

Abstract
Synapse elimination, also known as synaptic pruning, is a critical step in the maturation of neural circuits during brain development. Mounting evidence indicates that the complement cascade of the innate immune system plays an important role in synapse elimination. Studies indicate that excess synapses during development are opsonized by complement proteins and subsequently phagocytosed by microglia which expresses complement receptors. The process is regulated by diverse molecular signals, including complement inhibitors that affect the activation of complement, as well as signals that affect microglial recruitment and activation. These signals may promote or inhibit the removal of specific sets of synapses during development. The complement-microglia system has also been implicated in the pathogenesis of several developmental brain disorders, suggesting that the dysregulation of mechanisms of synapse pruning may underlie the specific circuitry defects in these diseases. Here, we review the latest evidence on the molecular and cellular mechanisms of complement-dependent and microglia-dependent synapse elimination during brain development, and highlight the potential of this system as a therapeutic target for developmental brain disorders.
This article is categorized under:
Neurological Diseases > Molecular and Cellular Physiology
Neurological Diseases > Stem Cells and Development
Immune System Diseases > Molecular and Cellular Physiology
Keywords: brain development, complement system, microglia, synapse elimination, synaptic pruning
Graphical Abstract
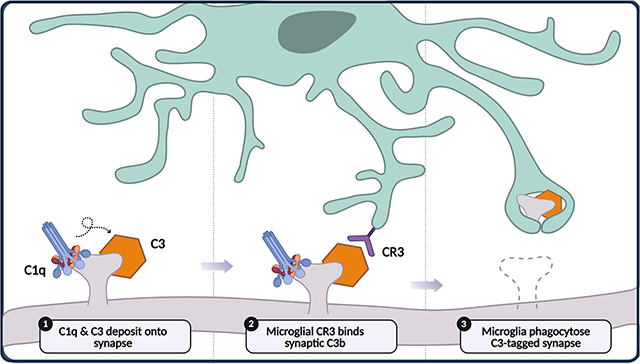
1 |. INTRODUCTION
1.1 |. Synapse elimination in brain development
Synapse elimination is a critical process through which the brain achieves proper maturation of brain circuits. The developing brain initially over-produces synapses, and then subsequently eliminates excess synapses in an activity-dependent manner (Changeux & Danchin, 1976) to achieve circuit specialization and remove redundant neural pathways (Figure 1a). The selective stabilization of active synapses and preferential elimination of inactive synapses provides a mechanism for early-life experiences to shape the developing brain. Brain development in humans has a protracted time course, with developmental synapse elimination occurring gradually over at least the first two decades of life (Huttenlocher, 1979), and possibly longer (Petanjek et al., 2011). Synapse elimination occurs with different time courses depending on the brain region (Huttenlocher & Dabholkar, 1997), typically with thalamic regions completing the process earlier than neocortical regions. Within the cortex, sensory and language-related cortical regions mature by early childhood, while higher-level regions such as the prefrontal cortex undergo synapse refinement throughout adolescence and into early adulthood (Figure 1b; Huttenlocher & Dabholkar, 1997; Thompson & Nelson, 2001). This stereotypical progression of synapse elimination has been observed in many other mammals, including in non-human primates (Bourgeois & Rakic, 1993; Rakic et al., 1986) and rodents (Pinto et al., 2013). The observation that developmental synapse elimination occurs in multiple brain regions and in multiple animals suggests that a conserved core mechanism operates to remove excess synapses during brain development. The prolonged period over which synapse elimination occurs, especially in the human prefrontal cortex, creates an extended window of vulnerability for deleterious environment–gene interactions, and perturbation in synapse elimination may be a significant factor in the pathogenesis of neurodevelopmental disorders such as schizophrenia and autism.
FIGURE 1.
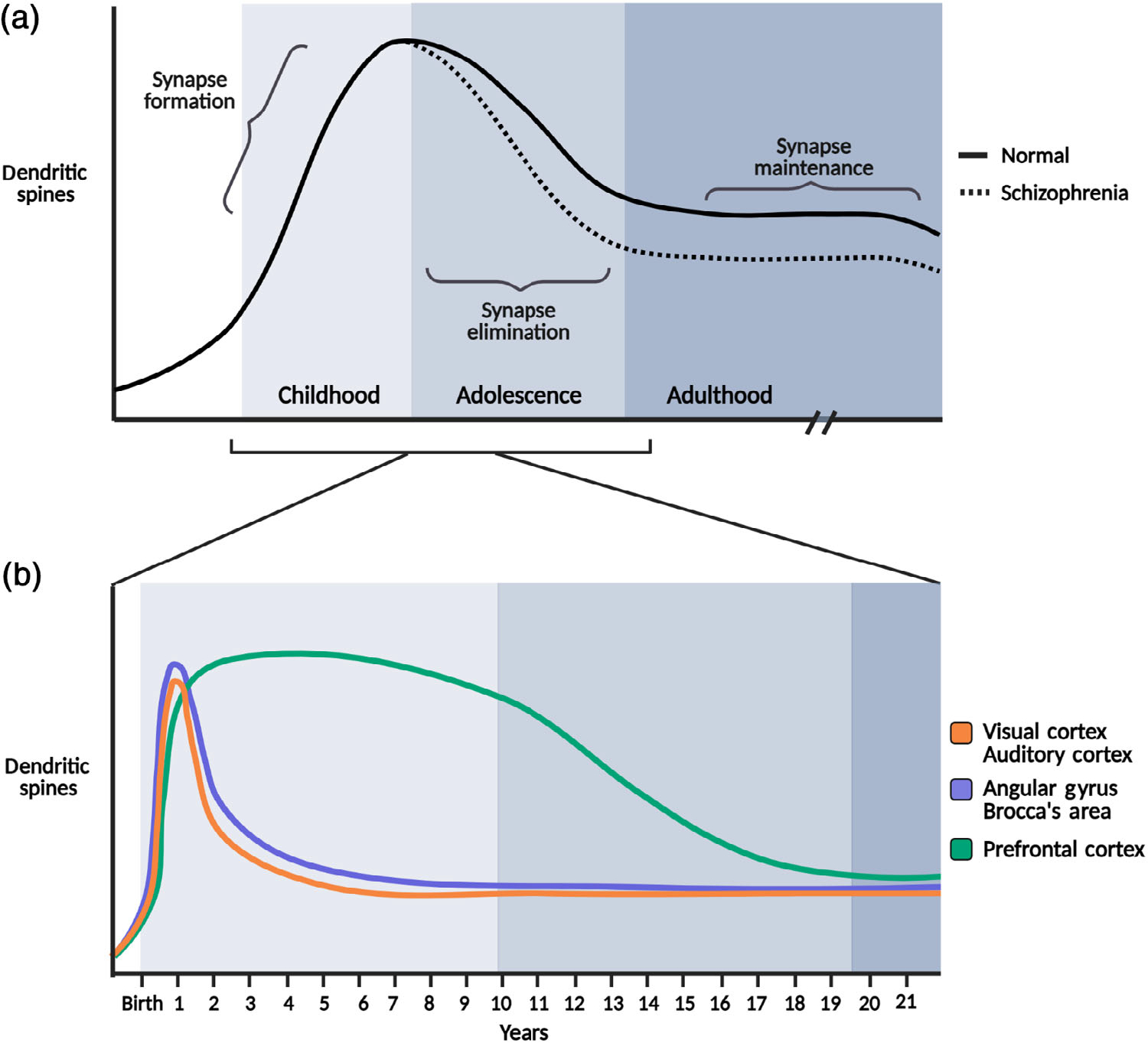
Developmental timeline for synapse formation and elimination. (a) Average dendritic spine density throughout the brain during normal development (solid line), autism (dotted line), and schizophrenia (dashed line). (b) Inset highlights regional temporal differences in synapse formation and elimination. The prolonged period of synapse elimination in the prefrontal cortex that extends from adolescence to adulthood (blue line). Top graph: (a) Reprinted with permission from Penzes et al. (2011); bottom graph: (b) Reprinted with permission from Thompson and Nelson (2001)
1.2 |. Complement and microglia involvement in synapse elimination
Complement-dependent microglial phagocytosis of synapses has recently been proposed as a core mechanism for synapse elimination in the developing mammalian brain. The complement system is composed of a series of serine proteases which, when activated, sequentially cleave downstream components in a self-amplifying cascade of reactions (Figure 2). The complement system can be activated through three pathways: the classical, lectin, and alternative pathways. These initiation pathways all lead to the proteolytic activation of the core complement effector molecule C3, which can also cleave itself in a self-amplifying loop. While the classical and lectin pathways rely on danger-sensing initiation molecules for activation, the alternative pathway is constitutively activated through the spontaneous hydrolysis of C3. These reactions eventually generate insoluble proteins which are deposited onto membrane surfaces, where they perform different functions. In host defense, the complement system deposits membrane attack complexes (MAC) and opsonins on the surface of invading pathogens, causing membrane lysis and tagging pathogens for phagocytosis by macrophages. The complement system also opsonizes and tags apoptotic cells and cellular debris for phagocytic clearance to maintain tissue homeostasis in the periphery (Bajic et al., 2015). In 2007, a seminal study revealed that complement proteins perform a similar function in the brain, mediating developmental synapse elimination. It was shown that mice lacking C1q or C3 show incomplete synapse elimination at the retinogeniculate synapse (Stevens et al., 2007). Electrophysiological data show that the supernumerary synapses in the complement knockout mice can release and sense synaptic vesicles in response to electrical stimulation, suggesting that complement-mediated synapse elimination does not merely clear away synaptic debris generated by some other mechanism, but is required for synapses to lose functionality (Stevens et al., 2007). Complement-mediated synapse elimination is believed to occur through microglial phagocytosis of weaker C3-tagged synapses via the C3 receptor (CR3) on microglia (Schafer et al., 2012). This basic model is supported by numerous studies examining both complement and microglia related genes in brain development (Table 1). Mice with genetic deletions in several phagocytosis-related or chemotaxis-related microglial proteins, such as the phagocytotic receptor TREM2 (Filipello et al., 2018) and the fractalkine receptor CX3CR1 (Paolicelli et al., 2011; Zhan et al., 2014), show supernumerary synapses in the brain. Conversely, mice lacking genes that inhibit either the complement system or microglial phagocytosis display reduced synapse densities, including mice lacking the microglial phagocytosis inhibitory factor CD47 (Lehrman et al., 2018) and the novel complement inhibitor SRPX2 (Cong et al., 2020). This model is also supported at the anatomical level by histological and live imaging studies showing microglial processes apposed to synaptic elements and presence of synaptic material within microglial lysosomes (Tremblay et al., 2010; Weinhard et al., 2018), as well as a proteomic study showing the existence of C1q-tagged synaptosomes (Györffy et al., 2018). Furthermore, human genetic studies have begun to implicate complement component genes and its regulators in the pathogenesis of neurodevelopmental disorders such as schizophrenia (Håvik et al., 2011; Sekar et al., 2016). While multiple neuron intrinsic and extrinsic molecular mechanisms have been shown to regulate synapse elimination (Riccomagno & Kolodkin, 2015), the totality of the evidence suggests that complement-mediated and microglia-mediated synapse elimination is a core mechanism in brain development. This review critically summarizes the evidence for complement and microglia involvement in synapse elimination during normal and pathological brain development, focusing on studies in humans and mammalian animal models.
