

Abstract
Long non-coding RNAs (lnRNAs) colorectal neoplasia differentially expressed (CRNDE) has been identified as a crucial regulator involved in tongue squamous cell carcinoma (TSCC). However, the molecular mechanism of CRNDE involved in TSCC progression is still unknown. In the study, qRT-PCR assay was used to detect the expression of CRNDE in TSCC tissues and cells. CCK-8 assay, colony formation assay, transwell assay and flow cytometric analysis were performed to determine cell proliferation ability, colony formation, migration and invasion capacities, and cell apoptosis, respectively. Western blot was employed to assess the activity of the PI3K/AKT/mTOR pathway. A xenograft mice model was performed to evaluate the role of CRNDE on tumor growth in vivo. The results showed CRNDE was upregulated in TSCC tissues and cell lines. CRNDE knockdown repressed the proliferation, colony formation, migration and invasion and promoted apoptosis in TSCC cells. Moreover, CRNDE regulated the PI3K/AKT/mTOR pathway in TSCC cells. Additionally, high levels of CRNDE inhibited tumor growth in vivo. In conclusion, high levels of CRNDE might promote TSCC progression at least partly through regulating the PI3K/AKT/mTOR pathway. Targeting CRNDE has potential to be used as a novel target of TSCC treatment.
Long non-coding RNAs (lnRNAs) colorectal neoplasia differentially expressed (CRNDE) has been identified as a crucial regulator involved in tongue squamous cell carcinoma (TSCC).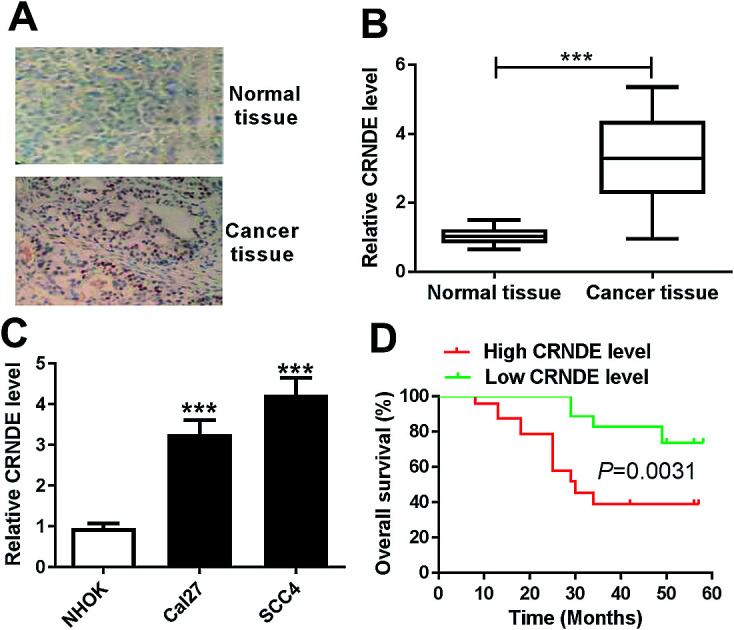
Introduction
The squamous cell carcinoma of the oral cavity (OSCC) is the most common malignancy of the head and neck and accounts for approximately 3% of all malignancies.1 Tongue squamous cell carcinoma (TSCC), one of the most common subtypes of OSCC, is well known for its high rate of proliferation and lymph nodal metastasis.2 In the USA, there are an estimated 16 400 new cases of TSCC diagnosed in 2017, leading to 2400 deaths.1 Although TSCC can be cured with appropriate treatment when diagnosed early, the 5 year overall survival rate of TSCC patients with tumor metastasis to lymph nodes is still very low.3 Therefore, identification of novel and improved markers is of importance for TSCC diagnosis and treatment.
