

Abstract
In all domains of life, multi-subunit RNA polymerases (RNAPs) catalyze both the extension of mRNA transcripts by nucleotide addition and the hydrolysis of RNA, which enables proofreading by removal of mis-incorporated nucleotides. A highly conserved catalytic module within RNAPs called the trigger loop (TL) functions as the key controller of these activities. The TL is proposed to act as a positional catalyst of phosphoryl transfer and transcript cleavage via electrostatic and steric contacts with substrates in its folded helical form. The function of a near-universally conserved TL histidine that contacts NTP phosphates is of particular interest. Despite its exceptional conservation, substitutions of the TL His with Gln support efficient catalysis in bacterial and yeast RNAPs. Unlike bacterial TLs, which contain a nearby Arg, the TL His is the only acid-base-catalyst candidate in the eukaryotic RNAPII TL. Nonetheless, replacement of the TL His with Leu is reported to support cell growth in yeast, suggesting that even hydrogen bonding and polarity at this position may be dispensable for efficient catalysis by RNAPII. To test how a TL His-to-Leu substitution affects enzymatic functions of RNAPII, we compared its rates of nucleotide addition, pyrophosphorolysis, and RNA hydrolysis to the wild-type RNAPII enzyme. The His-to-Leu substitution slightly reduced rates of phosphoryl transfer with little if any effect on intrinsic transcript cleavage. These findings indicate that the highly conserved TL His is neither an obligate acid–base catalyst nor polar contact for NTP phosphates but instead functions as a positional catalyst mainly through steric effects.
Keywords: transcription, RNA polymerase II, phosphoryl transfer, trigger loop, positional catalysis, RNA hydrolysis
Graphical Abstract
INTRODUCTION
Transcription of the information encoded in DNA into RNA is governed by multi-subunit RNA polymerases (RNAPs) in all domains of life. Prokaryotes and archaea use a single RNAP to perform all transcription, whereas eukaryotes use up to five distinct nuclear RNAPs. All eukaryotes make large rRNAs with RNAPI, mRNAs and many non-coding RNAs with RNAPII, and tRNAs and other sRNAs including 5S rRNA with RNAPIII; land plants and some algae additionally use RNAPIV and RNAPV in gene silencing pathways.1–5 Although the complex regulation of transcription by both protein and nucleic-acid cofactors varies, the structure and function of the active site in mRNA-synthesizing RNAPs is highly conserved from bacteria to mammals. This single active site catalyzes both the reversible phosphoryl transfer reaction that produces RNA and hydrolysis reactions that remove 3′ ribonucleotides for proofreading or reactivation of stalled RNAP.6–9 Both activities require two proton transfers (i.e., acid–base catalysis)10–12 and are aided by elongation and cleavage cofactors, respectively.
RNA synthesis by RNAP is described by an iterative nucleotide addition cycle (NAC, Figure 1A). RNAP moves (translocates) the DNA template in 1-bp steps, binds NTP substrate, catalyzes two-Mg2+-mediated phosphodiester bond formation, and releases pyrophosphate (PPi). The phosphoryl transfer step is rate-limiting for iterative nucleotide addition during RNA synthesis,10, 11 although the translocation step also can contribute to the overall rate.13 The NAC can be interrupted by reverse translocation of DNA (backtracking) by one or more nucleotides (nt), which removes the RNA 3′ hydroxyl (3′-OH) from the RNAP active site and precludes RNA synthesis. Backtracking can be caused by roadblocks to RNAP forward translocation on DNA such as a nucleosome or DNA damage,14 by misincorporation of an incorrectly paired ribonucleotide,15 or in response to a DNA-encoded pause signal.16 Hydrolysis, phosphorolysis, or pyrophosphorolysis of backtracked RNA forms a new 3′ end in the active site,17–19 allowing the NAC to resume. In bacterial RNAPs and eukaryotic RNAPII, hydrolysis can occur via an intrinsic cleavage mechanism at rates that vary among species, or by a fast factor-assisted mechanism in which cofactors (GreA/B in bacteria; TFS in archaea; TFIIS or Rpb9 paralogs in eukaryotes) stabilize second Mg2+ ion binding.22, 23 Co-transcriptional nucleotide excision after misincorporation plays an essential role in transcriptional fidelity.24
Figure 1. The active site of RNA polymerase.
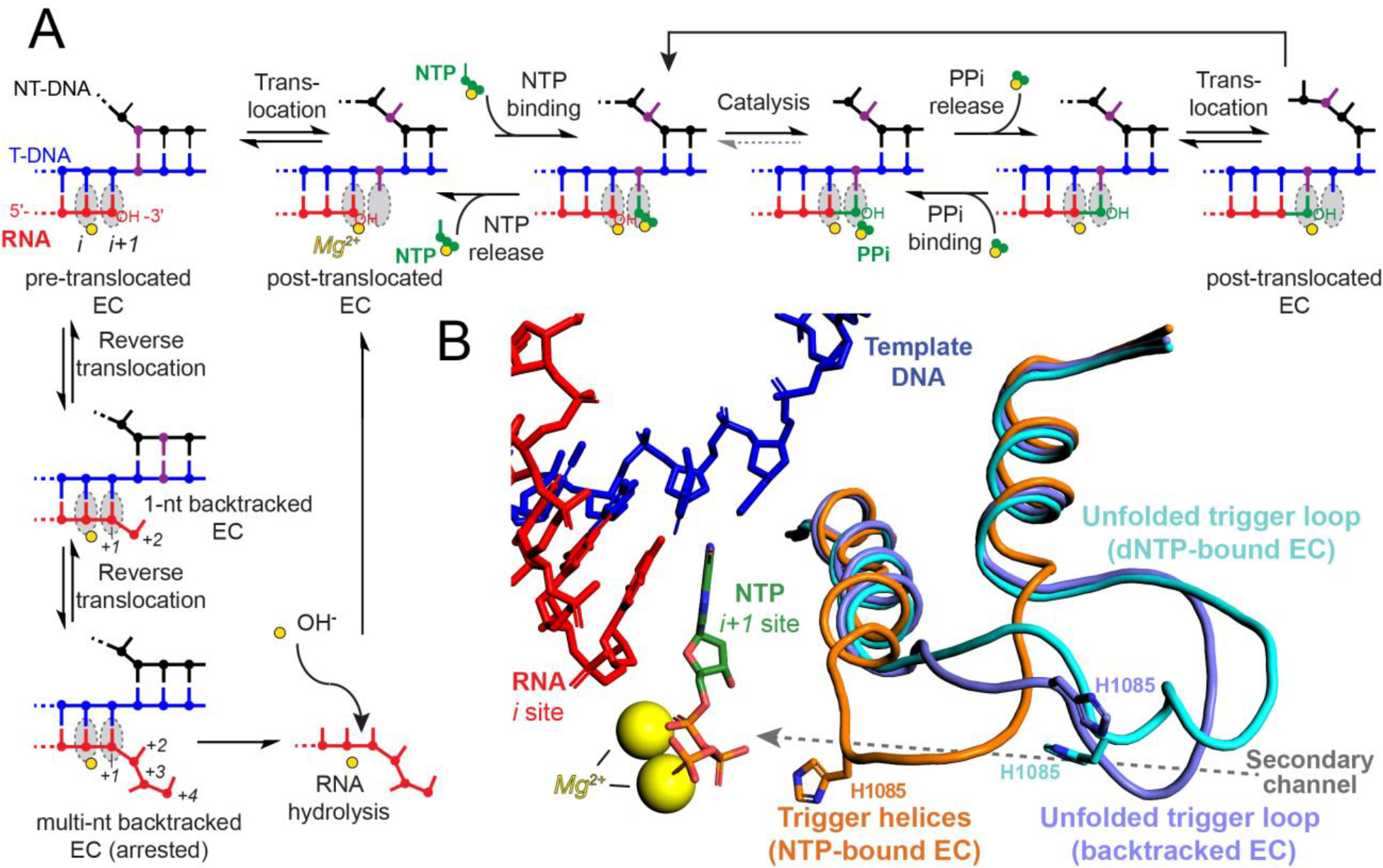
(A) The nucleotide addition cycle (NAC). Binding of NTP substrate to the i+1 site, catalysis of phosphodiester bond formation, pyrophosphate release, and translocation of the nucleic acids occur iteratively to extend the transcript. Reverse translocation (backtracking) can interrupt the NAC in response to misincorporation or a DNA-encoded pause signal. Backtracked ECs can hydrolyze the nascent RNA with or without the aid of extrinsic factors to resume the NAC. Template DNA is shown in blue, nontemplate DNA in black, RNA in red, incoming NTP in green, i and i+1 sites in grey, and magnesium ions in yellow. One base pair in the nontemplate and template DNA is colored in purple to indicate translocation throughout the events of the NAC. (B) Conformers of the yeast RNAPII TL. Nucleic acids and magnesium ions are colored as in panel A. The partially folded TH from S. cerevisiae RNAPII with 3′-deoxy RNA and GTP in the active site are shown in orange (PDB ID code 2E2H).20 The unfolded TL from the S. cerevisiae RNAPII EC with an incorrect dNTP substrate in the active site is shown in cyan (PDB ID code 2NVQ).20 An unfolded TL conformer from a 1-nt backtracked S. cerevisiae RNAPII EC is shown in magenta (PDB ID code 3GTG).21 The secondary channel through which substrates enter the active site is indicated with a grey arrow. For simplicity, only the nucleic-acid scaffold from 2E2H is depicted.
The trigger loop (TL) is a highly conserved module of the largest RNAP subunit (called Rpb1 in RNAPII) that plays crucial roles in the NAC. Folding of the TL into an α-helical hairpin dubbed the trigger helices (TH) closes the active site pore into the secondary channel used for NTP entry and PPi release and positions reactants for efficient catalysis (Figure 1B).12, 25–27 The TL–TH undergoes cycles of folding and unfolding during the NAC, with the folded TL accelerating nucleotide addition by a factor of ≥340.27, 28 TH unfolding into a fluctuating loop conformation allows translocation of the nucleic-acid template through RNAP, PPi release, and NTP entry.29, 30 Binding of an incorrect NTP or of a 2′-dNTP inhibits TL folding whereas TH-stabilizing alterations increase misincorporation, suggesting that TL–TH energetics modulate transcriptional fidelity.20, 31, 32
Sequence alignments of the Rpb1/β′ subunit reveal universal conservation of a TL histidine (TL His, S. cerevisiae Rpb1 H1085, E. coli β′ H936) that directly contacts the NTP substrate in structures of elongation complexes (ECs, Figure 1B).20, 27, 33 In structures of backtracked ECs, the TL assumes a partially unfolded conformer and the TL His is displaced from the active site.21, 34, 35 TL conformational dynamics may aid positioning of the backtracked nucleotide for hydrolysis, at least in E. coli.36 In yeast, inhibition of RNAPII by α-amanitin, which traps H1085 and the TL in an unfolded state,31, 37 fails to stop intrinsic RNA hydrolysis, suggesting intrinsic transcript cleavage occurs without TL assistance in RNAPII.38 The TL also does not participate directly in factor-assisted transcript hydrolysis, as binding of GreA/B or TFIIS sterically excludes the TL from the active site.21, 34, 39 However, H936 stabilizes early backtracked states in E. coli RNAP that are precursors to intrinsic and factor-assisted proofreading,12 thereby potentially affecting transcriptional fidelity. In yeast, substitutions of H1085 are inviable (Ala, Phe, Gly, Pro, Asn, Asp, Glu, Ser) or confer severe (Tyr, Arg, Lys, Trp) or modest (Gln, Leu) growth defects.31, 40, 41 The conserved TL His was initially proposed to act as a general acid–base during phosphoryl transfer and RNA hydrolysis.11, 20 However, more recent evidence suggests that it does not participate in acid–base chemistry but instead acts as a positional catalyst through electrostatic and steric contacts with the NTP substrate.12, 41
The viability of the H1085L substitution is surprising given that it is the only nonpolar substitution for the conserved TL His that supports S. cerevisiae.41 The other viable substitutions, including Gln which supports efficient phosphoryl transfer catalysis when substituted for the conserved TL His in E. coli RNAP,12 can form hydrogen bonds or charge-charge interactions with the NTP phosphates. However, TL H1085L RNAPII remains to be characterized biochemically. Biochemical characterization is needed to verify its apparent catalytic efficiency since other regulators or even genetic suppressors could give compensatory effects in vivo. Further, in vitro analysis of H1085L RNAPII can provide an unambiguous test of any possible role of the TL in acid–base catalysis.
