

Abstract
Mass spectrometry (MS)-based spatially resolved top-down proteomics (TDP) of tissues is crucial for understanding the roles played by microenvironmental heterogeneity in the biological functions of organs and for discovering new proteoform biomarkers of diseases. There are few published spatially resolved TDP studies. One of the challenges relates to the limited performance of TDP for the analysis of spatially isolated samples using, for example, laser capture microdissection (LCM) because those samples are usually mass-limited. We present the first pilot study of LCM-capillary zone electrophoresis (CZE)-MS/MS for spatially resolved TDP and used zebrafish brain as the sample. The LCM-CZE-MS/MS platform employed a non-ionic detergent and a freeze–thaw method for efficient proteoform extraction from LCM isolated brain sections followed by CZE-MS/MS without any sample cleanup step, ensuring high sensitivity. Over 400 proteoforms were identified in a CZE-MS/MS analysis of one LCM brain section via consuming the protein content of roughly 250 cells. We observed drastic differences in proteoform profiles between two LCM brain sections isolated from the optic tectum (Teo) and telencephalon (Tel) regions. Proteoforms of three proteins (npy, penkb, and pyya) having neuropeptide hormone activity were exclusively identified in the isolated Tel section. Proteoforms of reticulon, myosin, and troponin were almost exclusively identified in the isolated Teo section, and those proteins play essential roles in visual and motor activities. The proteoform profiles accurately reflected the main biological functions of the Teo and Tel regions of the brain. Additionally, hundreds of post-translationally modified proteoforms were identified.
Introduction
Cellular systems, such as the brain, are heterogeneous, producing specific microenvironments with unique molecular and functional characteristics for carrying out biological functions.1–6 To better understand the function of the brain, there is a need for spatially resolved omics analysis. Since proteins are the primary functional machines in cells, spatially resolved proteomics is crucial for pursuing a better fundamental understanding of the brain and brain-related diseases.
Some spatially resolved bottom-up proteomics studies of the brain or other tissues have been reported.1,3,4,6–15 However, these bottom-up studies provided limited information of spatial distribution of proteoforms across the brain because proteoforms were digested into peptides for mass spectrometry (MS) and tandem MS (MS/MS) analysis and the intact pictures of proteoforms were lost during the enzymatic digestion step. Proteoforms from the same gene could have different biological functions.16 Therefore, it is vital to characterize proteins in a proteoform-specific manner to understand their functions accurately.
Antibody-based approaches are commonly used for protein imaging in tissues with high spatial resolution and have been applied to protein imaging in brains.17,18 However, those approaches heavily depend on the availability and quality of antibodies. The number of proteins that can be measured in one experiment is limited. More importantly, it is still challenging to produce high-quality antibodies for proteoform-specific measurements. Top-down proteomics directly analyzes intact proteoforms using MS and MS/MS and could provide a bird’s eye view of proteoforms in a complex biological sample.19–21 The state-of-the-art top-down proteomics workflows have identified thousands of proteoforms from complex proteomes using reversed-phase liquid chromatography (RPLC)-electrospray ionization (ESI)-MS/MS.22–25 However, based on our best knowledge, there are still very few spatially-resolved top-down proteomics studies of the brain using RPLC-MS/MS. Delcourt et al. employed RPLC-MS/MS for top-down MS characterization of three different regions in the hippocampus, corpus callosum, and medulla oblongata (Bregma index lateral 1.90 mm) of a rat brain, and in total, 123 proteoforms were identified within a mass range of 1.6–21.9 kDa.2 The data revealed significant differences among the three brain regions analyzed regarding proteoform profiling. Park et al. performed quantitative top-down proteomic measurements of specific regions of female mouse brain that were isolated from mice trained for cocaine-conditioned place preference.26 After gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) pre-fractionation of roughly 140 μg proteins and further RPLC-MS/MS analyses, about 600 proteoforms were quantified from each brain region studied, revealing significant proteoform-level changes in female mouse brain after cocaine and estrogen treatments.
Some challenges exist for spatially-resolved top-down proteomics of brain or other tissues using RPLC-ESI-MS/MS. First, the amount of protein material isolated from the thin tissue sections is limited and careful sample preparation is required to achieve high sample recovery for following RPLC-MS/MS analysis. Using a small volume of lysis buffer compatible with MS for protein extraction and avoiding sample transfer and cleanup before MS analysis is critical for reducing sample loss due to adsorption to surfaces. Novel sample preparation methods for top-down proteomics of mass-limited samples are required. Recently, Zhou et al. employed nanoPOTS (nanodroplet processing in one pot for trace samples) and RPLC-MS/MS for top-down proteomics of 70 to 770 HeLa cells with the identification of 170 to 620 proteoforms.27 The nanoPOTS method processed samples using a sub-microliter volume of lysis buffer containing a nonionic detergent and urea for top-down proteomics. Second, the sensitivity of RPLC-ESIMS/MS needs to be boosted drastically for the analysis of spatially isolated tissue samples. Capillary zone electrophoresis (CZE)-ESI-MS/MS has been recognized as an alternative to RPLC-MS/MS for top-down proteomics due to its high efficiency for proteoform separation and high sensitivity for proteoform detection.28–32 Proteoforms have drastically lower diffusion coefficients compared to peptides and small molecules. RPLC has difficulties for highly efficient separations of proteoforms due to severe peak broadening from the resistance to mass transfer and strong interactions between proteoforms and RPLC beads, leading to significant loss of sensitivity of RPLC-MS/MS for proteoforms. Typically tens to hundreds of micrograms of protein materials are required to identify thousands of proteoforms from mammalian cells or tissues using RPLC-MS/MS.23,25,26 CZE does not employ a stationary phase for separation and only utilizes an open tubular capillary. It can produce high separation efficiency for large biomolecules (i.e., proteoforms) because of their low diffusion coefficients, resulting in drastically less peak broadening during the separation than RPLC. CZE-MS/MS has achieved a close to one million theoretical plates for proteoform separation and thousands of proteoform identifications from hundreds of nanograms of zebrafish brain proteins.32 Several studies have compared CZE-MS/MS and RPLC-MS/MS directly regarding their sensitivity for proteoform characterization, showing that CZE-MS/MS has much better sensitivity than RPLC-MS/MS, most likely due to its better separation efficiency for proteoforms.29,30 CZE-MS/MS has also been applied for spatially resolved top-down proteomics and revealed drastic differences between the whole zebrafish brain cerebellum and optic tectum regarding proteoform profiling.32
Here, we describe a CZE-MS/MS-based workflow for spatially resolved top-down proteomics of thin tissue sections. First, specific regions (~500 μm2) of zebrafish brain sections (20 μm in thickness) were isolated with laser capture microdissection (LCM). Second, the isolated brain tissues were lysed in a 5 μL lysis buffer containing a non-ionic detergent octyl glucopyrano-side (OG) via a repeated freeze–thaw protocol. After that, the samples were centrifuged, and the supernatants were directly analyzed by CZE-ESI-MS/MS. The detergent OG has shown great performance for the extraction of proteins even membrane proteins for proteomics.33 Using the workflow, hundreds of proteoforms were identified in single CZE-MS/MS runs from the LCM brain samples with consumption of cell lysate corresponding to roughly 250 cells. Drastic differences between different regions of the thin brain section [optic tectum (Teo) vs. telencephalon (Tel)] were observed regarding proteoform profiles.
Experimental
Materials and reagents
Acrylamide was purchased from Acros Organics (NJ, USA). Standard proteins, ammonium bicarbonate (NH4HCO3), urea, dithiothreitol (DTT), iodoacetamide (IAA) and 3-(trimethoxysilyl) propyl methacrylate were purchased from Sigma-Aldrich (St. Louis, MO). LC/MS grade water, acetonitrile (ACN), methanol, formic acid and HPLC-grade acetic acid were purchased from Fisher Scientific (Pittsburgh, PA). Aqueous mixtures were filtered with Nalgene Rapid-Flow Filter units (Thermo scientific) with 0.2 μm CN membrane and 50 mm diameter. Fused silica capillaries (50 μm i.d./360 μm o.d.) were obtained from Poly-micro Technologies (Phoenix, AZ).