Long non-coding RNAs (lnRNAs), a novel heterogeneous class of RNA molecule that is longer than 200 nucleotides, play important roles in the fundamental processes across every branch of life.4 By interacting with cellular macromolecules such as DNA, RNA and protein, lncRNAs can drive a series of vital tumor phenotypes.5 An increasing literature has suggested that aberrant expression of lncRNAs is involved in human cancers, highlighting current and potential future applications of lncRNAs research in the treatment of cancers.5 Some lncRNAs have been identified as a crucial regulator involved in TSCC, such as long intergenic non-coding RNA 152 (LINC00152)6 and actin filament associated protein 1 antisense RNA1 (AFAP1-AS1).7 A recent document reported that lncRNA colorectal neoplasia differentially expressed (CRNDE) was upregulated in TSCC and high CRNDE expression accelerated the proliferation, cell cycle and invasion of TSCC cells.8 However, the molecular mechanism of CRNDE involved in TSCC progression is still unknown.
In this study, our data supported that CRNDE was upregulated in TSCC tissues and cell lines. We also found that CRNDE knockdown repressed the proliferation, colony formation, migration and invasion and promoted the apoptosis in TSCC cells. Moreover, CRNDE regulated PI3K/AKT/mTOR pathway in TSCC cells. Taken together, our study suggested that CRNDE might promote TSCC progression at least partly through regulating PI3K/AKT/mTOR pathway.
Materials and methods
Tissue specimens
Between October 2011 and December 2012, 42 pairs TSCC tissues and adjacent noncancerous tongue tissues from TSCC patients who underwent curative resection without preoperative chemotherapy or radiotherapy were obtained from The First Affiliated Hospital of Jinzhou Medical University, with informed consent before surgery. All tissues were immediately frozen in liquid preservative RNAlater (Ambion, Austin, TX, USA) and kept at −80 °C until use. The latest follow-up was updated in May 2018 and the follow-up information was obtained from these patients' medical records. According to the median ration of CRNDE expression level in tumor tissues, the 42 TSCC patients were classified into two groups: high CRNDE level group (n = 21) and low CRNDE level group (n = 21). The overall survival was analyzed by Kaplan–Meier survival analysis and log-rank tests. The study was approved by the Ethical Committee of The First Affiliated Hospital of Jinzhou Medical University. This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH publication no. 85-23 rev. 1985) and was approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Jinzhou Medical University.
Cell culture, treatment and transfection
Primary normal human oral keratinocytes (NHOKs) were prepared from separated oral epithelial tissues as described previously.9 TSCC cell lines (Cal27 and SCC4) were procured from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). NHOKs were cultured in EpiLife™ serum-free medium (Gibco, Karlsruhe, Germany) containing Human Keratinocyte Growth Supplement (HKGS, Gibco) and TSCC cells were maintained in DMEM medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Life Technologies), 1% penicillin/streptomycin (Life Technologies) at 37 °C in a humidified atmosphere of 5% CO2.
To explore the role of PI3K/AKT/mTOR pathway in TSCC, Cal27 and SCC4 cells were treated with 10 μM of LY294002 (Sigma-Aldrich, St. Louis, MO, USA), a inhibitor of PI3K/AKT/mTOR pathway, for 48 h.
The CRNDE overexpression plasmid (pcDNA-CRNDE) was synthesized by GenePharma (Shanghai, China), with an empty plasmid (pcDNA) as negative control. To investigate the function of CRNDE in TSCC, cells were transfected with CRNDE-specific siRNA (si-CRNDE, GenePharma) or negative control siRNA (si-NC, GenePharma), pcDNA-CRNDE or pcDNA using Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) according to the protocol of manufacturers.
In situ hybridization (ISH)
ISH was performed in TSCC tissues using a ISH Detection kit (Dako, Glostrup, Denmark) according to the instruction of manufacturers. Formalin-fixed and paraffin-embedded specimens were cut into 4 μm thick sections and then deparaffinized and rehydrated with a graded ethanol and xylene. The sections were digested with proteinase K, and then were treated with a DIG-labeled LNA-CRNDE-based probe (Dako) for hybridization, followed by the incubation with the anti-DIG-AP antibody. After that, the sections were stained with NBT/BCIP substrates and CRNDE expression in tissues was detected based on the staining intensity using a microscope (Leica, Wetzlar, Germany).
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from tissues and cells using an RNeasy Mini kit (Qiagen, Hilden, Germany) according to the instruction of manufacturers. RNA extracts were reverse-transcribed into cDNA with a ReverTra Ace® qPCR RT Master Mix with gDNA Remover (Toyobo, Osaka, Japan). qRT-PCR was performed by using SYBR™ Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) on an Mx3000P qPCR system (Agilent Technologies, Santa Clara, CA, USA), with GAPDH as a housekeeping gene for normalization. The relative expression of CRNDE was calculated by using the 2−ΔΔCt method.
