

Abstract
A facile, highly efficient, and greener method for the synthesis of new 1,4-disubstituted-1,2,3-triazoles was conducted using [Et3NH][OAc] as a medium by the implementation of ultrasound irradiation via click chemistry, affording excellent yields. The present synthetic method exhibited numerous advantages such as mild reaction conditions, excellent product yields, minimal chemical waste, operational simplicity, shorter reaction time, and a wide range of substrate scope. The synthesized compounds were further evaluated for in vitro antifungal activity against five fungal strains, and some of the compounds displayed equivalent or greater potency than the standard drug. A molecular docking study against the modelled three-dimensional structure of cytochrome P450 lanosterol 14α-demethylase was also performed to understand the binding affinity and binding interactions of the enzyme. Furthermore, the synthesized compounds were evaluated for DPPH radical scavenging activity and antitubercular activity against Mycobacterium tuberculosis H37Rv strain.
A facile, highly efficient, and greener method for the synthesis of new 1,4-disubstituted-1,2,3-triazoles was conducted using [Et3NH][OAc] as a medium by the implementation of ultrasound irradiation via click chemistry, affording excellent yields.
1. Introduction
1,2,3-Triazole, a five-membered heterocyclic ring system, is a very well-known biologically active pharmacophore constructed by the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction, which is popular as a click chemistry reaction.1 Over the last decade, 1,2,3-triazole has become one of the key structural motifs and is used in numerous fields including polymer chemistry,2 material science,3 and drug discovery.4 1,2,3-triazole-based molecules display various biological activities such as anti-fungal,5,6 anti-bacterial,6 anti-tubercular,7 anti-inflammatory,8 anti-allergic,9 anti-HIV,10 anti-cancer,11 and anti-phytopathogenic.12 Some marketed drugs have the 1,2,3-triazole unit in their structure, and these include Cefatrizine (a broad-spectrum antibiotic), Tazobactam (an antibiotic), and Carboxyamidotriazole (CAI) (a calcium channel blocker) (Fig. 1).
Fig. 1. Structures of drugs containing the 1,2,3-triazole unit.

Azole drugs have broad-spectrum activities against most yeasts and filamentous fungi and are mostly used in antifungal chemotherapy.13 Some well-known antifungal agents including fluconazole, voriconazole, ravuconazole, and itraconazole contain a 1,2,4-triazole ring in their structure, as shown in Fig. 2. However, their clinical uses have been restricted by their relatively high risk of toxicity, pharmacokinetic deficiencies, emergence of drug resistance, and undesirable side effects. These antifungal drugs inhibit CYP51, a key enzyme in the biosynthesis of ergosterol, through a mechanism in which the antifungal agent having a triazole scaffold is positioned perpendicular to the porphyrin plane with a ring nitrogen atom (N-4 of triazole) coordinated with a heme iron atom.14 Over a couple of decades, the incidence of systemic fungal infections has increased due to cancer chemotherapy, organ transplantation, tuberculosis, and immunodeficiency virus (HIV) infection.15 However, the extensive use of antifungal drugs has led to an increase in the resistance to these drugs.16 Hence, there is an urgent need for developing new antifungal agents with effective activities, low toxicity, and minimum side effects.
Fig. 2. 1,2,4-Triazole-based marketed antifungal drugs.

Ionic liquids (ILs) are environment-friendly solvents because of their interesting properties and they can be used as alternatives to harmful organic solvents. Furthermore, they are useful in catalytic reactions17 and organic synthesis18 because of their unique properties, making them superior media for increasing selectivity, reactivity, and recyclability. Various ionic liquids have been significantly used in heterocyclic synthesis as solvents or catalysts.19 The copper-catalyzed 1,3-dipolar cycloaddition of organic azides and terminal alkynes has been reported by using ionic liquids such as [bmim][BF4],20a,b [Bmim]OH,20c (SNIL-Cu(ii)),20d ([Hmim]TFA),20e and [C8dabco][N(CN)2],20f and DBU-based ionic liquids.20g The ionic liquid [Et3NH][OAc] has been used extensively in various organic transformations.21
Considering the importance of ionic liquids in organic synthesis and in continuation of our work7d,22 as well as for the development of new synthetic methodologies using ionic liquids,23 herein, we reported the efficient synthesis of new 1,4-disubstituted-1,2,3-triazoles via the click chemistry approach using [Et3NH][OAc] as the medium under ultrasonic irradiation. Furthermore, the newly synthesized compounds were investigated for their antifungal, antioxidant, and antitubercular activities.
2. Results and discussion
2.1. Chemistry
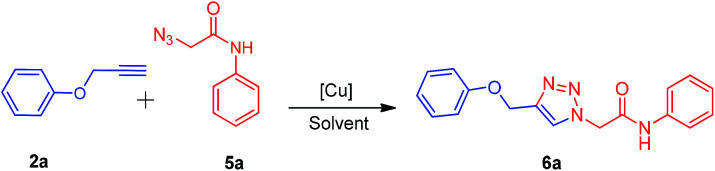
There are several reports on the synthesis of 1,4-disubstituted-1,2,3-triazoles bearing amide functionality and displaying various biological activities;24 recently, Ferroni et al. developed triazoles as non-steroidal anti-androgens for prostate cancer treatment.25 On the basis of these findings, we designed small 1,2,3-triazoles with amide linkage in their structures. Initially, the starting materials, i.e., (prop-2-yn-1-yloxy)benzenes 2a–c were prepared by a previously reported method.7d 2-Azido-N-phenylacetamides 5a–g were synthesized from their corresponding anilines via chloroacetylation using chloroacetyl chloride, followed by nucleophilic substitution with sodium azide in excellent yields (Scheme 1).
Scheme 1. Synthesis of alkynes 2a–c and 2-azido-N-phenylacetamides 5a–g. Reagents and conditions: (a) propargyl bromide, K2CO3, DMF, 2 h, 90–95%; (b) chloroacetyl chloride, NEt3, CH2Cl2, 0 °C to rt, 3–5 h, 85–95%; (c) NaN3, toluene, reflux, 5–7 h, 88–96%.

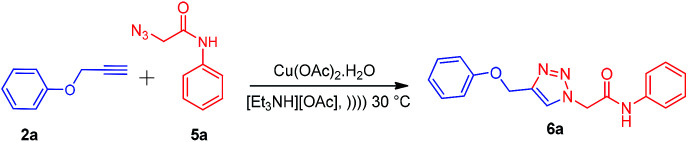
After the synthesis of alkynes 2a–c and azides 5a–g, we considered the model substrates (prop-2-yn-1-yloxy)benzenes 2a (1.0 mmol) and 2-azido-N-phenylacetamide 5a (1.0 mmol) for the click reaction (Scheme 2).
Scheme 2. Standard model reaction.

The literature survey reveals the use of t-BuOH–H2O, THF–H2O, and DMF–H2O as solvents in combination with copper salts for the click reaction. Accordingly, we performed the model reactions of 2a and 5a by using CuSO4·5H2O, Cu(OAc)2·H2O, CuI, and CuCl with or without sodium ascorbate as a reducing agent in solvents such as t-BuOH and THF (Table 1). In our initial attempt, the model reaction was carried out by using the conventional method, i.e., the reaction mixture was stirred at room temperature for 10 h using various copper salts in t-BuOH or THF as a solvent (Table 1, entries 2–7). The product 2-(4-(phenoxymethyl)-1H-1,2,3-triazol-1-yl)-N-phenylacetamide 6a was obtained in 28–72% yield. Furthermore, the model reaction was carried out using copper salts and t-BuOH/THF as a solvent under ultrasound irradiation (40 kHz, 30 °C) for 7 h, which resulted in the product 6a in 32–83% yield (Table 1, entries 2–7). These results indicate a longer reaction time with a lower yield of the product 6a.
Optimization of the reaction conditions for the synthesis of 6a.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Copper salt | Solvent/medium | Conventional method | US method | ||
| Timea | Yieldb (%) | Timea | Yieldb (%) | |||
| 1 | — | t-BuOH : H2O | 10 h | 0 | 6 h | 0 |
| 2 | CuSO4·5H2O | t-BuOH : H2O | 10 h | 28 | 6 h | 32 |
| 3 | CuSO4·5H2O + Na ascorbate | t-BuOH : H2O | 10 h | 70 | 6 h | 78 |
| 4 | Cu(OAc)2·H2O | t-BuOH : H2O | 10 h | 72 | 6 h | 82 |
| 5 | Cu(OAc)2·H2O + Na ascorbate | t-BuOH : H2O | 10 h | 72 | 6 h | 83 |
| 6 | CuI | THF | 10 h | 70 | 6 h | 80 |
| 7 | CuCl | THF | 10 h | 60 | 6 h | 65 |
| 8 | — | [Et3NH][OAc] | 10 h | 0 | 6 h | 0 |
| 9 | Cu(OAc)2·H2O | [Et3NH][OAc] | 7 h | 90 | 35 min | 95 |
| 10 | Cu(OAc)2·H2O + Na ascorbate | [Et3NH][OAc] | 7 h | 90 | 35 min | 95 |
| 11 | CuSO4·5H2O + Na ascorbate | [Et3NH][OAc] | 7 h | 81 | 35 min | 84 |
| 12 | Cu(OAc)2·H2O | [Et3NH][Cl] | 7 h | Trace | 35 min | 10 |
| 13 | Cu(OAc)2·H2O | [Et3NH][HSO4] | 7 h | Trace | 35 min | 12 |
| 14 | Cu(OAc)2·H2O | [HDBU][OAc] | 7 h | 84 | 45 min | 88 |
| 15 | CuSO4·5H2O + Na ascorbate | [HDBU][OAc] | 7 h | 81 | 45 min | 84 |
| 16 | Cu(OAc)2·H2O | [HDBU][HSO4] | 7 h | 10 | 45 min | 14 |
Reaction conditions: 2a (1 mmol), 5a (1 mmol), and 10 mol% [Cu], 1 mL solvent, 30 °C; reaction progress monitored by TLC.
Isolated yield, US: ultrasonication.