Tissue preparation
Zebrafish brains were collected from two mature female zebrafish (AB/Tuebingen line). The zebrafish were provided by Professor Jose Cibelli’s group at the department of Animal Science at Michigan State University. All the animal related experiments were performed using a protocol approved by the Institutional Animal Care and Use Committee of Michigan State University. After extraction, the brains were rinsed with phosphate buffered saline, placed into a cryostat block and covered with Optimal Cutting Temperature Compound (OCT compound) and frozen using dry ice. The brains in the OCT compound were stored at −80 °C until use. A cryostat was used to cut the zebrafish brain tissue to a thickness of 20 μm and placed onto PEN membrane slides. The brain tissues were then stained with Cresyl Violet Staining (CEV) to aid in the visualization and determination of the different brain regions. Briefly, the slide was washed with PBS for 30 s, then stained with CEV solution for 30 s followed by a 30 s PBS rinse and DEPC water rinse for 15 s. The slide was washed with three consecutive ethanol rinses and air-dried for 10 minutes.
Laser capture microdissection
Zeiss Palm MicroBeam IV laser capture microdissection system was employed to cut areas of the brain tissue. The PEN membrane slides were placed onto the microscope slide adaptor and square sections (500 μm2) at specific regions of interest on the brain tissue were cut by the laser. To collect the isolated tissue sections, we used adhesive caps to stock the tissue sections on the adhesive caps. Ten brain sections (500 μm2 × 20 μm each) were collected and stored at −80 °C before further sample preparation for CZE-MS/MS analysis.
Sample preparation for CZE-MS/MS
Octyl glucopyranoside, OG, detergent was dissolved in 100 mM NH4HCO3 (pH 8) for cell lysis and the OG concentration was 4.5% (w/v). The tissue samples from the LCM were re-suspended in 5 μL of the lysis buffer by placing the lysis buffer directly onto the adhesive cap. The lysis buffer was then pipetted up and down several times, and the sample in the lysis buffer was transferred from the cap to the bottom of the Eppendorf tube via a quick centrifugation. The samples were further processed by two different approaches, Fig. 1. In one approach, we employed an on-ice sonication for 20 minutes (Branson Sonifier 250, VWR Scientific, Batavia, IL) for cell lysis and protein extraction. We prepared three brain sections using the sonication approach. In the other approach, another four samples originating from the same brain slice as the sonication method went through a repeated freeze/thaw protocol; the samples were placed into liquid nitrogen and thawed at 37 °C for 2 minutes for a total of 6 rounds of the freeze/thaw. The seven samples were marked in Fig. 2. All the samples were then spun down. Each sample was diluted by a factor of two with water to get a sample containing 50 mM NH4HCO3 (pH 8) and about 2% OG for CZE-MS/MS analysis.
Fig. 1.
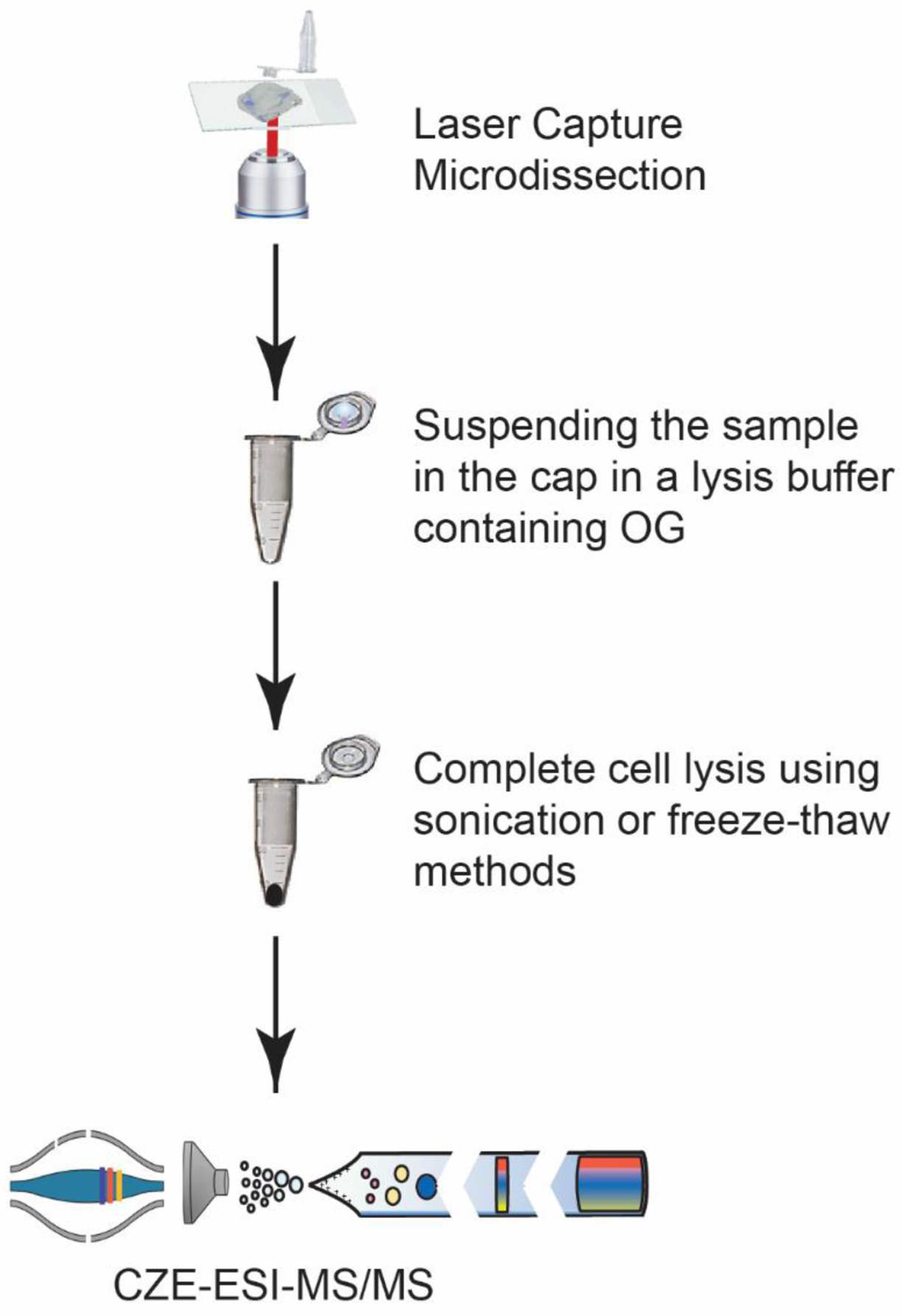
Schematic overview of the LCM-CZE-MS/MS workflow.
Fig. 2.
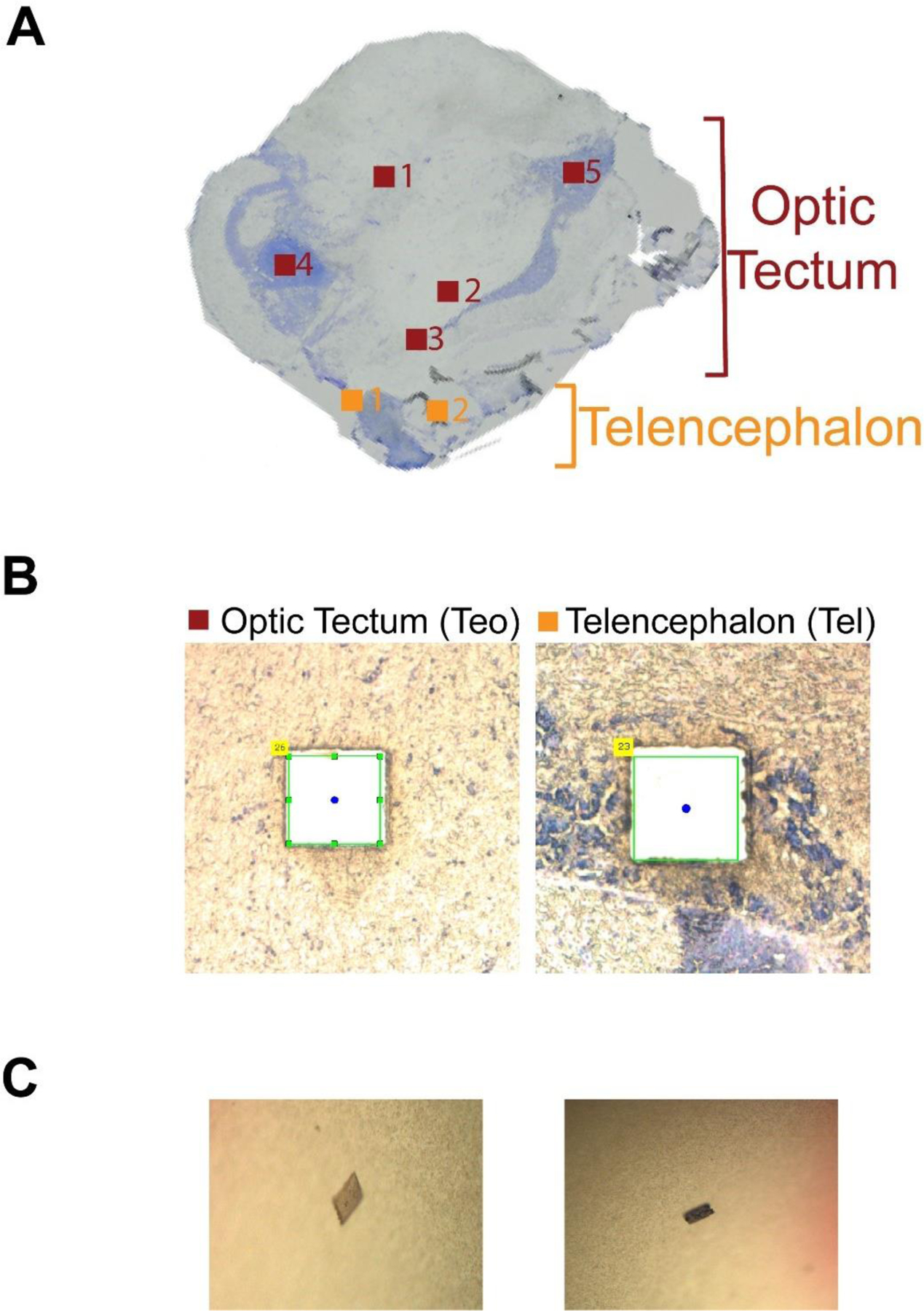
(A) An image of one 20 μm-thick zebrafish brain slice used in the study. The collected square regions of the zebrafish brain Optic Tectum (Teo) and Telencephalon (Tel) are highlighted. Each region has an area of 500 μm2. (B) The microscopic images of two square regions of Teo and Tel after the microdissection. (C) The corresponding tissue sections collected in (B) on the caps of Eppendorf tubes that were used for sample collection in the LCM experiment.