Cell proliferation assay
Cell proliferation ability was assessed with a Cell Counting Kit 8 (CCK-8, Dojindo, Kumamoto, Japan) according to the instruction of manufacturers. To be brief, at the indicated time period, 10 μl of CCK-8 solution was added into each well of 96-well plate and then incubated at 37 °C for 2 h, followed by the measurement of an absorbance at 450 nm using a microplate reader (Bio-Rad, Hercules, CA, USA).
Colony formation assay
Transfected or treated cells were seeded in cell-culture plate for colony formation assay. After 14 days of incubation, cells were stained with 0.1% crystal violet (Solarbio, Shanghai, China), and then the number of colonies was counted.
Transwell assay of cell migration and invasion
Cell migration and invasion capacities were determined by transwell assay using a 24-well transwell with 8 μm pore, 6.5 mm polycarbonate transwell filters (Corning, Cambridge, MA, USA). For migration assay, the cells resuspending in free-serum medium were seeded into the upper chamber of transwell with non-coated membrane. For invasion assay, following serum starvation, the cells were added into the upper chamber of transwell with Matrigel-coated membrane. In both assays, medium containing 10% FBS was added into the lower chamber as a chemo-attractant. After 24 h incubation, the migrated and invaded cells were fixed in 4% paraformaldehyde (Solarbio) and stained with 0.1% crystal violet. The number of migrated and invaded cells was counted in 5 different fields with a microscope (Leica).
Flow cytometric analysis
Cell apoptosis was detected by flow cytometric analysis with Annexin V-FITC/PI Apoptosis Detection Assay Kit (Dojindo). In brief, cells were resuspended in 1× Annexin V Binding Solution, followed by the incubation with Annexin V/FITC and PI. The apoptotic rate was analyzed using FACSCalibur flow cytometer (BD Biosciences, Heidelberg, Germany) with the CellQuest Pro software (BD Biosciences).
Caspase-3 activity assay
Caspase-3 activity was measured using the Caspase-3 Colorimetric Assay Kit (Abcam, Cambridge, UK) according to the standard protocols. Briefly, cell lysates were incubated with enzyme-specific substrates at 37 °C for 2 h. Then, the absorbance at 405 nm was determined using a microplate reader.
Western blot
Total protein extracts were resolved on a 10% SDS-PAGE and transferred onto a Hybond PVDF membrane (GE Healthcare, Little Chalfont, UK). Then, the membranes were probed with primary antibodies, followed by the incubation with horseradish peroxidase-conjugated secondary antibody (Abcam; dilution 1 : 10 000). The protein bands were quantified with the enhanced chemiluminescence solution (ECL, GE Healthcare) with an ImageJ software (National Institutes of Health, Bethesda, MD, USA). The primary antibodies were used: anti-MMP-9 (Abcam; dilution 1 : 1000), anti-MMP-2 (Abcam; dilution 1 : 2000), anti-Bcl-2 (Cell Signaling Technology, Danvers, MA, USA; dilution 1 : 1000), anti-cleaved caspase-3 (Cell Signaling Technology; dilution 1 : 1000), anti-Bax (Abcam; dilution 1 : 5000), anti-PI3K (Abcam; dilution 1 : 1000), anti-mTOR (Cell Signaling Technology; dilution 1 : 1000), anti-AKT (Abcam; dilution 1 : 10 000), anti-p-PI3K (Abcam; dilution 1 : 1000), anti-p-mTOR (Cell Signaling Technology; dilution 1 : 1000) and anti-p-AKT (Abcam; dilution 1 : 500). β-Actin was used as a protein control.
sh-CRNDE-transfected cells construct
Lentiviruses harbouring the sequences of CRNDE knock-out or negative control (sh-CRNDE or sh-NC) were commercially constructed by Genechem (Shanghai, China), and empty lentiviruses were used as blank control. Then, Cal27 cells were infected by lentiviruses with blank control, sh-CRNDE or sh-NC in medium containing polybrene. 72 h after infection, cells were treated with puromycin to select stable sh-CRNDE-transfected cells.
