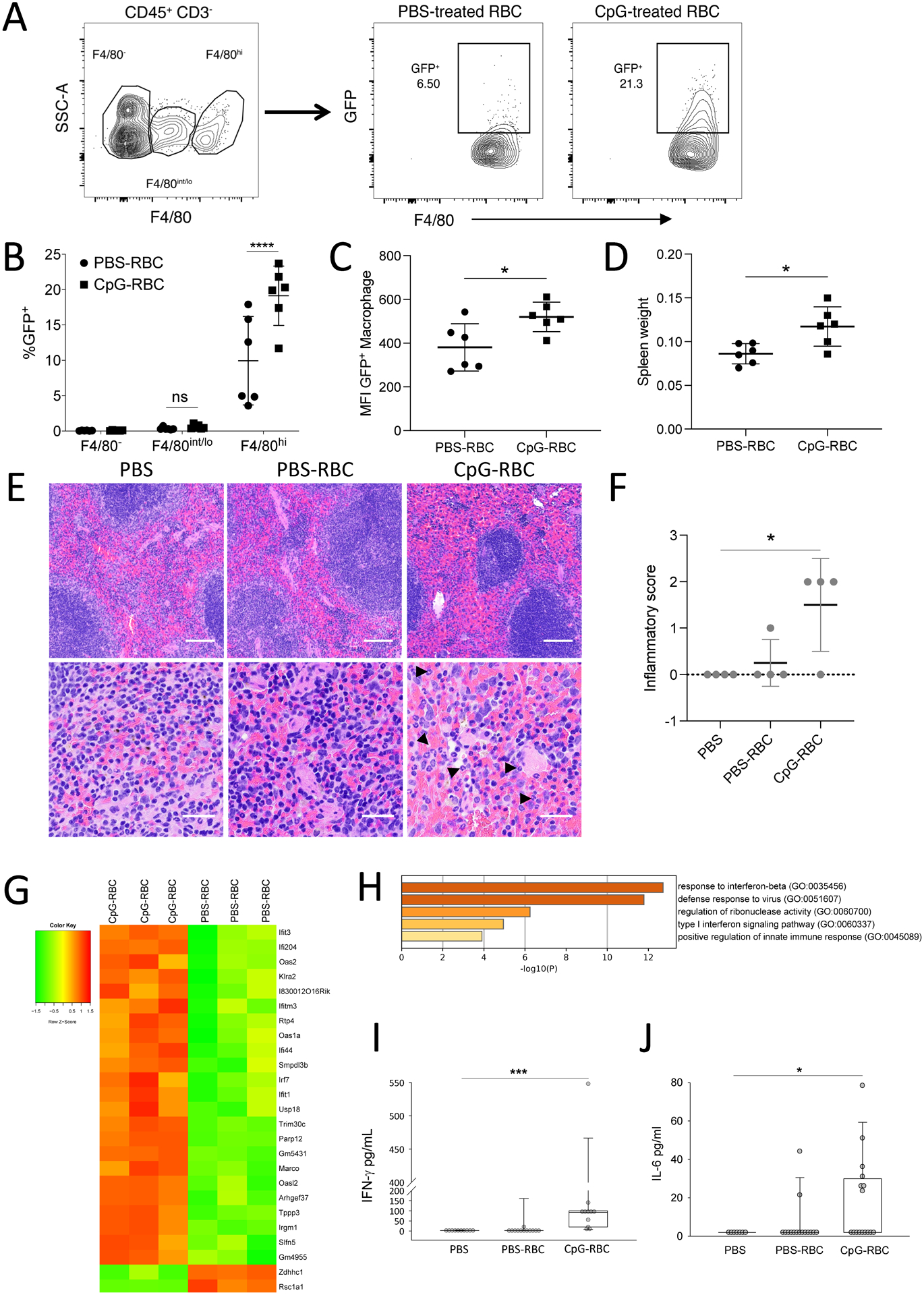
(A to D) GFP-expressing RBC were treated with PBS or CpG and transfused to WT mice. At 1 hour post-infusion, CD45+CD3− splenocytes were analyzed. Erythrophagocytosis of cells expressing indicated amount of F4/80 was measured (n=6 mice per group, two independent experiments). (A) Gating strategy for F4/80-positivity and subsequent GFP-positivity in target cells is shown. (B) The erythrophagocytic capacity of cells expressing F4/80 is shown. Splenic red pulp macrophages (RPMs) are CD45+CD3−F4/80hi. Data are presented as mean±SD. Data were analyzed by a two-way ANOVA with Sidak’s multiple comparisons test. (C) Mean Fluorescence Intensity (MFI) of GFP in RPMs is shown. Data are presented as mean±SD and analyzed by an unpaired t-test. (D) Spleen weights at 20 hours post-infusion are shown. Data are presented as mean±SD and analyzed by an unpaired t-test. (E to J) Physiological changes at 6 hours are shown for mice infused with PBS, PBS-treated RBC, or CpG-treated RBC. (E) H&E staining of spleens is shown, arrowheads indicate neutrophils in the CpG-RBC treated group. Scale bars represents 100μm for the top panel and 20μm for the bottom panel. (F) Quantification of splenic injury is shown (3 to 4 mice per group from two independent studies is shown). Data are presented as mean±SD; statistical analysis was done with a one-way ANOVA with a Holm-Sidak test for multiple comparisons. (G) A heatmap of RNA sequencing analysis of spleens from WT mice infused with PBS- or CpG-treated RBCs after 6 hours is shown. (H) GO-term analysis of the top 25 differentially expressed genes are shown. (I) Quantification of plasma IFN-γ is shown (n=10 to 12 mice per group). (J) Quantification of plasma IL-6 is shown (n=7 to 16 mice per group). J and I are analyzed by Kruskal-Wallis test with a Dunn’s correction for multiple comparisons. For all panels, *P<0.05, **P<0.01, ***P<0.005, and ****P<0.001 denote significant findings.
Given our findings of accelerated clearance of DNA-treated RBCs in mice, we asked whether mtDNA would differ between critically ill patients with anemia and sepsis and critically ill patients with sepsis who were not anemic. A hemoglobin threshold of 7 g/dL was used to define anemia based on the current standard of care and transfusion guidelines for critically ill patients with sepsis (35, 36). We observed that critically ill septic patients with anemia had higher RBC-associated mtDNA than non-anemic patients with sepsis, supporting the in vivo findings of accelerated erythrophagocytosis of DNA carrying RBCs (Table 1).
Table 1. Critically ill sepsis population stratified by clinical anemia threshold.
Data are shown as number observed (%) for categorical data, or by mean ± standard deviation or median (interquartile range) for continuous data, as appropriate to the data distribution. Comparisons shown were calculated by chi square tests (categorical data: sex, race) and Ranksum testing (continuous, non-normally distributed: age and RBC-bound mtDNA).
| Anemic Hb ≤ 7 | Not Anemic Hb > 7 | p-value | |
|---|---|---|---|
| (n=9) | (n=39) | ||
| Age | 59.3 ± 16 | 56.9 ± 16 | 0.82 |
| Female sex | 2 (22%) | 13 (33%) | 0.52 |
| Race | |||
| African American | 2 (22%) | 14 (36%) | 0.26 |
| Caucasian | 6 (67%) | 23 (59%) | |
| Asian | 0 | 1 (3%) | |
| Other or More than 1 | 1 (11%) | 1 (3%) | |
| RBC-bound mtDNA (x 107)/107 RBCs | 1.21 (0.27 – 3.47) | 0.24 (0.03 – 0.80) | 0.036 |
We next examined whether CpG-carrying RBCs would alter innate immune responses. To isolate the role of RBCs in initiating systemic inflammation, we utilized a reductionist model of CpG-carrying RBC infusion. Splenic histology 6 hours following CpG-RBC (but not PBS-treated RBCs) infusion also revealed increased neutrophil infiltration and enhanced red pulp congestion (Fig. 4E and F). To further characterize the immune response following CpG-RBCs, we performed RNA-seq on spleens from mice treated with RBCs or CpG-treated RBCs (Table 2 and Data File S1 and S2). CpG-RBCs elicited a transcriptomic response characterized by increased expression of interferon signaling pathway genes compared with PBS-treated RBCs (Fig. 4G and H). Given the central role of interferon (IFN)-γ in mediating hemophagocytic lymphohistiocytosis, a condition characterized by accelerated erythrophagocytosis, which is also often observed in sepsis and infection, we examined IFN-γ in the plasma after the administration of CpG-carrying RBCs (12, 14). Plasma IFN-γ and interleukin (IL)-6 were increased at 6 hours post-infusion of CpG-treated, but not PBS-treated, RBCs (Fig. 4I and J). Collectively, these data demonstrate that CpG-carrying RBCs undergo accelerated erythrophagocytosis and initiate local and systemic immune responses.