CZE-ESI-MS/MS
A CESI 8000 CE autosampler (Bruker) was used for automated CE operation. The CE autosampler was coupled to a Q-Exactive HF mass spectrometer (Thermo fisher scientific) through a commercialized electro-kinetically pumped sheath flow CE-MS interface (CMP Scientific, Brooklyn, NY).34,35 A fused silica capillary (50 μm i.d., 360 μm o.d., 75 cm in length) with a linear polyacrylamide (LPA) coating on the inner wall was used for the CZE separation. The outlet end of the capillary was etched with hydrofluoric acid to reduce the outer diameter. (Caution: use appropriate safety procedures while handling hydrofluoric acid solutions.) The CZE background electrolyte (BGE) was 10% acetic acid (pH ~ 2.2) and the sheath buffer 0.2% (v/v) formic acid containing 10% (v/v) methanol. ESI emitters were pulled to an opening of 30–40 μm using borosilicate glass capillaries (1.0 mm o.d., 0.75 mm i.d., and 10 cm length) with a Sutter P-1000 flaming/brown micropipette puller. Each CZE sample vial was first coated with 2 mg mL−1 bovine serum albumin (BSA) to reduce sample adsorption to the inner walls during analysis.36 The procedure is as follows: (1) 10 μL of 2 mg mL−1 BSA was placed into a sample vial, (2) the BSA solution was incubated at room temperature for 15 min, (3) the BSA solution was removed followed by three consecutive washes with 100 μL of 50 mM NH4HCO3 (pH 8), and (4) dried the vial with nitrogen. A blank run was done before the sample to ensure there was no residual BSA left within the sample vial. Sample injection was carried out by applying 5 psi for 70 s corresponding to ~370 nL of sample material injected onto the capillary. 30 kV was used for separation and was applied at the injection end of the capillary, while 2.0–2.2 kV was applied to the sheath buffer for ESI. After each CZE run, the capillary was taken out of the emitter and then flushed with the BGE by applying 20 psi pressure for 10 min to remove OG detergent from capillary and to ensure OG wasn’t being injected into the mass spectrometer. The capillary was then placed back into the emitter for the next CZE-MS/MS run.
A Q-Exactive HF mass spectrometer was operated in data-dependent acquisition mode; the top 5 most intense ions in one MS spectrum were sequentially isolated in the quadrupole and fragmented using higher-energy collision dissociation (HCD). Only charge states higher than 3 were selected for HCD fragmentation. MS was set to a mass resolution of 120 000 (at m/z 200), 3 microscans, 1 × 106 AGC target value, 50 ms maximum injection time, and 600–2000 m/z scan range. For the MS/MS, the mass resolution was 60 000, 3 microscans, AGC value of 1 × 105, 200 ms maximum injection time, 4 m/z isolation window, and a normalized collision energy (NCE) of 20%. The dynamic exclusion was set to 30 s, and the exclude isotopes function was on.
Data analysis
All samples were analyzed using Xcalibur software (Thermo fisher scientific) to get the intensity and migration time of proteoforms. The electropherograms were exported from Xcalibur and were formatted using Adobe Illustrator to report the final figures.
The LCM sample RAW files were analyzed using Top-down mass spectrometry based proteoform identification and characterization (TopPIC) platform for the proteoform identification and quantification.37 The RAW files were converted to mzML files using the MsConvert tool,38 and spectral deconvolution was done using the Top-down mass spectrometry feature detection (TopFD) to generate msalign files. The msalign files were used as the input for TopPIC (version 1.4.0) for database searching. UniProt databases for zebrafish (AUP000000437) were used for the database search. The maximum number of unexpected modifications was set to 2, and the maximum mass shift of unknown modification was set to 500 Da, while the precursor and fragment mass error tolerances were 15 ppm. The target decoy approach was used for the false discovery rate (FDR),39,40 and a 1% proteoform-spectrum match (PrSM) level FDR and a 5% proteoform level FDR were used to filter the PrSM and proteoform identifications. Label-free quantification for top-down proteomics was performed using the TopDiff tool integrated in the TopPIC platform.32
Results and discussion
Development of an OG-based sample preparation method for CZE-MS/MS-based top-down proteomics of mass-limited samples
In our previous work, we have shown that CZE-MS/MS identified hundreds to thousands of proteoforms in a single run with the consumption of nanograms of complex proteome samples, indicating its high potential for top-down proteomics of mass-limited samples.32 However, the sample preparation starting from cells or tissues needs to be improved substantially regarding sample recovery before CZE-MS/MS analysis. The traditional sample preparation procedure usually employs detergents for efficient proteoform extraction from cells or tissues. Because detergents are not compatible with MS measurement in general, some sample cleanup steps (e.g., buffer exchange and precipitation) are essential for removing the salts and detergents before MS analysis, which will lead to serious sample loss for mass-limited samples (e.g., laser capture microdissected tissue samples). Development of novel sample preparation procedures that can be coupled with CZE-MS/MS straightforwardly will be useful for advancing the top-down MS measurement of mass-limited samples.
CZE separates analytes according to their electrophoretic mobilities, which correspond to their charge-to-size ratios. When a separation capillary with neutral coating on its inner wall is used for CZE separation, the electro-osmotic flow in the capillary is eliminated. Under such conditions, the neutral analytes in the sample will not migrate under the electric field. Therefore, if a neutral detergent in a volatile salt buffer is used as a lysis buffer for cell lysis and proteoform extraction, the sample can be analyzed directly by CZE-MS/MS without sample cleanup because the neutral detergent molecules will not migrate towards the mass spectrometer during the CZE separation. The detergent can be flushed out of the capillary after each CZE-MS/MS run. This is an advantage of CZE-MS/MS over commonly used RPLC-MS/MS, because neutral detergents most likely will contaminate the RPLC-MS/MS system.
We tested the idea by using OG, a nonionic surfactant, because it has been successfully used for extraction of proteins even membrane proteins for proteomics.33 As shown in Fig. 1, a zebrafish brain section was isolated by LCM and collected by an adhesive lid of an Eppendorf tube. A 5 μL aliquot of the lysis buffer containing 4.5% (w/v) OG and 100 mM NH4HCO3 (pH 8.0) was added onto the lid to suspend the collected tissue section, which was then transferred to the bottom of the Eppendorf tube. We then employed sonication or freeze–thaw methods for complete cell lysis and proteoform extraction as described in the experimental part. After simple centrifugation, the supernatant was further diluted by a factor of two with water and analyzed by the dynamic pH junction-based CZE-MS/MS.41 About 370 nL of the sample was injected for CZE-MS/MS and the consumed sample amount was about 5% of the available sample. Each sample was analyzed in duplicate. To minimize sample loss due to the adsorption on the inner wall of the insert tube used for CZE-MS, we coated the inner wall with BSA according to our previous work.36 In total, seven zebrafish brain sections were collected by LCM from optic tectum (Teo, 5 sections) and telencephalon (Tel, 2 sections) of a 20 μm thick brain slice for the analysis, Fig. 2. Three samples (Teo3, Teo4, and Teo5) were prepared by the sonication method and four samples (Teo1, Teo2, Tel1, and Tel2) were processed by the freeze–thaw approach. Each LCM brain section had an area of 500 μm2, corresponding to roughly 5000 cells if we assume that each cell is about 10 μm in size.
