

Abstract
Recent evidence has revealed that small polypeptides (containing fewer than 100 amino acids) can be translated from noncoding RNAs (ncRNAs), which are usually defined as RNA molecules that do not encode proteins. However, studies on functional products translated from primary transcripts of microRNA (pri‐miRNA) are quite limited. Here, we describe a peptide termed miPEP31 that is encoded by pri‐miRNA‐31. miPEP31 is highly expressed in Foxp3+ regulatory T cells (Tregs) and significantly promotes the differentiation of Tregs without affecting their inhibitory ability. Our results show that miPEP31 is a cell‐penetrating peptide both in vitro and in vivo. miPEP31 downregulates miR‐31 expression, enhances peripheral Treg induction, and dramatically suppresses experimental autoimmune encephalomyelitis. Mechanistically, we show that miPEP31 acts as a transcriptional repressor inhibiting the expression of miRNA‐31, a negative regulator of Tregs. Our results reveal an indispensable role of miPEP31 in maintaining immune homeostasis by promoting Treg differentiation and also present a potential therapeutic peptide for modulating miRNA expression and treating autoimmune diseases.
Keywords: autoimmune disease, miPEP31, miR‐31, transcriptional repressor, Treg
Subject Categories: Chromatin, Transcription & Genomics; Immunology; RNA Biology
miPEP31, encoded by the primary transcript of miR‐31, is a cell‐penetrating peptide which promotes Treg differentiation. Synthesized miPEP31 enhances the induction of peripheral Tregs and suppresses experimental autoimmune encephalomyelitis.
Introduction
According to the current annotations from databases, 51.8% of the human genome is transcribed, but only 1.2% encodes proteins (Djebali et al, 2012; Ransohoff et al, 2018). Small open reading frames (sORFs) with fewer than 100 codons are generally considered not to be translated into annotated proteins with predictable functions (Bazzini et al, 2014). Millions of sORF sequences are found in eukaryotic genomes, and thousands can be mapped to transcripts and, in many cases, to putative long noncoding RNAs (lncRNAs) (Geisler & Coller, 2013). Ribosome profiling with deep sequencing (ribo‐seq) (Ingolia et al, 2009, 2011) and proteomic approaches (Saghatelian & Couso, 2015) have enabled the identification of functional sORF‐encoded micropeptides derived from noncoding loci in flies, mice, and humans (Savard et al, 2006; Galindo et al, 2007; Kondo et al, 2007; Hanyu‐Nakamura et al, 2008; Ingolia et al, 2011; Magny et al, 2013; Nelson et al, 2016; Niu et al, 2020). Noncoding ORF‐harboring loci may constitute up to 10% of the genome, and hundreds to thousands of putative micropeptides originate from genes currently annotated as noncoding (Savard et al, 2006; Galindo et al, 2007; Kondo et al, 2007; Hanyu‐Nakamura et al, 2008; Andrews & Rothnagel, 2014; Saghatelian & Couso, 2015; Plaza et al, 2017). Increasing evidence has shown that some lncRNAs encode proteins with biological functions (Anderson et al, 2015; Lauressergues et al, 2015; Nelson et al, 2016; Huang et al, 2017).
Peptide therapy can provide a safe, effective, and economically viable approach for disease‐modifying therapy compared with traditional antibody and small molecule medicine. Among the advantages of using peptides as drug candidates are their high potency, specificity, and good safety profile (Angell et al, 2018). These positive attributes of peptides along with advances in drug delivery technologies have generated renewed interest in the discovery, optimization, and development of peptides as therapeutics (Vlieghe et al, 2010). Target selection for peptide therapeutics is challenging due to the inherent properties of peptides (Du et al, 2015; Fosgerau & Hoffmann, 2015); therefore, identifying a clear differentiation strategy for a new peptide program over a small molecule or antibody program from the outset is critical for successful navigation of drug development hurdles. The newly identified peptides encoded by putative ncRNAs have inspired a new strategy for finding new potential peptide drugs. Exploration of the endogenously expressed functional ncRNA‐encoded peptides helps us identify candidate peptide drug characteristics with natural safety, stability, and biological efficiency, overcoming major challenges that could lead to failure in peptide drug development.
In our early research, we reported that the conditional deletion of miR‐31 in CD4+ T cells leads to enhanced induction of regulatory T cells (Treg) in the periphery and decreases the severity of experimental autoimmune encephalomyelitis (EAE), which highlights the critical role of miR‐31 in the regulation of Treg‐cell induction in autoimmune diseases (Zhang et al, 2015). Pri‐miR‐31, annotated as lncRNA in the database, also has mRNA‐like features, such as capping and polyadenylation (Guttman & Rinn, 2012), which indicates the coding potential of the sORF within it. miR‐31 is rapidly induced after T‐cell activation by anti‐CD3 plus anti‐CD28 dependent on calcium signaling (Moffett et al, 2017) and is known to inhibit the differentiation of CD4+ regulatory T cells in humans through direct regulation of the expression of the transcription factor Foxp3 and inhibits the differentiation of induced regulatory T cells in mice through repression of the G protein‐coupled receptor Gprc5a (Rouas et al, 2009; Zhang et al, 2015). However, whether pri‐miR‐31‐encoded peptides are potential immune regulators that can be used for therapeutic interventions in autoimmune diseases remains unknown.
In this study, we found that a 44‐amino‐acid peptide was encoded by pri‐miR‐31, and we named this peptide miPEP31. miPEP31 is endogenously and highly expressed in Foxp3+ Treg cells. miPEP31 promoted the differentiation of Treg cells by downregulating miR‐31 expression. Furthermore, synthetic miPEP31 dramatically suppresses experimental autoimmune encephalomyelitis (EAE). Mechanistically, we demonstrated that miPEP31 acts as a transcriptional repressor that inhibits miR‐31 transcription in a sequence‐dependent manner. Thus, miPEP31 is a novel endogenous peptide that maintains immune homeostasis by promoting the differentiation of Treg cells. Together, our study provides a strategy to investigate the specific function of sORFs nested in pri‐miRNAs and reveals indispensable roles of peptides encoded by sORFs nested in miRNA host genes. Our data highlight the significant therapeutic effect of miPEP31 on autoimmune diseases and also provide novel mechanistic insights into the role of miPEPs in the regulation of miRNAs.
Results
Endogenous expression of miPEP31 encoded by pri‐miR‐31
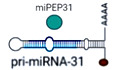
Because some noncoding RNAs encode peptides (Lauressergues et al, 2015; Nelson et al, 2016), we hypothesized that pri‐miR‐31 might also encode peptide(s) that could have biological functions. Using the open reading frame (ORF) finder, we discovered a putative sORF within pri‐miR‐31 (Fig 1A and B). The binding of ribosomes is a strong indicator of lncRNA translation (Ingolia et al, 2009). Pri‐miR‐31 was found in a public ribo‐seq study on NIH 3T3 cells (GSM2778762, SRR6023964) (Namkoong et al, 2018) (Fig 1C), which supports our evidence of the translation of the sORF. To identify whether the start codon of the putative ORF might be active, we fused the gfp tag (without ATG) to the 5′ leader sequence with ATG or with it mutated to ATT. We observed that ATG in the sORF drove the translation of gfp (Fig 1D). To further detect the endogenously produced peptide encoded by the sORF, we produced an antibody against that peptide. Western blotting showed that the antibody specifically detected the ORF‐GFP fusion protein (Fig 1E). The cytoplasmic localization of RNAs is required for the translation of the ORF within it. We found that pri‐miR‐31 mainly existed in the cytoplasm of CD4+ T cells (Fig 1F). Importantly, the pri‐miR‐31‐encoded peptide that we named miPEP31 is naturally highly expressed in immune‐related tissues, liver, and the intestine of wild‐type (WT) mice (Fig 1G). We further found that miPEP31 was not expressed in immune‐related tissues of miR‐31−/− mice, which confirms the specificity of the antibody (Fig 1H). Collectively, our data identified an endogenous peptide encoded by pri‐miR‐31.
Figure 1. miPEP31 is an endogenous peptide encoded by pri‐miR‐31.
- Schematic representation of miPEP31 translation. Pri‐miR‐31 transcribed from the miR‐31 host gene. sORF was found by the ORF Finder. The yellow box indicates the ATG of the sORF. The red line indicates the sequence of miR‐31. And the green line indicates the location of sORF on pri‐miR‐31.
- The nucleotide and amino acid sequences of the sORF.
- Ribo‐seq data of NIH 3T3 cells (SRR6023964) were analyzed and visualized by IGV. Ribo‐seq reads were found on pri‐miR‐31. The red box indicates the reads on partial of the coding region of miPEP31.
- Plasmid with 5′ leading sequence of the sORF in pri‐miR‐31 fused with gfp or which ATG mutated to ATT was transfected into NIH 3T3 cells. Cells were selected by G418 for 2 days. Analysis of the GFP expression by confocal microscopy or flow cytometry, scale bar 100 μm.
- Immunoblot analysis of GFP and miPEP31 in NIH 3T3 cells transfected by pEGFP‐N1‐miPEP31 or pEGFP‐N1; the red arrow indicates predicted size of miPEP31‐GFP band.
- Flow‐FISH was used to detect the localization of pri‐miR‐31. The location of pri‐miR‐31 was analyzed by confocal microscopy. Scale bar 5 μm.
- Immunoblot analysis of multiple mouse tissues with a specific antibody against miPEP31. Values are expressed as fold changes relative to controls and normalized to GAPDH, the red arrow indicate predicted size of miPEP31.
- Immunoblot analysis of spleen, lymph node, and thymus from WT or miR‐31−/− mice with miPEP31 antibody, the red arrow indicates predicted size of miPEP31.
Source data are available online for this figure.
miPEP31 is required for the differentiation of Treg cells
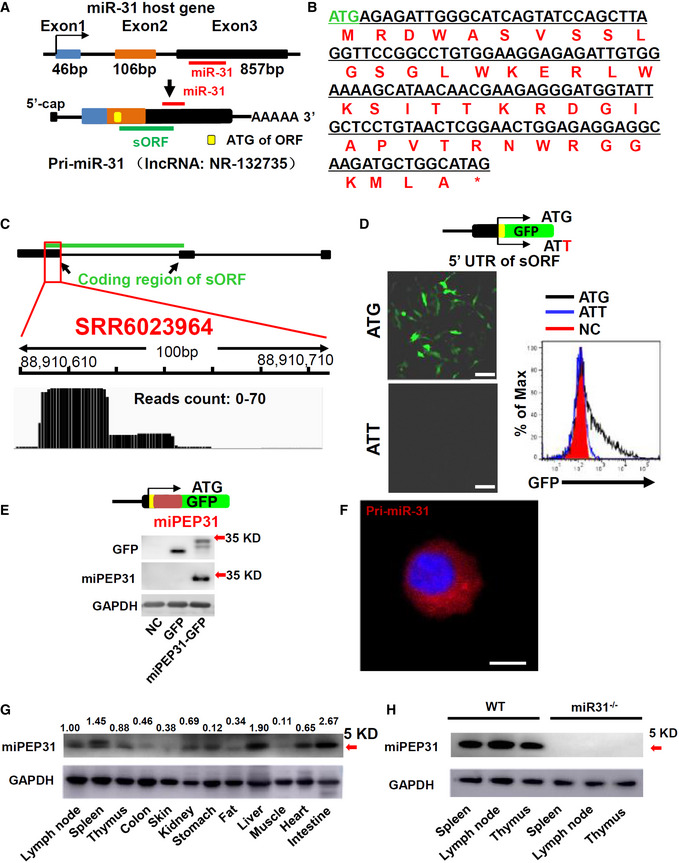
We previously reported that miR‐31 negatively regulates the generation of peripheral Treg (pTreg) cells (Zhang et al, 2015). We observed higher levels of miPEP31 in CD4+ FoxP3+ T cells than in CD4+ FoxP3− T cells (Fig 2A–C). Thus, we hypothesized that miPEP31 might favor the differentiation of Treg cells. To test this idea, we incubated CD4+ T cells with synthetic miPEP31. We differentiated naive T cells for the TGF‐β‐dependent generation of Treg cells in the presence of miPEP31. We observed that the differentiation of Treg cells was significantly promoted by miPEP31, while a scrambled peptide with the same number of amino acids (scPEP) had no effect on the induction of Treg‐cell differentiation (Fig 2D and E). Furthermore, we found that the proliferation marker Ki67 was expressed at higher levels in FoxP3+ cells treated with miPEP31 than in those treated with scPEP (Fig 2F). To determine whether miPEP31 alters the differentiation of Treg cells under inflammatory and noninflammatory conditions, we induced an EAE animal model of multiple sclerosis by administration of myelin oligodendrocyte glycoprotein peptide (MOG35‐55). We isolated splenocytes from EAE mice at Day 15 after MOG35‐55 immunization and then restimulated them with MOG35‐55 and different doses of miPEP31. The number of differentiated Treg cells was dose dependently increased. We also treated splenocytes from nonimmunized wild‐type mice with different doses of miPEP31, and the percentage of FoxP3+ cells was increased after miPEP31 treatment (Fig 2G and H).
Figure 2. miPEP31 promotes the differentiation of Treg cells.
-
AThe expression of miPEP31, FoxP3, and CD4 was shown in FoxP3+ and FoxP3− cells gated from CD4+ T cells by imaging flow cytometry. (B) Representative images of miPEP31 in FoxP3+ and FoxP3− cells gated in (A) were shown, scale bar 10 μm. (C) Quantification and statistical analysis of miPEP31 MFI in images of FoxP3+ and FoxP3− cells gated in (A) were shown. Data are representative of 12 images from 3 biological experiments (mean ± SEM). ***P < 0.001, unpaired two‐tailed Student’s t‐test.
-
D–FFlow cytometric analysis of Treg‐cell induction in the presence of 10 μM synthetic scPEP or miPEP31 (D). Data are presented as mean ± SEM of 11 biological replicates ****P < 0.0001, unpaired two‐tailed Student’s t‐test (E). The expression of Ki67 in FoxP3+ cells was analyzed by flow cytometry (F).