Xenograft mice assay in vivo
6–8 weeks BALB/c nude mice (male, 18–20 g) were purchased from Henan Research Center of Laboratory Animal (Zhengzhou, China) and maintained in free specific pathogen condition. Cal27 cells (approximately 1.0 × 107) stably transfected with, blank control, sh-CRNDE or sh-NC were subcutaneously injected into nude mice (n = 10). After 7 days infection, tumor volume was measured with a calliper every 7 days and was calculated by the formula: volume (mm3) = (length × width2)/2. On day 35 after cancer cell implantation, all mice were killed and the xenografts were excised. All animal experimental procedures were approved by the Animal Experimental Ethics Committee of The First Affiliated Hospital of Jinzhou Medical University and were performed according to the Guide for Care and Use of Laboratory Animals.
Statistical analysis
All data were analyzed with GraphPad Prism 7.0 software (GraphPad Software, La Jolla, CA, USA) and shown as mean ± S.E.M. Differences between two groups were compared with a Student's t-test or Mann-whitney U test. P value < 0.05 was considered significant. *P < 0.05 or **P < 0.01 or ***P < 0.001.
Results
CRNDE was upregulated in TSCC tissues and cell lines
To determine whether CRNDE was aberrantly expressed in TSCC, ISH and qRT-PCR assays were performed to detect the expression of CRNDE in TSCC tissues and adjacent noncancerous tissues. The data revealed a significant upregulation of CRNDE expression in TSCC tissues compared with noncancerous control (Fig. 1A and B). Subsequently, we assessed CRNDE expression in TSCC cells. These results presented that in comparison to normal human oral keratinocyte (NHOK), CRNDE was significantly upregulated in TSCC cells (Fig. 1C).
Fig. 1. CRNDE was upregulated in TSCC tissues and cell lines. The expression of CRNDE was detected by ISH (A) and qRT-PCR (B) in TSCC tissues and adjacent noncancerous tongue tissues. (C) CRNDE expression by qRT-PCR in TSCC cells (Cal27 and SCC4) and normal human oral keratinocyte (NHOK). (D) The correlation between CRNDE level and TSCC patients prognosis was determined by Kaplan–Meier survival analysis and log-rank tests. ***P < 0.001.

Then, to evaluate the correlation between CRNDE expression and TSCC patients prognosis, Kaplan–Meier survival analysis and log-rank tests using patient post-operative survival were carried out. The 42 TSCC patients were classified into high CRNDE level group (n = 21) and low CRNDE level group (n = 21) according to the median ration of CRNDE expression level. The Kaplan–Meier survival curves demonstrated that the patients in low CRNDE level group had a markedly higher survival rate than that in high CRNDE level group (Fig. 1D).
Knockdown of CRNDE repressed the proliferation and colony formation of TSCC cells
To investigate CRNDE involved in TSCC progression, CRNDE-specific siRNA (si-CRNDE) was transfected into Cal27 and SCC4 cells. As shown in Fig. 2A, si-CRNDE transfection resulted in about 65% reduction of CRNDE expression in Cal27 cells, and 58% reduction in SCC4 cells. Subsequent functional experiments revealed that compared with negative control, CRNDE knockdown repressed the proliferation and colony formation of Cal27 and SCC4 cells (Fig. 2B–E).
Fig. 2. CRNDE knockdown repressed the proliferation and colony formation of TSCC cells. Cal27 and SCC4 cells were transfected with si-CRNDE or si-NC, followed by the detection of CRNDE expression by qRT-PCR (A), cell proliferation ability by CCK-8 assay (B) and (C), cell colony formation by colony formation assay (D) and (E). **P < 0.01 or ***P < 0.001.

Knockdown of CRNDE repressed the migration, invasion and related proteins expression of TSCC cells
Next, we observed the role of CRNDE on the migration and invasion of TSCC cells by si-CRNDE transfection. Transwell assay demonstrated that transfection of si-CRNDE, but not a control sequence, inhibited the migration and invasion of Cal27 and SCC4 cells (Fig. 3A–D). Matrix metalloproteinase (MMP)-2 and MMP-9 were manifested to be involved in tumor cells migration and invasion processes.10,11 As expected, our data showed that si-CRNDE transfection led to a decrease of MMP-2 and MMP-9 levels in Cal27 and SCC4 cells in comparison to negative control (Fig. 3E and F), supporting the inhibitory role of CRNDE knockdown on TSCC cells migration and invasion.
