

Abstract
Adipocyte fatty acid-binding protein (A-FABP, also called FABP4, aP2) is an adipokine identified as a critical regulator of metabolic function due to its dual functions of fatty acid transport and pro-inflammation. Because of the high therapeutic potential of A-FABP inhibition for the treatment of metabolic diseases and related vascular complications, numerous inhibitors have been developed against A-FABP. However, none of these inhibitors have been approved for use in patients due to severe side effects. Here, we used a virtual screening (VS) strategy to identify potential inhibitors of A-FABP in the latest FDA-approved drug library (∼2600 compounds), aiming to explore the existing drugs with proven safety profiles. We firstly combined ligand-based machine learning and structure-based molecular docking to develop a screening pipeline for identifying A-FABP inhibitors. The screening of FDA-approved drugs identified four compounds (Cobimetinib, Larotrectinib, Pantoprazole, and Vildagliptin) with the highest scores, whose inhibitory effects on A-FABP were further assessed in cellular assays. Among the selected compounds, Cobimetinib significantly inhibited the activation of the JNK/c-Jun signaling pathway by A-FABP in mouse macrophages without causing obvious cytotoxicity. In summary, we present an integrated VS pipeline for A-FABP inhibitor screening, and identified Cobimetinib as a novel A-FABP inhibitor that may be repurposed for the treatment of metabolic diseases and associated vascular complications.
The integrated virtual screening pipeline was constructed to identify potential inhibitors of A-FABP in the latest FDA-approved drug library, aiming to explore the existing drugs with proven safety profiles.
1. Introduction
Adipocyte-type fatty acid-binding protein (A-FABP, also called as FABP4, aP2) is a member of the apolipoprotein family with a molecular weight of 14.6 kDa. It is mainly expressed in mature adipocytes, macrophages, and endothelial cells.1–3 A-FABP primarily mediates the storage and decomposition of fat in adipocytes as a carrier of free fatty acid molecules and regulates lipid accumulation.4 In addition, A-FABP can promote the expression of a variety of inflammatory factors in macrophages, including MCP-1, TNF-α, IL-6, and IL-1β.5,6 Interestingly, A-FABP can be secreted to the extracellular space and blood to promote inflammation and is strongly associated with a variety of metabolic diseases such as obesity, diabetes, lipid metabolism disorder, and atherosclerosis.7–9 Bone marrow transplantation from A-FABP knockout mice was shown to improve atherosclerosis without metabolic side effects; the mutation of A-FABP results in lower triglyceride levels in mice with reduced risk of cardiovascular disease and reduces obesity-induced type 2 diabetes.10,11 Related studies have also clarified the molecular mechanism by which A-FABP upregulates the expression of many inflammatory factors through the JNK/c-Jun/AP-1 signaling pathway in macrophages.12 It has been shown that the expression of A-FABP protein in brain tissue is increased after ischemic stroke, promotes the expression of inflammatory factors, and aggravates neuroinflammation after stroke.13,14 Therefore, A-FABP is a highly promising therapeutic target for the treatment of metabolic diseases and related cardio-cerebrovascular diseases.
In the past decades, hundreds of A-FABP inhibitors have been reported, including pyrazole derivatives,15 oxazole derivatives,16 imidazole derivatives,16,17 indole derivatives,18–20 benzimidazole derivatives,21 thiophene and thiazoles derivatives,22 pyrimidine, bicyclic pyridine and quinoxaline derivatives,23,24 urea, carbamoyl derivatives25 and miscellaneous compounds.26 Some of these inhibitors have shown efficacies in animal models of atherosclerosis, diabetes, and other related diseases.27 BMS309403, a potent small-molecule inhibitor of A-FABP, has been systematically studied in vitro and in vivo in various disease models.28 Although the inhibitory effect of BMS309403 on A-FABP has been well verified in animal disease models, no further clinical trials have been launched to date likely because of its cardiotoxicity,27,29–31 resulting in no therapeutic drugs targeting A-FABP being used or tested in the clinic.
In recent years, researchers have focused on natural products and US Food and Drug Administration (FDA)-approved drugs for the development of A-FABP inhibitors. The currently available screening approaches for A-FABP inhibitors are labor-extensive, time-consuming, and expense-costly.27 Therefore, it becomes necessary to improve the screening efficiency by applying virtual screening (VS) in the lead identification of novel A-FABP inhibitors. Therefore, computer-aided drug design techniques such as machine learning (ML) and molecular docking are widely used in drug design. Molecular docking is a commonly used computer-aided drug design technology based on the characteristics of receptors and the interaction between receptors and drug molecules.32 Wang et al. screened the natural compound library (∼5200 compounds)33 and the FDA drug library (∼1500 compounds)34 using molecular docking and found that hyperoside, quercetin, other flavonoids, and levofloxacin, an FDA-approved drug for the treatment of different bacterial infections, showed inhibitory effects on FABP4. Machine learning approaches possess robust abilities in separating inhibitors from non-inhibitors by building classification models adopting statistical algorithms, including naïve Bayesian (NB) models.35
Since each individual VS method has its own advantages and disadvantages, therefore, we combined the ligand-based machine learning with structure-based molecular docking in a sequential manner to form an integrated VS pipeline for lead identification of A-FABP inhibitors. In detail, each compound of FDA's latest drug list (∼2600 compounds) was subjected to activity prediction by a naïve Bayesian classification model, followed by molecular docking analysis. The identified candidate compounds were validated against A-FABP through cellular experiments. The results of this study will provide new ideas for the development of A-FABP inhibitors and FDA-approved drugs for the treatment of various metabolic diseases.
