

 This work is licensed under a
This work is licensed under a Abstract
Elucidating the mechanisms of regulation of β-cell proliferation is key to understanding the pathogenesis of diabetes mellitus. Txnip is a tumor suppressor that is upregulated in diabetes and plays an important role in the regulation of insulin sensitivity; however, its potential effect on pancreatic β-cell proliferation remains unclear. Here, we evaluated the role of Txnip in pancreatic β-cell compensatory proliferation by subjecting WT and Txnip knockout (KO) mice to a high-fat diet (HFD). Our results demonstrate that Txnip deficiency improves glucose tolerance and increases insulin sensitivity in HFD-induced obesity. The antidiabetogenic effect of Txnip deficiency was accompanied by increased β-cell proliferation and enhanced β-cell mass expansion. Furthermore, Txnip deficiency modulated the expression of a set of transcription factors with key roles in β-cell proliferation and cell cycle regulation. Txnip KO in HFD mice also led to activated levels of p-PI3K, p-AKT, p-mTOR and p-GSK3β, suggesting that Txnip may act via PI3K/AKT signaling to suppress β-cell proliferation. Thus, our work provides a theoretical basis for Txnip as a new therapeutic target for the treatment of diabetes mellitus.
Keywords: Txnip, pancreatic β cell, proliferation, PI3K/AKT signaling pathway
Introduction
Diabetes mellitus (DM) is a progressive and chronic metabolic disease with multiple etiologies. DM is characterized by defective glucose metabolism due to low levels or an absolute lack of insulin, which seriously threatens human physical and mental health. Diabetes is correlated with the occurrence and development of many diseases, including exacerbated effects of coronavirus (COVID-19) infection (1). Compared with non-diabetic individuals, diabetic patients require more medical interventions and have a higher rate of severe pneumonia, multiple organ injury and mortality (2, 3). As a result, DM has become a major threat to human health worldwide. In the pathogenesis of diabetes, for either type 1 diabetes mellitus (T1DM) or type 2 diabetes mellitus (T2DM), a deficiency in the functional pancreatic β-cell mass and decreased numbers of β cells cause insufficiency of insulin secretion, which constitutes the key mechanism of DM (4). Pancreatic β-cell replacement and regeneration therapies provide promising approaches to the treatment of diabetes. Therefore, a key goal in the treatment of diabetes is to promote the proliferation of β cells and expand the functional β-cell mass. However, the mechanism driving β-cell proliferation as an adaptive process to insulin deficiency remains unclear.
In healthy individuals, the number of β cells is maintained by neogenesis of endocrine progenitor cells, reprogramming of non-β cells or replication of pre-existing β cells (5, 6, 7). However, in adults, mature β cells rarely replicate and proliferate (8). β-cell neogenesis predominantly takes place during embryogenesis and neonatal life, whereas β-cell proliferation rates decrease dramatically with age (9). Nevertheless, in response to increased physiological and pathological metabolic demands in adults, such as insulin resistance, obesity, pregnancy or pancreatic injury, β cells may undergo compensatory proliferation that is critical for increasing insulin demands and maintaining glucose homeostasis (10, 11, 12, 13, 14). Previous studies have demonstrated that a high-calorie Western diet or a high-fat diet (HFD) stimulates mild β-cell proliferation and expansion of β-cell mass in humans and rodents (15, 16). However, the molecular mechanisms underlying this progression remain unclear. Understanding the mechanisms behind β-cell regeneration is crucial for identifying novel targets to prevent or revert disease. Numerous studies have demonstrated that pancreatic transcription factors, including Pdx1, Mafa and Foxo1, play vital roles in β-cell proliferation, development, differentiation, maturation and functional maintenance (8, 17, 18, 19). Insulin, glucose, growth factors, hormones and nutrients stimulate multiple pathways that promote β-cell replication, at least in part through the activation of pro-proliferative/pro-survival protein kinases, such as PI3K and AKT. The PI3K/AKT signaling pathway plays a critical role in regulating β-cell mass and function (20, 21). The function of AKT in stimulating β-cell replication may occur via the activation of cell cycle regulators, such as Cyclin D2, Cdk4 and p27Kip1 (22, 23, 24). In addition, the pancreatic transcription factor Foxo1 is a downstream target of AKT, and Pdx1 localization and protein levels are tightly regulated throughout the cell cycle (25, 26). Thus, the precise regulatory mechanisms of HFD-induced β-cell proliferation are likely to be multifaceted.
Thioredoxin-interacting protein (Txnip; also known as thioredoxin-binding protein-2/TBP-2) is a member of the α-arrestin family that was first identified as vitamin-D3-upregulated protein-1 (VDUP1) in the human promyelocytic leukemia cell line (HL-60) (27, 28). Txnip is a multifunctional protein that is ubiquitously expressed and is localized in the nucleus, mitochondria and cytoplasm. It is associated with the development of diabetes, cardiovascular disease, CNS diseases, cancer and other diseases (29, 30, 31). Recently, Txnip has been the focus of investigation due to its unique characteristic of specifically binding and regulating Trx activity (32). Txnip is closely associated with glucose and lipid metabolism and is a major regulator of glucose homeostasis (33, 34). Consistently, several studies have demonstrated that Txnip expression is increased in DM and that it plays an important role in the regulation of insulin sensitivity (35, 36, 37). Txnip-deficient HcB-19 (HcB) mice have been shown to have a three-fold increase in insulin levels (37), increased pancreatic β-cell mass and enhanced insulin secretion and sensitivity (36, 38). However, the associated mechanisms for Txnip in DM, including its potential role in β-cell proliferation, have not been fully characterized.
As a tumor suppressor, Txnip has been shown to be involved in cellular growth, with functions in suppressing cellular proliferation and arresting cell cycle progression (39, 40). Numerous studies have demonstrated that Txnip is downregulated in various cancers and human cancer cells, such as breast cancer, stomach cancer, lung cancer, colon cancer, cervical cancer, gastrointestinal cancer, prostate cancer, cutaneous T-cell lymphoma and adult T-cell leukemia (31, 41, 42, 43, 44, 45, 46). There is also increasing evidence that Txnip can inhibit the proliferation of tumor cells. Therefore, we hypothesized that Txnip may also modulate the proliferation of pancreatic β cells.
In this study, to uncover the mechanism of Txnip in regulating insulin sensitivity in DM, we explored the relationship between Txnip and pancreatic β-cell proliferation in HFD-induced mice. Our results provide insight into the mechanism of Txnip in regulating DM and suggest that it may serve as a novel target for clinical treatment.
Materials and methods
Animals and ethics
Experimental studies were performed on male mice on a C57BL/6J background. Txnip knockout mice (KO, catalog number: ENSMUST00000074519.12) were originally generated by GemPharmatech Co., Ltd (Nanjing, China) using CRISPR/Cas9 technology. In brief, Cas9 mRNA and gRNA (sequence, S1: 5’-ACACGGTGTGCTCCTAGCG-3’; S2: 5’-CAACCAATCAGCGAGGCCGC-3’; S3: 5’-GGTCTCCGGCTTCAAG-3’; S4: 5’-CTATGCCACAGTGCGGGCAC-3’) generated by in vitro transcription were injected into fertilized eggs of C57BL/6J mice via the microinjection technique. Then, Cas9 protein binds to the target site under the guidance of gRNA, resulting in DNA double-strand break, thus realizing the deletion of base sequence at the target site. Txnip KO homozygous mice and WT littermate mice were used. All mice were bred, reared and housed in our specific pathogen-free facility at the Shanxi Medical University. The mice were maintained under standardized conditions on a fixed 12 h light: 12 h darkness cycle with free access to food and water. When the mice were 4 weeks old, they were randomly divided into four groups: (1) WT group; (2) KO group; (3) WT-HFD group; (4) KO-HFD group. They were fed normal chow diet (NC) or high-fat diet (HFD) starting at age 4 weeks, with continuous feeding for 12 weeks. HFD was composed of 60% fat, 20% carbohydrate and 20% protein and was prepared by Beijing Vital River Laboratory Animal Technology Co., Ltd (D12492, Beijing, China). The mice were weighed weekly until the end of the experiment, at which time they were anesthetized with isoflurane and sacrificed by cervical dislocation.