To test the role of the TL His in RNAPII, we purified H1085L RNAPII and assayed its ability to catalyze phosphoryl transfer and hydrolysis reactions in vitro. We determined kinetics of nucleotide addition, pyrophosphorolysis, and factor-independent RNA hydrolysis reactions catalyzed by RNAPII for both the wild-type (WT) and H1085L mutant enzymes. We also created an alignment of 21,161 Rpb1/β′ homologs from all domains of life to explore whether the TL His is completely conserved. Together, our findings support a refined view of the TL as a positional catalyst that functions primarily via steric effects on substrate alignment in the active site of eukaryotic RNAPII rather than participating directly in proton transfer essential for catalysis.
MATERIALS AND METHODS
Proteins
Q5 high-fidelity DNA polymerase, T4 DNA ligase, T4 polynucleotide kinase, apyrase, and high-fidelity StyI were obtained from New England Biolabs. E. coli RNAP was prepared as described previously.12, 25 For RNAPII purification, yeast strain CKY283 and plasmids pCK859 and pCK1748, encoding WT RPO21 and H1085L rpo21 respectively, were kindly provided by Craig Kaplan (U. Pittsburgh). Yeast expressing Rpb1 (UniProt ID P04050) from these plasmids were generated using a plasmid shuffling assay into strain CKY283 (rpo21Δ with plasmid encoding RPO21 and URA3) as described.40, 41 The TL sequence was confirmed at each step by Sanger sequencing using forward primer #11529 and reverse primer #11530 (Table S1). WT and H1085L RNAPII were purified via the tandem-affinity tag on the Rpb3 subunit encoded in strain CKY283 as described.42, 43 Cells collected from 6 L of 2X YAPDT culture were lysed using a BioSpec BeadBeater in 2X lysis buffer (100 mM Tris-HCl pH 7.5, 300 mM KCl, 2 mM EDTA, 10% glycerol) supplemented with 5 mM DTT, 1 mM PMSF, 0.28 mg/L leupeptin, 1.37 mg/L pepstatin A, and 0.33 g/L benzamidine. The lysate was adjusted to 0.5 M ammonium sulfate (~13% of saturation) and separated by ultracentrifugation in a Beckman 50.2 Ti rotor (158,000 × g, 80 min, 4 °C). The supernatant (~180 mL) was then adsorbed with 2 mL IgG Sepharose 6 resin (GE Healthcare) prewashed with 1X lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM KCl, 1 mM EDTA, 5% glycerol) in a disposable chromatography column (Poly-Prep columns, Bio-Rad Laboratories). The column was washed with 40 mL 1X lysis buffer with 0.5 M ammonium sulfate supplemented with DTT and protease inhibitors as above, then 40 mL 1X lysis buffer with 5 mM ATP and 5 mM MgCl2 supplemented with DTT and protease inhibitors, and finally washed with 40 mL 1X lysis buffer supplemented with DTT only. RNAPII was eluted after overnight incubation with TEV protease, concentrated, and further purified by anion-exchange FPLC on a 1 mL MonoQ column (GE Healthcare). RNAPII-containing fractions were collected and transferred into RNAPII storage buffer (20 mM Tris-HCl, pH 8.0, 40 mM KCl, 5 mM MgCl2, 5 mM DTT, 10% glycerol) by dilution and centrifugal ultrafiltration (Amicon Ultra-4 100 kDa molecular weight cutoff; MilliporeSigma) to ~0.35 mg/mL. Aliquots were stored at −80 °C until use.
Nucleic Acids
NTPs and 32P-labeled NTPs were obtained from GE Healthcare Life Sciences and PerkinElmer, respectively. RNA and DNA oligonucleotides were from Integrated DNA Technologies and were gel-purified before use by electrophoresis through 8% (DNA) or 15% (RNA) polyacrylamide gels (19:1 acrylamide:bisacrylamide) containing 8 M urea and TBE buffer (1.25 mM Na2EDTA and 44.5 mM Tris-borate, pH 8.3). Gel pieces containing the oligos were excised and incubated overnight in low salt oligo purification buffer (10 mM Tris-Cl pH 7.5, 1 mM EDTA, 100 mM NaCl). The extracted nucleic acids were then purified on DEAE Sepharose resin (Bio-Rad), eluted with high salt oligo purification buffer (10 mM Tris-Cl pH 7.5, 1 mM EDTA, 2 M NaCl), and ethanol-precipitated. For the ligated-scaffold nucleotide addition assay, fragments of the rpoB gene and the RPO21 gene were PCR-amplified from plasmids pRL785 and pCK859 using forward primers #10551 and #12024, which encoded a StyI cleavage site, and reverse primers #10242 and #12027, respectively (Table S1). The fragments were purified by spermine precipitation44 and digested with StyI to create sticky ends suitable for ligation to the oligonucleotide scaffold. The products of the restriction digest were again spermine precipitated and stored in transcription buffer (TB; 20 mM Tris-HCl, pH 8.0, 40 mM KCl, 5 mM MgCl2, 1 mM DTT) at −20 °C.
Ligated-Scaffold Nucleotide Addition Assay
Template DNA (T-DNA, #10160 or #11991, 10 μM) was annealed to RNA (#8855, 5 μM) in TB in a thermocycler as described previously to form scaffold 1-rpoB or scaffold 1-RPO21, respectively.12, 45 RNAPII (600 nM) was incubated with the template DNA–RNA hybrid (200 nM) at 30 °C for 15 min in TB, then nontemplate DNA (NT-DNA; #10161, 600 nM) was added and incubated at 30 °C for 15 min to complete the EC reconstitution. The ECs (150 nM) were then radiolabeled by reaction with [α-32P]GTP for 5 min at 30 °C followed by incubation with ATP and GTP (100 μM each) for 5 min at 30 °C to form labeled A26 ECs. The dsDNA fragment derived from rpoB or RPO21 by PCR and restriction digestion (15 nM) was then ligated to the downstream end of the A26 EC–scaffold (5 nM) by incubation with T4 ligase (25 units/μL) and ATP (1 mM) for 1 h at 16 °C in TB. Heparin (100 μg/mL) was added to the ligated A26 ECs and incubated for 3 min at 30 °C before transcription was initiated by addition of all 4 NTPs (1 mM each final) in TB at 30 °C. Samples were withdrawn at 10, 20, 30, 40, 60, 300, and 600 s and mixed with an equal volume of 2X stop solution (8 M urea, 30 mM Na2EDTA, 90 mM Tris-borate buffer, and 0.05% each bromophenol blue and xylene cyanol). The time point samples were heated for 2 min at 95 °C and then separated by electrophoresis through an 8% polyacrylamide (19:1 acrylamide:bisacrylamide) gel in 8 M urea–TBE next to a radiolabeled pBR322 DNA ladder. The gel was exposed to a PhosphorImager screen, scanned with a Typhoon PhosphorImager, and quantified with ImageQuant software (GE Healthcare). Densitometry plots were generated by converting pixels to transcript length using a four-factor polynomial fitting of the pBR322 DNA ladder calculated by KaleidaGraph software as a reference. Average transcript length was determined by summing the product of each transcript length with its signal intensity and dividing this sum by the total intensity.46 Elongation rates were estimated by dividing the average transcript length by the reaction time. Rates for at least three independent samples were measured to assess experimental error. Statistical significance was assessed using the unpaired two-sample t-test in GraphPad Prism software.
Single-Nucleotide Addition Assay
ECs were reconstituted as described for the ligated-scaffold nucleotide addition assay using T-DNA (#10160), RNA, (#8855) and NT-DNA (#10161), but with 100 nM template DNA–RNA hybrid, 50 nM RNAPII, and 200 nM NT-DNA. ECs (10 nM) were then radiolabeled by reaction with [α-32P]GTP for 2 min at 30 °C. Extension of RNA to G19 was completed by reaction with unlabeled GTP (100 nM) for 3 min at 30 °C. Radiolabeled G19 ECs were reacted with 1 mM ATP for 2, 4, 6, 8, 12, 16, 32, 64, 128, 256, 512, and 1000 ms at 30 °C in a quench-flow instrument (KinTeK RQF-3). Reaction samples were quenched with 2 M hydrochloric acid, immediately neutralized to pH 8.0 by mixing with 3 M Tris base and 250 μg torula yeast RNA/mL. RNA products were then purified by extraction with phenol:chloroform (5:1; pH 4.3–4.7) followed by ethanol precipitation. Pellets of the ethanol-precipitated samples were resuspended in 1X stop buffer (4 M urea, 15 mM Na2EDTA, 45 mM Tris-borate buffer, and 0.05% each bromophenol blue and xylene cyanol) to constant 32P cpm/mL. The RNA samples were heated for 3 min at 95 °C and then separated by electrophoresis through a 15% polyacrylamide (19:1 acrylamide: bisacrylamide) gel in 8 M urea–TBE. The gel was exposed to a PhosphorImager screen, scanned with a Typhoon PhosphorImager, and quantified with NIH ImageJ software. The amount of RNA of a given length (G19, A20, and A21) at each time point was calculated as a fraction of all RNA species in the quenched reaction. The quench-flow data were fit with kinetic models using the program KinTek Explorer v6.1. To correct for incomplete reactivation of the halted ECs, an unreactive fraction of G19 RNA was included in the model. The fraction of unreactive G19 RNA and the rate of G19-A20 conversion were obtained first and then held constant to obtain the rate of the second step of AMP addition (A20 RNA extension to A21). Each quench-flow dataset was individually fit to obtain rate constants, and the average and error of the rate constants for three replicates were used to estimate experimental error. Statistical significance was assessed using the unpaired two-sample t-test in GraphPad Prism software.
Pyrophosphorolysis
RNA (#11001) was 5′-end radiolabeled by reaction with [γ-32P]ATP and T4 polynucleotide kinase. Labeled RNA (5 μM) was annealed simultaneously with T-DNA (#6355, 7.5 μM) and NT-DNA (#12028, 9.4 μM) to form scaffold 2 as described previously.47 Labeled U14 ECs were formed by incubation of each RNAPII (675 nM) with the DNA–RNA scaffold (225 nM) for 15 min at 30 °C in reconstitution buffer (RB, 20 mM Tris-acetate, pH 8.0, 20 mM potassium acetate, 0.1 mM EDTA). U14 ECs were incubated with washed magnetic Streptavidin M-280 Dynabeads for 15 min at room temperature and then washed twice with RB. The pyrophosphorolysis reaction was initiated by resuspending the bead-bound U14 ECs in RB supplemented with 5 mM magnesium acetate, 0.5 mM sodium pyrophosphate, and 1.5 units/mL apyrase. Samples were withdrawn at 3, 6, 10, 20, 40, 60, 90, 120, and 150 min, mixed with an equal volume of 2X stop buffer, and separated by electrophoresis as described above but using a 20% polyacrylamide gel (19:1 acrylamide: bisacrylamide). The fraction of U14 RNA remaining was fit to a single-exponential decay model using Igor software to obtain rate constants. Decay kinetics were determined for at least three independent samples to determine experimental error. Statistical significance was assessed using the unpaired two-sample t-test in GraphPad Prism software.