Fewer than 40 proteoforms were identified from the three Teo samples prepared by the sonication method using CZE-MS/MS, Fig. 3A, and hundreds of proteoforms were identified from the four samples processed by the freeze–thaw procedure, Fig. 3B. The data clearly indicate that the freeze–thaw approach provided much better sample recovery compared to the sonication method. The low sample recovery from sonication is most likely due to the protein adsorption onto the inner wall of the Eppendorf tube because of splashing caused by the vibrations. For the freeze–thaw method, the sample was well controlled at the bottom of the Eppendorf tube, significantly minimizing the surface area that the sample could interact with.
Fig. 3.
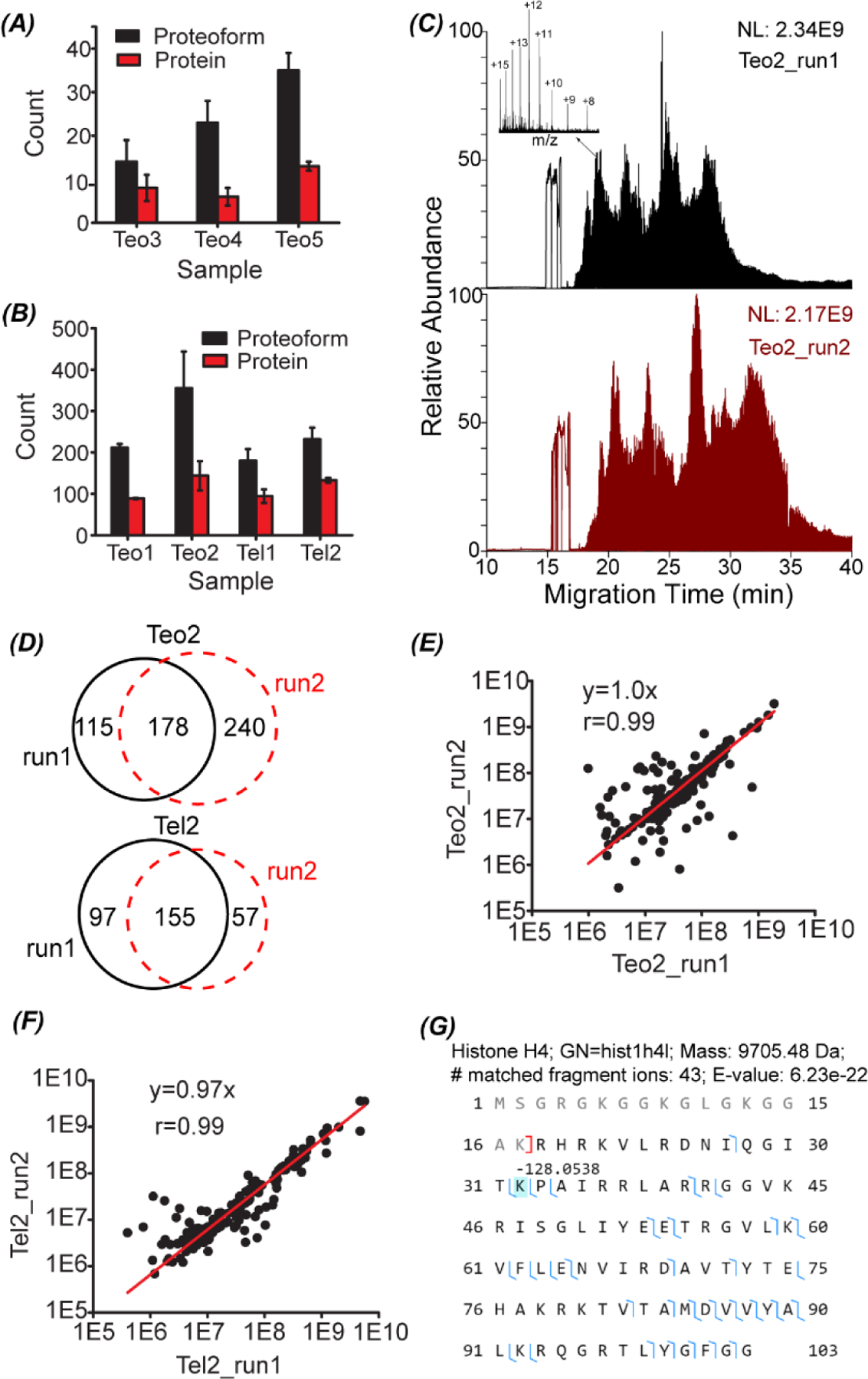
The number of proteoform and protein identifications from the LCM-CZE-MS/MS experiments using the OG-sonication sample preparation method (A) and OG-freeze–thaw method (B). The error bars represent the standard deviations of the number of protein and proteoform identifications from duplicate CZE-MS/MS runs. (C) Total ion current (TIC) electropherograms of the Teo2 sample prepared by the OG-freeze–thaw method and analyzed by CZE-MS/MS in duplicate. One example proteoform mass spectrum is shown in the inserted figure. (D) Overlaps of identified proteoforms between duplicate CZE-MS/MS analyses of Teo2 and Tel2 samples prepared by the OG-freeze–thaw method. Correlations of proteoform intensity between duplicate analyses of Teo2 (E) and Tel2 (F) samples. (G) Sequence and fragmentation pattern of one identified histone H4 proteoform from the Teo2 sample.
Fig. 3C shows the total ion current (TIC) electropherograms of the sample Teo2 after preparation with the freeze–thaw approach and analysis by CZE-MS/MS in duplicate. The dynamic pH junction-based CZE-MS/MS showed reasonably good reproducibility for the analyses of the Teo2 sample regarding the separation profile and TIC intensity. 356 ± 88 proteoforms were identified from the Teo2 sample and 43–60% of the proteoforms identified in one run were also identified in the other run, Fig. 3D. For the Tel2 sample, 232 ± 28 proteoforms were identified and about 50% of the identified proteoforms were overlapped between the duplicate runs, Fig. 3D. The proteoform intensities display nice linear correlations between the duplicate CZE-MS/MS analyses for the Teo and Tel samples, Fig. 3E and F. All the data suggest that the dynamic pH junction-based CZE-MS/MS system enabled the reproducible characterization of Teo2 and Tel2 samples even when the samples contained about 2% OG detergent. We also need to point out that we did not see clear signal of the detergent during the CZE runs because the detergent is neutral and did not migrate in the capillary. The CZE-MS/MS analyses achieved clear mass spectra of proteoforms and the insert figure in Fig. 3C shows an example mass spectrum with a clear charge state distribution from +8 to +16, which corresponds to one proteoform of histone H4 with the first 17 amino acid residues truncated and a −128 Da mass shift at the lysine residue at the position 32, Fig. 3G. The mass shift most likely corresponded to the deletion of the lysine residue. The proteoform was identified with 43 matched fragment ions and a 6.23 × 10e–22 E-value. The data indicate that the detergent OG did not interfere with the detection and high-confident delineation of proteoforms.
We need to highlight that our top-down proteomics workflow including the OG and freeze–thaw-based sample preparation method and dynamic pH junction-based CZE-MS/MS has extremely high overall sensitivity for proteoform identification from mass-limited samples. In our experiment, each LCM isolated brain section contained roughly 5000 cells based on the assumption that each cell is about 10 μm in size. About 5% of the sample was injected for CZE-MS/MS analysis, corresponding to the protein content of 250 cells. We identified up to 418 proteoforms in a single CZE-MS/MS run from the 250 cells in the Teo2. Based on our knowledge, the data represent one of the two examples of top-down proteomics of a small number of cells. In another example, Zhou et al. applied the NanoPOTS sample preparation method and nanoRPLC-MS/MS platform (Orbitrap lumos tribrid mass spectrometer) for top-down proteomics of hundreds of HeLa cells with the production of 170 to 620 proteoforms from 70 to 770 HeLa cells.27 We expect that our OG-freeze–thaw-CZE-MS/MS platform will become a useful tool for advancing top-down proteomics towards large-scale proteoform profiling of mass-limited samples.