FIGURE 2.
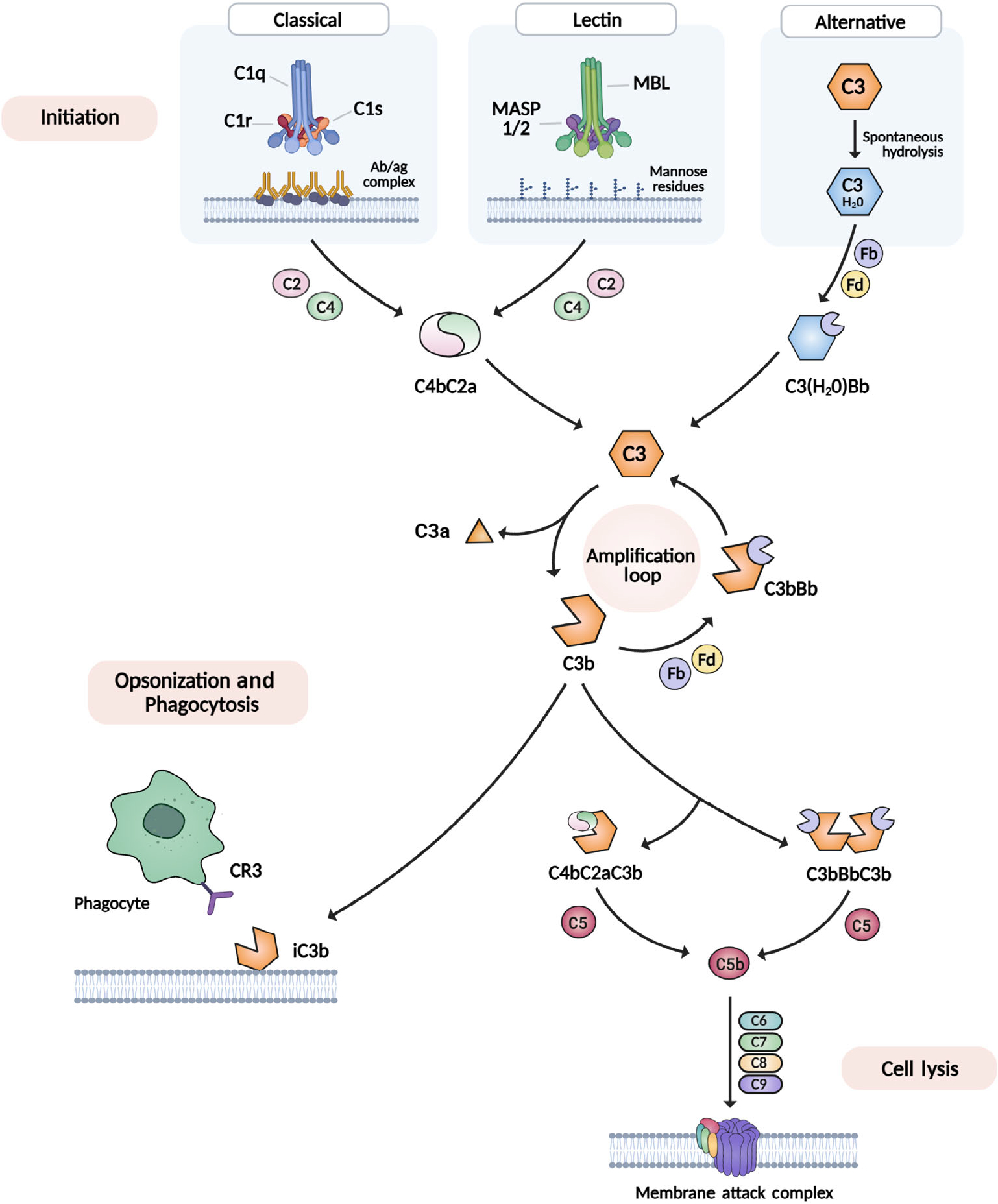
The complement cascade: classical, lectin, and alternative pathways. Initiation of the complement cascade occurs through three routes: the classical pathway (C1q binds antibody–antigen complex), the lectin pathway (MBL binds mannose-residues on pathogen) and the alternative pathway (C3 spontaneously hydrolyzes into C3·H20). Each route yields a C3 convertase (C4bC2a or C3[H20]Bb) which cleaves the downstream complement component C3 into a and b fragments. The signal is amplified by the generation of more C3 convertase from C3b fragments. Inactivated, membrane-bound C3b fragments interact with the C3 receptor (CR3). Robust C3b production enables the formation of C5 convertases (C4bC2aC3b and C3bBbC3b), leading cell lysis via insertion of the membrane attack complex (MAC)
TABLE 1.
Anatomical readouts in complement/microglia mouse models
| Author | Gene | Gene function | Region | Time point | Results |
|---|---|---|---|---|---|
| Stevens et al. (2007) | C3 KO, C1QA KO | Complement components | LGN | P10–P30 | Persistent deficit in eye-specific axon segregation |
| Schafer et al. (2012) | CR3/CD11b knockout | Complement receptor | LGN | P10–P30 | Persistent deficit in RGC axon segregation |
| Bialas and Stevens (2013) | Tgfbr2 KO | Regulator of C1q expression | LGN | P10 | Persistent deficit in RGC axon segregation |
| Lehrman et al. (2018) | CD47 | Phagocytosis inhibitor | LGN | P5–P60 | ↓ VGlut2+ retinogeniculate synapses |
| Cong et al. (2020) | SRPX2 | Complement inhibitor | LGN | P4–P30 | Transient deficit in RGC axon segregation, ↓ VGlut2+ retinogeniculate synapses |
| Paolicelli et al. (2011) | CX3CR1 | Fractalkine receptor | CA1 | P15 | ↓ Microglia cell density, ↑ synapse density |
| Zhan et al. (2014) | CX3CR1 | Fractalkine receptor | CA1 | P40 | ↓ Multisynapse bouton density |
| Filipello et al. (2018) | TREM2 | Phagocytosis receptor | CA1 | P18–P20 | ↓ Microglia cell density, ↑ synapse density |
| Tremblay et al. (2010) | V1 cortex | P28 | Microglia engulfment of synapses modulated by activity | ||
| Schecter et al. (2017) | CX3CR1 KO | V1 cortex | P28–P31 | No difference in ODP | |
| Welsh et al. (2020) | C1QA KO | V1 cortex | P10–P30 | No difference in L2/3 apical dendritic spines, no difference in ODP | |
| Ding et al. (2021) | Microglial SIRPα KO, CD47 KO | CA1, V1 cortex | P30 | ↓ VGlut1+ synapses | |
| Chu et al. (2010) | C1QA KO | Neocortex | P30 | Epileptiform activity, ↑ L4 and L5 synapses | |
| Cong et al. (2020) | SRPX2 KO | Complement inhibitor | LGN, SS cortex | P30–P90 | Reduction in VGlut2/PSD95 puncta and dendritic spine density |
| Kopec et al. (2018) | CR3 antagonist | Complement receptor | NAc | P20–P54 | ↓ D1R engulfment |
2 |. ROLE OF COMPLEMENT AND MICROGLIA IN DEVELOPMENTAL SYNAPSE ELIMINATION
2.1 |. Complement and complement inhibitor expression in the brain
Historically, the brain was believed to be devoid of complement activity because the blood brain barrier segregates it from blood-derived complement components. However, recent studies have shown that both neurons and glia express complement components in the developing brain. While studies have reported the presence of complement protein in the brain by immunostaining or blotting, the most definitive demonstration of local complement expression is the detection of mRNA transcripts in specific brain cells. Several studies have now reported the existence of low levels of complement component mRNA transcripts in neurons in the healthy post-mortem human brain (Sager et al., 2021; Shen et al., 1997), as well as in the mouse brain (Stevens et al., 2007). Studies in mice have also reported that microglia are the predominant source of C1q (Fonseca et al., 2017) and C3aR (Lian et al., 2016), while astrocytes are the predominant source of C3 (Clarke et al., 2018; Lian et al., 2016). These studies indicate that complement components are expressed endogenously in the brain, and may participate in normal physiological functions such as mediating microglia phagocytosis of excess synapses during development.