Fig. 3. CRNDE knockdown repressed the migration, invasion and related proteins expression of TSCC cells. Cal27 and SCC4 cells were transfected with si-NC or si-CRNDE, followed by the determination of cell migration and invasion abilities by transwell assay (A)–(D), MMP-2 and MMP-9 levels by Western blot (E) and (F). *P < 0.05 or **P < 0.01 or ***P < 0.001.

Knockdown of CRNDE regulated apoptosis and apoptosis-related proteins expression of TSCC cells
Further, we validated how CRNDE influenced the apoptosis of TSCC cells in si-CRNDE-transfected Cal27 and SCC4 cells. Flow cytometric analysis presented that the apoptotic rate of si-CRNDE-transfected Cal27 and SCC cells was higher than that of control (Fig. 4A and B). Caspase-3 activity assay revealed that CRNDE knockdown resulted in an increase of caspase-3 activity in Cal27 and SCC4 cells (Fig. 4C). Likewise, in comparison to control, CRNDE knockdown led to an increase of cleaved caspase-3 and Bax levels, as well as a decrease of Bcl-2 level in Cal27 and SCC4 cells (Fig. 4D and E). All these results suggested that CRNDE knockdown might promote TSCC cells apoptosis.
Fig. 4. Knockdown of CRNDE regulated apoptosis and apoptosis-related proteins expression of TSCC cells. Flow cytometric analysis for cell apoptosis (A) and (B), caspase-3 activity assay for caspase-3 activity (C), Western blot for Bcl-2, cleaved caspase-3 and Bax levels (D) and (E) in si-NC- or si-CRNDE-transfected Cal27 and SCC cells. **P < 0.01 or ***P < 0.001.

CRNDE regulated PI3K/AKT/mTOR pathway in TSCC cells
PI3K/AKT pathway was demonstrated to be involved in CRNDE-mediated pro-tumor effect in some cancers.12 Moreover, we confirmed that the inactivation of PI3K/AKT/mTOR pathway inhibited the proliferation, migration and invasion, and promoted apoptosis in TSCC cells (ESI Fig. 1†). As a result, we further explored the effect of CRNDE on PI3K/AKT/mTOR pathway in TSCC cells. These data revealed that transfection of pcDNA-CRNDE, but not pcDNA control, induced the activation of PI3K/AKT/mTOR pathway in Cal27 and SCC cells (Fig. 5A–D). In addition, si-CRNDE transfection in Cal27 and SCC cells blockaded PI3K/AKT/mTOR pathway (Fig. 5A–D).
Fig. 5. CRNDE regulated PI3K/AKT/mTOR pathway in TSCC cells. The expression levels of PI3K, mTOR, AKT, p-PI3K, p-mTOR and p-AKT were measured by Western blot in pcDNA-, pcDNA-CRNDE-, si-NC- or si-CRNDE-transfected Cal27 cells (A) and (B), and SCC4 cells (C) and (D). **P < 0.01 or ***P < 0.001.

Knockdown of CRNDE inhibited tumor growth in vivo
Given our data in vitro, we further evaluated the role of CRNDE on tumor growth in vivo. These results indicated that sh-CRNDE introduction significantly retarded tumor growth, presenting by the decrease of tumor volume and weight (Fig. 6A and B). Subsequently, qRT-PCR assay confirmed that sh-CRNDE transfection markedly inhibited CRNDE expression in xenograft tumor tissues (Fig. 6C). Western blot results revealed that CRNDE knockdown remarkably blockaded PI3K/AKT/mTOR pathway in xenograft tissues (Fig. 6D and E).