All animal procedures were approved by and performed in accordance with the Ethical Committee of Shanxi Medical University with the approval of the Institutional Animal Care and Use Committee. Concerted efforts were made to minimize the number of animals used and their suffering.
Bio-Plex assays
Mice were anesthetized with isoflurane, and the blood was collected from the retroorbital venous plexus using a microcapillary tube. Blood samples were collected and allowed to clot at room temperature for 30–45 min. Then, the samples were centrifuged at 1000 g for 15 min at 4°C, and the supernatant was transferred to a clean centrifuge tube. Centrifugation was repeated at 10,000 g for 10 min to completely remove platelets and other precipitates. Serum samples were then diluted 1:4 and stored at −80°C until assay.
For Mouse Bio-Plex Pro Assays, 25 μL of serum per sample was analyzed. The concentration levels of insulin, glucagon, glucagon-like peptide-1 (GLP1) and Leptin were measured using the Mouse Diabetes Panel 8-Plex Assay (catalog no. 171-F7001M, BIO-RAD) according to the manufacturer’s instructions.
Oral glucose tolerance test and insulin tolerance test
To perform the oral glucose tolerance test (OGTT), mice were fasted overnight for 16 h. Fasting blood glucose was measured from tail vein blood using the OneTouch Ultra glucose meter (Life Scan, Milpitas, USA). All mice were administered glucose (2 g/kg body weight) by oral gavage, and the blood glucose was measured at 15, 30, 60, 90 and 120 min after administration. For the insulin tolerance test (ITT), mice were fasted for 6 h. The fasting blood glucose was measured, and then, the mice were injected intraperitoneally with 0.075 U/mL human insulin in filter-sterilized PBS at 0.1 mL per 10 g body weight. Blood glucose levels were measured at 15, 30, 60, 90 and 120 min after injection.
β-cell mass and β-cell proliferation analysis
For morphometric analysis of the β-cell mass, the pancreas was removed, and fat and lymph nodes were cleared in saline solution. The whole body and the pancreas were weighed, and then, the pancreas was fixed in 4% paraformaldehyde for 48 h, embedded in paraffin and sectioned at a thickness of 5 μm. Seven to 11 sections of each pancreas were prepared approximately 200 μm apart. Then, the sections were immunostained for insulin (1:6400, CST, 3014s) and scanned at 40× using a Nano Zoomer-SQ Digital slide scanner (Hamamatsu, Japan). Images were analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA). The β-cell mass was determined by calculating the insulin-positive percent area of each pancreas and multiplying the pancreas weight. Five to six mice per group were analyzed.
β-cell proliferation was assessed by immunostaining for insulin (1:100, Abcam, ab7842) and either Ki67 (1:200, Abcam, ab15580) or Pcna (1:2400, CST, 2586S). Two to three slides per pancreas were used to calculate the average percentage of insulin/Ki67 or insulin/Pcna double-positive cells. At least 4000 cells were counted per animal. Images were captured using an Olympus FV3000 laser scanning confocal microscope (Olympus), and cells were counted using Image-Pro Plus 6.0 software. The percentage of proliferating β cells was calculated by dividing the total number of insulin/Ki67 or insulin/Pcna double-positive cells by the total number of insulin-positive cells.
Islet isolation
Islets were isolated by collagenase v (Sigma, 1 mg/mL) perfusion through the common hepatic bile duct into the pancreas. Then, the pancreas was dissected and digested for 15 min in a 37°C shaking water bath. Digestion was halted with 10% serum HBSS. The samples were then centrifuged at 1200 g for 15 min, and the supernatant was discarded. The islets were washed three times, observed under a light microscope and cultured in RPMI medium 1640 containing 10% fetal bovine serum and antibiotics.
RT-qPCR
Total RNA was extracted from islets using the RNAiso Plus (code no.9109) (Takara). The integrity of the RNA was verified by agarose gel electrophoresis, and the absorbance was determined by NanoDrop (NanoDrop Technologies, Wilmington, USA). The cDNA was synthesized using a PrimeScript™RT reagent Kit with gDNA Eraser (code no. RR047A) (Takara) according to the manufacturer’s protocol. The mRNA expression was quantified by RT-qPCR with TB Green®Premix Ex Taq™II (Takara) using a LightCycler® 96 System (Roche), with the data shown as the 2−ΔΔCt value. All mRNA expression levels are normalized to β-actin. The primer sequences are listed in Table 1.
Table 1.
Primer sequences.
| Gene | Primer sequences |
|---|---|
| β-actin | F: 5’CACTATTGGCAACGAGCGGTTCCG3’ |
| R: 5’ACGGATGTCAACGTCACACT3’ | |
| Txnip | F: 5’CAAGGGTCTCAGCAGTGCAAAC3’ |
| R: 5’AAGCTCGAAGCCGAACTTGTACTC3’ | |
| Ki67 | F: 5’GTGTCAAACAAACTTGAATCTGTGG3’, |
| R: 5’TCTGCAGATGCATCAAACTTGG3’ | |
| Pcna | F: 5’GAGAGCTTGGCAATGGGAACA3’, |
| R: 5’CAGAGCAAACGTTAGGTGAACAGG3’; | |
| Pdx1 | F: 5’GCCCGGGTGTAGGCAGTAC3’ |
| R: 5’CAGTGGGCAGGAGGTGCTTA3’ | |
| Mafa | F: 5’GGAGGTCATCCGACTGAAACA3’ |
| R: 5’GCACCTCTCGCTCTCCAGAAT3’ | |
| Mafb | F: 5’TGAGCTAGAGGGAGGAAGGA3’ |
| R: 5’CCGGGTTTCTCTAACTCTGC3’ | |
| Foxo1 | F: 5’GAGAAGAGGCTCACCCTGTC3’ |
| R: 5’ACAGATTGTGGCGAATTGAA3’ | |
| Foxm1 | F: 5’CACTTGGATTGAGGACCACTT3’ |
| R: 5’GTCGTTTCTGCTGTGATTCC3’ | |
| Nkx6.1 | F: 5’AGAGAGCACGCTTGGCCTATTC3’ |
| R: 5’GTCGTCAGAGTTCGGGTCCAG3’ | |
| Ins2 | F: 5’TCAACATGGCCCTGTGGAT3’ |
| R: 5’AAAGGTGCTGCTTGAAAAAGC3’ | |
| Glut2 | F: 5’CTCGTGGCGCTGATGCT3’ |
| R: 5’CTGGTTGAATAGTAAAATATCCCATTGA3’ | |
| Ccna2 | F: 5’TTGGCTGCACCAACAGTAAATCA3’ |
| R: 5’ACGGGTCAGCATCTATCAAACTCA3’ | |
| Ccnb1 | F: 5’CATGCTGGACTACGACATGGTG3’ |
| R: 5’ACATTCTTAGCCAGGTGCTGCATA3’ | |
| Ccnd1 | F: 5’GCGTACCCTGACACCAATCTC3’ |
| R: 5’CTCCTCTTCGCACTTCTGCTC3’ | |
| Ccnd2 | F: 5’GGCTAAGACAGGGTGGCTTTCA3’ |
| R: 5’GATTGCTACCTCCAGTTCCCACA3’ | |
| Ccne1 | F: 5’TGATCGTTACATGGCATCACAA3’ |
| R: 5’GCGCCATCTGTAACATAAGCAA3’ | |
| Cdk2 | F: 5’GGACTAGCAAGAGCCTTTGG3’ |
| R: 5’AAGAATTTCAGGTGCTCGGT3’ | |
| Cdk4 | F: 5’ACATGTGGAGCGTTGGCTGTA3’ |
| R: 5’CAACTGGTCGGCTTCAGAGTTTC3’ | |
| Cdkn2a(p16) | F: 5’CGTGAACATGTTGTTGAGGCTAGAG3’ |
| R: 5’ATCTGCACCGTAGTTGAGCAGAAG3’ | |
| Cdkn1a(p21) | F: 5’AGTCTCATGGTGTGGTGGAA3’ |
| R: 5’GACATCACCAGGATTGGACA3’ | |
| Cdkn1b(p27) | F: 5’AGTGTCCAGGGATGAGGAAG3’ |
| R: 5’CTTCTGTTCTGTTGGCCCTT3’ | |
| Cdkn1c(p57) | F: 5’AATCAGCCAGCCTTCGAC3’ |
| R: 5’ATCACTGGGAAGGTATCGCT3’ | |
| Rb | F: 5’CCCAACACATGCTTTGAACTGA3’ |
| R: 5’TGGGTTGTACTGTACTAGGGTCCTC3’ |
Western blot analysis
