

SUMMARY
Antibiotics are deployed against bacterial pathogens, but their targeting of conserved microbial processes means they also collaterally perturb the commensal microbiome. To understand acute and persistent effects of antibiotics on the gut microbiota of healthy adult volunteers, we quantify microbiome dynamics before, during, and 6 months after exposure to 4 commonly used antibiotic regimens. We observe an acute decrease in species richness and culturable bacteria after antibiotics, with most healthy adult microbiomes returning to pre-treatment species richness after 2 months, but with an altered taxonomy, resistome, and metabolic output, as well as an increased antibiotic resistance burden. Azithromycin delays the recovery of species richness, resulting in greater compositional distance. A subset of volunteers experience a persistent reduction in microbiome diversity after antibiotics and share compositional similarities with patients hospitalized in intensive care units. These results improve our quantitative understanding of the impact of antibiotics on commensal microbiome dynamics, resilience, and recovery.
In brief
How are robust microbiomes affected by antibiotics? Anthony et al. characterize “antibiotic scarring” in healthy volunteers, identifying universal compositional changes and treatment-specific effects on resistance burden and time to recovery of diversity. Three low-sdiversity microbiomes end compositionally similar to ICU patient microbiomes, highlighting the need for antibiotic stewardship in medicine.
Graphical Abstract
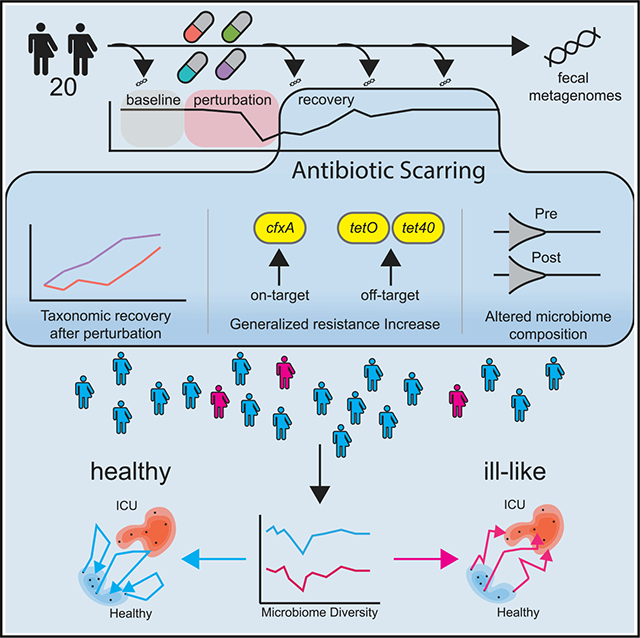
INTRODUCTION
Antibiotics are critical, lifesaving medications that have served as the primary treatment for bacterial infections since their clinical introduction in the 1940s (Nicolaou and Rigol, 2018; Ribeiro Da Cunha et al., 2019). However, antibiotic exposure can also result in significant unintended consequences, including acute and persistent changes in the commensal host microbiome (Wipperman et al., 2017; Dethlefsen et al., 2008; Ubeda et al., 2010; Palleja et al., 2018; De Gunzburg et al., 2018), as well as selection for antibiotic resistance (AR) genes in commensals and pathogens (Gasparrini et al., 2016, 2019). Immediately after antibiotic usage, changes in the microbiome can leave patients more susceptible to re-infection or infection from opportunistic pathogens, such as Clostridioides difficile (Stevens et al., 2011; Brown et al., 2013). Furthermore, taxonomic alterations to the microbiome due to antibiotics can lead to long-term consequences such as slower development of a diverse microbiome in preterm infants (Gasparrini et al., 2016, 2019; D’souza et al., 2019). Such gut microbiome dysbioses, commonly observed with broad-spectrum antibiotic treatment in patients hospitalized in intensive care units (ICUs) (Mcdonald et al., 2016; Ojima et al., 2016), have been linked to hospital-acquired infections (Gershuni and Friedman, 2019) and multiple organ dysfunction syndrome (Meng et al., 2017). Resistome selection and enrichment can also persist over long periods, as shown in studies in which preterm infants (Gasparrini et al., 2016, 2019; D’souza et al., 2019), nursing facility residents (Araos et al., 2019), and the elderly (Rampelli et al., 2020; Li et al., 2021) have increased AR organism and AR gene carriage, which has been theorized to be caused in part by repeated exposure to antibiotic treatment.
The bulk of this research on the impact of antibiotics on the human microbiome and resistome has been understandably performed with retrospective cohorts of severely ill and hospitalized individuals, who are at high risk for infections and who accordingly receive many courses of prophylactic and empiric antibiotic therapy (Sievert et al., 2013; Mcdonald et al., 2016; Ojima et al., 2016). Antibiotic exposure in these populations occurs in the context of diverse confounding factors, such as infection (Mcdonald et al., 2016; Ojima et al., 2016), drug exposure (D’souza et al., 2019), hospital environment (Ojima et al., 2016; Gasparrini et al., 2019), and potential immunocompromise (D’souza et al., 2019). Statistical modeling, which allows for some control of these confounders, demonstrates that the state of the microbiome pre-antibiotic exposure has some predictive power regarding the impact and severity of antibiotic exposure perturbation on the microbiome, but these models are still limited by the immense variation observed between individuals studied retrospectively (Gasparrini et al., 2019; Shetty et al., 2017). To separate the acute and persistent effects on microbiome composition and function from antibiotic exposure versus from the effects of illness and hospitalization, we must look to prospective studies that characterize the impact of antibiotics on healthy, un-hospitalized adults (Dethlefsen et al., 2008; Dethlefsen and Relman, 2011; Palleja et al., 2018; De Gunzburg et al., 2018). Furthermore, the healthy, adult microbiome is relatively stable in an individual and has been theorized to be robust and resilient to perturbation (Faith et al., 2013). However, adult microbiomes can vary considerably across individuals (Huse et al., 2012; Manor et al., 2020), and this variability may result in different levels of vulnerability to and severity of AR perturbation. Understanding these differences and defining the effects of specific antibiotics on healthy human microbiomes can help identify predictive factors that distinguish healthy and dysbiotic states, informing the development of future anti-infective treatments that minimize collateral microbiome damage (Shetty et al., 2017).
The existing literature explaining the effects of clinically relevant antibiotic exposure on the taxonomic and functional architecture of the healthy human microbiome is limited, as healthy individuals do not routinely need antibiotics. Two foundational studies assessed the effect of a short course of ciprofloxacin on the taxonomic structure of the microbiomes of 3 healthy individuals, using pyrosequencing of the 16S rRNA gene hypervariable region (Dethlefsen et al., 2008; Dethlefsen and Relman, 2011). The authors observed an almost universal acute decrease in taxonomic diversity after treatment and an incomplete recovery in some cases, but they were unable to assess changes in AR genes or other functional components of the microbiome because of the methodologies used. Similar findings have been reported for amoxicillin (De La Cochetière et al., 2005) and the amoxicillin-clavulanic acid combination (Mangin et al., 2012), which is indicative of an interplay between more universal signatures of bacterial selection and person-to-person variability of outcomes after antibiotic treatment. A few recent studies have applied shotgun metagenomics to this question, which can assess not only variation in taxonomic diversity but also resistome abundance and composition. These studies demonstrated an effect of moxifloxacin on bacterial gene richness within the first 5 weeks (De Gunzburg et al., 2018), a significant increase in tet resistance genes after 1 week in healthy volunteers given amoxicillin (Zaura et al., 2015), and a partial recovery of species richness after a 4-day course of a 3-drug cocktail in a cohort of 18 healthy young men (Palleja et al., 2018). While these studies have increased our understanding of taxonomic and resistance composition changes beyond what 16S rRNA sequencing can describe, they are still limited by the number of treatments and timepoints studied, and thus a cohesive understanding of both the acute and chronic effects of commonly prescribed antibiotics is still missing. The concept of ecological opportunity is used to describe environments with niche availability and discordance that drive persistence and adaption in community members (Wellborn and Langerhans, 2015; Simpson, 1984). Gut microbiomes have been theorized to be environments with great ecological opportunity (Scanlan, 2019; Mcdonald et al., 2020), especially during low-diversity events such as infant development (Ferretti et al., 2018; Angell and Rudi, 2020) and perturbation due to antibiotics or disease (Scanlan, 2019). There are open questions concerning the effect of time-induced drift during microbiome recovery: As the community begins the process of reassembly in the presence of external factors (antibiotics), do treatments vary in the time to recovery and net compositional differences? What genetic components and commensal bacteria define the ecological succession?
A complementary approach to prospectively investigate antibiotic perturbation of the microbiome is to use laboratory murine models, in which many of the confounders from a retrospective human cohort can be controlled for. Murine models have shown that antibiotic perturbation alongside diet can modulate compositional differences in the microbiome (Turnbaugh et al., 2009; Cabral et al., 2020) and host susceptibility to infection by Salmonella enterica (Sekirov et al., 2008) and C. difficile (Theriot et al., 2014; Laubitz et al., 2021). While these murine studies have served important roles in hypothesis testing and mechanistic exploration (Daillère et al., 2016; Gopalakrishnan et al., 2018), it is often difficult to translate these findings to human health and treatment because of the substantial differences in “natural” human and mouse microbiomes and host biology (Park and Im, 2020), and the idealized nature of experimental work in mice (inbred mouse lines often fed restricted, highly regulated diets) (Nguyen et al., 2015).
