

Abstract

High-quality ring strain energy (RSE) data for 1H-unsaturated (CH)2X parent rings, where X is a group 13–16 element, are reported in addition to the 2H-isomers of the pnictogenirene rings. RSE data are obtained from appropriate homosdesmotic reactions and calculated at the DLPNO-CCSD(T)/def2-TZVPP//B3LYP-D3/def2-TZVP(ecp) level. 1H-Tallirene and 1H-plumbirene have unique donor–acceptor structures between an acetylene π(CC) orbital and an empty p orbital of a metallylene subunit (a Dewar–Chatt–Duncanson description) and therefore cannot be described as proper rings but as pseudocyclic structures. Also, 1H-indirene and 1H-oxirene lack ring critical points and constitute borderline cases of pseudorings. 1H-Unsaturated rings exhibit enhanced RSE compared to their saturated homologues. The mechanism of ring strain relaxation by increasing the s character in the lone pair (LP) of group 15–16 elements is remarkable and increases on descending the groups. Furthermore, RSE is affected by the aromatic character of group 13 rings and certain aromatic or antiaromatic character in group 14 or 15–16 rings, respectively, which tend to vanish on descending the group as shown by NICS(1) values. 2H-Unsaturated rings were found only for group 15 elements (although only 2H-azirine shows a proper cyclic structure) and displayed lower RSE (higher stability) than the corresponding 1H-isomers.
Short abstract
Accurate ring strain energies (RSE) are calculated for three-membered 1H-unsaturated rings containing a 13−16 group element and group 15 2H-isomers. The presence or absence of lone pairs at the heteroatom and the existence of intrinsic or residual (anti)aromaticity were shown to affect the RSE.
Introduction
The chemistry of unsaturated three-membered heterocycles has been an important field of study for theoreticians and experimentalists for decades. Their peculiar geometrical characteristics lie in their inherent high ring strain and the presence of unsaturated bonds and heteroatoms, which introduce (de)stabilizing factors such as lone pair repulsion, σ-aromaticity, delocalization, and rehybridization.1 They are also a source of starting materials for the synthesis of a wide range of other more complex substances. For instance, the versatile cyclopropene is used as a reagent in the well-known Diels–Alder reactions2 or in its chiral version to produce enantiomerically enriched methylene- and alkylidenecyclopropane derivatives.3 Also, 2H-azirine is an important intermediate for the preparation of acyclic amino compounds4 and substituted aziridines,5 among other important reactions. And dimerization of borirene leads to 1,4-diboracyclohexadiene.6
In the study of three-membered unsaturated heterocycles containing (at least) a group 13–16 heteroatom, the essential concept of aromaticity (or antiaromaticity in case) in chemistry comes into play.7 Numerous studies are reported in the literature that try to elucidate how this condition affects the geometrical and electronic aspects of this type of rings. Although the combination of acetylene and silylene was suggested to be an appropriate way of describing silacyclopropane (silirane), the stability exhibited when successfully synthesized by Seyferth in 19728 hinted at considering that its unsaturated version, silacyclopropene (1H-silirene), might also be stable due to some predicted aromatic character,9 as a few years later demonstrated by the synthesis of the tetramethyl-1-silacycloprop-2-ene derivative by Conlin et al.10 The heavier analogues 1H-germirenes were reported to be stable species,11 and a high ring strain energy (RSE) of 44.4 and 47.6 kcal/mol were computed for the parent and 1-bromo-substituted rings,12 respectively, whereas a somewhat lower strain was obtained for the corresponding 1H-silirene (RSE = 40.6 and 42.0 kcal/mol, respectively). On the other hand, the rather unstable 1H-thiirene was the first formally antiaromatic (4π electron) heterocycle prepared,13 which tries to alleviate this destabilization by lengthening the C–S bonds and shortening the C=C bond. No less interesting is 1H-oxirene, which has given rise to considerable controversy as to whether the C2v-symmetric structure exists as a local minimum or a saddle point energy between two degenerated low-symmetry (Cs) minima, which greatly turned out to depend on the computational level used.14 However, it seems unanimous that its derivative dimethyloxirene does constitute a high-symmetry (C2v) energy minimum.15 As for the antiaromatic pnictogens group, the three-membered N- and P-containing unsaturated rings constitute exciting fields in heterocyclic chemistry. The chemistry of aziridines and azirines has been a tremendously exploited field since the review of Padwa and Woolhouse.16 Two types of azirines can be formed, namely, 1H-azirine and 2H-azirine, both exhibiting unique reactivity such as the ability to act as either nucleophile or electrophile,17 as well as the possibility of being used as precursors to more complex N-containing molecules.4 Their structures have been well studied recently.18 A 2H-azirine is the highly strained key intermediate in the Neber rearrangement that transforms ketoximes into α-amino-ketones.19 In contrast, the development of the chemistry of three-membered P-containing rings was somewhat delayed, starting with Wagner’s discovery of phosphirane in 1963.20 A few years later, 1H-phosphirene and 2H-phosphirene were discovered by Mathey21 and Regitz,22 respectively, both fields growing rapidly to become key building blocks in organophosphorus chemistry.23 But it is not all about antiaromaticity: in 1998, the remarkable stability of 2π-electron aromatic 1H-borirene was reported to be the most stable isomer of BC2H3, in contrast to its aluminum counterpart, which highlights the ability of the B atom to form stable rings.24
In the present work, high-quality benchmark RSE (ring strain energy) data are reported for parent (CH)2X rings, where X is a group 13–16 element (Figure 1), with its most characteristic covalency (3, 4, 3, and 2 for groups 13–16, respectively) completed by bonds with H, and is designated as 1El (“El” refers to the heavy element of group X). In addition, a discussion of possible factors affecting the ring strain as well as (anti)aromaticity in each case is presented. The 2H-isomers 2 were also studied, although only those with pnictogen atoms were found as stable species.
Figure 1.

1H- (1) and 2H-unsaturated (2) three-membered heterocycles studied.
Results and Discussion
1H-Unsaturated Ring Structures, 1
The main geometrical parameters for all computed (see the Experimental Section) energy minima of rings 1El (Figure 1) were collected (Table 1) to obtain an appropriate description of their structures.
Table 1. Calculated (B3LYP-D3/def2-TZVPecp) C=C Bond Distances (Å) and C–X–C Bond Angles (Degrees, in Parentheses) for Compounds 1El.
| group 13 | group 14 | group 15 | group 16 |
|---|---|---|---|
| B 1.346 (54.6) | C 1.287 (50.6) | N 1.269 (49.5) | O 1.269 (48.0) |
| Al 1.369 (43.3) | Si 1.330 (43.1) | P 1.292 (41.1) | S 1.271 (40.1) |
| Ga 1.356 (41.8) | Ge 1.319 (40.1) | As 1.287 (37.8) | Se 1.268 (36.8) |
| In 1.330 (36.1) | Sn 1.320 (36.3) | Sb 1.287 (34.3) | Te 1.267 (33.4) |
| Tl 1.201 (22.2) | Pb 1.203 (23.7) | Bi 1.280 (32.4) | Po 1.261 (31.5) |
The C=C bond distance maintains a general tendency to decrease on moving right within the row, probably due to the increasing electronegativity of the heteroatom, as also observed for the fully saturated counterparts and tested on simple acyclic X–CH2–CH2–X model species (X = BH2, CH3, NH2, OH).25 On descending within the same group, the C=C bond distance decreases regularly, reaching limiting values (also very acute C–X–C bond angles) for rings containing heteroatoms with the highest metallic character Tl and Pb. Thus, 1Tl* (1H-thallirene) and 1Pb* (1H-plumbirene) present a pseudocyclic structure (designated with the asterisk) according to the Dewar–Chatt–Duncanson (DCD) model,26 where the HCCH moiety displays a high triple-bond (acetylene) character (WBI = 2.936 and 2.868 for 1Tl* and 1Pb*, respectively). Hence, these pseudorings are better described by a dative bonding from a filled π(C≡C) orbital in an acetylene subunit to an empty p atomic orbital (AO) in the X (TlH or PbH2) subunit (Figure 2). The energies corresponding to this interaction, obtained from the second-order perturbation theory (SOPT) of the Fock matrix in the natural bond orbital (NBO) basis, for pseudo-thallirene and -plumbirene are 6.28 and 22.84 kcal/mol, respectively. Atoms-in-molecules (AIM) analysis27 also supports this description by lacking a ring critical point (RCP) and attributing a single bond critical point (BCP) between the organic and metallic fragments (see Figures 2 and S1). This different behavior exhibited by 1Tl* and 1Pb* could be attributed to the well-known inert pair effect, typical for this type of post-transition-metal elements, which affects the tendency of the pair of the outermost s electrons not to be shared, making more favorable a structure with DCD-type interaction between two subunits.