Table 2.
Differentially expressed genes.
| Top 25 DEG based on EdgeR | unadjusted p-value from EdgeR | adjusted p-values from EdgeR | unadjusted p-value from DEseq2 | adjusted p-values from Deseq2 |
|---|---|---|---|---|
| Upregulated genes | ||||
| Ifit3 | 3.53E-04 | 5.11E-01 | 7.84E-07 | 6.86E-04 |
| Ifi204 | 1.20E-04 | 5.08E-01 | 7.54E-10 | 3.58E-06 |
| Oas2 | 5.26E-04 | 5.36E-01 | 1.49E-09 | 5.30E-06 |
| Klra2 | 1.02E-03 | 5.89E-01 | 1.17E-05 | 5.94E-03 |
| Ifit3b | 6.97E-04 | 5.36E-01 | 1.46E-06 | 1.09E-03 |
| Ifitm3 | 9.84E-04 | 5.89E-01 | 1.68E-05 | 7.72E-03 |
| Rtp4 | 6.70E-04 | 5.36E-01 | 4.85E-07 | 4.61E-04 |
| Oas1a | 7.68E-04 | 5.36E-01 | 7.36E-06 | 4.77E-03 |
| Ifi44 | 7.94E-04 | 5.36E-01 | 3.09E-07 | 3.38E-04 |
| Smpdl3b | 8.51E-04 | 5.36E-01 | 4.43E-06 | 3.00E-03 |
| Irf7 | 4.85E-04 | 5.36E-01 | 1.66E-08 | 3.93E-05 |
| Ifit1 | 2.04E-04 | 5.08E-01 | 3.27E-08 | 6.65E-05 |
| Usp18 | 6.09E-04 | 5.36E-01 | 9.32E-07 | 7.37E-04 |
| Trim30c | 7.24E-04 | 5.36E-01 | 1.45E-05 | 6.87E-03 |
| Parp12 | 2.46E-04 | 5.08E-01 | 3.48E-07 | 3.54E-04 |
| Gm5431 | 4.67E-04 | 5.36E-01 | 2.48E-07 | 2.99E-04 |
| Marco | 1.68E-04 | 5.08E-01 | 4.05E-08 | 7.21E-05 |
| Oasl2 | 3.47E-05 | 4.56E-01 | 3.32E-15 | 4.73E-11 |
| Arhgef37 | 6.29E-05 | 4.56E-01 | 4.25E-10 | 3.03E-06 |
| Tppp3 | 2.21E-04 | 5.08E-01 | 4.18E-09 | 1.19E-05 |
| Irgm1 | 8.33E-04 | 5.36E-01 | 1.21E-05 | 5.94E-03 |
| Slfn5 | 3.50E-04 | 5.11E-01 | 1.43E-07 | 2.26E-04 |
| Ifi206 | 6.99E-04 | 5.36E-01 | 9.79E-06 | 5.16E-03 |
| Downregulated genes | ||||
| Zdhhc1 | 3.04E-04 | 5.11E-01 | 8.59E-01 | 1.00 |
| rsc1a1 | 5.39E-04 | 5.36E-01 | not found | not found |
Erythrophagocytosis of CpG-RBCs and CpG-induced inflammation are dependent on RBC-TLR9
The antibody mIAP301 blocks the antiphagocytic CD47 epitope on murine erythrocytes (37). We thus asked whether CpG-treatment would lead to a loss of CD47 detection using this antibody. CpG-treatment of erythrocytes from wild type (WT), but not TLR9 knockout (KO) mice, resulted in a TLR9-dependent loss of CD47 detection (Fig. 5A and B). Interestingly, TLR9 KO mice also exhibited a higher number of CD47 negative RBCs in the circulation. Recent studies have demonstrated a role for monocyte and macrophage TLR9 signaling in erythrophagocytosis; these findings may reflect decreased clearance of CD47 negative RBCs in TLR9 KO mice; alternatively, these findings may reflect fundamental differences in TLR9 KO and WT erythrocytes (12–14).
Figure 5. RBC-TLR9 mediates accelerated erythrophagocytosis following CpG binding.
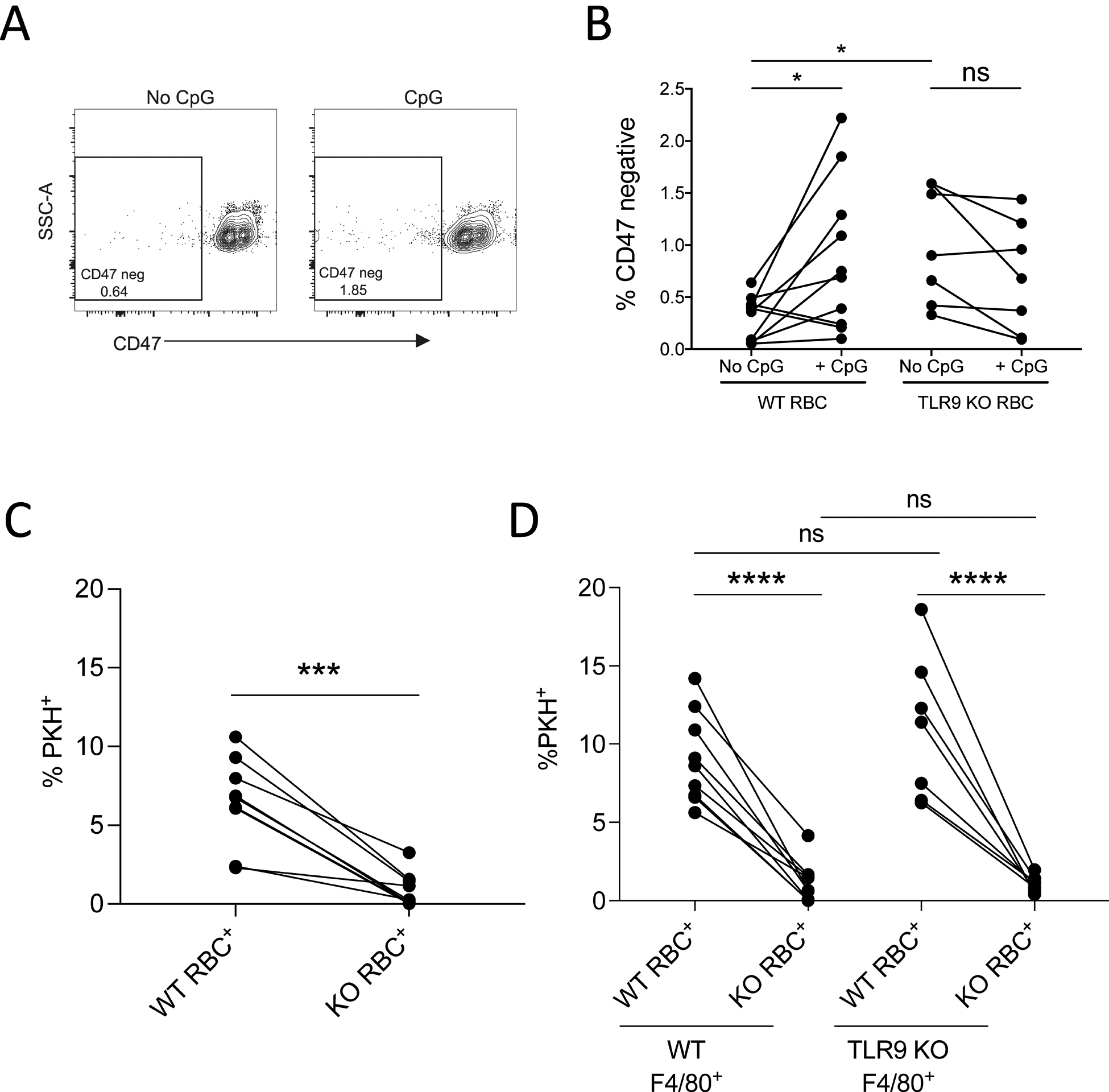
(A) CD47 detection (clone mIAP301) on WT murine RBC treated with 100nM CpG is shown (n=10, one representative study shown). (B) The proportion of CD47-negative RBCs from ex vivo CpG-treated WT or TLR9 KO RBCs was quantified. Each line represents RBCs from an individual mouse. A paired t-test was used to calculate significance within groups (WT or TLR9 KO) and Mann-Whitney U test was used to compare between groups. (WT versus TLR9 KO), n=6 to 9 mice per group, 3 independent experiments. (C and D) WT and TLR9 KO mice were separately labeled with PKH dyes and 1×108 cells from each were mixed and treated with PBS or 25nM CpG immediately before infusion into WT or TLR9 KO mice. Erythrophagocytosis was analyzed by measuring PKH positivity on CD45+CD3−F4/80+ cells. (C) The percent of splenic red pulp macrophages that phagocytosed PBS-treated RBCs is shown (n=6 or more per group). (D) The percent of splenic macrophages that phagocytosed RBCs in WT mice and CpG-treated RBCs isolated from WT or TLR9 KO mice is shown (n=6 or more per group). For (C and D), statistical analysis within group was done by paired t-test and analysis between groups were done with one-way ANOVA with Tukey’s correction Data shown are pooled data from three independent experiments. For all panels, *P<0.05, **P<0.01, ***P<0.005, and ****P<0.001 denote significant findings.