Intrinsic Transcript Cleavage
U16 ECs (150 nM) were formed on scaffold 3 in HEPES elongation buffer (HB, 25 mM HEPES-KOH, pH 8.0, 50 mM KCl, 5 mM MgCl2, 1 mM DTT, 25 μg acetylated BSA/mL, 5% glycerol) as described above for scaffold 1 in the ligated-scaffold nucleotide addition assay and then radiolabeled by reaction with [α-32P]CTP.12 Labeled C17 ECs were immobilized on washed magnetic Streptavidin M-280 Dynabeads by incubation for 15 min at room temperature, washed twice with HB, and resuspended in 50 μL HB. The “C17” sample was withdrawn and quenched with an equal volume of 2X stop solution. The immobilized C17 ECs were then pelleted, and the supernatant was replaced with 50 μL HB prewarmed to 30 °C containing CTP and UTP (10 μM each) and incubated at 30 °C for 10 min to form C21 ECs. The beads were washed four times with wash buffer (WB; same as HB but without MgCl2) to remove any cleaved RNA products and unincorporated CTP and UTP. The washed C21 ECs were resuspended in 105 μL WB, the “C21” sample was withdrawn and mixed with 2X stop solution. The C21 ECs were split into two 50 μL portions for replicate time courses. All WB was removed and the beads were resuspended in cleavage buffer (CB; 25 mM Tris-HCl, pH 9.0, 50 mM KCl, 20 mM MgCl2, 1 mM DTT, 25 μg acetylated BSA/mL, 5% glycerol) prewarmed to 30 °C to initiate the hydrolysis reaction. Samples were withdrawn at 3, 6, 10, 20, 40, 60, 120, and 150 min and were quenched with 2X stop solution and separated by gel electrophoresis as described for the pyrophosphorolysis assay. A cleavage reaction using E. coli RNAP allowed to run for 150 min was performed as described previously and used as a reference for the length of cleavage products.12, 48 The amount of starting C21 RNA and the reaction products (C17 RNA and the 5 nt cleavage product) were quantified by phosphorimaging and used to determine the fraction of each RNA species at each time point. The fractions were then plotted vs. time and fit to single-exponential models using Igor graphing software to obtain rate constants. Rates were calculated for at least three independent samples to obtain experimental error. Statistical significance was assessed using the unpaired two-sample t-test in GraphPad Prism software.
Creation of β′/Rpb1 homolog MSAs
MSAs were prepared using the BlaFA method as described.33 The BlaFA source code33 was edited to enable use of the Entrez Programming Utilities currently supported by NCBI. Pattern files to recognize split β′ (A in archaea) subunits found in cyanobacteria, plastids, and archaea and the fused β/β′ subunits found in several bacterial lineages were updated to ensure recognition of newly deposited sequences. All protein sequences corresponding to these four groups (cyanobacteria, plastid, archaea, and β/β′ fusion RNAPs) contained in the UniProt Knowledgebase protein family entitled the “RNA polymerase β′ chain family” were retrieved.49 MSAs for each group were prepared using ClustalW2 and visualized in Jalview to determine conserved sequence patterns.50, 51 The NCBI BLAST web server was then used to acquire β′/Rpb1 homolog lists as the NetBLAST tool used previously33 is no longer supported. The S. cerevisiae S288C Rpb1 protein sequence (UniProt ID P04050) was used as a query to generate the BLAST lists for all RNAP classes except for plastid RNAPs. The BLAST list for cyanobacteria protein sequences was generated and analyzed separately to capture both the β′ and β′′ subunits. BLAST lists for eukaryotic protein sequences from phyla containing plastids were generated and analyzed separately from phyla lacking plastids to enable facile separation of contaminating plastid RNAP sequences from the nuclear RNAPI, RNAPII, and RNAPIII MSA. The BLAST list for the plastid RNAPs was generated using a fused β′/β′′ sequence from the cyanobacterium Prochlorococcus marinus as the query sequence to search protein sequences from eukaryotic species containing plastids. BLAST lists were then processed using the modified BlaFA code (Table S2). The resulting groups of sequences were then aligned separately using ClustalW2 (SI MSA files) and MSAs were viewed and manually edited to remove partial and incorrect sequences in Jalview.50, 51 The number of sequences in the bacterial RNAP class (Table 1) is the sum of the number of sequences contained in the full bacteria and cyanobacteria MSAs whereas the number of sequences listed for the eukaryotic RNAPs class is the sum of the number of sequences contained in the separate MSAs for eukaryotic species with and without plastids. The phylogenetic tree for TL His variant sequences was created by the neighbor-joining method in Jalview.51
Table 1. Number of sequences in β′/Rpb1 MSAs.
Residue numbers correspond to S. cerevisiae Rpb1. Sub-categorization of the eukaryotic RNAP MSA is shown in italics.
| Number of variants at TL residue (identity of amino acid(s)) | |||||
|---|---|---|---|---|---|
| RNAP class | Number of sequences | T1077 | Q1078 | H1085 | G1088 |
| Bacterial | 10782 | 9 (V, M) | 0 | 8 (K, Q, N) | 0 |
| Plastid | 3550 | 0 | 0 | 1 (L) | 0 |
| Archaeal | 881 | 4 (S) | 1 (H) | 0 | 0 |
| Eukaryotic (RNAPI, RNAPII, RNAPIII) | 5787 | 2 (I, S) | 12 (K, S, L) | 1 (Q) | 4 (V, A, S) |
| RNAPI | 1037 | 1 (S) | 0 | 0 | 0 |
| RNAPII | 1294 | 1 (I) | 5 (K, L) | 0 | 0 |
| RNAPIII | 1108 | 0 | 4 (S) | 1 (Q) | 1 (V) |
| Unannotated | 2348 | 0 | 3 (K) | 0 | 3 (A, S, V) |
| Nuclear-cytoplasmic large double-stranded DNA viruses | 161 | 1 (A) | 0 | 5 (R) | 80 (E, K, A, V, S) |
| Total | 21161 | 16 (V, S, M, I, A) | 13 (K, S, H, L) | 15 (K, R, Q, N, L) | 84 (E, K, V, A, S) |
Homology modeling of the RNAPII TH
The homology model of the fully folded yeast RNAPII trigger loop was generated by aligning the S. cerevisiae RNAPII NTP-bound EC structure (PDB ID code 2E2H)20 with the T. thermophilus RNAP NTP-bound EC structure (PDB ID code 2O5J)27 using the Cα positions in the core double-Ψ β-barrel domains of both Rpb1–β′ and Rpb2–β and then replacing coordinates for S. cerevisiae Rpb1 residues 1075–1088 with the coordinates for the corresponding residues in the fully folded TH of T. thermophilus β′. Amino acid side chains that differed between S. cerevisiae RNAPII and T. thermophilus RNAP were replaced with the S. cerevisiae residues using the mutate function of PyMOL to select the rotamer that minimized steric clash. To generate the H1085L mutant structures, H1085 was mutated to Leu in PyMOL and the rotamer closest in position to H1085 was selected.
RESULTS
Substitution of Leu for the TL His only slightly slows transcript elongation
To test the effects of the H1085L substitution on processive, multi-round nucleotide addition, we used a ligated-scaffold transcription assay developed previously.12, 52, 53 This transcript elongation assay is advantageous because it does not require initiation at a promoter. We were thus able to measure the basal rate of transcription in the absence of extrinsic transcription factors such as TFIIS or Spt5 under physiologically relevant conditions (30 °C and 1 mM NTPs; see Methods). We assembled ECs on a 73-bp oligonucleotide scaffold in which the template DNA strand contained a phosphorylated 5′ overhang designed to be complementary to the sticky end generated by StyI digestion of a dsDNA to be ligated to the scaffold (Figure S1A). Halted, radiolabeled A26 ECs were generated by incubation with [α-32P]GTP and ATP (100 μM each; Figure 2A). A fragment (~2 kb) of the E. coli gene rpoB was then ligated to the ECs. Transcript elongation was then assayed at 30 °C by quantitation of samples halted at successive times after addition of NTPs to 1 mM each, separated by denaturing gel electrophoresis, and visualized by phosphorimaging (Figure 2B and Figure S1B).
Figure 2. Multi-round nucleotide addition by RNAPII.
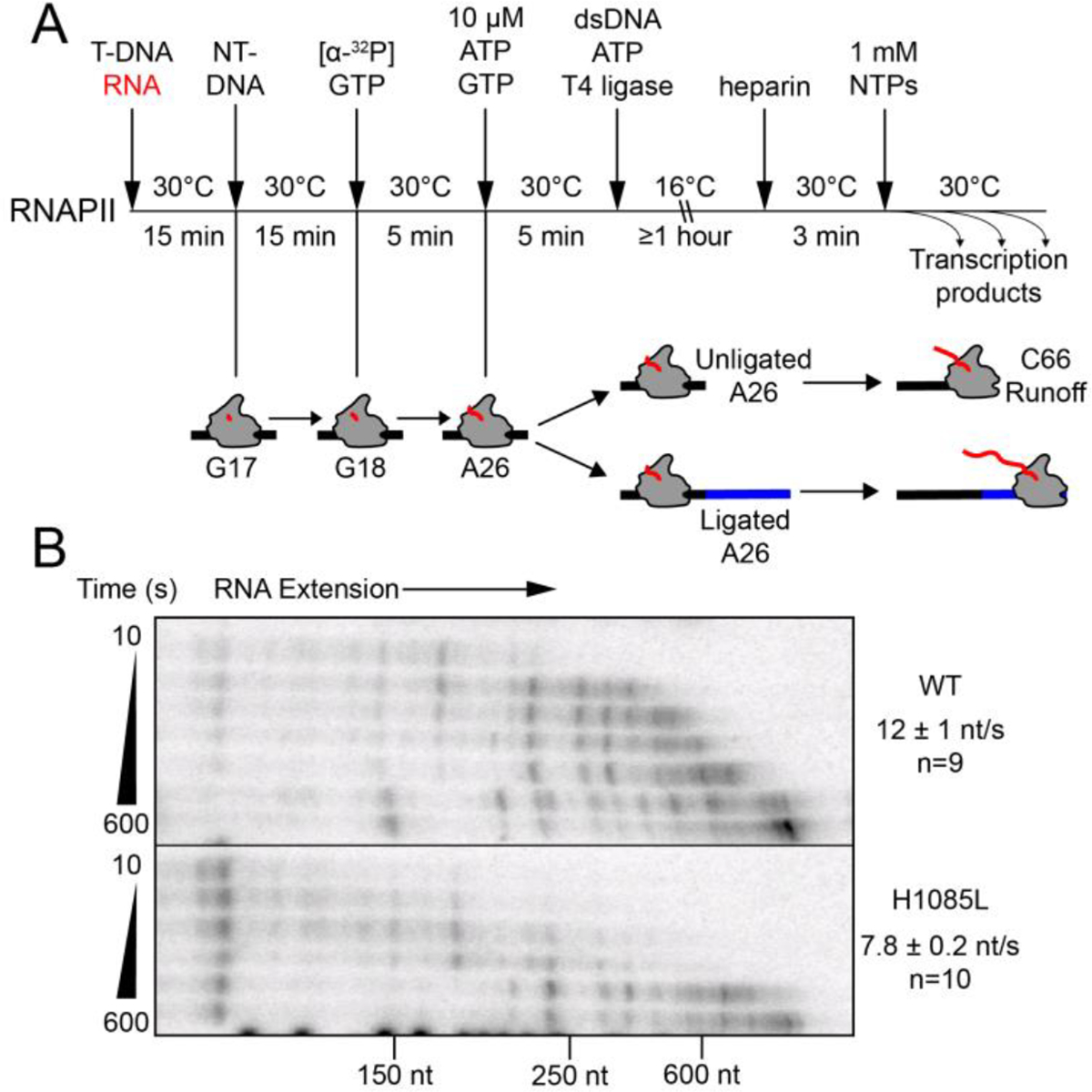
(A) Experimental overview for preparation of labeled ECs on scaffold 1 and ligation to a 2.3 kb fragment of the E. coli rpoB gene (see Figure S2A for the sequence of the nucleic acid scaffold). (B) Time course of transcription by WT and H1085L RNAPII. Average nucleotide addition rates were determined from the change in the average transcript length (see Methods). H1085L RNAPII elongated at about two-thirds the rate of WT RNAPII (unpaired two-sample t-test P<0.0001). An uncropped image of the gel is shown in Figure S1B.
