

Abstract
Cutaneous T‐cell lymphomas (CTCLs) are telomerase‐positive tumors expressing hTERT, although neither gene rearrangement/amplification nor promoter hotspot mutations could explain the hTERT re‐expression. As the hTERT promoter is rich in CpG, we investigated the contribution of epigenetic mechanisms in its re‐expression. We analyzed hTERT promoter methylation status in CTCL cells compared with healthy cells. Gene‐specific methylation analyses revealed a common methylation pattern exclusively in tumor cells. This methylation pattern encompassed a hypermethylated distal region from −650 to −150 bp and a hypomethylated proximal region from −150 to +150 bp. Interestingly, the hypermethylated region matches with the recently named TERT hypermethylated oncogenic region (THOR). THOR has been associated with telomerase reactivation in many cancers, but it has so far not been reported in cutaneous lymphomas. Additionally, we assessed the effect of THOR on two histone deacetylase inhibitors (HDACi), romidepsin and vorinostat, both approved for CTCL treatment and a DNA methyltransferase inhibitor (DNMTi) 5‐azacytidine, unapproved for CTCL. Contrary to our expectations, the findings reported herein revealed that THOR methylation is relatively stable under these epigenetic drugs' pressure, whereas these drugs reduced the hTERT gene expression.
Keywords: cutaneous T‐cell lymphomas, DNA methylation, DNMTi, HDACi, telomerase, TERT
Cutaneous T‐cell lymphomas (CTCLs) represent a group of lymphoproliferative disorders arising from the skin. CTCLs are characterized by hTERT gene expression despite the lack of hTERT amplifications or rearrangements. Here, we investigated hTERT promoter methylation and associated TERT hypermethylated oncogenic region (THOR) with hTERT reactivation in CTCL. Additionally, we evaluated THOR methylation and hTERT expression after treatment with epigenetic drugs. Altogether, our study offers a better understanding of the response to epigenetic drugs in patients with CTCL.
Abbreviations
- CTCLs
cutaneous T‐cell lymphomas
- DNMTi
DNA methyltransferase inhibitors
- E‐box
enhancer box
- HDACi
histone deacetylase inhibitors
- hTERT
human telomerase reverse transcriptase gene
- MZF‐2
myeloid zinc finger protein 2
- NTC
nontreated cells
- SS PDCs
Sézary syndrome patient‐derived cells
- SS
Sézary syndrome
- TBP
TATA‐box‐binding protein
- TF
transcription factor
- THOR
TERT hypermethylated oncogenic region
- T‐MF
transformed tumor stage mycosis fungoïdes
- TSS
transcription start site
- WT1
Wilms' tumor 1
1. Introduction
Cutaneous T‐cell lymphomas (CTCLs) encompass a heterogeneous group of rare T lymphoproliferative disorders, characterized by clonal proliferation of malignant T cells involving the skin as a primary site. They include cutaneous anaplastic large cell lymphoma (C‐ALCL), mycosis fungoïdes (MF), and Sézary syndrome (SS) [1]. While most C‐ALCL and MF have an indolent course, some MF may progress to a transformed tumor stage (T‐MF) of poor prognosis. SS can be developed in a patient affected many years with MF, but it arises more frequently as erythroderma associated with a frank leukemic variant [2]. Treatment of MF/SS can be very challenging, especially in the advanced stages of the disease. The choice of the therapeutic agent is stage‐dependent, including drugs such as bexarotene, methotrexate, interferon‐alpha, and histone deacetylases inhibitors (HDACi) or the recently introduced monoclonal antibodies such as mogamulizumab, brentuximab vedotin, or IPH4102. While chemotherapies only allow short‐lived responses, allogenic stem cell transplantation remains the only curative option [3, 4].
In cancer cells, replicative immortality can be acquired through telomerase reactivation driven by the human telomerase reverse transcriptase (hTERT) gene expression [5]. The hTERT gene can be upregulated either by genetic mechanisms (like promoter mutations and less frequently gene amplifications or rearrangements [6]) or by epigenetic mechanisms (DNA methylation, histone modifications, and noncoding RNA effects [7, 8, 9, 10, 11, 12]). The hTERT promoter methylation stimulated the interest of the scientific committee because it is ‘unusual’, comprising two oppositely methylated regions: A hypermethylated region located upstream the transcription start site (TSS) and a hypomethylated region flanking the TSS and exon 1 [7, 11, 13]. The upstream hypermethylated region was reported to be associated with hTERT expression in different types of tumors [7, 11, 14, 15] and was recently investigated in a large cohort of cancer patients and cell lines [16, 17]. Since this region is frequently observed hypermethylated and associated with telomerase reactivation in tumor cells, it was recently named TERT hypermethylated oncogenic region (THOR) [16]. Although this region was investigated in many cancers, it was not investigated in cutaneous lymphomas [16]. Within THOR, three transcription factors' binding sites are typically located: two transcriptional silencers WT1 (Wilms' tumor 1) and MZF‐2 (myeloid zinc finger 2), and one transcriptional enhancer c‐MYC that binds to an enhancer box (E‐box) [18].