2. Material and methods
2.1. Materials
High glucose Dulbecco's Modified Eagle's Medium (#11995065), fetal bovine serum (#10270) were purchased from Thermo Fisher Scientific (Gibco, Waltham, MA, USA). Thioglycolate medium Brewer (Lot #5243569) was purchased from Becton, Dickinson and Company (Sparks, MD, USA). RIPA Buffer (#R0020) was purchased from Solarbio (Beijing, China). Antibodies were as follows: Phospho-SAPK/JNK (Thr183/Tyr185) (81E11) Rabbit mAb (#4668), SAPK/JNK Antibody (#9252), β-Actin (8H10D10) Mouse mAb (#3700), Anti-rabbit IgG HRP-linked Antibody (#7074) and Anti-mouse IgG HRP-linked Antibody (#7076) were purchased from Cell Signaling Technology (Boston, MA, USA); Phospho-c-Jun-S63 Rabbit mAb (AP0105), JUN Rabbit pAb (#A16905) were purchased from ABclonal (Wuhan, China). Chemiluminescence substrate detection solution (#WBKLS0500) was purchased from MERCK (MilliPore, Boston, MA, USA). Cell Counting Kit-8 (CCK-8) (#BA00208) was obtained from Bioss (Beijing, China). Dimethylsulfoxide was purchased from MERCK (Sigma-Aldrich, St. Louis, MO, USA).
2.2. Data collection and preparation
A total of 2595 FDA-approved drugs were collected from the DrugBank database (https://go.drugbank.com). Compounds defined as active A-FABP inhibitors were collected from the ChEML database,36–38 with IC50 ≤ 10 μM as the selection criterion. It is noteworthy that the compounds collected from the ChEMBL database do not overlap with the FDA-approved drugs dataset. Corresponding decoys (defined as inactive) were automatically generated by DUD-E online database with the ratio of 4 : 1 to active compounds.39,40
Morgan fingerprints (4096 bits, radius = 2) were generated using the RDKit package in Python and then fed into the t-distributed stochastic neighbor embedding (t-SNE) algorithm to obtain 2-dimensional representations. To investigate whether the diversity and spatial distribution of the data set meet the modeling requirements of subsequent machine learning models.
Both the active datasets and inactive datasets were randomly distributed into the training set and test set with the ratio of 4 : 1 as shown in Table 1. The A-FABP inhibitors and decoys were respectively marked as “1” and “0” in all datasets. Before the calculation of molecular descriptors, all compounds were required with the addition of hydrogen atoms, deprotonation of strong acids, protonation of strong bases, generation of a valid 3D conformation, and energy minimization. The detailed information of the training sets and test sets were described in Table S1.†
Detailed statistical description of the entire data set.
| Model | Training set (ECFP_2) | Test set (ECFP_2) | Total |
|---|---|---|---|
| Inhibitors | 108 | 27 | 135 |
| Decoys | 432 | 108 | 540 |
| Total | 540 | 135 | 675 |
2.3. Naïve Bayesian model generation
Molecular descriptors are the basis for the combination of machine learning.41 Therefore, we used 542 descriptors calculated by Discovery Studio 2016 (DS 2016)42 and MOE 2014.9 (ref. 43) software to calculate three sets of two-dimensional (2D) descriptors for describing each compound. In addition, molecular fingerprints involved the SciTegic extended-connectivity fingerprints (FCFP and ECFP) and Daylight-style path-based fingerprints (FPFP and EPFP) were also calculated with DS 2016 in this paper.
In this study, Pearson correlation analysis was performed to find out the descriptors that were highly correlated with activity.44 Firstly, the descriptors whose values appeared in the high frequency of more than 50% were eliminated. Secondly, the descriptors whose correlation coefficients with activity less than 0.1 were excluded. If the absolute value of the correlation coefficients between two descriptors was higher than 0.9, the descriptor possessing a lower correlation coefficient with activity would be deleted. Finally, the remaining molecular descriptors were filtered by logistic regression, only the molecular descriptors kept in the regression equation were used for building naïve Bayesian models.
The quality of the models was assessed by 5-fold cross-validation and test set validation. Measurement parameters included true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN). Subsequently, sensitivity (SE), specificity (SP), positive predictive value (PPV), and Matthews correlation coefficient (MCC) were calculated by eqn (1)–(4). TP indicates the number of active compounds predicted to be active; TN indicates the number of inactive compounds predicted to be inactive; FP represents the number of inactive compounds predicted as active and FN represents the number of active compounds predicted to be inactive. Similarly, SE stands for the accuracy of prediction for active compounds and SP represents the accuracy of prediction for inactive compounds. PPV indicates the overall prediction accuracy for all compounds in the dataset. MCC signifies the most important indicator for the quality of binary classification and it is calculated to evaluate the predictive power of the model with values ranging from −1 to 1. In this study, the MCC value is considered the main evaluation index.
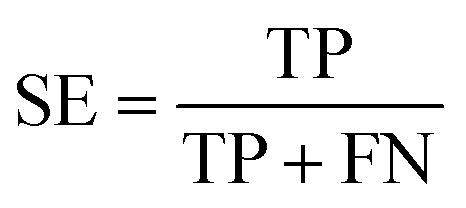 |
1 |
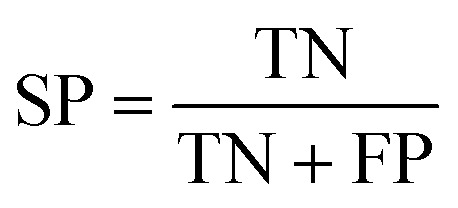 |
2 |
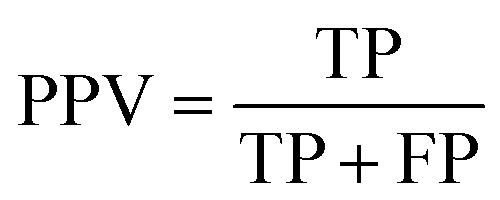 |
3 |
 |
4 |
2.4. Molecular docking
The crystal structure of A-FABP was retrieved from the Protein Data Bank (PDB ID: 2NNQ). The molecular docking studies were performed in MOE 2014.9. For the preparation of the protein, the water, and the co-crystallized ligand were removed and hydrogens were added and other important parameters were adapted by the MMFF94x force field. The A-FABP protein was then further processed with the prepared protein module to model missing loop regions, calculate protein ionization and protonate the protein structure. The prepared protein was defined as the receptor and the binding site was defined by generating the α-site spheres in the site finder. The docking inhibitors will attack the protein surfaces in its interior grooves, after 30 trials, till the most stable docking complexes are reached.