-
G, HFlow cytometric analysis of Treg cells in MOG35‐55 re‐challenged splenocytes of FoxP3EGFP mice 15 days postimmunization of MOG35‐55 (EAE) or splenocytes from non‐immunized wild‐type (Non‐Immu) mice in the presence of different doses of synthetic miPEP31. Data in (G, H) are presented as mean ± SEM of four biological replicates **P < 0.01, ****P < 0.0001 (0 μM vs. 0.1 μM or 1 μM or 10 μM), nonparametric one‐way ANOVA, Kruskal–Wallis test.
-
I, JWT mice were treated with synthetic scPEP or miPEP31 at day 8 postimmunization every 2 days for three injections. FoxP3EGFP+ Treg cells were sorted from splenocytes derived from mice treated with synthetic scPEP or miPEP31. The CellTrace™ Violet (CTV)‐labeled naive T cells (1 × 105) were cultured in 96‐well plates for 72 h together with a decreasing ratio of sorted Treg cells in the presence of anti‐CD3 plus γ‐irradiated antigen‐presenting cells (1 × 105). The suppressive function of Treg cells was determined by the proliferation of activated responder T cells (Tresp) on the basis of CTV dilution. Data in (J) are presented as mean ± SEM of four biological replicates. ns, not significant, (1:2 or 1:4 or 1:8 or 1:16 miPEP31 vs. scPEP), nonparametric one‐way ANOVA, Kruskal–Wallis test.
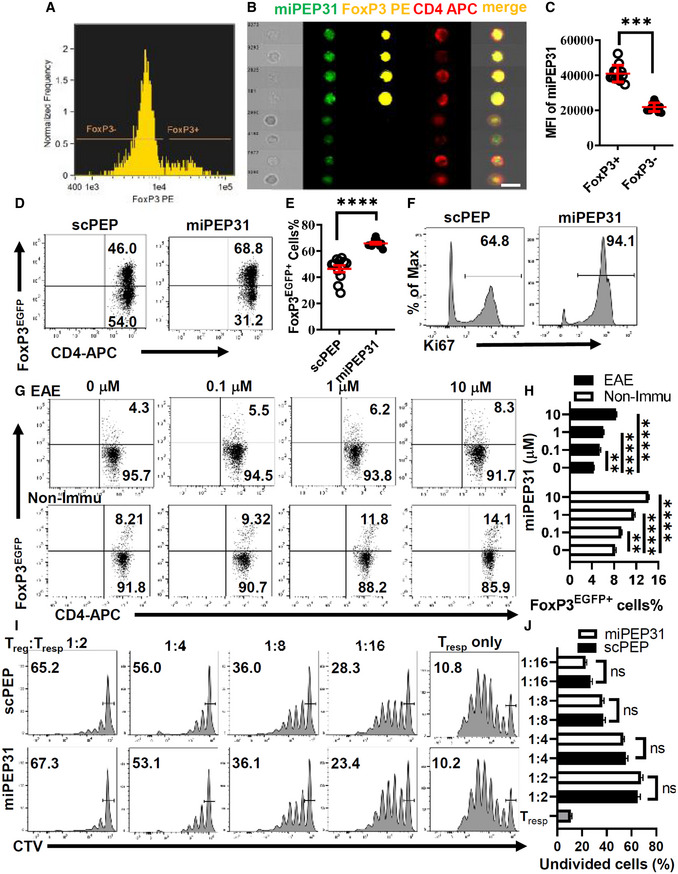
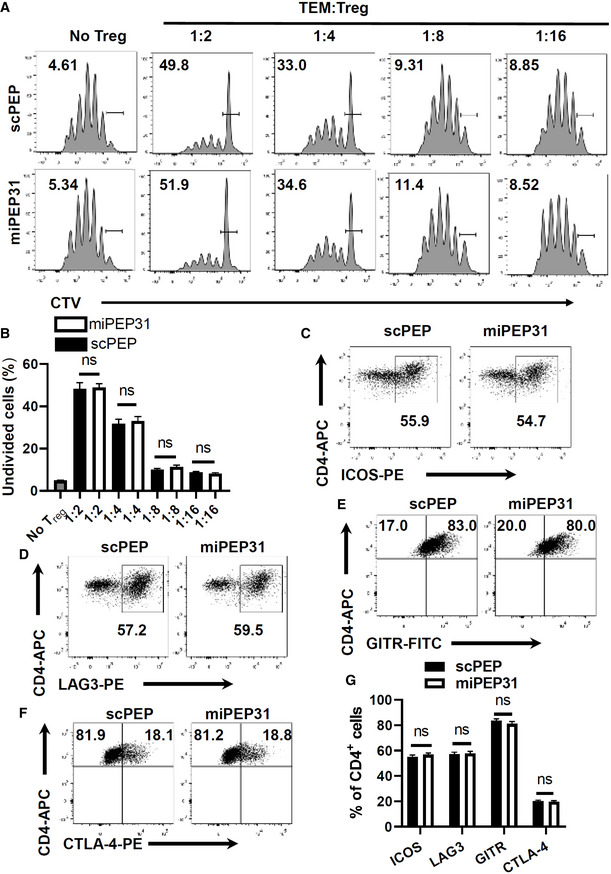
We further studied the suppressive capacity of Treg cells derived from miPEP31‐ or scPEP‐treated mice. By measuring the CellTrace™ Violet (CTV) fluorescence dilution, we found that miPEP31 had no impact on the suppressive function of Treg cells on naive Tconv cells (Fig 2I and J) and memory Tconv cells (Fig EV1A and B). We also detected the Treg‐cell suppression markers ICOS, LAG3, GITR, and CTLA‐4, and the expression of these suppression markers was not affected by miPEP31 (Fig EV1C–G). Altogether, these data show that miPEP31 can induce Treg‐cell differentiation but it does not affect Treg‐cell immunosuppression capacity.
Figure EV1. miPEP31 does not affect the inhibitory function of Treg cells.
-
A, BWT mice were treated with synthetic scPEP or miPEP31 at day 8 postimmunization every 2 days for three injections. FoxP3EGFP+ Treg cells were sorted from splenocytes derived from mice treated with synthetic scPEP or miPEP31. The CellTrace™ Violet (CTV)‐labeled memory Tconv cells (1 × 105) were cultured in 96‐well plates for 72 h together with a decreasing ratio of sorted Treg cells in the presence of anti‐CD3 plus γ‐irradiated antigen‐presenting cells (1 × 105). The suppressive function of Treg cells was determined by the proliferation of activated responder T cells (Tresp) on the basis of CTV dilution. Data in (B) are presented as mean ± SEM of four biological replicates, ns, not significant (1:2 or 1:4 or 1:8 or 1:16 miPEP31 vs. scPEP), nonparametric one‐way ANOVA, Kruskal–Wallis test.
-
C–GFlow cytometric analysis of ICOS (C), LAG3 (D), GITR (E), and CTLA‐4 (F) expression in Treg‐cell induction in the presence of 10 μM synthetic scPEP or miPEP31. Data in (G) are presented as mean ± SEM of six biological replicates. ns, not significant (scPEP vs. miPEP31), unpaired two‐tailed Student’s t‐test.
miPEP31 is a cell‐penetrating peptide both in vitro and in vivo
The cellular delivery of peptides into T cells has been a very difficult task. To clarify whether miPEP31 affects Treg cells through cell surface receptors or acts as an intracellular molecule, we synthesized fluorescein‐labeled miPEP31 (FAM‐miPEP31) and incubated it with anti‐CD3/28‐activated CD4+ T cells. We found that fluorescein‐labeled miPEP31 could enter CD4+ T cells, which suggests that miPEP31 is a cell‐penetrating peptide (CPP) (Fig 3A and B). We used histone H2A, β‐actin, and cytochrome C as proper controls to indicate the nuclear and cytoplasmic protein distribution in the cell fractions (Fig 3C). In total cell and nuclear lysate normalized to 1 μg protein per μl, the miPEP31 concentration was measured by ELISA in CD4+ T cells and Treg with or without 10 μM miPEP31. The elevated miPEP31 concentration seems unbiased in nucleus and cytoplasm in CD4+ T cells and Treg (Fig 3D). The penetration of miPEP31 into CD4+ T cells was further confirmed by imaging flow cytometry (Fig 3E). Treg cells play an important role in autoimmune diseases. To determine whether miPEP31 alters the differentiation of Treg cells under inflammatory conditions in EAE mice, we first examined the in vivo penetrating ability of miPEP31 and its control, scPEP. We intravenously injected FAM‐labeled miPEP31 and thereafter analyzed mouse spleen samples by imaging flow cytometry. Our in vivo study clearly showed that FAM‐labeled miPEP31 entered both CD4+ T cells and non‐CD4+ cells isolated from the spleen of the mice (Fig 3F and G).
Figure 3. miPEP31 is cell‐penetrating peptide both in vitro and in vivo .
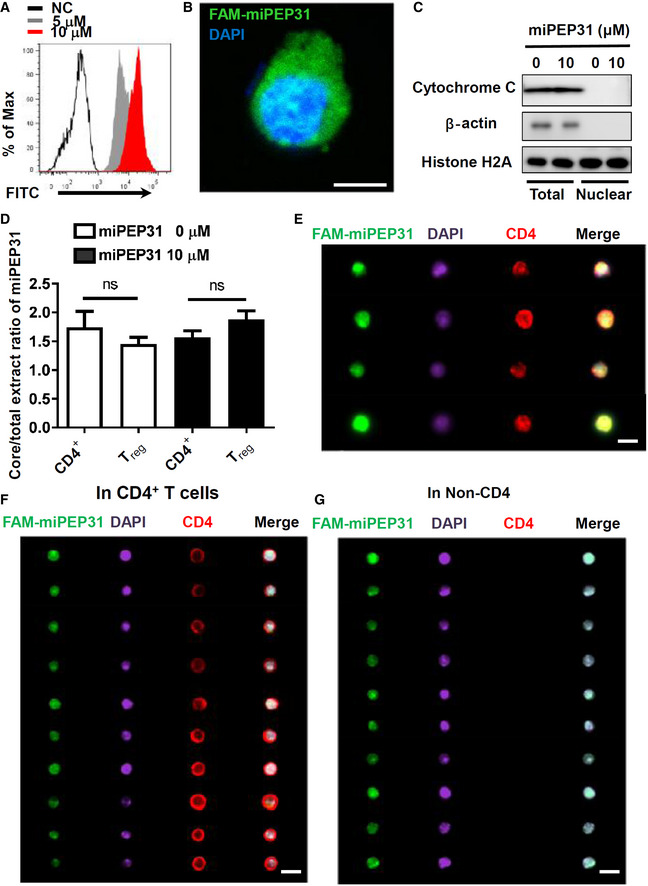
-
ACD4+ T cells were activated with anti‐CD3/28 and treated with different concentration of FAM‐miPEP31 for 24 h, the fluorescein was detected by flow cytometry.
-
BConfocal microscopy was used to analyze the fluorescence of FAM‐miPEP31 in anti‐CD3/28‐activated CD4+ T cells treated with 10 μM FAM‐miPEP31. Scale bar 5 μm.
-
C, DAnti‐CD3/28‐activated CD4+ T cells were cultured with 10 μM miPEP31 for 24 h. (C) Western blotting analysis of cytochrome C and histone H2A in total lysates and nuclear lysates. (D) ELISA to detect the concentration of miPEP31 in protein of total cell and nuclear extracts in CD4+ T and Treg cells treated with or without 10 μM miPEP31. Data in (D) are presented as mean ± SEM of four biological replicates. ns, not significant (CD4+ T vs. Treg), unpaired two‐tailed Student’s t‐test.
-
ENaive CD4+ T cells were isolated from WT C57BL/6 mice, and activated by anti‐CD3/28 for 48 h. Subsequently, FAM‐labeled miPEP31 was added. Imaging flow cytometry analysis of FAM‐labeled miPEP31 in CD4+ T cells. Scale bar 7 μm.
-
F, GC57BL/6 mice were injected intravenously with 100 μg FAM‐labeled miPEP31. Splenocytes were isolated and stained for CD4 and DAPI. Imaging flow cytometry analysis of miPEP31 in CD4+ T cells (F) and non‐CD4 splenocytes (G), scale bar 10 μm.
Source data are available online for this figure.
In most well‐known CPPs, multiple positively charged residues, such as arginine and lysine, play an important role in penetrating negatively charged lipid membranes. Cell membrane proteoglycans, especially heparan sulfate proteoglycans, can attract positively charged CPPs by electrostatic forces and act as primary binding sites to enhance their cargo uptake efficacy. Thus, we synthesized a series of FAM‐labeled mutant peptides based on miPEP31 with 2–5 arginine or lysine residues mutated to alanine residues (Fig 4A). The penetrating ability of these peptides decreased as the arginine or lysine residues were reduced (Fig 4B and C). Furthermore, we incubated CD4+ T cells with FAM‐labeled miPEP31 at 4°C or 37°C, and we found that miPEP31 enters the cytoplasm but not the nucleus at 4°C, while it enters both the cytoplasm and nucleus at 37°C (Fig 4D and E), which suggests that miPEP31 enters the cells through an energy‐independent direct penetration mechanism. Taken together, our data indicated that positively charged residues of miPEP31 are critical for its ability to penetrate CD4+ T cells.
Figure 4. Positive charged residues are critical for the penetrating ability of miPEP31.