Figure 2.
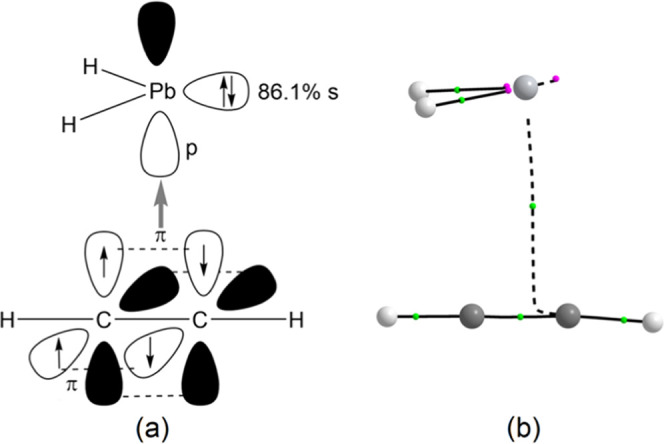
Sketched representation of DCD interaction (a) and computed (B3LYP/def2-TZVPPecp) structure with BCP (small green spheres), nuclear nonattractive critical points (NNACP, small pink spheres), and bond paths (b) for pseudo-plumbirene (1Pb*).
1H-Indirene (1In*) represents a borderline case as it does not show a typical DCD structure due to the double C–C bond character (Table 1) (WBI = 2.028; natural localized molecular orbitals NLMO-BO = 2.007), but it can be categorized as a pseudocycle as it lacks RCP, only displaying one BCP at the shorter C–In bond (2.063 Å) and being the other slightly elongated (2.210 Å) (see the Supporting Information).
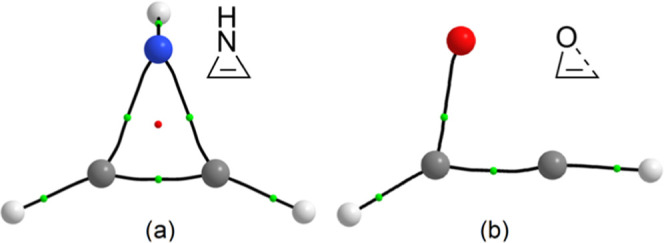
On the other hand, for the controversial oxirene 1O* geometry (vide supra), it was found that at the MP2(FU)/6-31G(d) level or using GGA functionals (BLYP, BP91, PBE, HCTH, HCTH147, and HCTH407), with various double- and triple-ζ quality basis sets with a variety of extra polarization functions, the C2v-symmetric 1O constitutes a transition state (TS) between two degenerate low-symmetry (Cs) minima, whereas at the HF/6-31G(d) level, C2v-1O is a true minimum; for hybrid functionals (B3LYP, B3P91, PBE0, B97, B97-1, and B3P86), a disparity of results was obtained.28 We have obtained similar results with the PBEh-3c29 functional to those with HF calculations. At the working level of theory used for optimizations throughout the current study (see the Computational Details section), oxirene 1O* has a Cs symmetry distorted structure with long (C1–O 1.673 Å) and short (C2–O 1.376 Å) bonds, interconverting through a very low-lying C2v-symmetric TS (ΔΔEZPE‡ = 0.28 kcal/mol). The weak and long C1–O bond is due to the fact that it is formed involving two almost pure p AO at carbon (94.3% p) and oxygen (96.6% p), as shown in the highest occupied molecular orbital (HOMO) – 1 isosurface, with HOMO and lowest unoccupied molecular orbital (LUMO) displaying π*(C–O) and π*(C–C) character, respectively (Figure 3). However, oxirene lacks a ring critical point (RCP) therefore being categorized as a pseudocyclic structure.
Figure 3.

Computed (B3LYP-D3/def2-TZVP) Kohn–Sham isosurfaces (0.07 au) for (a) HOMO – 1, (b) HOMO, and (c) LUMO of pseudo-oxirene (1O*).
Ring Strain Energy Values and Related Parameters for Compounds 1
Ring strain is one of the most remarkable aspects of small rings as it is closely linked to chemical reactivity and has been found to affect properties such as the stereochemical stability of σ3λ3-pnictogen ring atoms.30 It frames the driving force for a ring to be transformed into open-chain products.31 RSEs were calculated using homodesmotic reactions (reaction class 4 or “RC4”), which constitute the penultimate type in a hierarchy of increasingly accurate processes, due to the conservation of larger fragments, according to a recent classification and redefinition of reaction types used in thermochemistry.32 Good estimates for RSE concerning saturated25,31,33 and unsaturated12,34 three-membered heterocycles were obtained according to this type of thermochemical reaction schemes. The highest ranked hyperhomodesmotic reactions (reaction class 5 or “RC5”) were skipped because they have been shown to give rise to very similar RSE data to RC4,25 but being somewhat problematic in some cases by introducing undesired noncompensated interactions due to the presence of longer chains. Therefore, the RSE for unsaturated three-membered (CH)2X heterocycles with group 13–16 heteroelements 1El was calculated using the RC4 scheme (Scheme 1) and obtained as the average of the opposite energy (including the zero-point energy correction) for the three bond cleavage reactions corresponding to the three endocyclic bond-breaking reactions (one C=C and two C–X bonds) (Table 2). Although in principle, the Z-isomer of the homodesmotic C–X bond cleavage product should be the most suitable diastereomer according to its similarity with the endocyclic C=C bond, it introduces undesirable long-distance steric clashes, and hence only the E-isomer was used. The introduction of a C=C double bond in a small 3MR is expected to drastically increase the ring strain owing to the higher energy that should be required to compress an acyclic C=C–El bond angle from ca. 120° to the ca. 60° expected in the three-membered cyclic species, compared to the shorter bond angle compression required in a saturated C–C–E moiety. This is long time known since the first report of RSE for cyclopropene (53.2 kcal/mol)35a in 1976, compared to that of cyclopropane (27.9 kcal/mol).25
Scheme 1. Homodesmotic (RC4) Ring-Opening Reactions Used for the Estimation of RSE for 1El.

Table 2. RC4-Derived Calculated (DLPNO-CCSD(T)/def2TZVPPecp) RSE Values (kcal/mol) for Compounds 1El and 2Pn.
|
1El |
2Pn | |||
|---|---|---|---|---|
| group 13 | group 14 | group 15 | group 16 | group 15 |
| B 31.15 (68.836c) | C 53.97 (53.2,35a 55.0,35b 55.5,35c 54.1,35d 55.7,35e 54.1,35f 54.1,35g 56.037) | N 71.02 (77.337) | Ob 76.10 [71.31]a (74.9,35f 82.437) | N 41.19 (43.9,37 41.938) |
| Al 42.09 | Si 41.94 (45.5,35b 34.5,35cc 49.039) | P 38.03 (39.037) | S 51.51 | Pb 32.57 [31.38]a (34.337) |
| Ga 45.46 | Ge 46.62 (44.412) | As 35.89 | Se 45.25 | Asb 32.73 [32.95]a |
| Inb 46.22 [46.29]a | Sn 45.01 | Sb 30.95 | Te 38.15 | Sbb 30.91 [29.71]a |
| Bi 26.92 | Po 33.17 | Bib 28.12 [28.11]a | ||
Values calculated at the CASSCF(n,6)/MRACPF/def2-SVPD level are presented in square brackets (see the Supporting Information for the specific number of active orbitals used, n).
Pseudocyclic structures.
See text.
Data collected in Table 2 are in reasonable agreement with previously reported RSE data that were obtained using different computational levels and, in most cases, lower-quality thermochemical equations (e.g., isodesmic reactions). Worth is mentioning the erroneously reported value of 34.5 kcal/mol for the RSE of 1Si, which was referenced to a publication where this was not mentioned.35c On the other hand, the extremely high RSE value reported for the 1H-borirene 1B ring is based on the wrong assumption that an increase from the saturated counterpart borirane (reported RSE = 43.7 kcal/mol) could be estimated from the RSE difference (ΔRSE = 24.8 kcal/mol) between cyclopropene (1C) and cyclopropane.36
For those (pseudo)rings with a diagnostic T1, a parameter widely used to evaluate the multi- or single-reference character, close to 0.02 (no ring or pseudo-ring exceeds this value). the multireference RSE value has been calculated by selecting the appropriate number of n electrons and m active orbitals (Table S3) to analyze possible differences between the two methods. The lowest values of single-reference contribution are shown for 1H-unsaturated 1In* (76.2%) and 1O* (80.6%) both with the highest diagnostic value T1 (0.02), although the RSE values for the two methods are quite similar for 1In* (despite having considerable multireference character). The pseudo-ring 1O* has a marked multireference character (of almost 20%), and this is reflected in a difference of about 5 kcal/mol between the two calculation methods.