WT RNAPII elongated at an average rate of 12 ± 1 nt·s−1, in agreement with past in vitro rate measurements of this enzyme.13, 54 The H1085L substitution modestly slowed the average rate of nucleotide addition to about two-thirds the rate of WT RNAPII (P<0.0001, Figure 2B). We obtained similar results using a ~2-kb fragment of the S. cerevisiae gene RPO21 (encoding Rpb1), which is naturally transcribed by RNAPII (Figure S1C). These results suggest that processive transcript elongation is not greatly impaired in the H1085L mutant, providing a biochemical rationale for the mild effects on growth observed in vivo.
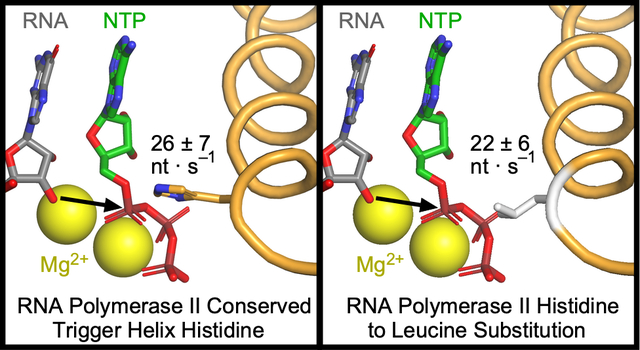
The multi-round transcript elongation assay does not distinguish effects of the TL substitution on different steps in the NAC; both catalysis and translocation contribute to the average rate of nucleotide addition on a long DNA template.13 To separate the effect of the H1085L substitution on these steps, we assayed the rates of single-nucleotide addition using a scaffold on which two successive rounds of AMP addition could be measured (Figure 3A).12 ECs were formed on this scaffold as described above for the unligated scaffold, radiolabeled with [α-32P]GTP, and then extended by two nucleotides with 1 mM ATP in a quench-flow instrument (Figure 3). We observed incomplete reactivation of the halted ECs, which is a known issue when RNAPII is halted on ECs long enough to conduct quench-flow experiments. However, we could correct for the unreactive fraction by including it in kinetic modeling of the reaction pathway (see Methods). Best fit pseudo-first order rates for the two steps of AMP addition were similar (~30 nt s−1 for WT RNAPII and ~23 nt s−1 for H1085L RNAPII, Figure 3C), and the difference was not statistically significant (P=0.4942 and P=0.1235 for A20 and A21 addition, respectively). The similarity of the rates for the two steps, only the second of which requires a translocation event, and the fact that these rates are not dramatically different than the average rates observed in the multi-round assay that includes pause events is consistent with prior reports that translocation makes only a partial contribution to overall transcription rate at saturating substrate concentrations.13 The results also suggest that H1085L does not dramatically alter translocation and that the slightly slower rates of nucleotide addition for this enzyme likely result from a modest reduction in the rate of phosphoryl transfer catalysis.
Figure 3. Single nucleotide addition steps by RNAPII.
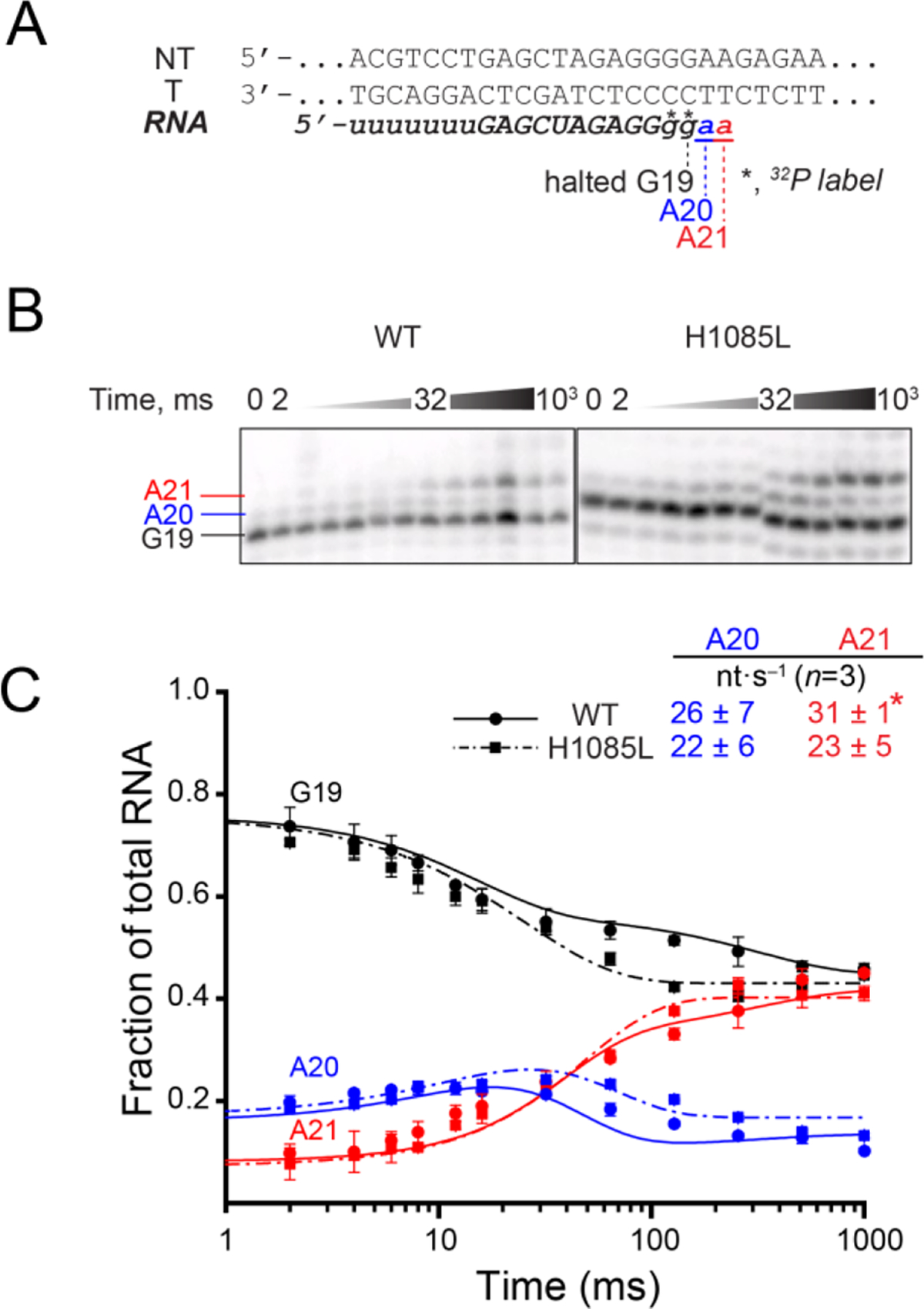
(A) RNA in the halted EC was radiolabeled by incorporation of [α-32P]GMP to form G19 RNA and then extended in the reaction with 1 mM ATP to form A20 and A21 RNAs. (B) The reactions were quenched at various time points from 2 to 1000 ms and RNA products were separated by gel electrophoresis. The discontinuity in the H1085L panel resulted from an air bubble in the gel and not from image editing. (C) Quantified single nucleotide addition data from three replicates per time points for WT (solid curve) and H1085L (dashed curve) RNAP closely matched. Quench-flow data from three replicates were used to obtain the rate constants for nucleotide addition by kinetic modeling (see Methods). Only the reactive portion of G19 contributes to the rate constants estimated by kinetic modeling. *, the rate constant of the second step of AMP addition for WT RNAP was modeled from duplicate data sets and reported as the mean ± the difference in the duplicate values.
The rate of pyrophosphorolysis is modestly reduced by the H1085L substitution
RNAPII can shorten the nascent transcript in 1-nt increments by pyrophosphorolysis, the reverse of nucleotide addition. This process typically requires higher concentrations of pyrophosphate than NTP concentrations needed for nucleotide addition and occurs when the EC is pretranslocated. Pyrophosphate reacts with the ribonucleotide in the i+1 site of the pretranslocated EC to produce NTP, leaving the EC in the posttranslocated state. To measure the rate of this reverse reaction for WT and H1085L RNAPII, we assembled ECs on a short oligonucleotide scaffold radioactively labeled with 32P on the 5′ end of the RNA (Figure 4A).This nucleic-acid scaffold contains 3′ GU in the RNA strand, which was previously shown to cause bacterial RNAP to favor the pretranslocated register.12, 47 RNAPII also was sensitive to pyrophosphate when bound to this scaffold, shortening the RNA by one nt in the presence of 0.5 mM pyrophosphate (Figure 4B). Apyrase (1.5 units/mL), which hydrolyzes the NTP generated by pyrophosphorolysis, was included in the reaction mixture to minimize the reincorporation of UTP and thus drive the pyrophosphorolysis reaction forward. H1085L RNAPII cleaved the RNA transcript at roughly one-third the rate of WT RNAPII (P=0.0037, Figure 4B and Figure S2A), confirming the modest effect of this substitution on the capacity of RNAPII to catalyze phosphoryl transfer (modest compared to the decreased rate of pyrophosphorolysis by a factor of 200 seen for an E. coli RNAP mutant defective for TL folding).26
Figure 4. Phosphoryl transfer in reverse.
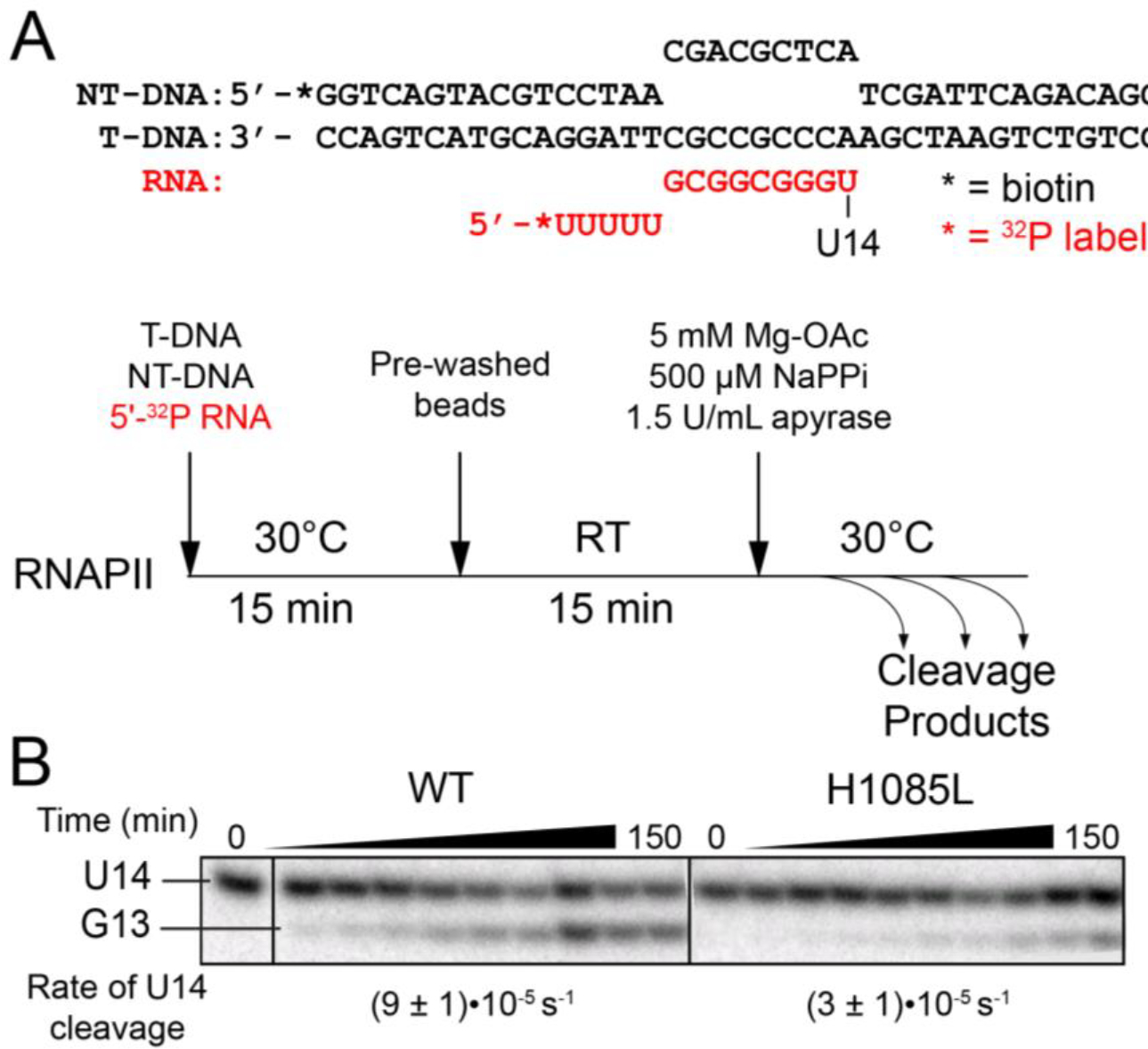
(A) Scaffold 2 and experimental overview of the pyrophosphorolysis assay. NT-DNA and T-DNA are noncomplementary in the region of the RNA-DNA hybrid. The RNA was 5′ end labeled in order to track the progress of the reaction. Apyrase was included to break down UTP released by the reaction and thus favor RNA cleavage. (B) Pyrophosphorolysis by WT and H1085L RNAPII. H1085L reduced the rate of U14 RNA decay by a factor of ~3 (unpaired two-sample t-test P=0.0037). See Figure S2 for an image of the full gel and kinetic fits of the data.