Fig. 6. Knockdown of CRNDE in xenograft tumor in vivo. Cal27 cells (1.0 × 107) stably transfected with blank control, sh-CRNDE or sh-NC were subcutaneously implanted into nude mice (n = 10). On day 35 after cancer cell implantation, all mice were killed. (A) Tumor volume was measured by a calliper every week after implantation for 7 days. (B) Tumor weight was calculated. (C) CRNDE expression was detected by qRT-PCR in xenograft tissues. (D) and (E) The expression levels of PI3K, mTOR, AKT, p-PI3K, p-mTOR and p-AKT were analyzed by Western blot in excised tumor tissues. *P < 0.05 or **P < 0.01 or ***P < 0.001.

Discussion
Over the past 10 years, lncRNAs have been demonstrated to be involved in the tumorigenesis and progression of human cancers, including TSCC. For example, Xiong et al.13 found that transcribed ultraconserved RNA 338 (TUC338) was upregulated in TSCC and it overexpression repressed cell growth and enhanced apoptosis of TSCC cells in vivo. Li and colleague14 manifested that taurine-upregulated gene 1 (TUG1) silencing inhibited TSCC cells proliferation and cell cycle progression. These literatures indicated some lncRNAs as potential oncogenes in TSCC.13,14 On the other hand, some lncRNAs were identified as tumor-suppressor in TSCC, for instance, tissue differentiation-inducing non-protein coding RNA (TINCR).15
CRNDE, located on the chromosome 16, was initially identified as a lncRNA whose expression was upregulated in colorectal cancer.16 CRNDE transcripts were thought to have promise as cancer biomarkers, potentially exhibiting high sensitivity and specificity for colorectal adenomas and cancers.16 Recent studies have identified CRNDE as oncomiR involved in carcinogenesis, such as non-small cell lung cancer,17 hepatocellular carcinoma,18 glioma19 and bladder cancer.20 Moreover, high CRNDE expression was demonstrated to promote cell proliferation, cell cycle and cell invasion in TSCC cell lines by negatively regulating miR-384.8 In the present study, we verified that CRNDE was upregulated in TSCC tissues and cell lines, in accordance with previous result.8 We also conformed that high CRNDE expression was associated with poor prognosis of TSCC patients. Furthermore, we found that CRNDE knockdown repressed the proliferation, colony formation, migration and invasion while enhanced the apoptosis in TSCC cells in vitro, in accordance with former work.21 Further, we evaluated the role of CRNDE on tumor growth by xenograft tumor model. Also, our data indicated that CRNDE knockdown inhibited tumor growth in vivo.
PI3K/AKT/mTOR signaling, one of the most important intracellular pathways, plays a crucial role in tumorigenesis and development of human cancers. PI3K/AKT/mTOR pathway is frequently activated in various cancers and regulates cell proliferation, differentiation and cell metabolism, leading to cancer cell survival and apoptosis.22 Previous literatures manifested that the activation of PI3K/AKT/mTOR pathway was capable of carcinogenesis in a large number of cancers, for instance, breast cancer,23 non-small cell lung cancer24 and prostate cancer.25 Additionally, targeting PI3K/AKT/mTOR pathway was generally proposed as anticancer therapy.26 Moreover, PI3K/AKT/mTOR pathway was reported to be the most frequently mutated oncogenic pathway in squamous cell carcinomas of the head and neck, suggesting targeting this pathway as a rational treatment approach.27 PI3K/AKT/mTOR pathway also was elucidated to participate in the regulation network of plumbagin on human TSCC cells.28 In this study, we manifested that the inactivation of PI3K/AKT/mTOR signaling inhibited the proliferation, colony formation, migration and invasion while promoted apoptosis in TSCC cells (ESI Fig. 1†).
Furthermore, in the present study, we found that CRNDE could regulate PI3K/AKT/mTOR pathway in TSCC cells in vitro and in vivo. The interrelation between CRNDE and PI3K/AKT/mTOR pathway had been identified in central metabolism.29 Similar with our findings, CRNDE regulated PI3K/AKT pathway, and thus exerted oncogenic function in cervical cancer.12 CRNDE promoted the progression of gastric cancer through regulating PI3K/AKT pathway.30 Moreover, CRNDE activated PI3K/AKT signaling to promote tumor cell proliferation and growth in non-small cell lung cancer, highlighting it role as a potential novel therapeutic target for this disease.31 Additionally, CRNDE was manifested to promote the progression of colorectal cancer cell through activating Ras/MAPK signaling pathway or epigenetically silencing DUSP5/CDKN1A expression.32,33 Besides, CRNDE was demonstrated to be correlated with insulin/IGF signaling in colorectal cancer cells.29
In conclusion, our study suggested that high level of CRNDE might promote TSCC progression at least partly through regulating PI3K/AKT/mTOR pathway. Targeting CRNDE is hopeful to be used as an novel target of TSCC treatment.