As described in the previous section, the pseudocyclic oxirene ring 1O* lacks an RCP and exhibits an extremely high RSE value (that should be taken with caution), paralleling the high exothermicity of its low barrier (ΔEZPE‡ = 3.63 kcal/mol) isomerization to ketene (ΔEZPE = −77.57 kcal/mol; reported: −80.02 kcal/mol40). Despite the similar high RSE value of 1H-azirine, 1N, its proper cyclic structure is supported by the existence of BCPs for every endocyclic bond (see Table S1), as well as an RCP (Figure 4) with a high value of Lagrange kinetic energy density per electron G(r)/ρ(r) (see Table S1), which have been shown to correlate with RSE.41 Moreover, calculation of the relaxed force constant,42 a numerically stable and fully transferable parameter related to the stiffness of bond lengths or bond angles, reveals that the C–C–El bond angle of pseudocyclic 1O* (k0 = 0.124 mdyn·Å) is below one-tenth of that for 1N (k0 = 1.572 mdyn·Å), which also points to an inherent difference between pseudocyclic and cyclic characters, respectively.
Figure 4.
Computed (B3LYP/def2-TZVPP) BCP (small green spheres), RCP (small red sphere), and bond paths for (a) 1N and (b) 1O*.
Unsaturated rings with heteroatoms belonging to groups 15 and 16 experience the lone pair (LP) s character-enhancing effect on the RSE (Figure 5),43 in line with the observed behavior of the already reported homologous saturated rings.25 This increase of the LP s character causes the increase in p character for the AOs used by heteroatom “El” in the endocyclic C–El bonds, resulting in an spn-type hybridization with n > 3 (i.e., beyond 75%p). As the s character of the LP AO increases on descending the group, the RSE for the corresponding heterocycle 1El decreases (Figure 5). The alternative plot of RSE against the p character of the AO used by the heteroatom “El” for its endocyclic C–El bonds, %p(El)C–El, shows a slightly worse correlation for groups 15 and 16 (R2 = 0.8692 and 0.9826, respectively) (see Figure S4), while for groups 13 and 14, the expected almost constant p character (around 72 and 81%, respectively) is observed due to the absence of LPs.
Figure 5.

Plot of RSE vs s character (%) of AO used by pnictogen and chalcogen heteroatoms “El” lone pairs (LPs) in 1.
Also, the relaxed force constants k0 computed for C–El bonds in heterocycles with heaviest group 13–16 elements 1El parallels the behavior of RSE, as k0C–El decreases (bond elasticity increases) with the increase of %p(El)C–El (Figure S2). The lighter elements (except C) fall out of the correlation, exhibiting unexpectedly low (for N and O) or high (for B) k0C–El values. For groups 13 and 14, the C–El bond stiffness is higher than for groups 15 and 16, and its decrease on moving down the group is much more pronounced. In the case of 1O* and 1In*, both %p(El)C–El and k0C–EI are averaged values for the two different C–El bonds in the pseudo-ring. Furthermore, due to the orthogonality of p AOs, an increase of p character should move the electron density of the σ(C–El) bond out of the hypothetical C–El straight bond line. Indeed, bond bending from NHO (natural hybrid orbital) directionality analysis shows a remarkable correlation between the deviation angle and the %p(El)C–El, only for group 15 and 16 elements (Figure S3). The angular deviation increases on moving down the group, being more pronounced (higher slope) for chalcogens than for pnictogens. This is indeed the opposite trend observed for RSE versus %p(El)C–El (Figure S4), which suggests the easiness of bond bending as a mechanism for relieving the ring strain. For groups 13 and 14, the %p(El)C–El values vary within a small range (ca. 70–74 and 79–82%, respectively) and, again, the angular deviation increases on descending the group (except for pseudocyclic 1In*).
However, despite the strain-relieving effect of the LP s character in group 15–16 unsaturated heterocycles 1El, the high RSE values displayed compared to their saturated counterparts (which represent the least strained rings within the series), and especially for 1N and 1O (also for 1S and 1Se), are due to their intrinsic antiaromatic character or the geometric deformation to partially mitigate this electronic destabilization. In fact, the observed elongation of the endocyclic C–El bonds (Table S1) compared to analogous C–El bonds in acyclic species (taking the homodesmotic ring-opening products as a reference; see Table S2) is more pronounced for 1N (9.7%) and 1O (11.6% in average) and decreases significantly from the third period onwards (1P 0.9%; 1As 1.2%; 1Sb 1.0; 1Bi 1.4%; 1S 5.8; 1Se 5.3; 1Te 4.6%; 1Po 4.6%), in accordance with the decreased antiaromaticity.
Although (anti)aromaticity is not an observable, several parameters have been described trying to quantify it from the geometrical, energetic, and magnetic perspectives.44 One of the most widely used parameters is the nucleus-independent chemical shift (NICS)45 which allows the aromatic character to be quantified so that the more negative the NICS value, the higher the aromatic character. As isotropic NICS values calculated at the ring centroid are strongly influenced by the σ-contributions,46 NICS(1) (computed at 1 Å above and below the ring centroid) are usually employed.47 The NICS(1) values for 1El were calculated at the standard B3LYP/6311+G(d,p) level47 for elements of the second to fourth rows and using the def2-TZVPP basis set for elements of the fifth and sixth rows. At first sight, a clear correlation between NICS(1) and RSE is observed (Figure 6), with almost linear character for elements having (groups 15–16) or not having LPs (groups 13–14), if 1N and 1O* are excluded. As expected, the most antiaromatic rings are those with the lightest group 15–16 elements (nitrogen and oxygen) due to the most efficient overlap of the heteroelements LP with the π(C=C) orbital. Hence, antiaromaticity considerably decreases on moving down in groups 15 and 16 and varying almost linearly with RSE. As foreseeable, 1B is the most aromatic ring of all herein described,48 therefore showing the most negative NICS(1) value and entailing a remarkable decrease in RSE (Figure 6). Again, on descending group 13, the heavier congeners of Al, Ga, and In use an empty p AO with increasing principal quantum number, which decreases the efficiency of the overlap with the C=C π-system, hence decreasing the aromaticity of the corresponding unsaturated heterocycles 1El and increasing their RSE.
Figure 6.

Plot of RSE vs computed (B3LYP/6311+G(d,p) or def2-TZVPP) NICS(1) (ppm) for compounds 1. Excluding 1N and 1O* from the linear correlation.