Total proteins were extracted from islets, and protein levels were measured by the BCA method. Equal quantities of protein were electrophoresed using a TGX Stain-Free FastCast Acrylamide Kit, 10% (BIO-RAD) and then transferred onto PVDF membranes (Millipore). The membranes were incubated in TBST buffer containing 5% BSA (Boster Biological Technology Co., Ltd, Wuhan, China) to block non-specific binding at room temperature and then were incubated with primary antibodies at 4°C overnight. On the next day, the membranes were washed in TBST buffer and then were incubated with secondary antibodies for 1 h at room temperature. Finally, the proteins bands were visualized using ECL Plus and the ChemiDocTM Imaging System (BIO-RAD). The band intensities were analyzed with ImageJ software (US National Institutes of Health). Protein levels were normalized to the β-actin protein, and phosphorylated forms were normalized to phosphorylation-independent levels of the same protein. Antibodies used in this study are listed in Table 2.
Table 2.
Antibodies used for Western blot analysis.
| Antibody | Catalog no. | Company | Dilution ratio |
|---|---|---|---|
| TXNIP | ab188865 | Abcam | 1:2000 |
| Cyclin D2 | ab230883 | Abcam | 1:2000 |
| CDK4 | ab137675 | Abcam | 1:3000 |
| p27 | ab92741 | Abcam | 1:5000 |
| p-PI3K | 17366s | CST | 1:1000 |
| PI3K | 4292s | CST | 1:1000 |
| p-AKT | 9271s | CST | 1:1000 |
| AKT | 9272s | CST | 1:1000 |
| p-mTOR | 5536s | CST | 1:1000 |
| mTOR | 2983s | CST | 1:1000 |
| p-GSK3β | 5558s | CST | 1:1000 |
| GSK3β | 9315s | CST | 1:1000 |
| β-actin | AP0060 | Bioworld | 1:10,000 |
Statistical analysis
Statistical analysis was performed using SPSS20.0 software (IBM). Data are expressed as the mean ± s.e.m. The Kolmogorov–Smirnov test was used to verify data normality. The independent samples t test was applied for comparisons between two groups, while one-way ANOVA was applied for multi-groups. P value <0.05 was considered statistically significant.
Results
Txnip deficiency and HFD feeding increase the pancreatic β-cell mass
To ensure that Txnip KO mice were eligible, Txnip protein expression level and mRNA expression level were measured. The results show that Txnip was almost undetectable of both protein and mRNA levels in Txnip KO mice (Fig. 1A and B). The proliferation ability of adult pancreatic β cells is normally very low; however, previous studies have demonstrated that high-fat feeding can induce pancreatic β cell compensatory proliferation (15). Thus, to induce mouse pancreatic β-cell proliferation, we fed mice NC or HFD. To evaluate the role of Txnip in the response to HFD, we treated both WT and KO mice. The mice were weighed weekly, and after 12 weeks, we observed that HFD resulted in substantially increased weight gain in the WT-HFD mice, whereas the weight gain of the KO-HFD mice was not significantly increased, thus supporting a protective effect of Txnip deficiency in preventing weight gain during HFD (Fig. 1C). Next, we measured the ratio of the pancreas to body weight. As shown in Fig. 1D, the ratios of the WT and KO groups were not significantly different, while the ratio of the KO-HFD group was significantly greater than the ratio of the WT-HFD group of mice. To verify these findings, we performed immunohistochemical staining to visualize the pancreatic islet morphology. The islet number and size were increased in both WT-HFD and KO-HFD mice (Fig. 1E). Furthermore, the β-cell mass was significantly increased in both WT-HFD and KO-HFD mice when compared to their control groups fed with normal chow, and the β-cell mass in the KO-HFD group was significantly increased compared with WT-HFD group mice (Fig. 1F). Collectively, these results indicate that Txnip KO mice fed on HFD have increased pancreatic β-cell mass in the absence of significant weight gain, which is indicative of productive pancreatic β-cell compensatory proliferation.
Figure 1.

Txnip deficiency and HFD accelerate the accumulation of β-cell mass. (A) The protein expression levels of TXNIP (n = 3). (B) The mRNA expression levels of Txnip (n = 3). (C) WT and Txnip knockout (KO) mice were fed normal chow diet (NC) or high-fat diet (HFD) starting at 4 weeks of age and continuing for 12 weeks. The mice were weighed weekly. **Significant difference in weight gain for WT-HFD vs KO-HFD mice. No significant differences were observed for the WT and KO groups of mice (n= 10). (D) The ratios of pancreas/body weight after 12 weeks. **Significant difference for KO-HFD vs WT-HFD mice. No significant differences were observed between the other three groups (n= 6). (E) Immunohistochemical staining of pancreatic tissue for insulin. The islet numbers and sizes were increased in KO-HFD mice. Scale bars, 1 mm (upper) and 0.5 mm (lower). (F) The β-cell mass in the four groups of mice. The β-cell mass was significantly greater in KO-HFD vs WT-HFD mice, while there were no significant differences between the WT and KO groups (n = 5). NS, not significant, *P < 0.05, **P < 0.01.
Txnip deficiency improves glucose tolerance and insulin sensitivity in HFD-induced obesity
To verify that Txnip KO improves glucose metabolism and insulin sensitivity in the HFD mouse model, we measured the serum insulin and fasting blood glucose levels of each group of mice. For both WT and KO mice, the serum insulin levels of HFD mice were significantly higher than those of NC mice. Furthermore, the insulin level of the KO-HFD group was greater than the insulin level of the WT-HFD group mice, whereas there was no significant difference between the insulin levels of the WT and KO groups of mice that were fed with NC (Fig. 2A). Furthermore, the fasting blood glucose levels of HFD mice were higher than those of NC mice, and the level of fasting blood glucose in the KO-HFD group was lower than that in the WT-HFD group (Fig. 2B). Double immunofluorescence staining of insulin/glucagon demonstrated that the pancreatic 𝛼 cells were evenly distributed around islets in the WT and KO groups of mice, while the number of pancreatic 𝛼 cells was higher in the WT-HFD and KO-HFD groups, and fewer cells were infiltrated into the middle of the islets (Fig. 2C). Furthermore, the glucagon levels were lower in the KO-HFD group compared to the WT-HFD group, though there was no observable difference between the WT and KO groups of mice (Fig. 2D). These results confirm that Txnip KO protects mice from the effects of HFD in inducing diabetes.