To determine if accelerated erythrophagocytosis was dependent on RBC-expressed TLR9, we examined erythrophagocytosis of untreated WT and TLR9 KO RBCs in naïve mice. WT and TLR9 KO RBCs were labeled with Paul Karl Horan (PKH) dye and mixed before transfusion to naïve WT mice (fig. S6B). Splenic phagocytes ingested more WT RBCs than TLR9 KO RBCs (Fig. 5C and D). This observation was consistent with our previous observations of higher amounts of endogenous mtDNA on WT RBCs than TLR9 KO RBCs(4). We next asked whether TLR9 KO RBCs underwent accelerated erythrophagocytosis following CpG treatment. TLR9 KO and WT RBCs were labeled with PKH dye and combined before incubation with CpG. The CpG-treated RBCs were administered to WT or global TLR9 KO mice. We observed that F4/80hi macrophages ingested higher amounts of WT than KO RBCs (Fig. 5D). This effect appears to be driven by the RBCs as macrophages from TLR9 KO mice ingested similar numbers of RBCs as WT macrophages.
To understand the role of erythrocyte TLR9 in the innate immune response in vivo, we generated erythrocyte TLR9 KO mice (Fig. 6A and B, Table 3). Erythroid cells from erythrocytetlr9−/− mice (Erytlr9−/−) did not express TLR9. To better define the role of cell-free nucleic acid-sensing and RBC-TLR9 in innate immunity, we subjected WT and Erytlr9−/− mice to a reductionist model of CpG-induced inflammation. CpG administration led to decreased circulating white blood cells and increased spleen weight in both WT and Erytlr9−/− mice (fig. S7A and B). IL-6 production in the spleen, liver, and plasma was attenuated in Erytlr9−/− mice (Fig. 6C to E). Erytlr9−/− mice also displayed attenuated liver endothelial activation and TLR9 upregulation but not IL-10, IFN-γ or IL12p70 production (fig. S7 C and D). Collectively, these findings suggest that RBC-TLR9 mediated CpG delivery regulates local and systemic IL-6 production.
Figure 6. Deletion of Erythrocyte TLR9 alters the host immune response.
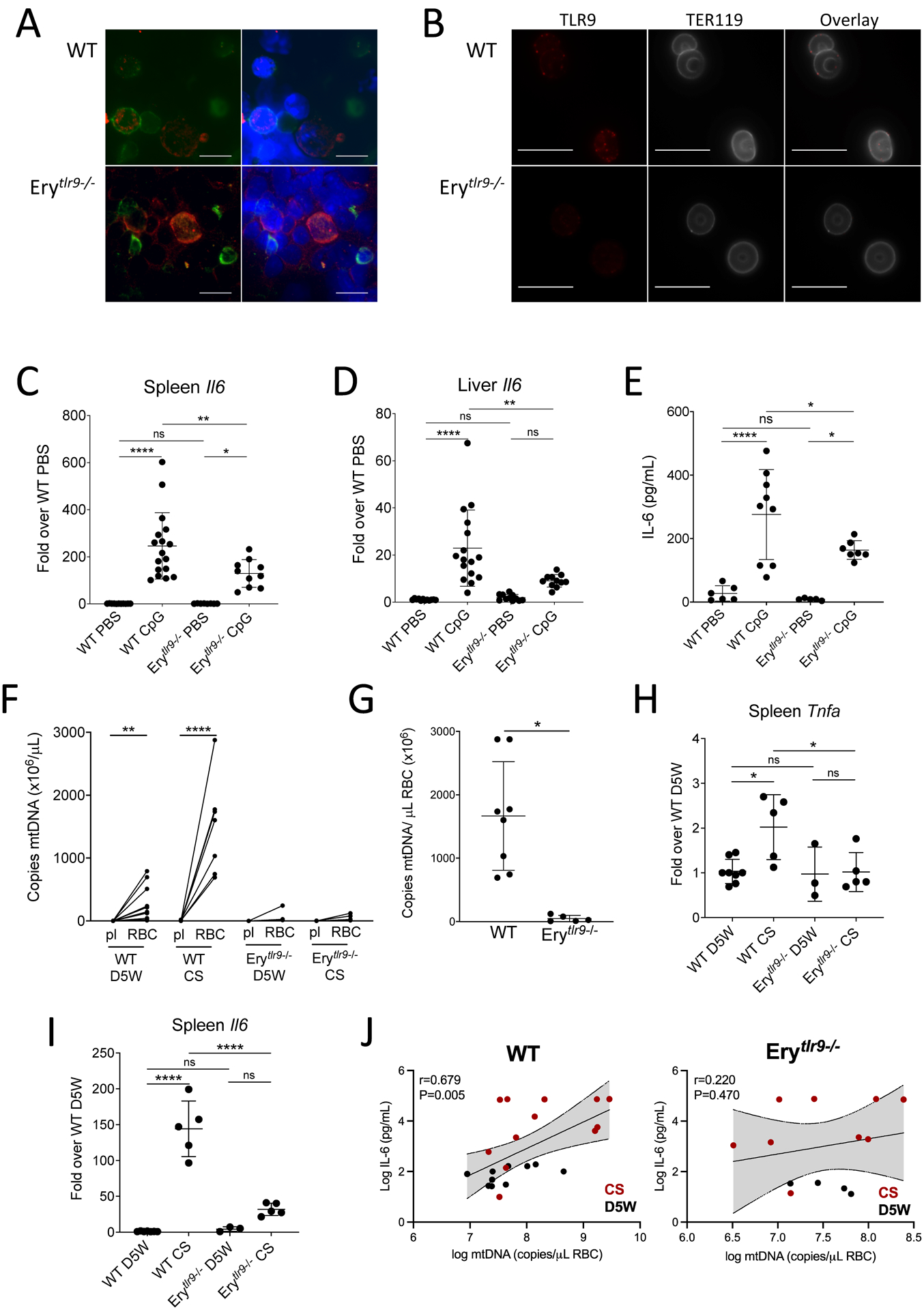
(A and B) TLR9 staining in a bone marrow smear (A) and mature RBCs (B) of WT and Erytlr9−/− mice are shown. The erythroid marker TER119 is used to identify erythroid cells. A representative image is shown. n=3 mice of each genotype were evaluated. Scale bars, 10μm. (C and D) Quantification of Il6 transcripts in spleens (C) and livers (D) of Erytlr9−/− mice following tail vein infusion with CpG (n = 8 or more per group). (E) Plasma IL-6 concentrations were measured in Erytlr9−/− mice following tail vein infusion with CpG (n=5 to 9 per group). In (C to E), data are presented as mean±SD and statistical analysis was done using a one-way ANOVA with Tukey’s correction for multiple comparisons. (F to I) Inflammatory responses of Erytlr9−/− mice were measured following cecal slurry (CS)-induced sepsis. (F) mtDNA on RBCs and in plasma (pl) of D5W and CS-treated mice was measured. Statistical analysis was done using a Kruskal-Wallis with a Dunn’s correction for multiple comparisons. This dataset is partially presented in Fig. 1H. (G) mtDNA on RBCs in CS-injected mice from (F) was compared. Statistical analysis was performed using a Kruskal-Wallis test followed by Dunn’s correction and data is presented as mean±SD. (H and I) Quantification of Tnfa (H) and Il6 (I) transcripts in spleens of injected mice is shown (n=3 to 8 mice per group, mean±SD). Statistical analysis was performed using a one-way ANOVA with a Holm-Sidak correction for multiple comparisons. (J) The correlation between plasma IL-6 and RBC-bound mtDNA is shown for control (D5W) and CS-treated WT and Erytlr9−/− mice. For all panels, *P<0.05, **P<0.01, ***P<0.005, and ****P<0.001 denote significant findings.