In addition to complement proteins, the brain also expresses complement inhibitors. In the periphery, nearly all cells express at least one membrane bound complement inhibitor to inhibit non-specific and spontaneous activation of blood-derived complement components. For example, many mammalian cells possess sialylated proteoglycans on the cell surface, which binds to the complement inhibitor Factor H (cFH), the predominant complement inhibitor in blood (Makou et al., 2013). However, brain cells do not detectably express most known conventional complement inhibitors (Cahoy et al., 2008). This raises the question of whether the brain possesses mechanisms to counteract complement activity to prevent “runaway” synapse elimination. We have recently shown that neurons express a novel complement inhibitor which protects synapses from elimination during brain development (Cong et al., 2020). Thus, the brain endogenously expresses both complement components and complement inhibitors to dynamically shape developing brain circuits.
2.2 |. Lateral geniculata nucleus of the thalamus
The involvement of the complement system in synapse elimination was first uncovered at the retinogeniculate synapse of the visual system (Stevens et al., 2007), a classic model for developmental synapse elimination. Several characteristics make this synapse experimentally attractive for these studies. In this model system, synapse elimination is activity-dependent and takes place over a short period immediately after birth (Huberman et al., 2008), allowing for rapid results. In the span of 2–4 weeks, retinal ganglion cell (RGC) axons from both eyes transition from initially evenly innervating the entire lateral geniculate nucleus (LGN), to eventually innervating distinct stereotypical surround-center regions, with synapses formed by axons from the ipsilateral eye being eliminated from the surround region, and synapses from the contralateral eye being eliminated from the center region (Figure 3a,b). Both populations of axons can be easily labeled and manipulated through intravitreal injection of the eyes. Intriguingly, before the discovery of the role of complement at this synapse, several immune-associated genes have already been shown to affect synapse elimination in the LGN, including the class I major histocompatibility complex (MHC) genes (Corriveau et al., 1998; Huh et al., 2000) and the neuronal pentraxins NP1/NP2 (Bjartmar et al., 2006). The earliest evidence for complement involvement in this process came from the Barres lab, which in the course of investigating astrocytic factors that affect neuronal function, found that coculturing RGCs with astrocytes caused a large increase in RGC C1q mRNA expression, and that C1q protein is localized to retinogeniculate synapses. Genetic deletion of both C1QA and C3 caused a persistent deficit in retinogeniculate synapse elimination, assessed both histologically and electrophysiologically, suggesting that the classical complement pathway plays a role in synapse elimination (Stevens et al., 2007). However, it should be noted that even in the C1QA and C3 KO mice, some synapse elimination still occurs in the LGN, suggesting that complement components operate in concert with other molecular pathways to regulate synapse elimination in this brain region. Subsequently, the Stevens lab identified TGF-β as the astrocyte-derived factor which increased C1q expression in RGCs, and showed that deletion of TGF-β receptor II (TGFβRII) specifically in RGCs phenocopied the LGN synapse elimination deficits found in the complement KO mice (Bialas & Stevens, 2013). Another study from the Stevens lab showed that the cell type that mediate complement-dependent synapse elimination is microglia, the tissue-resident professional phagocyte of the brain. Microglia were shown to engulf less active synapses in the LGN, and genetic deletion of both C3 and its receptor CR3/CD11b, which is only expressed by microglia in the brain, caused reductions in both microglial synapse engulfment and synapse elimination (Schafer et al., 2012). These foundational papers laid the basic molecular and cellular framework for complement-dependent synapse elimination, which has been validated by subsequent studies examining genes that antagonize this process. Deletion of “don’t eat me” signal CD47 or its receptor SIRPα, which inhibit peripheral macrophage phagocytosis, also increases microglial engulfment of retinogeniculate synapses and synapse elimination in the LGN (Lehrman et al., 2018), as does genetic deletion of SRPX2, a neuronal complement inhibitor which binds to and inhibits C1q (Cong et al., 2020). Together, these studies show that complement-mediated and microglia-mediated synapse elimination plays an important role in retinogeniculate synapse elimination during early postnatal development of the visual system (Figure 3c).
FIGURE 3.
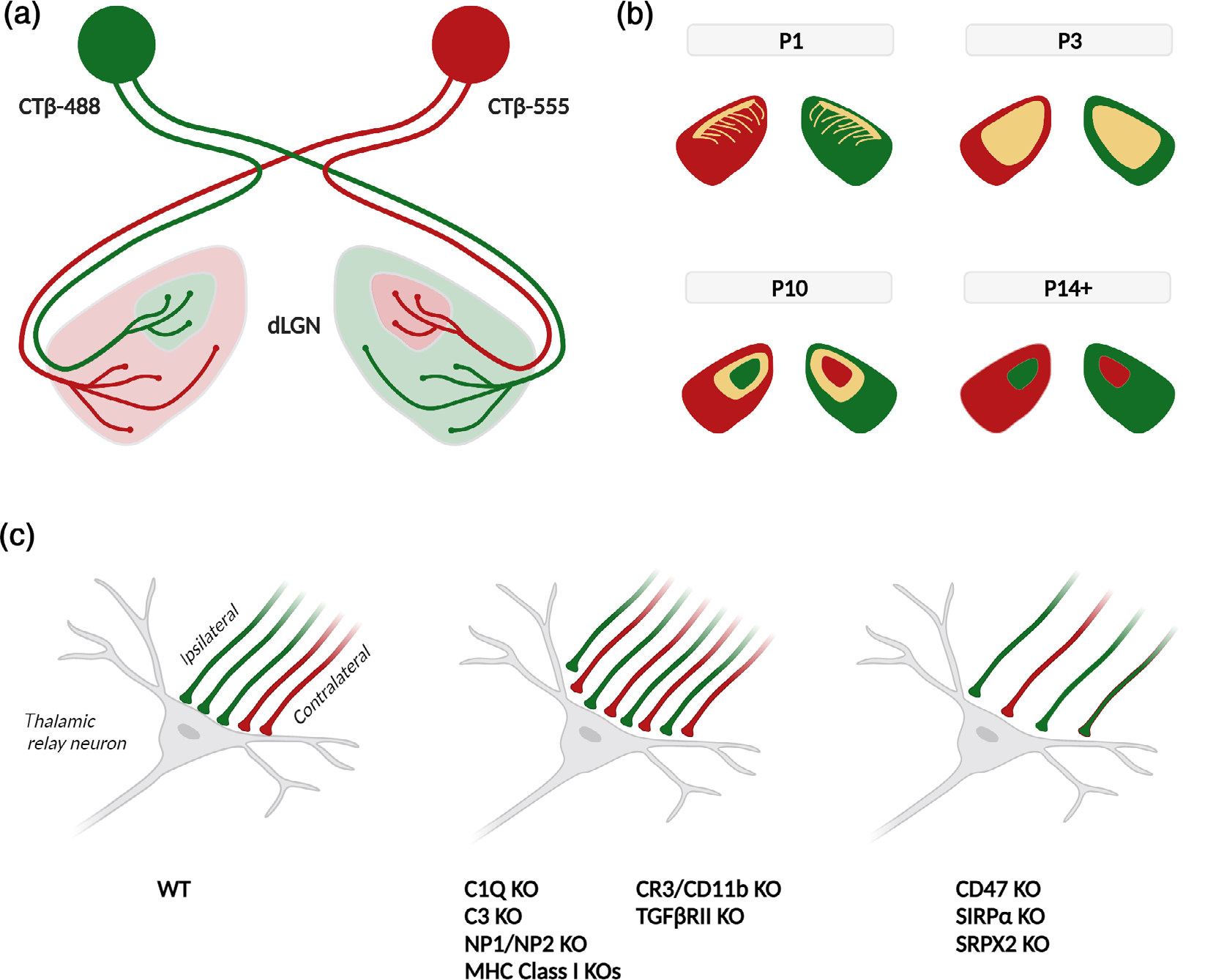
RGC axon segregation. (a) Schematic for the retinal ganglion cell (RGC) axon segregation assay for synapse refinement. Intraocular injections of anterograde tracer CTβ conjugated with A488 (green) or A55 (red) illuminate the ipsilateral and contralateral RGC axons innervating in the dLGN. (b) Segregation of CTβ-labeled axonal territories in the dLGN over the first two postnatal weeks of life. Overlapping inputs in yellow. (c) Binocular innervation of thalamic relay neurons in the developing dLGN by ipsilateral (green) and contralateral (red) RGC axons, at age P10 (left). Genetic deletion of several immune-related genes produces deficits in eye-specific axon segregation, causing increased bilateral innervation (middle). Deletion of “don’t eat me” signals results in excessive synapse elimination, causing decreased bilateral innervation (right)
2.3 |. Hippocampus
While developmental synapse elimination largely occurs during the early postnatal period in the thalamus, in the cortex, analysis of synapse elimination is more complicated due to the presence of different classes of synapses in diverse subregions, as well as the occurrence of synapse elimination over an extended time period. In the hippocampus, deletion of the fractalkine receptor CX3CR1, a chemotaxis receptor for the fractalkine CX3CL1 (Harrison et al., 1998), caused a transient reduction in microglia numbers in the hippocampal CA1 region at P15. Histology with both confocal microscopy and electron microscopy showed that microglia engulfed synapses in hippocampal CA1, and dendritic spine density and mEPSC frequency are both increased at P15 (Paolicelli et al., 2011), suggesting that microglia prune synapses in the hippocampus during early development. While spine density was shown to be normal by P40 (Paolicelli et al., 2011), a subsequent study reported that numbers of multisynapse boutons were persistently reduced even at P40 in the CX3CR1 KO mouse (Zhan et al., 2014), suggesting that a transient deficit in microglia synapse pruning can lead to persistent circuitry defects. In another study, genetic deletion of TREM2, a phagocytosis receptor expressed by microglia, led to a reduction in microglia numbers and increased synapses in the hippocampal CA1 region at P18–P20, along with a reduction in microglia synaptic engulfment in this region (Filipello et al., 2018). Finally, microglial-specific deletion of SIRPα, the CD47 “don’t eat me” signal receptor, led to a reduction in CA1 synapses at both P15 and P30 (Ding et al., 2021). Thus, manipulations of microglia in the hippocampal CA1 region in early development leads to perturbations of synapse elimination in this region.