Figure 2.

Txnip knockout increases insulin secretion, decreases fasting blood glucose and improves glucose tolerance and insulin sensitivity. (A) Bio-Plex assays showing serum insulin levels in four groups of mice. The serum insulin levels were significantly higher in HFD mice. Additionally, the serum insulin levels were higher in KO-HFD vs WT-HFD mice (n = 6). (B) Fasting blood glucose levels. The levels were significantly higher in HFD mouse groups. Additionally, the fasting blood glucose levels were lower in Txnip knockout vs WT mouse groups (n = 6). (C) Double immunofluorescence staining of insulin/glucagon. The number of pancreatic 𝛼 cells was higher in HFD mice, but there was no apparent difference between Txnip knockout and WT mice. Scale bar, 50 μm. (D) The glucagon levels were lower in KO-HFD vs WT-HFD mice (n = 6). (E) Oral glucose tolerance test (OGTT) showing changes in blood glucose levels. (F) The area under the curve (AUC) was calculated for the OGTT (n = 6). (G) Insulin tolerance test (ITT) showing changes in blood glucose levels. (H) The AUC of ITT is shown (n = 6). NS, not significant, *P < 0.05, **P < 0.01.
To further investigate β-cell mass functionality, we performed the OGTT and ITT before sacrificing the mice. The blood glucose levels in the OGTT were higher in HFD mice than in NC mice, while the post-load glucose levels were significantly lower in the Txnip KO groups than in their control groups mice (Fig. 2E and F). The results indicate that the impaired glucose tolerance in mice on HFD is improved by deletion of Txnip. Similarly, the blood glucose levels from the ITT were significantly lower in KO-HFD mice than in WT-HFD group mice, though there were no differences between the WT and KO groups of mice (Fig. 2G and H). Collectively, these results indicate that HFD impairs insulin tolerance and that deletion of Txnip can improve β-cell mass functionality and insulin sensitivity.
Txnip deficiency promotes β-cell proliferation in HFD-induced mice
To measure the effect of Txnip deficiency on β-cell proliferation, we performed double immunofluorescence staining of insulin and proliferation marker (Ki67) or insulin and proliferating cell nuclear antigen (Pcna). Ki67/insulin double-positive cells were barely detectable in NC-fed WT and KO mice; however, significantly more β-cell proliferation was observed in the HFD groups of mice (1.7- to 2.3-fold more Ki67+Ins+ cells). In addition, the percentages of Ki67+Ins+ cells were almost 1.4-fold greater in KO-HFD mice compared with WT-HFD mice, whereas there was no significant difference in the β-cell proliferation between WT and KO groups of mice (Fig. 3A and B). Similar results were obtained with Pcna. The percentage of Pcna+Ins+ cells in the HFD groups was 1.9- and 2.6-fold greater than in the NC-fed groups. Furthermore, the percentage of Pcna+Ins+ cells was almost 1.8-fold greater in KO-HFD mice than in WT-HFD mice (Fig. 3D and E). These results indicate that Txnip KO can promote β-cell proliferation in HFD-treated mice.
Figure 3.

Txnip deficiency and HFD feeding lead to increased β-cell accumulation. (A) Double immunofluorescence staining of insulin and Ki67. Scale bar, 50 μm. (B) The percentage of Ki67+Ins+ cells. The Ki67 positive cells number was increased by HFD and was significantly greater in KO-HFD vs WT-HFD mice. NS, no significant difference in WT vs KO mice(n = 3). (C) The mRNA expression levels of Ki67 were significantly higher in KO-HFD vs WT-HFD mice. NS, no significant difference in WT vs KO mice (n = 3). (D) Double immunofluorescence staining of insulin and Pcna. The Pcna-positive cell number was increased by HFD and was significantly greater in KO-HFD vs WT-HFD mice. NS, no significant difference in WT vs KO mice. Scale bar, 50 μm. (E) The percentage of Pcna+Ins+ cells (n = 3). (F) The mRNA expression levels of Pcna were significantly higher in KO-HFD vs WT-HFD mice. NS, no significant difference in WT vs KO mice (n = 3). NS, not significant, *P < 0.05, **P < 0.01.
To further confirm these differences, we evaluated the relative expression of Ki67 and Pcna mRNA levels. The results suggest that the mRNA levels of Ki67 and Pcna were significantly greater in the KO-HFD group compared with WT-HFD group mice. However, there were no differences between the WT and KO groups of mice (Fig. 3C and F). Thus, these results suggest that Txnip deficiency increases the β-cell proliferation caused by HFD.
Txnip deficiency modulates transcription factor expression in HFD mice
A variety of pancreatic transcription factors, including Pdx1 and Mafa, play crucial roles in the pancreas and function in the maintenance of mature β cells. To identify pancreatic transcription factors that are modulated in mice, we performed real-time RT-PCR. Our results show that the mRNA levels of Pdx1, a marker of mature β cells, were upregulated in the KO-HFD group compared with the WT-HFD group of mice, whereas there was no significant difference between the WT and KO groups of mice that were fed with NC (Fig. 4A). Similarly, the mRNA levels of Mafa, a β-cell-specific transcription factor that functions as a potent activator of insulin gene transcription (Fig. 4B), as well as Mafb, Foxm1 and Ins2 (Fig. 4C, E and G), were significantly more highly expressed in KO-HFD mice as compared with WT-HFD mice. By contrast, the mRNA levels of Foxo1, a key transcription factor in insulin signaling, were decreased in the KO-HFD group of mice (Fig. 4D), and the expression of Nkx6.1 or Glut2 mRNA was not significantly different (Fig. 4F and H). These results suggest that Txnip modulates a set of transcription factors with known roles in regulating gene expression in β cells.
Figure 4.

Txnip deficiency promotes changes in transcription factor mRNA levels in HFD-induced mice. (A, B, C, D, E, F, G and H) The mRNA expression levels of Pdx1, Mafa, Mafb, Foxo1, Foxm1, Nkx6.1, Ins2 and Glut2were detected by RT-qPCR. n = 3, *P < 0.05, **P < 0.01.
Txnip deficiency accelerates cell cycle progression to promote β-cell replication
To further dissect the molecular events underlying the enhanced β-cell proliferation in mice, we evaluated the levels of cyclins and Cdks that are known to be modulated during cell cycle progression in mammals. This includes cyclinD2, a key regulator of the cell cycle in adult β-cell growth; Cdk4, which forms a complex with cyclinD2 and is known to have a critical role in the regulation of β-cell mass; and p27Kip1, an inhibitor of Cdk activity that is also known to regulate β-cell mass. Western blot results showed that the protein levels of cyclin D2 and CDK4 were upregulated, and the protein levels of p27 were downregulated in the KO-HFD group compared with the WT-HFD group. Yet, there was no significant change between the WT and KO groups of mice that were fed with NC (Fig. 5A, B, C and D), which is consistent with the increased proliferation of β cells in Txnip-deficient mice. Furthermore, real-time quantitative PCR results were consistent with the Western blot results (Fig. 5E, F and G), suggesting that the expression of these proteins is regulated at the level of the mRNA. To further investigate the role of cell cycle regulators, we performed real-time quantitative PCR of Rb, cyclins and cyclin-dependent kinase inhibitors (CDKIs), each of which are key regulators of cell cycle progression. The mRNA expression levels of Rb, Cdkn2a and Cdkn1c were significantly decreased and Ccna2, Ccnb1, Ccnb2 and Cdk2 were significantly increased in the KO-HFD group. There was no significant change between the WT and KO groups. However, the mRNA expression levels of Ccnd1, Ccne1 and Cdkn1a were not affected by Txnip knockdown (Fig. 5H). Collectively, the above results suggest that Txnip KO may promote β-cell replication by accelerating cell cycle progression via transcriptional regulation of specific cell cycle regulators in HFD-induced mice.
