

Abstract
Background
Neoantigens, new immunogenic sequences arising from tumor mutations, have been associated with response to immunotherapy and are considered potential targets for vaccination. Hepatocellular carcinoma (HCC) is a moderately mutated tumor, where the neoantigen repertoire has not been investigated. Our aim was to analyze whether tumors in HCC patients contain immunogenic neoantigens suitable for future use in therapeutic vaccination.
Methods
Whole-exome sequencing and RNAseq were performed in a cohort of fourteen HCC patients submitted to surgery or liver transplant. To identify mutations, single-nucleotide variants (SNV) originating non-synonymous changes that were confirmed at the RNA level were analyzed. Immunogenicity of putative neoAgs predicted by HLA binding algorithms was confirmed by using in vitro HLA binding assays and T-cell stimulation experiments, the latter in vivo, by immunizing HLA-A*02.01/HLA-DRB1*01 (HHD-DR1) transgenic mice, and in in vitro, using human lymphocytes.
Results
Sequencing led to the identification of a median of 1217 missense somatic SNV per patient, narrowed to 30 when filtering by using RNAseq data. A median of 13 and 5 peptides per patient were predicted as potential binders to HLA class I and class II molecules, respectively. Considering only HLA-A*02.01- and HLA-DRB1*01-predicted binders, 70% demonstrated HLA-binding capacity and about 50% were immunogenic when tested in HHD-DR1 mice. These peptides induced polyfunctional T cells that specifically recognized the mutated but not the wild-type sequence as well as neoantigen-expressing cells. Moreover, coimmunization experiments combining CD8 and CD4 neoantigen epitopes resulted in stronger CD8 T cell responses. Finally, responses against neoantigens were also induced in vitro using human cells.
Conclusion
These results show that mutations in HCC tumors may generate immunogenic neoantigens with potential applicability for future combinatorial therapeutic strategies.
Keywords: antigens, liver neoplasms, immunotherapy, active, immunogenicity, vaccine
Introduction
Current therapies for advanced stage hepatocellular carcinoma (HCC), such as targeted therapies based on receptor tyrosin kinase inhibitors, provide a limited efficacy,1 indicating the necessity of more efficacious treatments. In this regard, immunomodulatory therapies may be useful in the treatment of HCC.2 The immune response, in addition to its initial role during cancer generation, can also provide a beneficial effect by controlling tumor growth. Thus, the presence of tumor infiltrating T cells is associated with a better survival.3–5 However, subsequent upregulation of immune-inhibitory mechanisms, finally leads to tumor escape.2 Thus, immune enhancing therapies like checkpoint inhibitors (CPI), antibodies against CTLA-4 and PD-1 were initially tested6 and approved as second-line therapy,7 8 and recent trials have led to approval of therapies combining blockade of PD-L1 + vascular endothelial growth factor (VEGF)9 and PD-1 +CTLA-4.10 However, as in other malignancies, the rate of response to CPI is still far from being satisfactory, with only 15%–35% of responder patients. Low tumor lymphocytic infiltration has been proposed as a mechanism explaining poor responses to CPI,11 suggesting that strategies aimed at ‘heating’ these tumors may be used to improve response rates to CPI. Neoantigens (neoAgs) are new sequences arising from tumor mutations that can be recognized by T-cells and become targets of antitumor immunity.12 13 Some responses mediated by T-cells reinvigorated by CPI therapies are directed against neoAgs.14 15 Moreover, tumor mutational burden (TMB), and presumably the concomitant neoAg load, are associated with the response rate to CPI.16 As opposed to heavily mutated tumors, like melanoma or lung cancer, HCC is a tumor with a low/intermediate TMB (five mutations/Mbase).17 Therefore, it is of interest to characterize whether this TMB would be sufficient for the generation of immunogenic neoAgs in HCC patients, allowing thus their use in future neoAg-based therapies, such as vaccination or adoptive T cell therapies with neoAg-specific transgenic TCRs.
With this aim, in a small cohort of HCC patients we analyzed the presence of mutations and their capacity to behave as neoAgs. By means of algorithms predicting HLA-binding capacity, and by using HLA-A*02.01 and HLA-DRB1*01 as representative HLA class I and class II alleles, we demonstrated in vitro binding of neoAgs to HLA, immunogenicity in vivo with HLA transgenic mice and in vitro with human T cells, and the capacity of neoAg-specific T cells to recognize tumor cells expressing mutated genes, suggesting that these sequences can be incorporated in antitumor vaccines.
Materials and methods
Patients and samples
Fourteen HCC patients classified as BCLC-A were studied. Samples of tumor (T) and non-tumor (NT) tissue were obtained at the time of surgery or transplant, kept in RNAlater (ThermoFisher) and frozen. Samples and data from patients 10,584 to 10,535 were provided by the Biobank of the University of Navarra and were processed following standard operating procedures approved by the Ethical and Scientific Committees. All samples were sent to Personal Genomics (Verona, Italy) for whole-exome sequencing (WES) and RNAseq experiments.
WES and RNAseq
DNA samples were processed using Twist Exome Kit and sequenced on Illumina platform generating 2×150bp-reads. All sequenced fragments were mapped to the reference hg38 using BWA-mem V.0.7.15 and variant calling was performed using MuTect2. RNA-seq library preparation was performed using NEBNext Ultra II Directional RNA Library Prep Kit and NebNext rRNA Depletion kit (NEB). Libraries were sequenced on an Illumina HiSeq 3000 machine using 150nt paired-end reads. Raw sequencing reads were quality filtered and trimming of Illumina adapter was performed. Then, filtered reads were mapped on human reference genome hg38 (Ensemble release 84) using HiSat2 V.2.1.0 and gene expression quantification was performed using StringTie V.1.3.3. Data can be accessed at GEO (GSE193567).
Identification of potential neoantigens
Mutations were filtered in the following order: (1) selection of missense single-nucleotide variants (SNV), without considering INDELs or splicing variants; (2) exclusion of mutations already annotated in databases (using dbSNP, CentoMD and Cosmic Mutant Export databases) and (3) selection of mutated sequences expressed only in tumor tissue and not in NT, according to RNAseq. Resulting mutations were manually reviewed using IGV 2.5. Next, we designed 29 mer peptides containing the mutation flanked by 14 residues at each side. Binding capacity to patients’ HLA alleles of all 9–11 mer peptides (for class I) and 15 mers (for class II) containing the mutation was tested in silico. Predictions were also carried out with the non-mutated peptide versions. We used NetMHCpan 4.0 for class I peptides and NetMHCIIpan 3.2 for class II peptides. Peptides were considered as binders when %Rank values<2 for class I and %Rank values<10 for class II.
Peptides and plasmids
Peptides (GeneCust; Boynes, France) used in initial screening of HLA binding and immunogenicity had >80% purity. Active peptides used in subsequent experiments had >90% purity. All peptides were solubilized in PBS containing 10% DMSO and preserved at −20°C.
Genes coding an elongated (e) version of KYV epitope or five epitopes arranged in tandem (eLKR/eALL/eQQW/eKVY/eEVT), flanked by the MHC class I signal peptide (SP) and trafficking signal domain (MITD) attached to the N and C terminus, respectively with a SP and a MITD attached to the N and C terminus, respectively,18 were synthesized (Genscript) and cloned in plasmid MSCV-IRES-Thy1.1 DEST,19 a gift from Anjana Rao (Addgene plasmid # 17442), to generate plasmids MSCV- SP-eKYV-MITD-IRES-Thy1.1 and MSCV-SP/neoAgs/MITD- IRES-Thy1.1, respectively.