Table 3. Baseline hematology labs from WT and Erythrocytetlr9−/− mice.
All data are presented as mean (standard deviation). Significance was determined using two tailed t-test for all parameters. NS, not significant.
| C57Bl/6J | Erythrocytetlr9−/− | p-value | |
|---|---|---|---|
| (n=8) | (n=5) | ||
| WBC (10/μL) | 458.0 (165.9) | 552.6 (234.9) | NS |
| RBC (104/μL) | 875.3 (83.9) | 842.8 (126.9) | NS |
| HGB (g/L) | 134.3 (10.5) | 135.8 (16.4) | NS |
| HCT (10−1 %) | 442.1 (41.8) | 432.6 (70.5) | NS |
| MCV (10−1 fL) | 505.5 (13.6) | 512.4 (10.9) | NS |
| MCH (10−1 pg) | 153.8 (4.9) | 161.8 (6.3) | p=0.026 |
| MCHC (g/L) | 304.1 (5.8) | 316.0 (16.3) | p=0.081 |
| PLT (103/μL) | 987.8 (259.7) | 1214.2 (301.5) | NS |
| RDW-SD (10−1 fL) | 238.4 (16.7) | 248.2 (10.2) | NS |
| RDW-CV (10−1 %) | 137.0 (8.2) | 141.2 (8.3) | NS |
| PCT (10−2 %) | 72.4 (17.6) | 89.6 (22.6) | NS |
Abbreviations: White blood cell (WBC), Red blood cell (RBC), Hemoglobin (HGB), Hematocrit (HCT), Mean cell volume (MCV), Mean cell hemoglobin (MCH), Mean cell hemoglobin concentration (MCHC), platelet (PLT), Red Cell Distribution Width-Standard Deviation (RDW-SD), Red Cell Distribution Width-Coefficient of Variation, (RDW-CV), Plateletcrit (PCT)
To determine whether RBC-nucleic acid-binding contributes to innate immune responses during sepsis, we subjected WT and Erytlr9−/− mice to a cecal slurry model of sepsis. Cecal slurry injection led to increased RBC-mtDNA sequestration in WT mice but not Erytlr9−/− mice (Fig. 6F and G). Because our data demonstrate accelerated clearance of DNA-bound RBCs and increased erythrophagocytosis in the spleen, we examined inflammatory responses in the spleen. Spleen IL-6 and TNF-α production was attenuated in the absence of RBC TLR9 during cecal-slurry-induced sepsis (Fig. 6H and I). Furthermore, plasma IL-6 correlated with RBC-bound mtDNA in WT but not Erytlr9−/− mice (Fig. 6J). These findings suggest that RBCs acquired CpG-DNA during sepsis, and RBC-TLR9 dependent DNA delivery drives local innate immune responses during sepsis.
Because plasma mtDNA is elevated in patients with COVID-19 and altered RBC rheology is reported in patients with COVID-19, we asked whether RBC-bound mtDNA was elevated in hospitalized patients with COVID-19 pneumonia. Table 4 shows baseline characteristics of the cohort (22, 38). We observed that RBC-bound mtDNA was elevated in patients with COVID-19 compared with healthy controls (Fig. 7A) and increased with disease severity (Fig. 7B). Given our findings of accelerated clearance of DNA-carrying RBCs and RBC-DNA mediated innate immune activation, we asked whether RBC-bound mtDNA was associated with hemoglobin concentration in patients with COVID-19. We found that day 7 RBC-bound mtDNA correlated with hemoglobin measured on day 7, and this association was driven by severe disease (Fig. 7C to E). When stratified by median hemoglobin, anemic patients with COVID-19 had higher RBC-associated mtDNA than non-anemic patients (Fig. 7 F to H), consistent with our in vivo findings of accelerated erythrophagocytosis of DNA carrying RBCs. Because we observed increased local and systemic IL-6 in mice correlated with RBC-bound mtDNA, we asked whether RBC-bound mtDNA correlated with disease severity as defined by the Apache III scoring system. We observed a correlation between RBC-bound mtDNA and disease severity (Fig. 7I and J). Consistent with our findings of accelerated RBC erythrophagocytosis and inflammation in mouse models, RBC-bound mtDNA in patients with COVID-19 correlated with anemia and severity of illness.
Table 4. Baseline characteristics of patients with COVID-19.
Data are shown as number observed (%) for categorical data, or by mean ± standard deviation or median (interquartile range) for continuous data, as appropriate to the data distribution.
| COVID-19 Patients (n=97) |
|
|---|---|
| Age | 57.6 ± 14.3 |
| Male | 49 (50.5%) |
| Race African American White Asian |
64 (66.0%) 31 (32.0%) 2 (2%) |
| APACHE III score | 44 (27–68) |
| Severity of COVID-19 Mild Severe |
49(50.5%) 48(49.5%) |
| Mortality (90 day) | 18 (18.6%) |
Figure 7. RBC-bound mtDNA is elevated in COVID-19.