Figure 5.

Txnip deficiency promotes β-cell replication by regulating the levels of cell cycle regulatory proteins. (A, B, C and D) The protein expression levels of cyclin D2, CDK4 and p27 were analyzed by Western blotting. (E, F and G) The mRNA expression levels of Ccnd2, Cdk4 and Cdkn1b were detected by RT-qPCR. (H) The mRNA expression levels of additional cell cycle regulators were detected by RT-qPCR. n = 3, *P < 0.05, **P < 0.01.
Txnip may regulate β-cell proliferation via the PI3K/AKT pathway
We next sought to explore the possible molecular mechanism by which Txnip deficiency may promote β-cell proliferation. A previous study demonstrated that the PI3K/AKT signaling pathway has the potential to regulate β-cell proliferation (22). Thus, we hypothesized that Txnip might regulate β-cell proliferation in the mouse DM model via the PI3K/AKT pathway. Western blot assays demonstrated that the phosphorylation of PI3K and AKT was increased significantly after Txnip KO in HFD fed mice (Fig. 6A, B and C). Furthermore, phosphorylation of the pivotal downstream mediators of the PI3K/AKT pathway, p-mTOR and p-GSK3β, were upregulated significantly in KO-HFD mice (Fig. 6D and E). Thus, these results suggest that the deletion of Txnip may promote β-cell proliferation via the activation of the PI3K/AKT pathway.
Figure 6.
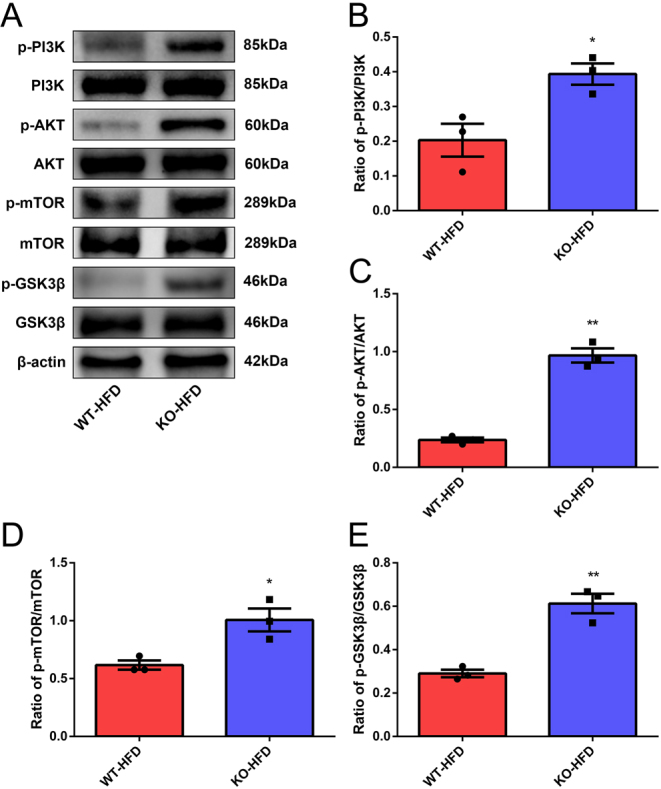
Txnip deficiency may activate the PI3K/AKT signaling pathway. (A) Western blot analysis was performed to determine PI3K/AKT signaling pathway-related protein expression and phosphorylation in WT-HFD vs KO-HFD mice. Representative images are shown. (B) The phosphorylation ratio of PI3K. (C) The phosphorylation ratio of AKT. (D) The phosphorylation ratio of mTOR. (E) The phosphorylation ratio of GSK3β. n = 3, *P < 0.05, **P < 0.01.
Discussion
Diabetes is a disease characterized by an absolute or relative deficiency of β cells that underlies both T1DM and T2DM. Loss of the functional β-cell mass is a key process leading to DM and understanding the molecular mechanisms behind β-cell failure is critical for preventing or reverting disease (47). Furthermore, replenishing the lost β-cell mass is a strategy that can alleviate some of the burdens of the disease. In this study, our results suggest that Txnip deficiency can improve glucose tolerance and insulin sensitivity by increasing β-cell mass. We found that Txnip deficiency can modulate the expression of transcription factors and cell cycle regulators and may activate PI3K/AKT signaling pathways to promote β-cell proliferation in HFD-induced obesity. Thus, our findings provide a molecular mechanism by which Txnip deficiency may provide a therapeutic target for DM.
The prevalence of obesity is increasing rapidly worldwide and is associated with peripheral insulin resistance and β-cell proliferation (16, 48). A commonly used model of obesity is HFD feeding in mice, which has been shown to promote β-cell proliferation and β-cell mass expansion (15, 49). In this study, we used HFD mice as a model for evaluating the mechanisms of β-cell compensatory proliferation, including the potential role of Txnip. After feeding for 12 weeks, the weight was increased in both WT-HFD and KO-HFD mice when compared to their control mice. Interestingly, the weight was significantly increased in WT-HFD mice compared to the other groups, while the weight of KO-HFD mice was not significantly increased. This suggests a mechanistic role of Txnip as a regulator of glucose and lipid metabolism and is consistent with previous studies demonstrating that Txnip-deficient (Hcb-19) mice have increased β-cell mass expansion and that this effect is more apparent when animals are stressed by multiple low-dose STZ administration or obesity (38, 50). Our data also show that Txnip KO mice have a greater pancreas to bodyweight ratio and significantly increased β-cell mass in KO-HFD mice. In contrast, these parameters were largely unchanged between WT and KO mice fed NC diet.
Aberrant glucagon production correlates with diabetes. Research has shown that blockade of glucagon signaling lowers glycemia in mouse models while enhancing the formation of functional β-cell mass (51). In our study, plasma glucagon levels significantly decreased in KO-HFD mice compared with WT-HFD group. This change may be related to the regulation of β cells by glucagon, while it also may be due to the conversion of α cells into β cells. However, the specific mechanism underlying this process still needs to be further investigated. Thus, our results suggest that HFD-induced obesity enhances β-cell mass expansion in Txnip KO mice. These findings are consistent with those of a previous study (38), though our results provide additional understanding of the ability of Txnip deficiency to improve glucose tolerance and insulin sensitivity in HFD-induced mice. Our results are also consistent with previous reports demonstrating that Txnip plays an important role in the regulation of insulin sensitivity (36, 52), thus providing a mechanism for Txnip in a DM model. We also measured the GLP1 and Leptin levels and found that the level of GLP1 was increased in KO-HFD mice, while the Leptin level was decreased in KO-HFD mice (Supplementary Fig. 1A, see section on supplementary materials given at the end of this article). Given that GLP1 has the ability to enhance β-cell proliferation and stimulate β-cell mass expansion in rodents and humans (53, 54), it is possible that Txnip may function in part by regulating the leptin/GLP-1balance. Leptin is additional negative regulator of β-cell mass expansion (55) and may be worth evaluating in the future to further enhance our understanding of the mechanism of Txnip deficiency in promoting β-cell mass expansion in HFD-induced obesity.