As for group 14, it is worth mentioning the remarkable aromatic character, according to its highly negative NICS(1) value, observed for the 1Si ring (Figure 6), which has been explained in terms of using empty low-lying 3dxz orbitals of silicon playing a similar role to the positive vacancy (2pz) in the C atom of cyclopropenyl cation.10 It should also be ascertained that the proposed10 DCD-type structure for the 1H-silirene ring 1Si, although suggested by the HOMO isosurface (Figure S5), must be ruled out, not only on the basis of its geometric features (Table 1) but also supported by the atoms-in-molecules (AIM) analysis,27 showing three BCPs for the three endocyclic bonds and an RCP (Figure 7), and not a single BCP between the hypothetical acetylene and silylene moieties, as is would be expected for a DCD-type structure. The aforementioned stabilization by d-orbital participation is supported by the NBO analysis, indicating ESOPT = 3.88 kcal/mol associated with an electron transfer from a filled π(C=C) to an almost empty Si orbital with almost pure (95.9%) d-character. This SOPT energy in 1Ge (1.24 kcal/mol, 90.7% d-character) and 1Sn rings (1.10 kcal/mol, 69.5% d-character) decrease due to participation of 4d and 5d orbitals, respectively, with increasing energy gap, whereas 1C lacks d orbitals, which justifies its nonaromatic character. However, the origin of the diatropic ring current (aromaticity) accounting for the observed highly negative NICS(1) value for 1Si can mostly arise from an energetically more important sacrificial hyperconjugation effect between the π(C=C) and the two σ*(Si–H) molecular orbitals, each amounting to ESOPT = 10.64 kcal/mol. The energy of the analogous double SOPT interaction decreases for the 1Ge and 1Sn rings (8.24 and 5.22 kcal/mol respectively), in line with their lower aromaticity. Similarly, for groups 15 and 16, whereas 1N and 1O* lack d-orbitals, a small yet significant electron transfer is observed for 1P and 1S from π(C=C) to empty d-orbitals (82.3 and 60.4% d-character, respectively), the SOPT interaction amounting to 5.89 and 5.74 kcal/mol, respectively. This suggests a certain (small) aromatic component, which has an effect on a decrease of RSE and a more negative NICS(1) resulting from a small diatropic current. This effect decreases (lower ESOPT) for heavier elements due to the increased energy gap between the donor π(C=C) orbital and the acceptor El-centered empty AO with increasingly lower d-character.49 The reason for the increase in the aromaticity-related NICS(1) descriptor as one moves down in groups 15 and 16 is not trivial but could be tentatively explained on the basis of the increasing size of the heavier atoms, whose inner-shell electrons could give rise to heteroatom-centered diatropic currents that could interfere in the overall NICS(1) value. The use of NICS(2) values (computed 2 Å away from the ring centroid) did not provide a clearer picture, and the same trends were observed when plotted against RSE (see Figure S6), with good linear correlations within groups and all rings displaying lower negative values than benzene.47 For group 15 rings, a decrease of sacrificial hyperconjugation of the π electron density with the σ*(El–H) molecular orbital on descending the group could also explain the decrease in aromaticity (ESOPT = 8.54, 6.40, 4.61, and 3.59 kcal/mol for 1P, 1As, 1Sb, and 1Bi, respectively).
Figure 7.
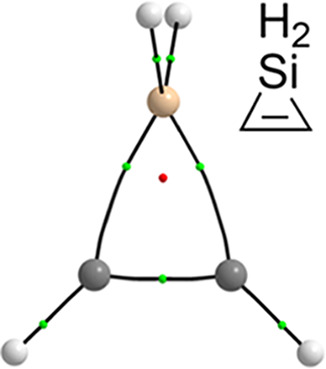
Computed (B3LYP/def2-TZVPP) BCP (small green spheres), RCP (small red sphere), and bond paths for 1Si.
Nevertheless, as a consequence of the very high energy of the d-orbitals from the third row onward, the above-described effect of aromatic components could be negligible in this type of rings50 and, instead, other substituent-depending hyperconjugation effects may come into play.
RSE may also be related to the strength of endocyclic bonds. Among bond-strength-related parameters, the widely used Wiberg bond index (WBI)51 as well as the bond order provided by the Natural Localized Molecular Orbitals (NLMO)52 analysis (NLMO-BO), less prone to overestimate the covalent character of the bond,53 were computed for all 1El heterocycles. The representation of the RSE versus both WBI (Figure 8) and NLMO-BO (see Figure S7) of the C=C bond show that for groups 14–16, as the ring strain decreases when moving down the group, the C=C bond order increases, but without reaching a value close to a genuine triple-bond character as in the case of DCD-type pseudorings (WBI/NLMO-BO = 2.934/2.943 and 2.867/2.902 for 1Tl* and 1Pb*, respectively). For 1In*, the WBI for the C=C bond (2.028) is clearly overestimated compared to the NLMO-BO (1.885), the latter falling out of the linear correlation of its group (Figure S7) probably because of its pseudocyclic character. 1H-Silirene 1Si is also markedly outside the linear correlation of group 14 (for both WBI and NLMO-BO), most likely due to its exceptional aromaticity with respect to the rest of the tetrel-based unsaturated rings, leading to a decrease in the RSE but without greatly affecting the strength of the C=C moiety. The most aromatic ring, 1H-borirene (1B), shows the lowest C=C bond orders (WBI = 1.720; NLMO-BO = 1.873) owing to the effective conjugation of the vacant p orbital in B to the C=C bond, drastically suppressed in the less aromatic and more strained 1Al and 1Ga rings (Figure 8).
Figure 8.

Plot of RSE vs WBIC=C for compounds 1, excluding 1Si, 1N, and 1O* from the linear correlation.
In all, despite the expected large difference in RSE between 1H-unsaturated 3MRs and their saturated analogues, this turned out to be true only for 1C. For the heaviest group 14 elements, this difference becomes less pronounced because of the aromatic stabilization of the unsaturated rings. By contrast, the aromatic stabilization of group 13 unsaturated rings results in a somewhat lower RSE than their saturated counterparts. In the case of group 15 and 16 heterocycles, the RSE of the unsaturated rings is much higher than that of their saturated counterparts due to the additional antiaromaticity destabilization effect.
2H-Unsaturated Three-Membered Rings 2
2H-Unsaturated three-membered heterocycles (2El) were only found as energy minima for the elements of group 15 (2Pn). The RSE data for these pnictogen-containing rings (Table 2) were calculated using the same type of homodesmotic reaction scheme as for the previously described rings (Scheme 2).
Scheme 2. Homodesmotic (RC4) Ring-Opening Reactions Used for the Estimation of RSE for 2Pn.

However, only the 2H-azirine ring, 2N, contains three BCPs corresponding to the three endocyclic bonds and a proper RCP (Figure 9a), while neither BCPs for the single C–El bond nor RCP were found for the rest of rings with heavier pnictogens. Taking 2P* as a case in point, the lack of RCP and BCP(P–C) can be observed (Figure 9b); furthermore, 2P* features a very elongated single C–P bond (1.906 Å) in comparison to the C–P single bond of the 1H-phosphirene 1P isomer (1.840 Å) or to that in phosphirane (1.876 Å).25 Consequently, except 2H-azirine (2N), all heavier 2H-pnictogenirenes 2Pn* must be considered pseudocyclic structures and their computed RSE used with caution. The analysis of the multireference RSE data calculated for these pseudorings (Table 2) shows that there is no significant difference with the single-reference (sr) data, even for 2Bi* (84.3% sr) and 2Sb* (86.5% sr) with a multireference contribution of about 15%.54
Figure 9.
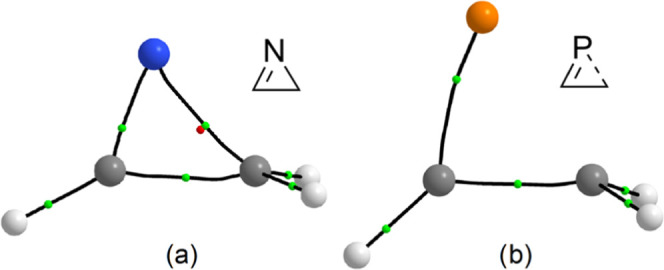
Computed (B3LYP/def2-TZVPP) BCP (small orange spheres) and bond paths for (a) 2N and (b) 2P*.
Recently, we have reported on the C=N bond-containing three-membered ring oxazirine.34a The latter could be considered either as oxa-analogue (replacement of −CH2– by −O−) of 2H-azirine (2N), compared to which exhibits a somewhat enhanced RSE (44.28 kcal/mol), or as aza-analogue (replacement of =CH– by =N−) of pseudo-1H-oxirene (1O*), displaying similar pseudocyclic structure due to the absence of RCP as a consequence of the elongated N–O bond (with no BCP).
It is worth mentioning the higher stability of the 2H-pnictogenirene rings 2Pn compared to the corresponding 1H-isomers 1Pn, as can be inferred from the variation in RSE (Table 2). This difference is very pronounced for the N-containing rings (ΔΔEZPE = 29.83 kcal/mol), mainly due to the antiaromatic character of 1N. For heavier pnictogens, this difference becomes less noticeable (ΔΔEZPE = 5.46, 3.16, 0.04, and −1.20 kcal/mol for P, As, Sb, and Bi, respectively), probably due to the decrease of the antiaromatic character.