Comparison of the proteoform profiles of zebrafish brain LCM sections isolated from Teo and Tel regions
The zebrafish Tel region is responsible for receiving and processing sensory information and directing behavior. It relates to the regulation of social behavior, memory, and emotion.42–44 The Teo region organizes the saccadic/microsaccadic eye movements of zebrafish and modulates the motor functions of prey-catching and avoidance behaviours.45–47 It plays a vital role in transforming visual information to motor activities. Because the significant functional difference between the Tel and Teo regions, we speculated that their proteoform profiles have drastic differences.
As shown in Fig. 3, we have achieved reasonably reproducible measurement of proteoforms from the Teo2 and Tel2 samples isolated by LCM using our OG-freeze–thaw-CZE-MS/MS method, which allowed us to compare their proteoform profiles with high confidence. The identified proteoforms from the Teo2 and Tel2 samples are listed in the ESI.† The MS raw data of Teo2 and Tel2 samples have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository48 with the data set identifier PXD027741. 309 and 533 proteoforms were identified from the Tel2 and Teo2 samples in duplicate CZE-MS/MS runs (technical replicates). Only 35 proteoforms were identified from both samples and the protein-level overlap was about 22% (60/279), Fig. 4A. The Tel2 and Teo2 samples also showed drastic differences in their electropherograms, Fig. 4B. The data clearly demonstrate the drastic proteome differences between the Teo and Tel regions.
Fig. 4.
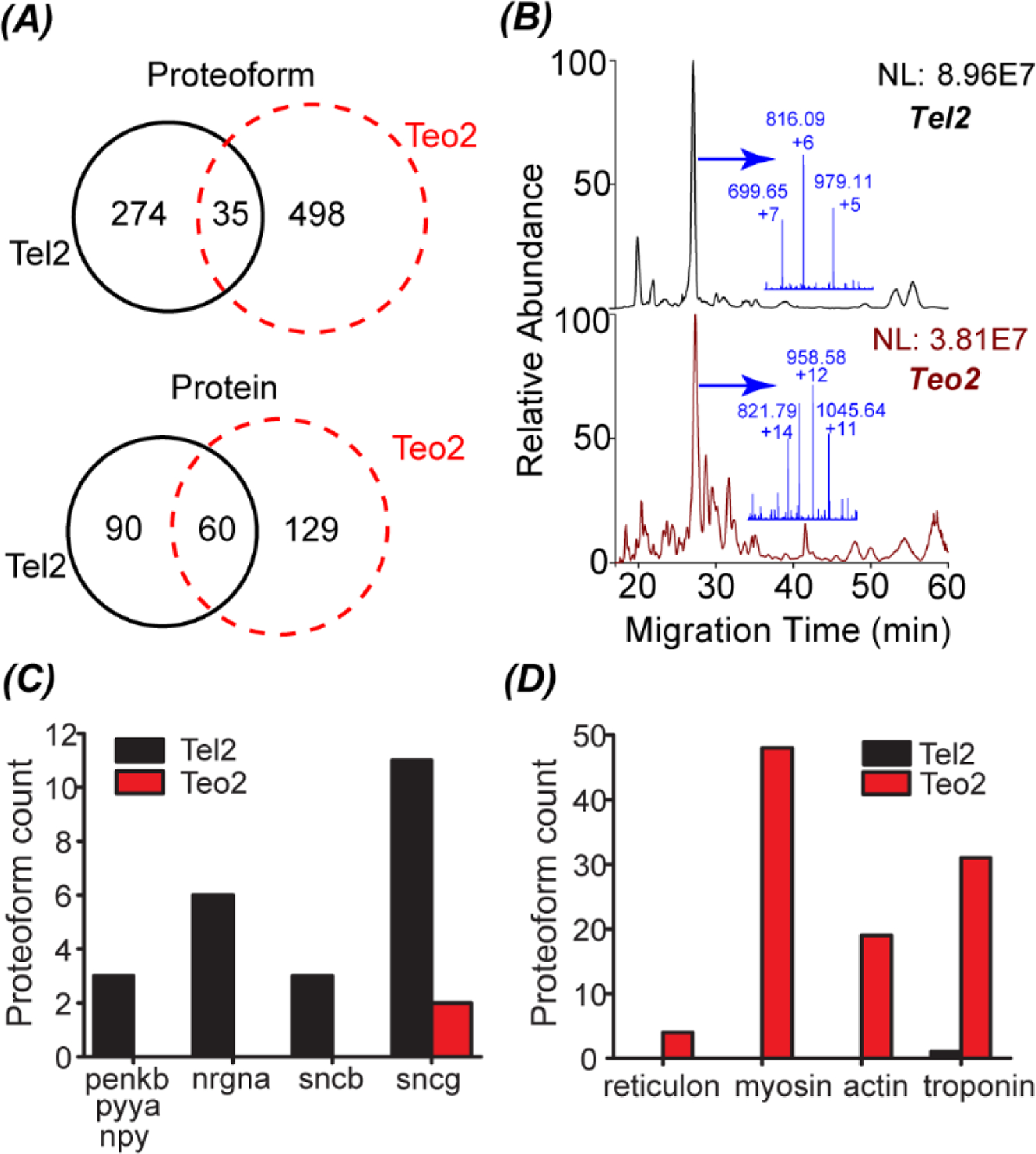
(A) Overlaps of identified proteoforms and proteins from Tel2 and Teo2 samples. The data was from the duplicate CZE-MS/MS analyses (technical replicates). (B) Base peak electropherograms of the Tel2 and Teo2 samples with the mass spectrum of the most abundant peak highlighted in the inserted figure. (C) The number of identified proteoforms of several genes from the Tel2 and Teo2 samples. For those genes, the Tel2 sample had much more proteoforms than the Teo2 sample. (D) The number of identified proteoforms of several genes from the Tel2 and Teo2 samples. For those genes, the Teo2 sample had much more proteoforms than the Tel2 sample.
We identified three proteoforms of three genes in neuropeptide signaling pathway in the Tel2 sample but not in the Teo2 sample. Those three proteins are Pro-neuropeptide Y (npy), Synenkephalin (penkb), and Peptide YY-A (pyya). They have neuropeptide hormone activity. The npy stimulates food intake of animals, e.g., decreasing latency or increasing the motivation to eat.49 Peptide YY also relates to the regulation of food intake.50 Synenkephalin could modulate the pain responses, anxiety and aggression in zebrafish based on the mice data.51 We also identified 6 proteoforms of neurogranin a (nrgna) from the Tel2 sample and did not identify any nrgna proteoforms in the Teo2 sample. It has been demonstrated that neurogranin plays important roles in memory formation and overall performance.52,53 The neuropeptide hormone and nrgna data (Fig. 4C) agree well with the function of Tel that relates to regulation of emotion and memory.
Synuclein proteins have been well studied for their roles in the neurodegenerative diseases, e.g., Parkinson’s disease (PD) and Alzheimer’s disease (AD).54,55 Patients with AD and late-stage PD both have difficulties in normal mental functions including memory. Interestingly, we identified 14 synuclein proteoforms in the Tel2 sample including 3 beta-synuclein (sncb) proteoforms and 11 gamma-synuclein (sncga and sncgb). We only identified 2 synuclein proteoforms (sncgb) from the Teo2 sample, Fig. 4C. Because zebrafish does not have the alpha-synuclein,56 we did not identify any alpha-synuclein proteoforms. Our data suggest the heterogenous distribution of synuclein proteoforms in the zebrafish brain.
The Teo region plays important roles in converting visual information to motor activities. We identified 4 proteoforms of reticulon exclusively in the Teo2 sample. Reticulon is crucial for the normal development of retina and Teo and influences the retinotectal projection drastically.57 We also identified 48 proteoforms of myosin in the Teo2 sample and did not identify any myosin proteoforms in the Tel2 sample. Myosin are well known as motor proteins. Actin filaments interact with myosin, generating many cellular movements. Nineteen actin proteoforms were only identified in the Teo2 sample. We detected 31 and 1 troponin proteoforms in the Teo2 and Tel2 sample, respectively. Troponin are a group of proteins regulate muscular contraction. The proteoform data mentioned are shown in Fig. 4D. The proteoform profile of the Teo2 sample agrees well with the basic functions of Teo.
As discussed above, our top-down MS data of the Tel2 and Teo2 samples enable the clear distinction of the Tel and Teo regions because of their drastic differences in proteoform profiles. Importantly, the proteoform profiles of Tel2 and Teo2 accurately reflect the basic functions of Tel and Teo of the zebrafish brain. This study represents the first example of coupling LCM to CZE-MS/MS for spatially resolved top-down proteomics.