2.5. Determination of A-FABP inhibitory activity of cell-based candidate compounds
2.5.1. Cell culture and treatment
RAW264.7 cells (mouse macrophage cell line) were cultured in high glucose Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum and 100 U ml−1 penicillin and 100 μg ml−1 streptomycin at 37 °C, 5% CO2 incubator. Cells were divided into three groups: (1) control group: no treatment, (2) model group: cells were treated with 1 μg ml−1 A-FABP protein (3) treatment group: candidate compounds were pretreated with 1 μg ml−1 A-FABP protein for 30 min; then cell culture with this at 37 °C, 5% CO2 incubator. Candidate compounds were diluted to two concentrations (10 μM, 100 μM).
2.5.2. Primary macrophage cells
This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee at the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences. We followed the methods of Layoun A. et al. (2015). 38 g thioglycolate medium Brewer was taken with 1000 ml of distilled water, boil the solution until it is completely dissolved that autoclave at 121 °C for 15 min. So, this solution was a 3.8% thioglycolate medium Brewer which was stored at 4 °C and protected from light for 3 months. The abdominal cavity of mice was injected with 1 ml of 3.8% thioglycolate medium Brewer and waited for 3 days; the mice were injected with 5 ml of pre-cooled D-PBS into the abdominal cavity of mice after being sacrificed by the cervical dislocation method; as massage gently in the abdominal cavity, then the liquid was aspirated and put in a 15 ml centrifuge tube and centrifuge at 400×g for 10 minutes at 4 °C. Discard the supernatant to obtain primary macrophages with greater purity. The primary macrophage cells were cultured in high glucose Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum and 100 U ml−1 penicillin and 100 μg ml−1 streptomycin at, 37 °C, 5% CO2 incubator for 24 hours, then changed the medium and the cells continued culturing for 24 hours. The primary macrophage cells were grouped and processed according to the above-mentioned cell grouping and processing methods.
2.5.3. Western blotting analysis
The RIPA Buffer was added in the cell culture dish to extract total cell protein, and the following processing: first, the protein sample were separated by SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes; secondly, membranes were blocked with 5% non-fat dry milk for 1 h at room temperature and then incubated overnight at 4 °C with primary antibodies diluted in TBS with 0.1% Tween 20 (TBST); membranes were washed in TBST and then incubated with secondary antibodies for 1 h at room temperature; finally, the membranes were exposed to enhanced chemiluminescence substrate detection solution and then detected by instrument (Gel view, GV6000). Image J software was used to quantify the band density.
2.5.4. The cytotoxicity of candidate compounds assays
Cell Counting Kit-8 was used to detect cytotoxicity. RAW264.7 cells were cultured according to the above requirements in 96-well plates at a density of 5 × 104 cells per ml, and experimental groups were established: (1) blank control group (medium + CCK8 reagent, cell-free) (2) reagent control group (cell + medium + CCK8 reagent) (3) solvent control group (DMSO 100 μM) (cell + medium + DMSO + CCK8 reagent) (4) A-FABP protein (1 μg ml−1) model group (cells + medium + A-FBP4 protein + CCK8 reagent) (5) A-FABP protein (1 μg ml−1) + candidate compounds treatment group (cells + medium + A-FBP4 protein + candidate compounds + CCK8 reagent). The candidate compounds were diluted to two concentrations (10 μM, 100 μM). In a 37 °C, 5% CO2 incubator, the candidate compounds were incubated with A-FABP protein for 30 minutes and then added to the cells for 30 minutes. Then the cells were added CCK8 reagent and treated for 2 h at 37 °C and 5% CO2 from light. Finally, the microplate reader was used to measure the absorbance (OD) at 450 nm.
2.6. Statistical analysis
SPSS 17.0 statistical software was used for the normality test. All data are expressed as mean ± standard error. For normally distributed data, one-way analysis of variance (One-Way ANOVA) is used: if the variances are uniform, the pairwise comparisons will use the least significant difference (LSD) method; if the variances are not uniform, Dunnett's T3 test is used. Data that do not conform to the normal distribution are counted by a non-parametric test. The statistical difference was set as *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
3. Results
3.1. Naïve Bayesian model generation
Generally, the performance of machine learning classification models is largely affected by the chemical space diversity of compounds included in the datasets for model training and testing. A wide chemical space diversity of training sets usually endows the classification model with high prediction accuracy and strong generalization ability. The chemical space diversity of the entire dataset was explored by t-SNE plot analysis. As we can see in the t-SNE plot (Fig. 1), remarkable colocalization of several active compound clusters was shown and well separated from inactive compounds. It should be noted that different active compound clusters may represent different chemotypes, which is worth further investigation in the future.
Fig. 1. The visual representation of active compound (red) and inactive compound (light green) based on A-FABP inhibition, generated using t-distributed stochastic neighbor embedding (t-SNE) based on Morgan fingerprint (4096 bits).

According to the molecular descriptor selection criteria, 39 discovery studio 2D descriptors (DS_2D) and 21 MOE 2D descriptors (MOE_2D) (Table 2) were finally chosen for Naïve Bayesian model construction. 5-Fold cross-validation and test set validation was then used to further evaluate the predictive power, as shown in Table 3. The Naïve Bayesian model emerged a presentable performance with the MCC value of 0.971 and 0.953 from 5-fold cross-validation and test set, respectively (Table 3).