The TL His is not necessary for intrinsic transcript cleavage
To characterize effects of H1085L on other catalytic activities, we examined the ability of H1085L RNAPII to cleave a backtracked RNA transcript hydrolytically. To probe the backtracked state, we used a bacterial pause sequence originally identified in a bglF antisense transcription unit at which E. coli RNAP was shown to backtrack by 4–6 nt following addition of cognate NTPs (Figure 5A).12, 48 Because backtracking is driven largely by the energetics of RNA and DNA base-pairing, we hypothesized that the bglF backtrack pause signal might function with RNAPII. To test this hypothesis, we formed RNAPII ECs on the same scaffold used to test E. coli RNAP12 and radiolabeled the nascent RNA by reaction with [α-32P]CTP to yield C17 complexes (Figure 5B). Labeled C17 ECs were then extended to the pause position (C21) by addition of CTP and UTP (10 μM each) and allowed to equilibrate among the different backtrack registers for 10 min. RNA hydrolysis from these various registers was initiated by adjusting the pH to 9.0 and the concentration of Mg2+ to 20 mM.
Figure 5. RNA hydrolysis by backtracked ECs.
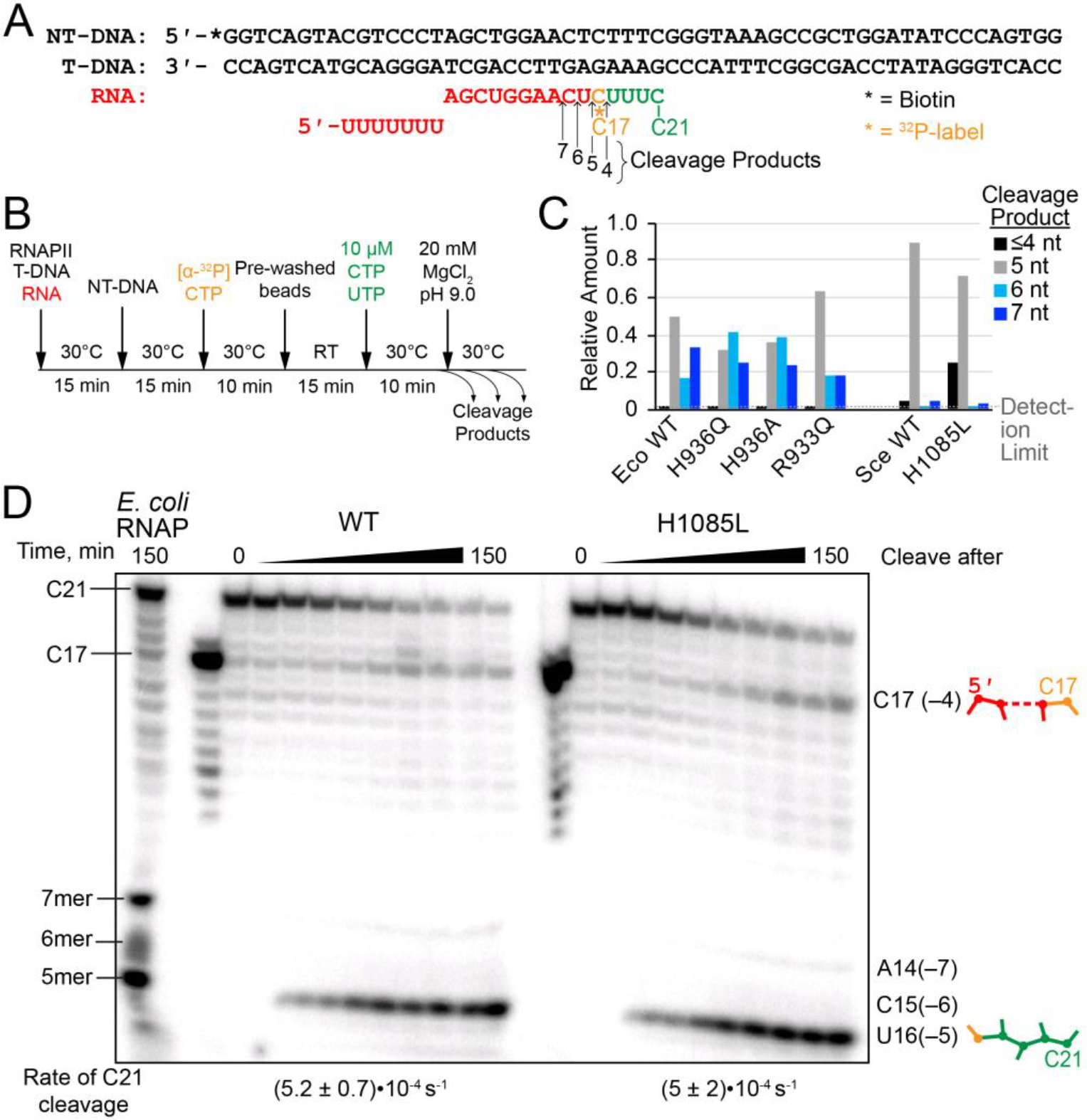
(A) Scaffold 3, encoding the E. coli bglF backtrack pause signal.12 (B) Experimental steps in the backtrack cleavage assay. RNA was labeled by incorporation of [α-32P]CMP and extended to the pause position, then allowed to equilibrate among various backtrack registers. Cleavage was initiated by addition of Mg2+ in pH 9.0 buffer. (C) Quantitation of relative amounts of cleavage products from backtracked ECs. The data for E. coli RNAP were published previously.12 The data for yeast RNAPII were quantified from the gel shown in panel D. (D) Intrinsic cleavage by WT and H1085L RNAPII. A reaction with E. coli RNAP was performed and allowed to run for 150 min in order to identify the species produced by RNAPII. The H1085L mutation did not affect the rate of cleavage from the C21 position (unpaired two-sample t-test P=0.9603), which is shown below the gel lanes. See Figure S3 for kinetic fits of the data.
On this scaffold, RNAPII backtracked primarily by 3–4 nt, as evidenced by the accumulation of a radiolabeled 17-nt RNA product (cleavage after C17 releases an unlabeled 4 nt cleavage product) and a radiolabeled 5-nt cleavage product (cleavage after U16) (Figure 5C,D). Thus, RNAPII responds to the bglF pause signal similarly to E. coli RNAP, but is less susceptible to cleavage in the –5 and –6 registers that yield 6- and 7-nt cleavage products, respectively (Figure 5C,D).12 For both RNAPII and E. coli RNAP, the major cleavage product is a 5-nt RNA resulting from cleavage after backtracking by 4 nt. In principle, the minor C17 RNA product that is more pronounced for RNAPII could be generated by iterative 1-nt cleavage events, but the lack of the appearance of intermediate products (i.e. U18, U19, and U20) suggests that C17 RNA was produced by a single hydrolysis event in –3 backtracked ECs. The H1085L substitution had no measurable effect on the rate of C21 transcript hydrolysis (P=0.9603) and only a slight but not statistically significant shift (P=0.5243) in the distribution of reaction products to the –3 backtracked register (Figure 5; Figure S3). This result indicates that the polar character of the TL His plays no major role in RNA cleavage in yeast RNAPII. Similar to bacterial RNAP, substitutions in the TL His modestly affect selection among backtrack registers.
The TL His is near-universally conserved
The TL His has been described as universally conserved among mRNA-synthesizing, cellular RNAPs based upon a multiple sequence alignment (MSA) of 1187 β′/Rpb1 homologs.33 Taken together with its position in the active site and accumulated biochemical data, it has been proposed to play an essential role in transcription. However, the relatively mild effects of replacement with Leu in yeast RNAPII both in vivo and in vitro and even more modest effects of Gln substitutions in both E. coli RNAP and yeast RNAPII leave unanswered why the TL His is so strongly conserved. In search for an answer, we re-visited the MSA performed in 2010.32 The number of protein sequences of β′/Rpb1 homologs contained in databases has increased considerably in the decade since this previous large-scale MSA of RNAPs. To ask whether the TL His remains invariant among available RNAP sequences, we aligned the amino-acid sequences of 21,161 β′/Rpb1 orthologs using the BLAST to FASTA file to alignment (BlaFA) method developed by Lane and Darst for the analysis of complex, evolutionarily diverse RNAP sequences.33
We created MSAs of five distinct classes of RNAP using BlaFA: bacterial RNAPs, archaeal RNAPs, eukaryotic RNAPs (including RNAPI, RNAPII, and RNAPIII), plastid RNAPs, and viral RNAPs, which are found in nuclear and cytoplasmic double-stranded DNA viruses (e.g., poxviruses and mimiviruses) and are related to eukaryotic RNAPs (Table 1). Although the results of the BlaFA analysis for eukaryotic RNAPs contained numerous RNAPIV and RNAPV large subunit sequences from plants, these RNAP IV and V sequences were removed from the final alignment because they do not align well with the TL region conserved for all other RNAP classes and appear to have much reduced catalytic activities.55, 56
Our MSAs reinforced the remarkable degree of sequence conservation of the TL, and in particular the TL His. Only 15 substitutions for the TL His were found in 21,161 sequences, corresponding to Lys, Arg, Gln, Asn, and Leu variants (Figure 6, Table 1). Other highly conserved TL residues (S. cerevisiae T1077, Q1078, and G1088) had a similar degree of conservation, although the region around G1088 appears to be highly divergent in large DNA viruses that encode their own RNAP. These MSAs also identified noteworthy features of RNAPs from evolutionary groups that were not contained in sequence databases at the time of the previous alignment. Lane and Darst identified a naturally-occurring bacterial β–β′ fusion protein in the Wolbachia family that had previously been described in the Helicobacteraceae family.33, 57 We also observed β–β′ fusion proteins in the Mycoplasma family, the Candidatus Magnetobacterium family in the Nitrospirae phylum, and a number of unclassified bacteria species, including members of the Patescibacteria group and the Candidatus Desantisbacteria and Candidatus Ratteibacteria families. Additionally, we observed Rpb1 homologs in pandoraviruses and molliviruses,58, 59 two giant virus families described subsequent to the previous study.
Figure 6. Conservation of the TL His.