Experimental Section
Density functional theory (DFT) calculations were performed with the ORCA program.55 All geometry optimizations were run in redundant internal coordinates in the gas phase, with tight convergence criteria, and using the B3LYP56 functional together with Ahlrichs segmented def2-TZVP basis set57 and the 2010 Grimme’s semiempirical atom-pairwise London dispersion correction (DFT-D3).58 From these geometries, all electronic data were obtained through single-point calculations (SP) using the same quality basis set but including additional polarization, def2-TZVPP.59 Energy values were corrected for the zero-point vibrational term at the optimization level and obtained by the newly developed DLPNO method60 for the “coupled-cluster” level with single, double, and triple perturbatively introduced excitations (CCSD(T)).61 It is worth mentioning that the RI-JK speeding up approximation for Coulomb and Hartree–Fock (HF) exchange (in HF step for post-HF calculations) gave rise to anomalous results for fifth-row elements even when using appropriate auxiliary basis sets.25 Therefore, this approach was skipped for all high-level RSE calculations. T1-Diagnostic values close to 0.02 prompted us to use dynamic correlation on top of the CASSCF(n,m) method62 employing a (6,6) active space for 1In* and 1O*, whereas (8,6) for 2As*, 2Sb* and 2Bi*, with the multireference average coupled-pair functional (MRACPF)63 approach and the def2-SVPD basis set.64 Analysis of the hybridization in the AO used for the endocyclic bonds was performed with the NBO method.65 Properties derived from the topological analysis of the electronic density were obtained with the Multiwfn program,66 whereas Figures 2b, 4, 7, and 9 were plotted with AIMall.67Figure 3 was drawn with Visual Molecular Dynamics (VMD).68
Conclusions
Accurate high-level (DLPNO-CCSD(T)/def2TZVPP//B3LYP-D3/def2TZVP) values were provided for the ring strain energy (RSE) in unsaturated three-membered rings containing only one 13–16 group element. A thorough exploration of the structural and electronic features of the 1H-unsaturated rings 1El resulted in the classification of those with the most metallic elements, 1Tl* and 1Pb*, as pseudocyclic structures because of their typical Dewar–Chatt–Duncanson-type bonding lacking a ring critical point (RCP), 1H-indirene (1In*) constituting a borderline case. At the working level of theory, the controversial C2v-symmetric 1H-oxirene, 1O, is a transition state (TS) between two low-symmetry (Cs) degenerate minima (1O*) with pseudocyclic character. Among the 2H-unsaturated three-membered rings, 2El, only found as a minimum for the pnictogen elements and presenting low RSE compared to the 1El isomers, just 2H-azirine (2N) presents a proper ring structure (i.e., featuring an RCP).
RSE in groups 15 and 16 containing 1H-unsaturated rings 1El is very much affected by the strain relaxation effect of the increased s character of one LP orbital of pnictogens and chalcogens, as already described for their saturated counterparts. The existence of an empty orbital in group 13 rings gives them the possibility of presenting a significant aromatic character, especially in the case of borirene 1B. The existence of empty d orbitals in 1H-tetrelirenes, allows their use to acquire some aromaticity, especially in the case of 1H-silirene 1Si, while moving down in the group the energy of these d orbitals increases, hence decreasing this aromatic contribution. On the other hand, the high antiaromaticity of 1O* and 1N, partially alleviated by geometric distortion, results in enhanced RSE. For the heavier group 15 and 16 elements, the antiaromaticity considerably decreases on descending the group, which together with the increase in heteroatom size and aromatic and/or hyperconjugative contributions result in a remarkable RSE decrease.
Acknowledgments
The authors thank the computation center at Servicio de Cálculo Científico (SCC—University of Murcia) for technical support and the computational resources used. Helpful discussions with Prof. L. Nyulászi are deeply acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c00067.
Plots of the relaxed force constants and of the RSE versus other parameters; additional tables with electronic, structural, and bond strength parameters; as well as Cartesian coordinates and energies for all calculated species (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Wiberg K. B. The Concept of Strain in Organic Chemistry. Angew. Chem., Int. Ed. 1986, 25, 312–322. 10.1002/anie.198603121. [DOI] [Google Scholar]; b Göller A.; Clark T. σ*-Aromaticity in Three-Membered Rings. J. Mol. Model. 2000, 6, 133–149. 10.1007/PL00010724. [DOI] [Google Scholar]; c Reactive Intermediate Chemistry; Moss R. A.; Platz M. S.; Jones M. Jr., Eds.; John Wiley & Sons, 2004. [Google Scholar]
- Liu F.; Paton R. S.; Kim S.; Liang Y.; Houk K. N. Diels-Alder Reactivities of Strained and Unstrained Cycloalkenes with Normal and Inverse-Electron-Demand Dienes: Activation Barriers and Distortion/Interaction Analysis. J. Am. Chem. Soc. 2013, 135, 15642–15649. 10.1021/ja408437u. [DOI] [PubMed] [Google Scholar]
- Marek I.; Simaan S.; Masarwa A. Enantiomerically Enriched Cyclopropene Derivatives: Versatile Building Blocks in Asymmetric Synthesis. Angew. Chem., Int. Ed. 2007, 46, 7364–7376. 10.1002/anie.200604774. [DOI] [PubMed] [Google Scholar]
- a Padwa A.; Dharan M.; Smolanof J.; Wetmore S. I. Photochemical transformations of small ring heterocyclic compounds. XLVI. Scope of the photoinduced 1,3-dipolar addition reactions of arylazirines. J. Am. Chem. Soc. 1973, 95, 1945–1954. 10.1021/ja00787a039. [DOI] [Google Scholar]; b Palacios F.; Aparicio D.; de Retana A. M. O.; de los Santos J. M.; Gil J. I.; Alonso J. M. Asymmetric Synthesis of 2H-Azirines Derived from Phosphine Oxides Using Solid-Supported Amines. Ring Opening of Azirines with Carboxylic Acids. J. Org. Chem. 2002, 67, 7283–7288. 10.1021/jo025995d. [DOI] [PubMed] [Google Scholar]
- Palacios F.; de Retana A. M. O.; de Marigorta E. M.; de los Santos J. M. Preparation, properties and synthetic applications of 2H-azirines a review. Org. Prep. Proced. Int. 2002, 34, 219–269. 10.1080/00304940209356770. [DOI] [Google Scholar]
- Budzelaar P. H. M.; Van der Kerk S. M.; Krogh-Jespersen K.; Schleyer P. v. R. Dimerization of Borirene to 1,4-Diboracyclohexadiene. Structures and Stabilities of (CH)4(BH)2 Molecules. J. Am. Chem. Soc. 1986, 108, 3960–3967. 10.1021/ja00274a017. [DOI] [Google Scholar]
- a Krogh-Jespersen K.; Cremer D.; Dill J. D.; Pople J. A.; Schleyer P. v. R. Aromaticity in Small Rings Containing Boron and Carbon ((CH)2(BH)n, n = 1, 2). Comparisons with Isoelectronic Carbocations. The Decisive Roles of Orbital Mixing and Nonbonded 1,3-Interactions in the Structures of Four-Membered Rings. J. Am. Chem. Soc. 1981, 103, 2589–2594. 10.1021/ja00400a018. [DOI] [Google Scholar]; b Gupta R.; Bansal R. K. Aromaticity/antiaromaicity of phospha-analoges of carbocyclic ions: A DFT investigation. Comput. Theor. Chem. 2016, 1076, 1–10. 10.1016/j.comptc.2015.11.020. [DOI] [Google Scholar]