Proteoforms with PTMs
In total, we identified 807 proteoforms from the Tel2 and Teo2 samples. 449 proteoforms have no mass shifts, indicating without any PTMs. 358 proteoforms have mass shifts, corresponding to some kinds of modifications, e.g., methylation (+14 Da), di-methylation (+28 Da), tri-methylation (+42 Da), acetylation (+42 Da), oxidation (+16 Da), and phosphorylation (+80 Da). We tried to match the identified proteoforms to the recently reported five-level classification system.58 We noted that some truncated proteoforms identified here could match to multiple protein sequences in the database because those proteins had similar amino acid sequences, for example, different isoforms of the same gene. Considering this ambiguity, about 60% of those 449 proteoforms without mass shifts are well characterized and belong to the Level-1 identifications.58 All other proteoforms belong to the Level-1 to Level-5 identifications depending on the ambiguity at the PTM, protein sequence, and gene levels.58
Fig. 5A summaries the identified proteoforms containing N-terminal acetylation, methylation (mono, di, and tri), oxidation, phosphorylation (phospho), and disulfide bond. We identified 211 N-terminal acetylated proteoforms, 12 methylated proteoforms, 25 oxidized proteoforms, 3 phosphorylated proteoforms, and 15 proteoforms containing one or two disulfide bonds. The unique advantage of top-down proteomics is the capability for characterizing the combination of PTMs on individual proteoforms. Some example proteoforms having multiple PTMs are shown in Fig. 5B–D. We identified one proteoform of Thymosin beta containing both acetylation and accurately localized phosphorylation, Fig. 5B. We also identified one proteoform of Calmodulin containing N-terminal acetylation and another 42 Da mass shift in the region of positions 67–89, Fig. 5D. The 42 Da mass shift corresponds to acetylation or tri-methylation PTM. Because Thymosin beta and Calmodulin play important roles in the organization of the cytoskeleton and regulating the control of many enzymes, ion channels, and other proteins through Ca2+, respectively, delineation of their proteoforms are critical for understanding their biological functions accurately. In the literature, there are no direct experimental evidence for the identified proteoforms of Thymosin beta and Calmodulin with the specific combinations of PTMs in zebrafish according to the information in the UniProt database. Here we provided confident experimental evidence for those zebrafish proteoforms for the first time. Additionally, we identified one proteoform of Mitochondrial import inner membrane translocase subunit Tim10 and the proteoform had both N-terminal acetylation and two disulfide bonds, Fig. 5C. The two disulfide bonds were determined on the four highlighted cysteine residues according to a −4 Da mass shift. The proteoform containing both N-terminal acetylation and disulfide bonds was not reported in the literature for zebrafish according to UniProt.
Fig. 5.
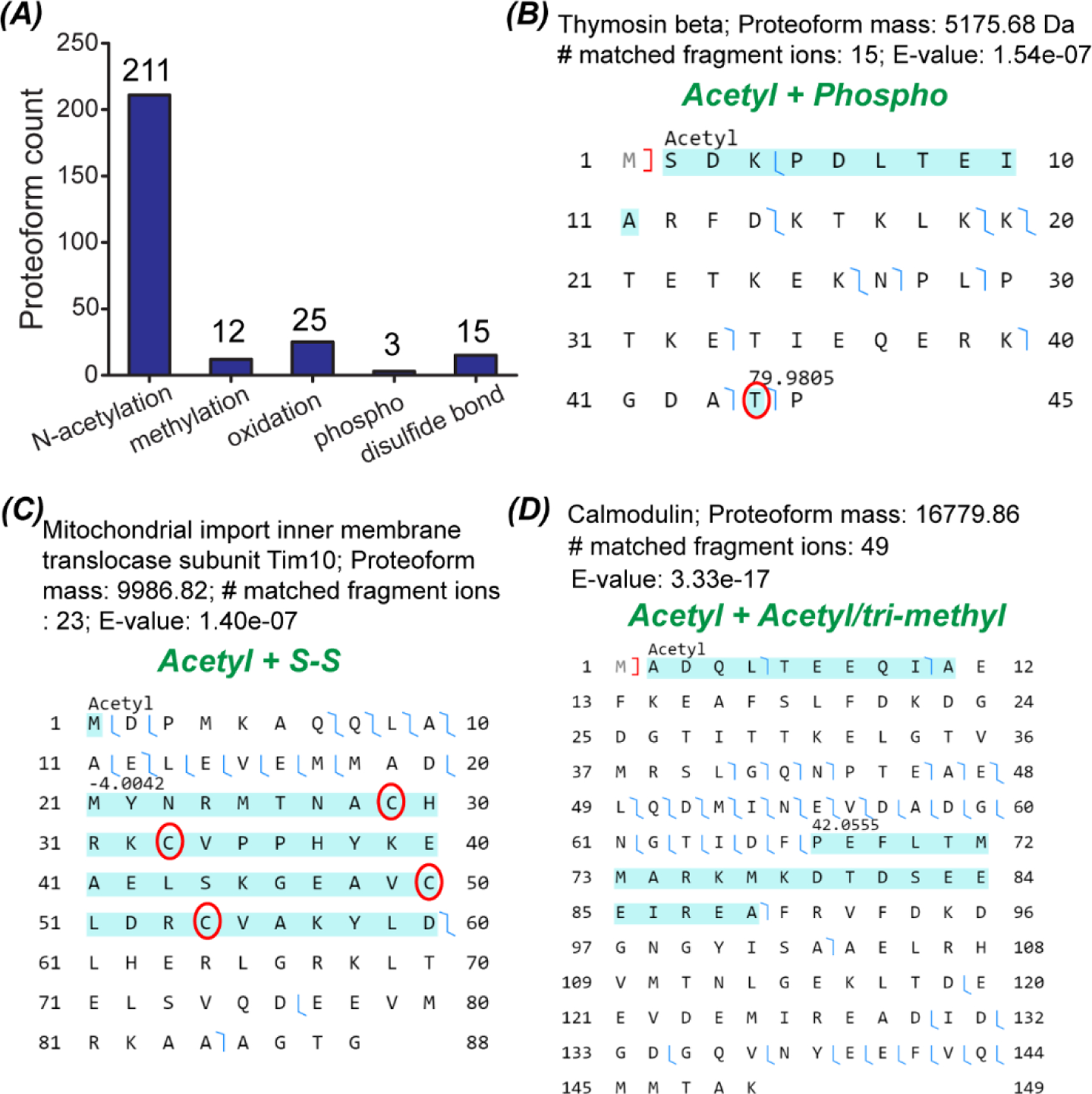
Data about proteoforms with PTMs. (A) The number of proteoforms with various PTMs identified from the Tel2 and Teo2 samples. (B–D) Sequences and fragmentation patterns of three proteoforms with combinations of various PTMs.
Conclusions
In this pilot study, we presented the first example of LCM-CZE-MS/MS for spatially resolved top-down proteomics. The LCMCZE-MS/MS utilized a non-ionic detergent OG and a freeze–thaw method for cell lysis and proteoform extraction from LCM isolated zebrafish brain sections followed by direct measurements by dynamic pH junction-based CZE-MS/MS. This workflow eliminated sample cleanup before MS and MS/MS analysis resulting in high overall sensitivity. For example, up to 418 proteoforms were identified in a single CZE-MS/MS analysis from roughly 250 cells in the zebrafish brain. The proteoform profiles of two LCM brain sections Teo2 and Tel2 accurately reflected the basic biological functions of Teo and Tel regions. Overall, the results demonstrate the great potential of the LCM-CZE-MS/MS workflow for qualitative and even quantitative top-down proteomics of mass-limited and spatially resolved protein samples. Coupling the OG and freeze–thaw-based sample preparation to CZE-MS/MS could also be a useful tool for top-down proteomics of a small number of cells.
Some further improvements to the LCM-CZE-MS/MS workflow need to be made. First, the non-ionic detergent and freeze–thaw-based sample preparation can be systematically optimized to maximize the proteoform extraction from cells. Second, the mass of proteoforms identified from the zebrafish brain LCM sections (Teo2 and Tel2) ranged from 1–18 kDa and over 80% of the identified proteoforms had masses lower than 10 kDa. The data demonstrates the challenge of our method for the identification of relatively large proteoforms from the LCM sections. Third, we noted that the migration time of proteoforms could have drastic changes across different CZE-MS/MS runs for the same sample prepared by the OG-freeze–thaw method. The phenomenon is most likely due to the fact that the detergent OG and the complex sample matrix (DNA, RNA, lipid, etc.) could influence the capillary inner-wall surface, leading to drastic changes of proteoforms’ migration time and even separation profile. Some efficient approaches for regular capillary inner wall cleanup need to be deployed. We expect that the optimized workflow, especially with a high-end orbitrap mass spectrometer, will be a powerful tool for spatially resolved top-down proteomics with high spatial resolution as well as for large-scale top-down proteomics of a small number of cells.