Ionic liquids have unique properties as solvents and as media; herein, we attempted to use various ionic liquids for the synthesis of new amide-linked 1,4-disubstituted-1,2,3-triazole 6avia the click chemistry approach. Freshly synthesized appropriate ionic liquids such as [Et3NH][OAc], [Et3NH][Cl], [Et3NH][HSO4], [HDBU][OAc], and [HDBU][HSO4] in combination with copper salts such as CuSO4·5H2O and Cu(OAc)2·H2O were used along with sodium ascorbate as a reducing agent to carry out the model reaction (Table 1, entries 9–16) at room temperature for 7 h. The maximum yield (90%) of the product 6a was obtained by using Cu(OAc)2·H2O as the catalyst and [Et3NH][OAc] as the solvent (Table 1, entries 9 and 10). In our further attempt, the model reaction was carried out under ultrasound irradiation for appropriate times (35 and 45 min) (Table 1, entries 9–16). The product 6a was obtained in 95% yield under ultrasound irradiation using Cu(OAc)2·H2O as the catalyst and [Et3NH][OAc] as the solvent (Table 1, entries 9 and 10). In short, it was observed from the optimization of the best reaction conditions that the Cu(OAc)2·H2O-catalyzed click reaction in [Et3NH][OAc] ionic liquid under ultrasound irradiation at 30 °C efficiently gives the desired 1,4-disubstituted-1,2,3-triazole derivative 6a as compared to the conventional method with respect to the reaction time and yield (Table 1, entry 9).
In order to evaluate the amount of the Cu(OAc)2·H2O catalyst for the model reaction for different concentrations such as 5, 10, 15, and 20 mol% (Table 2, entries 1–4), the reaction was carried out in the presence of 1 mL of [Et3NH][OAc] as a solvent at 30 °C under ultrasound irradiation. It was observed that 10 mol% of Cu(OAc)2·H2O was sufficient to achieve an excellent yield of the product 6a in the shortest reaction time (Table 2, entry 2), while an excess amount of the catalyst (up to 20 mol%) did not increase the yield of the product. The above results reveal that 10 mol% of Cu(OAc)2·H2O in [Et3NH][OAc] is the sufficient catalyst amount in terms of an excellent yield and short reaction time (Table 2, entry 2).
The influence of the amount of Cu(OAc)2·H2O on the synthesis of 6a.
| |||
|---|---|---|---|
| Entry | mol% | Time (min) | Isolated yield (%) |
| 1 | 5 | 55 | 70 |
| 2 | 10 | 35 | 95 |
| 3 | 15 | 35 | 95 |
| 4 | 20 | 35 | 95 |
The recyclability of the [Et3NH][OAc] solvent was also explored for the model reaction. The reaction was conducted for four successive cycles on the same recycled solvent. After completion of the first reaction with 95% yield, the reaction mass was poured into ice cold water to get the solid product, which was isolated by filtration. The filtrate was subjected to evaporation of water to get a viscous liquid, which on cooling gave the ionic liquid. Then, the recovered [Et3NH][OAc] was reused for four more successive cycles without significant loss in the yield of the product 6a. The yields achieved were 94%, 92%, 91%, and 88% after the successful runs (Fig. 3). The result demonstrates the high stability of the ionic liquid [Et3NH][OAc] under the reaction conditions, which could be recycled and reused without a significant loss in the product yield.
Fig. 3. Reusability of [Et3NH][OAc].

In a further attempt, we also examined a one-pot reaction for the selected model reaction with 2a (1.0 mmol), 4a (1.0 mmol), and sodium azide (1.5 mmol) for the synthesis of 6a, which was optimized for different copper sources (Table 3) for the one-pot, three-component click reaction in [Et3NH][OAc] as the solvent, thus producing 2-(4-(phenoxymethyl)-1H-1,2,3-triazol-1-yl)-N-phenylacetamide 6a (Scheme 3). The optimization result of this one-pot model reaction revealed that 15 mol% of Cu(OAc)2·H2O in [Et3NH][OAc] as the solvent was a sufficient catalyst loading amount to obtain an excellent yield. However, it required a longer reaction time and higher concentration of the Cu catalyst (Table 3, entry 2) than the two-component reaction, as discussed earlier.
The effect of the amount of copper salts for the one-pot synthesis of 6a.
| Entry | Catalyst | mol% | Time (h) | Isolated yield (%) |
|---|---|---|---|---|
| 1 | Cu(OAc)2·H2O | 10 | 17 | 90 |
| 2 | Cu(OAc)2·H2O | 15 | 16 | 95 |
| 3 | Cu(OAc)2·H2O | 20 | 16 | 95 |
| 4 | CuSO4·5H2O + Na ascorbate | 10 | 18 | 81 |
| 5 | CuSO4·5H2O + Na ascorbate | 15 | 17 | 87 |
| 6 | CuI | 10 | 18 | 50 |
| 7 | CuI | 15 | 17 | 61 |
| 8 | CuCl | 10 | 17 | 40 |
| 9 | CuCl | 15 | 17 | 45 |
Scheme 3. Synthesis of 1,4-disubstituted-1,2,3-triazole 6avia the one-pot three-component click reaction.

After optimization of the reaction parameters and to define the present methodology, as discussed earlier, the reaction was assessed with a variety of substituted alkynes 2a–c and azides 5a–g in the presence of Cu(OAc)2·H2O in [Et3NH][OAc] under ultrasound irradiation. The reaction conditions were favorable for the electron-donating and electron-withdrawing groups on alkynes 2a–c and azides 5a–g, resulting in the corresponding 1,4-disubstituted-1,2,3-triazole derivatives 6a–g, 7a–g, and 8a–g in excellent yields (Scheme 4).
Scheme 4. Synthesis of 1,4-disubstituted-1,2,3-triazole derivatives 6a–g, 7a–g, and 8a–g.

All the synthesized compounds, namely, 6a–g, 7a–g, and 8a–g were confirmed by physical data and spectral analysis.
2.2. Biological activity
2.2.1. Antifungal activity
All the newly synthesized compounds were screened for their in vitro antifungal activities against five different fungal strains, namely, Candida albicans, Fusarium oxysporum, Aspergillus flavus, Aspergillus niger, and Cryptococcus neoformans. The minimum inhibitory concentration (MIC, μg mL−1) values of all the newly synthesized compounds were determined by the standard agar dilution method as per the CLSI (formerly, NCCLS) guidelines26 (Table 4). DMSO was used as the negative control and Miconazole was used as the standard antifungal drug for the comparison of antifungal activities. Among the series, the triazole derivatives 6 and 7 derived from alkynes 2a and 2b (particularly from phenol and 4-methoxyphenol) did not show any activity against all the fungal strains. However, the triazoles derived from alkyne 2c (from 4-nitrophenol) displayed excellent antifungal activity against all the strains. The compounds 8b, 8c, and 8f having MIC of 12.5 μg mL−1 were found to be more potent; compounds 8a, 8d, and 8g having MIC of 25 μg mL−1 were found to be equipotent as compared to the standard drug Miconazole against the fungal strain Candida albicans. The compounds 8b, 8c, 8d, 8e, and 8g with MIC of 25 μg mL−1 were found to be equivalent to the standard drug Miconazole and 8f with MIC of 12.5 μg mL−1 was more potent than Miconazole against the fungal strain F. oxysporum. Compounds 8e and 8g with MIC of 12.5 μg mL−1 exhibited equivalent activities as compared to Miconazole against A. flavus. For the fungal strain A. niger, the compounds 8b, 8d, 8e, and 8g with MIC of 25 μg mL−1 displayed equivalent antifungal activities as compared to Miconazole. However, 8f having MIC of 12.5 μg mL−1 was more active than Miconazole. For the fungal strain C. neoformans, the compounds 8e, 8f, and 8g with MIC of 12.5 μg mL−1 were more potent than the standard drug and 8c with MIC of 25 μg mL−1 was equivalent to Miconazole.
In vitro biological evaluation of the synthesized compounds 6a–g, 7a–g, and 8a–g MIC (μg mL−1)a.
| Entry | Antifungal activity | Anti TB MTB H37Rv | DPPH IC50 (μg mL−1) | ||||
|---|---|---|---|---|---|---|---|
| CA | FO | AF | AN | CN | |||
| 6a | 87.5 | 100 | 175 | 125 | 187.5 | 25 | 55.37 |
| 6b | 150 | 125 | 150 | 150 | * | >25 | 26.64 |
| 6c | 175 | 200 | 150 | 175 | 200 | 25 | 20.54 |
| 6d | 187.5 | 162.5 | 187.5 | 200 | * | >25 | * |
| 6e | 200 | * | * | 187.5 | 175 | >25 | 29.64 |
| 6f | 150 | 175 | 162.5 | 200 | * | 25 | 74.28 |
| 6g | 162.5 | 175 | 175 | 187.5 | 150 | >25 | 74.41 |
| 7a | 137.5 | 125 | 100 | 137.5 | 125 | >25 | 11.44 |
| 7b | 175 | 200 | 100 | 175 | 200 | >25 | 16.79 |
| 7c | * | * | 200 | * | 175 | >25 | 12.30 |
| 7d | 200 | 175 | 150 | 150 | 200 | >25 | 19.46 |
| 7e | 175 | 175 | 200 | * | * | >25 | 70.33 |
| 7f | 100 | 150 | 100 | 100 | 75 | >25 | * |
| 7g | 75 | 100 | 87.5 | 100 | 200 | >25 | 49.60 |
| 8a | 25 | 37.5 | 25 | 75 | 75 | >25 | 97.29 |
| 8b | 12.5 | 25 | 25 | 25 | 37.5 | >25 | * |
| 8c | 12.5 | 25 | 25 | 50 | 25 | >25 | 48.02 |
| 8d | 25 | 25 | 37.5 | 25 | 50 | >25 | 22.17 |
| 8e | 50 | 25 | 12.5 | 25 | 12.5 | >25 | 62.01 |
| 8f | 12.5 | 12.5 | 25 | 12.5 | 12.5 | >25 | * |
| 8g | 25 | 25 | 12.5 | 25 | 12.5 | >25 | 16.99 |
| MA | 25 | 25 | 12.5 | 25 | 25 | NA | NA |
| INH | NA | NA | NA | NA | NA | 0.1 | NA |
| RIF | NA | NA | NA | NA | NA | 0.2 | NA |
| EMB | NA | NA | NA | NA | NA | 1.56 | NA |
| CIP | NA | NA | NA | NA | NA | 1.56 | NA |
| BHT | NA | NA | NA | NA | NA | NA | 16.47 |
CA: Candida albicans; FO: Fusarium oxysporum; AF: Aspergillus flavus; AN: Aspergillus niger and CN: Cryptococcus neoformans. MA: Miconazole, DPPH: 2,2-diphenyl-1-picrylhydrazyl; INH: Isoniazid; RIF: Rifampicin; EMB: Ethambutol; CIP: Ciprofloxacin; BHT: butylated hydroxytoluene; *no activity was observed, NA: not applicable.