We hypothesized that antibiotic regimens, which are routinely used for the treatment of bacterial infections, would cause acute perturbation to the microbiomes of healthy human volunteers by increasing the relative abundance of pathobionts and select for AR genes. We further hypothesized that while the taxonomic diversity of these microbiomes would largely recover to pre-perturbation states within weeks to a few months, the “scars” of perturbed taxonomic composition, AR gene enrichment, and compositional drift would persist over the same time scales. Thus, in this study, we designed and executed a prospective, longitudinal investigation of the impact of 4 different antibiotic regimens recommended for outpatient community-associated pneumonia on the taxonomy, resistome, and functional output of healthy volunteer microbiomes. This represents a patient population who would receive antibiotics for respiratory infections, which are frequently not bacterial in etiology. Thus, many antibiotics are frequently misused for that indication, and we were accordingly motivated to understand the effect of these common unwarranted treatments on a healthy human microbiome. In our study design, we collected multiple control samples per individual before antibiotic exposure to establish pre-perturbation baseline microbiome states. Using both quantitative microbiologic culture and metagenomic sequencing, we analyzed both the acute effect of antibiotic perturbation (AP) on healthy volunteer gut microbiomes and the putative persistent “antibiotic scarring” (defined as a statistically significant increase in AR compared to the pre-antibiotic state) observed up to 6 months after antibiotic exposure, and we compared the trajectories of these perturbations to the microbiomes of hospitalized, critically ill patients.
We found a remarkable similarity in the effect of each antibiotic treatment on species richness and viable colony forming unitss (CFU) concentrations with antibiotic-specific enrichment in taxonomy and resistance. Recovery after treatments containing azithromycin was slower, resulting in niche discordance and greater net compositional distance. Three volunteers already at low microbiome diversity seemed primed to large taxonomic perturbations after antibiotic treatment, increasing their taxonomic similarity to microbiomes from critically ill ICU patients. Even though most of the volunteers returned to a similar level of taxonomic diversity after treatment, significant increases in AR genes, changes to the functional output of the microbiome, and an altered taxonomic state imply significant long-term consequences. These data quantitatively illuminate how the healthy microbiome buffers antibiotic-specific changes to the relative abundance of specific taxa during perturbation, and identifies key areas of further research, such as identifying individuals at greater risk of perturbation.
RESULTS
To quantify the effect of antibiotics on the gut microbiota of healthy individuals over time, we recruited a study group of 20 volunteers from the St. Louis, MO, metropolitan area (Table S1). The cohort was then randomized into 1 of 4 antibiotic treatment groups: (1) azithromycin (AZM) dispensed as a standard oral dose pack with a 500-mg first dose on day 1 and 250 mg daily thereafter, (2) levofloxacin (LVX) dispensed as 750 mg, (3) cefpodoxime (CPD), dispensed as 250 mg twice per day, and (4) a combination of the azithromycin and cefpodoxime (CPD + AZM) treatments at the aforementioned individual doses. All of the treatments were administered orally for 5 days, consistent with Infectious Diseases Society of America/American Thoracic Society guidelines for community-acquired pneumonia in adults (Mandell et al., 2007). Fecal samples from volunteers were collected longitudinally at 15 timepoints encompassing periods before, during, and after antibiotic treatment (Table S2). The first 4 sampling points were taken before antibiotic administration to establish a robust baseline for the unperturbed microbiome of each patient over a time span similar to the period of acute perturbation after treatment (2 weeks). Antibiotic treatments started on day 0, and samples were taken on days 3, 6, 9, 12, and 19 after the start of antibiotics. After the first 30 days post-treatment, sampling intervals were increased to 1 month (days 35, 65, 95, 125), and finally 2 months (day 185) for the last interval. The participant retention rate was 100%, with a 96.3% sample submission rate, for a total of 289 fecal samples for analysis. A subset of 10 sampling points per individual underwent semi-quantitative microbiologic culture. The microbiomes of this healthy volunteer cohort were then compared to fecal samples from 26 ICU patients in the St. Louis, Missouri area who were being screened for C. difficile colonization. ICU patients are a patient population with a well-documented high AR burden (Kollef and Fraser, 2001; Macvane, 2016).
Antibiotics decrease microbiome bacterial load and richness and perturb microbial community structure
We analyzed gut microbiomes using complementary culturebased and culture-independent methods. To assess the effect of antibiotics on the bacterial load of the gut microbiome, we used semi-quantitative microbiologic cultures of viable bacteria in a subset of fecal samples. We found a significant mean reduction of 4.78 log-transformed CFU (95% confidence interval [CI] 2.89–6.36, p = 0.0074, paired Wilcoxon rank sum) and 2.90 (95% CI 1.90–3.91, p = 0.0047) in aerobic and anaerobic bacterial titers, respectively, between days 6 and −14 (Figures 1A and 1B; Tables S3 and S4). No significant differences were found between culture results from samples taken on days −14 and −1. The day after the end of treatment, bacterial species richness also decreased significantly when compared to day −14 (11.50 95% CI 7.99–16.00, p = 0.0052) (Figure 1C; Table S5). Again, there were no significant differences in species richness between any of the other 3 pre-antibiotic time points and the time point at day −14.
Figure 1. Healthy microbiomes are perturbed by antibiotic treatment.

(A) Longitudinal tracking of changes in semi-quantitative culturing of aerobic bacteria. All confidence intervals are bootstrapped 95% CI of the mean. Asterisks represent significantly different timepoints (See Tables S3–S5 for p values).
(B) Longitudinal tracking of changes in semi-quantitative culturing of anaerobic bacteria. All confidence intervals are bootstrapped 95% CI of the mean. Asterisks represent significantly different timepoints (See Tables S3–S5 for p values).
(C) Longitudinal tracking of metagenomic species richness. All confidence intervals are bootstrapped 95% CI of the mean. Asterisks represent significantly different timepoints (See Tables S3–S5 for p values.)
(D) The trajectory of the stool samples through the PCA space before and at the end of antibiotic administration. The PCA was generated using all of the sample points, with arrows illustrating volunteer microbiome movement through the PCA space starting at day 14 to day 6. Samples are color-coded by antibiotic—blue for CPD or CPD + AZM and green for AZM or LVX.
(E) A cladogram overlaying colors representing significant effect sizes found at each taxonomic level (blue for CPD/CPD + AZM and green for AZM/LVX). See Table S7 for the biomarker legend. Each node denotes a taxonomic unit within the bacterial hierarchy, and when colored, were found to be significantly enriched in the treatment group represented by that color. White nodes represent taxonomic units that were present, but not significantly enriched in either group.
See also Figures S1 and S2.
In addition to these broad trends, we also observed antibiotic-specific effects on the microbiome. Principal-component analysis (PCA) of species abundance for the full dataset (all volunteer time points and ICU group; Figure S1) was used to visualize the effect of antibiotic administration on volunteer microbiomes. We observed that volunteer microbiomes given specific treatments shared similar trajectories through PCA space after treatment, with the CPD and CPD + AZM treatment groups separating significantly from the LVX and AZM groups (p = 0.01 [PERMANOVA]) (Figures 1D and S2). Linear discriminant analysis (LDA) between the 2 groups at day 6 revealed an enrichment of the Bacteroidetes phylum (log average: 5.82, LDA effect size = 5.02, p <0.01) and the Clostridium genus (marked as B on Figure 2E) (log average: 4.43, LDA effect size = 3.99, p < 0.05) in the healthy volunteers given CPD or CPD + AZM. Volunteers given LVX or AZM were enriched for genera in the Firmicutes phylum (log average: 5.73, LDA effect size = 5.03, p < 0.01) such as Eubacterium (log average: 5.28, LDA effect size = 4.82, p < 0.01), Ruminococcus (log average: 4.73, LDA effect size = 4.34, p < 0.01), and Anaerostipes (log average: 3.22, LDA effect size = 3.91, p < 0.05) (Figure 1E). Notably, significant compositional differences between the groups were abrogated by day 185 (p = 0.98 [PERMANOVA]; Figure S2).
Figure 2. The microbiome recovers but is delayed by AZM.

(A) Longitudinal recovery of relative species richness after AP. Black confidence intervals in metagenomic data represent bootstrapped 95% confidence intervals for the average of CPD and LVX volunteers at each time point, and blue confidence intervals represent 95% bootstrapped confidence intervals for AZM and XPD + AZM volunteers at each timepoint. The dashed line represents the pre-treatment average species richness.
(B) p value significance tests over imputed intervals between LVX and CDP and the AZM and CPD + AZM groups after antibiotic administration. The dashed line represents 0.05 p value significance threshold.
See also Figures S3 and S4.
Azithromycin delays recovery of species richness and is associated with the relative abundance of 9 gut commensals and metabolic pathways
After treatment with antibiotics, viable aerobic and anaerobic bacteria cultured from volunteer fecal samples significantly decreased in concentration. We observed lower aerobic CFU/mL counts from fecal samples from days 6 (4.78 log-transformed CFU 95% CI [2.89–6.36], p = 0.0074 [adjusted Wilcoxon rank sum]) and 12 (4.56 log-transformed CFU 95% CI [2.47–6.86], p = 0.0074 [adjusted Wilcoxon rank sum]) than those recorded on day −14 (Table S3). Anaerobic CFU/mL counts exhibited a similar perturbation after treatment with reduced CFU counts on day 6 (2.90 95% CI [1.91–3.91], p = 0.0047 [Wilcoxon rank sum]) and 12 (2.83 95% CI [1.70–4.31], p = 0.0047 [Wilcoxon rank sum]; Table S4). Recovery to baseline was reestablished by day 19 after treatment for both aerobic and anaerobic cultures. The species number for all of the volunteers recovered by day 19 (Table S5). Microbiome diversity recovered in a treatment-specific manner, with volunteers given LVX or CPD recovering 12 days after antibiotic administration (3.00 95% CI [−9.99 to 10.00], p = 0.4 [Wilcoxon rank sum]) (Figures 2A and 2B). In contrast, volunteers given AZM or CPD + AZM appeared to deviate from the recovery shown by LVX or CPD volunteers.