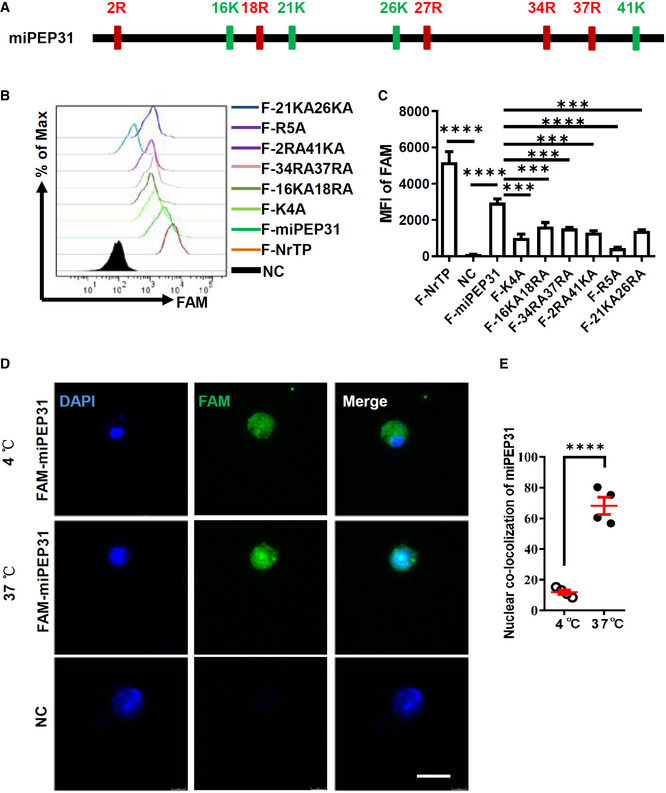
-
APositions of arginine (R) and lysine (K) on miPEP31.
-
B, CMutated FAM‐labeled peptides with two different position of positive charged residues mutated to A (F‐21KA26KA, F‐2RA41KA, F‐34RA37RA, and F‐16KA18RA) or all 5 R (F‐R5A) or all 4 K (F‐K4A) mutated to A were added into activated CD4+ T cells. Flow cytometry analysis of FAM in cells (B). MFI of FAM was shown in (C). Data in (C) are presented as mean ± SEM of four biological replicates. ***P < 0.001, ****P < 0.0001 (F‐NrTP vs. NC and F‐miPEP31 vs. NC or mutant F‐miPEP31), nonparametric one‐way ANOVA, Brown–Forsythe, and Welch's test.
-
D, ENaive CD4+ T cells were isolated from WT C57BL/6 mice, and activated by anti‐CD3/28 for 48 h. Subsequently, 10 μM FAM‐labeled miPEP31 was added for 20 min at 4°C or 37°C. Confocal microscopy analysis of the fluorescence. Scale bar 10 μm. The localization of miPEP31 with DAPI was shown in (E). Data are representative nuclear co‐localization of miPEP31 in four representative images from four biological experiments (mean ± SEM). ****P < 0.0001, unpaired two‐tailed Student’s t‐test.
miPEP31 suppresses EAE by promoting the differentiation of Treg cells
These observations led us to explore the potential therapeutic role of miPEP31 in Treg disorder‐mediated inflammatory immune pathogenesis in vivo. To investigate the role of miPEP31 in EAE development, we first analyzed the in vivo penetrating ability of miPEP31 and its uptake by immune cells in mice during the course of EAE. Interestingly, after intravenous injection of FAM‐miPEP31, the uptake of FAM‐miPEP31 by CD4+ T cells was confirmed by imaging flow cytometry (Fig 5A). Moreover, a single intravenous injection of 50 μg miPEP31 achieved maximal miPEP31 levels 6 h postinjection and it remained elevated for at least 2 days in CD4+ T cells (Fig 5B). The relative numbers of B220+, CD3+, CD4+, CD8+, CD11b+, and CD11c+ cells showed no significant difference in the splenocytes of miPEP31‐treated mice compared with controls, which indicates that there is a minimal impact of miPEP31 on other immune cells (Fig EV2A–E).
Figure 5. miPEP31 attenuates EAE by suppressing inflammation.
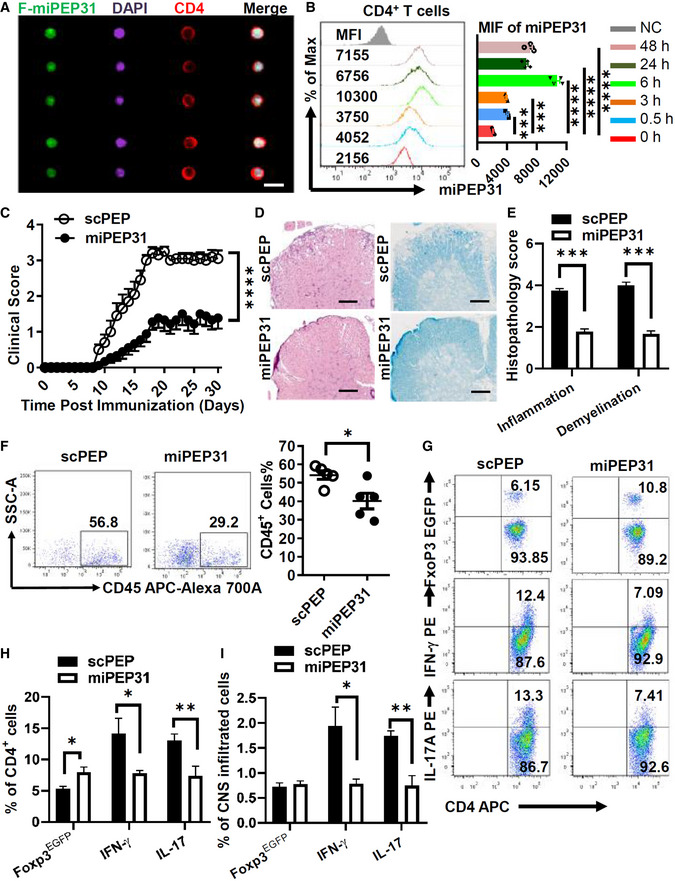
-
AEAE was induced by MOG35‐55. FAM‐miPEP31 (50 μg) was injected intravenously every 2 days starting from day 8 postimmunization. Splenocytes from FAM‐miPEP31‐treated EAE mice were isolated on day 15 postimmunization and analyzed by imaging flow cytometry. Representative images of CD4+ population were shown, scale bar 10 μm.
-
BmiPEP31 (50 μg) was injected intravenously into C57BL/6 mice at different time points. Splenocytes from miPEP31‐treated EAE mice were isolated and the level of miPEP31 in CD4+ T cells was analyzed by flow cytometry. MFI of miPEP31 was shown. Data in (B) are presented as mean ± SEM of four biological replicates. ***P < 0.001, ****P < 0.0001 (0 h vs. 0.5 h or 3 h or 6 h or 24 h or 48 h), nonparametric one‐way ANOVA, Kruskal–Wallis test.
-
C–EClinical scores (mean and SEM) of scPEP‐ or miPEP31 (50 μg)‐treated mice after the induction of EAE were assessed every day (C). scPEP or miPEP31 was injected intravenously every 3 days starting from day 8 postimmunization. (D, E) H&E and Fast blue staining of paraffin sections of spinal cords derived from scPEP‐ or miPEP31‐treated mice on day 15 after immunization. Histopathology scores of inflammation and demyelination were shown in (E). Scale bars, 70 μm. Data in (C, E) are presented as mean ± SEM of three independent experiments with 9 to 10 mice per group in each. ****P < 0.0001, repeated‐measures two‐way ANOVA and Tukey post‐hoc test (C) and ***P < 0.001 unpaired two‐tailed Student’s t‐test (E).
-
F–IEAE was induced by MOG35‐55 in FoxP3EGFP mice. scPEP or miPEP31 (50 μg) was injected intravenously every 2 days starting from day 8 postimmunization. Mononuclear cell infiltrates of scPEP‐ or miPEP31‐treated EAE mice were isolated from brains and spinal cords on day 15 postimmunization. (F) Flow cytometric analysis of CD45+ cells. (G, H) Flow cytometric analysis of FoxP3EGFP+ cells, IFN‐γ+ cells, and IL‐17+ cells in CD4+ T cells. (I) Percentage of FoxP3EGFP+ cells, IFN‐γ+ cells, and IL‐17+ cells in CNS infiltrated cells. Data in (F, H, and I) are presented as mean ± SEM of five (F) or three (H, I) biological replicates. *P < 0.05, unpaired two‐tailed Student’s t‐test (F) and *P < 0.05, **P < 0.01, nonparametric one‐way ANOVA, Kruskal–Wallis test.
Figure EV2. The role of miPEP31 in the induction of immune cells in EAE mice.
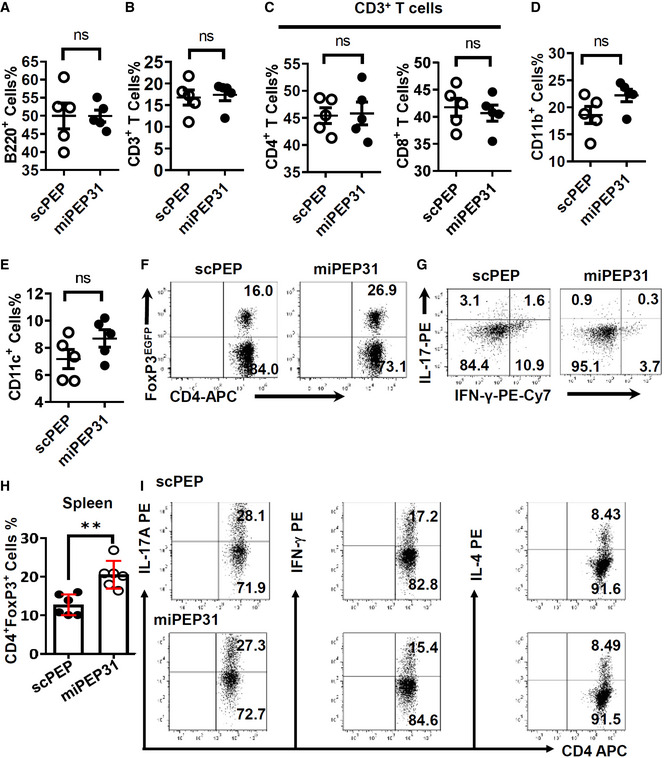
-
A–EEAE was induced by MOG35‐55. scPEP or miPEP31 (50 μg) was injected intravenously every 2 days starting from day 8 postimmunization. Splenocytes from scPEP‐ or miPEP31‐treated EAE mice were isolated on day 15 postimmunization and analyzed by flow cytometry. (A) B220 for total B lymphocytes. (B) CD3 for total T lymphocytes. (C) CD4 and CD8 for T‐helper cells and cytotoxic T cells (gate on CD3+ T cells). (D) CD11b for macrophages. (E) CD11c for dendritic cells. Data in (A–E) are presented as mean ± SEM of five biological replicates. ns, not significant, unpaired two‐tailed Student’s t‐test.
-
F–HEAE was induced by MOG35‐55 in FoxP3EGFP mice. scPEP or miPEP31 (50 μg) was injected intravenously every 2 days starting from day 8 postimmunization. Splenocytes of scPEP‐ or miPEP31‐treated EAE mice were isolated on day 15 postimmunization. (F,H) Flow cytometric analysis of FoxP3EGFP+ cells in CD4+ T cells. (G) Flow cytometric analysis of IFN‐γ+ cells and IL‐17+ cells in CD4+ T cells. Data in (H) are presented as mean ± SEM of five biological replicates. **P < 0.01, unpaired two‐tailed Student’s t‐test.
-
INaive CD4+ T cells were sorted from C57BL/6 mice and cultured in Th1, Th2, and Th17 polarizing medium for 3 days with 10 μM synthetic scPEP or miPEP31. The differentiation of Th1, Th2, and Th17 was analyzed by flow cytometry.
Next, we induced EAE with MOG35‐55 and treated EAE mice with miPEP31. Eight days after immunization, we intravenously injected 50 μg miPEP31 every 3 days into MOG35‐55‐induced EAE mice. miPEP31 significantly decreased the severity of EAE compared with scPEP (Fig 5C). Moreover, treatment with miPEP31 led to a marked decrease in the infiltration of inflammatory cells and reduced demyelination in the spinal cord (Fig 5D and E), which suggests that miPEP31 exhibits strong therapeutic potential for treating autoimmune diseases.
To obtain more details on how immune cells are affected by miPEP31, we treated EAE mice with scPEP or miPEP31 and analyzed their immune cells at Day 15 postimmunization. CD45, a marker defining hematopoietic cells, was used to distinguish infiltrating cells from resident cells in the central nervous system (CNS). We found that the numbers of infiltrated CD45+ cells in the CNS of EAE mice treated with miPEP31 were significantly decreased compared with those of mice treated with scPEP (Fig 5F). However, Treg cells were significantly induced not only in the spleen (Fig EV2H) but also in the CNS of miPEP31‐treated mice compared with scPEP‐treated controls. Accordingly, the numbers of IFN‐γ+ Th1 cells and IL‐17+ Th17 cells were significantly decreased in both the spleen and CNS of miPEP31‐treated mice (Figs 5G and H, and EV2F and G). The numbers of infiltrated Th17 and Th1 cells in the CNS were significantly decreased after miPEP31 treatment while the number of infiltrated Treg cells remained unchanged (Fig 5I). Taken together, we demonstrated a significant therapeutic effect of miPEP31 on autoimmunity. We showed that Treg cells from miPEP31‐treated mice inhibited responder T (Tresp)‐cell proliferation to the same extent as Treg cells from scPEP‐treated mice (Fig 2F). The in vitro assay showed that the differentiation of Th1, Th2, and Th17 cells was not affected by miPEP31 (Fig EV2I). Here, we provided strong evidence that promoting the induction of pTreg cells by miPEP31 is responsible for the improved phenotype.