The remaining compounds displayed moderate to low antifungal activities against all the tested fungal strains. The antifungal activity depends on the various substituents present on phenyl rings. Among the series, the compounds with nitro groups (8a–g) were the most active against all the tested fungal strains. Compounds 8f and 8g were the most active in the series as they possessed a nitro group at R and chloro groups at R2 and R3; the chloro group at the R1 position for 8e resulted in less activity against one fungal strain. The methyl group at the R1/R2/R3 position and the nitro group at the R position in 8b, 8c, and 8d resulted in good activity than that of the others in the series.
2.2.2. Antitubercular activity
All the newly synthesized 1,4-disubstituted-1,2,3-triazoles (6a–g, 7a–g, and 8a–g) were screened for their in vitro antitubercular activities against the MTB H37Rv (ATCC 27294) strain using the microplate Alamar Blue assay method with the determination of MIC in triplicate; the results are mentioned in Table 4. The newly synthesized 1,2,3-triazoles exhibited MICs ranging from 25 to ≥25 μg mL−1. Among the series, compounds 6a, 6c, and 6f exhibited good antitubercular activities with MIC of 25 μg mL−1 as compared to other compounds.
2.2.3. Antioxidant activity
The antioxidant activities of the synthesized compounds 6a–g, 7a–g, and 8a–g were screened using a 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assay.27 The DPPH radical scavenging assay is the most commonly used procedure for screening the antioxidant activities of different natural and synthetic compounds. A lower IC50 value demonstrates more antioxidant activity. The IC50 (concentration required to scavenge 50% of the radicals) values were calculated to assess the potential antioxidant activities. Butylated hydroxytoluene (BHT) was used as the standard drug for the comparison of antioxidant activities; the observed results are summarized in Table 4. According to the DPPH assay, compounds 7a and 7c exhibited excellent radical scavenging activities with IC50 values of 11.44 and 12.30 μg mL−1, respectively, compared to the synthetic antioxidant BHT. Similarly, compounds 7b, 7d, and 8g having IC50 values of 16.79, 19.46, and 16.99 μg mL−1, respectively, showed comparable antioxidant activities with that of BHT; in contrast, the compounds 6b, 6c, 6e, and 8d showed moderate activities with IC50 values of 26.64, 20.54, 29.64, and 22.17 μg mL−1, respectively.
2.2.4. Molecular docking
The molecular docking study of all the synthesized triazole derivatives 6a–g, 7a–g, and 8a–g was performed against the modelled three-dimensional structure of cytochrome P450 lanosterol 14α-demethylase of C. albicans to understand the binding affinity and binding interactions of the enzyme and the synthesized derivatives using the SYBEL 2.1.1 package following the standard procedures.28,29 The molecular docking results are presented in Table 5.
Molecular docking score for the compounds 6a–g, 7a–g, and 8a–g.
| Molecular docking score | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Total score (−log Ki) | Crash score | Polar score | Entry | Total score (−log Ki) | Crash score | Polar score |
| 6a | 7.0377 | −0.6933 | 1.9115 | 7e | 6.4127 | −1.2494 | 1.4225 |
| 6b | 6.4564 | −2.9621 | 0.0004 | 7f | 7.2492 | −1.4724 | 2.5093 |
| 6c | 6.8229 | −0.7726 | 2.0682 | 7g | 7.2394 | −1.4033 | 3.0192 |
| 6d | 6.0468 | −1.4517 | 1.3586 | 8a | 7.0868 | −1.1801 | 3.6314 |
| 6e | 6.5873 | −0.9241 | 2.8329 | 8b | 7.5441 | −2.8665 | 0.9019 |
| 6f | 6.7343 | −0.8582 | 3.0543 | 8c | 7.4658 | −1.0993 | 0.7391 |
| 6g | 7.0332 | −1.0463 | 1.6968 | 8d | 7.3945 | −1.5222 | 1.9024 |
| 7a | 6.0045 | −0.7865 | 2.8105 | 8e | 7.3314 | −0.9292 | 0.7808 |
| 7b | 6.6907 | −0.9183 | 4.5244 | 8f | 7.9971 | −1.9946 | 0.0031 |
| 7c | 6.5748 | −2.7834 | 0.0090 | 8g | 7.5972 | −1.3842 | 3.0509 |
| 7d | 7.0239 | −1.2982 | 3.2802 | MA | 6.4895 | −1.4009 | 1.0311 |
The docking results indicated that the triazole scaffold of the synthesized derivatives 6a–g, 7a–g, and 8a–g was incorporated appropriately in the active site of CYP450 and through various kinds of non-bonded interactions such as H-bond interactions and π bond interactions with the active sites of the amino acid residues. Among all the synthesized derivatives, 8f (7.9971), 8g (7.597), and 8b (7.544) were the most active triazole derivatives when compared with the standard Miconazole drug (6.4895). The most active triazole derivative 8f (7.9971) contained hydrophilic as well as hydrophobic amino acids in the active sites such as isoleucine, valine, methionine, cysteine, leucine, and phenylalanine. The hydrophobic amino acids Cys506 and Ile507 formed conventional hydrogen bonds with the oxygen (–O) of the nitro group on the phenyl ring with the distances of 1.95 and 2.02 Å, respectively. Met336 contains non-polar side chain forms conventional hydrogen bond interaction with a hydrogen atom (–H) attached to the nitrogen atom and methylene group with distance 2.41 and 2.45 Å, respectively. The hydrophobic amino acids such as Val166, Ile167, Ala182, Leu186, Leu240, Leu316, and Ala185 interacted with the π electron cloud to form weak π-interactions such as π-alkyl and alkyl interactions with the chloro-phenyl (–Cl) ring; the π electron cloud with the phenyl ring and the π electron cloud of the triazole ring are shown in Fig. 4a. The second most active triazole derivative 8g (7.597) interacted with the active site of amino acids by forming non-covalent bonded interactions such as H-bond interaction and π bond interaction. The hydrophobic amino acid Ala501 contains a non-polar side chain that interacts with the oxygen (–O) atom of the nitro group to form a conventional hydrogen bond interaction with distance of 2.02 Å. The polar aromatic amino acid Tyr168 forms hydrogen bond interactions with the nitrogen atom of the triazole ring with a distance of 1.84 Å.
Fig. 4. Binding pose and molecular interactions in the active sites of C. albicans lanosterol 14α-demethylase: (a) 8f and (b) 8g.

The amino acid Gly500 formed a carbon–hydrogen bond with the oxygen (–O) of the nitro group on the phenyl ring with a distance of 2.39 Å. The hydrophobic amino acid Try154 with an aromatic ring interacted with the triazole π cloud to form π–π stacked interactions, while the amino acids Leu412, Met544, Met413, and His546 interacted with the π cloud of the chloro group on the phenyl ring (–Cl) and the phenyl ring to form weak π-alkyl and alkyl interactions, as shown in Fig. 4b.
3. Conclusion
In conclusion, we demonstrated [Et3NH][OAc]- and ultrasound-mediated synthesis of new N-phenylacetamide-incorporated 1,2,3-triazole derivatives 6(a–g), 7(a–g), and 8(a–g) in excellent yields via the click chemistry approach for the first time. This safer method reduced the use of harmful solvents and reaction time. We also studied the recovery and reusability of [Et3NH][OAc] as a medium. The synthesized triazole derivatives were evaluated for their antifungal, antioxidant, and antitubercular activities. Among the series, the compounds 8a, 8b, 8c, 8d, 8e, 8f, and 8g displayed excellent antifungal activities as compared to the standard drug Miconazole with lower MIC values.
The in silico molecular docking study supported the experimental results and demonstrated that the 1,2,3-triazole derivatives 8f and 8g are the most active and have the potential to inhibit the survival of fungal species. The predictions of the pharmacokinetic parameters suggest that the synthesized derivatives have the potential to have high oral drug bioavailability.
4. Experimental
4.1. General methods
All the solvents and reagents were purchased from commercial suppliers, namely, Spectrochem Pvt. Ltd., Avra, Sigma Aldrich, and Alfa Aesar and were used without further purification. The progress of each reaction was monitored by thin layer chromatography (TLC) using silica gel F254 precoated TLC aluminium sheets (Merck, Germany) and by locating the spots using a UV light as the visualizing agent or iodine vapours. The melting points were obtained by the open capillary method and were uncorrected. The IR spectra were recorded on a Bruker FT-IR spectrometer. The 1H NMR spectra were recorded on Bruker AC-200 MHz and Bruker Advance III 500 MHz NMR spectrometers. The 13C NMR spectra were recorded on Bruker AC-50 MHz and Bruker Advance III 125 MHz NMR in DMSO-d6, CDCl3 + DMSO-d6, and CDCl3 + CD3OD using tetramethylsilane (TMS) as the internal standard; the chemical shifts (δ) were recorded in parts per million (ppm). The splitting pattern abbreviations are designed as singlet (s), doublet (d), double doublet (dd), triplet (t), quartet (q), and multiplet (m). High-resolution mass spectra (HRMS) were recorded on an Agilent 6520 (QTOF) ESI-HRMS instrument and Agilent Technologies 1200 series electron spray ionization (ESI) was used for the LC-MS data.