Many studies have demonstrated that Txnip comprises a major tumor suppressor that inhibits the proliferation of tumor cells (31, 40, 43, 44, 45, 46), which motivated us to speculate that Txnip may promote diabetes by inhibiting β-cell proliferation. To explore the relationship between Txnip and pancreatic β-cell proliferation, we performed immunofluorescence staining of proliferative antigens, such as Ki67 and Pcna. Our data show that in Txnip KO mice, Ki67+Ins+ and Pcna+Ins+ cells were significantly increased after 12 weeks of HFD. Thus, our results suggest that Txnip deficiency can promote pancreatic β-cell proliferation in HFD-induced obesity.
Numerous studies have identified a variety of pancreatic transcription factors that play crucial roles in β-cell regeneration and maintenance of mature β-cell function (8, 17, 18). To understand factors that regulate β-cell proliferation, we used real-time RT-PCR to examine the mRNA levels of specific transcription factors, such as Pdx1, Mafa, Foxo1 and Nkx6.1. Pdx1, which is a marker of β cells, is expressed in mature β cells and is required for β-cell differentiation and proliferation. Mafa also is expressed exclusively in β cells and functions as a potent activator of insulin gene transcription (17, 18, 19). Recently, Pdx1 has been shown to contribute to β-cell mass expansion and proliferation induced by the AKT pathway (56). Foxo1 is a key cell cycle entry factor that inhibits cell proliferation through the transcriptional regulation of cell cycle inhibitors and activators (57). Interestingly, Foxo1 is mutually exclusive with Pdx1 and promotes Pdx1 nuclear exclusion to counteract Pdx1 function in β cells (58, 59). Foxm1, on the other hand, promotes progression through the cell cycle by regulating genes important for the G1/S and G2/M transitions, as well as genes required for karyokinesis and cytokinesis (60, 61). Our results show that these pancreatic transcription factors are differentially and coordinately regulated at the level of mRNA in β cells, indicating that Txnip may inhibit β-cell proliferation by regulating pancreatic transcription factors, though the individual roles of these factors are still unclear and need further study.
Adult pancreatic β-cell regeneration mainly results from the proliferation of existing β cells (5). Thus, cell cycle molecules are likely to play a pivotal role in the proliferation of β cells. We have shown that TXNIP modulates the expression of a variety of genes involved in the cell cycle, including increases in the expression of Ccnd2 and Cdk4 and reduction in the expression of p27, at both the protein and mRNA levels. This is consistent with reports demonstrating that cyclin D2/CDK4 complex formation is essential for β-cell proliferation (62); and that, conversely, increased accumulation of p27 can inhibit β-cell proliferation (24, 63). Our data also show that Txnip deficiency leads to the activation of the PI3K/AKT pathway. This included the activation of mTOR as a downstream target of the PI3K/AKT pathway and increased phosphorylation of GSK3β. Previous work has shown that mTOR is an important regulator of the β-cell mass and β-cell proliferation in the pancreas. Furthermore, there is evidence that cyclin D2 stability acts downstream of mTOR signaling in pancreatic β cells (64). mTOR has been shown to regulate β-cell proliferation via cell cycle progression by modulating cyclin D2 and CDK4/cyclin D activity (64, 65). Interestingly, cyclin D2 has also been reported to act downstream of GSK3β (22). GSK3β inhibition promotes β-cell proliferation by regulating cell cycle activators and inhibitors, such as p27, cyclinD1, cyclinD2, CDK4 and CDK2 (66). Therefore, Txnip deficiency is likely to promote β-cell proliferation in HFD-induced obesity via PI3K/AKT signaling. However, the specific regulatory mechanism needs further investigation. We also demonstrated that the mRNA level of retinoblastoma protein (Rb) was reduced in KO-HFD mice, which is consistent with the possibility that Txnip deficiency may promote G1 to S transition to accelerate cell cycle progression and cell proliferation (67), though additional studies will be needed to verify this possibility. We examined the mRNA levels of other key cell cycle regulators that may influence cell cycle progression, including Ccna2, Ccnb1, Ccnb2, Ccnd1, Ccne1, Cdk2, Cdkn1a, Cdkn2a and Cdkn1c, most of which showed significant differences in KO-HFD vs WT-HFD mice. Thus, further studies may clarify the roles of these regulators in Txnip-mediated β-cell proliferation.
In conclusion, our results demonstrate that Txnip plays an important role in the regulation of β-cell mass expansion and β-cell proliferation. Txnip deficiency promotes β-cell proliferation in HFD-induced obesity, which may be mediated via activation of the PI3K/AKT signaling pathway. Our work thus provides a theoretical basis for Txnip as a new therapeutic target for the treatment of DM.
Supplementary Material
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by the Applied Basic Research Project of Natural Science Foundation of Shanxi Province (grant no. 20210302124377 and 201901D111192).
Acknowledgement
The authors thank LetPub (www.letpub.com) for linguistic assistance and pre-submission expert review.
References
- 1.Lim S, Bae JH, Kwon HS, Nauck MA. COVID-19 and diabetes mellitus: from pathophysiology to clinical management. Nature Reviews: Endocrinology 20211711–30. ( 10.1038/s41574-020-00435-4) [DOI] [Google Scholar]
- 2.Yu B, Li C, Sun Y, Wang DW. Insulin treatment is associated with increased mortality in patients with COVID-19 and type 2 diabetes. Cell Metabolism 20213365, .e2–77.e2. ( 10.1016/j.cmet.2020.11.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu L, She ZG, Cheng X, Qin JJ, Zhang XJ, Cai J, Lei F, Wang H, Xie J, Wang Wet al. Association of blood glucose control and outcomes in patients with COVID-19 and pre-existing Type 2 diabetes. Cell Metabolism 2020311068, .e3–1077.e3. ( 10.1016/j.cmet.2020.04.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 200554 (Supplement 2) S97–S107. ( 10.2337/diabetes.54.suppl_2.s97) [DOI] [PubMed] [Google Scholar]
- 5.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 200442941–46. ( 10.1038/nature02520) [DOI] [PubMed] [Google Scholar]
- 6.Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers Det al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008132197–207. ( 10.1016/j.cell.2007.12.015) [DOI] [PubMed] [Google Scholar]
- 7.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008455627–632. ( 10.1038/nature07314) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ackermann AM, Gannon M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. Journal of Molecular Endocrinology 200738193–206. ( 10.1677/JME-06-0053) [DOI] [PubMed] [Google Scholar]