Ethical statement
42 pair TSCC tissues and adjacent non-cancer tongue tissues from TSCC patients who underwent curative resection without preoperative chemotherapy or radiotherapy were obtained from The First Affiliated Hospital of Jinzhou Medical University, with informed consent before surgery. The study was approved by the Ethical Committee of The First Affiliated Hospital of Jinzhou Medical University. All animal experimental procedures were approved by the Animal Experimental Ethics Committee of The First Affiliated Hospital of Jinzhou Medical University and were performed according to the Guide for Care and Use of Laboratory Animals.
Conflicts of interest
No conflicts of interest is declared by all co-authors.
Supplementary Material
Acknowledgments
This study was supported by the Department of Science and Technology of Liaoning Province (Grant no. 20170540347).
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01321k
References
- Siegel R. L. Miller K. D. Jemal A. Ca-Cancer J. Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Chi A. C. Day T. A. Neville B. W. Ca-Cancer J. Clin. 2015;65:401–421. doi: 10.3322/caac.21293. [DOI] [PubMed] [Google Scholar]
- Marur S. Forastiere A. A. Head and neck squamous cell carcinoma: update on epidemiology, diagnosis, and treatment. Mayo Clin. Proc. 2016;91:386–396. doi: 10.1016/j.mayocp.2015.12.017. [DOI] [PubMed] [Google Scholar]
- Fatica A. Bozzoni I. Nat. Rev. Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- Spizzo R. Almeida M. I. Colombatti A. Calin G. A. Oncogene. 2012;31:4577–4587. doi: 10.1038/onc.2011.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J. Liu Y. Guo C. Zhang S. Gong Z. Tang Y. Yang L. He Y. Lian Y. Li X. Deng H. Liao Q. Li X. Li Y. Li G. et al. . J. Cancer. 2017;8:523–530. doi: 10.7150/jca.17510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. Y. Hu M. Dai M. H. Xiong J. Zhang S. Wu H. J. Zhang S. S. Gong Z. J. Mol. Cancer. 2018;17:3. doi: 10.1186/s12943-017-0752-2. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ren Y. He W. Chen W. Ma C. Li Y. Zhao Z. Gao T. Ni Q. Chai J. Sun M. J. Cell. Biochem. 2019;120:155–163. doi: 10.1002/jcb.27206. [DOI] [PubMed] [Google Scholar]
- Feng C. Bai M. Yu N. Z. Wang X. J. Liu Z. J. Cell. Mol. Med. 2017;21:278–285. doi: 10.1111/jcmm.12963. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lee A. Y. Fan C. C. Chen Y. A. Cheng C. W. Sung Y. J. Hsu C. P. Kao T. Y. Integr. Cancer Ther. 2015;14:484–490. doi: 10.1177/1534735415588930. [DOI] [PubMed] [Google Scholar]
- Yamada S. Yanamoto S. Naruse T. Matsushita Y. Takahashi H. Umeda M. Nemoto T. K. Kurita H. Pathol. Oncol. Res. 2016;22:625–632. doi: 10.1007/s12253-016-0049-6. [DOI] [PubMed] [Google Scholar]
- Yang H. Y. Huang C. P. Cao M. M. Wang Y. F. Liu Y. Neoplasma. 2018;65:872–880. doi: 10.4149/neo_2018_171225N841. [DOI] [PubMed] [Google Scholar]
- Ouyang K. X. Zou R. Liang J. Bai Z. B. Li Z. Q. Zhao J. J. J. Oral Maxillofac. Surg. 2017;75:423–428. doi: 10.1016/j.joms.2016.08.009. [DOI] [PubMed] [Google Scholar]
- Li Z. Q. Zou R. Ouyang K. X. Ai W. J. J. Oral Pathol. Med. 2017;46:956–960. doi: 10.1111/jop.12599. [DOI] [PubMed] [Google Scholar]