Molecular descriptors used in this study.
| Descriptor class | Number of descriptors | Descriptors |
|---|---|---|
| DS_2D | 39 | ast_violation_ext, b_1rotR, b_ar, b_max1len, density, diameter, GCUT_SLOGP_1, h_logP, h_pavgQ, h_pKb, h_pstates, opr_nring, opr_violation, PEOE_RPC+, PEOE_VSA-1, PEOE_VSA-5, PEOE_VSA-6, PEOE_VSA_FPPOS, PEOE_VSA_PPOS, petitjean, Q_RPC-, Q_VSA_FHYD, Q_VSA_FNEG, Q_VSA_FPOL, Q_VSA_FPOS, Q_VSA_FPPOS, radius, rings, RPC-, SlogP_VSA0, SlogP_VSA2, SlogP_VSA4, SlogP_VSA5, SMR_VSA0, SMR_VSA2, SMR_VSA4, SMR_VSA5, SMR_VSA7, vsa_acc |
| MOE_2D | 21 | ES_Count_aaN, ES_Count_dO, ES_Count_ssCH2, ES_Count_sssCH, ES_Sum_sOH, QED_ALERTS, QED_AROM, SAscore, SAscore_Fragments, HBA_Count, Num_AromaticRings, Num_Rings, Num_Rings5, Molecular_PolarSASA, BIC, CHI_3_CH, CHI_V_3_C, JX, JY, Kappa_3, Kappa_3_AM |
Performance of the Naïve Bayesian model.
| TP | FN | FP | TN | SE | SP | PPV | MCC | |
|---|---|---|---|---|---|---|---|---|
| 5-Fold-cross | 106 | 3 | 2 | 429 | 0.972 | 0.995 | 0.991 | 0.971 |
| External-test-set | 25 | 0 | 2 | 108 | 1.000 | 0.982 | 0.985 | 0.953 |
As a primary screening for the A-FABP inhibitors, virtual screening of the FDA-approved drug dataset was performed based on the Naïve Bayesian model. The Bayesian categorization model not only predicts “true” or “false”, but also gives the estimated probability (0 ≤ EstPGood ≤ 1) that a compound is in the active class. As a result, 317 compounds were predicted to be active A-FABP inhibitors (detailed information is available in Table S2†), of which 30 compounds have EstPGood values ≥ 0.9 (Table 4).
Prediction results of the top 30 drugs selected for docking validation.
| Drugbank ID | Generic name | EstPGood | Prediction | |
|---|---|---|---|---|
| 1 | DB06803 | Niclosamide | 0.997 | TRUE |
| 2 | DB00824 | Enprofylline | 0.996 | TRUE |
| 3 | DB12332 | Rucaparib | 0.996 | TRUE |
| 4 | DB00900 | Didanosine | 0.995 | TRUE |
| 5 | DB03585 | Oxyphenbutazone | 0.992 | TRUE |
| 6 | DB00670 | Pirenzepine | 0.987 | TRUE |
| 7 | DB01392 | Yohimbine | 0.987 | TRUE |
| 8 | DB08826 | Deferiprone | 0.978 | TRUE |
| 9 | DB00697 | Tizanidine | 0.974 | TRUE |
| 10 | DB06193 | Pixantrone | 0.973 | TRUE |
| 11 | DB04876 | Vildagliptin | 0.972 | TRUE |
| 12 | DB00998 | Frovatriptan | 0.971 | TRUE |
| 13 | DB00993 | Azathioprine | 0.959 | TRUE |
| 14 | DB00507 | Nitazoxanide | 0.958 | TRUE |
| 15 | DB00457 | Prazosin | 0.955 | TRUE |
| 16 | DB00889 | Granisetron | 0.949 | TRUE |
| 17 | DB09282 | Molsidomine | 0.946 | TRUE |
| 18 | DB00213 | Pantoprazole | 0.945 | TRUE |
| 19 | DB00315 | Zolmitriptan | 0.936 | TRUE |
| 20 | DB11071 | Phenyl salicylate | 0.930 | TRUE |
| 21 | DB05239 | Cobimetinib | 0.928 | TRUE |
| 22 | DB00310 | Chlorthalidone | 0.926 | TRUE |
| 23 | DB09343 | Tipiracil | 0.922 | TRUE |
| 24 | DB09151 | Flutemetamol (18F) | 0.920 | TRUE |
| 25 | DB04816 | Dantron | 0.916 | TRUE |
| 26 | DB04880 | Enoximone | 0.914 | TRUE |
| 27 | DB00744 | Zileuton | 0.913 | TRUE |
| 28 | DB00819 | Acetazolamide | 0.912 | TRUE |
| 29 | DB14723 | Larotrectinib | 0.909 | TRUE |
| 30 | DB00277 | Theophylline | 0.906 | TRUE |
3.2. Molecular docking study
Molecular docking has been widely used for structure-based virtual screening, as well as exploring the binding modes of small molecules within protein–ligand complexes. Herein, molecular docking analyses were performed for A-FABP by applying docking algorithm by MOE module. The docking process was executed between the 30 compounds selected for docking validation and 2NNQ PDB co-crystals of A-FABP, respectively. With BMS309403 as the reference, the 30 compounds were sorted by docking score (kcal mol−1) (as shown in Table S3†). Through the analysis of the protein–ligand interaction model, four compounds with the highest score were selected: Cobimetinib (currently approved for the treatment of melanoma), Larotrectinib (currently approved for the treatment of solid tumor), Pantoprazole (currently approved for the treatment of gastric ulcers, duodenal ulcers, etc.), and Vildagliptin (currently approved for the treatment of type-2 diabetes).
The X-ray structure of BMS309403 in complex with A-FABP (PDB code: 2NNQ) revealed that the aromatic at position 3 of the central core of the pyrazole binds to the hydrophobic pocket formed by Phe16, Met20, Met40, Ala75, Val25, Arg126 and Asp76. Another aromatic ring at position 4 is located in another hydrophobic pocket formed by Ala36, Phe57, Pro38, Ser55, Ser53, Asn39 and Pro38. The computational study showed that the binding poses of four compounds (Cobimetinib, Larotrectinib, Pantoprazole, and Vildagliptin) with A-FABP has a similar orientation to that of BMS309403. Notably, identical to BMS309403, two hydrogen bond interactions were identified with backbone amino acid residues Arg126 and Tyr128 in Cobimetinib, Larotrectinib and Vildagliptin. Fundamental data was exported (Table 5) and docking complexes images (interaction and surface maps), are presented as well in Fig. 2.