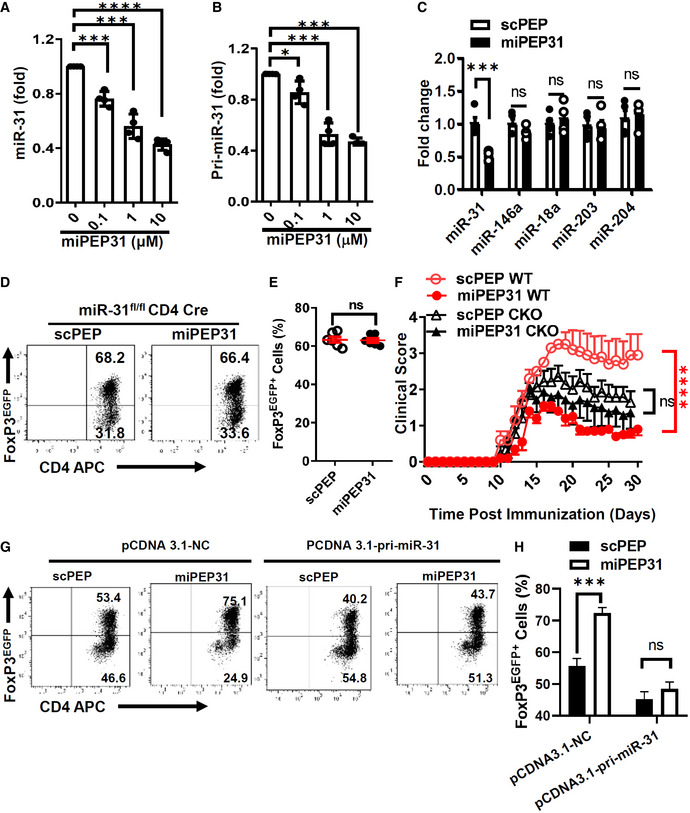
miPEP31 promotes the differentiation of Treg cells by inhibiting miR‐31 expression
In plants, peptides encoded by pri‐miRNAs can regulate their respective miRNAs (Lauressergues et al, 2015; Chen et al, 2020; Sharma et al, 2020). To investigate whether miPEP31 has a regulatory function on the expression of miR‐31, we next treated CD4+ T cells with different doses of synthetic miPEP31 and observed that miR‐31 expression was gradually decreased in a dose‐dependent manner (Fig 6A). The expression of pri‐miR‐31 was also inhibited by miPEP31 (Fig 6B). As a control for specificity, we tested four other miRNAs that were reported to control cell differentiation and/or proliferation (Yi et al, 2008; Lu et al, 2010; Courboulin et al, 2011; Jiang et al, 2011) and found that their expression was not affected by miPEP31 (Fig 6C). These data suggest that miPEP31 downregulates miR‐31 expression.
Figure 6. miPEP31 induces Treg‐cell differentiation through inhibiting miR‐31 expression.
-
AmiR‐31 expression in anti‐CD3/28‐activated CD4+ T cells treated by different dose of synthetic miPEP31. Data in (A) are presented as mean ± SEM of four biological replicates. ***P < 0.001, ****P < 0.0001 (0 μM vs. 0.1 μM or 1 μM or 10 μM), nonparametric one‐way ANOVA, and Kruskal–Wallis test.
-
BqPCR analysis of the expression of pri‐miR‐31 in anti‐CD3/28‐activated CD4+ T cells treated by different doses of miPEP31. Data in (B) are presented as mean ± SEM of four biological replicates. *P < 0.05, ***P < 0.001 (0 μM vs. 0.1 μM or 1 μM or 10 μM), nonparametric one‐way ANOVA, Kruskal–Wallis test.
-
CAnti‐CD3/28‐activated CD4+ T cells were treated by 10 μM miPEP31 or scPEP, the expression of miR‐31, miR‐146a, miR‐18a, miR‐203, and miR‐204 was detected by qPCR. Results are presented as the ratio of miRNA to the small nuclear RNA U6. Data in (C) are presented as mean ± SEM of four biological replicates. ns, not significant, ***P < 0.001 (scPEP vs. miPEP31), nonparametric one‐way ANOVA, Kruskal–Wallis test.
-
D, EFlow cytometric analysis of Treg cells polarized from naive CD4+ T cells of miR‐31fl/fl‐CD4‐Cre (cKO) mice in the presence of 10 μM synthetic scPEP or miPEP31. Data in (E) are presented as mean ± SEM of six biological replicates. ns, not significant, unpaired two‐tailed Student’s t‐test.
-
FClinical scores of scPEP‐ or miPEP31‐treated cKO or WT mice after the induction of EAE were assessed every day. scPEP or miPEP31 was injected intravenously into mice every 3 days starting from day 8 postimmunization. Data in (F) are presented as mean ± SEM of three independent experiments with 9 to 10 mice per group in each. ns, not significant, ****P < 0.0001, repeated measures two‐way ANOVA and Tukey post‐hoc test.
-
G, HCD4+ T cells were transduced with pCDNA 3.1‐NC or pCDNA 3.1‐pri‐miR‐31 and cultured in Treg polarizing condition in the presence of 10 μM synthetic scPEP or miPEP31 for 3 days. The Treg differentiation was analyzed by flow cytometry. Data in (H) are presented as mean ± SEM of four biological replicates. ns, not significant, ***P < 0.001, unpaired two‐tailed Student’s t‐test.
We previously reported that miR‐31 negatively regulates pTreg‐cell generation (Zhang et al, 2015). Thus, we hypothesized that miPEP31 might favor the differentiation of Treg cells by regulating miR‐31 expression. To test this idea, we incubated CD4+ T cells from miR‐31‐deficient mice with synthetic miPEP31. We found that miPEP31 failed to affect miR‐31‐deficient CD4+ Treg‐cell differentiation, emphasizing that miPEP31 induces Treg‐cell differentiation by downregulating miR‐31 expression in vitro (Fig 6D and E). In addition, we induced EAE in miR‐31fl / flCD4Cre (cKO) or WT mice and treated them with scPEP or miPEP31. The cKO mice showed no significant difference in the EAE severity between the scPEP‐ and miPEP31‐treated groups (Fig 6F).
Conditional deletion of miR‐31 in CD4+ T cells also leads to increased Treg‐cell differentiation (Zhang et al, 2015). To exclude the possibility that Treg cells in CKO CD4+ T cells are fully differentiated, we transduced plasmid that expressed pri‐miR‐31 under constant promoter into WT CD4+ T cells and polarized them into Treg cells with miPEP31 or scPEP. The promotion of Treg‐cell differentiation was impaired by the constant expression of miR‐31 (Fig 6G and H). Taken together, we demonstrated that the therapeutic effect of miPEP31 on autoimmunity occurs through regulating miR‐31.
miPEP31 inhibits the transcription of pri‐miR‐31 in a sequence‐dependent manner
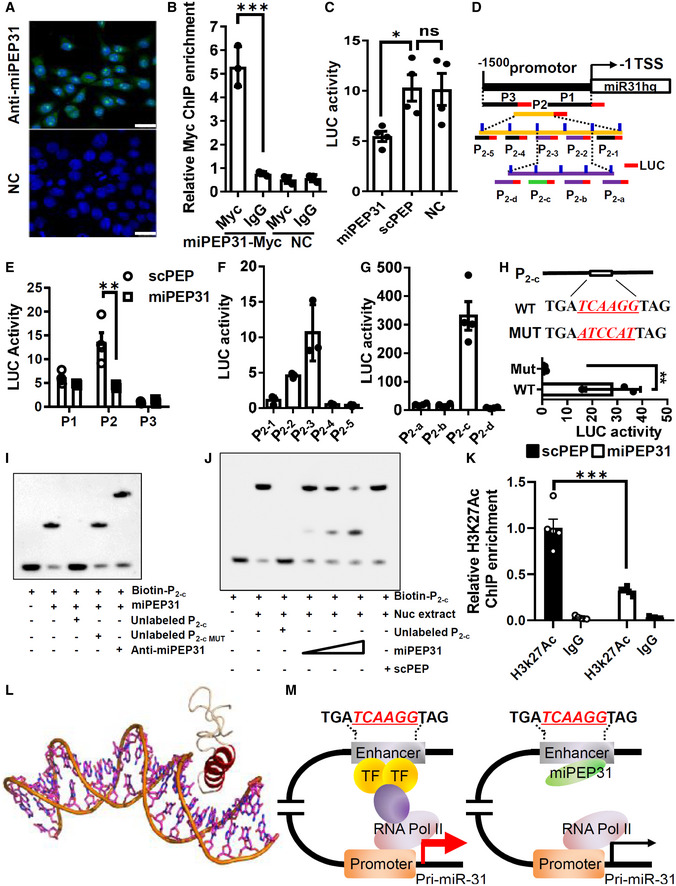
To determine how miPEP31 affects the expression of miR‐31, we used an antibody against miPEP31 to confirm its cellular location. We found that miPEP31 was localized to both the nucleus and cytoplasm but was primarily concentrated in the nucleus in most cells, which indicates that miPEP31 might regulate the production of miR‐31 at the transcriptional level (Fig 7A). To verify whether miPEP31 could bind to the promoter element of miR31hg, we performed chromatin immunoprecipitation (ChIP) assays. These assays showed that miPEP31 bound to the miR31hg promoter (Fig 7B). Luciferase reporter experiments demonstrated that miPEP31 inhibited miR31hg promoter activity (Fig 7C). Next, we divided the promoter into three regions, P1, P2, and P3, among which P2 had the strongest promoter activity, and miPEP31 inhibited the promoter activity of the P2 region (Fig 7D and E). Furthermore, we divided P2 into five regions, P2‐1 to P2‐5, and found that only P2‐2 and P2‐3 showed promoter activity (Fig 7D and F). Then, we narrowed down the promoter region into P2‐c, a 60‐bp fragment (Fig 7G). Interestingly, we found a highly conserved sequence in the human MIR31HG promoter with the same 6 bp core sequence of P2‐c (Fig EV3A), which indicates that miPEP31 or its homologous peptide could play important roles through a similar molecular mechanism. In agreement with this hypothesis, the addition of miPEP31 to human CD4+ T cells induced a marked increase in FoxP3‐expressing Treg cells under Treg‐cell polarizing conditions (Fig EV3B). Importantly, mutation of the 6 bp core sequence of P2‐c led to a complete loss of the promoter activity of P2‐c (Fig 7H). Accordingly, EMSA showed that miPEP31 directly bound to P2‐c; furthermore, a cold probe with a mutation in P2‐c could not compete with the binding of miPEP31, which indicates that the 6 bp core sequence is required for the binding of miPEP31 (Fig 7I). The shifted complex of P2‐c with nuclear extracts from NIH 3T3 cells was also competitively induced by miPEP31 in a dose‐dependent manner (Fig 7J). Chromatin immunoprecipitation analysis performed on the P2‐c region of the miR‐31 promoter after miPEP31 treatment revealed that miPEP31 promotes the deacetylation of histone H3K27 on the promoter of pri‐miR‐31 (Fig 7K), which is a well‐recognized marker for active enhancers (Shen et al, 2016).
Figure 7. miPEP31 inhibits the enhancer activity of pri‐miR‐31 in a sequence‐dependent manner.
-
AFluorescence images of the immunolocalization of the miPEP31 in NIH 3T3 cells. Results are representative of three independent experiments, scale bar 25 μm.
-
BpCDNA 3.1‐miPEP31‐myc or pCDNA 3.1‐NC was transfected into NIH 3T3 cells. Transfected cells were selected by G418 for 2 days. Myc was immunoprecipitated from the transfected cells. Immunoprecipitates were assayed for the expression levels of miR‐31 promoter. Data in (B) are presented as mean ± SEM of three biological replicates. ***P < 0.001, nonparametric one‐way ANOVA, Kruskal–Wallis test.
-
CLuciferase activity in NIH 3T3 cells transfected with luciferase reporter plasmids of pGL3 empty vector or miR‐31 promoter, treated with miPEP31 or scPEP. Results are presented as the ratio of firefly luciferase‐to‐renilla luciferase activity, relative to that of untreated NIH 3T3 cells transfected with pGL3 empty vector. Data in (C) are presented as mean ± SEM of four biological replicates. *P < 0.05, ns, not significant, nonparametric one‐way ANOVA, Kruskal–Wallis test.
-
D–GThree different regions (P1, P2, and P3) of miR‐31 promoter. The region P2 was divided into five sub‐regions P2‐1 to P2‐5 with 10 bp overlap with adjacent sub‐region as indicated. P2 (100‐310) (P2‐2 together with P2‐3) was divided into four sub‐regions P2‐a to P2‐d with 10 bp overlap with adjacent sub‐region. These regions were cloned into pGL3 empty vector and transfected into NIH 3T3 cells. Luciferase activity was measured in NIH 3T3 cells transfected with pGL3‐P1, P2, or P3 (E), P2‐1 to P2‐5 (F), and P2‐a to P2‐d (G). Results are presented as the ratio of firefly luciferase‐to‐renilla luciferase activity, relative to that of untreated NIH 3T3 cells transfected with pGL3 empty vector. Data in (E–G) are presented as mean ± SEM of four biological replicates. **P < 0.01, nonparametric one‐way ANOVA, Kruskal–Wallis test.
-
HWT and mutant sequence in P2‐c. pGL3‐P2‐c or pGL3‐P2‐c with mutation (in red) was transfected into NIH 3T3 cells. Luciferase activity was measured. Data in (H) are presented as mean ± SEM of three biological replicates. **P < 0.01, unpaired two‐tailed Student’s t‐test.
-
I, JEMSA assay to identify the interaction between miPEP31 and P2‐c or P2‐c with mutated (MUT). Anti‐miPEP31 was added to see the super shift. (J) EMSA assay to identify the interaction of P2‐c and NIH 3T3 cells nuclear extracts in the presence of different dose of miPEP31 or scPEP.
-
KChIP analysis of miR‐31 promoter by anti‐Histone H3K27Ac or IgG in NIH 3T3 cells treated with 10 μM synthetic scPEP or miPEP31. Data in (K) are presented as mean ± SEM of five biological replicates. ***P < 0.001 (scPEP vs. miPEP31), nonparametric one‐way ANOVA, Kruskal–Wallis test.
-
LCartoon representation of the modeled interaction between miPEP31 and DNA. Red helix is the DNA major groove binding helix from miPEP31; wheat chain is the coil.
-
MModel illustrating the mechanistic role of miPEP31.
Figure EV3. miPEP31 promotes the differentiation of human Treg cells.