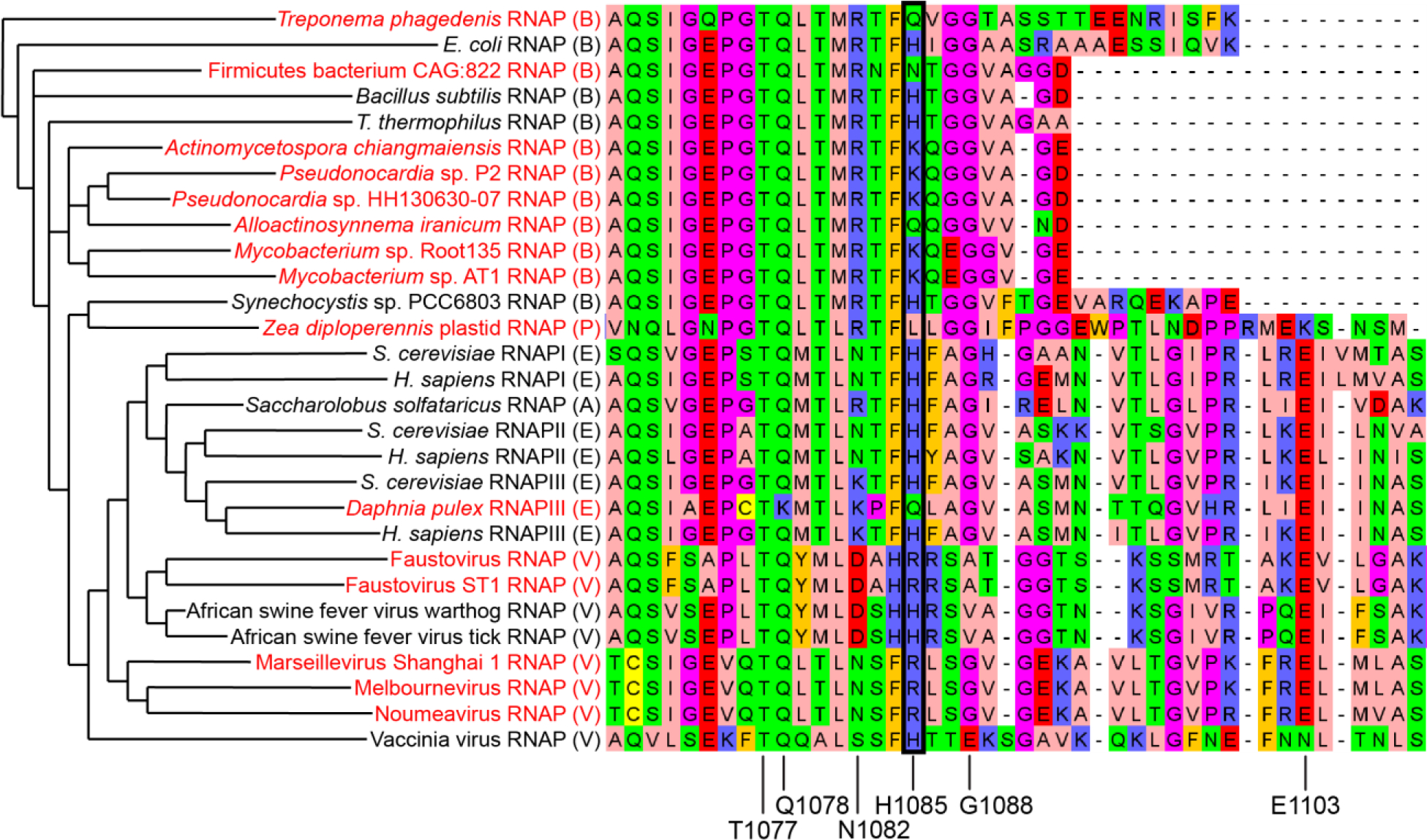
MSA of model RNAPs and sequences containing putative TL His variants occuring in 15 of the 21161 TL sequences examined. The names of variant sequences identified in this study are colored in red. The class of each sequence (Table 1) is indicated in parentheses after the name: A=archaeal, B=bacterial, E=eukaryotic, P=plastid, V=viral. RNAP sequences from model organisms in each class were included for comparison. The position corresponding to H1085 is highlighted with a black box. Key TL residues studied extensively in S. cerevisiae RNAPII are indicated at the bottom of the alignment. Sequences are clustered based on their similarity. The phylogenetic tree inferred from the MSA is shown on the left (horizontal distances are arbitrary). Amino acids are colored with aliphatic/hydrophobic residues in pink, aromatic residues in orange, positively charged residues in blue, negatively charged residues in red, hydrophilic residues in green, Gly and Pro in purple, and Cys in yellow.
DISCUSSION
The TL His is a steric catalyst of transcription
Our findings that H1085L only slightly impairs phosphoryl transfer catalysis strongly supports the positional catalyst model of TL–TH function21 and explain the viability of yeast in which this substitution was discovered.33 Because RNAPII lacks another ionizable TL residue that could contact substrate triphosphate, our results preclude an obligate, direct role for the TL in proton transfer during the phosphoryl transfer reaction and indicate that even polar interactions between the TL His and substrate are at least partially dispensable. Thus, the positional catalyst model of TL function explains the improved viability of yeast carrying the H1085L mutation relative to substitutions that preserve a capacity for proton transfer (Lys, Arg) or polar interactions (Ser, Asn) but not the steric volume of the amino-acid side chain.41
We observed a modest (~2-fold) effect of H1085L on the rate of transcription over hundreds of bp (Figure 2) but steps in addition to phosphoryl transfer catalysis such as translocation, pausing, and TL folding also can contribute to the overall rate of transcription.13, 54, 60 A quench-flow kinetic assay with ms resolution indicated kcat for H1085L was ~85% of WT and ~74% of WT for a second step that included translocation (Figure 3) compared to ~58% of WT for the overall transcription rate over hundreds of bp at saturating NTP concentration, even though the latter occurred by a factor of 2.5–3 more slowly for both the mutant and WT enzyme. These results strongly support the positional catalyst model of TL function and indicate that even the ~2-fold effect of H1085L on overall transcription rate may include effects on translocation, TL folding, or pausing rather than catalysis per se. This interpretation is consistent with modest effects of the substitution on the extent of backtracking evident in a transcript cleavage assay where the substitution had no effect on hydrolysis itself (Figure 5). It also may explain the larger effect of H1085L on pyrophosphorolysis (factor of ~3; Figure 4) since an effect on translocation state can change pyrophosphorolysis rates by more than an order of magnitude.47 However, we cannot exclude the possibility that the TL His may non-obligately aid proton abstraction during pyrophosphorolysis, a reaction thought to be unimportant in vivo due to rapid hydrolysis of PPi.
Our results with RNAPII H1085L also are important because the proposal of the positional catalyst model of TL function and absence of an obligate, direct role of the TL His in proton transfer role during catalysis11, 20 was based on studies of bacterial RNAP in which a TL Arg near the TL His (R933 near H936 in E. coli RNAP) might function as a back-up to proton-transfer by the TL His.12 A double R933Q, H936Q E. coli RNAP exhibited an ~2-fold defect in phosphoryl transfer catalysis suggesting any role in proton transfer is not essential but complicating the interpretation.12 In eukaryotic RNAPIIs, the TL Arg is replaced by Asn. Thus, the negligible effect of the H1085L substitution on kcat suggests that either direct donation of a proton to PPi by the TL during nucleotide addition is not necessary or that this mechanistic requirement is bypassed by H1085L RNAPII. Even in the latter case, our results argue that sterically driven catalysis by H1085L is nearly as efficient as direct TL-mediated proton transfer.
Our results also argue that even polar contacts between the TL His and substrates are not essential for TL function because the nonpolar side chain of Leu is capable of replacing the conserved His for catalysis of nucleotide addition and transcript cleavage. The effects of this substitution can be understood by examining the charge distribution of the TH relative to the bound NTP substrate (Figure 7). The fully folded TH are observed in crystal structures of substrate-bound transcription complexes containing an RNA 3′-OH whereas in structures of substrate-bound transcription complexes lacking the 3′-OH the TH are only partially formed (Figure 7A).20, 27, 39, 61 In the partially formed TH (observed for both yeast RNAPII and bacterial RNAP in complexes lacking a 3′-OH), the TL His is shifted 6.5Å away from its location in the fully formed TH and both the substrate γ phosphate and the associated Mg2+ ion are shifted away from the TL His.
Figure 7. Effect of H1085L on contacts to substrate NTP in the RNAPII active site.

(A) Comparison of four structures of cognate NTPs bound in the active site of a multi-subunit RNAP. In two structures, one of yeast RNAPII (PDB ID code 2E2H)20 and one of E. coli RNAP (PDB ID code 6RH3),39 a 3′-deoxy RNA is present in reconstituted ECs to block reaction with bound NTP. In these structures, the TH are incompletely formed, the TL His is nearest the β phosphate oxygen, and the γ phosphate and Mg2+II are shifted relative to their positions in structures containing an RNA 3′-OH. The 3′-OH-containing structures, one with T. thermophilus RNAP and bound AMPCPP to block reaction (PDB ID code 2O5J)27 and one with E. coli RNAP captured after nucleotide addition but before PPi release (PDB ID code 5IPL),62 reveal fully formed TH with the TL His shifted 6.5 Å toward the center of catalysis relative to the 3′-deoxy structures. In the TH position, the TL His helps position the triphosphate for reactive alignment necessary to achieve the trigonal bipyramidal transition state for phosphoryl transfer. (B) The open position of the TL, shown here for a complete S. cerevisiae RNAPII EC in which the TL could be resolved (PDB ID code 5C4X),63 positions the TL His far from the active site and creates a large solvent-exposed cavity that connects the active site with the surrounding solution (green volume) that allows NTP entry and PPi release. (C) TH charge distribution and space between TH and NTP in the S. cerevisiae RNAPII EC containing 3′-deoxy RNA (PDB ID code 2E2H).20 The inset shows the cavity between the TH and substrate NTP in which water may potentially reside (green volume partially behind the TH volume in this view). (D) TH charge distribution and space between TH and NTP in the S. cerevisiae RNAPII EC containing 3′-deoxy RNA and the H1085L substitution. (E) TH charge distribution and space between TH and NTP in a model of the S. cerevisiae RNAPII EC containing 3′-OH RNA and a fully folded TH based on the TH Cα positions observed the T. thermophilus EC (PDB ID code 2O5J).27 (F) TH charge distribution and space between TH and NTP in the model shown in panel E but instead containing H1085L.
To visualize the effects of the TL His-to-Leu change, we modeled the yeast RNAPII TH based on the fully folded T. thermophilus TH and compared the TH surface charge of the wild-type and mutant TH in both the fully folded and partially folded conformations. These non-energy-minimized models illustrate the modest effects of the TL His-to-Leu change on active site configurations but should be regarded as illustrative and hypothetical rather than as structural predictions. Upon TH formation a large water-filled cavity that connects the active site to solution (green cavity in Figure 7B) is closed such that the active site become relatively dehydrated with little free space surrounding the substrate (Figures 7C–F).64, 65 Although the TH becomes slightly more neutral upon Leu substitution for the TL His, the minimal remaining volume between the TH and the substrate NTP is largely unchanged in either the model of the fully folded RNAPII TH (Figure 7C and D) or the incompletely folded TH conformation observed in the 3′-deoxy S. cerevisiae RNAPII EC (Figure 7E and F).20 Thus, both the steric effect of the TH on substrate NTP positioning and dehydration of the active site are minimally changed by the Leu substitution, consistent with a modest effect on catalysis. It is possible the TL Leu could even enhance the dehydration effect of TH formation since Leu is more hydrophobic than the TL His. This effect may explain its near-normal function in the positional catalyst model of TH function and the improved viability of yeast carrying the H1085L mutation relative to substitutions that preserve the capacity for proton transfer (Lys, Arg) or polar interactions (Gln, Asn) and occupy similar steric volumes.41, 66, 67 Strikingly, a Gln substitution, which retains the polar character of His, exhibited a stronger loss-of-function in S. cerevisiae than the Leu substitution.41 Obtaining a structure of the RNAPII TH in NTP-bound ECs containing a nascent RNA 3′-OH, possibly aided by the E1103G RNAPII mutant that biases TL conformation towards the folded state,68, 69 would help interpret these biochemical data.