Mice
HHD-DR1 mice (B2mtm1Unc H2-Ab1tm1Doi Tg(HLA-A/H2-D/B2M)1Bpe Tg(HLA-DR1)/Orl), encoding human HLA-A*02.01 and HLA-DRB1*0120 were obtained from Dr. F. Lemonnier (Paris, France) and breed in our facilities. After study approval by the review committee (ref# 045–19), mice were maintained in pathogen-free conditions and treated according to guidelines of our institution.
Cell lines
HLA-A*02.01+ T2 cells21 and HLA-DRB1*01+ HOM2 cells22 were used for in vitro binding assays. They were grown in complete medium (RPMI-1640 (Lonza) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin). HEK293 (ECACC) and HEP-G2 cells (obtained from Dr. M. Ávila, CIMA, Spain), were grown in DMEM (Lonza) with 10% FBS and antibiotics. Transient expression of neoAgs in HEK293 cells was obtained after transfection with Lipofectamine 2000 (ThermoFisher) and MSCV- SP-eKYV-MITD-IRES-Thy1.1 plasmid. HEP-G2 cells expressing neoantigens were generated by retroviral (RV) infection with a RV generated with plasmid MSCV-SP/neoAgs/MITD- IRES-Thy1.1 using PLATE cells (American Type Culture Collection) as virus packaging cells. They were routinely tested for mycoplasma.
Peptide binding assays
Binding to HLA molecules was determined as described.21 22 Briefly, in the case of HLA-A*02.01, T2 cells were cultured with different peptide concentrations for 24 hours and the mean fluorescence intensity (MFI) of HLA expression was determined by flow cytometry, using an anti-HLA-A*02 antibody (GeneTex, Ref#GTX75804). Binding was expressed as Fluorescence Index (FI): (MFI with peptide – MFI without peptide)/MFI without peptide.
For HLA-DRB1*01 binding, HOM2 cells were cultured with the biotinylated control peptide APK (APKYVKQNTLKLATG) (10 µM) plus experimental peptides (100 µM). One day later, after washing and incubating with streptavidin-PE (BioLegend #Ref:405204), peptide binding was indirectly measured by flow cytometry, as loss of fluorescence in competition with control peptide. Peptide inhibition was calculated as: 100 – (MFI with peptide – MFI without peptide)/(MFI biot APK – MFI no peptide), and Binding Score (BS) corresponded to: % inhibition with experimental peptide/% inhibition with unbiotinylated APK. Peptides with FI and BS values >0.5 were considered as binders.
Mice immunization
Eight weeks HHD-DR1 mice were vaccinated with individual peptides (100 nmoles/mouse) combined with polyI:C (Amersham) and anti-CD40 (FGK4.5; Bioxcell), both being administered at 50 µg/mouse. Class I-restricted peptides were administered subcutaneously, whereas class II-restricted peptides were administered intravenously and were also given in a boost 1 week later to increase their immunogenicity. Seven days after the last immunization, mice were sacrificed, and spleens obtained.
Analysis of murine T cell responses
T cell responses induced by vaccination were determined using an IFN-γ ELISPOT Set from BD-Biosciences as described.23 Briefly, after spleen homogenization and erythrocyte lysis, cells (8×105/well) were stimulated with different peptide concentrations (10–0.01 µM) or irradiated tumor cells (8×104 cells/well) for 24 hours. The number of spot-forming cells was enumerated with an ImmunoSpot automated counter using Immunospot Image Acquisition 4.5 and Inmunospot V.3 software. Results obtained in control wells containing cells in the absence of peptide were subtracted from antigen-stimulated wells.
In some cases, responses were determined by flow cytometry. Splenocytes were stimulated for 4 hours with 10 µM of the peptide in the presence of GolgiStop and GolgiPlug (BD-Biosciences) and antiCD107a-FITC (1D4B Ref#553793; BDPharmingen). Then cells were surface stained with the following antibodies: CD3ε-Percp-Cy5 (145-2 C11 Ref#100327), CD4-FITC (RM4-5 Ref#100510), CD8-BV421 (53-6.7 Ref#10753) from BioLegend. Next, cells were fixed and permeabilized using BD Cytofix/Cytoperm Fixation/Permeablization Kit and intracellularly stained with IFNγ-PE (XMG1.2 Ref#562020) and TNFα-BV510 (BioLegend, clone MP6-XT22, Ref#506339) antibodies. Dead cells were excluded from the analysis using Maleimide (PromoKine). Samples were acquired with a FACSCantoII (Becton Dickinson) flow cytometer for binding assays and with Cytoflex (Beckman Coulter) for stimulation assays. All data were analyzed using FlowJo software (Tree Star).
Analysis of human T cell responses
Eight HLA-A*02:01 positive healthy individuals were recruited at the Freiburg University Medical Center, Germany. HLA typing was performed by next-generation sequencing. Written informed consent was obtained from all participants and the study was conducted according to federal guidelines, local ethics committee regulations (Albert-Ludwigs-Universität, Freiburg, Germany; no. 322/20) and the Declaration of Helsinki (1975). Venous blood samples were collected in ethylenediaminetetraacetic acid-anticoagulated tubes. PBMCs were isolated with lymphocyte separation medium density gradients (Pancoll separation medium, PAN Biotech) and resuspended in RPMI 1640 medium supplemented with 10% FCS, 1% penicillin/streptomycin and 1.5% HEPES buffer 1 mol/L (complete medium; all additives from Thermo Scientific). Monocyte-derived dendritic cells (MD-DCs) were generated from freshly isolated CD14+ PBMCs. In particular, CD14+ monocytes were separated by CD14 microbeads (Miltenyi) according to the manufacturer’s instruction. The flow-through (CD14- cells) was frozen for the subsequent isolation of CD8 T cells. Approximately 1–2 million CD14+ monocytes were cultured for 24 hours in complete RPMI culture medium containing rhGM-CSF (Peprotech; 10 ng/mL) and rhIL-4 (Peprotech; 2 ng/mL). After 24 hours, rhTNF (R&D; 10 ng/mL), IL-1β (Peprotech; 10 ng/mL) and PGE2 (Sigma-Aldrich; 1 µM) were added to each well for maturation of MD-DCs. Cells were cultured for additional 24 hours before cells were collected and loaded with peptide (10 µg/mL) for 1 hour, washed three times and then used for MD-DCs/T-cell co-culture. CD8+ T cells were isolated from the CD14- flow-through by CD8 microbeads (Miltenyi) according to the manufacturer’s instruction. Peptide-loaded MD-DCs and isolated CD8+ T cells were co-cultured for 21 days in a 24-well plate at a ratio of 1:30 in complete RPMI culture medium containing anti-human CD28 mAb (eBioscience; 0.5 µg/mL). The co-culture was fed on d3, 7, 10, 14 and 17 with new RPMI culture medium containing IL-2 (Stemcell Technologies, 20 U/mL). NeoAg-specific CD8 +T cells were detected by intracellular cytokine production. NeoAg-derived peptides (10 µg/mL) were added to the cells for 1 hour at 37°C. Then, brefeldin A (GolgiPlug, 0.5 µL/mL) and monensin (GolgiStop, 0.5 µL/mL) (all BD Biosciences, Germany) were added for additional 5 hours, followed by CD8 and intracellular IFNγ staining were carried out. Stimulation with PMA (50 ng/mL) and Ionomycin (1 µg/mL) (all Sigma, Germany) was performed as positive control. Unstimulated controls were used for background subtraction.