While microglia-oriented studies in the hippocampus have focused on time points before the first month, complement expression in the hippocampus is generally low at early postnatal ages (Shi et al., 2015; Stephan et al., 2013), and studies involving complement genes have focused on later time points when complement expression is more robust. A study using the C1QA KO mouse revealed no deficit in dentate gyrus granule cell spine density even at 18 months, when C1QA expression is relatively high in the region (Stephan et al., 2013), and a comprehensive study with the C3 KO mouse showed that VGlut2 synapse densities are similar in KO and WT mice at 1 month, but in the KO mouse, these synapses fail to show an age-dependent decline seen in WT mice (Shi et al., 2015). Intriguingly, this effect is CA3-specific, and CA1 synapse densities are similar in KO and WT at all ages (Shi et al., 2015). Thus, complement and microglia involvement in synapse elimination in the hippocampus varies depending on both the region and the time point of analysis.
2.4 |. Neocortex
Studies in the neocortex have generally found involvement of either microglia or complement in synapse elimination in late, but not early postnatal development. For example, in the primary visual (V1) cortex, baseline layer 2/3 apical spine density at P10–P30 is largely unchanged in the C1QA KO mouse (Welsh et al., 2020), and synapse elimination due to ocular dominance plasticity induced by monocular deprivation at P28–P30 does not require either C1QA (Welsh et al., 2020) or CX3CR1 (Schecter et al., 2017). However, at P30, a serial section EM and two-photon live imaging study in L2 of the V1 cortex found microglial processes apposed to synapses, and light deprivation and reexposure stimulated microglia to contact and engulf synapses more frequently (Tremblay et al., 2010). In addition, microglial deletion of SIRPα as well as global KO of the SIRPα ligand CD47 both caused a decrease in VGlut1 synapses in the V1 cortex at P30 but not earlier (Ding et al., 2021). Epileptiform activity was also detected at multiple regions in the neocortex of the C1QA KO mouse but not in the WT, and increased EPSCs can be recorded from L5 pyramidal neurons when glutamate is uncaged in L4 and L5, suggesting that more synapses are present in these layers in the C1QA KO mouse (Chu et al., 2010). In the somatosensory (SS) cortex, synapse elimination induced by whisker lesioning at P10 requires CX3CR1 but not CR3 (Gunner et al., 2019). At later time points, developmental spine pruning in the SS cortex occurs over 1–3 months (Bian et al., 2015), and is decreased in the C3 KO and increased by the deletion of the complement inhibitor SRPX2 (Cong et al., 2020). In addition, VGlut2 but not VGlut1 synapses are decreased in L4 of the SS cortex of the SRPX2 KO mouse (Cong et al., 2020), suggesting that the complement system specifically regulates thalamocortical synapses in the cortex. Taken together, these studies suggest that complement and microglia contribute to synapse elimination in the sensory cortices in the juvenile/adolescent mouse, at age 1 month or later.
2.5 |. Other brain regions
In addition to the LGN, hippocampus, and sensory cortices, microglia and/or complement involvement in synapse elimination has also been reported in other brain regions. In the nucleus accumbens of the adolescent rat, at P20–P54, dopamine D1 receptor (D1r) synapses are associated with C3 and microglial processes, and blocking C3-CR3 interaction with an inhibitory peptide increased D1r synapses in male but not female rats (Kopec et al., 2018). Another study also showed that conditional astrocytic deletion of interleukin-33 (IL33), a cytokine which has been implicated in tissue homeostasis in the periphery, also reduces microglial engulfment of synapses and increases synapse density in both the thalamus and spinal cord at P12–P30 (Vainchtein et al., 2018). These observations were phenocopied in the conditional microglial deletion of the IL33 receptor IL1RL1 (Vainchtein et al., 2018). These studies suggest that complement- and microglia-mediated synapse elimination may take place in nearly all brain regions.
3 |. ROLE OF COMPLEMENT - AND MICROGLIA-MEDIATED SYNAPSE ELIMINATION IN NEURODEVELOPMENTAL DISEASES
3.1 |. Schizophrenia
The strongest evidence for complement involvement in human neurological function comes from studies of schizophrenia. A large GWAS study of schizophrenia, involving tens of thousands of schizophrenic patients, has previously shown that the MHC locus on chromosome 6 harbors the strongest genetic risk for schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). In 2016, a landmark study deconstructed the risk on chromosome 6, and showed that the risk is associated with the C4 gene within the MHC locus, which has an unusually complex structure (Sekar et al., 2016). The human C4 gene exists as two paralogous isotypes, C4A and C4B, each of which can be present in multiple copies at the C4 locus. C4A and C4B are 99% identical, but differ at a critical domain which determines their substrate specificity, with C4A binding more efficiently to proteins, and C4B binding more efficiently to carbohydrates (Law & Dodds, 1997). C4A alleles are also expressed at higher levels than C4B alleles. The study found that schizophrenia risk increases with increasing C4A allele copy number (Sekar et al., 2016), and indicating that C4 confers the greatest schizophrenia risk of any known gene. The same study also examined the C4 KO mouse, and found that LGN synapse elimination was impaired in the mice. Remarkably, excessive synapse elimination has been proposed as the cause of schizophrenia as early as 1982 (Feinberg, 1982), and loss of gray matter (Cannon et al., 2002, 2015) and dendritic spines (Garey et al., 1998; Glantz & Lewis, 2000; Sweet et al., 2009) are some of the most consistent anatomical findings in schizophrenia patients. Schizophrenia symptoms also tend to emerge in adolescence or early adulthood, a time frame when synapse elimination is occurring in the neocortex. Other complement-related genes implicated in schizophrenia include CSMD1/CSMD2 (Håvik et al., 2011; Liu et al., 2017), which are complement inhibitors that are highly expressed in the brain (Escudero-Esparza et al., 2013; Kraus et al., 2006). More modern research techniques have also been utilized to probe the synapse elimination-schizophrenia hypothesis. Microglia derived from schizophrenic patients using iPSC-like techniques show increased neuronal complement deposition and synapse phagocytosis (Sellgren et al., 2019), and radioligand-based PET imaging for TSPO, a protein elevated in activated microglia and astrocytes, show increased levels of TSPO in patients at high risk for schizophrenia (Bloomfield et al., 2016). Therefore, taken together, current human studies strongly suggest that synapse elimination is a major underlying biological mechanism for schizophrenia pathogenesis.
To probe the complement-related mechanisms underlying schizophrenia pathogenesis, several mouse models have been created. Overexpression of C4 in the mouse prefrontal cortex using in utero electroporation caused decreased spine densities, increased microglia synapse engulfment, and impaired social behavior (Comer et al., 2020). Another elegant study utilized transgenic expression of the human C4A or C4B gene in the C4 knockout mouse, thereby replacing the murine C4 gene with the human version (Yilmaz et al., 2021). The study found that the human C4A gene can substitute for the murine gene in rescuing synapse elimination deficits in the LGN, but not the human C4B gene, suggesting that C4A may be more efficient at supporting synapse elimination due to its binding bias for a protein target at the synapse. Overexpression of human C4A by increasing transgenic gene dosage reduced synapse density at the prefrontal cortex at P60, increased synaptic uptake by microglia, and produced several behavioral deficits including reduced sociability, increased anxiety, and spatial working memory defects (Yilmaz et al., 2021). These studies suggest that C4 overexpression in mice produces behavioral deficits resembling negative symptoms in schizophrenia. Prefrontal cortex miswiring was also found in a two-hit mouse model which combines genetic deletion of Disrupted-In-Schizophrenia 1 (DISC1) with the environmental intervention of maternal immune activation (MIA). In addition to disorganized oscillatory entrainment of prefrontal neurons, the two-hit model caused decreased synapse density and increased microglial synapse engulfment, as well as impaired working memory (Chini et al., 2020). These deficits were reversed by administration of minocycline, an antibiotic which may act through regulating microglia (Chini et al., 2020). This study suggests that environmental factors can contribute to the pathogenesis of schizophrenia, and that targeting microglia is a possible therapeutic strategy.