The TL His of RNAPII is not necessary for intrinsic transcript cleavage
The effect of H1085L on cleavage of backtracked RNA revealed several intriguing results. The bacterially derived pause signal sequence on which we assayed backtracking and cleavage resembles U-rich sequences on which RNAPII is observed to undergo backtrack-induced transcriptional arrest upon incorporation of cognate NTPs that can be rescued by TFIIS.16, 70 Our results provide a second example in which a eukaryotic RNAP recognizes a bacterial pause signal, the first being the consensus elemental pause signal that is evolutionarily conserved across domains of life.71–76 In the present case, RNAPII responds similarly to bacterial RNAP (i.e., by backtracking), although the extent of backtracking was less for RNAPII (Figure 5C).12
Substitution of the TL His had no effect on the rate of RNA hydrolysis from the backtracked register and only modest but not statistically significant effects on the distribution of cleavage products. A similar lack of effect of individual substitutions in the TL was seen previously for E. coli RNAP, suggesting that the absence of a catalytic role for the TL His in transcript hydrolysis is widely conserved.21, 34, 38 We and others have noted effects of the TL on backtracked RNA positioning,12,35 but these effects on positioning are distinct from the lack of effect of the TL on hydrolytic catalysis per se. A notable difference between RNAPII and E. coli RNAP was evident only in which backtrack registers were preferred. Yeast RNAPII exhibited less tendency to occupy longer backtrack registers giving rise to 6- and 7-nt cleavage products (Figure 5C).12 For E. coli RNAP and to a lesser extent yeast RNAPII, however, substitutions of the TL His affected the extent of backtracking as indicated by altered distributions of cleavage products. A Tyr substitution of H1085 in yeast RNAPII was found previously to enhance RNA hydrolysis while inhibiting elongation,31, 42 which could reflect enhancement of backtracking but may also involve a gain-of-function effect on RNA hydrolysis. The varying effects of TL substitutions on backtracking in diverse RNAPs suggest that the TL may have evolved to play slightly differing roles in different organisms.12, 40, 41, 77
Conservation of the TL His
Our alignment of β′/Rpb1 orthologs from all domains of life confirmed the high degree of conservation of the TL His. Although certain substitutions are tolerated experimentally with only small effects on transcriptional activities, these deficiencies are enough to maintain a strong selective advantage in diverse environments and cell lineages. The small number of TL His variants we discovered (only 0.07% of sequences examined were not His) raises the possibility that these variants result from sequencing errors in deposited data.
Sequencing errors are distinctly possible for the His-to-Asn and His-to-Gln changes that require only a single nucleotide change. However, the recorded His-to-Arg, His-to-Lys, and His-to-Leu changes involve two nucleotide changes (to AGA or CGC, to AAG, or to CTA, respectively). Furthermore, the His-to-Lys changes were reported independently for closely related Actinobacteria (Mycobacteria, Actinomycetospora, and Pseudonocardia species in Figure 6). These His-to-Lys changes are thus likely to reflect viable substitutions, although of note these species all contain Arg at the position equivalent to E. coli β′933. The single instance of a His-to-Leu (CTA in Zea diploperennis plastid RNAP) change is ambiguous since it could arise by reversal of the 2nd and 3rd codon positions).
Two TL His-to-Arg changes identified in Faustovirus strains are highly divergent in the surrounding TL sequences (Figure 6) relative to other β′/Rpb1 homologs including those containing other potential TL His variants (e.g., Noumeavirus, Melbournevirus, and Marseillevirus RNAPs). Even highly divergent Asfarviruses Rpb1 homolog sequences, which were withheld from a previous analysis for being too divergent to align with confidence,33 contain the TL His in our alignment despite strongly resembling the divergent Faustovirus sequences in flanking TL sequences (Figure 6). Resequencing of the TL region from species exhibiting TL His variants and possibly biochemical analysis of their RNAPs may give insight into the functional consequences of rare natural substitutions of the TL His.
Conclusion
Our study of the H1085L RNAPII substitution confirms the positional catalyst model of TL function12 for eukaryotic RNAPII and provides a biochemical basis for the observed phenotype of yeast bearing this substitution. The TL His functions primarily as a steric not acid–base catalyst of transcription. These findings leave unanswered how proton transfer is accomplished during phosphoryl transfer reactions catalyzed by RNAPII. Future genetic, biochemical, and structural studies should address this question.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Craig Kaplan for generously sharing materials, protocols, and advice; Jess Vera for help in sequence analysis of Rpb1 orthologs; and members of our research group for their valuable comments and feedback throughout the conduct of this study and preparation of the manuscript.
Funding
The work was supported by a grant from the NIH Institute of General Medical Sciences (R01 GM038660) to R.L.
Abbreviations:
- EC
elongation complex
- NAC
nucleotide addition cycle
- NTP
nucleoside triphosphate
- nt
nucleotide
- PPi
pyrophosphate
- RNAP
RNA polymerase
- RNAPII
RNA polymerase II
- TFIIS
transcription factor IIS
- TH
trigger helices
- TL
trigger loop
- WT
wild type
Footnotes
The authors declare no competing interests.
ASSOCIATED CONTENT
Supporting Information Available: Table of strain, plasmids, and oligonucleotides used in this study; Scaffold sequence used in elongation rate assays and complete electrophoretic gel image of assay results; Uncropped electrophoretic gel image of pyrophosphorolysis assay result and plot of rate of pyrophosphorolysis (additional data related to Figure 4); Plot of rates of hydrolytic transcript cleavage (additional data related to Figure 5) (PDF).
Table of results for Rpb1/β′ ortholog BlaFA search (XLSX).
FASTA files of sequences of β′/Rpb1 orthologs used for sequence alignments (ZIP).
UniProt Protein Accession IDs
| RNAPII subunit | Uniprot ID | Uniprot name |
|---|---|---|
| RPB1 | P04050 | RPB1_YEAST |
| RPB2 | P08518 | RPB2_YEAST |
| RPB3 | P16370 | RPB3_YEAST |
| RPB4 | P20433 | RPB4_YEAST |
| RPB5 | P20434 | RPAB1_YEAST |
| RPB6 | P20435 | RPAB2_YEAST |
| RPB7 | P34087 | RPB7_YEAST |
| RPB8 | P20436 | RPAB3_YEAST |
| RPB9 | P27999 | RPB9_YEAST |
| RPB10 | P22139 | RPAB5_YEAST |
| RPB11 | P38902 | RPB11_YEAST |
| RPB12 | P40422 | RPAB4_YEAST |
REFERENCES
- [1].Cramer P (2002) Multisubunit RNA polymerases, Curr Opin Struct Biol 12, 89–97. [DOI] [PubMed] [Google Scholar]
- [2].Herr AJ, Jensen MB, Dalmay T, and Baulcombe DC (2005) RNA polymerase IV directs silencing of endogenous DNA, Science 308, 118–120. [DOI] [PubMed] [Google Scholar]
- [3].Pontier D, Yahubyan G, Vega D, Bulski A, Saez-Vasquez J, Hakimi MA, Lerbs-Mache S, Colot V, and Lagrange T (2005) Reinforcement of silencing at transposons and highly repeated sequences requires the concerted action of two distinct RNA polymerases IV in Arabidopsis, Genes Dev 19, 2030–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Onodera Y, Haag JR, Ream T, Costa Nunes P, Pontes O, and Pikaard CS (2005) Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation, Cell 120, 613–622. [DOI] [PubMed] [Google Scholar]
- [5].Kanno T, Huettel B, Mette MF, Aufsatz W, Jaligot E, Daxinger L, Kreil DP, Matzke M, and Matzke AJ (2005) Atypical RNA polymerase subunits required for RNA-directed DNA methylation, Nat Genet 37, 761–765. [DOI] [PubMed] [Google Scholar]
- [6].Orlova M, Newlands J, Das A, Goldfarb A, and Borukhov S (1995) Intrinsic transcript cleavage activity of RNA polymerase, Proc Natl Acad Sci U S A 92, 4596–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tschochner H (1996) A novel RNA polymerase I-dependent RNase activity that shortens nascent transcripts from the 3’ end, Proc Natl Acad Sci U S A 93, 12914–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Weilbaecher RG, Awrey DE, Edwards AM, and Kane CM (2003) Intrinsic transcript cleavage in yeast RNA polymerase II elongation complexes, J Biol Chem 278, 24189–24199. [DOI] [PubMed] [Google Scholar]
- [9].Whitehall SK, Bardeleben C, and Kassavetis GA (1994) Hydrolytic cleavage of nascent RNA in RNA polymerase III ternary transcription complexes, J Biol Chem 269, 2299–2306. [PubMed] [Google Scholar]
- [10].Castro C, Smidansky E, Maksimchuk KR, Arnold JJ, Korneeva VS, Gotte M, Konigsberg W, and Cameron CE (2007) Two proton transfers in the transition state for nucleotidyl transfer catalyzed by RNA- and DNA-dependent RNA and DNA polymerases, Proc Natl Acad Sci U S A 104, 4267–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Castro C, Smidansky ED, Arnold JJ, Maksimchuk KR, Moustafa I, Uchida A, Gotte M, Konigsberg W, and Cameron CE (2009) Nucleic acid polymerases use a general acid for nucleotidyl transfer, Nat Struct Mol Biol 16, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mishanina TV, Palo MZ, Nayak D, Mooney RA, and Landick R (2017) Trigger loop of RNA polymerase is a positional, not acid-base, catalyst for both transcription and proofreading, Proc Natl Acad Sci U S A 114, E5103–E5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dangkulwanich M, Ishibashi T, Liu S, Kireeva ML, Lubkowska L, Kashlev M, and Bustamante CJ (2013) Complete dissection of transcription elongation reveals slow translocation of RNA polymerase II in a linear ratchet mechanism, eLife 2, e00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kireeva ML, Hancock B, Cremona GH, Walter W, Studitsky VM, and Kashlev M (2005) Nature of the nucleosomal barrier to RNA polymerase II, Mol Cell 18, 97–108. [DOI] [PubMed] [Google Scholar]
- [15].Zenkin N, Yuzenkova Y, and Severinov K (2006) Transcript-assisted transcriptional proofreading, Science 313, 518–520. [DOI] [PubMed] [Google Scholar]
- [16].Komissarova N, and Kashlev M (1997) Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3’ end of the RNA intact and extruded, Proc. Natl. Acad. Sci. U. S. A 94, 1755–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gottesman ME, and Mustaev A (2018) Inorganic phosphate, arsenate, and vanadate enhance exonuclease transcript cleavage by RNA polymerase by 2000-fold, Proc Natl Acad Sci U S A 115, 2746–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sosunov V, Sosunova E, Mustaev A, Bass I, Nikiforov V, and Goldfarb A (2003) Unified two-metal mechanism of RNA synthesis and degradation by RNA polymerase, The EMBO journal 22, 2234–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rudd MD, Izban MG, and Luse DS (1994) The active site of RNA polymerase II participates in transcript cleavage within arrested ternary complexes, Proc. Natl. Acad. Sci. U. S. A. 91, 8057–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang D, Bushnell DA, Westover KD, Kaplan CD, and Kornberg RD (2006) Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis, Cell 127, 941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang D, Bushnell DA, Huang X, Westover KD, Levitt M, and Kornberg RD (2009) Structural basis of transcription: backtracked RNA polymerase II at 3.4 angstrom resolution, Science 324, 1203–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yuzenkova Y, Roghanian M, and Zenkin N (2012) Multiple active centers of multi-subunit RNA polymerases, Transcription 3, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ruan W, Lehmann E, Thomm M, Kostrewa D, and Cramer P (2011) Evolution of two modes of intrinsic RNA polymerase transcript cleavage, J Biol Chem 286, 18701–18707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Imashimizu M, Oshima T, Lubkowska L, and Kashlev M (2013) Direct assessment of transcription fidelity by high-resolution RNA sequencing, Nucleic acids research 41, 9090–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Windgassen TA, Mooney RA, Nayak D, Palangat M, Zhang J, and Landick R (2014) Trigger-helix folding pathway and SI3 mediate catalysis and hairpin-stabilized pausing by Escherichia coli RNA polymerase, Nucleic acids research 42, 12707–12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang J, Palangat M, and Landick R (2010) Role of the RNA polymerase trigger loop in catalysis and pausing, Nature structural & molecular biology 17, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vassylyev DG, Vassylyeva MN, Zhang J, Palangat M, Artsimovitch I, and Landick R (2007) Structural basis for substrate loading in bacterial RNA polymerase, Nature 448, 163–168. [DOI] [PubMed] [Google Scholar]