We discovered that AZM and CPD + AZM volunteer microbiomes, hereafter referred to as the slow recovery group (SRG), exhibited a significant delay in recovery between days 24 and 30 when compared to the microbiomes of volunteers given LVX or CPD, referred to as the fast recovery group (FRG) (p < 0.05) (Figure 2B). The average species richness of the AZM and CPD + AZM groups continued to be lower than pre-antibiotic levels until day 65 (5.00 95% CI [2.20 × 10−5e−5−1.49e−1], p < 0.051 [Wilcoxon rank sum]; Figures 2A and 2B). Eight commensal bacteria were identified to have recovered significantly slower in volunteer microbiomes given AZM or CPD + AZM than in microbiomes given LVX and CPD: Alistepes putridensis, Bifidobacterium longum, Collinsella aerofaciens, Eubacterium eligens, Dorea longicatena, and Barnseilla intestinihominis (p < 0.05 [ZIBR, zero-inflated β random effect model]). The average abundance of Methanobrevibacter smithii exhibited 2 short blooms on days 12 and 35 in AZM and CPD + AZM. Bacteroides massiliensis was the only species identified as being enriched in AZM and CPD + AZM after treatment (p < 0.05 [ZIBR]).
Functional analysis confirmed that the slow recovery of Bifidobacterium longumwas associated with the decreased abundance of the alternative, non-mevalonate 2-C-methyl-d-erythritol 4-phosphate/1-deoxy-d-xylulose 5-phosphate pathway (MEP/DOXP pathway), a metabolic pathway belonging to the Bifidobacterium genera.
Similar to the recovery over time in species number, the cumulative distance through the first 10 dimensions of the PCA did not differ for both the FRG and SRG. However, the SRG had a significantly higher distance through 10-dimensional Euclidean distance during recovery (p = 0.022, t test on log-transformed distances; Figure S3).
The resistance reservoir increases in healthy volunteer microbiomes over time after antibiotic perturbation
After AP, AR gene burden increased significantly for the volunteers receiving CPD, AZM, and CPD + AZM treatments when compared to the average of their pre-antibiotic sampling points (Figures 3A and 3B; Table S6). Conversely, volunteers receiving LVX exhibited no significant changes over time (adjusted p < 0.05, paired Wilcoxon rank sum). Only 3 resistance elements were identified as changing significantly over time in the CPD, AZM, and CPD + AZM treatments. Three resistance genes, cfxA, tetO, and tet40, increased significantly (adjusted p < 0.05 [Splinectomer], Figures 3C–3E). tetO and tet40 are often found in Firmicutes (Zilhao et al., 1988; Kazimierczak et al., 2008), and we observed a concomitant increase in the average relative abundance of Firmicutes for all of the volunteers beginning at day 65 (Figure S4).
Figure 3. Resistance gene burden increases up to 2 months after antibiotic perturbation.

(A) Resistance gene burden measured as total reads per kilobase of transcript, per million mapped reads (RPKM) of all resistance markers increases significantly immediately after antibiotic perturbation and then again at day 65. All confidence intervals are bootstrapped 95% CI of the mean.
(B) p value compared to first sample point at all time points. Long-term increases in tetracycline resistance after all of the treatments. The dashed line represents the 0.05 p value threshold (see Table S6 for p values). The black line represents the p value over time for AZM-, AZM+CPD-, and CPD-treated volunteers. The purple line represents the p value over time for LVX-treated volunteers.
(C–E) Average RPKM with bootstrapped 95% confidence intervals of the genes found to be significantly increased over time. The y axis represents average RPKM.
See also Figure S4.
The healthy volunteer resistome after perturbation is distinct from the resistome of ICU patients
We next identified significant differences in AR gene content between the healthy volunteer microbiomes the day after the end of antibiotic treatment (day 6) and microbiomes from 26 ICU patient fecal samples (Figure 4A). The microbiomes of the ICU patients were characterized by the enrichment of AR genes encoding multi-drug resistance (log average: 5.07, effect size = 4.75, p < 6.46e−7 [LDA]), specifically efflux pump classes such as ABC class efflux (log average: 3.30, effect size = 3.13, p < 0.009 [LDA]), and SMR class efflux (log average: 3.26, effect size = 3.14, p < 0.009 [LDA]). Healthy volunteer microbiomes were enriched for the β-lactam resistance gene cfxA (log average: 5.57, effect size = 5.12, p < 0.001 [LDA]) and macrolide resistance genes acting on the 23S ribosomal RNA methyltransferase (log average: 5.42, effect size = 4.59, p < 0.02 [LDA]), both targeting antibiotic treatments just administered.
Figure 4. The healthy volunteer resistome after AP is distinct from the ICU patient resistome.

(A) Cladogram of the antibiotic resistance genes found to be discriminatory between healthy volunteers at day 6 and ICU remnant microbiomes. The ICU microbiomes were highly enriched for multi-drug resistance and efflux pump complexes. The healthy microbiomes were enriched for cfxA resistance and the 23S ribosomal RNA methyltransferase mechanism of macrolide resistance (Au). See Table S8 for the biomarker legend.
(B) Heatmap of the 70 most highly variable resistance markers for the same sample set. The healthy volunteer samples cluster together and are largely depleted of the markers representative of the ICU microbiomes. The ICU microbiomes have 4 distinct clusters, which were dominated by distinct sets of variable markers. The scale represents log-transformed RPKM values.
We identified the 70 most highly variable and abundant AR genes in averaged acute post-antibiotic healthy volunteers and the ICU dataset. Hierarchical clustering identified 5 clusters, with the healthy volunteers forming 1 distinct cluster (Figure 4B). The ICU cohort was split into 4 distinct groups: Cluster 3 was largely depleted of the set of highly variable genes; cluster 2 shared multiple genes overlapping with the averaged acute post-antibiotic healthy volunteer profile; cluster 4 contained glycopeptide resistance genes that confer vancomycin resistance commonly found in vancomycin-resistant Enterococcus (VRE); and finally, cluster 5 contained genes encoding mostly β-lactam efflux pumps.
Most healthy volunteers remain inside the healthy PCA space and some enter the ICU PCA space after antibiotics
To compare the effect of AP on healthy volunteer microbiomes with the presumed dysbiotic microbiome state of the ICU patients, healthy volunteer metagenomes were mapped through the first 2 dimensions of the PCA ordination space at days −14, 6, and 185 (Figure 5A). Pre-antibiotic samples were used to estimate a density contour within the first 2 axes of the PCA, which we defined as the area of the pre-antibiotic “healthy state.” Another density contour was estimated from ICU microbiomes to characterize the dysbiotic state of critically ill patients. After perturbation, healthy volunteer microbiomes experienced a shift in their taxonomic composition toward a diversity minimum, where the majority of ICU patients were clustered (Figure 5A). Most of the volunteers (17 of 20) then showed a reversal of this trajectory, returning to an area near where they began, with most never leaving the healthy state area. However, 3 volunteer microbiomes experienced large decreases in diversity and richness, traversed long paths through PCA space over time, and entered the density contour demarcated by critically ill ICU microbiomes. The PCA distance had no positive enrichment of any functional pathways. However, of the top 20 most significant negatively enriched pathways, 18 were from the genus Eubacterium. While the other volunteers lost on average 10.28 species on day 6 after antibiotic exposure, the 3 “ill-like” volunteers lost an average of 23.67. For these 3 individuals, the recovery of Shannon’s diversity index was incomplete and was significantly different from the rest of the volunteers (linear regression, p < 4.66e−6). By the end of the study, they had only 63.4% of the species they started with, as opposed to a 99.1% recovery in the rest (Figure 5B). These individuals contained a significantly lower abundance of 9 metabolic pathways originating from the Eubacterium genera and were instead enriched for lysine biosynthesis pathways from the genera Clostridiodes and Erysipelotrichaceae. Interestingly, their resistance gene burden did not differ from that of the rest of the volunteers (Figure 5C). This analysis indicates that a subset of the healthy volunteers was at greater risk of AP of their microbiome taxonomic diversity, and their return to a healthy state was incomplete.
Figure 5. A subset of ill-like individuals were heavily perturbed during the study.

(A) A 6-month longitudinal analysis of healthy volunteer microbiomes through the PCA space. The red dots are the ICU remnant microbiomes, while the blue dots represent the starting locations of all of the volunteer microbiomes. The arrows represent passage over time of the microbiomes, with the starting point samples at day −14, the inflection point is their location at day 6, and the arrowhead is their location at day 185. All of the volunteer trajectories are present, but the 3 that ended in ICU space are bold. The blue density contour was estimated using the starting coordinates of the volunteer microbiomes and overlaid onto the PCA space, and the magenta contour represents the density of the ICU microbiomes.
(B) Longitudinal analysis of overall resistance burden for the ICU-like subset of 3 healthy volunteers (represented in purple) and the rest of the volunteers. The error bar confidence intervals represent 95% bootstrapped confidence intervals of the mean, and a linear model estimate was fit to both groups starting after the end of antibiotic administration. Shaded regions represent 95% confidence interval for the linear fit for each group.