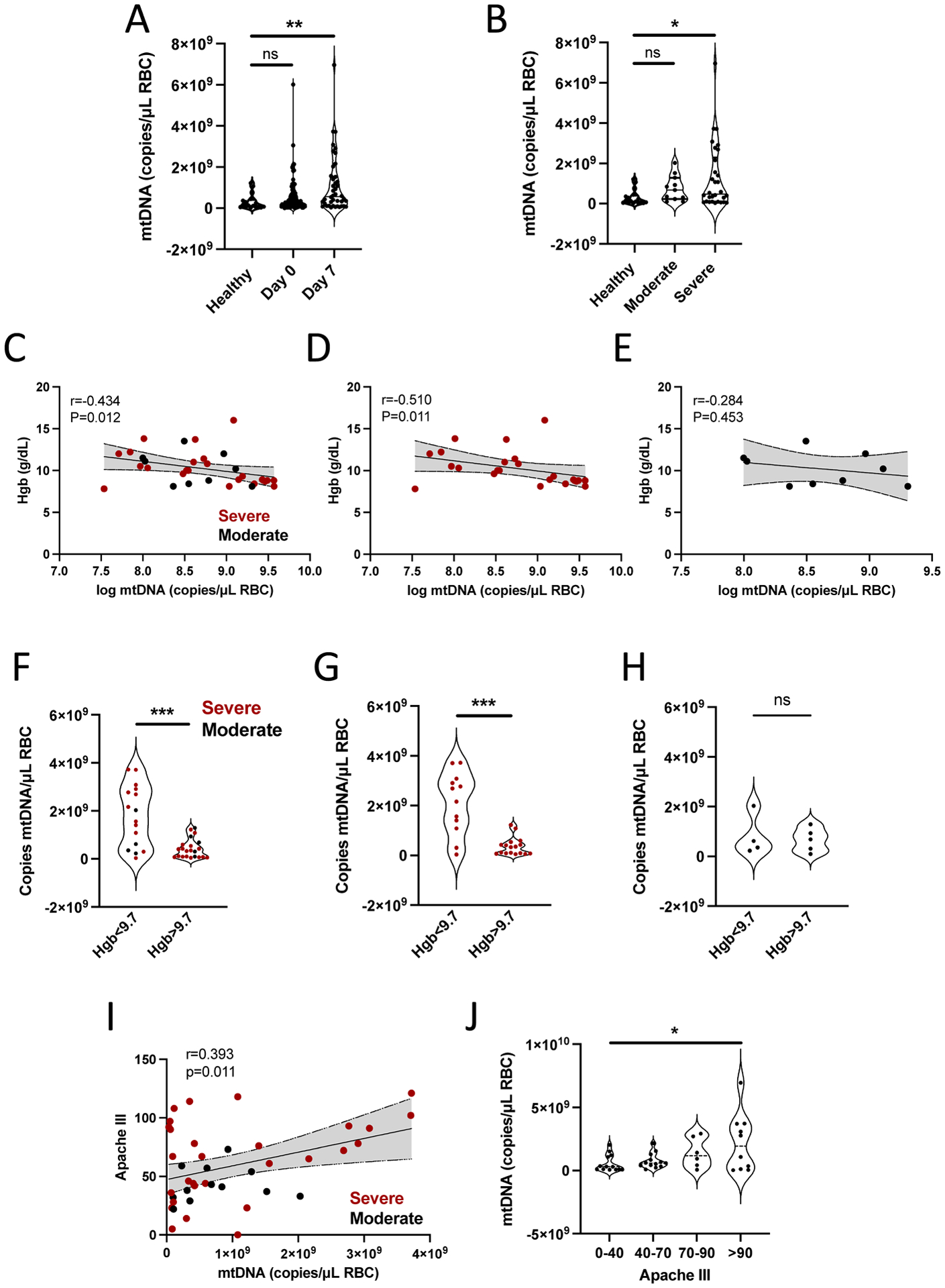
(A) RBC-bound mtDNA was measured in healthy donors and patients hospitalized with COVID-19 at day 0 and day 7 of study enrollment. Data were analyzed by a Kruskal-Wallis test with Dunn’s correction for multiple comparisons. (B) RBC-bound mtDNA in patients was stratified by disease severity. Data were analyzed using a Kruskal Wallis test with Dunn’s correction for multiple comparisons. (C to E) The correlation between RBC-bound mtDNA and hemoglobin for all patients (C), patients with severe disease (D) and patients with moderate disease (E) are shown. Statistical analysis was done using Spearman rank correlation. (F to H) RBC-bound mtDNA was evaluated in patients stratified by median hemoglobin. Data are shown for all patients (F), patients with severe disease (G), and patients with moderate disease (H). Data in (F to H) were analyzed by Mann-Whitney tests. (I) A correlation between Apache III score and RBC-bound mtDNA is shown for COVID-19 patients. Statistical analysis was done using a Pearson’s correlation (J) RBC-bound mtDNA was measured in COVID-19 patients stratified into quartiles based on Apache III score. Data were analyzed by a one-way ANOVA. For all panels, *P<0.05, **P<0.01, ***P<0.005, and ****P<0.001 denote significant findings.
DISCUSSION
In this study, we identify a new role of RBCs in the immune response to infection. We show that mammalian RBCs express surface TLR9 that can bind CpG-containing cell-free DNA. RBC-bound mtDNA is elevated in human sepsis and COVID-19 and associated with anemia. Under basal conditions with low circulating cell-free DNA concentrations, RBCs bind CpG DNA and act as a “sink” without undergoing overt morphological changes (4). However, when plasma CpG DNA is high, such as during sepsis, pneumonia, or malarial infection, TLR9-dependent CpG DNA binding leads to fundamental alterations of RBC morphology, functional loss of CD47 on a subset of RBCs, accelerated erythrophagocytosis, and innate immune activation driven by RBC-DNA delivery with clearance.
Our data show that DNA binding to RBCs results in accelerated erythrophagocytosis. Surprisingly, CpG-binding to RBCs was sufficient to cause accelerated clearance in naïve mice and was dependent on RBC, not phagocyte, TLR9. This may have implications for anemia pathogenesis in multiple infectious and non-infectious inflammatory diseases since excess CpG-DNA in the plasma may result in the removal and destruction of RBCs. Indeed, a recent study of Plasmodium berghei-infected mice showed that disruption of CD47-Sirpα binding led to accelerated erythrophagocytosis, thus implicating this interaction in the host response to malaria (39, 40). Malarial anemia and non-malarial infectious anemia may also arise by developing inflammatory hemophagocytes or the generation of other erythrophagocytic macrophage subsets. CpG-TLR9 interactions have been shown in mice to promote inflammatory anemia during P. yoelli blood-stage infection, and nucleic acid-sensing TLRs promote anemia in a hemophagocytic lymphohistiocytosis model; however, to our knowledge, this is the first report of RBCs driving accelerated erythrophagocytosis (13, 14).
Anemia is common in sepsis, and accelerated clearance of erythrocytes is a defining feature of anemia during sepsis that is poorly understood. Our findings provide one mechanism by which RBCs undergo accelerated clearance in sepsis as we observed RBC-TLR9 dependent accelerated clearance of CpG-carrying RBCs in vivo and an association between RBC-bound mtDNA and hemoglobin in non-COVID-19 sepsis and COVID-19 viral sepsis. Accordingly, targeting RBC-TLR9 with blocking antibodies or antagonistic small molecule inhibitors may be a viable option to combat inflammatory anemia. This could potentially eliminate the enhanced CpG-TLR9 mediated RBC phagocytosis without interfering with CpG-TLR9 signaling in classical immune cells essential for host defense.
In addition to ingestion by macrophages, CpG-carrying RBCs may also be taken up by dendritic cells or other antigen-presenting cells, which may alter antigen presentation and acquired immune responses. Previous studies demonstrated that even a small fraction (0.5%) of CD47-negative RBCs could activate splenic dendritic cells and CD4+ T cells (41–43). Our findings of increased interferon production in mice receiving CpG-RBCs suggest that RBCs can present CpG to immune cells. Indeed, we detected CpG-containing mtDNA on RBCs during parasitic infection, pneumonia, and polymicrobial sepsis in mice and during sepsis and SARS-CoV2 infection in patients. In vivo, CpG-carrying RBCs induced an innate immune response in naïve mice as the administration of CpG-carrying RBCs led to transcriptomic changes in the spleen characterized by upregulation of host response to virus, innate immune response, and interferon signaling pathways.
In the absence of RBC-TLR9, CpG-induced IL-6 was attenuated in local and systemic compartments in a model of CpG-induced inflammation. RBC-TLR9 regulated spleen IL-6 and TNF production, and plasma IL-6 correlated with RBC-bound mtDNA in a murine model of polymicrobial sepsis. Thus, how RBC-TLR9 contributes to immune dysregulation may be context-dependent, depending on the inciting stimuli. RBC-TLR9 may serve as a delivery mechanism of CpG to driving local and systemic IL-6 production.