- Zhuang Z. Cheng W. Nan X. Yue W. Hou J. Liu X. Huang H. Cancer Res. 2016;76:985. [Google Scholar]
- Graham L. D. Pedersen S. K. Brown G. S. Ho T. Kassir Z. Moynihan A. T. Vizgoft E. K. Dunne R. Pimlott L. Young G. P. Lapointe L. C. Molloy P. L. Genes Cancer. 2011;2:829–840. doi: 10.1177/1947601911431081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H. Xia H. Qian M. Lv X. Biomed. Pharmacother. 2018;110:825–833. doi: 10.1016/j.biopha.2018.12.024. [DOI] [PubMed] [Google Scholar]
- Wang H. Ke J. Guo Q. Barnabo Nampoukime K. P. Yang P. Ma K. J. Cell. Mol. Med. 2018;22:5862–5876. doi: 10.1111/jcmm.13856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Wang Y. Li J. Zhang Y. Yin H. Han B. Cancer Lett. 2015;367:122–128. doi: 10.1016/j.canlet.2015.03.027. [DOI] [PubMed] [Google Scholar]
- Cheng J. Chen J. Zhang X. Mei H. Wang F. Cai Z. Biomed. Pharmacother. 2018;99:638–644. doi: 10.1016/j.biopha.2017.12.055. [DOI] [PubMed] [Google Scholar]
- Dai J. Mu J. W. Mu H. Oncol. Lett. 2019;17:3330–3340. doi: 10.3892/ol.2019.9978. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Martini M. De Santis M. C. Braccini L. Gulluni F. Hirsch E. Ann. Med. 2014;46:372–383. doi: 10.3109/07853890.2014.912836. [DOI] [PubMed] [Google Scholar]
- Jeong Y. J. Choi Y. Shin J. M. Cho H. J. Kang J. H. Park K. K. Choe J. Y. Bae Y. S. Han S. M. Kim C. H. Chang H. W. Chang Y. C. Food Chem. Toxicol. 2014;68:218–225. doi: 10.1016/j.fct.2014.03.022. [DOI] [PubMed] [Google Scholar]
- Yip P. Y. Transl. Lung Cancer Res. 2015;4:165–176. doi: 10.3978/j.issn.2218-6751.2015.01.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlind M. P. Hsieh A. C. Asian J. Androl. 2014;16:378–386. doi: 10.4103/1008-682X.122876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polivka Jr J. Janku F. Pharmacol. Ther. 2014;142:164–175. doi: 10.1016/j.pharmthera.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Simpson D. R. Mell L. K. Cohen E. E. Oral Oncol. 2015;51:291–298. doi: 10.1016/j.oraloncology.2014.11.012. [DOI] [PubMed] [Google Scholar]
- Pan S. T. Qin Y. Zhou Z. W. He Z. X. Zhang X. Yang T. Yang Y. X. Wang D. Qiu J. X. Zhou S. F. Drug Des., Dev. Ther. 2015;9:1601–1626. doi: 10.2147/DDDT.S76057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis B. C. Graham L. D. Molloy P. L. Biochim. Biophys. Acta. 2014;1843:372–386. doi: 10.1016/j.bbamcr.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Du D. X. Lian D. B. Amin B. H. Yan W. Eur. Rev. Med. Pharmacol. Sci. 2017;21:5392–5398. doi: 10.26355/eurrev_201712_13925. [DOI] [PubMed] [Google Scholar]
- Liu X. X. Xiong H. P. Huang J. S. Qi K. Xu J. J. Clin. Exp. Pharmacol. Physiol. 2017;44:895–902. doi: 10.1111/1440-1681.12780. [DOI] [PubMed] [Google Scholar]
- Jiang H. Wang Y. Ai M. Wang H. Duan Z. Wang H. Zhao L. Yu J. Ding Y. Wang S. Cell Death Discovery. 2017;8:e2862. doi: 10.1038/cddis.2017.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J. Li J. Wang H. Tian Y. Xie M. He X. Ji H. Ma Z. Hui B. Wang K. Ji G. Cell Death Discovery. 2017;8:e2997. doi: 10.1038/cddis.2017.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.