Statistical analysis
Statistical analyses (Student’s t test and one-way analysis of variance with Bonferroni’s multiple comparison test) were performed with GraphPad Prism (GraphPad) software V.7. A p<0.05 was taken to represent statistical significance.
Results
Analysis of mutations in tumors from HCC patients
To identify immunogenic neoAgs arising from mutations found in HCC patients, we obtained samples from a cohort of 14 patients with HCC (BCLC-A) of different etiologies. Data from WES and RNAseq experiments were used for mutation screening. According to our annotation criteria, focused only on SNV (without considering INDELs or splicing variants), WES data showed a median value of 1217 mutations/patient, narrowed to a median of 351 in the case of missense mutations. Moreover, since we were only interested in new neoAgs not previously described and annotated in databases, the number decreased to a median of 253 (range 190–1053). Finally, an additional filter to confirm expression of the mutated sequence only in tumor tissue was applied to 13 patients with available RNAseq data, resulting in a median of 30 mutations/patient (range 9–94) (table 1 and online supplemental table S1).
Table 1.
Number of mutations and potential neoantigens in HCC patients included in the study
| Patient | Mutations | Neoantigens | ||||||
| Total MUT | Missense | No data base | RNA in T | No RNA in NT | Predicted HLA-I Binders* | Predicted HLA-II Binders* | Total Predicted Binders* | |
| 10 584 | 1217 | 350 | 263 | 16 | 15 | 7 | 3 | 10 |
| 10 594 | 1115 | 267 | 196 | 19 | 18 | 8 | 3 | 11 |
| 10 615 | 4018 | 1247 | 273 | 41 | 38 | 10 | 4 | 14 |
| 10 619 | 1112 | 276 | 196 | 21 | 21 | 12 | 7 | 19 |
| 10 622 | 984 | 270 | 190 | 26 | 22 | 13 | 4 | 17 |
| 10 627 | 1083 | 328 | 235 | 14 | 13 | 7 | 1 | 8 |
| 10 628 | 1218 | 296 | 200 | 10 | 9 | 6 | 1 | 7 |
| 10 632 | 1204 | 353 | 243 | 33 | 31 | 14 | 5 | 19 |
| 10 634 | 1103 | 274 | 205 | – | – | – | – | – |
| 10 635 | 1308 | 380 | 296 | 30 | 30 | 17 | 14 | 31 |
| HLA063 | 5185 | 1409 | 1093 | 101 | 94 | 42 | 26 | 65 |
| HLA066 | 4318 | 1061 | 819 | 60 | 42 | 26 | 9 | 35 |
| HLA069 | 2862 | 735 | 544 | 40 | 34 | 20 | 7 | 27 |
| HLA078 | 5118 | 1560 | 718 | 38 | 32 | 19 | 5 | 24 |
| Median | 1217.5 | 351.5 | 253 | 30 | 30 | 13 | 5 | 19 |
*Number of mutations originating peptides with in silico predicted capacity to bind to HLA class I or class II molecules.
HCC, hepatocellular carcinoma; NT, non-tumor.
jitc-2021-003978supp001.xlsx (208.2KB, xlsx)
After filtering the mutations with all the criteria explained above, we used MHC binding algorithms to predict binding of mutated peptides to patients’ own HLA molecules. We selected only those peptides whose WT sequences were predicted as non-binders. This yielded a median of 19 (range 7–68) potential predicted neoAgs per patient (13 for class I and 5 for class II molecules) (table 1 and online supplemental table S2). Analyses of mutations included in predicted neoAgs showed that only six mutations (1.5% of the total selected) were shared by more than one patient, in all cases being present only in two patients (online supplemental table S1). All of them were included in peptides predicted as potential neoAgs, presented at least by one HLA molecule. However, only one mutation originated a neoAg potentially presented by the same HLA allele in two patients (online supplemental table S2).
jitc-2021-003978supp002.xlsx (149.9KB, xlsx)
Interestingly, about 21% of mutations were contained in peptide cores that could be presented by more than one class I HLA allele (online supplemental table S2), and in other cases, different peptide cores containing the same mutation could be presented by the same HLA allele, increasing thus the possibility of neoAg generation from the same mutation.
TMB has been associated in other tumors with lymphocytic infiltrate. By using CIBERSORT software, a tool previously used in HCC samples to estimate relative contribution of immune subsets in the tumor infiltrate,24 we determined lymphocytic infiltration from RNAseq data in paired T and NT samples (online supplemental figure S1A) in a subgroup of nine patients. There were no statistically significant differences between T and NT samples, except in the proportion of plasma cells (p=0.004) and NK cells (p=0.04), predominant in NT, and M0 macrophages (p=0.04), over-represented in tumors (online supplemental figure S1B). No correlations were observed when analyzing total mutations, missense mutations and class I or class II predicted binder peptides with respect proportions of estimated T cells, either total or CD8 and CD4 T cells (online supplemental figure S1C).
jitc-2021-003978supp003.pdf (168.5KB, pdf)
Identification of HLA-A*02.01-restricted neoAgs: binding capacity and immunogenicity
Availability of in vitro and in vivo tools in the HLA-A*02.01 setting allowed us to use this HLA allele as a model in the immunological characterization of mutated peptides predicted as binders in seven HLA-A*02.01+ patients. We selected a set of 27 9-10 mer peptides (median of 4 peptides/patient) (figure 1A). Eleven mers were not tested since most of them contained their corresponding 9–10 mers. In vitro assays confirmed binding to HLA-A*02.01 of 19 of these peptides (70%) with different affinities (figure 1B, C), where six out of seven patients had peptides binding to this HLA allele. Immunization of HHD-DR1 transgenic mice with these peptides plus poly (I:C) and antiCD40 adjuvants revealed that 12 out of 19 confirmed binder peptides (66%) were immunogenic in IFN-γElispot assays (figure 2A). WT peptides (except for peptide GLS), were not recognized by T cells induced by the corresponding mutated peptide. Indeed, WT versions of immunogenic peptides (with the exception of peptides KYV and ALL), were unable to bind (online supplemental figure S2), in agreement with predictions. Moreover, with the exception of WT KYV, none of the WT versions of three other representative highly immunogenic neoAg peptides (RIE, ALL and FLE) was immunogenic (online supplemental figure S2). Regarding non-binder peptides, none of them elicited an immune response. T cells from mice immunized with stronger peptides could still recognize them at the nanomolar range (figure 2B) and these CD8 T cells were polyfunctional, producing not only IFN-γ, but also TNF-α and the cytotoxicity-associated molecule CD107 (figure 2C). Of note, some mutations were present in overlapping peptides containing different lengths. Peptides sharing the same binding core sequence (CQQ and QQW; RAL and ALF) showed equivalent immunogenicity, as well as those with different binding cores (ALL and FLE), allowing the potential design of longer polyepitopic peptides.
Figure 1.

Binding of mutated peptides to HLA-A*02.01 molecules. (A) Sequences of mutated peptides predicted as binders to HLA-A*02.01 molecules with their corresponding WT version. (B) Mutated peptides were tested in binding assays at 100 µM using T2 cells. Results are expressed as Fluorescence Index (FI). Peptides above the 0.5 FI threshold (horizontal bar) were considered positive. Results correspond to the mean of three to six different assays. (C) Selected binder peptides were titrated with decreasing peptide concentrations.