TLR9 is expressed on nucleated erythrocytes in other vertebrates, including fish (44). Birds express the avian homolog of TLR9, TLR21, on their erythrocytes (44). Here, we demonstrate the presence of TLR9 and DNA binding by human, chimpanzee, and murine RBCs and show a role for RBCs in sensing CpG DNA, a potent activator of the innate immune system. Humans produce over 2 million RBCs each second and are at risk for exposure to large amounts of DNA during mitophagy and nuclear expulsion. Thus, it is tempting to speculate that TLR9 is retained on erythrocytes to protect RBCs during erythroid maturation by scavenging mitochondrial DNA that escapes mitophagy. Indeed, recent studies have shown that loss of mitophagy leads to RBC destruction and anemia, and other studies have demonstrated that mtDNA that escapes mitophagy leads to cell-autonomous TLR9-mediated inflammation (45, 46). Alternatively, given the inflammatory response observed following administration of CpG-treated RBCs to naïve mice, it is plausible to speculate that retention of TLR9 on RBCs promoted host survival by allowing for propagation of local signals remotely and early innate detection of cell-free DNA released during infection or following trauma. Although further studies will be required to elucidate the potential role of TLR9 in erythroid development, our findings of RBC-mediated nucleic acid sensing confirm a role for TLR9 on mature erythrocytes in regulating the immune response during acute inflammation.
Consistent with our discovery of RBC-immune function, nearly two decades of research has solidified the role of another enucleated cell, platelets, in innate and adaptive immunity (18, 47, 48). Future studies examining the cooperation of platelets, RBCs, and coagulation in the innate immune response will be needed to truly understand innate immunity in the vascular compartment. Our findings of elevated RBC-bound mtDNA in patients with COVID-19 that correlate with disease severity and anemia are not surprising in light of studies reporting altered RBC rheology, disruption of vascular homeostasis, and elevated plasma mtDNA in COVID-19 (22, 28, 38). However, these observations underscore the need for further studies into RBC-immune function, which will be essential for a comprehensive understanding of the innate immune response to pathogen and sterile insults. If CpG-delivery by RBCs drives IL-6 production in various inflammatory diseases, targeting RBC-TLR9 may be an effective way to treat cytokine storm without concomitant immune suppression known to occur with monoclonal anti-cytokine antibody therapies. Alternatively, RBC-mediated CpG delivery can be exploited in the development of vaccines and immunotherapy.
Our studies have several limitations. Further in vivo studies will be required to obtain a comprehensive understanding of how RBC-TLR9 modulates innate inflammatory responses following erythrophagocytosis. Although we observe masking of CD47 and morphologic changes following CpG-binding, additional mechanisms may contribute to accelerated erythrophagocytosis of RBCs during sepsis. Analysis of the erythrophagocytic cells in the spleen, liver, and bone marrow on a single-cell level will be necessary to determine the exact mechanisms of innate immune regulation by CpG-carrying RBCs. Additionally, although we do not observe cell-intrinsic cytoplasmic signaling within the RBC upon CpG binding, CpG-induced functional and morphologic changes occurring through TLR9 may be a result of indirect signaling events occurring at the RBC membrane. Further in-depth structural analysis of RBC-TLR9 will be important in the development of specific inhibitors or therapies targeting RBC-TLR9. Although the detrimental effects of severe acute anemia (Hgb < 7) and of blood transfusion are well established in the context of sepsis and critical illness, the implications of more modest anemia and accelerated RBC clearance are not well elucidated. Our animal models demonstrate that RBC clearance drives local and systemic cytokine production and our observations in humans support this paradigm, yet we are not designed to establish causation in the human cohort. Lastly, because our blood collection ended during the acute hospitalization, our cohort studies did not allow us to determine if RBC-bound mtDNA associates with anemia post-COVID recovery in the long term. Given the recent studies demonstrating anemia as a major feature of recovered patients with COVID-19, longitudinal studies will be necessary for a complete understanding of the effects of RBC-DNA binding and clearance on sustained anemia and immune recovery (49, 50).
Our data demonstrate that RBCs serve as DNA sensors through surface expression of TLR9, which appears to be beneficial during quiescent states, where it promotes scavenging of trace CpG to prevent non-specific inflammation (4). However, during conditions characterized by excess circulating CpG, such as sepsis and COVID-19, binding of CpG by RBC-TLR9 leads to accelerated clearance and inflammation. This innate immune mechanism may be beneficial in the clearance of damaged RBCs and likely contributes to systemic inflammation and the development of anemia during pathologic states where cell-free DNA is elevated. Thus, DNA recognition by TLR9 on RBCs provides bona fide evidence for RBCs as immune sentinels.
MATERIALS AND METHODS
Study Design
We designed a series of studies to examine the mechanisms of RBC-DNA binding and test the hypothesis that RBCs function as DNA sensors and couriers capable of altering the host response during inflammatory states. We first designed an observational study of human and chimpanzee RBCs to establish the presence of TLR9 and CpG-DNA on RBCs. We next designed an observational cohort study of mice subjected to different models of infectious challenge (cecal slurry sepsis, pneumonia or parasite infection) to confirm the acquisition of CpG-DNA during infection. We then proceeded with prospective cohort studies in mice to determine if TLR9 on RBCs altered erythrophagocytosis and systemic and local inflammatory responses following CpG challenge or cecal slurry induced sepsis. Mice were randomized to respective treatment groups. Spleen pathology quantification, RNA-Seq analysis and cytokine assays were performed by blinded investigators. Analysis of experimental data in mice was not blinded. Observational cohort studies of critically ill sepsis patients and hospitalized COVID-19 patients were then designed to determine the association between RBC-bound mtDNA and anemia and disease severity.
Animal studies were conducted in accordance with the Institutional Animal Care and Use Committee at the University of Pennsylvania. Studies involving human subjects were approved by the University of Pennsylvania Institutional Review Board. Healthy volunteers, patients, or their surrogates gave written informed consent prior to inclusion.
Sepsis Cohort
RBCs from the day of ICU admission and 7 days later were obtained from human subjects enrolled in the Molecular Epidemiology of SepsiS in the ICU (MESSI) cohort study at the University of Pennsylvania, IRB # 808542(51, 52). Patients were eligible if they presented to the medical ICU with strongly suspected or confirmed infection and new or worsening organ dysfunction in accordance with historic Sepsis-2 “severe sepsis” criteria (53), since enrollment preceded publication of Sepsis-3(54). Exclusion criteria included primary reason for ICU admission unrelated to infection, admission from a long-term acute-care hospital which might select for sepsis survivors, or desire for exclusively palliative measures on ICU admission. Human subjects or their proxies provided informed consent. The cohort of patients with COVID-19 were MESSI subjects enrolled between March 23, 2020 and November 2, 2020 who tested positive for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by PCR(55). Patients with SARS-CoV-2 were admitted to the hospital ward or the ICU. COVID-19 severity was classified by the WHO ordinal scale at enrollment and every 7 days until day 28, and subjects were considered to have moderate disease if hospitalized and requiring low flow oxygen or no oxygen, and severe disease if requiring high flow oxygen (greater than 40 liters per minute), non-invasive ventilation, or invasive ventilation with or without additional organ support (56, 57). Hemoglobin parameters were collected from the electronic medical record complete blood count on day 0 and day 7, selecting the lowest value from the day if more than one measurement was available per day.
Experimental animals
C57BL/6 mice were purchased from the Charles River Laboratories Inc. TLR9 knockout mice were produced by S. Akira and provided by Dr. Edward Behrens (Children’s Hospital of Philadelphia). Mice lacking TLR9 in erythroid compartment are generated by crossing ErGFPcre mice (a generous gift from Dr. Ursula Klingmüller, German Cancer Research Center) and TLR9 KO Conditional (TLR9 flx obtained from the European Mutant Mouse Archive)(58). Genotype was confirmed through PCR amplification with the primers listed in table S1. All experimental procedures were performed on 8 to 12-week-old mice.
Statistics
Differences between groups were compared using a t-test, Mann-Whitney U test, or one-way analysis of variance (ANOVA), as appropriate based on tests for normality (Shapiro-Wilk). All statistical analyses were performed using Sigma Plot 13 software (Systat Software Inc) and GraphPad Prism version 9 for Windows. A P value<0.05 was considered significant for all analyses. Individual level data is included in Data File S3.