Molecular docking score.
| Drugbank ID | Generic name | S (kcal mol−1) | Hydrogen bond | π–π interaction |
|---|---|---|---|---|
| BMS309403 | −7.704 | Arg126, Tyr128 | Phe16, Pro38 | |
| DB05239 | Cobimetinib | −6.931 | Arg126, Tyr128, Arg106 | |
| DB14723 | Larotrectinib | −6.868 | Arg126, Tyr128 | Ile104 |
| DB00213 | Pantoprazole | −6.791 | Tyr128 | Phe16 |
| DB04876 | Vildagliptin | −6.416 | Arg126, Tyr128, Ala75 |
Fig. 2. The docking complex images and the two-dimensional diagram of the binding mode of BMS309402 (A), Cobimetinib (B), Larotrectinib (C), Pantoprazole (D), and Vildagliptin (E).

3.3. The effect of candidate compounds on the biological activity of A-FABP
To explore the inhibitory effect of the candidate compounds on A-FABP, we used western blotting to detect the phosphorylation state of the JNK/c-Jun signaling pathway in RAW264.7 cells (mouse macrophage cell line) and mouse primary macrophage cells after treatment of A-FABP protein without or with the above four candidate compounds. The A-FABP protein was added into RAW264.7 cells and mouse primary macrophages culture medium. Compared with the blank control group, the A-FABP protein enhanced the phosphorylation of JNK/c-Jun (Fig. 3). We found that Cobimetinib can significantly inhibit the phosphorylation of JNK/c-Jun by A-FABP, while Larotrectinib, Pantoprazole, and Vildagliptin have no obvious effect on inhibiting the activity of A-FABP (Fig. 3). Therefore, we further set the concentration gradient of Cobimetinib to (1 μM, 10 μM, 30 μM, 100 μM) and adopted the same method described above to detect the changes in the phosphorylation of the JNK/c-Jun signaling pathway in RAW264.7 cells. We found that Cobimetinib significantly decreased the phosphorylation of JNK/c-Jun at concentrations higher than 1 μM, demonstrating potent inhibition of the JNK/c-Jun signaling by Cobimetinib in RAW264.7 cells (Fig. 4).
Fig. 3. Effects of candidate compounds on phosphorylation of JNK/c-Jun signaling pathway activated by A-FABP. JNK and c-Jun phosphorylation (p-JNK and p-c-Jun) was assessed in RAW264.7 (A), (C) and (E) and primary macrophages (B), (D) and (F) by western blotting using phosphospecific antibodies. Data were expressed as mean ± SEM. **P < 0.01 compared to the A-FABP group.

Fig. 4. Inhibition of Cobimetinib on phosphorylation of JNK/c-Jun signaling pathway activated by A-FABP. RAW264.7 was treated with Cobimetinib at various dosages (A), and cell lysates were analyzed by western blot with antibodies against p-JNK/JNK (B) and p-c-Jun/c-Jun (C). Data were expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.005 compared to the A-FABP group.

3.4. Toxic effects of candidate compounds on cells
To ensure the candidate compounds can inhibit the activation of A-FABP without causing toxic effects on cells, we detected the cytotoxicity by CCK8 assay. Compared with the absorbance of the control group, the solvent DMSO has no obvious toxic effect on RAW264.7 cells; compared with the solvent control group and the A-FABP protein + candidate compound treatment group, it was found that the 10 μM/100 μM concentration of candidate compounds except Cobimetinib (100 μM) had no obvious toxic effects on RAW264.7 cells, while the low concentration of Cobimetinib (10 μM) had no obvious toxic effect on the cells. Further analysis of the concentration gradients of Cobimetinib (1 μM, 10 μM, 30 μM, 100 μM) demonstrated that except for 100 μM, lower concentrations of Cobimetinib have no obvious toxic effects on RAW264.7 cells (Fig. 5).
Fig. 5. Toxic effects of candidate compounds on RAW264.7 cells (A), and toxic effects of Cobimetinib at various dosages on RAW264.7 (B). Data were expressed as mean ± SEM. ###P < 0.005 compared to the control group.

4. Discussion
Over the past two decades, there has been a dramatic increase in research and development expenditures on drugs for the treatment of metabolic and related cardiovascular and cerebrovascular diseases.45,46 With the progress of computer-aided drug design technology, the application of machine learning and molecular docking in drug development has increased rapidly. Machine learning integrates massive data resources to solve biomedical problems with the combination of computer science and statistics and has the ability to deal with large and complex data.47,48 In terms of drug development and evaluation, machine learning has become an indispensable tool for drug designers to mine chemical information from large compound databases and design drugs with important biological properties.49,50 For example, Sean Ekins et al. used machine learning to screen new compounds that may treat Mycobacterium tuberculosis;51 Yang et al. used machine learning to identify and evaluate neuroprotective agents.52 Molecular docking is computer technology for studying drug–target interactions and drug design based on structure.53,54 For example, DesJarlais RL et al. have successfully used molecular docking to design a new type of HIV-1 protease inhibitors;55 Zhao et al. used molecular docking to identify JAK2 inhibitors;56 Mirza SB et al. used molecular docking to screen non-structural protein 3 (NS3) against dengue virus and found five inhibitors that could reduce virus titers in HUH7 cells.57 Therefore, the virtual screening method that we combine both the ligand-based machine learning method and the structure-based molecular docking is of great significance for the discovery and application of drugs.