4.2. General experimental procedure for the synthesis of N-phenylacetamide-incorporated 1,2,3-triazole derivatives (6a–g, 7a–g, and 8a–g)
The mixture of appropriate (prop-2-yn-1-yloxy) benzenes 2a–c (1 mmol), substituted 2-azido-N-phenylacetamide 5a–g (1 mmol) and Cu(OAc)2·H2O (10 mol%) in [NEt3H][OAc] (1 mL) was placed in a round bottom flask. The mixture was sonicated (40 kHz) at 30 °C for an appropriate time until the completion of the reaction. The progress of the reaction was monitored by TLC. After the completion of the reaction, the mixture was quenched with crushed ice. The obtained solid was filtered and washed with water. The crude solid was crystallized in ethanol to afford the corresponding pure product. The synthesized compounds 6a–g, 7a–g, and 8a–g were characterized by IR, 1H NMR, 13C NMR, and mass spectroscopy.
4.2.1. 2-(4-(Phenoxymethyl)-1H-1,2,3-triazol-1-yl)-N-phenylacetamide (6a)
The compound 6a was obtained via a 1,3-dipolar cycloaddition reaction between azide 5a and alkyne 2a in 35 min as a white solid; mp: 166–168 °C; FT-IR (cm−1): 3263 (N–H stretching), 1667 (C O stretching); 1H NMR (200 MHz, DMSO-d6, δ ppm): 5.17 (s, 2H, –NCH2CO–), 5.36 (s, 2H, –OCH2), 6.96 (t, J = 8.0 Hz, 1H, Ar–H), 7.08 (t, J = 8.0 Hz, 3H, Ar), 7.28–7.38 (m, 4H, Ar–H), 7.59 (d, J = 8.0 Hz, 2H, Ar–H), 8.26 (s, 1H, triazole), and 10.49 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6, δ ppm): 51.8, 60.5, 114.3, 118.9, 120.5, 123.5, 125.9, 128.6, 129.2, 138.0, 142.2, 157.7, and 163.8; HRMS calcd for C17H17N4O2 [M + H]+: 309.1346 and found 309.1340.
4.2.2. 2-(4-(Phenoxymethyl)-1H-1,2,3-triazol-1-yl)-N-(o-tolyl)acetamide (6b)
The compound 6b was obtained via a 1,3-dipolar cycloaddition reaction between azide 5b and alkyne 2a in 37 min as a white solid; mp: 184–186 °C; FT-IR (cm−1): 3271 (N–H stretching), 1665 (C O stretching); 1H NMR (200 MHz, DMSO-d6, δ ppm): 2.24 (s, 3H, –CH3), 5.17 (s, 2H, –NCH2CO–), 5.41 (s, 2H, –OCH2), 6.95 (t, J = 8.0 Hz, 1H, Ar–H), 7.05 (d, J = 8.0 Hz, 2H, Ar–H), 7.10–7.35 (m, 5H, Ar–H), 7.43 (d, J = 8.0 Hz, 1H, Ar–H), 8.26 (s, 1H, triazole), and 9.84 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6, δ ppm): 17.5, 51.3, 60.5, 114.3, 120.4, 124.8, 125.8, 129.1, 130.1, 131.2, 135.1, 142.2, 157.7, and 164.0; HRMS calcd for C18H19N4O2 [M + H]+: 323.1508 and found 323.1510.
4.2.3. 2-(4-(Phenoxymethyl)-1H-1,2,3-triazol-1-yl)-N-(m-tolyl)acetamide (6c)
The compound 6c was obtained via a 1,3-dipolar cycloaddition reaction between azide 5c and alkyne 2a in 38 min as a yellow solid; mp: 130–132 °C; FT-IR (cm−1): 3302 (N–H stretching), 1676 (C O stretching); 1H NMR (500 MHz, CDCl3 + DMSO-d6, δ ppm): 2.21 (s, 3H, –CH3), 5.10 (s, 2H, –NCH2CO–), 5.13 (s, 2H, –OCH2), 6.79–6.93 (m, 4H, Ar–H), 7.08 (d, J = 8.0 Hz 1H), 7.13–7.30 (m, 3H, Ar–H), 7.34 (s, 1H, Ar–H), 7.85 (s, 1H, triazole), and 9.70 (s, 1H, NH); 13C NMR (125 MHz, CDCl3 + DMSO-d6, δ ppm): 21.1, 52.7, 61.4, 114.3, 116.7, 120.2, 124.8, 124.9, 128.3, 129.1, 137.3, 138.4, 143.7, 157.8, and 163.0; LC-MS for C18H18N4O2: [M]+ 323.1.
4.2.4. 2-(4-(Phenoxymethyl)-1H-1,2,3-triazol-1-yl)-N-(p-tolyl)acetamide (6d)
The compound 6d was obtained via a 1,3-dipolar cycloaddition reaction between azide 5d and alkyne 2a in 36 min as a yellow solid; mp: 202–204 °C; FT-IR (cm−1): 3271 (N–H stretching), 1665 (C O stretching).
4.2.5. 2-(4-(Phenoxymethyl)-1H-1,2,3-triazol-1-yl)-N-(2-chlorophenyl)-acetamide (6e)
The compound 6e was obtained via a 1,3-dipolar cycloaddition reaction between azide 5e and alkyne 2a in 36 min as a white solid; mp: 178–180 °C; FT-IR (cm−1): 3267 (N–H stretching), 1671 (C O stretching); 1H NMR (500 MHz, CDCl3 + DMSO-d6, δ ppm): 5.19 (s, 2H, –NCH2CO–), 5.31 (s, 2H, –OCH2), 6.94 (d, J = 8.0 Hz, 3H, Ar–H), 7.05 (d, J = 8.0 Hz, 1H, Ar–H), 7.16–7.34 (m, 4H, Ar–H), 7.89 (s, 1H, triazole), 8.02 (d, J = 8.0 Hz, 1H, Ar–H), and 9.01 (s, 1H, NH); 13C NMR (125 MHz, CDCl3 + DMSO-d6, δ ppm): 52.8, 61.3, 114.3, 120.8, 123.1, 124.3, 125.5, 127.1, 129.0, 133.7, 141.9, 157.7, and 162.6. LC-MS for C17H15ClN4O2: [M]+ 343.
4.2.6. 2-(4-(Phenoxymethyl)-1H-1,2,3-triazol-1-yl)-N-(3-chlorophenyl)-acetamide (6f)
The compound 6f was obtained via a 1,3-dipolar cycloaddition reaction between azide 5f and alkyne 2a in 37 min as a white solid; mp: 140–142 °C; FT-IR (cm−1): 3257 (N–H stretching), 1672 (C O stretching); 1H NMR (500 MHz, CDCl3 + DMSO-d6, δ ppm): 5.45 (s, 2H, –NCH2CO–), 5.46 (s, 2H, –OCH2), 7.15–7.57 (m, 7H Ar–H), 7.67 (s, 1H, Ar–H), 7.92 (s, 1H, triazole), 8.15 (s, 1H, Ar–H), and 10.16 (s, 1H, NH). 13C NMR (125 MHz, CDCl3 + DMSO-d6, δ ppm): 53.1, 61.7, 114.7, 117.9, 120.0, 121.2, 124.5, 129.5, 129.9, 134.4, 138.9, 158.1, and 163.5; LC-MS for C17H15ClN4O2: [M]+ 343.1.
4.2.7. 2-(4-((4-Methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-phenylacetamide (7a)
The compound 7a was obtained via a 1,3-dipolar cycloaddition reaction between azide 5a and alkyne 2b in 37 min as a white solid; mp: 180–182 °C; FT-IR (cm−1): 3271 (N–H stretching), 1674 (C O stretching); 1H NMR (500 MHz, CDCl3, δ ppm): 3.76 (s, 3H, OCH3), 5.19 (s, 2H, –NCH2CO–), 5.20 (s, 2H, –OCH2), 6.83 (d, J = 8.0 Hz, 2H, Ar–H), 6.92 (d, J = 8.0 Hz, 2H, Ar–H), 7.14 (t, J = 8.0 Hz, 1H, Ar–H), 7.31 (t, J = 8.0 Hz, 2H, Ar–H), 7.43 (d, J = 8.0 Hz, 2H, Ar–H), 7.81 (s, 1H, triazole), and 7.87 (s, 1H, NH); 13C NMR (125 MHz, CDCl3, δ ppm): 53.9, 55.7, 62.5, 114.8, 115.9, 120.3, 125.4, 129.1, 137.0, 139.3, 152.1, and 162.3; LC-MS for C18H18N4O3: [M]+ 339.1.
4.2.8. 2-(4-((4-Methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(o-tolyl)acetamide (7b)
The compound 7b was obtained via a 1,3-dipolar cycloaddition reaction between azide 5b and alkyne 2b in 35 min as a white solid; mp: 176–178 °C; FT-IR (cm−1): 3266 (N–H stretching), 1674 (C O stretching); 1H NMR (500 MHz, CDCl3, δ ppm): 2.10 (s, 3H, CH3), 3.75 (s, 3H, –OCH3), 5.21 (s, 2H, –NCH2CO–), 5.22 (s, 2H, –OCH2), 6.82 (d, J = 8.0 Hz, 2H, Ar–H), 6.91 (d, J = 8.0 Hz, 2H, Ar–H), 7.08 (t, J = 8.0 Hz, 1H, Ar–H), 7.15 (d, J = 8.0 Hz, 1H, Ar–H), 7.20 (t, J = 8.0 Hz, 1H, Ar–H), 7.80 (s, 1H, triazole), and 7.82 (s, 1H, NH); 13C NMR (125 MHz, CDCl3, δ ppm): 17.4, 53.9, 55.7, 62.4, 114.8, 115.9, 122.3, 124.5, 125.9, 126.9, 127.9, 137.6, and 162.8; LC-MS for C19H20N4O3: [M]+ 353.1.