Figure 2.
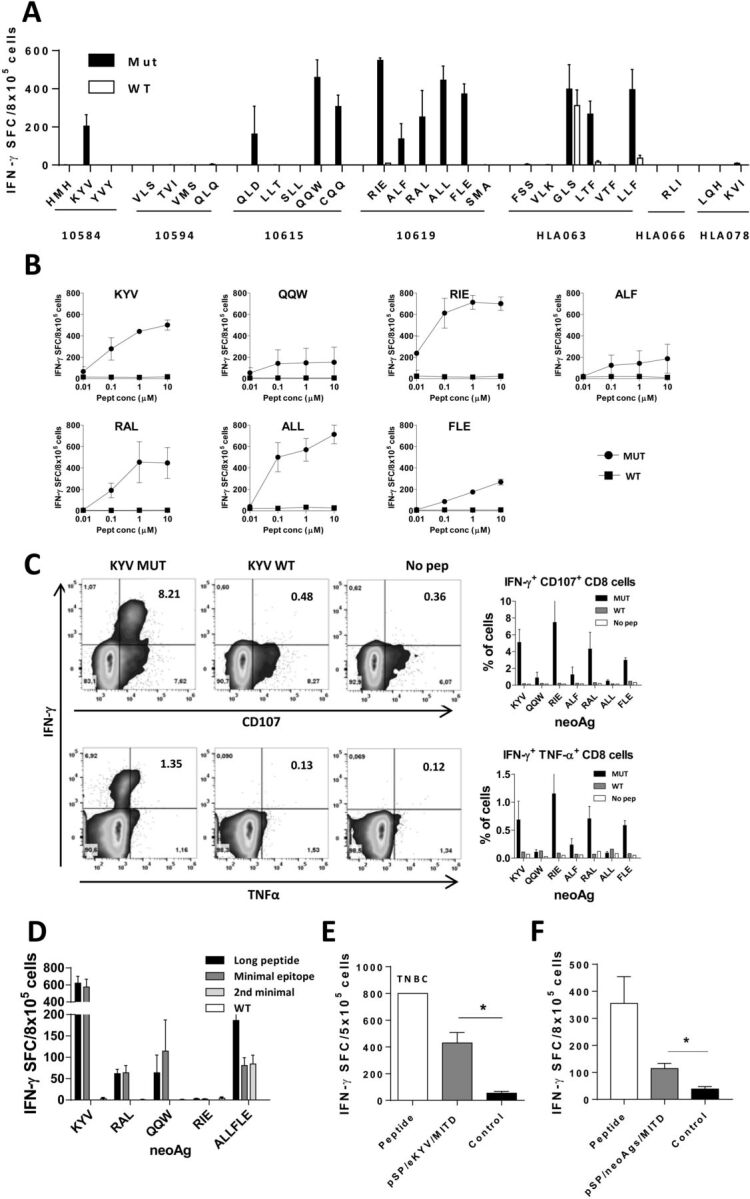
Immunogenicity of mutated HLA-A*02.01-restricted peptides in HHD-DR1 mice. (A) HHD-DR1 mice (n=3–6/group) were immunized with individual peptides plus poly(I:C) and anti-CD40 adjuvants. Ten days later splenocytes were stimulated with the mutated or WT version of each peptide and responses measured by ELISPOT. (B) Selected immunogenic peptides (n=3 mice/peptide) were also tested against decreasing concentrations of mutated and WT peptides. (C) Splenocytes from the same immunized mice were stimulated with the mutated and WT version of the peptides and expression of IFN-γ, TNF-α and CD107 in CD8 T cells were measured by flow cytometry. Representative example of peptide KYV (left panels) and summarized results (right panels) are shown. (D) HHD-DR1 mice (n=4/group) were immunized with longer versions (containing four extra amino acids at each end) of neoAg peptides and splenocytes were stimulated in vitro with the long peptide and the minimal epitope. (E) Splenocytes from mice immunized with peptide KYV (n=4) were stimulated with the peptide or with HEK-293 cells transfected with a plasmid encoding an elongated version of KYV (pSP/eKYV/MITD) or mock transfected (control). (F) Splenocytes from mice (n=4) immunized with peptide KYV were stimulated with the peptide or with HepG2 cells transfected with a plasmid encoding an elongated version of neoAgs LKR, ALL, QQW, KVY and EVT (pSP/neoAgs/MITD) or mock transfected (control). TNBC, too numerous to be counted. *p< 0.05.
Immunogenicity of human peptides in mice may depend on species-specific differences. To discard this possibility, we checked the human/murine homology of all immunogenic neoAg peptides. Most immunogenic neoAgs had a homology with the murine counterpart above 80% and three of them (RIE, FLE and GLS) were 100% identical (online supplemental figure S3A). Moreover, peptides corresponding to the murine version of KYV and QQW (with 80% and 67% to the human sequence), modified to contain the mutated amino acid found in HCC patients, were also immunogenic in mice, showing a higher specificity for the mutated sequence. However, nil or very low immunogenicity was observed when using as immunogens the WT murine version (online supplemental figure S3B). These results suggest that immunogenicity depends more on the presence of tumor-specific mutations than on differences between human and murine sequences. In summary, our results confirmed in the HLA-A*02.01 setting that 12 out of 27 mutated peptides (44%) from HCC samples predicted by HLA binding algorithms bind to HLA and are immunogenic in vivo in the murine setting.
Once immunogenicity was demonstrated, we tested processing of some identified neoAgs to confirm their generation by antigen presenting cells (APC) and tumor cells. In a first approach in vivo, we immunized with longer peptides spanning the minimal epitope plus four natural flanking residues at both ends. Except peptide RIE, longer versions of the peptides were immunogenic, inducing responses that recognized both the longer version and the minimal epitope, but not the minimal WT version. In the case of the longer version containing the overlapping epitopes ALL and FLE, it induced responses against both minimal epitopes (figure 2D).
In addition, using peptide KYV as model, we transfected HEK293 cells (HLA-A*02.01+) with a plasmid encoding the longer version of this peptide. KYV-specific T-cells recognized in vitro neoAg-expressing cells, but not control cells (figure 2E). The same was true for recognition of HepG2 cells transfected with a plasmid expressing this neoAg (figure 2F). Globally, these experiments carried out in vivo, with long peptides, and in vitro, with neoAg-transfected cells, suggest that both APC and non-APC generate these epitopes for T cell priming and recognition, respectively. Moreover, since HEK293 and HepG2 cells are of human origin, our results indicate that this peptide is generated not only by murine, but also by the human processing machinery.
Binding capacity and in vivo immunogenicity of HLA-DRB1*01-restricted peptides
We next analyzed class II-restricted peptides by using as model HLA-DRB1*01 molecules. Potential binder peptides were selected using data from the three HLA-DRB1*01+ patients (figure 3A). Since several overlapping peptides containing the same mutation were predicted as binders, and HLA class II molecules are open at both sides, peptides spanning 15–21 amino acids were tested. Binding was experimentally confirmed for 8 out of 10 (80 %) predicted peptides (figure 3B). Binding of the 15 mer versions contained in peptides with longer sequences (LKR and RLV; 21 and 17 mer, respectively) was also determined, confirming the binding capacity of most 15 mers predicted by algorithms (online supplemental figure S4A). Vaccination experiments demonstrated that 6 out of 10 predicted peptides were immunogenic in HHD-DR1 mice, with 4 of them showing prominent responses (figure 3C). Non-binder peptides SQL and SPE had null or poor immunogenicity. T cells obtained after immunization with mutated peptides showed a null (EHP and EVT) or lower (EHP and RLV) degree of cross-recognition of the WT sequence (figure 3D). Regarding binding of these WT versions, only those corresponding to LKR y RLV, did bind to HLA-DRB1*01 molecules (online supplemental figure S4B).