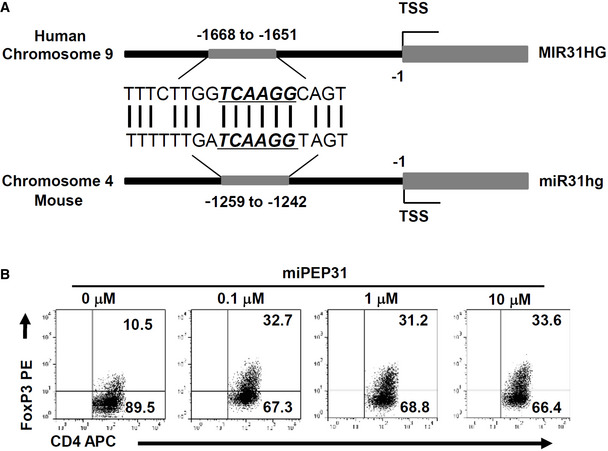
- Homologous alignment of miPEP31‐binding site in human and mouse miR‐31 host gene promoter.
- Flow cytometric analysis of human Treg‐cell induction from CD4+ T cells isolated from peripheral blood mononuclear cells in the presence of different doses of miPEP31. Shown in (B) is representative of three independent experiments.
Although pri‐miR‐31 is not fully conserved in the human genome, the exon that mainly codes for miPEP31 was found to be conserved in the human genome (Fig EV4A). We further generated cell lysates of the human HCT116 cell line and performed anti‐miPEP31 immunoprecipitation. Subsequently, purified fractions were subjected to mass spectrometry (Fig EV4B). Strikingly, we found a fragment that mapped uniquely to the predicted miPEP31 ORF‐encoded protein with more than 54% total protein coverage on mouse miPEP31 (Fig EV4C). The highly conserved miPEP31‐binding sequence in the human MIR31HG promoter also suggests that miPEP31 could share the same functional effect and mechanism in both mouse and human T cells.
Figure EV4. Existence of the homologous peptide of miPEP31 in human HCT 116 cells.
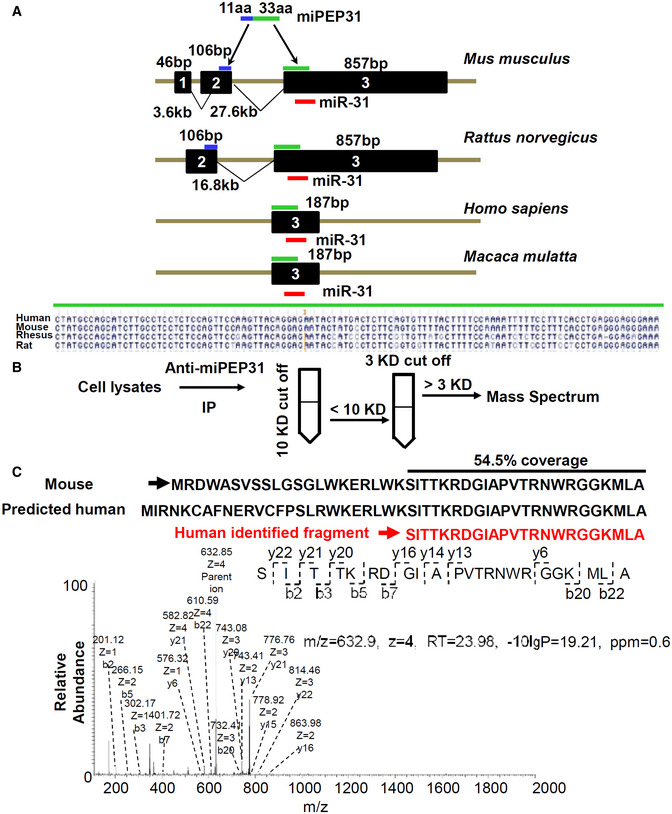
- miR‐31 host gene in Mus musculus, Rattus norvegicus, Homo sapiens, and Macaca mulatta. The coding sequence of miPEP31 located on Exon 2 (106 bp, in blue) and Exon 3 (857 bp, in green) of mouse miR‐31 host gene. The coding sequence of miPEP31 (under the green line) on Exon 3 is conserved across the four species.
- Workflow of mass spectrometry analysis to detect small peptides in HCT 116 cells. Protein lysates of HCT 116 cells were subjected to anti‐miPEP31 immunoprecipitation. Purified lysates were subject to mass spectrometry analysis.
- Representative secondary mass spectrometry analysis of fragment. Identified fragment ions (632.85 Da, charge = 4) are indicated below the peptide sequence. Shown in (C) is representative of three independent experiments.
Study of the miPEP31 structure might help us to understand the characteristics and function of miPEP31. Small peptides are difficult to crystallize because the coil tails of peptides are usually too flexible. Therefore, we used circular dichroism to investigate the secondary structure of miPEP31 and scPEP. An α‐helix structure was found in miPEP31 and scPEP in the covalent solvent 50% TFE with 59.2% helical residue in miPEP31 and 50.5% in scPEP (Fig EV5A–C). With I‐TASSER (Yang et al, 2015), we predicted the structural model of miPEP31, which is consistent with the circular dichroism results. The N‐terminal half (2–26) of miPEP31 has a highly helical trend, while the C‐terminus (27–44) has a high coil trend (Fig EV5D). The predicted structure of miPEP31 is an α‐helix head followed by a coiled tail, which is quite similar to the DNA‐binding domains in transcription factor (Figs 7L and EV5E). With similar physicochemical parameters, we found that scPEP could also penetrate into CD4+ T cells in vitro and in vivo (Fig EV5F–H). Altogether, our data suggest that miPEP31 regulates miR‐31 expression by competitively binding to the enhancer region of pri‐miR‐31 (Fig 7M).
Figure EV5. Secondary structures of scPEP and miPEP31.
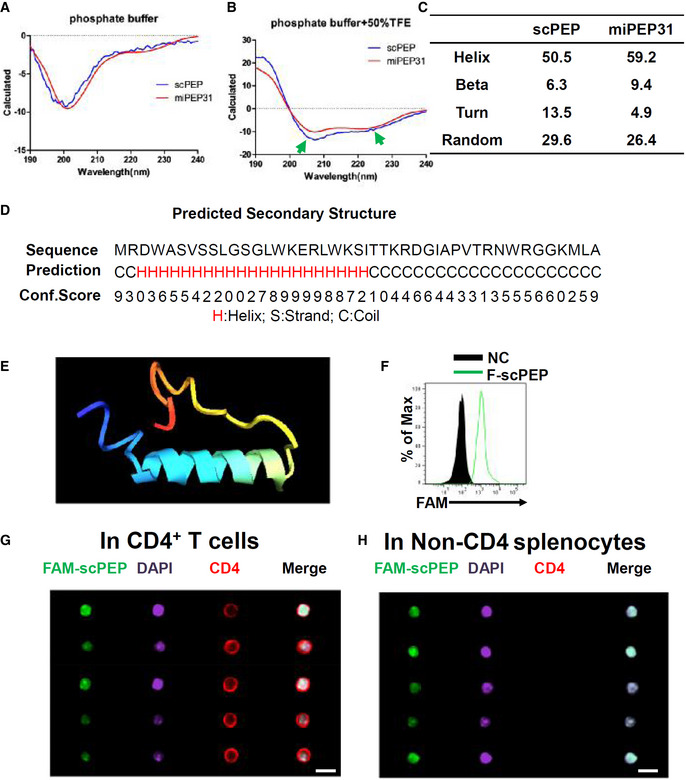
-
A, BmiPEP31 or scPEP with the concentration of 100 μg/ml in phosphate buffer (A) or phosphate buffer with 50% TFE (B) was analyzed by circular dichroism. The ratio between molar ellipticity at 222 nm and 207 nm was used to confirm an α‐helical structure of peptides. The arrows indicate the α‐helical structure of miPEP31 and scPEP.
-
CSecondary structures of miPEP31 and scPEP.
-
DThe secondary structure of miPEP31 was predicted by I‐TASSER. H, S, and C stands for helix, stand, or coil tend of different residues.
-
EThe structure model of miPEP31 predicted by I‐TASSER.
-
FCD4+ T cells were activated with anti‐CD3/28 and treated with 10 μM FAM‐scPEP for 24 h, the fluorescein was detected by flow cytometry.
-
G, HC57BL/6 mice were injected intravenously with 100 μg FAM‐labeled scPEP. Splenocytes were isolated and stained for CD4 and DAPI. Imaging flow cytometry analysis of FAM‐scPEP in CD4+ T cells (G) and non‐CD4 cells (H). Shown in (G, H) are representative of two independent experiments, scale bar 10 μm.
Discussion
Recent transcriptomic and bioinformatic studies suggest that thousands of sORFs map to ncRNAs (Cheng et al, 2005; The, 2007; Washietl et al, 2007). sORFs were found in these ncRNAs, which are usually considered to be nontranslated. Nevertheless, small peptides encoded by so‐called "noncoding RNAs" were recently identified in plants and mammals (Hanada et al, 2007; Ingolia et al, 2011; Magny et al, 2013; Lauressergues et al, 2015; Nelson et al, 2016). The encoded products function differently from the transcripts of ncRNAs (Huang et al, 2017). Emerging evidence has demonstrated that miRNAs play a vital role in the regulation of immunological functions and the prevention of autoimmunity (Johnnidis et al, 2008; Xiao & Rajewsky, 2009; Zhang et al, 2015). However, the role of pri‐miRNA‐encoded micropeptides in immune homeostasis remains unknown. In this study, we found that pri‐miR‐31‐encoded miPEP31 was capable of suppressing the transcription of pri‐miR‐31, promoting the peripheral differentiation of Treg cells and attenuating the development of EAE in mice. We further identified that miPEP31 interacts with the promoter of pri‐miR‐31 and promotes its deacetylation of histone H3K27, thus suppressing its transcription. Our data revealed that the peptide encoded by pri‐miR‐31 plays an important role in immune homeostasis and provides new insight into the regulation of miRNA and a potential therapeutic approach to autoimmune diseases.
The existence of pri‐miRNA‐encoded peptides in mammalian cells is still a debated question. Unlike lncRNAs, pri‐miRNAs are generally thought to be sub‐localized in the nucleus and processed by Drosha, a protein that cleaves RNA to form the microprocessor complex, making pri‐miRNA spatially unable to be translated (Lee et al, 2003). The recruitment of ribosomes to pri‐miRNAs requires an "open state"; however, the hairpin structure is essential for the production of miRNAs (Lee et al, 2003). Although spliced pri‐miRNAs excluding the miRNAs have been reportedly allowed to be exported into the cytoplasm (Chang et al, 2015), the mechanism of how pri‐miRNAs are translated is still not clear. Recently, ribosome footprint profiling and bioinformatic analyses showed that ribosomes were able to bind to sORFs on pri‐miRNAs, which indicates the coding potential of pri‐miRNAs (Andrews & Rothnagel, 2014; Bazzini et al, 2014). These missing coding genes nested in miRNA host genes could be systematically and erroneously omitted by conventional annotation algorithms. Interestingly, pri‐miR‐31 was found in the ribo‐seq research dataset (GSE102354) (Namkoong et al, 2018). We also confirmed the cytoplasmic location of pri‐miR‐31 in CD4+ T cells, which supports our evidence of the translation of miPEP31. Furthermore, we detected the expression of miPEP31 in immune‐related tissues. Taken together, we clearly showed the existence of miPEP31.
The immune system faces a particularly difficult challenge to maintain a state of tolerance toward dietary antigens and commensal organisms, which requires a delicate balance between regulatory and effector T cells (Xu et al, 2018). The penetrating ability of miPEP31 makes it more likely to be secreted by mucosal epithelial cells, which highly express miR‐31 (Yan et al, 2015; Tian et al, 2020); this expression contributes to a unique immune microenvironment, making Treg cells present at higher frequencies in the gut lamina propria and colon. Our results demonstrated that pri‐miR‐31‐encoded miPEP31 plays an important role in maintaining immune homeostasis. In addition to miPEP31, Ruaidhrí Jackson and his colleagues found the translation of noncanonical open reading frames from an ncRNA‐regulated mucosal immunity (Jackson et al, 2018). Thus, small peptides encoded by ncRNAs or pri‐miRNAs could play important roles in the regulation of the immune system. Treg cells have also been suggested to be capable of limiting central nervous system (CNS) autoimmunity in several CD4+ T‐cell‐driven EAE animal models (Reddy et al, 2005; Huarte et al, 2016; Lee et al, 2017). Importantly, decreased proliferative potential and cloning frequency of Treg cells were reported in patients with MS (Viglietta et al, 2004; Carbone et al, 2014). Although in vitro‐induced Treg cells might not be sufficiently stable and functional due to epigenetic modification (Zhou et al, 2009), our results showed that under inflammatory conditions, miPEP31 can induce the differentiation of Treg cells challenged by MOG. In vivo regulators of Treg‐cell induction are considered to have valuable potential for clinical applications in the treatment of autoimmune diseases. In our previous study, we reported that miR‐31 expression in CD4+ T cells was triggered by TCR signaling, and its conditional deletion substantially enhanced Treg‐cell induction and ameliorated disease severity in the EAE model (Zhang et al, 2015). The discovery of miPEP31 encoded by pri‐miR‐31 inhibiting miR‐31 expression demonstrates the in vivo functions of miPEP in mammalian animals and potentially defines a new therapeutic strategy for autoimmune disorders. Our findings advance the noncoding RNA field by clearly linking noncoding RNA‐encoded peptides to peptide‐mediated miRNA regulation. The cell‐penetrating nature of miPEP31 and its clear target qualify miPEP31 to be an ideal candidate for the treatment of Treg‐cell–mediated autoimmunity.