4.2.9. 2-(4-((4-Methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(m-tolyl)acetamide (7c)
The compound 7c was obtained via a 1,3-dipolar cycloaddition reaction between azide 5c and alkyne 2b in 35 min as a yellow solid; mp: 140–142 °C; FT-IR (cm−1): 3271 (N–H stretching), 1665 (C O stretching); 1H NMR (500 MHz, CDCl3 + DMSO-d6, δ ppm): 2.18 (s, 3H, CH3), 3.62 (s, 3H, OCH3), 5.01 (s, 2H, –NCH2CO–), 5.10 (s, 2H, –OCH2), 6.68 (d, J = 6 Hz, 2H, Ar–H), 6.79 (d, J = 8.0 Hz, 2H, Ar–H), 7.04 (t, J = 8.0 Hz, 1H, Ar–H), 7.18 (d, J = 8.0 Hz, 1H, Ar–H), 7.26 (d, J = 8.0 Hz, 1H, Ar–H), 7.31 (s, 1H, Ar–H), 7.79 (s, 1H, triazole), and 9.73 (s, 1H, NH); 13C NMR (125 MHz, CDCl3 + DMSO-d6, δ ppm): 21.3, 52.9, 55.5, 62.4, 114.5, 115.7, 116.8, 120.4, 124.4, 125.1, 128.5, 137.6, 138.5, 144.1, 152.2, 153.9, and 163.3. LC-MS for C19H20N4O3: [M]+ 353.1.
4.2.10. 2-(4-((4-Methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(p-tolyl)acetamide (7d)
The compound 7d was obtained via a 1,3-dipolar cycloaddition reaction between azide 5d and alkyne 2b in 36 min as a white solid; mp: 196–198 °C; FT-IR (cm−1): 3273 (N–H stretching), 1675 (C O stretching); 1H NMR (500 MHz, CDCl3 + DMSO-d6, δ ppm): 2.10 (s, 3H, CH3), 3.55 (s, 3H, OCH3), 4.94 (s, 2H, –NCH2CO–), 5.02 (s, 2H, –OCH2), 6.62 (d, J = 8.0 Hz, 2H, Ar–H), 6.72 (d, J = 8.0 Hz, 2H, Ar–H), 6.89 (d, J = 8.0 Hz, 2H, Ar–H), 7.24 (d, J = 8.0 Hz, 2H, Ar–H), 7.73 (s, 1H, triazole), and 9.68 (s, 1H, NH); 13C NMR (125 MHz, CDCl3 + DMSO-d6, δ ppm): 20.4, 52.6, 55.2, 62.1, 114.2, 115.4, 119.5, 124.2, 128.9, 133.5, 134.9, 151.9, 153.6, and 162.8; LC-MS for C19H20N4O3: [M]+ 353.1.
4.2.11. N-(2-Chlorophenyl)-2-(4-((4-methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (7e)
The compound 7e was obtained via a 1,3-dipolar cycloaddition reaction between azide 5e and alkyne 2b in 35 min as a white solid; mp: 168–170 °C; FT-IR (cm−1): 3260 (N–H stretching), 1683 (C O stretching); 1H NMR (500 MHz, CDCl3 + DMSO-d6, δ ppm): 3.89 (s, 3H, OCH3), 5.32 (s, 2H, –NCH2CO–), 5.47 (s, –OCH2), 6.96 (d, J = 8.0 Hz, 2H, Ar–H), 7.06 (d, J = 8.0 Hz, 2H, Ar–H), 7.23 (t, J = 8.0 Hz, 1H, Ar–H), 7.39 (t, J = 8.0 Hz, 1H, Ar–H), 7.50 (d, J = 8.0 Hz, 1H, Ar–H), 8.03 (s, 1H, triazole), 8.26 (d, J = 8.0 Hz, 1H, Ar–H), and 8.98 (s, 1H, NH); 13C NMR (125 MHz, CDCl3 + DMSO-d6, δ ppm): 53.0, 55.4, 62.3, 114.4, 115.6, 122.8, 124.3, 125.6, 127.3, 129.1, 133.4, 144.7, 152.0, 153.9, and 163.3; LC-MS for C18H17ClN4O3: [M]+ 373.
4.2.12. N-(2-Chlorophenyl)-2-(4-((4-methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (7f)
The compound 7f was obtained via a 1,3-dipolar cycloaddition reaction between azide 5f and alkyne 2b in 35 min as a green solid; mp: 160–162 °C; FT-IR (cm−1): 3266 (N–H stretching), 1675 (C O stretching); 1H NMR (500 MHz, CDCl3 + DMSO-d6, δ ppm): 3.57 (s, 3H, OCH3), 4.96 (s, 2H, –NCH2CO–), 5.05 (s, 2H, –OCH2), 6.63 (d, J = 8.0 Hz, 2H, Ar–H), 6.73 (d, J = 8.0 Hz, 2H, Ar–H), 6.87 (d, J = 8.0 Hz, 1H, Ar–H), 7.03 (t, J = 8.0 Hz, 1H, Ar–H), 7.24 (d, J = 8.0 Hz, 1H, Ar–H), 7.53 (s, 1H, Ar–H), 7.73 (s, 1H, triazole), and 10.04 (s, 1H, NH); 13C NMR (125 MHz, CDCl3 + DMSO-d6, δ ppm): 52.4, 55.1, 62.0, 114.1, 115.27, 117.3, 119.3, 123.7, 124.1, 129.4, 133.8, 138.7, 151.8, 153.5, and 163.2; LC-MS for C18H17ClN4O3 [M + H]+ 373.
4.2.13. N-(4-Chlorophenyl)-2-(4-((4-methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (7g)
The compound 7g was obtained via a 1,3-dipolar cycloaddition reaction between azide 5g and alkyne 2b in 36 min as a white solid; mp: 192–194 °C; FT-IR (cm−1): 3266 (N–H stretching), 1672 (C O stretching).
4.2.14. 2-(4-((4-Nitrophenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-phenylacetamide (8a)
The compound 8a was obtained via a 1,3-dipolar cycloaddition reaction between azide 5a and alkyne 2c in 40 min as a yellow solid; mp: 180–182 °C; FT-IR (cm−1): 3250 (N–H stretching), 1682 (C O stretching); 1H NMR (500 MHz, CDCl3 + CD3OD, δ ppm): 5.18 (s, 2H, –NCH2CO–), 5.27 (s, 2H, –OCH2), 7.05 (d, J = 8.0 Hz, 3H, Ar–H), 7.27 (t, J = 8.0 Hz, 3H, Ar–H), 7.49 (d, J = 8.0 Hz, 2H, Ar–H), 7.98 (s, 1H, triazole), and 8.17 (d, J = 8 Hz, 2H, Ar–H); 13C NMR (125 MHz, CDCl3 + CD3OD, δ ppm): 52.7, 61.9, 114.5, 125.1, 125.7, 128.8, 137.5, 141.7, and 162.8; LC-MS for C17H15N5O4: [M]+ 354.
4.2.15. 2-(4-((4-Nitrophenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(o-tolyl)acetamide (8b)
The compound 8b was obtained via a 1,3-dipolar cycloaddition reaction between azide 5b and alkyne 2c in 39 min as a white solid; mp: 178–180 °C; FT-IR (cm−1): 3257 (N–H stretching), 1668 (C O stretching); 1H NMR (500 MHz, CDCl3 + CD3OD, δ ppm): 2.15 (s, 3H, CH3), 5.23 (s, 2H, –NCH2CO–), 5.25 (s, 2H, –OCH2), 7.03–7.13 (m, 6H, Ar–H), 7.47 (s, 1H, NH), 7.98 (s, 1H, triazole), and 8.14 (s, 2H, Ar–H); 13C NMR (125 MHz, CDCl3 + CD3OD, δ ppm): 17.2, 52.6, 62.1, 114.9, 124.9, 125.4, 125.9, 126.4, 126.5, 127.4, 130.7, 131.2, and 163.7; LC-MS for C18H17N5O4 [M]+ 368.1.
4.2.16. 2-(4-((4-Nitrophenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(m-tolyl)acetamide (8c)
The compound 8c was obtained via a 1,3-dipolar cycloaddition reaction between azide 5c and alkyne 2c in 38 min as a white solid; mp: 168–170 °C; FT-IR (cm−1): 3280 (N–H stretching), 1662 (C O stretching); 1H NMR (200 MHz, DMSO-d6, δ ppm): 2.28 (s, 3H, CH3), 5.36 (s, 4H, –NCH2CO, –OCH2), 6.91 (d, J = 8.0 Hz, 1H, Ar–H), 7.19 (d, J = 8.0 Hz, 1H, Ar–H), 7.27 (t, J = 8 Hz, 1H, Ar–H), 7.33 (d, J = 8.0 Hz, 2H, Ar–H), 7.42 (s, 1H, Ar–H), 8.24 (d, J = 8.0 Hz, 2H, Ar–H), 8.32 (s, 1H, triazole), and 10.42 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6, δ ppm): 21.1, 52.2, 61.9, 115.3, 116.4, 119.8, 124.5, 125.9, 126.7, 128.7, 138.1, 138.3, 141.0, 141.5, 163.3, and 164.0; HRMS calcd for C18H18N5O4 [M + H]+: 368.1359 and found 368.1364.
4.2.17. 2-(4-((4-Nitrophenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(p-tolyl)acetamide (8d)
The compound 8d was obtained via a 1,3-dipolar cycloaddition reaction between azide 5d and alkyne 2c in 40 min as a white solid; mp: 210–212 °C; FT-IR (cm−1): 3260 (N–H stretching), 1696 (C O stretching); 1H NMR (500 MHz, CDCl3 + CD3OD, δ ppm): 2.26 (s, 3H, CH3), 5.14 (s, 2H, –NCH2CO–), 5.26 (s, 2H, –OCH2), 7.0 (d, J = 8.0 Hz, 2H, Ar–H), 7.07 (d, J = 8.0 Hz, 2H, Ar–H), 7.35 (d, J = 8.0 Hz, 2H, Ar–H), 7.96 (s, 1H, triazole), and 8.16 (d, J = 8.0 Hz, 2H, Ar–H); 13C NMR (125 MHz, CDCl3 + CD3OD, δ ppm): 17.7, 53.1, 62.6, 115.4, 124.8, 125.9, 126.5, 126.9, 131.2, 143.5, 152.0, and 164.2; LC-MS for C18H17N5O4: [M + H]+ 368.1.