Figure 3.

Binding of mutated peptides to HLA-DRB1*01 molecules and immunogenicity. (A) Sequences of mutated peptides predicted as binders to HLA-DRB1*01 molecules with their corresponding WT version. (B) Mutated peptides were tested in binding assays at 100 µM using HOM2 cells. Results are expressed as Binding Score (BS). Peptides above the 0.5 BS threshold (horizontal bar) were considered positive. Results correspond to the mean of three different assays. (B) Mutated peptides were used to immunize HHD-DR1 mice (n=3 mice/peptide) and responses were measured by ELISPOT. (C) Immunogenic mutated peptides (n=3 mice/peptide) were tested against decreasing peptide concentrations of the mutated and WT versions.
Identification of new neoAgs based on mutations pointing to putative TCR contact residues
In our first screening approach, selection criteria included mutated peptides with clear HLA binding capacity whose corresponding WT sequences were predicted as non-binders, avoiding thus presentation and recognition of WT sequences on NT cells. However, analysis of mutated positions in some peptides suggested that these amino acids might not have a prominent role on HLA binding but on TCR recognition, indicating that improved HLA presentation may not be the cause of their enhanced immunogenicity. To increase thus the set of potential neoAgs, we searched for binder peptides with mutations generating significant size and/or chemical changes and located at putative TCR contact sites. A new set of neoAgs potentially non-cross reactive with WT sequences restricted by HLA-A2.01 and HLA-DRB1*01 was predicted (online supplemental table S3), some of them were selected and tested (figure 4A). Despite predicted as binders, only two out of five HLA-A2.01-restricted peptides demonstrated binding capacity (figure 4B). However, none of them was immunogenic (figure 4C). For HLA-DRB1*01-restricted peptides, three epitopes were predicted (figure 4A). Interestingly, two of them demonstrated clear HLA binding (figure 4D), which correlated with their immunogenicity (figure 4C), mainly recognizing the mutated but not the WT sequence (figure 4D). These results suggest that by using this second screening strategy a new set of immunogenic neoAgs can be identified.
Figure 4.
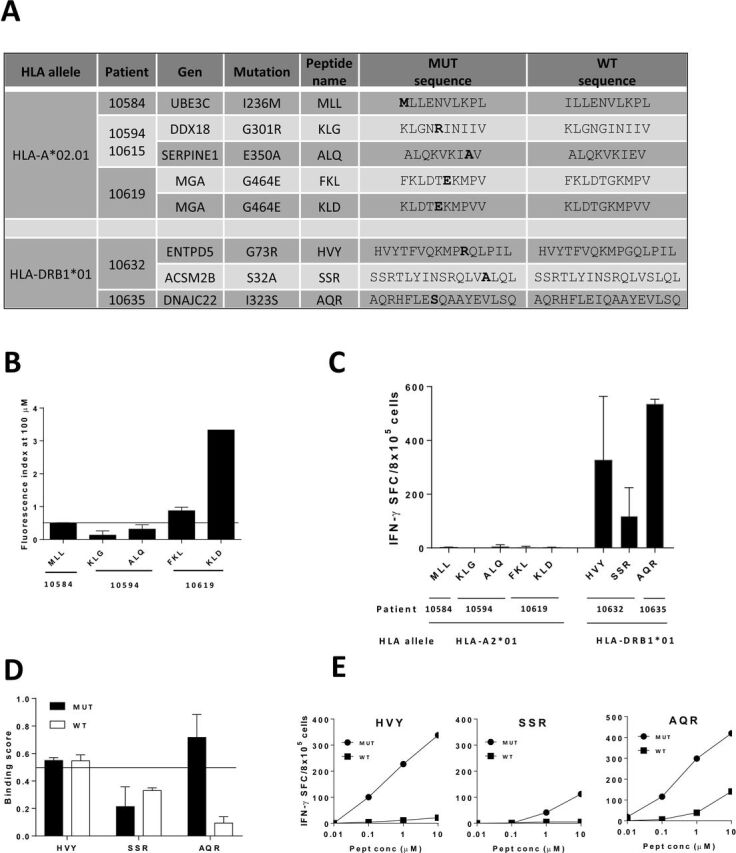
Identification of a new set of immunogenic neoAgs. (A) Sequences of mutated peptides with changes at non-anchor positions and predicted as binders to HLA-A*02.01 or HLA-DRB1*01 molecules, with their corresponding WT version. (B) Mutated peptides with predicted binding capacity to HLA-A*02.01 were tested in binding assays with T2 cells. Results are expressed as Fluorescence Index (FI) and peptides with FI >0.5 were considered positive. (C) HHD-DR1 mice (n=3 mice/peptide) were immunized with mutated peptides with changes at non-anchor positions and responses were evaluated by Elispot. (D) Mutated peptides with predicted binding capacity to HLA-DRB1*01 were tested in binding assays with HOM2 cells. Results are expressed as Binding Score (BS) and peptides with BS >0.5 were considered positive. (E) Immunogenic peptides (n=3 mice/peptide) were also tested against decreasing concentrations of mutated and WT peptides.
jitc-2021-003978supp004.xlsx (246.7KB, xlsx)
Help provided by neoAg-specific CD4 T cells enhances CD8 T cell responses
Due to the direct killing capacity of CD8 T cells, traditionally considered as the main antitumor effector subset, vaccines have mainly relied on CD8 epitopes. However, CD4 T cells play also a key role through their antitumor effects and by providing help for CD8 T cell priming and maintenance.25 To confirm this role in a neoAg-based vaccine, we immunized with the CD8 neoAg epitope KYV plus poly(I:C) adjuvant, with or without the CD4 neoAg epitope EVT. AntiCD40 antibodies were initially omitted to avoid confounding factors provided by this adjuvant, since they may mimic some functions provided by CD4 T cells.26 27 These experiments showed that immune responses induced by CD8 epitope KYV were enhanced when co-administered with the CD4 epitope EVT (figure 5A).
Figure 5.

Enhancement of CD8 T cell responses by help provided by CD4 T cells. HHD-DR1 mice (n=4 mice/peptide) were immunized with CD8 neoAg epitope KYV and poly(I:C), in combination with CD4 neoAg epitope EVT, antiCD40 antibodies or both. Responses against KYV (A) and EVT (B) were measured by Elispot. Results correspond to a representative experiment out of two carried out independently. Equivalent immunization experiments (n=4 mice/group) were carried out using other epitope combinations, sharing either the CD8 (C) or the CD4 (D) epitopes used in panels A, B. * p<0.05; ** p<0.01; *** p<0.001. NT, not tested.