Supplementary Material
Acknowledgements
We thank Prof. Jose Cibelli and Mr Billy Poulos at the Department of Animal Science of Michigan State University for their help on collecting zebrafish brains for the project. We would also like to thank Ms Amy Porter from the Histology lab at Michigan State University for her help with the cryosection of the Zebrafish brain and Dr Melinda Frame from the Center for Advanced Microscopy from Michigan State University for her help with the microdissecting of the brain tissue using LCM. We thank Dr Xiaowen Liu at the Tulane University for valuable discussions about proteoform identifications. We thank the support from the National Institute of General Medical Sciences (NIGMS) through Grant R01GM125991 and the National Science Foundation through Grant DBI1846913 (CAREER Award).
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mo00335f
Conflicts of interest
There are no conflicts of interest to declare.
Data availability
The MS raw data of Teo2 and Tel2 samples have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD027741.
References
- 1.Davis S, Scott C, Ansorge O and Fischer R, Development of a Sensitive, Scalable Method for Spatial, Cell-Type-Resolved Proteomics of the Human Brain, J. Proteome Res, 2019, 18(4), 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delcourt V, Franck J, Quanico J, Gimeno J-P, Wisztorski M, Raffo-Romero A, Kobeissy F, Roucou X, Salzet M and Fournier I, Spatially-Resolved Top-down Proteomics Bridged to MALDI MS Imaging Reveals the Molecular Physiome of Brain Regions, Mol. Cell. Proteomics, 2018, 17(2), 357–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu Y, Dou M, Piehowski PD, Liang Y, Wang F, Chu RK, Chrisler WB, Smith JN, Schwarz KC, Shen Y, Shukla AK, Moore RJ, Smith RD, Qian W-J and Kelly RT, Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets, Mol. Cell. Proteomics, 2018, 17(9), 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clair G, Piehowski PD, Nicola T, Kitzmiller JA, Huang EL, Zink EM, Sontag RL, Orton DJ, Moore RJ, Carson JP, Smith RD, Whitsett JA, Corley RA, Ambalavanan N and Ansong C, Spatially-Resolved Proteomics: Rapid Quantitative Analysis of Laser Capture Microdissected Alveolar Tissue Samples, Sci. Rep, 2016, 6, 39223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satija R, Farrell JA, Gennert D, Schier AF and Regev A, Spatial Reconstruction of Single-Cell Gene Expression Data, Nat. Biotechnol, 2015, 33(5), 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hosp F, Gutiérrez-Ángel S, Schaefer MH, Cox J, Meissner F, Hipp MS, Hartl F-U, Klein R, Dudanova I and Mann M, Spatiotemporal Proteomic Profiling of Huntington’s Disease Inclusions Reveals Widespread Loss of Protein Function, Cell Rep, 2017, 21(8), 2291–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mao Y, Wang X, Huang P and Tian R, Spatial Proteomics for Understanding the Tissue Microenvironment, Analyst, 2021, 146(12), 3777–3798. [DOI] [PubMed] [Google Scholar]
- 8.Carlyle BC, Kitchen RR, Kanyo JE, Voss EZ, Pletikos M, Sousa AMM, Lam TT, Gerstein MB, Sestan N and Nairn AC, A Multiregional Proteomic Survey of the Postnatal Human Brain, Nat. Neurosci, 2017, 20(12), 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch U-K, Philips M-A, Rossner MJ, Mann M and Simons M, Cell Type- and Brain Region-Resolved Mouse Brain Proteome, Nat. Neurosci, 2015, 18(12), 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKetney J, Runde RM, Hebert AS, Salamat S, Roy S and Coon JJ, Proteomic Atlas of the Human Brain in Alzheimer’s Disease, J. Proteome Res, 2019, 18(3), 1380–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doll S, Dreßen M, Geyer PE, Itzhak DN, Braun C, Doppler SA, Meier F, Deutsch M-A, Lahm H, Lange R, Krane M and Mann M, Region and Cell-Type Resolved Quantitative Proteomic Map of the Human Heart, Nat. Commun, 2017, 8(1), 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dyring-Andersen B, Løvendorf MB, Coscia F, Santos A, Møller LBP, Colaço AR, Niu L, Bzorek M, Doll S, Andersen JL, Clark RA, Skov L, Teunissen MBM and Mann M, Spatially and Cell-Type Resolved Quantitative Proteomic Atlas of Healthy Human Skin, Nat. Commun, 2020, 11(1), 5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu R, Tang J, Deng Q, He W, Sun X, Xia L, Cheng Z, He L, You S, Hu J, Fu Y, Zhu J, Chen Y, Gao W, He A, Guo Z, Lin L, Li H, Hu C and Tian R, Spatial Resolution Cell Type Proteome Profiling of Cancer Tissue by Fully Integrated Proteomics Technology, Anal. Chem, 2018, 90(9), 5879–5886. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Y, Dou M, Piehowski PD, Liang Y, Wang F, Chu RK, Chrisler WB, Smith JN, Schwarz KC, Shen Y, Shukla AK, Moore RJ, Smith RD, Qian WJ and Kelly RT, Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets, Mol. Cell. Proteomics, 2018, 17(9), 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piehowski PD, Zhu Y, Bramer LM, Stratton KG, Zhao R, Orton DJ, Moore RJ, Yuan J, Mitchell HD, Gao Y, Webb-Robertson B-JM, Dey SK, Kelly RT and Burnum-Johnson KE, Automated Mass Spectrometry Imaging of over 2000 Proteins from Tissue Sections at 100 Mm Spatial Resolution, Nat. Commun, 2020, 11(1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith LM and Kelleher NL, Proteoforms as the next Proteomics Currency, Science, 2018, 359(6380), 1106–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo H, Hernandez R, Hong H, Graves SA, Yang Y, England CG, Theuer CP, Nickles RJ and Cai W, Noninvasive brain cancer imaging with a bispecific antibody fragment, generated via click chemistry, Proc. Natl. Acad. Sci. U. S. A, 2015, 112(41), 12806–12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maric D, Jahanipour J, Li XR, Singh A, Mobiny A, Nguyen HV, Sedlock A, Grama K and Roysam B, Whole-brain tissue mapping toolkit using large-scale highly multiplexed immunofluorescence imaging and deep neural networks, Nat. Commun, 2021, 12(1), 1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toby TK, Fornelli L and Kelleher NL, Progress in Top-down Proteomics and the Analysis of Proteoforms, Annu. Rev. Anal. Chem, 2016, 9(1), 499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tucholski T, Cai W, Gregorich ZR, Bayne EF, Mitchell SD, McIlwain SJ, de Lange WJ, Wrobbel M, Karp H, Hite Z, Vikhorev PG, Marston SB, Lal S, Li A, Dos Remedios C, Kohmoto T, Hermsen J, Ralphe JC, Kamp TJ, Moss RL and Ge Y, Distinct Hypertrophic Cardiomyopathy Genotypes Result in Convergent Sarcomeric Proteoform Profiles Revealed by Top-down Proteomics, Proc. Natl. Acad. Sci. U. S. A, 2020, 117(40), 24691–24700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cupp-Sutton KA and Wu S, High-throughput quantitative top-down proteomics, Mol. Omics, 2020, 16(2), 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM and Kelleher NL, Mapping Intact Protein Isoforms in Discovery Mode Using Top-down Proteomics, Nature, 2011, 480(7376), 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durbin KR, Fornelli L, Fellers RT, Doubleday PF, Narita M and Kelleher NL, Quantitation and Identification of Thousands of Human Proteoforms below 30 KDa, J. Proteome Res, 2016, 15(3), 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Y, Tolić N, Piehowski PD, Shukla AK, Kim S, Zhao R, Qu Y, Robinson E, Smith RD and Paša-Tolić L, High-Resolution Ultrahigh-Pressure Long Column Reversed-Phase Liquid Chromatography for Top-down Proteomics, J. Chromatogr. A, 2017, 1498, 99–110. [DOI] [PubMed] [Google Scholar]
- 25.Anderson LC, DeHart CJ, Kaiser NK, Fellers RT, Smith DF, Greer JB, LeDuc RD, Blakney GT, Thomas PM, Kelleher NL and Hendrickson CL, Identification and Characterization of Human Proteoforms by Top-down LC-21 Tesla FT-ICR Mass Spectrometry, J. Proteome Res, 2017, 16(2), 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park HM, Satta R, Davis RG, Goo YA, LeDuc RD, Fellers RT, Greer JB, Romanova EV, Rubakhin SS, Tai R, Thomas PM, Sweedler JV, Kelleher NL, Patrie SM and Lasek AW, Multidimensional Top-Down Proteomics of Brain-Region-Specific Mouse Brain Proteoforms Responsive to Cocaine and Estradiol, J. Proteome Res, 2019, 18(11), 3999–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou M, Uwugiaren N, Williams SM, Moore RJ, Zhao R, Goodlett D, Dapic I, Paša-Tolić L and Zhu Y, Sensitive Top-down Proteomics Analysis of a Low Number of Mammalian Cells Using a Nanodroplet Sample Processing Platform, Anal. Chem, 2020, 92(10), 7087–7095. [DOI] [PubMed] [Google Scholar]