(C) The changes over time in species diversity for the ICU-like volunteers and the rest of the volunteers, with a linear model fit to both starting after the end of antibiotic administration. The error bar confidence intervals represent 95% bootstrapped confidence intervals of the mean. Shaded regions represent 95% confidence interval for the linear fit for each group.
See also Figure S1.
The taxonomy and resistance composition of the healthy volunteer microbiome is altered after recovery
We next wanted to ascertain whether post-antibiotic selection resulted in effects large enough to significantly alter the composition of the microbiome, similar to what was observed with diversity. The compositional paired-mean difference between an average of the pre-antibiotic composition and the first (pre-perturbation) healthy volunteer microbiome sample was compared to the dissimilarity between an average of the pre-antibiotic composition and all of the other time points from the same volunteer via Bray-Curtis dissimilarity. Bray-Curtis dissimilarity between all of the pre-antibiotic time points was low, and no time points were significantly different from the starting composition (Figures 6A and 6B). However, after the start of antibiotic treatment, the Bray-Curtis dissimilarity between the microbiome samples of a volunteer to his or her first sample increased dramatically and remained elevated for both taxonomy and AR genes (Figures 6C and 6D). This contrasts with microbial richness, which by day 40 was no longer significantly lower than pre-treatment levels (Figure 2B).
Figure 6. Antibiotic perturbation in the healthy volunteer microbiome.

(A and C) Bray-Curtis dissimilarity of each time point to the median of the pre-antibiotic composition. Within-patient Bray-Curtis dissimilarity for all healthy volunteers was compared to the first time point. All time points for the pre-antibiotic period exhibit low dissimilarity, but this increases immediately starting with antibiotic administration and remains high until the end of the study (day 185). This analysis was conducted for species composition (A) and for resistance gene composition (C).
(B and D) Bootstrapped 95% confidence intervals generated for data at each time point.
DISCUSSION
Antibiotic exposure may result in acute and persistent changes in the commensal host microbiome (Wipperman et al., 2017; Dethlefsen et al., 2008; Ubeda et al., 2010; Stevens et al., 2011). We found a significant reduction in viable bacterial titers in aerobic and anaerobic microbiologic cultures and a reduction in metagenomic species richness. These results are consistent with previous studies involving antibiotics in healthy individuals, which support our initial hypothesis that antibiotics can cause acute perturbations in the gut microbiome of healthy volunteers (Palleja et al., 2018; De Lastours et al., 2017). This reduction was first observed on day 6, the first day after the 5-day treatment. This was likely due to our study design, which collected time points the first day of treatment, before the antibiotic perturbation had time to reach the stool. The small number of volunteers and 4 different antibiotic treatment groups may have included too much variation to detect the beginning of decreased species richness and CFUs at the next sampling point, which was day 3 of the 5-day treatment. Palleja et al. (2018) found a significant decrease in metagenomic operational taxonomic units 1 day after the end of a 4-day treatment with a 3-drug cocktail, which correlates well with our observation of a significant decrease 1 day after the end of the 5-day treatment in our study. Given the information from this previous study, it seems reasonable that had we collected samples on day 4 of the 5-day treatment, we may have seen a significant difference in species richness or culture CFUs.
Furthermore, we observed a direct relationship between species relative abundance and resistance elements. Immediately after treatments containing CPD, there was a relative enrichment of Bacteroidetes. Bacteriodes spp. have a well-documented resistance to β-lactams, and the cfxA gene produced phenotypic resistance to cefoxitin in Bacteriodes species (Edwards, 1997; Ferreira et al., 2007; García et al., 2008). cfxA increased in relative abundance after CPD or CPD + AZM treatment, suggesting that this is a primary form of resistance for enteric Bacteriodes spp. as well. Previous studies had found that cefprozil, a second-generation cephalosporin, induced increases in Enterobacter cloacae in healthy volunteers with low-diversity, Bacteroides-dominated microbiomes (Raymond et al., 2016). Our results indicate that Bacteroides species survive CPD treatment, likely via cfxA, resulting in a low-diversity, high-Bacteroides environment, generating an opportunity for the expansion of pathogens such as Enterobacter spp. In comparison, the AZM and LVX groups were enriched for multiple Gram-positive genera within the Firmicutes phylum. The increases correlate well with observed increases in the Bacteriodes:Firmicutes ratio in mice (Li et al., 2017). These taxonomic changes were implicated in increased adipogenesis, altered microbiome short-chain fatty acid production, and other risk factors for obesity.
The recovery of species richness after exposure varied by treatment, similar to previous studies (Korpela et al., 2016; Palleja et al., 2018). The SRG, which consisted of volunteers given AZM or AZM + CPD, experienced a 6-day period of extended lower diversity, approximately the duration of the terminal half-life for this drug of 5 days (Drew and Gallis, 1992). This is in sharp contrast to the much shorter half-life of CPD and LVX, which are reported to be 2–3 and 6–8 h, respectively; this could explain in part the slower recovery of species richness in the SRG (Borin, 1991; Fish and Chow, 1997). Previous analyses using 16S rRNA gene amplicon data reported a reduction in species richness 1 year after children were treated with the macrolides clarithromycin and erythromycin, as well as a reduction in the abundance of Bifidobacterium and Collinsella genera (Korpela et al., 2016). Our time series data in adults identified both B. longum and C. aerofaciens as well as E. eligens and D. longicatena recovered more slowly after AZM. B. longum has been shown to be reduced in abundance in both the elderly (Woodmansey, 2007; Rampelli et al., 2020) and the critically ill (Shimizu et al., 2011; Ojima et al., 2016). A recent study found a negative correlation between increased abundance of the Bacteriodes and Methanobrevibacter genera and the healthy microbiome (Wilmanski et al., 2021), and the change over time of the 2 species significantly associated with slow recovery hail from these genera. The 2 recovery groups had similar cumulative distances through the PCA space, but the SRG also had a significantly higher distance between their first and last samples. The stability of the post-antibiotic microbiome is well documented (Jernberg et al., 2007; Moya and Ferrer, 2016), but the effect of time-induced drift associated with 1 antibiotic versus another is not well understood. Our results confirm that antibiotic treatment produces ecological opportunity in healthy individuals, reshaping even robust fecal microbiomes through increased resistance gene content and altered taxonomic composition. The increased net compositional distance exhibited by the SRG is likely due to the longer bioavailability of AZM driving prolonged niche discordance and ecological opportunity. Increased intrapatient temporal variation in stool and skin microbiomes has been significantly associated with adverse infectious outcomes after patients with acute myeloid leukemia underwent induction chemotherapy (Galloway-Peña et al., 2017). Co-option of the community instability inherent in ecological opportunity by pathogenic organisms has been theorized to drive pathogen virulence and adaptation (Scanlan, 2019). The SRG was also associated with lower counts of the Bacteriodes pentose phosphate pathway, as well as the alternative, MEP/DOXP pathway from the genus Bifidobacterium (of which B. longum, one of the species identified as recovering slower in this group, is a part) (Milani et al., 2014). This pathway is also present in many pathogens (Hale et al., 2012), and is currently being investigated as a target for antibiotic development due to its absence in humans and the known ability of fosmidomycin to inhibit 1-deoxy-D-xylulose 5-phosphate reductoisomerase, a key enzyme in the pathway (Zinglé et al., 2010). It is hypothesized to be a method to modulate host response and lower the chance of an immunogenic reaction to commensal bacteria (Eberl et al., 2003).
When we compared antibiotic resistance composition, we found that immediately after a 5-day course of antibiotics, the AR gene burden in healthy microbiomes increased and remained elevated for the length of the study in volunteers receiving CPD, AZM, and CPD + AZM. This initial increase in AR gene burden was not unexpected, and similar increases have been described (Palleja et al., 2018). The length of elevated AR burden varied from previous studies; Palleja et al. (2018) reported no significant increases in total AR burden by 45 days in adults; however, D’souza et al. (2019) reported significant increases in AR prevalence by 6 months in HIV-exposed, uninfected infants receiving weekly clotrimoxazole prophylaxis. The reason for these differences in severity and duration of AR elevation is likely to be governed by the maturity, stability, and health of the microbiomes under study, and the effects of each specific antibiotic treatment, as LVX did not increase resistance gene burden.
While AR gene burden was in general higher in ICU patient microbiomes, certain classes of AR genes in healthy volunteers were enriched compared to ICU patient microbiomes, including AR gene classes specific to the antibiotics given during the study, such as the β-lactam resistance gene cfxA (CPD) and 23S ribosomal RNA methyltransferase resistance (AZM). ICU patients were instead enriched for multi-drug resistance. In the generalist-specialist game theory model of ecological succession, generalists initially dominate environments with high ecological opportunity during population expansion (Angell and Rudi, 2020). Remodeling of the healthy individual resistome after antibiotic exposure resulted in increases in 3 genes, tetO, cfxA, and tet40, 2 of which do not convey resistance to any of the antibiotics given in the study, confirming that antibiotic perturbation creates opportunities for species with generally broad antibiotic resistance to dominate transiently. While some AR genes can reliably be used as signatures of specific bacteria, such as ampC for Enterobacteriaceae (Jacoby, 2009; Pitout, 2008), tetO may instead be an indicator of previous antibiotic exposure and subsequent microbiome-wide increase in AR gene burden and selection (Forsberg et al., 2015). Short courses of antibiotics could trigger the acquisition or entrenchment of diverse resistance genes, leading to increased AR seen in some ICU patients and the elderly (Shimizu et al., 2011; Choy and Freedberg, 2020). The persistent increases in overall AR burden, in tandem with reduced microbiome diversity and similarity to ICU microbiomes of a subset of volunteers, could be used as biomarkers for future adverse reactions to antibiotic treatment, or for the higher risk of hospital-acquired infection as has been proposed for other diseases (Zhou et al., 2019).