In addition to CD40L, that induces DC maturation, CD4 T cells provide other factors for CD8 activity. To demonstrate whether CD4 neoAg epitopes could trigger these additional mechanisms, we repeated co-administration experiments, but in this case in the presence of antiCD40 antibodies. Although responses were higher than when using KYV alone, antiCD40 did not improve responses over those induced by KYV plus EVT (figure 5A), suggesting that antiCD40 can be replaced by CD4 epitopes in terms of help for CD8 activation. Finally, we tested responses against EVT. Interestingly, the poor responses against EVT induced by a suboptimal single immunization, were enhanced by antiCD40 antibodies (figure 5B). In addition to this particular CD8 and CD4 epitope combination, we tested new peptide combinations sharing either the CD8 or CD4 peptides, observing equivalent enhancement of CD8 responses when including a T helper epitope (figure 5C, D). These results confirm the helper capacity of neoAg-based CD4 epitopes, suggesting their inclusion in multiepitopic vaccines.
In vitro induction of T cell responses against neoAgs in humans
After demonstrating neoAg immunogenicity in a humanized murine model, potential differences in the T cell repertoire between mice and humans prompted us to study its immunogenicity in humans. Since no cells were available from HCC patients, we used cells from HLA-A*02.01+ healthy donors (donor HLA typing data is shown in online supplemental table S4). By repeated in vitro stimulation with mutant peptides KYV (figure 6A) and ALL (figure 6B) T cells were obtained that recognized better these neoAgs than the corresponding WT sequences. These results were observed in 25% (two out of eight) donors tested (figure 6C), demonstrating first their immunogenicity in humans and second, confirming the selective recognition of the mutated version. Interestingly, WT versions of peptides KYV and ALL were immunogenic in donor #1, always recognizing better the WT peptide (figure 6A, B).
Figure 6.
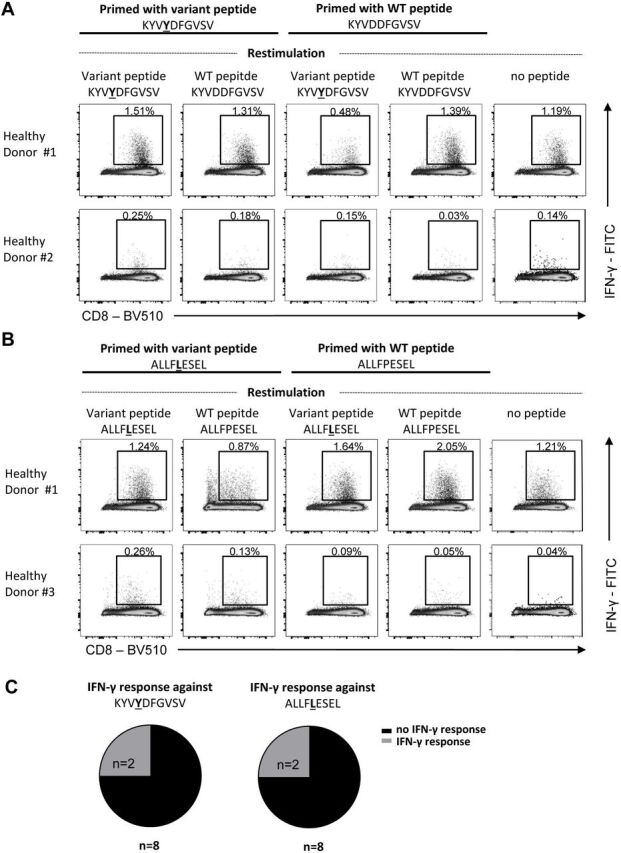
In vitro induction of human CD8 T cell responses against neoAgs in healthy donors. CD8 T cells form HLA-A*02.01 healthy donors were stimulated in vitro with monocyte-derived dendritic cells pulsed with neoAg CD8 epitope peptides KYV (A) or ALL (B) and expanded. (A, B) Dot plots showing IFN-γ production of in vitro primed CD8 +T cells after mutant or WT peptide-specific restimulation on day 21 (gated on bulk CD8 +T cells). Frequencies of IFN-γ+ cells within the CD8 +T cell population after subtracting values obtained in the absence of peptide restimulation are depicted. (C) Proportions of healthy donors showing an IFN-γ production after restimulation with the mutant peptides are depicted, respectively. Numbers of analyzed healthy donors are indicated.
jitc-2021-003978supp005.xlsx (9.8KB, xlsx)
Discussion
Despite promising initial results obtained in HCC patients when using CPI (anti-CTLA-4 or anti-PD-1)6–8 and the increased response rates observed with combinations,9 10 these therapies are still far from being optimal. Thus, new strategies are necessary to improve these results. Vaccination has been proposed to enhance antitumor immunity, and the use of neoAgs would promote tumor-specific responses in combination with CPI, as shown in other tumors.28 HCC has a low/medium tumor mutational load17 and the presence of mutations generating neoAgs suitable for immunization is unknown. In the present work, we have identified mutations in a cohort of HCC patients (BCLC-A) and characterized their potential as immunogenic neoAgs. TMB values obtained in our patient cohort are in line with those previously reported in the literature for HCC,16 far below those observed in patients with melanoma and lung cancer, which have been pioneer examples in the use of neoAg-based vaccines.28–30 It has been proposed that TMB would associate with tumor antigenicity, and concomitantly with lymphocyte infiltration and the possibility of therapeutic benefit when using CPI.14 31 In our cohort, we have not found any correlation between TMB or the number of predicted neoAgs and lymphocyte infiltration as determined with CIBERSORT. Although this lack of association might be true for tumors with low/medium mutational load, this may also depend on the small sample size used in our study, which would not allow concluding in this respect. Moreover, we have only considered non previously annotated SNV, and not indels or frameshift mutations, which would have modified these results. Different results have been reported regarding TMB/neoAgs and immune infiltration in HCC. A correlation has been observed between TP53-neoAgs and longer overall survival.32 Also, the ratio of observed/expected neoAgs is higher in HCC tumors with low copy-number alterations,33 associated with the highly inflamed ‘immune class HCC’.34 Moreover, high affinity neoAgs correlate better with OS than TMB, associated with the presence of neoAg-specific CD8 in the tumor.35 On the contrary, we have reported lack of correlation between neoAg burden with immune infiltration in patients not undergoing immunotherapy.36 Although conflicting, the majority of these results highlight the relevance of neoAgs in tumor inflammation and their potential implication in the control of HCC progression, suggesting their importance as targets for antitumor immunity.
After identification of mutations and applying different filters we found a median of 30 mutations per patient, narrowed down to a median of 19 putative neoAgs after using HLA binding prediction algorithms. Finally, taking as models HLA-A*02.01 and HLA-DRB1*01 alleles, approximately half of the tested peptides were immunogenic, a result in line with previous reports of immunogenicity of predicted neoantigen epitopes.30 37 Important efforts have been done during the last years to develop reliable algorithms to predict epitope immunogenicity. Different factors, including HLA binding capacity,38 dissimilarity with the self-proteome,39 gene expression, binding stability, recognition by T-cells40 or antigen processing41 among others, have been considered for neoAg screening, and some are being tested in patients. Deployment of these tools in the clinical setting will help to define the best options to optimize neoAg prediction for vaccine design.