4.2.18. N-(2-Chlorophenyl)-2-(4-((4-nitrophenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (8e)
The compound 8e was obtained via a 1,3-dipolar cycloaddition reaction between azide 5e and alkyne 2c in 39 min as a red solid; mp: 169–171 °C; FT-IR (cm−1): 3243 (N–H stretching), 1672 (C O stretching); 1H NMR (500 MHz, CDCl3 + CD3OD, δ ppm): 5.31 (s, 2H, –NCH2CO–), 5.33 (s, 2H, –OCH2), 7.08 (d, J = 8.0 Hz, 3H, Ar–H), 7.29 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 1H, Ar–H), 7.96 (s, 1H, triazole), 8.15 (s, 1H, NH), and 8.20 (d, J = 8.0 Hz, 2H, Ar–H); 13C NMR (125 MHz, CDCl3 + CD3OD, δ ppm): 53.2, 62.1, 114.8, 125.1, 126.0, 126.1, 127.7, 129.4, 133.5, 136.5, 146.4, 152.3, and 163.0; LC-MS for C17H14ClN5O4: [M]+ 388.
4.2.19. N-(3-Chlorophenyl)-2-(4-((4-nitrophenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (8f)
The compound 8f was obtained via a 1,3-dipolar cycloaddition reaction between azide 5f and alkyne 2c in 40 min as a white solid; mp: 190–192 °C; FT-IR (cm−1): 3327 (N–H stretching), 1683 (C O stretching); 1H NMR (200 MHz, DMSO-d6, δ ppm): 5.36 (s, 2H, –NCH2CO–), 5.40 (s, 2H, –OCH2), 7.15 (d, J = 8.0 Hz, 1H, Ar–H), 7.29 (d, J = 8.0 Hz, 2H, Ar–H), 7.42 (d, J = 8.0 Hz, 2H, Ar–H), 7.78 (s, 1H, Ar–H), 8.23 (d, J = 8.0 Hz, 2H, Ar–H), 8.33 (s, 1H, triazole), and 10.81 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6, δ ppm): 52.3, 61.9, 115.4, 117.7, 118.9, 123.6, 125.9, 126.8, 130.7, 133.3, 139.9, 141.1, 141.7, 163.4, and 164.7; HRMS calcd for C17H15ClN5O4 [M + H]+: 388.0813 and found 388.0814.
4.2.20. N-(4-Chlorophenyl)-2-(4-((4-nitrophenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (8g)
The compound 8g was obtained via a 1,3-dipolar cycloaddition reaction between azide 5g and alkyne 2c in 37 min as a white solid; mp: 188–190 °C; FT-IR (cm−1): 3243 (N–H stretching), 1672 (C O stretching); 1H NMR (200 MHz, DMSO-d6, δ ppm): 5.36 (s, 4H, –NCH2CO, –OCH2), 7.29 (d, J = 8.0 Hz, 2H, Ar–H), 7.39 (d, J = 8.0 Hz, 2H, Ar–H), 7.61 (d, J = 8.0 Hz, 2H, Ar–H), 8.23 (d, J = 8.0 Hz, 2H, Ar–H), 8.32 (s, 1H, triazole), and 10.64 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6, δ ppm): 52.2, 61.9, 115.3, 120.8, 125.8, 126.7, 127.4, 128.8, 137.3, 141.0, 141.6, 163.3, and 164.3; HRMS calcd for C17H15ClN5O4 [M + H]+: 388.0813 and found 388.0819.
4.3. Experimental protocol for biological activity
4.3.1. Antifungal activity
Antifungal activity was determined by the standard agar dilution method as per the CLSI (formerly, NCCLS) guidelines.26 The newly synthesized compounds were screened for their in vitro antifungal activity against five human pathogenic fungal strains including Candida albicans (NCIM 3471), Fusarium oxysporum (NCIM 1332), Aspergillus flavus (NCIM 539), Aspergillus niger (NCIM 1196), and Cryptococcus neoformans (NCIM 576). The synthesized compounds and the standard Miconazole were dissolved in DMSO. The medium yeast nitrogen base was dissolved in phosphate buffer of pH 7 and was autoclaved at 110 °C for 10 min. With each set, a growth control without the antifungal agent and solvent control DMSO were included. The fungal strains were freshly subcultured onto sabouraud dextrose agar (SDA) and incubated at 25 °C for 72 h. The fungal cells were suspended in sterile distilled water and diluted to get 105 cells per mL. Ten μL of the standardized suspension was inoculated onto the control plates and the media incorporated with the antifungal agents. The inoculated plates were incubated at 25 °C for 48 h. The readings were taken at the end of 48 and 72 h. MIC is the lowest concentration of the drug preventing the growth of macroscopically visible colonies on the drug-containing plates when there is visible growth on the drug-free control plates.
4.3.2. In vitro Mtb MABA assay
The newly synthesized compounds were screened for their in vitro antitubercular activity against Mtb H37Rv (ATCC 27294) by using the Microplate Alamar Blue Assay (MABA)30 for the determination of MIC in triplicate. The MIC (in μg mL−1) was recorded as the lowest concentration/highest dilution of the compounds/control drugs that completely inhibited the growth of the Mtb cultures. The MIC values of the compounds were compared with those of the standard drugs (Isoniazid, Rifampicin, Ethambutol, and Ciprofloxacin). The experimental method for antitubercular activity is briefly described as follows.
Initially, the inoculum was prepared from a fresh LJ medium re-suspended in the 7H9-S medium (7H9 broth, 0.1% casitone, 0.5% glycerol, supplemented oleic acid, albumin, dextrose, and catalase [OADC]), adjusted to a McFarland tube no. 1, and diluted 1 : 20; 100 μL was used as the inoculum. Each drug stock solution was thawed and diluted in 7H9-S at four-fold the final highest concentration tested. Serial two-fold dilutions of each drug were prepared directly in a sterile 96-well microtiter plate using 100 μL 7H9-S. A growth control containing no antibiotic and a sterile control was also prepared on each plate. Sterile water was added to all perimeter wells to avoid evaporation during incubation. The plate was covered, sealed in plastic bags, and incubated at 37 °C in normal atmosphere. After 7 days incubation, 30 μL of alamar blue solution was added to each well, and the plate was re-incubated overnight. A change in the colour from blue (oxidised state) to pink (reduced) indicated the growth of bacteria, and the MIC was defined as the lowest concentration of the drug that prevented this change in colour.
4.3.3. Antioxidant activity (DPPH radical scavenging)
The hydrogen atom or electron donation ability of some of the compounds was measured from the bleaching of the purple coloured methanol solution of 1,1-diphenyl-1-picrylhydrazyl (DPPH). The spectrophotometric assay uses the stable radical DPPH as a reagent. One mL of various concentrations of the test compounds (5, 10, 25, 50, and 100 μg mL−1) in methanol was added to 4 mL of 0.004% IJ w/v methanol solution of DPPH. The reaction mixture was incubated at 37 °C. The scavenging activity on DPPH was determined by measuring the absorbance at 517 nm after 30 min. All tests were performed in triplicate, and the mean values were obtained. The percent of inhibition of free radical production from DPPH was calculated by using the following equation:% of scavenging = [(Acontrol − Asample)/IJAsample × 100)],where Acontrol is the absorbance of the control (DPPH radical without test sample) and Asample is the absorbance of the test sample (DPPH radical with test sample). The control contained all the reagents except the test samples.