This study uses molecular descriptors to construct a binary classification model of A-FABP inhibitors. The naïve Bayesian model with good predictive power was used to screen potential A-FABP inhibitors in FDA-approved drugs. After combining machine learning with molecular docking, four potential A-FABP inhibitors (Cobimetinib, Larotrectinib, Pantoprazole, Vildagliptin) were identified. Previous studies have found that A-FABP promotes the expression of inflammatory factors by activating the JNK/c-Jun signaling pathway,12,14 thereby exacerbating the inflammatory response in related tissues. We found that Cobimetinib can significantly inhibit the phosphorylation of JNK/c-Jun by A-FABP, while Larotrectinib, Pantoprazole, and Vildagliptin have no obvious effect on inhibiting the activity of A-FABP. To ensure the candidate compounds can inhibit the activation of A-FABP without causing toxic effects on cells, we detected the cytotoxicity by CCK8 assay. This result indicates that Cobimetinib shows potential cytotoxicity at high concentrations (100 μM). However, the inhibitory effect of Cobimetinib at lower concentrations (1 μM, 10 μM, 30 μM) on JNK/c-Jun phosphorylation shown in Fig. 3 and 4 was not due to cytotoxicity. In summary, Cobimetinib is an effective and novel inhibitor of A-FABP.
Cobimetinib (GDC-0973) is a MEK (mitogen-activated protein kinase)-specific small molecule inhibitor, which has an IC50 of 4.2 nM and a half-life of ∼44 h, and a molecular weight of 531.58 It is FDA-approved for BRAF-mutated melanoma in combination with vemurafenib and atezolizumab.59 Cobimetinib is taken orally together with atezolizumab for the treatment of melanoma. The dose regimen is 60 mg/1 time/day on the 1st to 21st day of each 28 day treatment cycle (compared with atezolizumab and vemurafenib combined) until the disease progresses or unacceptable toxicity occurs. Adverse reactions of Cobimetinib include nausea, vomiting, fever, bleeding, etc.59–61 At present, Cobimetinib is mainly used in cancer-related research, such as the treatment of melanoma or other types of cancers, including neuroblastoma, colorectal cancer, and acute myeloid leukemia.62–64 Here we demonstrate that Cobimetinib can be potentially repurposed as a “new drug” for the treatment of metabolic diseases and cardiovascular and cerebrovascular diseases through targeted inhibition of A-FABP.
In summary, the current study identified several potential novel A-FABP inhibitors through virtual screening methods based on machine learning and molecular docking, and confirmed the inhibitory effect of Cobimetinib on A-FABP-activated JNK/c-Jun phosphorylation in cellular assays. Further animal and clinical studies are required to determine the therapeutic efficacy and adverse effects of Cobimetinib on metabolic diseases and associated cardio-cerebrovascular diseases.
Ethical statement
The feeding and experimental procedures of all animals were in accordance with the guidelines of the Experimental Animal Ethics Committee of the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences. The animal experiment protocol in this article was approved by the Institutional Animal Care and Use Committee at the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences (SIAT-IACUC-20210112-YYS-DBYWZX-YSL-A0956-01).
Author contributions
SY performed the virtual screening. SL performed the cellular experiments. SY and SL analyzed the data and wrote the manuscript. JC conceived the project, interpreted the data and revised the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
The work was funded by the National Natural Science Foundation of China (32170985, 82104167), National Key Research and Development Program of China (2021YFA0910000), Science Technology and Innovation Commission of Shenzhen Municipality (SGLH20180625142404672, JCYJ20210324115800003, JCYJ20210324101401004), International collaboration project of Chinese Academy of Sciences (172644KYSB20200045), CAS-Croucher Funding Scheme for Joint Laboratories, and Guangdong Innovation Platform of Translational Research for Cerebrovascular Diseases.
Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01057g
References
- Xu A. et al., Adipocyte fatty acid-binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clin. Chem. 2006;52(3):405–413. doi: 10.1373/clinchem.2005.062463. [DOI] [PubMed] [Google Scholar]
- Furuhashi M. Hotamisligil G. S. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discovery. 2008;7(6):489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuseya T. et al., Ectopic Fatty Acid-Binding Protein 4 Expression in the Vascular Endothelium is Involved in Neointima Formation after Vascular Injury. J. Am. Heart Assoc. 2017;6(9):e006377. doi: 10.1161/JAHA.117.006377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.-S. et al., Fatty acid-binding protein 4 regulates fatty infiltration after rotator cuff tear by hypoxia-inducible factor 1 in mice. Journal of Cachexia, Sarcopenia and Muscle. 2017;8(5):839–850. doi: 10.1002/jcsm.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A. Vanhoutte P. M. Adiponectin and adipocyte fatty acid binding protein in the pathogenesis of cardiovascular disease. Am. J. Physiol.: Heart Circ. Physiol. 2012;302(6):H1231–H1240. doi: 10.1152/ajpheart.00765.2011. [DOI] [PubMed] [Google Scholar]
- Makowski L. et al., The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor gamma and IkappaB kinase activities. J. Biol. Chem. 2005;280(13):12888–12895. doi: 10.1074/jbc.M413788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamounier-Zepter V. et al., Adipocyte fatty acid-binding protein suppresses cardiomyocyte contraction: a new link between obesity and heart disease. Circ. Res. 2009;105(4):326–334. doi: 10.1161/CIRCRESAHA.109.200501. [DOI] [PubMed] [Google Scholar]
- Cao H. et al., Adipocyte lipid chaperone AP2 is a secreted adipokine regulating hepatic glucose production. Cell Metab. 2013;17(5):768–778. doi: 10.1016/j.cmet.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burak M. F. et al., Development of a therapeutic monoclonal antibody that targets secreted fatty acid-binding protein aP2 to treat type 2 diabetes. Sci. Transl. Med. 2015;7(319):319ra205. doi: 10.1126/scitranslmed.aac6336. [DOI] [PubMed] [Google Scholar]