- 28.Shen X, Yang Z, McCool EN, Lubeckyj RA, Chen D and Sun L, Capillary Zone Electrophoresis-Mass Spectrometry for Top-down Proteomics, Trends Anal. Chem, 2019, 120(115644), 115644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han X, Wang Y, Aslanian A, Fonslow B, Graczyk B, Davis TN and Yates JR, In-Line Separation by Capillary Electrophoresis Prior to Analysis by Top-down Mass Spectrometry Enables Sensitive Characterization of Protein Complexes, J. Proteome Res, 2014, 13(12), 6078–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCool EN and Liangliang S, Comparing Nanoflow Reversed-Phase Liquid Chromatography-Tandem Mass Spectrometry and Capillary Zone Electrophoresis-Tandem Mass Spectrometry for Top-down Proteomics, Se Pu., 2019, 37(8), 878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen D, McCool EN, Yang Z, Shen X, Lubeckyj RA, Xu T, Wang Q and Sun L, Recent Advances (2019–2021) of Capillary Electrophoresis-mass Spectrometry for Multilevel Proteomics, Mass Spectrom. Rev, 2021, DOI: 10.1002/mas.21714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lubeckyj RA, Basharat AR, Shen X, Liu X and Sun L, Large-Scale Qualitative and Quantitative Top-down Proteomics Using Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry with Nanograms of Proteome Samples, J. Am. Soc. Mass Spectrom, 2019, 30(8), 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shevchenko G, Musunuri S, Wetterhall M and Bergquist J, Comparison of Extraction Methods for the Comprehensive Analysis of Mouse Brain Proteome Using Shotgun-Based Mass Spectrometry, J. Proteome Res, 2012, 11(4), 2441–2451. [DOI] [PubMed] [Google Scholar]
- 34.Sun L, Zhu G, Zhang Z, Mou S and Dovichi NJ, Third-Generation Electrokinetically Pumped Sheath-Flow Nanospray Interface with Improved Stability and Sensitivity for Automated Capillary Zone Electrophoresis-Mass Spectrometry Analysis of Complex Proteome Digests, J. Proteome Res, 2015, 14(5), 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wojcik R, Dada OO, Sadilek M and Dovichi NJ, Simplified Capillary Electrophoresis Nanospray Sheath-Flow Interface for High Efficiency and Sensitive Peptide Analysis: Capillary Electrophoresis Electrospray Interface, Rapid Commun. Mass Spectrom, 2010, 24(17), 2554–2560. [DOI] [PubMed] [Google Scholar]
- 36.Yang Z, Shen X, Chen D and Sun L, Improved Nanoflow RPLC-CZE-MS/MS System with High Peak Capacity and Sensitivity for Nanogram Bottom-up Proteomics, J. Proteome Res, 2019, 18(11), 4046–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kou Q, Xun L and Liu X, TopPIC: A Software Tool for Top-down Mass Spectrometry-Based Proteoform Identification and Characterization, Bioinformatics, 2016, 32(22), 3495–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kessner D, Chambers M, Burke R, Agus D and Mallick P, ProteoWizard: Open Source Software for Rapid Proteomics Tools Development, Bioinformatics, 2008, 24(21), 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keller A, Nesvizhskii AI, Kolker E and Aebersold R, Empirical Statistical Model to Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search, Anal. Chem, 2002, 74(20), 5383–5392. [DOI] [PubMed] [Google Scholar]
- 40.Elias JE and Gygi SP, Target-Decoy Search Strategy for Increased Confidence in Large-Scale Protein Identifications by Mass Spectrometry, Nat. Methods, 2007, 4(3), 207–214. [DOI] [PubMed] [Google Scholar]
- 41.Lubeckyj RA, McCool EN, Shen X, Kou Q, Liu X and Sun L, Single-Shot Top-down Proteomics with Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry for Identification of Nearly 600 Escherichia Coli Proteoforms, Anal. Chem, 2017, 89(22), 12059–12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinozuka K and Watanabe S, Effects of Telencephalic Ablation on Shoaling Behavior in Goldfish, Physiol. Behav, 2004, 81(1), 141–148. [DOI] [PubMed] [Google Scholar]
- 43.Teles MC, Cardoso SD and Oliveira RF, Social Plasticity Relies on Different Neuroplasticity Mechanisms across the Brain Social Decision-Making Network in Zebrafish, Front. Behav. Neurosci, 2016, 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stednitz SJ, McDermott EM, Ncube D, Tallafuss A, Eisen JS and Washbourne P, Forebrain Control of Behaviorally Driven Social Orienting in Zebrafish, Curr. Biol, 2018, 28(15), 2445–2451.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen JD and Castro-Alamancos MA, Neural Correlates of Active Avoidance Behavior in Superior Colliculus, J. Neurosci, 2010, 30(25), 8502–8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krauzlis RJ, Lovejoy LP and Zénon A, Superior Colliculus and Visual Spatial Attention, Annu. Rev. Neurosci, 2013, 36, 165–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunn TW, Gebhardt C, Naumann EA, Riegler C, Ahrens MB, Engert F and Del Bene F, Neural Circuits Underlying Visually Evoked Escapes in Larval Zebrafish, Neuron, 2016, 89(3), 613–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz S, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A and Vizcaíno JA, The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data, Nucleic Acids Res, 2019, 47(D1), 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beck B, Neuropeptide Y in Normal Eating and in Genetic and Dietary-Induced Obesity, Philos. Trans. R. Soc., B, 2006, 361(1471), 1159–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan P, Jia J, Yang G, Wang D, Sun C and Li W, Duplication of Neuropeptide Y and Peptide YY in Nile Tilapia Oreochromis Niloticus and Their Roles in Food Intake Regulation, Peptides, 2017, 88, 97–105. [DOI] [PubMed] [Google Scholar]
- 51.König M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ and Zimmer A, Pain Responses, Anxiety and Aggression in Mice Deficient in Pre-Proenkephalin, Nature, 1996, 383(6600), 535–538. [DOI] [PubMed] [Google Scholar]
- 52.Casaletto KB, Elahi FM, Bettcher BM, Neuhaus J, Bendlin BB, Asthana S, Johnson SC, Yaffe K, Carlsson C, Blennow K, Zetterberg H and Kramer JH, Neurogranin, a Synaptic Protein, Is Associated with Memory Independent of Alzheimer Biomarkers, Neurology, 2017, 89(17), 1782–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gribaudo S, Saraulli D, Nato G, Bonzano S, Gambarotta G, Luzzati F, Costanzi M, Peretto P, Bovetti S and De Marchis S, Neurogranin Regulates Adult-Born Olfactory Granule Cell Spine Density and Odor-Reward Associative Memory in Mice, Int. J. Mol. Sci, 2021, 22(8), 4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Twohig D and Nielsen HM, α-Synuclein in the Pathophysiology of Alzheimer’s Disease, Mol. Neurodegener, 2019, 14(1), 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meade RM, Fairlie DP and Mason JM, Alpha-Synuclein Structure and Parkinson’s Disease - Lessons and Emerging Principles, Mol. Neurodegener, 2019, 14(1), 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toni M and Cioni C, Fish Synucleins: An Update, Mar. Drugs, 2015, 13(11), 6665–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinzón-Olejua A, Welte C, Abdesselem H, Málaga-Trillo E and Stuermer CA, Essential Roles of Zebrafish Rtn4/Nogo Paralogues in Embryonic Development, Neural Dev., 2014, 9(1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith LM, Thomas PM, Shortreed MR, Schaffer LV, Fellers RT, LeDuc RD, Tucholski T, Ge Y, Agar JN, Anderson LC, Chamot-Rooke J, Gault J, Loo JA, Paša-Tolić L, Robinson CV, Schlüter H, Tsybin YO, Vilaseca M, Vizcaíno JA, Danis PO and Kelleher NL, A Five-Level Classification System for Proteoform Identifications, Nat. Methods, 2019, 16(10), 939–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS raw data of Teo2 and Tel2 samples have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD027741.