- 9.Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 2009581312–1320. ( 10.2337/db08-1651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abouna S, Old RW, Pelengaris S, Epstein D, Ifandi V, Sweeney I, Khan M. Non-beta-cell progenitors of beta-cells in pregnant mice. Organogenesis 20106125–133. ( 10.4161/org.6.2.10374) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010532167–2176. ( 10.1007/s00125-010-1809-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, Sharma A. beta-Cell adaptation and decompensation during the progression of diabetes. Diabetes 200150 (Supplement 1) S154–S159. ( 10.2337/diabetes.50.2007.s154) [DOI] [PubMed] [Google Scholar]
- 13.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. Journal of Clinical Investigation 20071172553–2561. ( 10.1172/JCI32959) [DOI] [Google Scholar]
- 14.Cox AR, Lam CJ, Rankin MM, King KA, Chen P, Martinez R, Li C, Kushner JA. Extreme obesity induces massive beta cell expansion in mice through self-renewal and does not alter the beta cell lineage. Diabetologia 2016591231–1241. ( 10.1007/s00125-016-3922-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golson ML, Misfeldt AA, Kopsombut UG, Petersen CP, Gannon M. High fat diet regulation of beta-cell proliferation and beta-cell mass. Open Endocrinology Journal 20104. ( 10.2174/1874216501004010066) [DOI] [Google Scholar]
- 16.Linnemann AK, Baan M, Davis DB. Pancreatic beta-cell proliferation in obesity. Advances in Nutrition 20145278–288. ( 10.3945/an.113.005488) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaneto H, Matsuoka TA. Role of pancreatic transcription factors in maintenance of mature beta-cell function. International Journal of Molecular Sciences 2015166281–6297. ( 10.3390/ijms16036281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balakrishnan S, Dhavamani S, Prahalathan C. beta-Cell specific transcription factors in the context of diabetes mellitus and beta-cell regeneration. Mechanisms of Development 2020163103634. ( 10.1016/j.mod.2020.103634) [DOI] [PubMed] [Google Scholar]
- 19.Kaneto H, Miyatsuka T, Kawamori D, Yamamoto K, Kato K, Shiraiwa T, Katakami N, Yamasaki Y, Matsuhisa M, Matsuoka TA. PDX-1 and MafA play a crucial role in pancreatic beta-cell differentiation and maintenance of mature beta-cell function. Endocrine Journal 200855235–252. ( 10.1507/endocrj.k07e-041) [DOI] [PubMed] [Google Scholar]
- 20.Elghazi L, Balcazar N, Bernal-Mizrachi E. Emerging role of protein kinase B/Akt signaling in pancreatic beta-cell mass and function. International Journal of Biochemistry and Cell Biology 200638157–163. ( 10.1016/j.biocel.2005.08.017) [DOI] [PubMed] [Google Scholar]
- 21.Stewart AF, Hussain MA, Garcia-Ocana A, Vasavada RC, Bhushan A, Bernal-Mizrachi E, Kulkarni RN. Human beta-cell proliferation and intracellular signaling: part 3. Diabetes 2015641872–1885. ( 10.2337/db14-1843) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fatrai S, Elghazi L, Balcazar N, Cras-Meneur C, Krits I, Kiyokawa H, Bernal-Mizrachi E. Akt induces beta-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes 200655318–325. ( 10.2337/diabetes.55.02.06.db05-0757) [DOI] [PubMed] [Google Scholar]
- 23.Georgia S, Hinault C, Kawamori D, Hu J, Meyer J, Kanji M, Bhushan A, Kulkarni RN. Cyclin D2 is essential for the compensatory beta-cell hyperplastic response to insulin resistance in rodents. Diabetes 201059987–996. ( 10.2337/db09-0838) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uchida T, Nakamura T, Hashimoto N, Matsuda T, Kotani K, Sakaue H, Kido Y, Hayashi Y, Nakayama KI, White MFet al. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nature Medicine 200511175–182. ( 10.1038/nm1187) [DOI] [Google Scholar]
- 25.Kitamura T.The role of FOXO1 in beta-cell failure and type 2 diabetes mellitus. Nature Reviews: Endocrinology 20139615–623. ( 10.1038/nrendo.2013.157) [DOI] [Google Scholar]
- 26.Zhu X, Oguh A, Gingerich MA, Soleimanpour SA, Stoffers DA, Gannon M. Cell cycle regulation of the Pdx1 transcription factor in developing pancreas and insulin-producing beta-cells. Diabetes 202170903–916. ( 10.2337/db20-0599) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen KS, DeLuca HF. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochimica et Biophysica Acta 1994121926–32. ( 10.1016/0167-4781(9490242-9) [DOI] [PubMed] [Google Scholar]
- 28.Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. Journal of Biological Chemistry 199927421645–21650. ( 10.1074/jbc.274.31.21645) [DOI] [Google Scholar]
- 29.Hu J, Yu Y. The function of thioredoxin-binding protein-2 (TBP-2) in different diseases. Oxidative Medicine and Cellular Longevity 201820184582130. ( 10.1155/2018/4582130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsubaki H, Tooyama I, Walker DG. Thioredoxin-interacting protein (TXNIP) with focus on brain and neurodegenerative diseases. International Journal of Molecular Sciences 2020219, 35, 7. ( 10.3390/ijms21249357) [DOI] [Google Scholar]
- 31.Masutani H.Thioredoxin-interacting protein in cancer and diabetes. Antioxidants and Redox Signaling 2021. [epub]. ( 10.1089/ars.2021.0038) [DOI] [Google Scholar]
- 32.Patwari P, Higgins LJ, Chutkow WA, Yoshioka J, Lee RT. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. Journal of Biological Chemistry 200628121884–21891. ( 10.1074/jbc.M600427200) [DOI] [Google Scholar]
- 33.Yoshihara E.TXNIP/TBP-2: a master regulator for glucose homeostasis. Antioxidants 20209765. ( 10.3390/antiox9080765) [DOI] [Google Scholar]
- 34.Chutkow WA, Birkenfeld AL, Brown JD, Lee HY, Frederick DW, Yoshioka J, Patwari P, Kursawe R, Cushman SW, Plutzky Jet al. Deletion of the alpha-arrestin protein Txnip in mice promotes adiposity and adipogenesis while preserving insulin sensitivity. Diabetes 2010591424–1434. ( 10.2337/db09-1212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Omar DF, Kamal MM, El-Hefnawy MH, El-Mesallamy HO. Serum vitamin D and its upregulated protein, thioredoxin interacting protein, are associated with beta-cell dysfunction in adult patients with type 1 and type 2 diabetes. Canadian Journal of Diabetes 201842588–594. ( 10.1016/j.jcjd.2018.02.012) [DOI] [PubMed] [Google Scholar]
- 36.Oka S, Yoshihara E, Bizen-Abe A, Liu W, Watanabe M, Yodoi J, Masutani H. Thioredoxin binding protein-2/thioredoxin-interacting protein is a critical regulator of insulin secretion and peroxisome proliferator-activated receptor function. Endocrinology 20091501225–1234. ( 10.1210/en.2008-0646) [DOI] [PubMed] [Google Scholar]
- 37.Hui TY, Sheth SS, Diffley JM, Potter DW, Lusis AJ, Attie AD, Davis RA. Mice lacking thioredoxin-interacting protein provide evidence linking cellular redox state to appropriate response to nutritional signals. Journal of Biological Chemistry 200427924387–24393. ( 10.1074/jbc.M401280200) [DOI] [Google Scholar]
- 38.Chen J, Hui ST, Couto FM, Mungrue IN, Davis DB, Attie AD, Lusis AJ, Davis RA, Shalev A. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB Journal 2008223581–3594. ( 10.1096/fj.08-111690) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida T, Kondo N, Oka S, Ahsan MK, Hara T, Masutani H, Nakamura H, Yodoi J. Thioredoxin-binding protein-2 (TBP-2): its potential roles in the aging process. BioFactors 20062747–51. ( 10.1002/biof.5520270105) [DOI] [PubMed] [Google Scholar]