- Tuncman G. et al., A genetic variant at the fatty acid-binding protein aP2 locus reduces the risk for hypertriglyceridemia, type 2 diabetes, and cardiovascular disease. Proc. Natl. Acad. Sci. U. S. A. 2006;103(18):6970–6975. doi: 10.1073/pnas.0602178103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M. et al., Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447(7147):959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui X. et al., Adipocyte fatty acid-binding protein modulates inflammatory responses in macrophages through a positive feedback loop involving c-Jun NH2-terminal kinases and activator protein-1. J. Biol. Chem. 2010;285(14):10273–10280. doi: 10.1074/jbc.M109.097907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso A. W. et al., Serum adipocyte fatty acid-binding protein associated with ischemic stroke and early death. Neurology. 2011;76(23):1968–1975. doi: 10.1212/WNL.0b013e31821e54b3. [DOI] [PubMed] [Google Scholar]
- Liao B. et al., Adipocyte fatty acid-binding protein exacerbates cerebral ischaemia injury by disrupting the blood-brain barrier. Eur. Heart J. 2020;41(33):3169–3180. doi: 10.1093/eurheartj/ehaa207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. Y. et al., Chronic administration of BMS309403 improves endothelial function in apolipoprotein E-deficient mice and in cultured human endothelial cells. Br. J. Pharmacol. 2011;162(7):1564–1576. doi: 10.1111/j.1476-5381.2010.01158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulsky R. et al., Potent and selective biphenyl azole inhibitors of adipocyte fatty acid binding protein (aFABP) Bioorg. Med. Chem. Lett. 2007;17(12):3511–3515. doi: 10.1016/j.bmcl.2006.12.044. [DOI] [PubMed] [Google Scholar]
- Kurian E. Kirk W. R. Prendergast F. G. Affinity of fatty acid for (r)rat intestinal fatty acid binding protein: further examination. Biochemistry. 1996;35(12):3865–3874. doi: 10.1021/bi952589y. [DOI] [PubMed] [Google Scholar]
- Lehmann F. et al., Discovery of inhibitors of human adipocyte fatty acid-binding protein, a potential type 2 diabetes target. Bioorg. Med. Chem. Lett. 2004;14(17):4445–4448. doi: 10.1016/j.bmcl.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Barf T. et al., N-Benzyl-indolo carboxylic acids: design and synthesis of potent and selective adipocyte fatty-acid binding protein (A-FABP) inhibitors. Bioorg. Med. Chem. Lett. 2009;19(6):1745–1748. doi: 10.1016/j.bmcl.2009.01.084. [DOI] [PubMed] [Google Scholar]
- Tagami U. et al., Interaction Analysis of FABP4 Inhibitors by X-ray Crystallography and Fragment Molecular Orbital Analysis. ACS Med. Chem. Lett. 2016;7(4):435–439. doi: 10.1021/acsmedchemlett.6b00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H. et al., Novel fatty acid binding protein 4 (FABP4) inhibitors: virtual screening, synthesis and crystal structure determination. Eur. J. Med. Chem. 2015;90:241–250. doi: 10.1016/j.ejmech.2014.11.020. [DOI] [PubMed] [Google Scholar]
- Hertzel A. V. et al., Identification and characterization of a small molecule inhibitor of fatty acid binding proteins. J. Med. Chem. 2009;52(19):6024–6031. doi: 10.1021/jm900720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringom R. et al., Substituted benzylamino-6-(trifluoromethyl)pyrimidin-4(1H)-ones: a novel class of selective human A-FABP inhibitors. Bioorg. Med. Chem. Lett. 2004;14(17):4449–4452. doi: 10.1016/j.bmcl.2004.06.058. [DOI] [PubMed] [Google Scholar]
- Makowski L. Hotamisligil G. S. The role of fatty acid binding proteins in metabolic syndrome and atherosclerosis. Curr. Opin. Lipidol. 2005;16(5):543–548. doi: 10.1097/01.mol.0000180166.08196.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H. et al., Discovery of highly selective inhibitors of human fatty acid binding protein 4 (FABP4) by virtual screening. Bioorg. Med. Chem. Lett. 2010;20(12):3675–3679. doi: 10.1016/j.bmcl.2010.04.095. [DOI] [PubMed] [Google Scholar]
- Lan H. et al., Small-molecule inhibitors of FABP4/5 ameliorate dyslipidemia but not insulin resistance in mice with diet-induced obesity. J. Lipid Res. 2011;52(4):646–656. doi: 10.1194/jlr.M012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresta G. et al., Adipocyte fatty acid binding protein 4 (FABP4) inhibitors. A comprehensive systematic review. Eur. J. Med. Chem. 2017;138:854–873. doi: 10.1016/j.ejmech.2017.07.022. [DOI] [PubMed] [Google Scholar]
- Furuhashi M. et al., Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447(7147):959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al., Adipocyte Fatty Acid-Binding Protein Promotes Palmitate-Induced Mitochondrial Dysfunction and Apoptosis in Macrophages. Front. Immunol. 2018;9:81. doi: 10.3389/fimmu.2018.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosquet A. et al., FABP4 inhibitor BMS309403 decreases saturated-fatty-acid-induced endoplasmic reticulum stress-associated inflammation in skeletal muscle by reducing p38 MAPK activation. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2018;1863(6):604–613. doi: 10.1016/j.bbalip.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Lee M. Y. et al., Chronic administration of BMS309403 improves endothelial function in apolipoprotein E-deficient mice and in cultured human endothelial cells. Br. J. Pharmacol. 2011;162(7):1564–1576. doi: 10.1111/j.1476-5381.2010.01158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzi L. Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019;20(18):4331. doi: 10.3390/ijms20184331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al., Using molecular docking screening for identifying hyperoside as an inhibitor of fatty acid binding protein 4 from a natural product database. J. Funct. Foods. 2016;20:159–170. [Google Scholar]
- Wang Y. et al., Discovery of FDA-approved drugs as inhibitors of fatty acid binding protein 4 using molecular docking screening. J. Chem. Inf. Model. 2014;54(11):3046–3050. doi: 10.1021/ci500503b. [DOI] [PubMed] [Google Scholar]