4.3.4. Homology modeling
The three-dimensional homology model structure of cytochrome P450 lanosterol 14α-demethylase of C. albicans was predicted using the theoretical methods of protein structure prediction, i.e., comparative modelling using the Biopredicta module of VLifeMDS 4.3 ProModel. The protein sequence of cytochrome P450 lanosterol 14α-demethylase for C. albicans was retrieved from the protein knowledge database Universal Protein Resource (UniProtKB) (http://www.uniprot.org/) (accession code: P10613) and its homologous template was identified from the protein structural database, i.e., Protein Data Bank (PDB) using Basic Local Alignment Search Tool for proteins (BLASTp). The homologous template identified using BLASTp solved the three-dimensional crystal structure of human lanosterol 14α-demethylase (CYP51) complex with azole (PDB ID: 3LD6). To construct a basic framework and to build the loop regions of C. albicans cytochrome P450 lanosterol 14α-demethylase, pairwise sequence alignment and loop refinement of CA-CYP51 (P10613) and human CYP51 (3LD6_B) were performed. Three-dimensional modelled protein structures were subjected to structural quality check programs such as PROCHECK and its other structural validation parameters.31
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
One of the authors, SVA, is very much grateful to the Council of Scientific and Industrial Research (CSIR), New Delhi for the award of research fellowship. Authors are also thankful to the Head, Department of Chemistry, Dr Babasaheb Ambedkar Marathwada University Aurangabad for providing laboratory facility.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03425k
Notes and references
- (a) Dheer D. Singh V. Shankar R. Bioorg. Chem. 2017;71:30. doi: 10.1016/j.bioorg.2017.01.010. [DOI] [PubMed] [Google Scholar]; (b) Massarotti A. Aprile S. Mercalli V. Grosso E. D. Grosa G. Sorba G. Tron G. C. ChemMedChem. 2014;9:2497. doi: 10.1002/cmdc.201402233. [DOI] [PubMed] [Google Scholar]; (c) Agalave S. G. Maujan S. R. Pore V. S. Chem.–Asian J. 2011;6:2696. doi: 10.1002/asia.201100432. [DOI] [PubMed] [Google Scholar]; (d) Whiting M. Muldoon J. Lin Y. C. Silverman S. M. Lindstrom W. Olson A. J. Kolb H. C. Finn M. G. Sharpless K. B. Elder J. H. Fokin V. V. Angew. Chem., Int. Ed. 2006;45:1435. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]; (e) Rostovtsev V. V. Green L. G. Fokin V. V. Sharpless K. B. Angew. Chem., Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (f) Tornoe C. W. Christensen C. Meldal M. J. Org. Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- (a) Kempe K. Krieg A. R Becer C. Schubert U. S. Chem. Soc. Rev. 2012;41:176. doi: 10.1039/C1CS15107J. [DOI] [PubMed] [Google Scholar]; (b) Akeroyd N. Klumperman B. Eur. Polym. J. 2011;47:1207. doi: 10.1016/j.eurpolymj.2011.02.003. [DOI] [Google Scholar]
- (a) Bryant J. J. Bunz U. H. F. Chem.–Asian J. 2013;8:1354. doi: 10.1002/asia.201300260. [DOI] [PubMed] [Google Scholar]; (b) Hanni K. D. Leigh D. A. Chem. Soc. Rev. 2010;39:1240. doi: 10.1039/B901974J. [DOI] [PubMed] [Google Scholar]
- (a) Tron G. C. Pirali T. Billington R. A. Canonico P. L. Sorba G. Genazzani A. A. Med. Res. Rev. 2008;28:278. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]; (b) Moorhouse A. D. Moses J. E. ChemMedChem. 2008;3:715. doi: 10.1002/cmdc.200700334. [DOI] [PubMed] [Google Scholar]; (c) Lutz J. F. Zarafshani Z. Adv. Drug Delivery Rev. 2008;60:958. doi: 10.1016/j.addr.2008.02.004. [DOI] [PubMed] [Google Scholar]
- (a) Fu N. Wang S. Zhang Y. Zhang C. Yang D. Weng L. Zhao B. Wang L. Eur. J. Med. Chem. 2017;136:596. doi: 10.1016/j.ejmech.2017.05.001. [DOI] [PubMed] [Google Scholar]; (b) Chen H. J. Jiang Y. J. Zhangc Y. Q. Jingb Q. W. Wang N. L. Y. Zhangb W. N. Shenga C. Q. Chin. Chem. Lett. 2017;28:913. doi: 10.1016/j.cclet.2016.11.027. [DOI] [Google Scholar]; (c) Dai Z. C. Chen Y. F. Zhang M. Li S. K. Yang T. T. Shen L. Wang J. X. Qian S. S. Zhu H. L. Ye Y. H. Org. Biomol. Chem. 2015;13:477. doi: 10.1039/C4OB01758G. [DOI] [PubMed] [Google Scholar]; (d) Jiang Z. Gu J. Wang C. Wang S. Liu N. Jiang Y. Dong G. Wang Y. Liu Y. Yao J. Miao Z. Zhang W. Sheng C. Eur. J. Med. Chem. 2014;82:490. doi: 10.1016/j.ejmech.2014.05.079. [DOI] [PubMed] [Google Scholar]; (e) Aher N. G. Pore V. S. Mishra N. N. Kumar A. Shukla P. K. Sharma A. Bhat M. K. Bioorg. Med. Chem. Lett. 2009;19:759. doi: 10.1016/j.bmcl.2008.12.026. [DOI] [PubMed] [Google Scholar]
- (a) Zhang B. Eur. J. Med. Chem. 2019;168:357. doi: 10.1016/j.ejmech.2019.02.055. [DOI] [PubMed] [Google Scholar]; (b) Wang X. L. Wan K. Zhou C. H. Eur. J. Med. Chem. 2010;45:4631. doi: 10.1016/j.ejmech.2010.07.031. [DOI] [PubMed] [Google Scholar]
- (a) Raju K. S. AnkiReddy S. Sabitha G. Krishna V. S. Sriram D. Reddy K. B. Sagurthi S. R. Bioorg. Med. Chem. Lett. 2019;29:284. doi: 10.1016/j.bmcl.2018.11.036. [DOI] [PubMed] [Google Scholar]; (b) Zhan S. Xu Z. Gao C. Ren Q. C. Chang L. Lv Z. S. Feng L. S. Eur. J. Med. Chem. 2017;138:501. doi: 10.1016/j.ejmech.2017.06.051. [DOI] [PubMed] [Google Scholar]; (c) Xu Z. Song X. F. Hu Y. Q. Qiang M. Lv Z. S. Eur. J. Med. Chem. 2017;138:66. doi: 10.1016/j.ejmech.2017.05.057. [DOI] [PubMed] [Google Scholar]; (d) Shaikh M. H. Subhedar D. D. Nawale L. Sarkar D. Khan F. A. K. Sangshetti J. N. Shingate B. B. Med. Chem. Commun. 2015;6:1104. doi: 10.1039/C5MD00057B. [DOI] [Google Scholar]; (e) Patpi S. R. Pulipati L. Yogeeswari P. Sriram D. Jain N. Sridhar B. Murthy R. Anjana D. T. Kalivendi S. V. Kantevar S. J. Med. Chem. 2012;55:3911. doi: 10.1021/jm300125e. [DOI] [PubMed] [Google Scholar]
- (a) Rao P. S. Kurumurthy C. Veeraswamy B. Kumar G. S. Poornachandra Y. Kumar C. G. Vasamsetti S. B. Kotamraju S. Narsaiah B. Eur. J. Med. Chem. 2014;80:184. doi: 10.1016/j.ejmech.2014.04.052. [DOI] [PubMed] [Google Scholar]; (b) Simone R. D. Chini M. G. Bruno I. Riccio R. Mueller D. Werz O. Bifulco G. J. Med. Chem. 2011;54:1565. doi: 10.1021/jm101238d. [DOI] [PubMed] [Google Scholar]
- Buckle D. R. Outred D. J. Rockell C. J. M. Smith H. Spicer B. A. J. Med. Chem. 1983;26:251. doi: 10.1021/jm00356a025. [DOI] [PubMed] [Google Scholar]
- (a) Tian Y. Liu Z. Liu J. Huang B. Kang D. Zhang H. Clercq E. D. mans D. D. Pannecouque C. Lee K. H. Chen C. H. Zhan P. Liu X. Eur. J. Med. Chem. 2018;151:339. doi: 10.1016/j.ejmech.2018.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu G. Zalloum W. A. Meuser M. E. Jing L. Kang D. Chen C. Tian Y. Zhang F. Cocklin S. Lee K. H. Liu X. Zhan P. Eur. J. Med. Chem. 2018;158:478. doi: 10.1016/j.ejmech.2018.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Giffin M. J. Heaslet H. Brik A. Lin Y. C. Cauvi G. Wong C. H. McRee D. E. Elder J. H. Stout C. D. Torbett B. E. J. Med. Chem. 2008;51:6263. doi: 10.1021/jm800149m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Allam M. Bhavani A. K. D. Mudiraj A. Ranjan N. Thippana M. Babu P. P. Eur. J. Med. Chem. 2018;156:43. doi: 10.1016/j.ejmech.2018.06.055. [DOI] [PubMed] [Google Scholar]; (b) Prachayasittikul V. Pingaew R. Anuwongcharoen N. Worachartcheewan A. Nantasenamat C. Prachayasittikul S. Ruchirawat S. Prachayasittikul V. SpringerPlus. 2015;4:571. doi: 10.1186/s40064-015-1352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Duan Y. C. Ma Y. C. Zhang E. Shi X. J. Wang M. M. Ye X. W. Liu H. M. Eur. J. Med. Chem. 2013;62:11. doi: 10.1016/j.ejmech.2012.12.046. [DOI] [PubMed] [Google Scholar]; (d) Zheng Y. C. Duan Y. C. Ma J. L. Xu R. M. Zi X. Lv W. L. Wang M. M. Ye X. W. Zhu S. Mobley D. Zhu Y. Y. Wang J. W. Li J. F. Wang Z. R. Zhao W. Liu H. M. J. Med. Chem. 2013;56:8543. doi: 10.1021/jm401002r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Dai Z. C. Chen Y. F. Cao L. L. Yan W. Li S. K. Wang J. X. Zhang Z. G. Ye Y. H. Eur. J. Med. Chem. 2017;126:171. doi: 10.1016/j.ejmech.2016.10.006. [DOI] [PubMed] [Google Scholar]
- Cha R. Sobel J. D. Expert Rev. Anti-Infect. Ther. 2004;2:357. doi: 10.1586/14787210.2.3.357. [DOI] [PubMed] [Google Scholar]
- Georgopapadakou N. H. Walsh T. J. Antimicrob. Agents Chemother. 1996;40:279. doi: 10.1128/AAC.40.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch D. A. Ludlam H. A. Brown N. M. J. Med. Microbiol. 2006;55:809. doi: 10.1099/jmm.0.46548-0. [DOI] [PubMed] [Google Scholar]