- Lambert R. L. Jr.; Seyferth D. Substituted 7-siladispiro(2.0.2.1)heptanes. The first stable silacyclopropanes. J. Am. Chem. Soc. 1972, 94, 9246–9248. 10.1021/ja00781a055. [DOI] [Google Scholar]
- Barthelat J.-C.; Trinquier G.; Bertrand G. Theoretical investigations on some C2SiH4 isomers. J. Am. Chem. Soc. 1979, 101, 3785–3789. 10.1021/ja00508a015. [DOI] [Google Scholar]
- Conlin R. T.; Gaspar P. P. Tetramethylsilacyclopropene. J. Am. Chem. Soc. 1976, 98, 3715–3716. 10.1021/ja00428a059. [DOI] [Google Scholar]
- a Mizuhata Y.; Sasamori T.; Tokitoh N. Stable Heavier Carbene Analogues. Chem. Rev. 2009, 109, 3479–3511. 10.1021/cr900093s. [DOI] [PubMed] [Google Scholar]; b Meiners F.; Saak W.; Weidenbruch M. A Germacyclopropene with Electronegative Groups at the Ring Carbon Atoms. Z. Anorg. Allg. Chem. 2002, 628, 2821–2822. . [DOI] [Google Scholar]; c Ishida S.; Iwamoto T.; Kira M. Addition of a stable dialkylsilylene to carbon–carbon unsaturated bonds. Heteroat. Chem. 2011, 22, 432–437. 10.1002/hc.20705. [DOI] [Google Scholar]; d Lai T. Y.; Guo J.-D.; Fettinger J. C.; Nagase S.; Power P. P. Facile insertion of ethylene into a group 14 element-carbon bond: effects of the HOMO–LUMO energy gap on reactivity. Chem. Commun. 2019, 55, 405–407. 10.1039/C8CC08488B. [DOI] [PubMed] [Google Scholar]
- Sugahara T.; Espinosa Ferao A.; Rey Planells A.; Guo J.-D.; Aoyama S.; Igawa K.; Tomooka K.; Sasamori T.; Hashizume D.; Nagase S.; Tokitoh N. 1,2-Insertion reactions of alkynes into Ge–C bonds of arylbromogermylene. Dalton Trans. 2020, 49, 7189–7196. 10.1039/D0DT01223H. [DOI] [PubMed] [Google Scholar]
- Hess B. A. Jr.; Schaad L. J.; Ewig C. S. An ab Initio SCF Study of the Structure and Vibrational Spectrum of Thiirene. J. Am. Chem. Soc. 1980, 102, 2507–2508. 10.1021/ja00527a085. [DOI] [Google Scholar]
- a Vacek G.; Colegrove B. T.; Schaefer H. F. III Does oxirene exist? A theoretical inquiry involving the coupled-cluster method. Chem. Phys. Lett. 1991, 177, 468–470. 10.1016/0009-2614(91)85085-B. [DOI] [Google Scholar]; b Vacek G.; Galbraith J. M.; Yamaguchi Y.; Schaefer H. F. III; et al. Oxirene: To Be or Not To Be?. J. Phys. Chem. A 1994, 98, 8660–8665. 10.1021/j100086a013. [DOI] [Google Scholar]
- Fowler J. E.; Galbraith J. M.; Vacek G.; Schaefer H. F. III Substituted Oxirenes (X2C2O, X = BH2, CH3, NH2, OH, F): Can They Be Made?. J. Am. Chem. Soc. 1994, 116, 9311–9319. 10.1021/ja00099a057. [DOI] [Google Scholar]
- Padwa A.; Woolhouse A. D.. Comprehensive Heterocyclic Chemistry; Katritzky A. R.; Rees C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 7. [Google Scholar]
- Padwa A. Azirine Photochemistry. Acc. Chem. Res. 1976, 9, 371–378. 10.1021/ar50106a005. [DOI] [Google Scholar]
- Császár A. G.; Demaison J.; Rudolph H. D. Equilibrium Structures of Three-, Four-, Five-, Six-, and Seven-Membered Unsaturated N-Containing Heterocycles. J. Phys. Chem. A 2015, 119, 1731–1746. 10.1021/jp5084168. [DOI] [PubMed] [Google Scholar]
- a Neber P. W.; Friedolsheim Av. Über eine neue Art der Umlagerung von Oximen. Justus Liebigs Ann. Chem. 1926, 449, 109–134. 10.1002/jlac.19264490108. [DOI] [Google Scholar]; b O’Brien C. The Rearrangement of Ketoxime O-Sulfonates to Amino Ketones (The Neber Rearrangement). Chem. Rev. 1964, 64, 81–89. 10.1021/cr60228a001. [DOI] [Google Scholar]
- a Wagner R. I. U.S. Patent US3086053 and 3086056.; b Wagner R. I.; Freeman L. V. D.; Goldwhite H.; Rowsell D. G. Phosphiran. J. Am. Chem. Soc. 1967, 89, 1102–1104. 10.1021/ja00981a013. [DOI] [Google Scholar]
- a Marinetti A.; Mathey F.; Fischer J.; Mitschler A. Generation and trapping of terminal phosphinidene complexes. Synthesis and x-ray crystal structure of stable phosphirene complexes. J. Am. Chem. Soc. 1982, 104, 4484–4485. 10.1021/ja00380a029. [DOI] [Google Scholar]; b Marinetti A.; Mathey F.; Fischer J.; Mitschler A. Synthesis and X-Ray Crystal Structure of 1,2,3-Triphenylphosphirene. J. Chem. Soc., Chem. Commun. 1984, 45–46. 10.1039/c39840000045. [DOI] [Google Scholar]
- Wagner O.; Maas G.; Regitz M. Das erste 2H-Phosphiren. Angew. Chem. 1987, 99, 1328–1330. 10.1002/ange.19870991237. [DOI] [Google Scholar]; The first 2H-Phosphirene. Angew. Chem. Int. Ed. 1987, 26, 1257 - 1259.10.1002/anie.198712571
- a Mathey F. Phosphaorganische Chemie: Panorama und Perspektiven. Angew. Chem. 2003, 115, 1616–1643. 10.1002/ange.200200557. [DOI] [Google Scholar]; Mathey F. Phospha-Organic Chemistry: Panorama and Perspectives. Angew. Chem., Int. Ed. 2003, 42, 1578–1604. 10.1002/anie.200200557. [DOI] [PubMed] [Google Scholar]; b Mathey F. Comparisons between the chemistry of phosphirenes and silirenes. Pure Appl. Chem. 1987, 59, 993–998. 10.1351/pac198759080993. [DOI] [Google Scholar]; c Mathey F. Chemistry of 3-Membered Carbon-Phosphorus Heterocycles. Chem. Rev. 1990, 90, 997–1025. 10.1021/cr00104a004. [DOI] [Google Scholar]; d Mathey F.; Regitz M.. Comprehensive Heterocyclic Chemistry I;Katritzky A. R.; Rees C. W.; Scriven E. F. V., Eds.; Pergamon, 1997; Vol. 1A, pp 277–304. [Google Scholar]; e Regitz M.Phosphorus-Carbon Heterocyclic Chemistry: The Rise of a New Domain; Mathey F., Ed.; Pergamon, 2001; pp 17–55. [Google Scholar]; f Streubel R.; Ostrowski A.; Wilkens H.; Ruthe F.; Jeske J.; Jones P. G. Überraschende intramolekulare Folgereaktionen intermediärer Phosphacarbonyl-Ylid-Wolframkomplexe. Angew. Chem. 1997, 109, 409–413. 10.1002/ange.19971090427. [DOI] [Google Scholar]; Streubel R.; Ostrowski A.; Wilkens H.; Ruthe F.; Jeske J.; Jones P. G. Unexpected Intramolecular Reactions of Intermediate Phosphacarbonyl Ylide Tungsten Complexes. Angew. Chem., Int. Ed. 1997, 36, 378–381. 10.1002/anie.199703781. [DOI] [Google Scholar]; g Streubel R.; Kusenberg A.; Jeske J.; Jones P. G. Thermisch induzierte Ringspaltung eines 2H-1,2-Azaphosphiren-Wolframkomplexes. Angew. Chem. 1994, 106, 2564–2566. 10.1002/ange.19941062330. [DOI] [Google Scholar]; Streubel R.; Kusenberg A.; Jeske J.; Jones P. G. Thermally Induced Ring Cleavage of a 2H-1,2-Azaphosphirene Tungsten Complex. Angew. Chem., Int. Ed. 1995, 33, 2427–2428. 10.1002/anie.199424271. [DOI] [Google Scholar]
- Galland N.; Hannachi Y.; Lanzisera D. V.; Andrews L. Theoretical study of structures, energetics and vibrational properties of BC2H3 species. Chem. Phys. 1998, 230, 143–151. 10.1016/S0301-0104(98)00050-0. [DOI] [Google Scholar]
- Rey Planells A.; Espinosa Ferao A. Accurate Ring Strain Energy Calculations on Saturated Three-Membered Heterocycles with One Group 13–16 Element. Inorg. Chem. 2020, 59, 11503–11513. 10.1021/acs.inorgchem.0c01316. [DOI] [PubMed] [Google Scholar]
- Chatt J.; Duncanson L. A. Olefin Co-ordination Compounds. J. Chem. Soc. 1953, 2939–2947. 10.1039/jr9530002939. [DOI] [Google Scholar]