3.2 |. Autism spectrum disorders
Autism spectrum disorders (ASDs) are a class of neurodevelopmental diseases characterized by a triad of symptoms: impairment in social interactions, language and communication deficits, and repetitive and restrictive behavior. While large GWAS studies have not yielded any genes obviously associated with complement or microglia, several small candidate gene and familial gene linkage studies have provided suggestive evidence of complement involvement. The C4B null allele, in particular, has been associated with both ASD (Mostafa & Shehab, 2010; Odell et al., 2005; Warren et al., 1994, 1995) and autoimmune diseases (Fielder et al., 1983; Mostafa & Shehab, 2010). In addition, a recent study has also found decreases in C1q, C4 and C3 mRNA levels in the frontal cortex of ASD patients (Fagan et al., 2017). Anatomical studies have found increased microglia activation (Hammond et al., 2018) in ASD patients. Increased dendritic spine density has also been seen in the cortex of ASD patients (Hutsler & Zhang, 2010), as well as in fragile X patients who show a syndromic form of autism (Irwin et al., 2001). Furthermore, cortical neurons cultured from iPSCs derived from ASD patients bearing mutations in the SHANK2 susceptibility gene show increased synapse densities and hyperconnectivity (Zaslavsky et al., 2019). However, functional brain imaging studies in humans have generally found underconnectivity between brain regions (Geschwind & Levitt, 2007; Just et al., 2004). This has led to suggestion that underconnectivity between brain regions and overconnectivity in local brain circuits can simultaneously cause impairment in some behavioral domains and enhancement in other domains (Geschwind & Levitt, 2007), accounting for the heterogeneity in ASD patients.
ASD is both heterogeneous and highly heritable, and GWAS studies suggest that a diverse set of genes affecting many different biological processes are involved in ASD pathogenesis (De Rubeis et al., 2014; Grove et al., 2019). In mouse models, both underpruning and overpruning of synapses have resulted in various behavioral abnormalities that resemble ASD symptoms (Table 2). Mouse models that show impaired social behavior and/or repetitive behavior in the context of reduced complement activity or microglial activation include the CX3CR1 fractalkine receptor KO mouse (Zhan et al., 2014), the TREM2 phagocytosis receptor KO mouse (Filipello et al., 2018), the autophagy-related ATG7 microglial conditional KO mouse (Kim et al., 2017), the HOXB8 microglial conditional KO mouse (Chen et al., 2010; Nagarajan et al., 2018), and the C3 prefrontal cortex knockdown mouse (Fagan et al., 2017). On the other hand, several mouse models showing increased complement activation in the brain also show social defects. Specifically, both C4 overexpression mouse models show social interaction deficits (Comer et al., 2020; Yilmaz et al., 2021), and deletion of the neuronal complement inhibitor SRPX2 results in normal sociability but impaired social memory (Soteros et al., 2018). Thus, it is possible that a complex set of brain circuits is required for social behavior, and that these circuits are sensitive to both overpruning and underpruning of synapses.
TABLE 2.
Behavioral readouts in complement/microglia mouse models
| Author | Genetic | Gene function | Behavior |
|---|---|---|---|
| Fagan et al. (2017) | C3 knockdown | Complement component | Social interaction deficit, repetitive behavior |
| Perez-Alcazar et al. (2014) | C3 knockout | Complement component | Enhanced place and reversal learning |
| Stephan et al. (2013) | C1QA knockout | Complement component | Enhanced spatial memory in aged mouse |
| Shi et al. (2015) | C3 knockout | Complement component | Enhanced memory, less anxiety |
| Chini et al. (2020) | PolyIC/DISC1 knockout | Two hit SZ model | Working memory deficit |
| Yilmaz et al. (2021) | Human C4 overexpression | Complement component | Social interaction deficit, increased anxiety, spatial memory deficit |
| Chu et al. (2010) | C1QA knockout | Complement component | Spontaneous epileptiform activity |
| Comer et al. (2020) | C4 IUE overexpression | Complement component | Social interaction deficit |
| Steen et al. (2013) | CSMD1 | Complement inhibitor | Increased anxiety |
| Distler et al. (2012) | CSMD1 | Complement inhibitor | Normal PPI, social interaction |
| Zhu et al. (2020) | SUSD4 | Complement inhibitor | Increased anxiety, impaired motor function |
| Kim et al. (2017) | ATG7 | Autophagy | Social interaction deficit |
| Filipello et al. (2018) | TREM2 | Phagocytosis receptor | Social interaction deficit, repetitive behavior |
3.3 |. Epilepsy
Recent research suggests that some forms of epilepsy may be caused by circuitry miswiring during brain development. Epilepsy is broadly divided into generalized and focal epilepsies. While the genetic basis of generalized epilepsy is well established and is commonly due to mutations in ion channels (Oyrer et al., 2018), focal epilepsy has long been considered an acquired condition, such as occurring as a result of miswiring after traumatic brain injury. Only recently has there been an appreciation that developmental processes and genes can also contribute to focal epilepsies (Thomas & Berkovic, 2014). This concept is exemplified by protocadherin 19 (PCDH19) epilepsy, caused by mutations in the X-linked PCDH19 gene. Unusually, the disease manifests in heterozygous female carriers, but not homozygous females or hemizygous males (Dibbens et al., 2008). A defect in synapse formation and brain wiring as a result of mismatched synaptic cadherins has been proposed as a mechanism for PCDH19 epilepsy (Hoshina et al., 2021). Evidence for complement involvement in human epilepsy derives largely from studies examining brain samples resected from patients with refractory epilepsy (RE) which are resistant to drug treatment, which has found elevated levels of C1q and C3 protein levels in RE samples when compared with samples resected from patients with non-epileptic lesions (Aronica et al., 2007; Wyatt et al., 2017). Genetic support for complement involvement also comes from studies of the human SRPX2 gene. SRPX2 is a neuronal complement inhibitor (Cong et al., 2020), and several SRPX2 mutations have been linked to rolandic epilepsy and language impairments (Chen et al., 2017; Roll et al., 2006; Schirwani et al., 2018). SRPX2 expression is also regulated by the language-associated transcription factor FoxP2 (Roll et al., 2010; Sia et al., 2013). Mutations in another potential complement inhibitor, CSMD3, has also been reported to be associated with epilepsy (Shimizu et al., 2003). These studies suggest that complement-mediated synapse elimination is a plausible mechanisms for some forms of epilepsy, although more research is required to determine whether complement activation is a causal factor for epilepsy, or a consequence of seizures.
4 |. CONCLUSION: FUTURE WORK
Despite tremendous progress in uncovering the role of the innate immune system in sculpting developing brain circuits, many important mechanistic details remain obscure. Below, we list several major questions in the field that remain unanswered.
How are synapses selected for complement-mediated and microglia-mediated elimination? Synapse elimination is believed to be due to competition between neighboring axons for postsynaptic partners based on differences in activity levels (Huberman et al., 2008; Sanes & Lichtman, 1999). While microglia have been shown to selectively phagocytose less active synapses (Schafer et al., 2012), it is unclear how they recognize and phagocytose less active synapses while sparing more active synapses. One possible mechanism is that more active synapses may selectively express or retain a complement- or microglial inhibitory protein, thereby protecting active synapses from elimination. Candidates for such a protein include the microglia inhibitory protein CD47, which has been shown to be colocalized with more active inputs in the LGN (Lehrman et al., 2018), and the neuronal complement inhibitor SRPX2 (Cong et al., 2020). Another possibility is a punishment-based mechanism whereby less active synapses are selected for elimination. One such mechanism may involve long term depression (LTD), a physiological process that serves to selectively weaken specific synapses. LTD has long been known to cause morphological changes such as spine shrinkage and loss (Bastrikova et al., 2008; Nägerl et al., 2004), and recently, some forms of LTD has been shown to involve the activation of caspase-3 (Li et al., 2010), the central regulator of apoptosis. Caspase activation in apoptotic cells is known to cleave flippases and lead to the externalization of phosphatidylserine (PS) in the cell membrane (Nagata et al., 2016). In the periphery, this acts as a signal for phagocytosis of apoptotic cells by phagocytes. A recent proteomic study has shown that C1q-tagged synaptosomes contain cleaved caspase-3, and is also enriched in annexin V, which binds PS (Györffy et al., 2018). Furthermore, microglia contain several receptors for PS, including TREM2 (Shirotani et al., 2019; Wang et al., 2015) and GPR56 (Li et al., 2020), and PS may be required for microglial synaptic pruning (Li et al., 2020; Scott-Hewitt et al., 2020). While this is a plausible mechanism, more research is required to understand whether protection-based and/or punishment-based mechanisms operate to achieve activity-regulated synapse elimination by microglia.
What cell types are involved in coordinating complement-mediated and microglia-mediated synapse elimination? The neuronal synapse exists in close proximity to surrounding astrocytic processes, forming the tripartite synapse. The astrocyte is therefore uniquely positioned to sense the activity state of both presynaptic and postsynaptic boutons. Astrocytes have long been known to be able to sense various signals from neurons, including neurotransmitters and signaling molecules (Fields & Stevens-Graham, 2002). Astrocytes can also secrete factors to regulate neuronal complement expression, such as TGF-β (Bialas & Stevens, 2013), as well as secrete factors to regulate microglial phagocytosis of synapses, such as IL-33 (Vainchtein et al., 2018). Astrocytes also express receptors for C1q (Iram et al., 2016), and can directly phagocytose synapses through the MEGF10/MERTK pathway (Chung et al., 2013). Finally, neurons can communicate directly with microglia, such as by the release of extracellular ATP (Badimon et al., 2020). Despite the recent progress, it is likely that we have only begun to scratch the surface in elucidating the full set of communications between all the different cell types that are necessary for the proper orchestration of synapse elimination.