- Stephenson N. et al., Survey of Machine Learning Techniques in Drug Discovery. Curr. Drug Metab. 2019;20(3):185–193. doi: 10.2174/1389200219666180820112457. [DOI] [PubMed] [Google Scholar]
- Mendez D. et al., ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47(D1):D930–D940. doi: 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento A. P. et al., The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42:D1083–D1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A. et al., ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaput L. et al., Benchmark of four popular virtual screening programs: construction of the active/decoy dataset remains a major determinant of measured performance. J. Cheminf. 2016;8:56. doi: 10.1186/s13321-016-0167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M. et al., Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012;55(14):6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips K. A. et al., High-throughput screening of chemicals as functional substitutes using structure-based classification models. Green Chem. 2017;19(4):1063–1074. doi: 10.1039/C6GC02744J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester S. J. et al., Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018;98(3):1627–1738. doi: 10.1152/physrev.00038.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar S. Cozza G. Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008;8(18):1555–1572. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- Wang L. et al., Using self-organizing map (SOM) and support vector machine (SVM) for classification of selectivity of ACAT inhibitors. Mol. Diversity. 2013;17(1):85–96. doi: 10.1007/s11030-012-9404-z. [DOI] [PubMed] [Google Scholar]
- Zlokovic B. V. et al., Vascular contributions to cognitive impairment and dementia (VCID): a report from the 2018 National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke Workshop. Alzheimer's Dementia. 2020;16(12):1714–1733. doi: 10.1002/alz.12157. [DOI] [PubMed] [Google Scholar]
- Petersen J. et al., Designing Poly-agonists for Treatment of Metabolic Diseases: Challenges and Opportunities. Drugs. 2019;79(11):1187–1197. doi: 10.1007/s40265-019-01153-6. [DOI] [PubMed] [Google Scholar]
- Camaggi C. M. et al., Serum albumin-bound proteomic signature for early detection and staging of hepatocarcinoma: sample variability and data classification. Clin. Chem. Lab. Med. 2010;48(9):1319–1326. doi: 10.1515/CCLM.2010.248. [DOI] [PubMed] [Google Scholar]
- Cabitza F. Banfi G. Machine learning in laboratory medicine: waiting for the flood? Clin. Chem. Lab. Med. 2018;56(4):516–524. doi: 10.1515/cclm-2017-0287. [DOI] [PubMed] [Google Scholar]
- Lo Y.-C. et al., Machine learning in chemoinformatics and drug discovery. Drug discovery today. 2018;23(8):1538–1546. doi: 10.1016/j.drudis.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. et al., From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug discovery today. 2017;22(11):1680–1685. doi: 10.1016/j.drudis.2017.08.010. [DOI] [PubMed] [Google Scholar]
- Ekins S. et al., Combining Metabolite-Based Pharmacophores with Bayesian Machine Learning Models for Mycobacterium tuberculosis Drug Discovery. PLoS One. 2015;10(10):e0141076. doi: 10.1371/journal.pone.0141076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. et al., Evaluation and Identification of the Neuroprotective Compounds of Xiaoxuming Decoction by Machine Learning: A Novel Mode to Explore the Combination Rules in Traditional Chinese Medicine Prescription. BioMed Res. Int. 2019;2019:6847685. doi: 10.1155/2019/6847685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzi L. Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019;20(18):4331. doi: 10.3390/ijms20184331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikia S. Bordoloi M. Molecular Docking: Challenges, Advances and its Use in Drug Discovery Perspective. Curr. Drug Targets. 2019;20(5):501–521. doi: 10.2174/1389450119666181022153016. [DOI] [PubMed] [Google Scholar]
- DesJarlais R. L. Dixon J. S. A shape- and chemistry-based docking method and its use in the design of HIV-1 protease inhibitors. J. Comput.-Aided Mol. Des. 1994;8(3):231–242. doi: 10.1007/BF00126742. [DOI] [PubMed] [Google Scholar]
- Zhao C. et al., Computer-aided discovery of aminopyridines as novel JAK2 inhibitors. Bioorg. Med. Chem. 2015;23(5):985–995. doi: 10.1016/j.bmc.2015.01.016. [DOI] [PubMed] [Google Scholar]
- Mirza S. B. et al., Discovery of selective dengue virus inhibitors using combination of molecular fingerprint-based virtual screening protocols, structure-based pharmacophore model development, molecular dynamics simulations and in vitro studies. J. Mol. Graphics Modell. 2018;79:88–102. doi: 10.1016/j.jmgm.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Signorelli J. Shah Gandhi A. Cobimetinib. Ann. Pharmacother. 2017;51(2):146–153. doi: 10.1177/1060028016672037. [DOI] [PubMed] [Google Scholar]
- Gutzmer R. et al., Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;395(10240):1835–1844. doi: 10.1016/S0140-6736(20)30934-X. [DOI] [PubMed] [Google Scholar]
- Hoeflich K. P. et al., Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012;72(1):210–219. doi: 10.1158/0008-5472.CAN-11-1515. [DOI] [PubMed] [Google Scholar]
- Larkin J. et al., Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014;371(20):1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- Singh A. et al., Targeted inhibition of MEK1 by cobimetinib leads to differentiation and apoptosis in neuroblastoma cells. J. Exp. Clin. Cancer Res. 2015;34(1):104. doi: 10.1186/s13046-015-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S. et al., Efficacy of the MEK Inhibitor Cobimetinib and its Potential Application to Colorectal Cancer Cells. Cell. Physiol. Biochem. 2018;47(2):680–693. doi: 10.1159/000490022. [DOI] [PubMed] [Google Scholar]
- Seipel K. et al., The Cellular p53 Inhibitor MDM2 and the Growth Factor Receptor FLT3 as Biomarkers for Treatment Responses to the MDM2-Inhibitor Idasanutlin and the MEK1 Inhibitor Cobimetinib in Acute Myeloid Leukemia. Cancers. 2018;10(6):170. doi: 10.3390/cancers10060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.