The primary sequences of sORFs are less conserved than protein‐coding genes but more conserved than introns (Ransohoff et al, 2018). In mice, pri‐miR‐31 is located on chromosome 4 and contains three exons. Only exon 3 is conserved in Mus musculus, Rattus norvegicus, Macaca mulatta, and Homo sapiens. The region encoding miPEP31 lies on both exon 2 and exon 3, which means that miPEP31 is not conserved across all species, and the coding region of miPEP31 is mainly located in conserved exon 3. By mass spectrometry, we identified a fragment of miPEP31 in the anti‐miPEP31‐purified fractions, which strongly indicates that a functional homologous peptide is encoded by the human miR‐31 host gene. Surprisingly, we found a highly homologous 21‐base‐pair region with the same "TCAAGG" miPEP31‐binding site in the promoter of the human miR‐31 host gene. Similar to other transcriptional repressors, miPEP31 is more likely to act at the genome scale and not just target miR‐31. However, with limited information about the miPEP31‐interacting transcriptional factors and DNA matrix, we have difficulties in investigating how miPEP31 globally regulates gene expression at the genome level, which needs further research work. miPEP31 could also promote the differentiation of human Treg cells. These results strongly indicate that although miPEP31 lacks full conservation, homologous peptides or proteins are highly likely to be found in Homo sapiens.
The penetration ability of peptides is one of the major challenges that limits the “druggability” of peptide drugs. There are five arginine residues and four lysine residues on miPEP31. These positively charged residues contribute to its penetrating ability across the negatively charged bilayer lipid membranes. The penetration of miPEP31 across the cell membrane appears to be independent of the energy because, at 4°C, FAM‐miPEP31 could also enter the cell, while the nuclear transportation of miPEP31 was energy dependent. Structure prediction by I‐TASSER and circular dichroism both indicated that miPEP31 has an α‐helix structure, which is a common element in CPPs and transcription factors. miPEP31 can function differently from the typical inhibitors of the DNA‐binding family, such as helix–loop–helix‐like peptides, which are 119–168 amino acids in length (Murre et al, 1994). The small size of miPEP31 indicates that it cannot form large globular proteins with a buried domain; however, it could be perfect for interfering with larger proteins. The α‐helix, which is the critical domain for DNA binding, is highly unstable and more likely to be in a random coil state in solution. Interaction with larger proteins can stabilize the conformation of the α‐helix. miPEP31 could interact with other scaffold proteins and form complicated machinery to regulate gene expression, which requires further study.
Peptide drugs display unparalleled advantages, such as ready synthesis, quick optimization, fewer immune responses, fewer toxic side effects, and rapid clearance (Fosgerau & Hoffmann, 2015). Dozens of peptide‐based drugs have been approved by the U.S. Food and Drug Administration (FDA) over the past decade, the majority for the treatment of metabolic disorders, including diabetes, with many more under clinical and preclinical studies, among which insulin is the most popular (Kaspar & Reichert, 2013; Zhang & Eiden, 2019). Compared with small molecule drugs, the time required for peptides to move from clinical trials to FDA approval is much shorter, and peptides are two times more likely to pass clinical trials (de la Torre & Albericio, 2018). Despite the significant progress that has been made in this field, we still face considerable challenges in developing peptide‐based drugs, owing to not only traditional problems such as instability, easy degradation, and difficulty in crossing the cell membrane but also mass production technology to reduce the production costs (Fosgerau & Hoffmann, 2015; Angell et al, 2018; Erak et al, 2018). Luckily, miPEP31 did not show significant toxicity or side effects at a dose of 50 mg/kg and had no adverse impact on the immune cell composition. Considering that the expression of miPEP31 is not restricted to lymphoid tissue, more toxicology study is required to clarify the possible adverse effects it might have. Although further modification of its stability and efficiency is still needed, the cell‐penetrating nature of miPEP31 and its clear target qualify miPEP31 as a potentially ideal candidate for the treatment of Treg‐cell–mediated autoimmunity.
In conclusion, the function of miRNAs in controlling the expression of diverse genes in various autoimmune diseases makes miRNAs ideal candidates for therapeutic applications. However, miRNA selective modulation via antisense inhibition or replacement requires technological advances. Our study demonstrates that an endogenous peptide encoded by pri‐miR‐31 regulates its respective miR‐31 as a transcriptional repressor and consequently inhibits the development of experimental autoimmunity by promoting Treg‐cell induction. These findings may provide an approach for modulating miRNA expression and treating autoimmune diseases.
Materials and Methods
Identification of conserved small open reading frames
The sequence of the primary transcription of RNAs was obtained from NCBI Gene. ORFs were found by ORF finder with the standard genetic codon, and “ATG” was used as the ORF start only.
Plasmid constructs
The 5' untranslated region (5' UTR) of the primary transcription of miR‐31 with the ATG (or mutated to ATT) sites of ORF and the sequence of gfp without the ATG site were constructed into pCDNA 3.1+ with the pEASY‐Uni Seamless Cloning and Assembly kit (Transgen, CU101‐03). Briefly, two fragments of interest were amplified with the respective primers, which have a 15 nt homologous arm with the adjacent fragment. The two fragments and the linearized plasmid were assembled by homologous recombination. The primers were used as follows: Primer Vec‐F, TTGGTACCGAGCTCGACGTAACCTAAAGCTAACAG; Primer GFP‐R, GCCCTCTAGACTCGATTACTTGTACAGCTCGTCCA; Primer ORF‐ATG‐R, CTCGCCCTTGCTCACTGCCAGCATCTTGCCTCCTC; Primer O‐ATG‐R, CTCGCCCTTGCTCACCATTTCAACTCTTCAACAAC; Primer O‐ATT‐R, CTCGCCCTTGCTCACAATTTCAACTCTTCAACAAC; Primer O‐ATG‐GFP‐F, TGAAGAGTTGAAATGGTGAGCAAGGGCGAGGAGCT; and Primer O‐ATT‐GFP‐F, TGAAGAGTTGAAATTGTGAGCAAGGGCGAGGAGCT. The sequence of miPEP31 with myc was synthesized by Generay and cloned into pCDNA 3.1+. The sequence of miPEP31 was synthesized by Generay and cloned into pEGFP‐N1. The sequences of the different regions of the miR‐31 promoter were synthesized by Generay and cloned into PGL3 basic. The sequence of pri‐miR‐31 was synthesized by Generay and cloned into PCDNA 3.1. The sequences are listed in Table EV1.
RNA reverse transcription and real‐time quantitative PCR
Total RNA was isolated using Trizol (Invitrogen, 15596018) according to the manufacturer's instructions. To measure mature miRNAs levels, 50 ng of total RNA was reverse transcribed using the TaqMan miRNA reverse transcription kit (4366597), miRNAs RT primers, and U6 snRNA (Applied Biosystems). The cDNAs were then analyzed by qPCR using the TaqMan probes for miRNAs (Applied Biosystems). Quantification of the relative miRNA expression was measured by the comparative CT (critical threshold) method, normalized to endogenous U6 expression and determined by the formula 2−ΔΔCt.
Flow cytometry
Cytokines, transcription factors, and surface markers were evaluated by flow cytometry with a FACSCanto II (BD Biosciences). To detect the intracellular expression of IL‐17A and IFN‐γ in CD4+ T cells, splenocytes and lymphocytes were first treated with 750 ng ml−1 ionomycin (Sigma, 55779‐48‐1), 50 ng ml−1 phorbol 12‐myristate 13‐acetate (PMA) (Sigma, P1585), and GolgiPlug (BD Biosciences, 555029) for 4–6 h at 37°C. TF fixation/permeabilization buffer (ebiosceince, 00‐5523‐00) was used for the permeabilization of nuclei. After nuclei permeabilization, BD Cytofix/Cytoperm (BD Biosciences, 554722) was used to maintain the penetrating state of nuclei and intracellular staining. After being washed, the stained cells were assayed with a BD Biosciences FACSCanto II flow cytometer, and the data were analyzed with FlowJo software. For flow cytometry, the monoclonal antibodies against CD4 (clone GK1.5, 48‐0041‐82), CD8 (clone 53‐6.7, 47‐0081‐80), CD11b (clone M1/70, 17‐0112‐82), CD11c (clone N418, 25‐0114‐82), CD45 (clone 2D1, 11‐9459‐42), CD62L (clone MEL‐14, RM4317), CD44 (clone IM, 17‐0441‐82), CD25 (clone PC61.5, 17‐0441‐82), B220 (clone RA3‐6B2, A15399), IL‐17A (clone eBio17B7, 12‐7177‐81), IFN‐γ (clone XMG1.2, 25‐7311‐82), Icos (clone ISA‐3, A14767), LAG3 (clone 3DS223H, 12‐2239‐42), GIRT (clone eBioAITR, 46‐5875‐41), CTLA‐4 (clone 14D3, 13‐1529‐82), and FoxP3 (clone FJK‐16s, 17‐5773‐82) were from eBioscience, and CD3 (clone 145‐2C11, 100311) was purchased from Biolegend.
Imaging flow cytometry
After being stained, cells were washed and fixed and then were analyzed at a magnification of ×40 on ImageStream flow cytometer (Amnis) and The IDEAS analysis software (Amnis). In each sample, 60,000 events were collected and imaged in the extended depth of field mode (EDF). Digital spectral compensation was performed on a pixel‐by‐pixel basis using single‐stained controls. Acquired cellular imagery was analyzed for the degree of miPEP31 nuclear translocation using the similarity feature, as described in IDEAS V.6.2 documentation.
Western blot analysis
The lysates were prepared by pulverizing the snap‐frozen tissues in liquid nitrogen and then homogenizing them in RIPA buffer (150 mM NaCl; 1% v/v Igepal CA‐630; 50 mM Tris–Cl, pH 8.3; 0.5% w/v sodium deoxycholate; and 0.1% w/v sodium dodecyl sulfate), with protease inhibitors added (complete ULTRA mini tablet, Roche, 04693116001), on ice using a “tight” glass dounce homogenizer. The protein concentrations were determined using a BCA Protein Assay kit (Pierce, 23225). For miPEP31, the samples were separated on a 16.5% tricine buffered polyacrylamide gel (BioRad, 161‐1180). The samples for other experiments were separated on Any kD™ tris‐glycine buffered polyacrylamide gels (BioRad, 456‐1083). The transfer was performed in phosphate buffer for 1 h at 200 mA, and the membrane was incubated for 45 min at room temperature in 0.2% (v/v) glutaraldehyde. The membranes were blocked for 1 h at room temperature in 5% w/v nonfat dry milk in TBST. Primary antibody hybridization was carried out overnight at 4°C with the following antibodies: miPEP31 (GL Biochem), 1:1,000; total GFP (GF28R; Invitrogen, MA5‐15256‐BTIN), 1:1,000; GAPDH (D4c6R; Cell Signaling Technology, 97166), 1:10,000; cytochrome c (136F3; Cell Signaling Technology, 4280), 1:1,000, and histone H2A (L88A6; Cell Signaling Technology, 3636), 1:1,000. The blots were washed five times for 5 min each in TBST and were then incubated with HRP‐conjugated secondary antibodies (BioRad, 1706516; 1721011) at 1:20,000. The signal was detected with the Pierce ECL Western Blotting Substrate (Thermo Scientific, 32106) and the GE ImageQuant LAS 4000 (GE Healthcare).
Peptide assays
The peptides were synthesized by ChinaPeptides® and were dissolved at 10 mM in sterile water. The molecular weight was confirmed by a mass spectrometer, and the purity was over 95%.
Peptide sequences
miPEP31: MRDWASVSSLGSGLWKERLWKSITTKRDGIAPVTRNWRGGKMLA; scrambled miPEP31: GWRTKDWSISPLKAVLGWIRTSGGDTRMNKLARLAKESWVRSMG; and NrTP: YKQCHKKGGKKGSGGLRKRLRKFRNK.
Antibody production
A custom polyclonal antibody was derived against miPEP31 by GL Biochem. Rabbits were immunized with a synthetic miPEP31 conjugated with KLH. Sera were collected and affinity purified against the peptide immunogen.
Mass spectrometry analysis
HCT 116 cells (1‐2 × 108) were lysed and subjected to anti‐miPEP31 immunoprecipitation. The purified protein lysates were eluted by 0.2 M glycine (pH = 2.6), then subjected to Amicon ultra centrifugal filters to segregate ingredients ≤ 10 kD and > 3 kD. The segregated peptides were treated with 1% trifluoroacetic acid (TFA), purified using the C18 Ziptips, and eluted with 0.1% TFA in 50–70% acetonitrile. All mass spectrometric experiments were performed on a QE‐Plus mass spectrometer connected to an Easy‐nLC 2000 via an Easy Spray (ThermoFisher Scientific). All MS/MS ion spectra were analyzed using PEAKS 8.0 (Bioinformatics Solutions) for processing, de novo sequencing, and database searching. Resulting sequences were searched through the UniProt Proteome database (downloaded on May 5th, 2018) with mass error tolerance set at 10 ppm and 0.02 Da for parent and fragment, respectively. FDR estimation was enabled. Peptides were filtered for log10p R 15, and the proteins were filtered for log10p R 15 plus one unique peptide. For all experiments, these settings gave an FDR of < 1% at the peptide‐spectrum match level.
Circular dichroism spectroscopy
All peptides were analyzed in 10 mM potassium phosphate buffer (pH = 7.0) with or without the addition of TFE (1:1 dilution) using a peptide concentration of 20 μM. Peptides were measured in a 0.1 cm quartz cuvette with a sensitivity of 100 mdeg in the range from 260 to 190 nm in 0.5 nm intervals. The scanning mode was continuous and a scanning speed of 50 nm/min was chosen. The results of pure buffer were subtracted from the spectra of the peptides. The ratio between the molar ellipticity at 222 nm and 207 nm was used to confirm an α‐helical structure of peptides.
T‐cell isolation and sorting
Peripheral T cells were obtained from the spleen and lymph nodes of 6‐week‐old mice. Naive CD4+ T cells (CD4+CD25−CD62Lhigh) were sorted by FACSAria III (BD Biosciences) after enrichment of the CD4+ T cells by the mouse CD4+ T Cell Isolation kit (Miltenyi, 130‐104‐454). For the isolation of the naive CD4+ T cells and the Treg cells from the FoxP3EGFP mouse, CD4+CD25−EGFP− CD62Lhigh cells and CD4+CD25+EGFP+ cells were isolated, respectively. The cell purity was over 94% as determined by flow cytometry. CNS‐infiltrating mononuclear cells from EAE mice were prepared by Percoll (GE Healthcare, 17‐5445‐01) gradient separation.