A potential limitation of our study could be related to testing immunogenicity of human mutated sequences in a murine model, due to differences between murine and human antigens. However, a high homology was observed for most neoAgs, and in several examples of peptides with at least 70% homology, the mutated murine sequences were also immunogenic, suggesting that immunogenicity was mainly due to the presence of the mutation. In addition, peptides KYV and QQW (not identical in humans and mice) demonstrated their immunogenicity in humans, confirming their capacity to activate neoAg-specific T cells. T cells from healthy donors were used to test immunogenicity in humans, due to the lack of available lymphocytes from patients. Although neoAg-specific T-cells identified in patients help to confirm immunogenicity in the human setting,35 immunity in these individuals may be subjected to immunosuppressive elements, precluding the detection of responses in some cases, which may mask potential immunogenicity of these antigens. Moreover, most of these immunogenic peptides were processed in vivo, and neoAg-specific T-cells preferentially recognized the mutated but not the WT peptide, presumably due to the initial neoAg selection criteria used for, based on candidates with no binding capacity in the WT version. Interestingly, WT peptides of KYV and ALL neoAgs demonstrated experimental binding to HLA molecules, as opposed to WT versions of other neoAgs, and were immunogenic in HHD-DR1 mice (KYV) and in a healthy donor (KYV and ALL), although their immunogenicity was usually lower. These peptides were mutated at positions not preferentially involved in anchoring to HLA molecules,41 as opposed to other neoAgs. This led us to search for mutated peptides with HLA binding capacity and bearing changes at TCR contacting residues as a new source of immunogenic neoAgs.42 Although we did not detect immunogenic CD8 T cell epitopes in this small group, new CD4 T cell epitopes were found, suggesting that this strategy could be useful to widen the repertoire of neoAgs. In this regard, a recent comprehensive study using several murine tumors reported that neoAg immunogenicity depends on the position of the mutated amino acid.43 For MHC anchoring residues the relative affinity for MHC of the mutated vs the WT peptide determines immunogenicity, whereas for non-anchoring positions (presumably those pointing to the TCR) the absolute affinity of the mutant peptide is the best predictor, in agreement with our two screening strategies.
Analyses of immunogenicity showed that in 7 out of 10 patients who were either HLA-A*02.01+ (patients 10584, 10615, 10 619 and HLA063) (figure 2A) or HLA-DRB1*01+,(patients 10,632, 10,635 and HLA069) (figure 3C), some of the predicted peptides were immunogenic. Although this may suggest a low patient coverage for vaccine development, it has to be considered that, regarding HLA repertoire, our work only reflects neoAgs restricted by a single HLA allele, not considering remaining HLA genes (HLA-B or HLA-C for class I and HLA-DP and HLA-DQ for class II) and additional alleles in heterozygous individuals. Moreover, we have selected only SNV, omitting mutations already reported in databases, which in some cases have been already reported as neoAgs (eg, TP53-related neoAgs).32 Therefore, we believe that HCC patients harbor a sufficient number of neoAgs suitable for vaccine design.
In addition to the number of neoAgs, their expression levels and processing in the tumor context have to be taken into account. A recent report with a multiomics approach in HCC samples did not detect by mass spectrometry peptide/HLA complexes corresponding to those neoAgs identified using WES, while TAA-related peptides where detected,44 suggesting the low expression/processing levels of this type of antigens. However, despite this lack of detection using proteomics, sensitive immunological assays testing recognition of neoAg peptides or tumor cells by neoAg-specific T cells would be also useful to confirm the existence of immunogenic neoAgs in HCC. Indeed, T-cell responses against neoAgs have been recently reported in HCC patients, demonstrating the presence of immunogenic neoAgs in the pool of predicted high affinity neoAgs, which correlate with patient survival.35 Moreover, our results using tumor infiltrating lymphocytes from HCC patients have allowed us to detect neoAg-specific T cells.45
Although many efforts have been traditionally done to develop vaccination strategies aimed at activating CD8 T-cells, CD4 T-cells have emerged as important players, by mediating direct and indirect antitumor immunity,25 46 suggesting the inclusion of CD4 epitopes in polyvalent vaccines. Unfortunately, we did not have any patient expressing HLA-A*02.01 and HLA-DRB1*01. Hence, for the proof of concept of combined vaccines we used epitopes from two patients. Responses induced by a vaccine containing a CD8 epitope plus poly(I:C) adjuvant, a common strategy in neoAg-based clinical trials,29 47 were improved when a CD4 epitope was included, reaching levels similar to those obtained when using the CD8 epitope in combination with poly(I:C) and antiCD40 adjuvants. These results reinforce the use of CD4 epitopes in neoAg vaccines, not only for the direct function of CD4 T cells, but also for their helper role on other effector cells.
In summary, we have shown that mutations identified in HCC patients may originate immunogenic neoAgs able to activate CD8 and CD4 T cells, as demonstrated in two representative HLA alleles. Despite the low/medium mutational load observed in these individuals, the number of neoAgs found would allow the preparation of personalized vaccines targeting these sequences, potentially useful to increase the response rate of currently available HCC immunotherapies.
Acknowledgments
We particularly acknowledge the patients for their participation and the Biobank of the University of Navarra for its collaboration. Authors also thank Virginia Villar for her help with sample management.
Footnotes
Contributors: DR, LB, MH, RT and PS conceived and designed experiments. MI, BS and LB: patient recruitment. DR, SH-S, BS and PS analyzed data and wrote the manuscript. PS supervised the project. DR, MR, DL, LS, BA, JE, IT-U, EV, JG-B, CC, MT, AM, BC, CR, KH and CT performed the in silico, in vitro and in vivo studies. LB, MH, RT, BS and PS: funding. PS is responsible for the overall content as the guarantor. All authors reviewed and approved the manuscript.
Funding: This work was supported by grants from ERANET-TRANSCAN-2 within HORIZON 2020 (TRS−2016-00000383: www.hepamut.eu) to BS and LB, Instituto de Salud Carlos III, Fondo Europeo de Desarrollo Regional ‘Una manera de hacer Europa’ (PI20/00260) and the ‘Murchante contra el cáncer’ initiative to PS, Plan Estatal de I+D+I 2013-2016 and cofinanced by ISCIII-Subdirección General de Evaluación y Fomento de la investigación and Fondo Europeo de Desarrollo Regional (FEDER) (PI19/00742) to BS, German Research Foundation (DFG) (CRC/TRR17 to RT and CR)–Project ID 272983813 and CRC1479 (to MH)–Project ID: 441891347, and Gobierno de Navarra [Departamento de Salud (045-2017)] co-financed (50%) with FEDER funds (UE, FEDER 2014-2020 ‘Una manera de hacer Europa’) to SH-S.
Competing interests: BS reports consultancy fees from Adaptimmune, Astra Zeneca, Bayer, BMS, Boston Scientific, BTG, Eisai, Eli Lilly, H3 Biomedicine, Ipsen, Novartis, Merck, Roche, Sirtex Medical, Terumo; speaker fees from Astra Zeneca, Bayer, BMS, BTG, Eli Lilly, Ipsen, Novartis, Merck, Roche, Sirtex Medical, Terumo; research grants (to Institution) from BMS and Sirtex Medical.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
Tissue samples were obtained after written informed consent was obtained from each patient. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the institution's human research committee (ref # 2017.074).