Which synapses, circuits, and behaviors are affected by complement-mediated and microglia-mediated synapse elimination? It is clear that not all forms of synapse elimination in the brain are dependent on complement and microglia. ODP in the V1 cortex, for example, does not require either C1q (Welsh et al., 2020) or CX3CR1 (Schecter et al., 2017), while whisker-lesioning induced synapse elimination in the SS cortex is dependent on CX3CR1 but not CR3 (Gunner et al., 2019). In addition, C3-dependent developmental synapse elimination in the SS cortex in juvenile mice appears to target only thalamocortical synapses, while sparing cortico-cortical synapses (Cong et al., 2020; Soteros et al., 2018). The specific set of synapses targeted by complement/microglia-mediated synapse elimination may also differ depending on sex. It has long been known that blood complement levels are lower in females (Gaya da Costa et al., 2018; Lachmann, 2010), and there is evidence that complement genes contribute to sex-biased vulnerability to autoimmune diseases (Kamitaki et al., 2020). A study in rats also showed that blocking C3–CR3 interaction abolishes male/female differences in social play and microglial engulfment of D1R (Kopec et al., 2018). There are also differences in the sensitivity of various behaviors to complement-mediated and microglia-mediated synapse elimination. For example, memory-associated behaviors appear to be asymmetrically sensitive to complement deletion, being relatively intact in complement deficient mouse models and impaired in complement over-activation mouse models, whereas social behavior appear to be impaired in both complement under-activation and over-activation models (Table 2). Understanding which synapses are selectively vulnerable to complement/microglia elimination, and how this vulnerability translates to impaired behavior, will lead to a deeper understanding of how activity-dependent synapse elimination shapes behavior in health and disease.
Recent research has led to major conceptual advances in the role of neuroimmune processes in the healthy brain. Complement-mediated and microglia-mediated synapse elimination in particular has been shown to play a critical role in normal brain development as well as in the pathogenesis of neurodevelopmental diseases. Furthermore, aberrant activation of complement in the adult and aged brain has also been linked to a number of neurodegenerative diseases (Gomez-Arboledas et al., 2021; Presumey et al., 2017) Solving these major unanswered questions will deepen our understanding of this important process, and may lead to new treatment strategies for intractable neurodevelopmental and neurodegenerative diseases (Boxes 1 and 2).
BOX 1. The complement system in tissue homeostasis.
During normal development, the complement system helps maintain tissue homeostasis by tagging dead cells and other unwanted cellular material for phagocytic clearance. Proper maintenance of tissue structure is essential to prevent adverse consequences, such as disruption of tissue function or the emergence of auto-immunity. For example, mice lacking C1q, the initiating component of the classical complement cascade, are only mildly immunocompromised, but die of glomerulonephritis with large numbers of apoptotic bodies in their kidneys and elevated levels of auto-antibodies in their blood (Botto et al., 1998). In humans, loss-of-function mutations in the C1q, C2, and C4 lead to a paradoxical increase in auto-antibodies and inflammation, and are major genetic risk factors for the autoimmune disease systemic lupus erythematosus (SLE; Lewis & Botto, 2006; Macedo & Isaac, 2016; Moser et al., 2009). Therefore, the complement system plays a critical role in tissue homeostasis, and a certain amount of complement activation is normal and necessary for the proper development of many organs.
BOX 2. Complement deficiencies in humans.
Inherited complement deficiencies in humans are rare, although the large number of complement genes may contribute to under-diagnosis (Grumach & Kirschfink, 2014). Homozygous deficiencies in complement components with complex gene structures, such as the C4 gene, have been detected in as much as 1–10% of the human population (Liesmaa et al., 2018). Furthermore, complement deficiencies can also be acquired, typically through liver disease. While rare, complete complement deficiencies in all complement pathways exist in humans and are not fatal. Genetic deficiency of any early component of the classical pathway (C1q, C1r/s, C2, and C4) leads to autoimmune diseases such as SLE (Lewis & Botto, 2006), whereas patients deficient for components in the lectin and alternative pathways (MBL, MASPs, and C3) and components in the terminal pathway (C5 to C9) are susceptible to recurrent bacterial infections (Mayilyan, 2012). Deficiencies in peripheral complement regulatory proteins leads to over-activation of complement and tissue damage. For example, mutations in Factor H are linked to atypical hemolytic uremic syndrome (aHUS) and age-related macular degeneration (AMD; Mayilyan, 2012). Interestingly, complete complement deficiencies appear neurologically innocuous in humans. This is in agreement with data from complement KO mice, which despite their described anatomical brain abnormalities show mild behavioral phenotypes. Indeed, unusually for knockout mice, the C3 KO mice appears to perform better than wildtype mice in several memory tasks (Perez-Alcazar et al., 2014; Shi et al., 2015), especially when comparing aged animals. Current evidence therefore suggests that complement over-activation is more likely to lead to neurological deficits in humans.
ACKNOWLEDGMENTS
All figures created with BioRender.com.
Funding information
National Institute of Neurological Disorders and Stroke, Grant/Award Number: R01NS112389; William and Ella Owens Medical Research Foundation
Footnotes
CONFLICT OF INTEREST
The authors have declared no conflicting interests.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Aronica E, Boer K, van Vliet EA, Redeker S, Baayen JC, Spliet WGM, van Rijen PC, Troost D, Lopes da Silva FH, Wadman WJ, & Gorter JA (2007). Complement activation in experimental and human temporal lobe epilepsy. Neurobiology of Disease, 26, 497–511. [DOI] [PubMed] [Google Scholar]
- Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, Hwang P, Chan AT, Graves SM, Uweru JO, Ledderose C, Kutlu MG, Wheeler MA, Kahan A, Ishikawa M, Wang YC, Loh YHE, Jiang JX, Surmeier DJ, … Schaefer A (2020). Negative feedback control of neuronal activity by microglia. Nature, 586, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic G, Degn SE, Thiel S, & Andersen GR (2015). Complement activation, regulation, and molecular basis for complement-related diseases. The EMBO Journal, 34, 2735–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastrikova N, Gardner GA, Reece JM, Jeromin A, & Dudek SM (2008). Synapse elimination accompanies functional plasticity in hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America, 105, 3123–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas AR, & Stevens B (2013). TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nature Neuroscience, 16, 1773–1782. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bian W-J, Miao W-Y, He S-J, Qiu Z, & Yu X (2015). Coordinated spine pruning and maturation mediated by inter-spine competition for cadherin/catenin complexes. Cell, 162, 808–822. [DOI] [PubMed] [Google Scholar]
- Bjartmar L, Huberman AD, Ullian EM, Rentería RC, Liu X, Xu W, Prezioso J, Susman MW, Stellwagen D, Stokes CC, Cho R, Worley P, Malenka RC, Ball S, Peachey NS, Copenhagen D, Chapman B, Nakamoto M, Barres BA, & Perin MS (2006). Neuronal pentraxins mediate synaptic refinement in the developing visual system. The Journal of Neuroscience, 26, 6269–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, Bloomfield MA, Bonoldi I, Kalk N, Turkheimer F, McGuire P, de Paola V, & Howes OD (2016). Microglial activity in people at ultra high risk of psychosis and in schizophrenia: An [(11)C]PBR28 PET brain imaging study. The American Journal of Psychiatry, 173, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto M, Dell'Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, & Walport MJ (1998). Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nature Genetics, 19, 56–59. [DOI] [PubMed] [Google Scholar]
- Bourgeois JP, & Rakic P (1993). Changes of synaptic density in the primary visual cortex of the macaque monkey from fetal to adult stage. The Journal of Neuroscience, 13, 2801–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, & Barres BA (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. The Journal of Neuroscience, 28, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon TD, Thompson PM, van Erp TGM, Toga AW, Poutanen V-P, Huttunen M, Lonnqvist J, Standerskjold-Nordenstam C-G, Narr KL, Khaledy M, Zoumalan CI, Dail R, & Kaprio J (2002). Cortex mapping reveals regionally specific patterns of genetic and disease-specific gray-matter deficits in twins discordant for schizophrenia. Proceedings of the National Academy of Sciences of the United States of America, 99, 3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon TD, Chung Y, He G, Sun D, Jacobson A, van Erp TGM, McEwen S, Addington J, Bearden CE, Cadenhead K, Cornblatt B, Mathalon DH, McGlashan T, Perkins D, Jeffries C, Seidman LJ, Tsuang M, Walker E, Woods SW, … North American Prodrome Longitudinal Study Consortium. (2015). Progressive reduction in cortical thickness as psychosis develops: A multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biological Psychiatry, 77, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux J-P, & Danchin A (1976). Selective stabilisation of developing synapses as a mechanism for the specification of neuronal networks. Nature, 264, 705–712. [DOI] [PubMed] [Google Scholar]
- Chen S-K, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, & Capecchi MR (2010). Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell, 141, 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XS, Reader RH, Hoischen A, Veltman JA, Simpson NH, Francks C, Newbury DF, & Fisher SE (2017). Next-generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Scientific Reports, 7, 46105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini M, Pöpplau JA, Lindemann C, Carol-Perdiguer L, Hnida M, Oberländer V, Xu X, Ahlbeck J, Bitzenhofer SH, Mulert C, & Hanganu-Opatz IL (2020). Resolving and rescuing developmental miswiring in a mouse model of cognitive impairment. Neuron, 105, 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, & Prince DA (2010). Enhanced synaptic connectivity and epilepsy in C1q knockout mice. PNAS, 107, 7975–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, & Barres BA (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature, 504, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, & Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proceedings of the National Academy of Sciences of the United States of America, 115, E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer AL, Jinadasa T, Sriram B, Phadke RA, Kretsge LN, Nguyen TPH, Antognetti G, Gilbert JP, Lee J, Newmark ER, Hausmann FS, Rosenthal SA, Liu Kot K, Liu Y, Yen WW, Dejanovic B, & Cruz-Martín A (2020). Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PLoS Biology, 18, e3000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Q, Soteros BM, Wollet M, Kim JH, & Sia G-M (2020). The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nature Neuroscience, 23, 1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, & Shatz CJ (1998). Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron, 21, 505–520. [DOI] [PubMed] [Google Scholar]