In vitro CD4+ T‐cell differentiation
All cultures of T cells used RPMI‐1640 medium (Gibco) supplemented with 10% heat‐inactivated fetal bovine serum (Gibco), 2 mM L‐glutamine (Gibco), 100 IU ml−1 penicillin, 100 mg ml−1 streptomycin, 10 mM HEPES (Gibco), and 5 mM b‐mercaptoethanol (Gibco). The naive CD4+CD25−FoxP3gfp−CD62Lhi T cells were activated with plate‐bound anti‐CD3 (5 mg ml−1; 145‐2C11; BD Biosciences) plus soluble anti‐CD28 (2 mg ml−1; 37.51; BD Biosciences). Th1‐cell differentiation conditions included 10 ng ml−1 rmIL‐12 (R&D Systems) and 10 mg ml−1 anti‐IL‐4 (11B11; Biolegend). The Th17‐cell differentiation conditions included 20 ng ml−1 rmIL‐6 (R&D Systems), 3 ng ml−1 rmTGF‐b1 (R&D Systems), 10 mg ml−1 anti‐IL‐4 (11B11; Biolegend), and 10 mg ml−1 anti‐IFN‐g (XMG1.2; eBioscience). The Treg‐cell differentiation conditions included 5 ng ml−1 rmTGF‐b1 (R&D Systems) and 10 ng ml−1 rmIL‐2 (R&D Systems).
Mice
C57BL/6J mice (stock number: 000664), B6.Cg‐FoxP3tm2Tch/J mice (stock number: 006772, designated FoxP3EGFP), and CD4Cre mice (stock number: 017336) were purchased from The Jackson Laboratory (Bar Harbor, ME). The mice were kept under specific pathogen‐free conditions in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals with the approval (SYXK‐2003‐0026) of the Scientific Investigation Board of Shanghai Jiao Tong University School of Medicine, Shanghai, China. To ameliorate any suffering in the mice observed throughout these experimental studies, the mice were euthanized by CO2 inhalation.
Generation of miR‐31fl/fl, miR‐31−/−, and miR‐31fl/flCD4Cre mice
The miR‐31 locus (mmu‐mir31 ENSMUSG00000065408, http://www.ensembl.org/index.html) is on chromosome 4 (Mus musculus) and encodes miR‐31. To create loxP‐miR‐31‐loxP mice, a targeting vector was designed and a frt‐flanked PGK‐neo cassette and a loxP site upstream of miR‐31 were inserted, and a second loxP site was downstream. The loxP site is a 34 bp length DNA sequence that is recognized by Cre recombinase catalyzes. If two loxP sites are introduced in the same orientation into a genomic locus, the expression of Cre results in the deletion of the loxP‐flanked DNA sequence. After linearization, the vector was electroporated into 129S6‐derived embryonic stem (ES) cells. The collected ES cells were screened with 300 mg ml−1 G418 and 2 mM Gan C for 8 days and ascertained by PCR. The ES cells with the correct homologous recombination were injected into the blastocyst. After birth, the chimeric mice were bred with 129S mice to generate the heterozygotes. At this point, the mutant mice were bred with Flp recombinase‐expressing mice to remove the frt‐flanked neo cassette. The resulting loxP‐miR‐31‐loxP mice were backcrossed into a C57BL/6J background for eight generations and were bred with CreER or CD4Cre transgenic mice to get miR‐31−/− or miR‐31fl/flCD4Cre mice. P1 and P2 were used to genotype the miR‐31 floxed allele (1,195 bp) and the miR‐31 deleted allele (474 bp). P1, 5′‐TTTAAGGGCTCATGGAGCAA‐3′; and P2, 5′‐TGAGGACTTGCAAACGTCAG‐3′. The excision by CD4Cre was complete for all the pups used in the experiments. These mice were further crossed with FoxP3EGFP mice to generate mice that expressed enhanced green fluorescent protein (EGFP) in their Treg cells.
Analysis of Ribo‐seq datasets from SRA
Sequence Read Archive (SRA) Toolkit was used to download Ribo‐seq fastq files from SRA. The downloaded files were then applied for quality control and adapter trimming to fastq files using Trim Galore version 0.6.0. Sequences were further aligned to the Ensembl GRCm38.p6 genome using STAR version 2.7.0, and a gene annotation file from Ensembl. Ribo‐seq data of NIH 3T3 cells (GSM2778762) were analyzed. Data visualization was performed by the Integrative Genomics Viewer (IGV).
Induction of EAE
EAE was induced by complete Freund's adjuvant (CFA)‐MOG35–55 peptide immunization (China Peptides Biotechnology) and was scored daily. Briefly, C57BL/6J mice were injected subcutaneously into the base of the tail with a volume of 200 μl containing 300 μg MOG35‐55 peptide emulsified in CFA (Sigma‐Aldrich, F5881). The mice were also injected intravenously with 200 μl of pertussis toxin (Merck‐Calbiochem, 516562) on days 0 and 2 postimmunization. All the reagents used for the in vivo experiments were free of endotoxin. The mice were monitored daily for the development of disease, which was scored according to the following scale: 0, no symptoms; 0.5, partially limp tail; 1, completely limp tail; 1.5, impaired righting reflex; 2, hind limb paresis; 2.5, hind‐limb paralysis; 3, forelimb weakness; 4, complete paralysis; and 5, moribund or death. For some experiments, 50 μg of miPEP31 or scrambled miPEP31, dissolved at 0.5 mg ml−1 in water, was intravenously injected on day 8 postimmunization and every 3 days after that. Inflammation and demyelination scores were calculated as previously described. Briefly, spinal cords stained with H&E were used to score inflammation (0, no inflammatory cells; 1, a few scattered inflammatory cells; 2, organization of inflammatory infiltrates around blood vessels; and 3, extensive perivascular cuffing with extension into adjacent parenchyma, or parenchymal infiltration). Spinal cords stained with luxol fast blue were used for demyelination scoring (0, no demyelination; 1, a few scattered naked axons; 2, small groups of naked axons; 3, large groups of naked axons; and 4, confluent foci of demyelination). The scores were ascribed by one well‐trained, board‐certified histopathologist blinded to the groups of mice.
ELISA
Cell lysates or diluted serum were coated in 96‐well plates overnight. Then, the plates were washed for three times with wash buffer, followed with incubation of block buffer for 1 h. Anti‐miPEP31 was added to the plates and incubated for 1 h at room temperature. After washing for three times, the plates were incubated with HRP‐conjugated anti‐rabbit antibody for 1 h at room temperature. Substrate was added into plates after washing for three times and plates were read on an ELISA plate reader. Different concentrations of synthetic miPEP31 were used to make the standard curves. For detection of the concentration of miPEP31 in lysate from cells, tissue, or nucleus, the concentration of protein was normalized to 1 μg per μl.
Immunohistochemistry
For all staining, tissues were routinely processed, and antigen retrieval was performed using citrate buffer (pH = 6) or Tris EDTA buffer (pH = 9), using the Decloaking Chamber from Biocare. After exhaustion of endogenous peroxidase with hydrogen peroxide, slides were blocked with Protein Block Solution (Dako) for 10 min at room temperature and incubated with primary antibody overnight at 4°C. Anti‐CD3 (Abcam, Ab16669, 1:800) was used for the staining.
In vitro Treg‐cell suppression assay
Treg cells were isolated from the spleen of the mice by sorting with flow cytometry based on cell surface markers (CD4+, CD25+, and FoxP3EGFP+). Naive CD4+ T cells were isolated from the spleen of the mice by sorting with flow cytometry based on cell surface markers (CD4+, CD25−, CD62Lhi, and FoxP3EGFP−). Memory Tconv cells were isolated from the spleen of the mice by sorting with flow cytometry based on cell surface markers (CD4+, CD25−, CD45Rblow, and FoxP3EGFP −). Splenocytes from RAG2−/− mice lacking mature T and B lymphocytes were used as the antigen presenting cells. The purified naive CD4+ T cells were labeled for 15 min at 37°C with 10 mM CTV (Life Technologies, C34557), and the CTV‐labeled T cells (1 × 105) were cultured in 96‐well plates for 72 h together with an increasing ratio of sorted Treg cells in the presence of anti‐CD3 (1 mg ml−1) plus γ‐irradiated antigen‐presenting cells (1 × 105). The suppressive function of the Treg cells was determined by measuring the proliferation of the activated CD4+ effector T cells on the basis of the CTV dilution.
Chromatin immunoprecipitation assay
ChIP assays were performed using the SimpleChIP Enzymatic Chromatin Immunoprecipitation kit (Cell Signaling Technology, 9003) according to the manufacturer's protocol with minor modifications. In brief, the cells transfected with the miPEP31‐myc or control plasmid were collected and cross‐linked with 1% (v/v) formaldehyde for 10 min at room temperature. Subsequently, the nuclei were isolated by lysing the cytoplasmic fraction, and the chromatin was digested into fragments of 150–900 bp by a micrococcal nuclease (400 gel units) for 20 min at 37°C, followed by ultrasonic disruption of the nuclear membrane using a standard microtip and a Branson W250D Sonifier (four pulses, 60% amplitude, and duty cycle 40%). The sonicated nuclear fractions were divided for input control and were incubated overnight at 4°C with 5 µg of either an anti‐Myc antibody (9B11; Cell Signaling Technology), anti‐Histone H3K27ac (acetyl Lys27) antibody (GeneTex, GTX128944), or the negative control IgG (Cell Signaling Technology) or anti‐H3 (D2B12; Cell Signaling Technology, 4620) as a positive control. After incubation with 30 μl of ChIP grade protein G‐agarose beads for 2 h at 4°C, the antibody–protein–DNA complexes were then eluted from the beads and digested with proteinase K (40 mg ml−1) for 2 h at 65°C, followed by spin column‐based purification of the DNA. Finally, the genomic DNA recovered from the ChIP assays was qPCR amplified with primers specific to the miR‐31 promoter region. Three pairs of primers were used for the detection of miR‐31 promoter. Sequences were as follows: P1 forward, CTGGCCTGCTGGTCAAGAAT and P1 reverse, AAGGTTCGCGACAGTCACC; P2 forward, CATGTCTCTGCTTGTACGCT and P2 reverse, AGCGTACAAGCAGAGACATG; P3 forward, ATCAACCTTGAGAGTTTTCAG and P3 reverse, TATCCATAGAAAACCCAAAA. The specificity of each primer set was verified by analyzing the dissociation curve of each gene‐specific PCR product.
Luciferase assays
NIH 3T3 cells were maintained in DMEM (HyClone, SH30022.01) supplemented with 10% fetal bovine serum (HyClone, SH30088.02), 2 mM glutamine, 100 IU ml−1 penicillin, and 0.1 mg ml−1 streptomycin. NIH 3T3 cells were seeded into 24‐well plates (1 × 105 cells per well) 1 day before transfection, and then, each well was transfected with a mixture of 100 ng pGL3‐promoter luciferase reporter vector and 10 ng Renilla TK. Twenty‐four hours posttransfection, the cells were lysed. Then, the luciferase activity was measured using the Dual‐Luciferase Reporter Assay System (Promega, E1910) and a Lumat3 LB 9508 Single Tube Luminometer instrument (Berthold Technologies). Each experiment was performed in triplicate. The ratio of Firefly luciferase to Renilla luciferase was calculated for each well.
Human CD4+ T‐cell differentiation and detection
CD4+ T cells were isolated from peripheral blood mononuclear cells and differentiated in polarizating medium. For the Tre‐cell differentiation, the cells were cultured in the presence of 2 μg ml−1 plate‐bound anti‐human CD3 (OKT3, eBioscience, 16‐0037‐81), 2 μg ml−1 anti‐human CD28 (CD28.2, eBioscience, 16‐0289‐81), and 10 ng ml−1 TGF‐β (eBioscience, PHG9202). Anti‐human CD4 (OKT4, Biolegend, 317420) and anti‐FoxP3 (PCH101, eBioscience, 12‐4776‐42) were used to detect the differentiation of the Treg cells by flow cytometry. Ethical approval was obtained from the Shanghai Jiaotong University School of Medicine Ethics Review Board.
Nuclear extraction and EMSA
Nuclear extracts were prepared from NIH 3T3 cells using the NE‐PER Nuclear and Cytoplasmic Extraction kit (Thermo Fisher Scientific, 78835) according to the manufacturer's instructions. The gel shift assays were performed with the LightShift Chemiluminescent EMSA kit (Thermo Fisher Scientific, 20148). Complementary biotinylated oligonucleotides, consisting of the P2‐c promoter region or P2‐c with mutation (seen in Table EV1), were annealed. The respective labeled oligonucleotides were added to a 20 µl reaction mix consisting of 1–3 µg of the nuclear extracts or 20–200 pmol miPEP31, DNA‐binding buffer, poly (dI‐dC), 1% NP‐40, and MgCl2 at concentrations based on the manufacturer's recommendations. For the competition assays, unlabeled oligonucleotides were allowed to bind the nuclear extracts or miPEP31 (30 min at room temperature) before the addition of the labeled probes. Anti‐miPEP31 was added to perform super shift reaction. The reactions were incubated for an additional 20 min with the respective labeled probes, followed by loading onto a 6% native DNA polyacrylamide gel. The gel was prerun and run with 0.5% Tris‐Borate‐EDTA and processed according to the manufacturer's instruction.
Peptide structure prediction and molecular docking
The secondary structure and 3D model were predicted by I‐TASSER server (https://zhanglab.ccmb.med.umich.edu/I‐TASSER/). FASTA format of miPEP31 was submitted to the I‐TASSER server. Secondary structure, solvent accessibility, normalized B‐factor, and top 5 final models were predicted by I‐TASSER. Higher score means more confident prediction of secondary structure. The job ID was S474608. Molecular docking of miPEP31 and DNA was performed using the AutoDock 4.2 program.
Statistical analysis
The data were analyzed with GraphPad Prism 8 and are presented as the mean ± SEM. Student's t‐test was used when two conditions were compared, and analysis of variance (ANOVA) with Bonferroni or Newman–Keuls correction was used for multiple comparisons. Probability values of < 0.05 were considered significant; two‐sided Student's t‐tests or ANOVA was performed. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant. Error bars depict SEM.