- a Bader R. F. W.Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, U.K., 1990. [Google Scholar]; b Bader R. F. W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. 10.1021/cr00005a013. [DOI] [Google Scholar]; c The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Matta C. F.; Boyd R. J., Eds.; Wiley-VCH: New York, 2007; pp 1–34. [Google Scholar]; d Biegler-König F.; Schönbohm J.; Bayles D. Software news and updates-AIM2000-A program to analyze and visualize atoms in molecules. J. Comput. Chem. 2001, 22, 545–559. . [DOI] [Google Scholar]; e Biegler-König F.; Schönbohm J. Update of the AIM2000-program for atoms in molecules. J. Comput. Chem. 2002, 23, 1489–1494. 10.1002/jcc.10085. [DOI] [PubMed] [Google Scholar]
- a Ma N. L.; Wong M. W. A Theoretical Study of the Properties and Reactivities of Ketene, Thioketene, and Selenoketene. Eur. J. Org. Chem. 2000, 2000, 1411–1421. . [DOI] [Google Scholar]; b Mawhinney R. C.; Goddard J. D. Assessment of density functional theory for the prediction of the nature of the oxirene stationary point. J. Mol. Struct.: THEOCHEM 2003, 629, 263–270. 10.1016/S0166-1280(03)00198-2. [DOI] [Google Scholar]
- Grimme S.; Brandenburg J. G.; Bannwarth C.; Hansen A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 054107 10.1063/1.4927476. [DOI] [PubMed] [Google Scholar]
- Espinosa Ferao A.; García Alcaraz A. Benchmarking the inversion barriers in σ3λ3-phosphorus compounds: a computational study. New J. Chem. 2020, 44, 8763–8770. 10.1039/D0NJ01237H. [DOI] [Google Scholar]
- Espinosa Ferao A.; Rey Planells A.; Streubel R. Between oxirane and phosphirane: the spring loaded oxaphosphirane ring. Eur. J. Inorg. Chem. 2021, 2021, 348–353. 10.1002/ejic.202000881. [DOI] [Google Scholar]
- Wheeler S. E.; Houk K. N.; v R Schleyer P.; Allen W. D. A Hierarchy of Homodesmotic Reactions for Thermochemistry. J. Am. Chem. Soc. 2009, 131, 2547–2560. 10.1021/ja805843n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Krahe O.; Neese F.; Streubel R. The Quest for Ring Opening of Oxaphosphirane Complexes: A Coupled-Cluster and Density Functional Study of CH3PO Isomers and Their Cr(CO)5 Complexes. Chem. - Eur. J. 2009, 15, 2594–2601. 10.1002/chem.200801494. [DOI] [PubMed] [Google Scholar]; b Espinosa A.; Streubel R. Computational studies on azaphosphiridines and the quest of how to effect ring-opening processes via selective bond activation. Chem. - Eur. J. 2011, 17, 3166–3178. 10.1002/chem.201002565. [DOI] [PubMed] [Google Scholar]; c Espinosa A.; Gómez C.; Streubel R. Single electron transfer-mediated selective endo- and exocyclic bond cleavage processes in azaphosphiridine chromium(0) complexes: a computational study. Inorg. Chem. 2012, 51, 7250–7256. 10.1021/ic300522g. [DOI] [PubMed] [Google Scholar]; d Albrecht C.; Schneider E.; Engeser M.; Schnakenburg G.; Espinosa A.; Streubel R. Synthesis and DFT Calculations of Spirooxaphosphirane Complexes. Dalton Trans. 2013, 42, 8897–8906. 10.1039/c3dt50556a. [DOI] [PubMed] [Google Scholar]; e Espinosa A.; de las Heras É.; Streubel R. Oxaphosphirane-borane complexes: Ring strain and migratory insertion reactions. Inorg. Chem. 2014, 53, 6132–6140. 10.1021/ic500536h. [DOI] [PubMed] [Google Scholar]; f Villalba Franco J. M.; Sasamori T.; Schnakenburg G.; Espinosa Ferao A.; Streubel R. Going for strain: synthesis of the first 3-imino-azaphosphiridine complexes and their surprising conversion into oxaphosphirane complex valence isomers. Chem. Commun. 2015, 51, 3878–3881. 10.1039/C4CC10266E. [DOI] [PubMed] [Google Scholar]; g Espinosa Ferao A.; Streubel R. Thiaphosphiranes and their complexes: systematic study on ring strain and ring cleavage reactions. Inorg. Chem. 2016, 55, 9611–9619. 10.1021/acs.inorgchem.6b01322. [DOI] [PubMed] [Google Scholar]; h Espinosa Ferao A. Kinetic energy density per electron as quick insight into ring strain energies. Tetrahedron Lett. 2016, 57, 5616–5619. 10.1016/j.tetlet.2016.10.115. [DOI] [Google Scholar]
- a Espinosa Ferao A.; Rey Planells A.. CHNO isomers and derivatives - A computational overview New J. Chem. 2022, 46, 5771−5778 10.1039/D1NJ05752A. [DOI]; b Rey A.; Espinosa Ferao A.; Streubel R. Quantum Chemical calculations on CHOP derivatives–spanning the chemical space of phosphinidenes, phosphaketenes, oxaphosphirenes and COP– isomers. Molecules 2018, 23, 3341 10.3390/molecules23123341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzelaar P. H. M.; Kos A. J.; Clark T.; von Rague Schleyer P. Effects of Boron substituents in Borirenes, Boriranes and Boranes. The Energies of B-X bonds. Organometallics 1985, 4, 429–437. 10.1021/om00122a001. [DOI] [Google Scholar]
- a George P.; Trachtman M.; Bock C. W.; Brett A. M. An alternative approach to the problem of assessing destabilization energies (strain energies) in cyclic hydrocarbons. Tetrahedron 1976, 32, 317–323. 10.1016/0040-4020(76)80043-9. [DOI] [Google Scholar]; b Gordon M. S. Ring Strain in Cyclopropane, Cyclopropene, Silacyclopropane, and Silacyclopropene. J. Am. Chem. Soc. 1980, 102, 7419–7422. 10.1021/ja00545a002. [DOI] [Google Scholar]; c Naruse Y.; Ma J.; Inagaki S. Relaxation of ring strain by introduction of a double bond. Tetrahedron Lett. 2001, 42, 6553–6556. 10.1016/S0040-4039(01)01281-3. [DOI] [Google Scholar]; d Bach R. D.; Dmitrenko O. Strain Energy of Small Ring Hydrocarbons. Influence of C–H Bond Dissociation Energies. J. Am. Chem. Soc. 2004, 126, 4444–4452. 10.1021/ja036309a. [DOI] [PubMed] [Google Scholar]; e Khoury P. R.; Goddard J. D.; Tam W. Ring strain energies: substituted rings, norbornanes, norbornenes and norbornadienes. Tetrahedron 2004, 60, 8103–8112. 10.1016/j.tet.2004.06.100. [DOI] [Google Scholar]; f Bach R. D.; Dmitrenko O. The Effect of Carbonyl Substitution on the Strain Energy of Small Ring Compounds and Their Six-Member Ring Reference Compounds. J. Am. Chem. Soc. 2006, 128, 4598–4611. 10.1021/ja055086g. [DOI] [PubMed] [Google Scholar]; g Bach R. D. Ring Strain Energy in the Cyclooctyl System. The Effect of Strain Energy on (3+2) Cycloaddition Reactions with Azides. J. Am. Chem. Soc. 2009, 131, 5233–5243. 10.1021/ja8094137. [DOI] [PubMed] [Google Scholar]
- Goumans T. P. M.; Ehlers A. W.; Lammertsma K.; Würthwein E.-U. Endo/exo Preferences for Double Bonds in Three-Membered Rings Including Phosphorus Compounds. Eur. J. Org. Chem. 2003, 2003, 2941–2946. 10.1002/ejoc.200300120. [DOI] [Google Scholar]
- Gómez-Zavaglia A.; Kaczor A.; Cardoso A. L.; Pinho e Melo T. M. V. D.; Fausto R. Substituent effects on the photolysis of methyl 2-carboxylate substituted aliphatic 2H-azirines. J. Mol. Struct. 2007, 834–836, 262–269. 10.1016/j.molstruc.2006.12.027. [DOI] [Google Scholar]
- Gordon M. S.; Koob R. D. Relative Stability of Multiple Bonds to Silicon: An ab Initio Study of C2SiH4 Isomers. J. Am. Chem. Soc. 1981, 103, 2939–2944. 10.1021/ja00401a005. [DOI] [Google Scholar]
- Wilson P. J.; Tozer D. J. A Kohn-Sham study of the oxirene-ketene potential energy surface. Chem. Phys. Lett. 2002, 352, 540–544. 10.1016/S0009-2614(01)01496-8. [DOI] [Google Scholar]
- Espinosa Ferao A. Kinetic energy density per electron as quick insight into ring strain energies. Tetrahedron Lett. 2016, 57, 5616–5619. 10.1016/j.tetlet.2016.10.115. [DOI] [Google Scholar]