Although antibiotics caused an average decrease in species richness, volunteer microbiomes returned to a pre-antibiotic level within 2 months. This resilience is similar to previous studies, which reported that antibiotic exposure yielded only a short-term effect on species richness (Dethlefsen et al., 2008; Dethlefsen and Relman, 2011). Despite this general trend of resilience, the microbiome of 3 individuals recovered slower than the rest of the volunteers. Instead, they exhibited substantial movement over time through the PCA space, ending the observation window within the PCA space dominated by ICU patient microbiomes, and had significant changes to their metabolic output, with reduced pathway abundance from the Eubacterium genera, an important gut commensal, but increased abundance of pathways from the genus Erysipelotrichaceae. Erysipelotrichaceae species are known to be highly immunogenic (Palm et al., 2014), linked to inflammatory bowel disease (IBD) and an increase in relative abundance post-antibiotics (Zhao et al., 2013). The PCA distance between the first and last sample of a volunteer was negatively correlated with 18 Eubacterium metabolic pathways, which suggests that functional pathways originating from that genus could be important to reducing the large changes over PCA space that the 3 ill-like volunteers underwent. The concept of antibiotic scarring has been used in previous work (Gasparrini et al., 2019) to describe long-term AR gene accumulation in pediatric microbiomes; we propose a modified definition in which scarring is characterized as a significantly altered, perturbed taxonomic composition with increased AR burden (a generalized response containing on- and off-target resistance elements) after antibiotic exposure. We further identify that antibiotic scarring pushes some low-diversity microbiomes toward an ill-like phenotype, demonstrating that long-term microbial community perturbation can occur from a single dose of antibiotics in healthy individuals. This definition has the potential to be integrated into patient care models optimized for selecting antibiotic treatment personalized to the unique microbiome composition of an individual, an important goal of antibiotic stewardship.
Limitations of the study
Although this is a pilot study and is limited in scope, we believe that it creates a usable framework for studying the universal effects of antibiotics on the healthy human microbiome, as well as for identifying microbiome compositions that may be more prone to potential antibiotic-induced dysbiosis. One limitation of short-read shotgun metagenomic DNA sequencing lies in its inability to determine what microbiota functions are actively being expressed. While our data suggest that relatively few AR genes increase in abundance long term after AP, future studies leveraging metagenomic RNA (cDNA) sequencing or long-read sequencing could determine whether this correlates with increased gene expression or horizontal transmission within the microbial community, respectively (Anthony et al., 2020; Leggett et al., 2020). The small treatment group size and 6-month study window of our study also limit the ability to discern whether the observed alterations to the taxonomy and resistome persist to longer intervals, although other studies have recorded similar trends years after antibiotic exposure (Dethlefsen and Relman, 2011; Gasparrini et al., 2019). Regardless, our results illustrate a dynamic but somewhat incomplete recovery process that is dependent on the specific antibiotic regimen and highlight the potential intrinsic resilience of particular gut microbial community architectures. Strain-level dynamics during antibiotic perturbation provide an extra layer of complexity and may help explain the remarkable change in the composition of the resistome (Yassour et al., 2016). The ICU comparator group was collected during routine C. difficile surveillance testing, and thus no clinical metadata were collected that could help identify patient covariates or further explain the variation in antibiotic perturbation in healthy volunteers. Although any observation of differential resistance elements between ICU patients and healthy volunteers is impossible to correlate to previous ICU antibiotic exposure without extensive knowledge of their clinical history, this comparator group serves as an example of the low-diversity, high-MDR taxonomic state found in ill patients, and thus of a general dysbiosis phenotype (Mcdonald et al., 2016).
In conclusion, our findings indicate that short courses of antibiotics commonly used for the treatment of bacterial infections can cause both short- and longer-term perturbations and scarring of the microbiome in healthy human volunteers, resulting in the prolonged increase in AR in healthy microbiomes. We further refine the definition of antibiotic scarring, identifying resistance genes that could be used to detect previous perturbation through the expansion of broadly AR-resistant organisms. Finally, we observed that AZM administration resulted in delayed recovery and greater compositional distance by the end of the study.
Further study and interventions are necessary to mitigate the development of AR and better identify individuals at risk of developing long-term negative effects after treatment. The approaches described in this work may be applicable for measuring the impact of existing and newly developed antibiotics on the gut microbiome and resistome, with such perturbation measures incorporated into antibiotic lead compound selection and development (Jia et al., 2008; Andremont et al., 2021). The long-term increase in resistance burden observed in this study is an example of events that can push a low AR resistome toward higher AR. With the continued development of AR, novel methods to understand and prevent AR are necessary, and these data form a resource for studying both the short- and long-term effects of antibiotic perturbation on the healthy microbiome and resistome.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Dr. Jennie H. Kwon (j.kwon@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Sequence data is stored as short-read metagenomic sequences in the Sequence Read Archive (SRA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Biological samples | ||
|
| ||
| Healthy Volunteer fecal samples | this paper | none |
|
| ||
| Deposited data | ||
|
| ||
| Short read metagenomic DNA from Microbiome Samples – Healthy Volunteer Data | Sequence Read Archive (SRA) | bioproject: PRJNA664754 |
| Short read metagenomic DNA from Microbiome Samples – ICU comparator group | Sequence Read Archive (SRA) | bioproject: PRJNA703034 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Blood Agar Plate, 5% Sheep Blood in Tryptic Soy Agar (TSA) Base, 15 × 100mm plate | Hardy Diagnostics | Cat no. A10 |
| BBE Agar, Bacteroides Bile Esculin, reducible, for anaerobes, 15 × 100mm plate | Hardy Diagnostics | Cat no. G05 |
| MoBio (now Qiagen) DNeasy PowerSoil Pro Kit | Qiagen | Cat no. 47016 |
| Nextera library prep | Illumina | Cat no. 20018704 |
| Agencourt AMPure XP system | Beckman Coulter | Cat no. A63880 |
| Quant-it PicoGreen dsDNA assay | Invitrogen | Cat no. P11496 |
|
| ||
| Software and algorithms | ||
|
| ||
| The R Project for Statistical Computing | https://www.r-project.org/ | Version 4.0 |
| MetaPhlAn | Segata et al. (2012) | Version 2; RRID:SCR_004915 |
| HUMAnN | Franzosa et al. (2018) | Version 2; RRID:SCR_014620 |
| ShortBRED | Kaminski et al. (2015) | Version 2 |
| LEfSe | Segata et al. (2011) | Version 1; RRID:SCR_014609 |
| MaAsLin | Mallick et al. (2021) | Version 2 |
| Zibr | Chen and Li (2016) | Version 0.1 |
| Dabestr | Ho et al. (2019) | Version 0.2.5 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Institutional review of sample collection
Stool specimen collection was reviewed and approved by the Human Research Protection Office of Washington University in St. Louis under IRB ID #: 201610071. ICU fecal specimens was reviewed and received a non-human subjects determination by the Institutional Review Board of Washington University in St. Louis IRB ID #: 201509022.
Subject recruitment and microbiome sampling
Twenty healthy adults from the St. Louis, MO metropolitan area were recruited for the study. Subjects were eligible for the study if they were between the ages of 21 and 60 and provided written, informed consent (Table S1). Subjects were not eligible if they met any of the following exclusion criteria: history of allergic reaction or contraindication to any of the study antibiotics; not able to provide regular fecal samples; any systemic antibiotic exposure in the previous 6 months; tube feeds in the previous 6 months; pregnant, risk of becoming pregnant, or breastfeeding during the study period; gastroenteritis in previous 3 months; non-elective hospitalization in previous 12 months; incontinent of stool; known colonization with a MDRO; anticipated change in diet or medications during the study period; elective surgery planned during the study period; history of an intestinal disorder; or inability to provide written, informed consent. Care was taken to recruit a volunteer cohort with equal sex parity (see Table S1).
After enrollment, subjects were interviewed about their medical history and underwent a physical exam. Subjects were randomly assigned to receive a 5-day course of one of the following antibiotics: AZM, LVX, CPD, or CPD + AZM. They were directed to begin the antibiotic on the appropriate day. Subjects sent back their empty pill bottles to confirm that all doses were taken. Study personnel involved in patient recruitment, specimen collection and processing were blinded to treatment group.
Study personnel provided subjects with supplies for collecting clinical specimens. Subjects submitted fecal specimens to study personnel at 15 time points throughout the study period (Table S2). Fecal samples from pre-specified time points underwent microbiologic culture upon receipt of the specimen (Table S2). All other specimens and remnant fecal specimens were frozen on the same day they were provided. At each specimen submission point, subjects also completed questionnaires on bowel movement consistency and frequency, diet, medications, and changes in medical history. This study received approval from the Washington University Human Research Protection Office. The ICU fecal specimens were convenience samples of remnant stool from the Clinical Microbiology Laboratory collected from 26 ICU inpatients who had diagnostic testing for Clostridioides difficile at Barnes-Jewish Hospital, thus no clinical metadata was collected.