- [28].Wang W, Walmacq C, Chong J, Kashlev M, and Wang D (2018) Structural basis of transcriptional stalling and bypass of abasic DNA lesion by RNA polymerase II, Proc Natl Acad Sci U S A 115, E2538–E2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Da LT, Wang D, and Huang X (2012) Dynamics of pyrophosphate ion release and its coupled trigger loop motion from closed to open state in RNA polymerase II, Journal of the American Chemical Society 134, 2399–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Feig M, and Burton ZF (2010) RNA polymerase II with open and closed trigger loops: active site dynamics and nucleic acid translocation, Biophys J 99, 2577–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kaplan CD, Larsson KM, and Kornberg RD (2008) The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by alpha-amanitin, Mol Cell 30, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yuzenkova Y, Bochkareva A, Tadigotla VR, Roghanian M, Zorov S, Severinov K, and Zenkin N (2010) Stepwise mechanism for transcription fidelity, BMC Biol 8, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lane WJ, and Darst SA (2010) Molecular evolution of multisubunit RNA polymerases: sequence analysis, J Mol Biol 395, 671–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cheung AC, and Cramer P (2011) Structural basis of RNA polymerase II backtracking, arrest and reactivation, Nature 471, 249–253. [DOI] [PubMed] [Google Scholar]
- [35].Sekine S, Murayama Y, Svetlov V, Nudler E, and Yokoyama S (2015) The ratcheted and ratchetable structural states of RNA polymerase underlie multiple transcriptional functions, Mol Cell 57, 408–421. [DOI] [PubMed] [Google Scholar]
- [36].Mosaei H, and Zenkin N (2021) Two distinct pathways of RNA polymerase backtracking determine the requirement for the Trigger Loop during RNA hydrolysis, Nucleic acids research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brueckner F, and Cramer P (2008) Structural basis of transcription inhibition by alpha-amanitin and implications for RNA polymerase II translocation, Nat Struct Mol Biol 15, 811–818. [DOI] [PubMed] [Google Scholar]
- [38].Rudd MD, and Luse DS (1996) Amanitin greatly reduces the rate of transcription by RNA polymerase II ternary complexes but fails to inhibit some transcript cleavage modes, J Biol Chem 271, 21549–21558. [DOI] [PubMed] [Google Scholar]
- [39].Abdelkareem M, Saint-Andre C, Takacs M, Papai G, Crucifix C, Guo X, Ortiz J, and Weixlbaumer A (2019) Structural basis of transcription: RNA polymerase backtracking and its reactivation, Molecular cell 75, 298–309 e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kaplan CD, Jin H, Zhang IL, and Belyanin A (2012) Dissection of Pol II trigger loop function and Pol II activity-dependent control of start site selection in vivo, PLoS genetics 8, e1002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Qiu C, Erinne OC, Dave JM, Cui P, Jin H, Muthukrishnan N, Tang LK, Babu SG, Lam KC, Vandeventer PJ, Strohner R, Van den Brulle J, Sze SH, and Kaplan CD (2016) High-resolution phenotypic landscape of the RNA polymerase II trigger loop, PLoS genetics 12, e1006321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Čabart P, Jin H, Li L, and Kaplan CD (2014) Activation and reactivation of the RNA polymerase II trigger loop for intrinsic RNA cleavage and catalysis, Transcription 5, e28869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, and Séraphin B (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification, Methods 24, 218–229. [DOI] [PubMed] [Google Scholar]
- [44].Hoopes BC, and McClure WR (1981) Studies on the selectivity of DNA precipitation by spermine, Nucleic Acids Res. 9, 5493–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kyzer S, Ha KS, Landick R, and Palangat M (2007) Direct versus limited-step reconstitution reveals key features of an RNA hairpin-stabilized paused transcription complex, J Biol Chem 282, 19020–19028. [DOI] [PubMed] [Google Scholar]
- [46].Haft RJ, Keating DH, Schwaegler T, Schwalbach MS, Vinokur J, Tremaine M, Peters JM, Kotlajich MV, Pohlmann EL, Ong IM, Grass JA, Kiley PJ, and Landick R (2014) Correcting direct effects of ethanol on translation and transcription machinery confers ethanol tolerance in bacteria, Proc Natl Acad Sci U S A 111, E2576–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hein PP, Palangat M, and Landick R (2011) RNA transcript 3’-proximal sequence affects translocation bias of RNA polymerase, Biochemistry 50, 7002–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kotlajich MV, Hron DR, Boudreau BA, Sun Z, Lyubchenko YL, and Landick R (2015) Bridged filaments of histone-like nucleoid structuring protein pause RNA polymerase and aid termination in bacteria, Elife 4, e04970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].The UniProt Consortium. (2017) UniProt: the universal protein knowledgebase, Nucleic acids research 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, and Higgins DG (2007) Clustal W and Clustal X version 2.0, Bioinformatics 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- [51].Waterhouse AM, Procter JB, Martin DM, Clamp M, and Barton GJ (2009) Jalview Version 2--a multiple sequence alignment editor and analysis workbench, Bioinformatics 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Komissarova N, Kireeva ML, Becker J, Sidorenkov I, and Kashlev M (2003) Engineering of elongation complexes of bacterial and yeast RNA polymerases, Methods Enzymol 371, 233–251. [DOI] [PubMed] [Google Scholar]
- [53].Palangat M, Larson MH, Hu X, Gnatt A, Block SM, and Landick R (2012) Efficient reconstitution of transcription elongation complexes for single-molecule studies of eukaryotic RNA polymerase II, Transcription 3, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Larson MH, Zhou J, Kaplan CD, Palangat M, Kornberg RD, Landick R, and Block SM (2012) Trigger loop dynamics mediate the balance between the transcriptional fidelity and speed of RNA polymerase II, Proc Natl Acad Sci U S A 109, 6555–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Haag JR, Ream TS, Marasco M, Nicora CD, Norbeck AD, Pasa-Tolic L, and Pikaard CS (2012) In vitro transcription activities of Pol IV, Pol V, and RDR2 reveal coupling of Pol IV and RDR2 for dsRNA synthesis in plant RNA silencing, Mol Cell 48, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Landick R (2009) Functional divergence in the growing family of RNA polymerases, Structure 17, 323–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zakharova N, Hoffman PS, Berg DE, and Severinov K (1998) The largest subunits of RNA polymerase from gastric helicobacters are tethered, J Biol Chem 273, 19371–19374. [DOI] [PubMed] [Google Scholar]
- [58].Philippe N, Legendre M, Doutre G, Couté Y, Poirot O, Lescot M, Arslan D, Seltzer V, Bertaux L, Bruley C, Garin J, Claverie JM, and Abergel C (2013) Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes, Science 341, 281–286. [DOI] [PubMed] [Google Scholar]
- [59].Legendre M, Lartigue A, Bertaux L, Jeudy S, Bartoli J, Lescot M, Alempic JM, Ramus C, Bruley C, Labadie K, Shmakova L, Rivkina E, Couté Y, Abergel C, and Claverie JM (2015) In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba, Proc Natl Acad Sci U S A 112, E5327–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mejia YX, Nudler E, and Bustamante C (2015) Trigger loop folding determines transcription rate of Escherichia coli’s RNA polymerase, Proc Natl Acad Sci U S A 112, 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zuo Y, and Steitz TA (2015) Crystal structures of the E. coli transcription initiation complexes with a complete bubble, Mol Cell 58, 534–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liu B, Zuo Y, and Steitz TA (2016) Structures of E. coli σS-transcription initiation complexes provide new insights into polymerase mechanism, Proc Natl Acad Sci U S A 113, 4051–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Barnes CO, Calero M, Malik I, Graham BW, Spahr H, Lin G, Cohen AE, Brown IS, Zhang Q, Pullara F, Trakselis MA, Kaplan CD, and Calero G (2015) Crystal Structure of a Transcribing RNA Polymerase II Complex Reveals a Complete Transcription Bubble, Molecular cell 59, 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Seibold SA, Singh BN, Zhang C, Kireeva M, Domecq C, Bouchard A, Nazione AM, Feig M, Cukier RI, Coulombe B, Kashlev M, Hampsey M, and Burton ZF (2010) Conformational coupling, bridge helix dynamics and active site dehydration in catalysis by RNA polymerase, Biochim Biophys Acta 1799, 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang B, Predeus AV, Burton ZF, and Feig M (2013) Energetic and structural details of the trigger-loop closing transition in RNA polymerase II, Biophys J 105, 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Krigbaum WR, and Komoriya A (1979) Local interactions as a structure determinant for protein molecules: II, Biochim Biophys Acta 576, 204–248. [DOI] [PubMed] [Google Scholar]
- [67].Pontius J, Richelle J, and Wodak SJ (1996) Deviations from standard atomic volumes as a quality measure for protein crystal structures, J Mol Biol 264, 121–136. [DOI] [PubMed] [Google Scholar]
- [68].Malagon F, Kireeva ML, Shafer BK, Lubkowska L, Kashlev M, and Strathern JN (2006) Mutations in the Saccharomyces cerevisiae RPB1 gene conferring hypersensitivity to 6-azauracil, Genetics 172, 2201–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kireeva ML, Nedialkov YA, Cremona GH, Purtov YA, Lubkowska L, Malagon F, Burton ZF, Strathern JN, and Kashlev M (2008) Transient reversal of RNA polymerase II active site closing controls fidelity of transcription elongation, Mol Cell 30, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Izban MG, and Luse DS (1992) The RNA polymerase II ternary complex cleaves the nascent transcript in a 3’--−−5’ direction in the presence of elongation factor SII, Genes Dev 6, 1342–1356. [DOI] [PubMed] [Google Scholar]
- [71].Kang JY, Mishanina TV, Bellecourt MJ, Mooney RA, Darst SA, and Landick R (2018) RNA Polymerase Accommodates a Pause RNA Hairpin by Global Conformational Rearrangements that Prolong Pausing, Mol Cell 69, 802–815.e801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Guo X, Myasnikov AG, Chen J, Crucifix C, Papai G, Takacs M, Schultz P, and Weixlbaumer A (2018) Structural Basis for NusA Stabilized Transcriptional Pausing, Mol Cell 69, 816–827.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Vos SM, Farnung L, Urlaub H, and Cramer P (2018) Structure of paused transcription complex Pol II-DSIF-NELF, Nature 560, 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bochkareva A, Yuzenkova Y, Tadigotla VR, and Zenkin N (2012) Factor-independent transcription pausing caused by recognition of the RNA-DNA hybrid sequence, EMBO J 31, 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Larson MH, Mooney RA, Peters JM, Windgassen T, Nayak D, Gross CA, Block SM, Greenleaf WJ, Landick R, and Weissman JS (2014) A pause sequence enriched at translation start sites drives transcription dynamics in vivo, Science 344, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gajos M, Jasnovidova O, van Bommel A, Freier S, Vingron M, and Mayer A (2021) Conserved DNA sequence features underlie pervasive RNA polymerase pausing, Nucleic acids research 49, 4402–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Yuzenkova Y, and Zenkin N (2010) Central role of the RNA polymerase trigger loop in intrinsic RNA hydrolysis, Proc Natl Acad Sci U S A 107, 10878–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.