- (a) Lepesheva G. I. Zaitseva N. G. Nes W. D. et al. . J. Biol. Chem. 2006;281:3577. doi: 10.1074/jbc.M510317200. [DOI] [PubMed] [Google Scholar]; (b) Jeu L. Piacenti F. J. Lyakhovetskiy A. G. Fung H. B. Clin. Ther. 2003;25:1321. doi: 10.1016/S0149-2918(03)80126-1. [DOI] [PubMed] [Google Scholar]; (c) Pfaller M. A. Messer S. A. Hollis R. J. Jones R. N. Antimicrob. Agents Chemother. 2001;45:2862. doi: 10.1128/AAC.45.10.2862-2864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Greaves T. L. Drummond C. J. Chem. Rev. 2008;108:206. doi: 10.1021/cr068040u. [DOI] [PubMed] [Google Scholar]; (b) Sheldon R. Chem. Commun. 2001:2399. doi: 10.1039/B107270F. [DOI] [PubMed] [Google Scholar]; (c) Wasserscheid P. Keim W. Angew. Chem., Int. Ed. 2000;39:3772. doi: 10.1002/1521-3773(20001103)39:21<3772::AID-ANIE3772>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (d) Welton T. Chem. Rev. 1999;99:2071. doi: 10.1021/cr980032t. [DOI] [PubMed] [Google Scholar]
- (a) Rub C. Konig B. Green Chem. 2012;14:2969. doi: 10.1039/C2GC36005E. [DOI] [Google Scholar]; (b) Miao W. Chan T. H. Org. Lett. 2003;5:5003. doi: 10.1021/ol035977y. [DOI] [PubMed] [Google Scholar]
- Martins M. A. P. Frizzo C. P. Tier A. Z. Moreira D. N. Zanatta N. Bonacorso H. G. Chem. Rev. 2014;114:PR1–PR70. doi: 10.1021/cr500106x. [DOI] [PubMed] [Google Scholar]
- (a) Ali A. A. Konwar M. Chetia M. Sarma D. Tetrahedron Lett. 2016;57:5661. doi: 10.1016/j.tetlet.2016.11.014. [DOI] [Google Scholar]; (b) Tavassoli M. Isfahani A. L. Moghadam M. Tangestaninejad S. Mirkhani V. Baltork I. M. ACS Sustainable Chem. Eng. 2016;4:1454. doi: 10.1021/acssuschemeng.5b01432. [DOI] [Google Scholar]; (c) Dabiri M. Movahed S. K. MaGee D. I. Res. Chem. Intermed. 2015;41:3335. doi: 10.1007/s11164-013-1436-1. [DOI] [Google Scholar]; (d) Singh H. Sindhu J. Khurana J. M. Sharma C. Aneja K. R. Eur. J. Med. Chem. 2014;77:145. doi: 10.1016/j.ejmech.2014.03.016. [DOI] [PubMed] [Google Scholar]; (e) Ahmady A. Z. Heidarizadeh F. Keshavarz M. Synth. Commun. 2013;43:2100. doi: 10.1080/00397911.2012.687424. [DOI] [Google Scholar]; (f) Marra A. Vecchi A. Chiappe C. Melai B. Dondoni A. J. Org. Chem. 2008;73:2458. doi: 10.1021/jo7026454. [DOI] [PubMed] [Google Scholar]; (g) Zhao Y. B. Yan Z. Y. Liang Y. M. Tetrahedron Lett. 2006;47:1545. doi: 10.1016/j.tetlet.2006.01.004. [DOI] [Google Scholar]
- (a) Parmar N. J. Barad H. A. Pansuriya B. R. Talpada N. P. RSC Adv. 2013;3:8064. doi: 10.1039/C3RA00068K. [DOI] [Google Scholar]; (b) Parmar N. J. Pansuriya B. R. Barad H. A. Parmar B. D. Kant R. Gupta V. K. Monatsh. Chem. 2013;144:865. doi: 10.1007/s00706-012-0873-7. [DOI] [Google Scholar]; (c) Parmar N. J. Patel R. A. Parmar B. D. Talpada N. P. Bioorg. Med. Chem. Lett. 2013;23:1656. doi: 10.1016/j.bmcl.2013.01.079. [DOI] [PubMed] [Google Scholar]; (d) Attri P. Pal M. Green Chem. Lett. Rev. 2010;3:249. doi: 10.1080/17518251003749361. [DOI] [Google Scholar]; (e) Sutariya T. R. Labana B. M. Parmar N. J. Kant R. Gupta V. K. Plata G. B. Padroón J. M. New J. Chem. 2015;39:2657. doi: 10.1039/C4NJ02308K. [DOI] [Google Scholar]; (f) Verma A. K. Attri P. Chopra V. Tiwari R. K. Chandra R. Monatsh. Chem. 2008;139:1041. doi: 10.1007/s00706-008-0886-4. [DOI] [Google Scholar]
- (a) Danne A. B. Choudhari A. S. Chakraborty S. Sarkar D. Khedkar V. M. Shingate B. B. Med. Chem. Commun. 2018;9:1114. doi: 10.1039/C8MD00055G. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Khare S. P. Deshmukh T. R. Sangshetti J. N. Krishna V. S. Sriram D. Khedkar V. M. Shingate B. B. ChemistrySelect. 2018;3:13113. doi: 10.1002/slct.201801859. [DOI] [Google Scholar]; (c) Danne A. B. Choudhari A. S. Sarkar D. Sangshetti J. N. Khedkar V. M. Shingate B. B. Res. Chem. Intermed. 2018;44:6283. doi: 10.1007/s11164-018-3490-1. [DOI] [Google Scholar]; (d) Shaikh M. H. Subhedar D. D. Arkile M. Khedkar V. M. Jadhav N. Sarkar D. Shingate B. B. Bioorg. Med. Chem. Lett. 2016;26:561. doi: 10.1016/j.bmcl.2015.11.071. [DOI] [PubMed] [Google Scholar]; (e) Shaikh M. H. Subhedar D. D. Shingate B. B. Khan F. A. K. Sangshetti J. N. Khedkar V. M. Nawale L. Sarkar D. Navale G. R. Shinde S. S. Med. Chem. Res. 2016;25:790. doi: 10.1007/s00044-016-1519-9. [DOI] [Google Scholar]; (f) Shaikh M. H. Subhedar D. D. Khan F. A. K. Sangshetti J. N. Shingate B. B. Chin. Chem. Lett. 2016;27:295. doi: 10.1016/j.cclet.2015.11.003. [DOI] [Google Scholar]; (g) Shaikh M. H. Subhedar D. D. Khedkar V. M. Jha P. C. Khan F. A. K. Sangshetti J. N. Shingate B. B. Chin. Chem. Lett. 2016;27:1058. doi: 10.1016/j.cclet.2016.03.014. [DOI] [Google Scholar]
- (a) Subhedar D. D. Shaikh M. H. Shingate B. B. Nawale L. Sarkar D. Khedkar V. M. Khan F. A. K. Sangshetti J. N. Eur. J. Med. Chem. 2017;125:385. doi: 10.1016/j.ejmech.2016.09.059. [DOI] [PubMed] [Google Scholar]; (b) Subhedar D. D. Shaikh M. H. Khan F. A. K. Sangshetti J. N. Khedkar V. M. Shingate B. B. New J. Chem. 2016;40:3047. doi: 10.1039/C6NJ00021E. [DOI] [Google Scholar]; (c) Subhedar D. D. Shaikh M. H. Shingate B. B. Nawale L. Sarkar D. Khedkar V. M. Med. Chem. Commun. 2016;7:1832. doi: 10.1039/C6MD00278A. [DOI] [PubMed] [Google Scholar]; (d) Subhedar D. D. Shaikh M. H. Nawale L. Yeware A. Sarkar D. Khan F. A. K. Sangshetti J. N. Shingate B. B. Bioorg. Med. Chem. Lett. 2016;26:2278. doi: 10.1016/j.bmcl.2016.03.045. [DOI] [PubMed] [Google Scholar]; (e) Subhedar D. D. Shaikh M. H. Arkile M. A. Yeware A. Sarkar D. Shingate B. B. Bioorg. Med. Chem. Lett. 2016;26:1704. doi: 10.1016/j.bmcl.2016.02.056. [DOI] [PubMed] [Google Scholar]; (f) Shaikh M. H. Subhedar D. D. Kalam Khan F. A. Sangshetti J. N. Shingate B. B. Res. Chem. Intermed. 2016;42:5115. doi: 10.1007/s11164-015-2348-z. [DOI] [Google Scholar]; (g) Subhedar D. D. Shaikh M. H. Nawale L. Yeware A. Sarkar D. Shingate B. B. Res. Chem. Intermed. 2016;42:6607. doi: 10.1007/s11164-016-2484-0. [DOI] [Google Scholar]
- (a) Dhumal S. T. Deshmukh A. R. Kharat K. R. Sathe B. R. Chavan S. S. Mane R. A. New J. Chem. 2019;43:7663. doi: 10.1039/C9NJ00377K. [DOI] [Google Scholar]; (b) Kaushik C. P. Luxmi R. Kumar M. Singh D. Kumar K. Pahwa A. Synth. Commun. 2019;49:118. doi: 10.1080/00397911.2018.1544371. [DOI] [Google Scholar]; (c) Wang G. Peng Z. Wang J. Li X. Li J. Eur. J. Med. Chem. 2017;125:423. doi: 10.1016/j.ejmech.2016.09.067. [DOI] [PubMed] [Google Scholar]; (d) Neeraja P. Srinivas S. Mukkanti K. Dubey P. K. Pal S. Bioorg. Med. Chem. Lett. 2016;26:5212. doi: 10.1016/j.bmcl.2016.09.059. [DOI] [PubMed] [Google Scholar]; (e) Wang G. Peng Z. Wang J. Li J. Li X. Bioorg. Med. Chem. Lett. 2016;26:5719. doi: 10.1016/j.bmcl.2016.10.057. [DOI] [PubMed] [Google Scholar]
- Ferroni C. Pepe A. Sang Kim Y. Lee S. Guerrini A. Parenti M. D. Tesei A. Zamagni A. Cortesi M. Zaffaroni N. De Cesare M. Beretta G. L. Trepel J. B. Malhotra S. V. Varchi G. J. Med. Chem. 2017;60:3082. doi: 10.1021/acs.jmedchem.7b00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Khan K., In vitro and In vivo Screening Techniques for Bioactivity Screening and Evaluation, Proc. Int. Workshop UNIDO-CDRI, 1997, p. 210 [Google Scholar]; (b) Collins A. H., Microbiological Methods, Butterworth, London, 2nd edn, 1976 [Google Scholar]
- Burits M. Bucar F. Phytother. Res. 2000;14:323. doi: 10.1002/1099-1573(200008)14:5<323::AID-PTR621>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Li S. Li D. Xiao T. Zhang S. Song Z. Ma H. J. Agric. Food Chem. 2016;23:8927. doi: 10.1021/acs.jafc.6b03464. [DOI] [PubMed] [Google Scholar]
- S Dofe V. Sarkate A. P. Lokwani D. K. Kathwate S. H. Gill C. H. Res. Chem. Intermed. 2017;43:15. doi: 10.1007/s11164-016-2602-z. [DOI] [Google Scholar]
- Franzblau S. G. Witzig R. S. McLaughlin J. C. J. Clin. Microbiol. 1998;36:362. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Sangshetti J. N. Kalam Khan F. A. Chouthe R. S. Damale M. G. Shinde D. B. Chin. Chem. Lett. 2014;25:1033. doi: 10.1016/j.cclet.2014.04.003. [DOI] [Google Scholar]; (b) Hooft R. W. Vriend G. Sander C. Abola E. E. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.