METHOD DETAILS
Semi-quantitative culturing
To provide an overall assessment of the aerobic and anaerobic microbial burden in the samples serial dilutions were plated to general purpose aerobic and anaerobic culture media. For aerobic culture, ~1g or 1mL of fecal waste was placed in a 2mL Nunc tube. An equal amount of 1 × PBS (1:1 dilution) was added and vortexed well to homogenize the sample. 10, 10-fold dilutions into PBS with sample were then made. BAP plates (Hardy Diagnostics) were inoculated with 10μL and 100μL of each dilution respectively; a cross streak pattern was used for the 10μL plate and a quadrant streak was used for the 100μL plate, and the plates were incubated in air overnight at 35°C. After incubation, colonies on the 10μL plate were counted, and anything over 50 was recorded as “>50 colonies”. For the 10μL plate a semi-quantitative amount of growth was recorded (0,+1,+2,+3,+4) and a general physical description of the colonies was recorded (e.g. mixed enteric flora, mixed gram positive flora, etc.). Sweeps of the plate were frozen in molecular grade water and in TSB-glycerol. Samples were then stored at −80°C.
For anaerobic culture, a dilution series was created from fecal samples as just described. Similar aliquots of dilutions were inoculated onto 2 pre-reduced BBA (Hardy Diagnostics) plates and incubated at 35°C in an anaerobic chamber or anaerobic bag overnight. Streaking methods were the same as just described.
DNA extraction and sequencing
Fecal samples were kept on dry ice while samples were chipped off and weighed to standardize the amount of fecal matter to be extracted. Total gDNA was extracted from samples using a PowerSoil DNA isolation kit (MoBio). Lysis was conducted using a Minibeadbeater (Biospec Products). gDNA was stored at −20°C until sequencing library creation. Shotgun metagenomic sequencing libraries were created using gDNA diluted to 0.5 ng/μL and the modifications to the Nextera library prep lot (Illumina) detailed in Baym et al. (Baym et al., 2015). This generated ~450 bp DNA fragments which were purified using the Agencourt AMPure XP system (Beckman Coulter) and quantified using the Quant-it PicoGreen dsDNA assay (Invitrogen). Samples were pooled onto lanes to ensure ~3M (2 × 150bp) reads per sample, and three technical replicates of this pooling process were quantified using the Qubit dsDNA BR assay and then combined in equimolar concentrations to reduce stochastic error in read distribution during the sequencing process. Samples were sequenced in Illumina NextSeq High-output sequencing machines at the Edison Family Center for Genome Sciences and System Biology at Washington University School of Medicine in St. Louis.
Processing of illumina sequence data
Total reads were demultiplexed by barcode into individual sample sequence bins. The adapters and barcodes were removed by Trimmomatic and simultaneously filtered for quality. Human reads were filtered using DeconSeq by mapping reads to the human genome (GrCh38) (Schmieder and Edwards, 2011; Bolger et al., 2014). A minimum sample read depth of 2.5M was determined using species and community resistance rarefaction curves generated with MetaPhlAn2 and ShortBRED (Kaminski et al., 2015; Truong et al., 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Community taxonomy and resistome quantification and downstream analysis
Species composition was predicted using MetaPhlAn2 (Segata et al., 2012). Resistome composition in RPKM (reads per kilobase of reference sequence per million sample reads) was estimated using ShortBRED based off a custom-built marker database including the CARD database (Kaminski et al., 2015). Functional output of the microbiome was estimated using HUMAnN 2 (Franzosa et al., 2018) and significant pathway enrichment was conducted using MaAsLin2 (Mallick et al., 2021). Species richness and diversity were calculated using the R package vegan (version 2.5–6). Abundance and prevalence thresholding was conducted to exclude low abundance, and thus low confidence, species. PCA ordination of patient trajectory trends over time computed on the euclidean distance of species abundance tables. We used an abundance threshold of .01% and a prevalence threshold of 20% (of all healthy volunteer samples) and 19% (of all ICU samples) to result in a median loss of less than 10% overall relative species abundance abundance in all taxonomic samples. Lefse analysis was conducted on the full, unthresholded metaphlan taxonomic abundance tables.
For the resistance analysis, we used an abundance threshold of .5% and a prevalence threshold of 20% (of all healthy volunteer samples) and 5% (of all ICU samples) resulting in 70 highly variable genes across both datasets. Significance for longitudinal data analysis of semi-quantitative culture, taxonomy, and resistome analysis was conducted using the paired Wilcoxon rank sum test in R. Loess fitting of species richness recovery for antibiotic treatments was estimated using the base r loess function, and 100 intervals were then imputed along the fit. The area under the curve was compared to a null distribution made from 999 random permutations of the data using the SplinctomeR package (Shields-Cutler et al., 2018). Increases in specific resistance genes was tested similarly using the SplinectomeR package, comparing the loess regression of a specific gene to a null distribution computed from regressions of all other genes over time. Significance tests begin after first 5% of the regression to account for flying tails. Patient samples were normalized to an average of their pre-antibiotic samples for each metric studied. 2D kernel density estimation of ill and healthy density contours in PCA space were estimated using the MASS (version 7.3.51.6) R package and visualized in ggplot2 (version 3.3.1). Principle component analysis was conducted without Patient 2, as they did not submit most of their samples. LDA effect size analysis was conducted in LEfSe (Segata et al., 2011) using standard parameters, antibiotic as a class, per sample normalization, and one-against-all multiclass analysis. Results visualized using using LEfSe (Galaxy module) and GraPhlAn v0.9, respectively (Segata et al., 2011; Asnicar et al., 2015). Commensals significantly associated with AZM delayed microbiome restoration were identified using a zero-inflated two part mixed effects model using the zibr (version 0.1) R package (Chen and Li, 2016). Zibr requires a complete dataset (no missing datapoints) so missing datapoints (5 of 209 datapoints) were imputed using the average abundance of each species for the entire dataset (this imputation method was also used for the same 5 samples to create a complete dataset for paired-mean compositional difference comparisons using dabestr). Patient 2, which did not submit most of the fecal samples, was not included. Heatmap of differences in ICU vs. healthy volunteer resistome was created using the pheatmap (version 1.0.12) package in R using the 70 most highly variable, log-transformed AR gene abundances in an averaged acute post-antibiotic healthy volunteer profile (three time points immediately after cessation of antibiotics) and the ICU dataset, after filtering to remove efflux pumps, low abundance markers, or genes not targeting one of the major antibiotic categories. Patient 2 was also removed from this analysis due to lack of samples. Cluster analysis of samples used the ward.D2 clustering algorithm (Murtagh and Legendre, 2014) on the Euclidean distance computed from log transformed RPKM abundances of each resistance gene for all samples and then sorted using the dendsort (version 0.3.3) R package. The longitudinal species diversity of the microbiomes of the three patients who transitioned to an ill-like microbiome state was fit to a linear model and compared to the rest of the healthy patients; visualization was created using ggplot2 (version 3.3.1). Longitudinal analysis of taxonomic and community resistance composition of within-patient bray-curtis (computed using R package vegan version 2.5–6) sample dissimilarity was computed for all of a patient’s samples to to an average of their pre-antibiotic microbial compositions. Then the paired mean difference of bootstrapped confidence intervals between all timepoints was compared to the first using the dabestr (version 0.2.5) R package (Ho et al., 2019). Sequence data is stored as short-read metagenomic sequences in the Sequence Read Archive (SRA); Healthy volunteer data is stored in bioproject: PRJNA664754 while the ICU metagenomes are stored in bioproject: PRJNA703034.
Supplementary Material
Highlights.
The microbiome recovers lost diversity after short courses of antibiotics
Azithromycin delays recovery and increases net compositional differences
Altered diversity, resistance, and composition redefine “antibiotic scarring”
Healthy microbiomes end compositionally similar to ICU microbiomes
ACKNOWLEDGMENTS
This work was supported in part by a US Agency for International Development award (award no. 3220–29047) to C.-A.D.B. and G.D. W.E.A. was supported by a T32 NIH Ruth L. Kirschstein National Research Training Grant Fellowship (5T32GM007067–44). K.V.S. was supported by the Society for Healthcare Epidemiology of America Research Scholar Award. A.W.D. was supported by the Institutional Program Unifying Population and Laboratory-Based Sciences Burroughs Welcome Fund grant to Washington University. This work is supported by the Centers for Disease Control and Prevention (OADS BAA 2016-N-17812) contract no. 200–2016-90962 to J.H.K. J.H.K. is also supported by 1K23AI137321–01A1 from the National Institute of Allergy and Infectious Diseases. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The authors thank members of the Dantas lab for helpful comments on the manuscript. The authors thank the Edison Family Center for Genome Sciences & Systems Biology at Washington University School of Medicine in St. Louis staff (Eric Martin, Brian Koebbe, Jessica Hoisington-López, and MariaLynn Crosby) for technical support in high-throughput sequencing and computing.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110649.