- de Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, … Buxbaum JD (2014). Synaptic, transcriptional, and chromatin genes disrupted in autism. Nature, 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibbens LM, Tarpey PS, Hynes K, Bayly MA, Scheffer IE, Smith R, Bomar J, Sutton E, Vandeleur L, Shoubridge C, Edkins S, Turner SJ, Stevens C, O'Meara S, Tofts C, Barthorpe S, Buck G, Cole J, Halliday K, … Gécz J (2008). X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nature Genetics, 40, 776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Wang J, Huang M, Chen Z, Liu J, Zhang Q, Zhang C, Xiang Y, Zen K, & Li L (2021). Loss of microglial SIRPα promotes synaptic pruning in preclinical models of neurodegeneration. Nature Communications, 12, 2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler MG, Opal MD, Dulawa SC, & Palmer AA (2012). Assessment of Behaviors Modeling Aspects of Schizophrenia in Csmd1 Mutant Mice. PLOS ONE, 7, e51235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero-Esparza A, Kalchishkova N, Kurbasic E, Jiang WG, & Blom AM (2013). The novel complement inhibitor human CUB and sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. The FASEB Journal, 27, 5083–5093. [DOI] [PubMed] [Google Scholar]
- Fagan K, Crider A, Ahmed AO, & Pillai A (2017). Complement C3 expression is decreased in autism spectrum disorder subjects and contributes to behavioral deficits in rodents. Molecular Neuropsychiatry, 3, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg I (1982). Schizophrenia: Caused by a fault in programmed synaptic elimination during adolescence? Journal of Psychiatric Research, 17, 319–334. [DOI] [PubMed] [Google Scholar]
- Fielder AH, Walport MJ, Batchelor JR, Rynes RI, Black CM, Dodi IA, & Hughes GR (1983). Family study of the major histocompatibility complex in patients with systemic lupus erythematosus: Importance of null alleles of C4A and C4B in determining disease susceptibility. British Medical Journal (Clinical Research Ed.), 286, 425–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, & Stevens-Graham B (2002). New insights into neuron-glia communication. Science, 298, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, Piccio L, Cignarella F, Perrucci F, Tamborini M, Genua M, Rajendran L, Menna E, Vetrano S, Fahnestock M, … Matteoli M (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity, 48, 979–991. [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Chu S-H, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, & Tenner AJ (2017). Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. Journal of Neuroinflammation, 14, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TR, & Hirsch SR (1998). Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. Journal of Neurology, Neurosurgery, and Psychiatry, 65, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaya da Costa M, Poppelaars F, van Kooten C, Mollnes TE, Tedesco F, Würzner R, Trouw LA, Truedsson L, Daha MR, Roos A, & Seelen MA (2018). Age and sex-associated changes of complement activity and complement levels in a healthy Caucasian population. Frontiers in Immunology, 9, 2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, & Levitt P (2007). Autism spectrum disorders: Developmental disconnection syndromes. Current Opinion in Neurobiology, 17, 103–111. [DOI] [PubMed] [Google Scholar]
- Glantz LA, & Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Archives of General Psychiatry, 57, 65–73. [DOI] [PubMed] [Google Scholar]
- Gomez-Arboledas A, Acharya MM, & Tenner AJ (2021). The role of complement in synaptic pruning and neurodegeneration. ImmunoTargets and Therapy, 10, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, Awashti S, Belliveau R, Bettella F, Buxbaum JD, Bybjerg-Grauholm J, Bækvad-Hansen M, Cerrato F, Chambert K, Christensen JH, … Børglum AD (2019). Identification of common genetic risk variants for autism spectrum disorder. Nature Genetics, 51, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumach AS, & Kirschfink M (2014). Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Molecular Immunology, 61, 110–117. [DOI] [PubMed] [Google Scholar]
- Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, Nagy MA, Liu L, Bemiller SM, Kim K-W, Lira SA, Lamb BT, Tapper AR, Ransohoff RM, Greenberg ME, Schaefer A, & Schafer DP (2019). Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nature Neuroscience, 22, 1075–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györffy BA, Kun J, Török G, Bulyaki É, Borhegyi Z, Gulyássy P, Kis V, Szocsics P, Micsonai A, Matkó J, Drahos L, Juhász G, Kékesi KA, & Kardos J (2018). Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proceedings of the National Academy of Sciences of the United States of America, 115, 6303–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Robinton D, & Stevens B (2018). Microglia and the brain: Complementary partners in development and disease. Annual Review of Cell and Developmental Biology, 34, 523–544. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, & Feng L (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proceedings. National Academy of Sciences. United States of America, 95, 10896–10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håvik B, Le Hellard S, Rietschel M, Lybæk H, Djurovic S, Mattheisen M, Mühleisen TW, Degenhardt F, Priebe L, Maier W, Breuer R, Schulze TG, Agartz I, Melle I, Hansen T, Bramham CR, Nöthen MM, Stevens B, Werge T, … Steen VM (2011). The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biological Psychiatry, 70, 35–42. [DOI] [PubMed] [Google Scholar]
- Hoshina N, Johnson-Venkatesh EM, Hoshina M, & Umemori H (2021). Female-specific synaptic dysfunction and cognitive impairment in a mouse model of PCDH19 disorder. Science, 372, eaaz3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman AD, Feller MB, & Chapman B (2008). Mechanisms underlying development of visual maps and receptive fields. Annual Review of Neuroscience, 31, 479–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, & Shatz CJ (2000). Functional requirement for class I MHC in CNS development and plasticity. Science, 290, 2155–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, & Zhang H (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Research, 1309, 83–94. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR (1979). Synaptic density in human frontal cortex - developmental changes and effects of aging. Brain Research, 163, 195–205. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR, & Dabholkar AS (1997). Regional differences in synaptogenesis in human cerebral cortex. The Journal of Comparative Neurology, 387, 167–178. [DOI] [PubMed] [Google Scholar]
- Iram T, Ramirez-Ortiz Z, Byrne MH, Coleman UA, Kingery ND, Means TK, Frenkel D, & El Khoury J (2016). Megf10 is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. The Journal of Neuroscience, 36, 5185–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, & Greenough WT (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: A quantitative examination. American Journal of Medical Genetics, 98, 161–167. [DOI] [PubMed] [Google Scholar]
- Just MA, Cherkassky VL, Keller TA, & Minshew NJ (2004). Cortical activation and synchronization during sentence comprehension in high-functioning autism: Evidence of underconnectivity. Brain, 127, 1811–1821. [DOI] [PubMed] [Google Scholar]
- Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, Taylor KE, Whelan CW, Tombleson P, Loohuis LMO, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Boehnke M, Kimberly RP, Kaufman KM, Harley JB, Langefeld CD, Seidman CE, Pato MT, Pato CN, … SA MC (2020). Complement genes contribute sex-biased vulnerability in diverse disorders. Nature, 582, 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-J, Cho M-H, Shim WH, Kim JK, Jeon E-Y, Kim D-H, & Yoon S-Y (2017). Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Molecular Psychiatry, 22, 1576–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec AM, Smith CJ, Ayre NR, Sweat SC, & Bilbo SD (2018). Microglial dopamine receptor elimination defines sex-specific nucleus accumbens development and social behavior in adolescent rats. Nature Communications, 9, 3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus DM, Elliott GS, Chute H, Horan T, Pfenninger KH, Sanford SD, Foster S, Scully S, Welcher AA, & Holers VM (2006). CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. Journal of Immunology, 176, 4419–4430. [DOI] [PubMed] [Google Scholar]
- Lachmann PJ (2010). Preparing serum for functional complement assays. Journal of Immunological Methods, 352, 195–197. [DOI] [PubMed] [Google Scholar]
- Law SK, & Dodds AW (1997). The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Science, 6, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, Walker AJ, Heller MD, Umemori H, Chen C, & Stevens B (2018). CD47 protects synapses from excess microglia-mediated pruning during development. Neuron, 100, 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MJ, & Botto M (2006). Complement deficiencies in humans and animals: Links to autoimmunity. Autoimmunity, 39, 367–378. [DOI] [PubMed] [Google Scholar]
- Li T, Chiou B, Gilman CK, Luo R, Koshi T, Yu D, Oak HC, Giera S, Johnson-Venkatesh E, Muthukumar AK, Stevens B, Umemori H, & Piao X (2020). A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. The EMBO Journal, 39, e104136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia J-M, Lo S-C, Whitcomb DJ, Jiao S, Cho K, & Sheng M (2010). Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell, 141, 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, Litvinchuk A, Chiang AC-A, Aithmitti N, Jankowsky JL, & Zheng H (2016). Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer's disease. The Journal of Neuroscience, 36, 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesmaa I, Paakkanen R, Järvinen A, Valtonen V, & Lokki M-L (2018). Clinical features of patients with homozygous complement C4A or C4B deficiency. PLoS One, 13, e0199305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Liu F, Xu X, & Bai Y (2017). Replicated association between the European GWAS locus rs10503253 at CSMD1 and schizophrenia in Asian population. Neuroscience Letters, 647, 122–128. [DOI] [PubMed] [Google Scholar]
- Macedo ACL, & Isaac L (2016). Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Frontiers in Immunology, 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makou E, Herbert AP, & Barlow PN (2013). Functional anatomy of complement factor H. Biochemistry, 52, 3949–3962. [DOI] [PubMed] [Google Scholar]