References
- 1.Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2021;7:6. 10.1038/s41572-020-00240-3 [DOI] [PubMed] [Google Scholar]
- 2.Sangro B, Sarobe P, Hervás-Stubbs S, et al. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2021;18:525–43. 10.1038/s41575-021-00438-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unitt E, Marshall A, Gelson W, et al. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J Hepatol 2006;45:246–53. 10.1016/j.jhep.2005.12.027 [DOI] [PubMed] [Google Scholar]
- 4.Ding W, Xu X, Qian Y, et al. Prognostic value of tumor-infiltrating lymphocytes in hepatocellular carcinoma: a meta-analysis. Medicine 2018;97:e13301. 10.1097/MD.0000000000013301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gabrielson A, Wu Y, Wang H, et al. Intratumoral CD3 and CD8 T-cell densities associated with relapse-free survival in HCC. Cancer Immunol Res 2016;4:419–30. 10.1158/2326-6066.CIR-15-0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol 2013;59:81–8. 10.1016/j.jhep.2013.02.022 [DOI] [PubMed] [Google Scholar]
- 7.El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–502. 10.1016/S0140-6736(17)31046-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol 2018;19:940–52. 10.1016/S1470-2045(18)30351-6 [DOI] [PubMed] [Google Scholar]
- 9.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894–905. 10.1056/NEJMoa1915745 [DOI] [PubMed] [Google Scholar]
- 10.Yau T, Kang Y-K, Kim T-Y, et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): results from CheckMate 040. JCO 2019;37:4012. 10.1200/JCO.2019.37.15_suppl.4012 [DOI] [Google Scholar]
- 11.Teng MWL, Ngiow SF, Ribas A, et al. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res 2015;75:2139–45. 10.1158/0008-5472.CAN-15-0255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74. 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 13.Yarchoan M, Johnson BA, Lutz ER, et al. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 2017;17:209–22. 10.1038/nrc.2016.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189–99. 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGranahan N, Furness AJS, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–9. 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 2017;377:2500–1. 10.1056/NEJMc1713444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kreiter S, Selmi A, Diken M, et al. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol 2008;180:309–18. 10.4049/jimmunol.180.1.309 [DOI] [PubMed] [Google Scholar]
- 19.Wu Y, Borde M, Heissmeyer V, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 2006;126:375–87. 10.1016/j.cell.2006.05.042 [DOI] [PubMed] [Google Scholar]
- 20.Pascolo S, Bervas N, Ure JM, et al. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J Exp Med 1997;185:2043–51. 10.1084/jem.185.12.2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarobe P, Pendleton CD, Akatsuka T, et al. Enhanced in vitro potency and in vivo immunogenicity of a CTL epitope from hepatitis C virus core protein following amino acid replacement at secondary HLA-A2.1 binding positions. J Clin Invest 1998;102:1239–48. 10.1172/JCI3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruiz M, Llopiz D, Zabaleta A, et al. Engineered promiscuous T helper peptides for the induction of immune responses. Mol Immunol 2007;44:2205–12. 10.1016/j.molimm.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 23.Llopiz D, Ruiz M, Silva L, et al. Inhibition of adjuvant-induced TAM receptors potentiates cancer vaccine immunogenicity and therapeutic efficacy. Cancer Lett 2021;499:279–89. 10.1016/j.canlet.2020.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. 10.1038/nmeth.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res 2010;70:8368–77. 10.1158/0008-5472.CAN-10-1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoenberger SP, Toes RE, van der Voort EI, et al. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998;393:480–3. 10.1038/31002 [DOI] [PubMed] [Google Scholar]
- 27.Bennett SR, Carbone FR, Karamalis F, et al. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998;393:478–80. 10.1038/30996 [DOI] [PubMed] [Google Scholar]
- 28.Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017;547:222–6. 10.1038/nature23003 [DOI] [PubMed] [Google Scholar]
- 29.Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217–21. 10.1038/nature22991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carreno BM, Magrini V, Becker-Hapak M, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015;348:803–8. 10.1126/science.aaa3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H, Sun L, Guan A, et al. Unique TP53 neoantigen and the immune microenvironment in long-term survivors of hepatocellular carcinoma. Cancer Immunol Immunother 2021;70:667–77. 10.1007/s00262-020-02711-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bassaganyas L, Pinyol R, Esteban-Fabró R, et al. Copy-number alteration burden differentially impacts immune profiles and molecular features of hepatocellular carcinoma. Clin Cancer Res 2020;26:6350–61. 10.1158/1078-0432.CCR-20-1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sia D, Jiao Y, Martinez-Quetglas I, et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 2017;153:812–26. 10.1053/j.gastro.2017.06.007 [DOI] [PubMed] [Google Scholar]
- 35.Liu T, Tan J, Wu M, et al. High-affinity neoantigens correlate with better prognosis and trigger potent antihepatocellular carcinoma (HCC) activity by activating CD39+CD8+ T cells. Gut 2021;70:1965–77. 10.1136/gutjnl-2020-322196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mauriello A, Zeuli R, Cavalluzzo B. High somatic mutation and neoantigen burden do not correlate with decreased progression-free survival in HCC patients not undergoing immunotherapy. Cancers 1824;2019:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vitiello A, Zanetti M. Neoantigen prediction and the need for validation. Nat Biotechnol 2017;35:815–7. 10.1038/nbt.3932 [DOI] [PubMed] [Google Scholar]
- 38.Paul S, Croft NP, Purcell AW, et al. Benchmarking predictions of MHC class I restricted T cell epitopes in a comprehensively studied model system. PLoS Comput Biol 2020;16:e1007757. 10.1371/journal.pcbi.1007757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richman LP, Vonderheide RH, Rech AJ. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst 2019;9:375–82. 10.1016/j.cels.2019.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wells DK, van Buuren MM, Dang KK, et al. Key parameters of tumor epitope immunogenicity revealed through a Consortium approach improve neoantigen prediction. Cell 2020;183:834.e13:818–34. 10.1016/j.cell.2020.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abelin JG, Keskin DB, Sarkizova S, et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 2017;46:315–26. 10.1016/j.immuni.2017.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinkawa T, Tokita S, Nakatsugawa M, et al. Characterization of CD8+ T-cell responses to non-anchor-type HLA class I neoantigens with single amino-acid substitutions. Oncoimmunology 2021;10:1870062. 10.1080/2162402X.2020.1870062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capietto A-H, Jhunjhunwala S, Pollock SB, et al. Mutation position is an important determinant for predicting cancer neoantigens. J Exp Med 2020;217 10.1084/jem.20190179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Löffler MW, Mohr C, Bichmann L, et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med 2019;11:28. 10.1186/s13073-019-0636-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vercher E, Tamayo I, Mancheño U, et al. Identification of neoantigen-reactive T cells in hepatocellular carcinoma: implication in adoptive T cell therapy. J Hepatol 2020;73:S39–40. 10.1016/S0168-8278(20)30630-9 [DOI] [Google Scholar]
- 46.Baxevanis CN, Voutsas IF, Tsitsilonis OE, et al. Tumor-specific CD4+ T lymphocytes from cancer patients are required for optimal induction of cytotoxic T cells against the autologous tumor. J Immunol 2000;164:3902–12. 10.4049/jimmunol.164.7.3902 [DOI] [PubMed] [Google Scholar]
- 47.Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019;565:234–9. 10.1038/s41586-018-0792-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2021-003978supp001.xlsx (208.2KB, xlsx)
jitc-2021-003978supp002.xlsx (149.9KB, xlsx)
jitc-2021-003978supp003.pdf (168.5KB, pdf)
jitc-2021-003978supp004.xlsx (246.7KB, xlsx)
jitc-2021-003978supp005.xlsx (9.8KB, xlsx)
Data Availability Statement
All data relevant to the study are included in the article or uploaded as online supplemental information.