- a Brandhorst K.; Grunenberg J. How strong is it? The interpretation of force and compliance constants as bond strength descriptors. Chem. Soc. Rev. 2008, 37, 1558–1567. 10.1039/b717781j. [DOI] [PubMed] [Google Scholar]; b Büschel S.; Jungton A.-K.; Bannenberg T.; Randoll S.; Hrib C. G.; Jones P. G.; Tamm M. Secondary Interactions in Phosphane-Functionalized Group 4 Cycloheptatrienyl–Cyclopentadienyl Sandwich Complexes. Chem. - Eur. J. 2009, 15, 2176–2184. 10.1002/chem.200802518. [DOI] [PubMed] [Google Scholar]; c Espinosa A.; de las Heras É.; Streubel R. Oxaphosphirane-Borane Complexes: Ring Strain and Migratory Insertion/Ring-Opening Reactions. Inorg. Chem. 2014, 53, 6132–6140. 10.1021/ic500536h. [DOI] [PubMed] [Google Scholar]
- Naruse Y.; Inagaki S.. Relaxation of Ring Strains. In Orbitals in Chemistry; Springer, 2009; Vol. 289, pp 265–291. [DOI] [PubMed] [Google Scholar]
- a Pauling L.; Wheland G. W. The Nature of the Chemical Bond. V. The Quantum-Mechanical Calculation of the Resonance Energy of Benzene and Naphthalene and the Hydrocarbon Free Radicals. J. Chem. Phys. 1933, 1, 362–374. 10.1063/1.1749304. [DOI] [Google Scholar]; b Fishtik I.; Datta R. Aromaticity vs Stoichiometry. J. Phys. Chem. A 2003, 107, 10471–10476. 10.1021/jp036768y. [DOI] [Google Scholar]; c Mo Y.; von Ragué Schleyer P. An Energetic Measure of Aromaticity and Antiaromaticity Based on the Pauling–Wheland Resonance Energies. Chem. - Eur. J. 2006, 12, 2009–2020. 10.1002/chem.200500376. [DOI] [PubMed] [Google Scholar]; d Lazzeretti P. Ring currents. Prog. Nucl. Magn. Reson. Spectrosc. 2000, 36, 1–88. 10.1016/S0079-6565(99)00021-7. [DOI] [Google Scholar]; e Mitchell R. H. Measuring Aromaticity by NMR. Chem. Rev. 2001, 101, 1301–1316. 10.1021/cr990359+. [DOI] [PubMed] [Google Scholar]; f Bird C. W. A new aromaticity index and its application to five-membered ring heterocycles. Tetrahedron 1985, 41, 1409–1414. 10.1016/S0040-4020(01)96543-3. [DOI] [Google Scholar]; g Kruszewski J.; Krygowski T. M. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 13, 3839–3842. 10.1016/S0040-4039(01)94175-9. [DOI] [Google Scholar]; h Krygowski T. M. Crystallographic Studies of Inter- and Intramolecular Interactions Reflected in Aromatic Character of π-Electron Systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. 10.1021/ci00011a011. [DOI] [Google Scholar]
- von Ragué Schleyer P.; Maerker C.; Dransfeld A.; Jiao H. J.; van Eikema Hommes N. J. R. Nucleus-independent chemical shifts: a simple and efficient aromaticity probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. 10.1021/ja960582d. [DOI] [PubMed] [Google Scholar]
- Alonso M.; Poater J.; Solà M. Aromaticity changes along the reaction coordinate connecting the cyclobutadiene dimer to cubane and the benzene dimer to hexaprismane. Struct. Chem. 2007, 18, 773–783. 10.1007/s11224-007-9240-4. [DOI] [Google Scholar]
- a Espinosa A.; Frontera A.; García R.; Soler M. A.; Tárraga A. Electrophilic behaviour of 3-methyl-2-methylthio-1,3,4-thiadiazolium salts: A multimodal theoretical approach. ARKIVOC 2005, 2005, 415–437. 10.3998/ark.5550190.0006.935. [DOI] [Google Scholar]; b Gese A.; Akter M.; Schnakenburg G.; García Alcaraz A.; Espinosa Ferao A.; Streubel R. P-Functionalized tetrathiafulvalenes from 1,3-dithiole-2-thiones?. New J. Chem. 2020, 44, 17122–17128. 10.1039/D0NJ02984J. [DOI] [Google Scholar]
- Compare with NICS(1) = −10.22 ppm and NICS(2) = −4.91 ppm computed for benzene at the same level.
- 1As (3.00 kcal/mol, 71.9% d-character), 1Sb (2.00 kcal/mol, 65.0% d-character), 1Bi (1.36 kcal/mol, 49.0% d-character) 1Se (3.56 kcal/mol, 48.4% d-character), 1Te (2.99 kcal/mol, 47.3% d-character) and 1Po (2.11 kcal/mol, 39.1% d-character).
- a Nyulászi L.; Benkő Z.. Three-Membered Rings with two Heteroatoms Including Selenium or Tellurium; Three-Membered Rings with Three Heteroatoms. In Comprehensive Heterocyclic Chemistry III; Elsevier, 2008. [Google Scholar]; b Nyulászi L.; Benkő Z.. Aromatic Phosphorus Heterocycles. In Topics in Heterocyclic Chemistry; Springer, 2009; Vol. 19, pp 27–81. [Google Scholar]; c Hajgató B.; De Proft F.; Szieberth D.; Tozer D. J.; Deleuze M. S.; Geerlings P.; Nyulászi L. Bonding in negative ions: the role of d orbitals in the heavy analogues of pyridine and furan radical anions. Phys. Chem. Chem. Phys. 2011, 13, 1663–1668. 10.1039/C0CP01151G. [DOI] [PubMed] [Google Scholar]
- Wiberg K. B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. 10.1016/0040-4020(68)88057-3. [DOI] [Google Scholar]
- Reed A. E.; Weinhold F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. 10.1063/1.449360. [DOI] [Google Scholar]
- Outeiral C.; Vincent M. A.; Pendás Á. M.; Popelier P. L. A. Revitalizing the concept of bond order through delocalization measures in real space. Chem. Sci. 2018, 9, 5517–5529. 10.1039/C8SC01338A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The multi-reference RSE calculation for 2As* was obtained only from the C=El and C-C bond cleavage homodesmotic reactions, due to convergence problems in the other (C-El bond cleavage) homodesmotic reaction.
- Neese F.ORCA - An Ab initio, DFT and Semiempirical SCF-MO Package, version 4.2.1; Max Planck Institute for Bioinorganic Chemistry, 2019.
- a Lee C.; Yang W.; Parr R. G. Development of the Colle- Salvetti correlation energy formula into a functional of the electron density. Phys. Rev. B. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]; b Becke A. D. Density-functional thermochemistry. III. The role of exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- a Schafer A.; Huber C.; Ahlrichs R. J.. Chem. Phys. 1994, 100, 5829–5835, basis sets may be obtained from the Basis Set Exchange (BSE) software and the EMSL Basis Set Library: https://bse.pnl.gov/bse/portal.; b Feller D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comput. Chem. 1996, 17, 1571–1586. . [DOI] [Google Scholar]
- Riplinger C.; Sandhoefer B.; Hansen A.; Neese F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101 10.1063/1.4821834. [DOI] [PubMed] [Google Scholar]
- Pople J. A.; Head-Gordon M.; Raghavachari K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. 10.1063/1.453520. [DOI] [Google Scholar]
- Roos B. O.The Complete Active Space Self-Consistent Field Method and its Applications in Electronic Structure Calculations. In Advances in Chemical Physics; John Wiley & Sons, 1987; pp 399–445. [Google Scholar]
- Gdanitz R. J.; Ahlrichs R. The averaged coupled-pair functional (ACPF): A size-extensive modification of MR CI(SD). Chem. Phys. Lett. 1988, 143, 413–420. 10.1016/0009-2614(88)87388-3. [DOI] [Google Scholar]
- Rappoport D.; Furche F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105 10.1063/1.3484283. [DOI] [PubMed] [Google Scholar]
- a Reed A. E.; Weinhold F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. 10.1063/1.445134. [DOI] [Google Scholar]; b Reed A. E.; Weinstock R. B.; Weinhold F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. 10.1063/1.449486. [DOI] [Google Scholar]
- Lu T.; Chen F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Keith T. A.AIMAll, Ver. 14.06.21; TK Gristmill Software: Overland Park, Kansas, U.S.A., aim.tkgristmill.com, 2014.
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.