- Mayilyan KR (2012). Complement genetics, deficiencies, and disease associations. Protein & Cell, 3, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser KL, Kelly JA, Lessard CJ, & Harley JB (2009). Recent insights into the genetic basis of systemic lupus erythematosus. Genes and Immunity, 10, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafa GA, & Shehab AA (2010). The link of C4B null allele to autism and to a family history of autoimmunity in Egyptian autistic children. Journal of Neuroimmunology, 223, 115–119. [DOI] [PubMed] [Google Scholar]
- Nagarajan N, Jones BW, West PJ, Marc RE, & Capecchi MR (2018). Corticostriatal circuit defects in Hoxb8 mutant mice. Molecular Psychiatry, 23, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S, Suzuki J, Segawa K, & Fujii T (2016). Exposure of phosphatidylserine on the cell surface. Cell Death & Differentiation, 23, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, & Bonhoeffer T (2004). Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron, 44, 759–767. [DOI] [PubMed] [Google Scholar]
- Odell D, Maciulis A, Cutler A, Warren L, McMahon WM, Coon H, Stubbs G, Henley K, & Torres A (2005). Confirmation of the association of the C4B null allelle in autism. Human Immunology, 66, 140–145. [DOI] [PubMed] [Google Scholar]
- Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, & Reid CA (2018). Ion channels in genetic epilepsy: From genes and mechanisms to disease-targeted therapies. Pharmacological Reviews, 70, 142–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, & Gross CT (2011). Synaptic pruning by microglia is necessary for normal brain development. Science, 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen J-E, & Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nature Neuroscience, 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Alcazar M, Daborg J, Stokowska A, Wasling P, Björefeldt A, Kalm M, Zetterberg H, Carlström KE, Blomgren K, Ekdahl CT, Hanse E, & Pekna M (2014). Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Experimental Neurology, 253, 154–164. [DOI] [PubMed] [Google Scholar]
- Petanjek Z, Judaš M, Šimic G, Rašin MR, Uylings HBM, Rakic P, & Kostović I (2011). Extraordinary neoteny of synaptic spines in the human prefrontal cortex. PNAS, 108, 13281–13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto JGA, Jones DG, & Murphy KM (2013). Comparing development of synaptic proteins in rat visual, somatosensory, and frontal cortex. Frontiers in Neural Circuits, 7, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presumey J, Bialas AR, & Carroll MC (2017). Complement system in neural synapse elimination in development and disease. Advances in Immunology, 135, 53–79. [DOI] [PubMed] [Google Scholar]
- Rakic P, Bourgeois JP, Eckenhoff MF, Zecevic N, & Goldman-Rakic PS (1986). Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science, 232, 232–235. [DOI] [PubMed] [Google Scholar]
- Riccomagno MM, & Kolodkin AL (2015). Sculpting neural circuits by axon and dendrite pruning. Annual Review of Cell and Developmental Biology, 31, 779–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll P, Rudolf G, Pereira S, Royer B, Scheffer IE, Massacrier A, Valenti MP, Roeckel-Trevisiol N, Jamali S, Beclin C, Seegmuller C, Metz-Lutz MN, Lemainque A, Delepine M, Caloustian C, Martin AS, Bruneau N, Depétris D, Mattéi MG, … Szepetowski P (2006). SRPX2 mutations in disorders of language cortex and cognition. Human Molecular Genetics, 15, 1195–1207. [DOI] [PubMed] [Google Scholar]
- Roll P, Vernes SC, Bruneau N, Cillario J, Ponsole-Lenfant M, Massacrier A, Rudolf G, Khalife M, Hirsch E, Fisher SE, & Szepetowski P (2010). Molecular networks implicated in speech-related disorders: FOXP2 regulates the SRPX2/uPAR complex. Human Molecular Genetics, 19, 4848–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager REH, Walker AK, Middleton F, Robinson K, Webster MJ, & Weickert CS (2021). Trajectory of change in brain complement factors from neonatal to young adult humans. Journal of Neurochemistry, 157, 479–493. [DOI] [PubMed] [Google Scholar]
- Sanes JR, & Lichtman JW (1999). Development of the vertebrate neuromuscular junction. Annual Review of Neuroscience, 22, 389–442. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, & Stevens B (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schecter RW, Maher EE, Welsh CA, Stevens B, Erisir A, & Bear MF (2017). Experience-dependent synaptic plasticity in V1 occurs without microglial CX3CR1. The Journal of Neuroscience, 37, 10541–10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirwani S, McConnell V, Willoughby J, Study DDD, & Balasubramanian M (2018). Exploring the association between SRPX2 variants and neurodevelopment: How causal is it? Gene, 685, 50–54. [DOI] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, Carey A, Faggiani E, Schuetz LT, Mason S, Tamborini M, Bizzotto M, Passoni L, Filipello F, Jahn R, Stevens B, & Matteoli M (2020). Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. The EMBO Journal, 39, e105380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, van Doren V, Genovese G, Rose SA, Handsaker RE, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Daly MJ, Carroll MC, Stevens B, & McCarroll SA (2016). Schizophrenia risk from complex variation of complement component 4. Nature, 530, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, Fu T, Worringer K, Brown HE, Wang J, Kaykas A, Karmacharya R, Goold CP, Sheridan SD, & Perlis RH (2019). Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nature Neuroscience, 22, 374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Li R, McGeer EG, & McGeer PL (1997). Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Research, 769, 391–395. [DOI] [PubMed] [Google Scholar]
- Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, Frost JL, Le KX, Li S, Dodart J-C, Caldarone BJ, Stevens B, & Lemere CA (2015). Complement C3-deficient mice fail to display age-related hippocampal decline. The Journal of Neuroscience, 35, 13029–13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu A, Asakawa S, Sasaki T, Yamazaki S, Yamagata H, Kudoh J, Minoshima S, Kondo I, & Shimizu N (2003). A novel giant gene CSMD3 encoding a protein with CUB and sushi multiple domains: A candidate gene for benign adult familial myoclonic epilepsy on human chromosome 8q23.3-q24.1. Biochemical and Biophysical Research Communications, 309, 143–154. [DOI] [PubMed] [Google Scholar]
- Shirotani K, Hori Y, Yoshizaki R, Higuchi E, Colonna M, Saito T, Hashimoto S, Saito T, Saido TC, & Iwata N (2019). Aminophospholipids are signal-transducing TREM2 ligands on apoptotic cells. Scientific Reports, 9, 7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia GM, Clem RL, & Huganir RL (2013). The human language-associated gene SRPX2 regulates synapse formation and vocalization in mice. Science, 342, 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soteros BM, Cong Q, Palmer CR, & Sia G-M (2018). Sociability and synapse subtype-specific defects in mice lacking SRPX2, a language-associated gene. PLoS One, 13, e0199399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai H-H, Huang EJ, Rowitch DH, Berns DS, Tenner AJ, Shamloo M, & Barres BA (2013). A dramatic increase of C1q protein in the CNS during normal aging. The Journal of Neuroscience, 33, 13460–13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM, & Barres BA (2007). The classical complement cascade mediates CNS synapse elimination. Cell, 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Steen VM, Nepal C, Ersland KM, Holdhus R, Nævdal M, Ratvik SM, Skrede S, & Håvik B (2013). Neuropsychological deficits in mice depleted of the schizophrenia susceptibility gene CSMD1. PLoS One, 8, e79501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet RA, Henteleff RA, Zhang W, Sampson AR, & Lewis DA (2009). Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology, 34, 374–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RH, & Berkovic SF (2014). The hidden genetics of epilepsy: A clinically important new paradigm. Nature Reviews. Neurology, 10, 283–292. [DOI] [PubMed] [Google Scholar]
- Thompson RA, & Nelson CA (2001). Developmental science and the media: Early brain development. American Psychologist, 56, 5–15. [DOI] [PubMed] [Google Scholar]
- Tremblay M-È, Lowery RL, & Majewska AK (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biology, 8, e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, Akil O, Joshita S, Barres BA, Paz JT, Molofsky AB, & Molofsky AV (2018). Astrocyte-derived Interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science, 359, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, & Colonna M (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell, 160, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RP, Burger RA, Odell D, Torres AR, & Warren WL (1994). Decreased plasma concentrations of the C4B complement protein in autism. Archives of Pediatrics & Adolescent Medicine, 148, 180–183. [DOI] [PubMed] [Google Scholar]
- Warren RP, Yonk J, Burger RW, Odell D, & Warren WL (1995). DR-positive T cells in autism: Association with decreased plasma levels of the complement C4B protein. Neuropsychobiology, 31, 53–57. [DOI] [PubMed] [Google Scholar]
- Weinhard L, Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, Exiga M, Vadisiute A, Raggioli A, Schertel A, Schwab Y, & Gross CT (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nature Communications, 9, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh CA, Stephany C-É, Sapp RW, & Stevens B (2020). Ocular dominance plasticity in binocular primary visual cortex does not require C1q. The Journal of Neuroscience, 40, 769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt SK, Witt T, Barbaro NM, Cohen-Gadol AA, & Brewster AL (2017). Enhanced classical complement pathway activation and altered phagocytosis signaling molecules in human epilepsy. Experimental Neurology, 295, 184–193. [DOI] [PubMed] [Google Scholar]
- Yilmaz M, Yalcin E, Presumey J, Aw E, Ma M, Whelan CW, Stevens B, McCarroll SA, & Carroll MC (2021). Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nature Neuroscience, 24, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslavsky K, Zhang W-B, McCready FP, Rodrigues DC, Deneault E, Loo C, Zhao M, Ross PJ, El Hajjar J, Romm A, Thompson T, Piekna A, Wei W, Wang Z, Khattak S, Mufteev M, Pasceri P, Scherer SW, Salter MW, & Ellis J (2019). SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nature Neuroscience, 22, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, & Gross CT (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nature Neuroscience, 17, 400–406. [DOI] [PubMed] [Google Scholar]
- Zhu H, Meissner LE, Byrnes C, Tuymetova G, Tifft C, & Proia LA (2020). The Complement Regulator Susd4 Influences Nervous-System Function and Neuronal Morphology in Mice. IScience, 23, 100957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.