REFERENCES
- Andremont A, Cervesi J, Bandinelli P-A, Vitry F, and De Gunzburg J (2021). Spare and repair the gut microbiota from antibiotic-induced dysbiosis: state-of-the-art. Drug Discov. Today 26, 2159–2163. [DOI] [PubMed] [Google Scholar]
- Angell IL, and Rudi K (2020). A game theory model for gut bacterial nutrient utilization strategies during human infancy. Proc. R. Soc. B: Biol. Sci. 287, 20200824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony WE, Burnham C-AD, Dantas G, and Kwon JH (2020). The gut microbiome as A reservoir for antimicrobial resistance. J. Infect. Dis. 223, S209–S213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araos R, Battaglia T, Ugalde JA, Rojas-Herrera M, Blaser MJ, and D’agata EMC (2019). Fecal microbiome characteristics and the resistome associated with acquisition of multidrug-resistant organisms among elderly subjects. Front. Microbiol. 10, 2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnicar F, Weingart G, Tickle TL, Huttenhower C, and Segata N (2015). Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3, e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, and Kishony R (2015). Inexpensive multiplexed library preparation for mega-base-sized genomes. PLoS One 10, e0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borin MT (1991). A Review of the pharmacokinetics of cefpodoxime proxetil. Drugs 42, 13–21. [DOI] [PubMed] [Google Scholar]
- Brown KA, Khanafer N, Daneman N, and Fisman DN (2013). Meta-analysis of antibiotics and the risk of community-associated Clostridium difficile infection. Antimicrob. Agents Chemother. 57, 2326–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral DJ, Wurster JI, Korry BJ, Penumutchu S, and Belenky P (2020). Consumption of a western-style diet modulates the response of the murine gut microbiome to ciprofloxacin. mSystems 5, e00317–e00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EZ, and Li H (2016). A two-part mixed-effects model for analyzing longitudinal microbiome compositional data. Bioinformatics 32, 2611–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy A, and Freedberg DE (2020). Impact of microbiome-based interventions on gastrointestinal pathogen colonization in the intensive care unit. Ther. Adv. Gastroenterol. 13, 1756284820939447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’souza AW, Moodley-Govender E, Berla B, Kelkar T, Wang B, Sun X, Daniels B, Coutsoudis A, Trehan I, and Dantas G (2019). Cotrimoxazole Prophylaxis Increases Resistance Gene Prevalence and α-Diversity but Decreases β-Diversity in the Gut Microbiome of Human Immunodeficiency Virus–Exposed, Uninfected Infants. Clin. Infect. Dis. 71, 2858–2868. 10.1093/cid/ciz1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daillère R, Vétizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, Duong CPM, Flament C, Lepage P, Roberti MP, et al. (2016). Enterococcus hirae and barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 45, 931–943. [DOI] [PubMed] [Google Scholar]
- De Gunzburg J, Ghozlane A, Ducher A, Le Chatelier E, Duval X, Ruppé E, Armand-Lefevre L, Sablier-Gallis F, Burdet C, Alavoine L, et al. (2018). Protection of the human gut microbiome from antibiotics. J. Infect. Dis. 217, 628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cochetière MF, Durand T, Lepage P, Bourreille A, Galmiche JP, and Doré J (2005). Resilience of the dominant human fecal microbiota upon short-course antibiotic challenge. J. Clin. Microbiol. 43, 5588–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lastours V, Maugy E, Mathy V, Chau F, Rossi B, Guérin F, Cattoir V, Fantin B, and Group FTCS (2017). Ecological impact of ciprofloxacin on commensal enterococci in healthy volunteers. J. Antimicrob. Chemother. 72, 1574–1580. [DOI] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, and Relman DA (2008). The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6, e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, and Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. U S A 108, 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew RH, and Gallis HA (1992). Azithromycin—spectrum of activity, pharmacokinetics, and clinical applications. Pharmacother. J. Hum. Pharmacol. Drug Ther. 12, 161–173. [PubMed] [Google Scholar]
- Eberl M, Hintz M, Reichenberg A, Kollas A-K, Wiesner J, and Jomaa H (2003). Microbial isoprenoid biosynthesis and human γδ T cell activation. FEBS Lett. 544, 4–10. [DOI] [PubMed] [Google Scholar]
- Edwards R (1997). Resistance to β-lactam antibiotics in Bacteroides spp. J. Med. Microbiol. 46, 979–986. [DOI] [PubMed] [Google Scholar]
- Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. (2013). The long-term stability of the human gut microbiota. Science 341, 1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira LQ, Avelar KES, Vieira JMBD, De Paula GR, Colombo APV, Domingues RMCP, and Ferreira MCS (2007). Association between the cfxA gene and transposon Tn4555 in Bacteroides distasonis strains and other Bacteroides species. Curr. Microbiol. 54, 348–353. [DOI] [PubMed] [Google Scholar]
- Ferretti P, Pasolli E, Tett A, Asnicar F, Gorfer V, Fedi S, Armanini F, Truong DT, Manara S, Zolfo M, et al. (2018). Mother-to-Infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24, 133–145.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish DN, and Chow AT (1997). The clinical pharmacokinetics of levofloxacin. Clin. Pharmacokinet. 32, 101–119. [DOI] [PubMed] [Google Scholar]
- Forsberg KJ, Patel S, Wencewicz TA, and Dantas G (2015). The tetracycline destructases: a novel family of tetracycline-inactivating enzymes. Chem. Biol. 22, 888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Mciver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, and Huttenhower C (2018). Species-level functional profiling of metagenomes and meta-transcriptomes. Nat. Methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway-Peña JR, Smith DP, Sahasrabhojane P, Wadsworth WD, Fellman BM, Ajami NJ, Shpall EJ, Daver N, Guindani M, Petrosino JF, et al. (2017). Characterization of oral and gut microbiome temporal variability in hospitalized cancer patients. Genome Med. 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García N, Gutiérrez G, Lorenzo M, García JE, Píriz S, and Quesada A (2008). Genetic determinants for cfxA expression in Bacteroides strains isolated from human infections. J. Antimicrob. Chemother. 62, 942–947. [DOI] [PubMed] [Google Scholar]
- Gasparrini AJ, Crofts TS, Gibson MK, Tarr PI, Warner BB, and Dantas G (2016). Antibiotic perturbation of the preterm infant gut microbiome and resistome. Gut microbes 7, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparrini AJ, Wang B, Sun X, Kennedy EA, Hernandez-Leyva A, Ndao IM, Tarr PI, Warner BB, and Dantas G (2019). Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol. 4, 2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershuni VM, and Friedman ES (2019). The microbiome-host interaction as a potential driver of anastomotic leak. Curr. Gastroenterol. Rep. 21, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale I, O’neill PM, Berry NG, Odom A, and Sharma R (2012). The MEP pathway and the development of inhibitors as potential anti-infective agents. MedChemComm 3, 418–433. [Google Scholar]
- Ho J, Tumkaya T, Aryal S, Choi H, and Claridge-Chang A (2019). Moving beyond P S: data analysis with estimation graphics. Nat. Methods 16, 565–566. [DOI] [PubMed] [Google Scholar]
- Huse SM, Ye Y, Zhou Y, and Fodor AA (2012). A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One 7, e34242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby GA (2009). AmpC beta-lactamases. Clin. Microbiol. Rev. 22, 161–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernberg C, Löfmark S, Edlund C, and Jansson JK (2007). Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 1, 56–66. [DOI] [PubMed] [Google Scholar]
- Jia W, Li H, Zhao L, and Nicholson JK (2008). Gut microbiota: a potential new territory for drug targeting. Nat. Rev. Drug Discov. 7, 123–129. [DOI] [PubMed] [Google Scholar]
- Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, and Huttenhower C (2015). High-specificity targeted functional profiling in microbial communities with ShortBRED. PLoS Comput. Biol. 11, e1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazimierczak KA, Rincon MT, Patterson AJ, Martin JC, Young P, Flint HJ, and Scott KP (2008). A new tetracycline efflux gene, tet(40), is located in tandem with tet(O/32/O) in a human gut firmicute bacterium and in metagenomic library clones. Antimicrob. Agents Chemother. 52, 4001–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollef MH, and Fraser VJ (2001). Antibiotic resistance in the intensive care unit. Ann. Intern. Med. 134, 298–314. [DOI] [PubMed] [Google Scholar]
- Korpela K, Salonen A, Virta LJ, Kekkonen RA, Forslund K, Bork P, and De Vos WM (2016). Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat. Commun. 7, 10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubitz D, Typpo K, Midura-Kiela M, Brown C, Barberán A, Ghishan FK, and Kiela PR (2021). Dynamics of gut microbiota recovery after antibiotic exposure in young and old mice (A pilot study). Microorganisms 9, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett RM, Alcon-Giner C, Heavens D, Caim S, Brook TC, Kujawska M, Martin S, Peel N, Acford-Palmer H, Hoyles L, et al. (2020). Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat. Microbiol. 5, 430–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Si H, Du H, Guo H, Dai H, Xu S, and Wan J (2021). Comparison of gut microbiota structure and Actinobacteria abundances in healthy young adults and elderly subjects: a pilot study. BMC Microbiol. 21, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Wang H, Shi Q, Wang N, Zhang Z, Xiong C, Liu J, Chen Y, Jiang L, and Jiang Q (2017). Effects of oral florfenicol and azithromycin on gut microbiota and adipogenesis in mice. PLoS One 12, e0181690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macvane SH (2016). Antimicrobial resistance in the intensive care unit: a focus on gram-negative bacterial infections. J. Intensive Care Med. 32, 25–37. [DOI] [PubMed] [Google Scholar]