Author contributions
Hong Zhou: Resources; Data curation; Software; Formal analysis; Investigation; Methodology; Writing—original draf; Project administration. Fangzhou Lou: Data curation; Methodology. Jing Bai: Formal analysis. Yang Sun: Methodology. Wei Cai: Methodology. Libo Sun: Formal analysis. Zhenyao Xu: Methodology. Zhaoyuan Liu: Methodology. Lingyun Zhang: Methodology. Qianqian Yin: Data curation. Junxun Zhang: Formal analysis. Yuanyuan Gao: Methodology. Zhikai Wang: Methodology. Liman Niu: Methodology. Xiaojie Cai: Data curation. Siyu DENG: Methodology. Hong Wang: Formal analysis. Li Xia: Data curation; Formal analysis. Florent Ginhoux: Writing—review & editing. Qun Li: Conceptualization; Funding acquisition; Writing—review & editing. Honglin Wang: Conceptualization; Supervision; Funding acquisition; Investigation; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
HZ and HW designed the research and analyzed the data. HW supervised the research and wrote the manuscript. HZ conducted most of the experiments. WC helped with the EAE and colitis experiments. FL helped with in vitro Treg‐cell suppression assay. YS offered help western blot of miPEP31in tissues. YG, WC, LS, JZ, and QY helped with cell isolation and flow cytometry. JB, LZ, ZL, ZX, ZW, DS, and HW helped with the experimental details. LX conducted the MS experiments. FG, QL, LN, XC, JB, and LZ helped with data discussion.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Source Data for Figure 1
Source Data for Figure 3
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81930088, 82050009, 82070509, 82101909, 81725018, 82073428, 81901593, and 82103719), MOST | National Key Research and Development Program of China Stem Cell and Translational Research (2020YFA0112900), Clinical Research Plan of Shanghai Shenkang Hospital Development Center (SHDC2020CR3061B), the Nature Science Foundation of Shanghai Science and Technology Committee (20ZR1447400), SJTU Trans‐med Awards Research (20210102), Integrated innovation fund of Shanghai Jiaotong University (2021JCPT04), and Innovative Research Team of High‐Level Local Universities in Shanghai.
EMBO reports (2022) 23: e53475.
See also: C Dozier & S Plaza (May 2022)
Contributor Information
Qun Li, Email: liqun@sibs.ac.cn.
Honglin Wang, Email: honglin.wang@sjtu.edu.cn.
Data availability
The sequence of pri‐miR‐31 was deposited in NCBI Gene (accessions: NR_132734.1). Mass spectrometry data: Proteomics Identifications Database PXD029131. The deposited data can be achieved by the link: https://www.ebi.ac.uk/pride/archive/projects/PXD029131. All data are available from the corresponding author upon reasonable request.
References
- Anderson D, Anderson K, Chang C‐L, Makarewich C, Nelson B, McAnally J, Kasaragod P, Shelton J, Liou J, Bassel‐Duby R et al (2015) A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160: 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SJ, Rothnagel JA (2014) Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet 15: 193–204 [DOI] [PubMed] [Google Scholar]
- Angell Y, Holford M, Moos WH (2018) Building on success: a bright future for peptide therapeutics. Protein Pept Lett 25: 1044–1050 [DOI] [PubMed] [Google Scholar]
- Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, Obermayer B, Fleming ES, Vejnar CE, Lee MT, Rajewsky N, Walther TC et al (2014) Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J 33: 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone F, De Rosa V, Carrieri PB, Montella S, Bruzzese D, Porcellini A, Procaccini C, La Cava A, Matarese G (2014) Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat Med 20: 69–74 [DOI] [PubMed] [Google Scholar]
- Chang TC, Pertea M, Lee S, Salzberg SL, Mendell JT (2015) Genome‐wide annotation of microRNA primary transcript structures reveals novel regulatory mechanisms. Genome Res 25: 1401–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q‐J, Deng B‐H, Gao J, Zhao Z‐Y, Chen Z‐L, Song S‐R, Wang L, Zhao L‐P, Xu W‐P, Zhang C‐X et al (2020) A miRNA‐encoded small peptide, vvi‐miPEP171d1, regulates adventitious root formation. Plant Physiol 183: 656–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G et al (2005) Transcriptional maps of 10 human chromosomes at 5‐nucleotide resolution. Science (New York, NY) 308: 1149–1154 [DOI] [PubMed] [Google Scholar]
- Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Côté J et al (2011) Role for miR‐204 in human pulmonary arterial hypertension. J Exp Med 208: 535–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F et al (2012) Landscape of transcription in human cells. Nature 489: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du QS, Xie NZ, Huang RB (2015) Recent development of peptide drugs and advance on theory and methodology of peptide inhibitor design. Med Chem 11: 235–247. [DOI] [PubMed] [Google Scholar]
- Erak M, Bellmann‐Sickert K, Els‐Heindl S, Beck‐Sickinger AG (2018) Peptide chemistry toolbox ‐ transforming natural peptides into peptide therapeutics. Bioorg Med Chem 26: 2759–2765 [DOI] [PubMed] [Google Scholar]
- Fosgerau K, Hoffmann T (2015) Peptide therapeutics: current status and future directions. Drug Discovery Today 20: 122–128 [DOI] [PubMed] [Google Scholar]
- Galindo MI, Pueyo JI, Fouix S, Bishop SA, Couso JP (2007) Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol 5: e106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Coller J (2013) RNA in unexpected places: long non‐coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol 14: 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Rinn JL (2012) Modular regulatory principles of large non‐coding RNAs. Nature 482: 339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Zhang X, Borevitz JO, Li WH, Shiu SH (2007) A large number of novel coding small open reading frames in the intergenic regions of the Arabidopsis thaliana genome are transcribed and/or under purifying selection. Genome Res 17: 632–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanyu‐Nakamura K, Sonobe‐Nojima H, Tanigawa A, Lasko P, Nakamura A (2008) Drosophila Pgc protein inhibits P‐TEFb recruitment to chromatin in primordial germ cells. Nature 451: 730–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J‐Z, Chen M, Chen DE, Gao X‐C, Zhu S, Huang H, Hu M, Zhu H, Yan G‐R (2017) A peptide encoded by a putative lncRNA HOXB‐AS3 suppresses colon cancer growth. Mol Cell 68: 171–184.e176 [DOI] [PubMed] [Google Scholar]
- Huarte E, Jun S, Rynda‐Apple A (2016) Regulatory T cell dysfunction acquiesces to BTLA+ regulatory B cells subsequent to oral intervention in experimental autoimmune encephalomyelitis. J Immunol 196: 5036–5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS (2009) Genome‐wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS (2011) Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R, Kroehling L, Khitun A, Bailis W, Jarret A, York AG, Khan OM, Brewer JR, Skadow MH, Duizer C et al (2018) The translation of non‐canonical open reading frames controls mucosal immunity. Nature 564: 434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Li C, Olive V, Lykken E, Feng F, Sevilla J, Wan Y, He L, Li QJ (2011) Molecular dissection of the miR‐17‐92 cluster's critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood 118: 5487–5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnnidis JB, Harris MH, Wheeler RT, Stehling‐Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, Camargo FD (2008) Regulation of progenitor cell proliferation and granulocyte function by microRNA‐223. Nature 451: 1125–1129 [DOI] [PubMed] [Google Scholar]
- Kaspar AA, Reichert JM (2013) Future directions for peptide therapeutics development. Drug Discov Today 18: 807–817 [DOI] [PubMed] [Google Scholar]
- Kondo T, Hashimoto Y, Kato K, Inagaki S, Hayashi S, Kageyama Y (2007) Small peptide regulators of actin‐based cell morphogenesis encoded by a polycistronic mRNA. Nat Cell Biol 9: 660–665 [DOI] [PubMed] [Google Scholar]
- Lauressergues D, Couzigou JM, Clemente HS, Martinez Y, Dunand C, Becard G, Combier JP (2015) Primary transcripts of microRNAs encode regulatory peptides. Nature 520: 90–93 [DOI] [PubMed] [Google Scholar]
- Lee PW, Severin ME, Lovett‐Racke AE (2017) TGF‐beta regulation of encephalitogenic and regulatory T cells in multiple sclerosis. Eur J Immunol 47: 446–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S et al (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419 [DOI] [PubMed] [Google Scholar]
- Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY (2010) Function of miR‐146a in controlling Treg cell‐mediated regulation of Th1 responses. Cell 142: 914–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magny EG, Pueyo JI, Pearl FM, Cespedes MA, Niven JE, Bishop SA, Couso JP (2013) Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science 341: 1116–1120 [DOI] [PubMed] [Google Scholar]
- Moffett HF, Cartwright ANR, Kim H‐J, Godec J, Pyrdol J, Äijö T, Martinez GJ, Rao A, Lu J, Golub TR et al (2017) The microRNA miR‐31 inhibits CD8(+) T cell function in chronic viral infection. Nat Immunol 18: 791–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C, Bain G, van Dijk MA, Engel I, Furnari BA, Massari ME, Matthews JR, Quong MW, Rivera RR, Stuiver MH (1994) Structure and function of helix‐loop‐helix proteins. Biochim Biophys Acta 1218: 129–135 [DOI] [PubMed] [Google Scholar]
- Namkoong S, Ho A, Woo YM, Kwak H, Lee JH (2018) Systematic characterization of stress‐induced RNA granulation. Mol Cell 70: 175–187.e178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET et al (2016) A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351: 271–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L, Lou F, Sun Y, Sun L, Cai X, Liu Z, Zhou H, Wang H, Wang Z, Bai J et al (2020) A micropeptide encoded by lncRNA MIR155HG suppresses autoimmune inflammation via modulating antigen presentation. Sci Adv 6: eaaz2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaza S, Menschaert G, Payre F (2017) In search of lost small peptides. Annu Rev Cell Dev Biol 33: 391–416 [DOI] [PubMed] [Google Scholar]
- Ransohoff JD, Wei Y, Khavari PA (2018) The functions and unique features of long intergenic non‐coding RNA. Nat Rev Mol Cell Biol 19: 143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy J, Waldner H, Zhang X, Illes Z, Wucherpfennig KW, Sobel RA, Kuchroo VK (2005) Cutting edge: CD4+CD25+ regulatory T cells contribute to gender differences in susceptibility to experimental autoimmune encephalomyelitis. J Immunol 175, 5591–5595 [DOI] [PubMed] [Google Scholar]
- Rouas R, Fayyad‐Kazan H, El Zein N, Lewalle P, Rothé F, Simion A, Akl H, Mourtada M, El Rifai M, Burny A et al (2009) Human natural Treg microRNA signature: role of microRNA‐31 and microRNA‐21 in FOXP3 expression. Eur J Immunol 39: 1608–1618 [DOI] [PubMed] [Google Scholar]
- Saghatelian A, Couso JP (2015) Discovery and characterization of smORF‐encoded bioactive polypeptides. Nat Chem Biol 11: 909–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savard J, Marques‐Souza H, Aranda M, Tautz D (2006) A segmentation gene in tribolium produces a polycistronic mRNA that codes for multiple conserved peptides. Cell 126: 559–569 [DOI] [PubMed] [Google Scholar]
- Sharma A, Badola PK, Bhatia C, Sharma D, Trivedi PK (2020) Primary transcript of miR858 encodes regulatory peptide and controls flavonoid biosynthesis and development in Arabidopsis . Nat Plants 6: 1262–1274 [DOI] [PubMed] [Google Scholar]
- Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, He C, Zheng L, Hu X, Hu Z et al (2016) Suppression of enhancer overactivation by a RACK7‐histone demethylase complex. Cell 165: 331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The EPC (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, She Z, Gao X, Wang W, Tian H (2020) MicroRNA‐31 regulates dental epithelial cell proliferation by targeting Satb2. Biochem Biophys Res Comm 532: 321–328 [DOI] [PubMed] [Google Scholar]
- de la Torre BG, Albericio F (2018) The pharmaceutical industry in 2017. An analysis of FDA drug approvals from the perspective of molecules. Molecules 23: 533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viglietta V, Baecher‐Allan C, Weiner HL, Hafler DA (2004) Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 199: 971–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M (2010) Synthetic therapeutic peptides: science and market. Drug Discovery Today 15: 40–56 [DOI] [PubMed] [Google Scholar]
- Washietl S, Pedersen JS, Korbel JO, Stocsits C, Gruber AR, Hackermüller J, Hertel J, Lindemeyer M, Reiche K, Tanzer A et al (2007) Structured RNAs in the ENCODE selected regions of the human genome. Genome Res 17: 852–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Rajewsky K (2009) MicroRNA control in the immune system: basic principles. Cell 136: 26–36 [DOI] [PubMed] [Google Scholar]
- Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, Galan C, Belkaid Y, Bonneau R, Littman DR (2018) c‐MAF‐dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554: 373–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Xu Z, Lou F, Zhang L, Ke F, Bai J, Liu Z, Liu J, Wang H, Zhu H et al (2015) NF‐κB‐induced microRNA‐31 promotes epidermal hyperplasia by repressing protein phosphatase 6 in psoriasis. Nat Commun 6: 7652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I‐TASSER Suite: protein structure and function prediction. Nat Methods 12: 7–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R, Poy MN, Stoffel M, Fuchs E (2008) A skin microRNA promotes differentiation by repressing 'stemness'. Nature 452: 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Eiden LE (2019) Progress in regulatory peptide research. Ann N Y Acad Sci 1455: 5–11 [DOI] [PubMed] [Google Scholar]
- Zhang L, Ke F, Liu Z, Bai J, Liu J, Yan S, Xu Z, Lou F, Wang H, Zhu H et al (2015) MicroRNA‐31 negatively regulates peripherally derived regulatory T‐cell generation by repressing retinoic acid‐inducible protein 3. Nat Commun 6: 7639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Chong MM, Littman DR (2009) Plasticity of CD4+ T cell lineage differentiation. Immunity 30: 646–655 [DOI] [PubMed] [Google Scholar]