Supplementary Material
Acknowledgments:
We thank Peggy Zhang for her excellent technical assistance. We thank Dorothy E. Loy and Dana Hodge for preparing P. falciparum culture. We thank Jane Fontenot, Melany Musso and Francois Villinger at the New Iberia Research Center for providing leftover blood samples from captive chimpanzees. We thank Caroline Ittner, Ariel Weissman, Leticia Kuri-Cervantes, and M. Betina Pampena for providing samples from sepsis and COVID-19 patients. We thank the Shin lab (Mark Boyer, Jessica Doerner, and Xin Liu) for providing samples from Legionella-infected mice. Illustrations were created with BioRender.
Funding:
The research was supported by the following grants: National Institutes of Health grant R01 HL126788 (to NSM), National Institutes of Health grant R01 AI 091595 (to BHH), National Institutes of Health grant UM1 AI126620 (to BHH) Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award No. W81XWH-15-1-0363 (to NSM).
Competing interests:
NJM receives funding to her institution for clinical trials sponsored by Quantum Leap Healthcare Collaborative, Athersys Inc, BioMarck Inc, and the Marcus Foundation. CH is a member of the SAB of Surface Oncology, Rubius Therapeutics, Xilio Therapeutics, AnaptysBio and Synthekine.
NSM is an inventor on “Methods for Detection of Pathogenic Infections using Red Blood Cell-containing Patient Samples”. US Provisional Patent Application No. 63/022,181. PCT application No. PCT/US2021/031323.
Footnotes
Data and materials availability:
All data associated with this study are in the paper or supplementary materials.
References
- 1.Neote K, Darbonne W, Ogez J, Horuk R, Schall TJ, Identification of a promiscuous inflammatory peptide receptor on the surface of red blood cells. J Biol Chem 268, 12247–12249 (1993). [PubMed] [Google Scholar]
- 2.Darbonne WC, Rice GC, Mohler MA, Apple T, Hebert CA, Valente AJ, Baker JB, Red blood cells are a sink for interleukin 8, a leukocyte chemotaxin. J Clin Invest 88, 1362–1369 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson RA Jr., The immune-adherence phenomenon; an immunologically specific reaction between microorganisms and erythrocytes leading to enhanced phagocytosis. Science 118, 733–737 (1953). [DOI] [PubMed] [Google Scholar]
- 4.Hotz MJ, Qing D, Shashaty MGS, Zhang P, Faust H, Sondheimer N, Rivella S, Worthen GS, Mangalmurti NS, Red Blood Cells Homeostatically Bind Mitochondrial DNA through TLR9 to Maintain Quiescence and to Prevent Lung Injury. Am J Respir Crit Care Med 197, 470–480 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S, A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL, Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med 202, 1131–1139 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ, Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ, Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22, 146–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cossarizza A, Pinti M, Nasi M, Gibellini L, Manzini S, Roat E, De Biasi S, Bertoncelli L, Montagna JP, Bisi L, Manzini L, Trenti T, Borghi V, Mussini C, Increased plasma levels of extracellular mitochondrial DNA during HIV infection: a new role for mitochondrial damage-associated molecular patterns during inflammation. Mitochondrion 11, 750–755 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA, Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y, Lawler LA, Christie JD, Meyer NJ, Mc Causland FR, Waikar SS, Waxman AB, Chung RT, Bueno R, Rosas IO, Fredenburgh LE, Baron RM, Christiani DC, Hunninghake GM, Choi AM, Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 10, e1001577; discussion e1001577 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, Kambayashi T, Koretzky GA, Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest 121, 2264–2277 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akilesh HM, Buechler MB, Duggan JM, Hahn WO, Matta B, Sun X, Gessay G, Whalen E, Mason M, Presnell SR, Elkon KB, Lacy-Hulbert A, Barnes BJ, Pepper M, Hamerman JA, Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science 363, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang A, Pope SD, Weinstein JS, Yu S, Zhang C, Booth CJ, Medzhitov R, Specific sequences of infectious challenge lead to secondary hemophagocytic lymphohistiocytosis-like disease in mice. Proc Natl Acad Sci U S A 116, 2200–2209 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Onji M, Kanno A, Saitoh S, Fukui R, Motoi Y, Shibata T, Matsumoto F, Lamichhane A, Sato S, Kiyono H, Yamamoto K, Miyake K, An essential role for the N-terminal fragment of Toll-like receptor 9 in DNA sensing. Nat Commun 4, 1949 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Eaton-Bassiri A, Dillon SB, Cunningham M, Rycyzyn MA, Mills J, Sarisky RT, Mbow ML, Toll-like receptor 9 can be expressed at the cell surface of distinct populations of tonsils and human peripheral blood mononuclear cells. Infect Immun 72, 7202–7211 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ewaschuk JB, Backer JL, Churchill TA, Obermeier F, Krause DO, Madsen KL, Surface expression of Toll-like receptor 9 is upregulated on intestinal epithelial cells in response to pathogenic bacterial DNA. Infect Immun 75, 2572–2579 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, Ni H, Lazarus AH, Freedman J, Semple JW, Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 107, 637–641 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT, Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A 104, 1919–1924 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang JZ, Liu Z, Liu J, Ren JX, Sun TS, Mitochondrial DNA induces inflammation and increases TLR9/NF-kappaB expression in lung tissue. Int J Mol Med 33, 817–824 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, Mehal WZ, Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest 126, 859–864 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scozzi D, Cano M, Ma L, Zhou D, Zhu JH, O’Halloran JA, Goss CW, Rauseo AM, Liu Z, Sahu SK, Peritore V, Rocco M, Ricci A, Amodeo R, Aimati L, Ibrahim M, Hachem RR, Kreisel D, Mudd PA, Kulkarni HS, Gelman AE, Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donadello K, Piagnerelli M, Reggiori G, Gottin L, Scolletta S, Occhipinti G, Zouaoui Boudjeltia K, Vincent JL, Reduced red blood cell deformability over time is associated with a poor outcome in septic patients. Microvasc Res 101, 8–14 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Piagnerelli M, Boudjeltia KZ, Vanhaeverbeek M, Vincent JL, Red blood cell rheology in sepsis. Intensive Care Med 29, 1052–1061 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Reggiori G, Occhipinti G, De Gasperi A, Vincent JL, Piagnerelli M, Early alterations of red blood cell rheology in critically ill patients. Crit Care Med 37, 3041–3046 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Bateman RM, Sharpe MD, Singer M, Ellis CG, The Effect of Sepsis on the Erythrocyte. Int J Mol Sci 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baskurt OK, Gelmont D, Meiselman HJ, Red Blood Cell Deformability in Sepsis. American Journal of Respiratory and Critical Care Medicine 157, 421–427 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Diorio C, Henrickson SE, Vella LA, McNerney KO, Chase J, Burudpakdee C, Lee JH, Jasen C, Balamuth F, Barrett DM, Banwell BL, Bernt KM, Blatz AM, Chiotos K, Fisher BT, Fitzgerald JC, Gerber JS, Gollomp K, Gray C, Grupp SA, Harris RM, Kilbaugh TJ, John ARO, Lambert M, Liebling EJ, Paessler ME, Petrosa W, Phillips C, Reilly AF, Romberg ND, Seif A, Sesok-Pizzini DA, Sullivan KE, Vardaro J, Behrens EM, Teachey DT, Bassiri H, Multisystem inflammatory syndrome in children and COVID-19 are distinct presentations of SARS–CoV-2. The Journal of clinical investigation 130, 5967–5975 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DC, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, Espevik T, Golenbock DT, Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol 8, 772–779 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Bruce LJ, Beckmann R, Ribeiro ML, Peters LL, Chasis JA, Delaunay J, Mohandas N, Anstee DJ, Tanner MJA, A band 