- 40.Han SH, Jeon JH, Ju HR, Jung U, Kim KY, Yoo HS, Lee YH, Song KS, Hwang HM, Na YSet al. VDUP1 upregulated by TGF-beta1 and 1,25-dihydorxyvitamin D3 inhibits tumor cell growth by blocking cell-cycle progression. Oncogene 2003224035–4046. ( 10.1038/sj.onc.1206610) [DOI] [PubMed] [Google Scholar]
- 41.Zhang P, Gao J, Wang X, Wen W, Yang H, Tian Y, Liu N, Wang Z, Liu H, Zhang Yet al. A novel indication of thioredoxin-interacting protein as a tumor suppressor gene in malignant glioma. Oncology Letters 2017142053–2058. ( 10.3892/ol.2017.6397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stolearenco V, Levring TB, Nielsen HM, Lindahl L, Fredholm S, Kongsbak-Wismann M, Willerslev-Olsen A, Buus TB, Nastasi C, Hu Tet al. The thioredoxin-interacting protein TXNIP is a putative tumour suppressor in cutaneous T-cell lymphoma. Dermatology 2021237283–290. ( 10.1159/000509159) [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Ning J, Cao W, Wang S, Du T, Jiang J, Feng X, Zhang B. Research progress of TXNIP as a tumor suppressor gene participating in the metabolic reprogramming and oxidative stress of cancer cells in various cancers. Frontiers in Oncology 202010568574. ( 10.3389/fonc.2020.568574) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie M, Xie R, Xie S, Wu Y, Wang W, Li X, Xu Y, Liu B, Zhou Y, Wang Tet al. Thioredoxin interacting protein (TXNIP) acts as a tumor suppressor in human prostate cancer. Cell Biology International 2020442094–2106. ( 10.1002/cbin.11418) [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Tian X, Yin H, Xiao S, Yi S, Zhang Y, Zeng F. TXNIP induced by MondoA, rather than ChREBP, suppresses cervical cancer cell proliferation, migration and invasion. Journal of Biochemistry 2020167371–377. ( 10.1093/jb/mvz105) [DOI] [PubMed] [Google Scholar]
- 46.Nishinaka Y, Nishiyama A, Masutani H, Oka S, Ahsan KM, Nakayama Y, Ishii Y, Nakamura H, Maeda M, Yodoi J. Loss of thioredoxin-binding protein-2/vitamin D3 up-regulated protein 1 in human T-cell leukemia virus type I-dependent T-cell transformation: implications for adult T-cell leukemia leukemogenesis. Cancer Research 2004641287–1292. ( 10.1158/0008-5472.can-03-0908) [DOI] [PubMed] [Google Scholar]
- 47.Eizirik DL, Pasquali L, Cnop M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nature Reviews: Endocrinology 202016349–362. ( 10.1038/s41574-020-0355-7) [DOI] [Google Scholar]
- 48.Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metabolism: Clinical and Experimental 2019926–10. ( 10.1016/j.metabol.2018.09.005) [DOI] [PubMed] [Google Scholar]
- 49.Mosser RE, Maulis MF, Moulle VS, Dunn JC, Carboneau BA, Arasi K, Pappan K, Poitout V, Gannon M. High-fat diet-induced beta-cell proliferation occurs prior to insulin resistance in C57BL/6J male mice. American Journal of Physiology: Endocrinology and Metabolism 2015308E573–E582. ( 10.1152/ajpendo.00460.2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Masson E, Koren S, Razik F, Goldberg H, Kwan EP, Sheu L, Gaisano HY, Fantus IG. High beta-cell mass prevents streptozotocin-induced diabetes in thioredoxin-interacting protein-deficient mice. American Journal of Physiology: Endocrinology and Metabolism 2009296E1251–E1261. ( 10.1152/ajpendo.90619.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang MY, Dean ED, Quittner-Strom E, Zhu Y, Chowdhury KH, Zhang Z, Zhao S, Li N, Ye R, Lee Yet al. Glucagon blockade restores functional beta-cell mass in type 1 diabetic mice and enhances function of human islets. PNAS 2021118. ( 10.1073/pnas.2022142118) [DOI] [Google Scholar]
- 52.Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJet al. TXNIP regulates peripheral glucose metabolism in humans. PLOS Medicine 20074 e158. ( 10.1371/journal.pmed.0040158) [DOI] [Google Scholar]
- 53.Dai C, Hang Y, Shostak A, Poffenberger G, Hart N, Prasad N, Phillips N, Levy SE, Greiner DL, Shultz LDet al. Age-dependent human beta cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. Journal of Clinical Investigation 20171273835–3844. ( 10.1172/JCI91761) [DOI] [Google Scholar]
- 54.Stoffers DA, Kieffer TJ, Hussain MA, Drucker DJ, Bonner-Weir S, Habener JF, Egan JM. Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein IDX-1 and increase islet size in mouse pancreas. Diabetes 200049741–748. ( 10.2337/diabetes.49.5.741) [DOI] [PubMed] [Google Scholar]
- 55.Marroqui L, Gonzalez A, Neco P, Caballero-Garrido E, Vieira E, Ripoll C, Nadal A, Quesada I. Role of leptin in the pancreatic beta-cell: effects and signaling pathways. Journal of Molecular Endocrinology 201249R9–R17. ( 10.1530/JME-12-0025) [DOI] [PubMed] [Google Scholar]
- 56.Jara MA, Werneck-De-Castro JP, Lubaczeuski C, Johnson JD, Bernal-Mizrachi E. Pancreatic and duodenal homeobox-1 (PDX1) contributes to beta-cell mass expansion and proliferation induced by Akt/PKB pathway. Islets 20201232–40. ( 10.1080/19382014.2020.1762471) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, Furuyama K, Thorel F, Gribble FM, Reimann Fet al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature 2014514503–507. ( 10.1038/nature13633) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawamori D, Kaneto H, Nakatani Y, Matsuoka TA, Matsuhisa M, Hori M, Yamasaki Y. The forkhead transcription factor FoxO1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. Journal of Biological Chemistry 20062811091–1098. ( 10.1074/jbc.M508510200) [DOI] [Google Scholar]
- 59.Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A, Accili D. Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. Journal of Clinical Investigation 2006116775–782. ( 10.1172/JCI24967) [DOI] [Google Scholar]
- 60.Laoukili J, Alvarez M, Meijer LA, Stahl M, Mohammed S, Kleij L, Heck AJ, Medema RH. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Molecular and Cellular Biology 2008283076–3087. ( 10.1128/MCB.01710-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wierstra I, Alves J. FOXM1, a typical proliferation-associated transcription factor. Biological Chemistry 20073881257–1274. ( 10.1515/BC.2007.159) [DOI] [PubMed] [Google Scholar]
- 62.Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. Journal of Clinical Investigation 2004114963–968. ( 10.1172/JCI22098) [DOI] [Google Scholar]
- 63.Georgia S, Bhushan A. p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes 2006552950–2956. ( 10.2337/db06-0249) [DOI] [PubMed] [Google Scholar]
- 64.Balcazar N, Sathyamurthy A, Elghazi L, Gould A, Weiss A, Shiojima I, Walsh K, Bernal-Mizrachi E. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. Journal of Biological Chemistry 20092847832–7842. ( 10.1074/jbc.M807458200) [DOI] [Google Scholar]
- 65.Balcazar Morales N, Aguilar de Plata C. Role of AKT/mTORC1 pathway in pancreatic beta-cell proliferation. Colombia Medica 201243235–243. [PMC free article] [PubMed] [Google Scholar]
- 66.Stein J, Milewski WM, Hara M, Steiner DF, Dey A. GSK-3 inactivation or depletion promotes beta-cell replication via down regulation of the CDK inhibitor, p27 (Kip1). Islets 2011321–34. ( 10.4161/isl.3.1.14435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene 2006255220–5227. ( 10.1038/sj.onc.1209615) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.