- Mallick H, Rahnavard A, Mciver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, et al. (2021). Multivariable association discovery in population-scale meta-omics studies. Preprint at bio-Rxiv 17, e1009442. 10.1371/journal.pcbi.1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell GD, Dean NC, Dowell SF, File TM Jr., Musher DM, Niederman MS, et al. (2007). Infectious diseases society of America/American thoracic society consensus guidelines on the management of community-acquired pneumonia in adults. Clin. Infect. Dis. 44, S27–S72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangin I, Lévêque C, Magne F, Suau A, and Pochart P (2012). Long-term changes in human colonic Bifidobacterium populations induced by a 5-day oral amoxicillin-clavulanic acid treatment. PLoS One 7, e50257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manor O, Dai CL, Kornilov SA, Smith B, Price ND, Lovejoy JC, Gibbons SM, and Magis AT (2020). Health and disease markers correlate with gut microbiome composition across thousands of people. Nat. Commun. 11, 5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcdonald D, Ackermann G, Khailova L, Baird C, Heyland D, Kozar R, Lemieux M, Derenski K, King J, Vis-Kampen C, et al. (2016). Extreme dysbiosis of the microbiome in critical illness. mSphere 1, e00199–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcdonald JE, Marchesi JR, and Koskella B (2020). Application of ecological and evolutionary theory to microbiome community dynamics across systems. Proc. R. Soc. B: Biol. Sci. 287, 20202886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng M, Klingensmith NJ, and Coopersmith CM (2017). New insights into the gut as the driver of critical illness and organ failure. Curr. Opin. Crit. Care 23, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C, Lugli GA, Duranti S, Turroni F, Bottacini F, Mangifesta M, Sanchez B, Viappiani A, Mancabelli L, Taminiau B, et al. (2014). Genomic encyclopedia of type strains of the genus Bifidobacterium. Appl. Environ. Microbiol. 80, 6290–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya A, and Ferrer M (2016). Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol. 24, 402–413. [DOI] [PubMed] [Google Scholar]
- Murtagh F, and Legendre P (2014). Ward’s hierarchical agglomerative clustering method: which algorithms implement ward’s criterion? J. Classif. 31, 274–295. [Google Scholar]
- Nguyen TLA, Vieira-Silva S, Liston A, and Raes J (2015). How informative is the mouse for human gut microbiota research? Dis. Models Mech 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou KC, and Rigol S (2018). A brief history of antibiotics and select advances in their synthesis. J. Antibiot. 71, 153–184. [DOI] [PubMed] [Google Scholar]
- Ojima M, Motooka D, Shimizu K, Gotoh K, Shintani A, Yoshiya K, Nakamura S, Ogura H, Iida T, and Shimazu T (2016). Metagenomic analysis reveals dynamic changes of whole gut microbiota in the acute phase of intensive care unit patients. Dig. Dis. Sci. 61, 1628–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palleja A, Mikkelsen KH, Forslund SK, Kashani A, Allin KH, Nielsen T, Hansen TH, Liang S, Feng Q, Zhang C, et al. (2018). Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 3, 1255–1265. [DOI] [PubMed] [Google Scholar]
- Palm NW, De Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JC, and Im S-H (2020). Of men in mice: the development and application of a humanized gnotobiotic mouse model for microbiome therapeutics. Exp. Mol. Med. 52, 1383–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitout JDD (2008). Multiresistant Enterobacteriaceae: new threat of an old problem. Expert Rev. Anti Infect. Ther. 6, 657–669. [DOI] [PubMed] [Google Scholar]
- Rampelli S, Soverini M, D’amico F, Barone M, Tavella T, Monti D, Capri M, Astolfi A, Brigidi P, Biagi E, et al. (2020). Shotgun metagenomics of gut microbiota in humans with up to extreme longevity and the increasing role of xenobiotic degradation. mSystems 5, e00124–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond F, Ouameur AA, Déraspe M, Iqbal N, Gingras H, Dridi B, Leprohon P, Plante P-L, Giroux R, Bérubé È, et al. (2016). The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 10, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro Da Cunha B, Fonseca LP, and Calado CRC (2019). Antibiotic discovery: where have we come from, where do we go? Antibiotics (Basel) 8, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan PD (2019). Microbial evolution and ecological opportunity in the gut environment. Proc. Biol. Sci. 286, 20191964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, and Edwards R (2011). Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6, e17288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, and Huttenhower C (2012). Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9, 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirov I, Tam NM, Jogova M, Robertson ML, Li Y, Lupp C, and Finlay BB (2008). Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect. Immun. 76, 4726–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty SA, Hugenholtz F, Lahti L, Smidt H, and De Vos WM (2017). Intestinal microbiome landscaping: insight in community assemblage and implications for microbial modulation strategies. FEMS Microbiol. Rev. 41, 182–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields-Cutler RR, Al-Ghalith GA, Yassour M, and Knights D (2018). SplinectomeR enables group comparisons in longitudinal microbiome studies. Front. Microbiol. 9, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K, Ogura H, Hamasaki T, Goto M, Tasaki O, Asahara T, Nomoto K, Morotomi M, Matsushima A, Kuwagata Y, and Sugimoto H (2011). Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig. Dis. Sci. 56, 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, and Fridkin S (2013). Antimicrobial-resistant pathogens associated with healthcare-associated infections summary of data reported to the national Healthcare safety network at the Centers for disease control and prevention, 2009–2010. Infect. Control. Hosp. Epidemiol. 34, 1–14. [DOI] [PubMed] [Google Scholar]
- Simpson GG (1984). Tempo and Mode in Evolution (Columbia University Press; ). [Google Scholar]
- Stevens V, Dumyati G, Fine LS, Fisher SG, and Van Wijngaarden E (2011). Cumulative antibiotic exposures over time and the risk of Clostridium difficile infection. Clin. Infect. Dis. 53, 42–48. [DOI] [PubMed] [Google Scholar]
- Theriot CM, Koenigsknecht MJ, Carlson PE Jr., Hatton GE, Nelson AM, Li B, Huffnagle GB, Z Li J, and Young VB (2014). Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 5, 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, and Gordon JI (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 1, 6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, Van Den Brink MRM, Kamboj M, and Pamer EG (2010). Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J. Clin. Invest. 120, 4332–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellborn GA, and Langerhans RB (2015). Ecological opportunity and the adaptive diversification of lineages. Ecol. Evol. 5, 176–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmanski T, Diener C, Rappaport N, Patwardhan S, Wiedrick J, Lapidus J, Earls JC, Zimmer A, Glusman G, Robinson M, et al. (2021). Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat. Metab. 3, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipperman MF, Fitzgerald DW, Juste MAJ, Taur Y, Namasivayam S, Sher A, Bean JM, Bucci V, and Glickman MS (2017). Antibiotic treatment for Tuberculosis induces a profound dysbiosis of the microbiome that persists long after therapy is completed. Sci. Rep. 7, 10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodmansey EJ (2007). Intestinal bacteria and ageing. J. Appl. Microbiol. 102, 1178–1186. [DOI] [PubMed] [Google Scholar]
- Yassour M, Vatanen T, Siljander H, Hämäläinen A-M, Härkönen T, Ryhänen SJ, Franzosa EA, Vlamakis H, Huttenhower C, Gevers D, et al. (2016). Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 8, 343ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaura E, Brandt BW, Teixeira De Mattos MJ, Buijs MJ, Caspers MPM, Rashid M-U, Weintraub A, Nord CE, Savell A, Hu Y, et al. (2015). Same exposure but two radically different responses to antibiotics: resilience of the salivary microbiome versus long-term microbial shifts in feces. mBio 6, e01693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wu J, Li JV, Zhou N-Y, Tang H, and Wang Y (2013). Gut microbiota composition modifies fecal metabolic profiles in mice. J. Proteome Res. 12, 2987–2999. [DOI] [PubMed] [Google Scholar]
- Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, Zhang MJ, Rao V, Avina M, Mishra T, et al. (2019). Longitudinal multiomics of host–microbe dynamics in prediabetes. Nature 569, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilhao R, Papadopoulou B, and Courvalin P (1988). Occurrence of the Campylobacter resistance gene tetO in Enterococcus and Streptococcus spp. Antimicrob. Agents Chemother. 32, 1793–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinglé C, Kuntz L, Tritsch D, Grosdemange-Billiard C, and Rohmer M (2010). Isoprenoid biosynthesis via the methylerythritol phosphate pathway: structural variations around phosphonate anchor and spacer of fosmidomycin, a potent inhibitor of deoxyxylulose phosphate reductoisomerase. J. Org. Chem. 75, 3203–3207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data is stored as short-read metagenomic sequences in the Sequence Read Archive (SRA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Biological samples | ||
|
| ||
| Healthy Volunteer fecal samples | this paper | none |
|
| ||
| Deposited data | ||
|
| ||
| Short read metagenomic DNA from Microbiome Samples – Healthy Volunteer Data | Sequence Read Archive (SRA) | bioproject: PRJNA664754 |
| Short read metagenomic DNA from Microbiome Samples – ICU comparator group | Sequence Read Archive (SRA) | bioproject: PRJNA703034 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Blood Agar Plate, 5% Sheep Blood in Tryptic Soy Agar (TSA) Base, 15 × 100mm plate | Hardy Diagnostics | Cat no. A10 |
| BBE Agar, Bacteroides Bile Esculin, reducible, for anaerobes, 15 × 100mm plate | Hardy Diagnostics | Cat no. G05 |
| MoBio (now Qiagen) DNeasy PowerSoil Pro Kit | Qiagen | Cat no. 47016 |
| Nextera library prep | Illumina | Cat no. 20018704 |
| Agencourt AMPure XP system | Beckman Coulter | Cat no. A63880 |
| Quant-it PicoGreen dsDNA assay | Invitrogen | Cat no. P11496 |
|
| ||
| Software and algorithms | ||
|
| ||
| The R Project for Statistical Computing | https://www.r-project.org/ | Version 4.0 |
| MetaPhlAn | Segata et al. (2012) | Version 2; RRID:SCR_004915 |
| HUMAnN | Franzosa et al. (2018) | Version 2; RRID:SCR_014620 |
| ShortBRED | Kaminski et al. (2015) | Version 2 |
| LEfSe | Segata et al. (2011) | Version 1; RRID:SCR_014609 |
| MaAsLin | Mallick et al. (2021) | Version 2 |
| Zibr | Chen and Li (2016) | Version 0.1 |
| Dabestr | Ho et al. (2019) | Version 0.2.5 |