3–based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood 101, 4180–4188 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Salomao M, Zhang X, Yang Y, Lee S, Hartwig JH, Chasis JA, Mohandas N, An X, Protein 4.1R-dependent multiprotein complex: New insights into the structural organization of the red blood cell membrane. Proceedings of the National Academy of Sciences 105, 8026–8031 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brittain JE, Mlinar KJ, Anderson CS, Orringer EP, Parise LV, Integrin-associated protein is an adhesion receptor on sickle red blood cells for immobilized thrombospondin. Blood 97, 2159–2164 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Burger P, Hilarius-Stokman P, de Korte D, van den Berg TK, van Bruggen R, CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood 119, 5512–5521 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP, Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Holst LB, Haase N, Wetterslev J, Wernerman J, Guttormsen AB, Karlsson S, Johansson PI, Aneman A, Vang ML, Winding R, Nebrich L, Nibro HL, Rasmussen BS, Lauridsen JR, Nielsen JS, Oldner A, Pettila V, Cronhjort MB, Andersen LH, Pedersen UG, Reiter N, Wiis J, White JO, Russell L, Thornberg KJ, Hjortrup PB, Muller RG, Moller MH, Steensen M, Tjader I, Kilsand K, Odeberg-Wernerman S, Sjobo B, Bundgaard H, Thyo MA, Lodahl D, Maerkedahl R, Albeck C, Illum D, Kruse M, Winkel P, Perner A, Lower versus higher hemoglobin threshold for transfusion in septic shock. N Engl J Med 371, 1381–1391 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, Rochwerg B, Rubenfeld GD, Angus DC, Annane D, Beale RJ, Bellinghan GJ, Bernard GR, Chiche JD, Coopersmith C, De Backer DP, French CJ, Fujishima S, Gerlach H, Hidalgo JL, Hollenberg SM, Jones AE, Karnad DR, Kleinpell RM, Koh Y, Lisboa TC, Machado FR, Marini JJ, Marshall JC, Mazuski JE, McIntyre LA, McLean AS, Mehta S, Moreno RP, Myburgh J, Navalesi P, Nishida O, Osborn TM, Perner A, Plunkett CM, Ranieri M, Schorr CA, Seckel MA, Seymour CW, Shieh L, Shukri KA, Simpson SQ, Singer M, Thompson BT, Townsend SR, Van der Poll T, Vincent JL, Wiersinga WJ, Zimmerman JL, Dellinger RP, Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Crit Care Med 45, 486–552 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, Xu H, Peng H, Fu YX, Xu MM, CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med 21, 1209–1215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renoux C, Fort R, Nader E, Boisson C, Joly P, Stauffer E, Robert M, Girard S, Cibiel A, Gauthier A, Connes P, Impact of COVID-19 on red blood cell rheology. British journal of haematology 192, e108–e111 (2021). [DOI] [PubMed] [Google Scholar]
- 39.Banerjee R, Khandelwal S, Kozakai Y, Sahu B, Kumar S, CD47 regulates the phagocytic clearance and replication of the Plasmodium yoelii malaria parasite. Proc Natl Acad Sci U S A 112, 3062–3067 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ayi K, Lu Z, Serghides L, Ho JM, Finney C, Wang JCY, Liles WC, Kain KC, CD47-SIRPalpha Interactions Regulate Macrophage Uptake of Plasmodium falciparum-Infected Erythrocytes and Clearance of Malaria In Vivo. Infect Immun 84, 2002–2011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yi T, Li J, Chen H, Wu J, An J, Xu Y, Hu Y, Lowell CA, Cyster JG, Splenic Dendritic Cells Survey Red Blood Cells for Missing Self-CD47 to Trigger Adaptive Immune Responses. Immunity 43, 764–775 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu J, Wu H, An J, Ballantyne CM, Cyster JG, Critical role of integrin CD11c in splenic dendritic cell capture of missing-self CD47 cells to induce adaptive immunity. Proc Natl Acad Sci U S A 115, 6786–6791 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van den Berg TK, van Bruggen R, Loss of CD47 Makes Dendritic Cells See Red. Immunity 43, 622–624 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Morera D, Roher N, Ribas L, Balasch JC, Donate C, Callol A, Boltana S, Roberts S, Goetz G, Goetz FW, MacKenzie SA, RNA-Seq reveals an integrated immune response in nucleated erythrocytes. PLoS One 6, e26998 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K, Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK, Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A 107, 832–837 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Pare A, Rousseau M, Naika GS, Levesque T, Laflamme C, Marcoux G, Lambeau G, Farndale RW, Pouliot M, Hamzeh-Cognasse H, Cognasse F, Garraud O, Nigrovic PA, Guderley H, Lacroix S, Thibault L, Semple JW, Gelb MH, Boilard E, Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 124, 2173–2183 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Semple JW, Italiano JE, Freedman J, Platelets and the immune continuum. Nature Reviews Immunology 11, 264–274 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Al-Aly Z, Xie Y, Bowe B, High-dimensional characterization of post-acute sequalae of COVID-19. Nature, (2021). [DOI] [PubMed] [Google Scholar]
- 50.Mao J, Dai R, Du R-C, Zhu Y, Shui L-P, Luo X-H, Hematologic changes predict clinical outcome in recovered patients with COVID-19. Annals of Hematology 100, 675–689 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faust HE, Reilly JP, Anderson BJ, Ittner CAG, Forker CM, Zhang P, Weaver BA, Holena DN, Lanken PN, Christie JD, Meyer NJ, Mangalmurti NS, Shashaty MGS, Plasma Mitochondrial DNA Levels Are Associated With ARDS in Trauma and Sepsis Patients. Chest, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reilly JP, Wang F, Jones TK, Palakshappa JA, Anderson BJ, Shashaty MGS, Dunn TG, Johansson ED, Riley TR, Lim B, Abbott J, Ittner CAG, Cantu E, Lin X, Mikacenic C, Wurfel MM, Christiani DC, Calfee CS, Matthay MA, Christie JD, Feng R, Meyer NJ, Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: evidence from Mendelian randomization and mediation analysis. Intensive Care Med 44, 1849–1858 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G, 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 31, 1250–1256 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC, The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). Jama 315, 801–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, Alanio C, Kuri-Cervantes L, Pampena MB, D’Andrea K, Manne S, Chen Z, Huang YJ, Reilly JP, Weisman AR, Ittner CAG, Kuthuru O, Dougherty J, Nzingha K, Han N, Kim J, Pattekar A, Goodwin EC, Anderson EM, Weirick ME, Gouma S, Arevalo CP, Bolton MJ, Chen F, Lacey SF, Ramage H, Cherry S, Hensley SE, Apostolidis SA, Huang AC, Vella LA, Betts MR, Meyer NJ, Wherry EJ, Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldman JD, Lye DCB, Hui DS, Marks KM, Bruno R, Montejano R, Spinner CD, Galli M, Ahn MY, Nahass RG, Chen YS, SenGupta D, Hyland RH, Osinusi AO, Cao H, Blair C, Wei X, Gaggar A, Brainard DM, Towner WJ, Muñoz J, Mullane KM, Marty FM, Tashima KT, Diaz G, Subramanian A, Remdesivir for 5 or 10 Days in Patients with Severe Covid-19. N Engl J Med 383, 1827–1837 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect Dis 20, e192–e197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heinrich AC, Pelanda R, Klingmüller U, A mouse model for visualization and conditional mutations in the erythroid lineage. Blood 104, 659–666 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Kirsten Moll AK, Scherf Arthur and Wahlgren Mats Methods in Malaria Research. Kirsten Moll AK, Scherf Arthurand Wahlgren Mats Ed., (Manassas, VA, USA, 2013). [Google Scholar]
- 60.Casson CN, Doerner JL, Copenhaver AM, Ramirez J, Holmgren AM, Boyer MA, Siddarthan IJ, Rouhanifard SH, Raj A, Shin S, Neutrophils and Ly6Chi monocytes collaborate in generating an optimal cytokine response that protects against pulmonary Legionella pneumophila infection. PLoS Pathog 13, e1006309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeLaney AA, Berry CT, Christian DA, Hart A, Bjanes E, Wynosky-Dolfi MA, Li X, Tummers B, Udalova IA, Chen YH, Hershberg U, Freedman BD, Hunter CA, Brodsky IE, Caspase-8 promotes c-Rel-dependent inflammatory cytokine expression and resistance against Toxoplasma gondii. Proc Natl Acad Sci U S A 116, 11926–11935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or supplementary materials.