In a previous work, our team reported that hTERT is expressed in CTCL despite the lack of hTERT amplifications or rearrangements [19]. In a complementary study, we stated the absence of hTERT hot spot promoter mutations in these types of tumors (A. Ropio, M. Prochazkova‐Carlotti, R. Batista, A. Pestana, J. Ferrer, A. Chebly, Y. Idrissi, D. Cappellen, C. Durães, P. Boaventura, J. Vinagre, L. Azzi‐Martin, J. Cabeçadas, M. Campos, M. Beylot‐Barry, M. Sobrinho‐Simões, J. P. Merlio, P. Soares, & E. Chevret, In preparation). Since little is known about the mechanisms underlying the methylation changes during tumorigenesis [20] and since no hTERT promoter epigenetic investigation was reported in CTCL, we present herein a pioneer exploration in this rare pathology. We evaluate THOR methylation status in CTCL cell lines and in SS patient‐derived cells in comparison with healthy cells (CD34+ and CD4+ lymphocytes). We explore THOR methylation under the pressure of a demethylating agent, unapproved for CTCL; we describe the effect of clinically approved HDAC inhibitors on THOR methylation status.
2. Materials and methods
2.1. Cell lines, SS patient‐derived cells, and cell culture
Five CTCL cell lines were studied: Myla (T‐MF) (kindly provided by K. Kaltoft, Denmark), HuT78 (SS) (ATCC, Molsheim, France), and Mac1, Mac2A, and Mac2B (C‐ALCL) (DSMZ, Braunschweig, Germany). Cells were cultured in RPMI 1640 medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% of fetal bovine serum (Eurobio, Les Ulis, France) and 100 U·mL−1 of penicillin and streptomycin (Gibco).
Four SS patient‐derived cells (PDCs) obtained from four SS patients (patients 1 to 4) were also investigated. They were cultured as recently described by Poglio et al. [21].
Cell lines and PDC cultures were incubated at 37 °C in a humidified incubator with 5% CO2.
2.2. SS patients and tumor cell isolation
Ten SS patients, eight females and two males, with a median age of 69.5 years (range: 52–93), were recruited for this study. The diagnosis was established in accordance with the criteria of the WHO‐EORTC (World Health Organization and the European Organization for Research and Treatment of Cancer) [1]. All of them presented a B2 stage; eight, a T4 stage; one, a T3 stage; and one, a T2b stage. Samples from patients 1 to 4 were used to establish PDC as mentioned above. Samples from patients 5 to 10 were used to obtain fresh SS cells. Peripheral blood mononuclear cells (PBMCs) were isolated using Pancoll (Pan Biotech, Aidenbach, Germany). Clonal TCRvβ was determined using IOTest® Beta Mark TCRVβ Repertoire Kit (Beckman Coulter, Villepinte, France). Tumor cells were sorted either according to the TCRvβ using a BD FACSAria™ II Cell Sorter (BD Biosciences, Le Pont de Claix, France) or according to the CD4+, as mentioned in Ref. [21] (Fig. S1 shows the evaluation of tumor cells' proportion before and after cell sorting). This study was approved by the local ethics committee and was carried out in accordance with the standards set by the Declaration of Helsinki. All SS patients included in this study signed an informed consent.
2.3. Controls and healthy donors
Seven healthy age‐matched donors were recruited from the Etablissement Français du Sang (EFS) in Bordeaux (DC 2015 2412‐18PLER012). PBMCs were isolated from peripheral blood samples, using Pancoll (Pan Biotech). CD4+ cells were manually sorted using CD4 MicroBeads Human Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and separated into two pools: A (three donors) and B (four donors). Progenitor/stem cells (CD34+) were collected from 20 healthy donors at the EFS and pooled together.
2.4. Chemicals
Drugs included in this study were two HDACi used to treat CTCL: romidepsin and vorinostat (Euromedex, Souffelweyersheim, France) and a DNA methyltransferase inhibitor (DNMTi): 5‐azacytidine not approved for CTCL treatment. Based on previous reports [22, 23], 1 × 106 SS PDCs (1, 2, 3, and 4) were exposed to 10 nm of romidepsin or 3 µm of vorinostat during 48 h. The Hut78 cell line and SS PDCs 2 and 3 were exposed to 3, 1.7, and 2.3 nm of 5‐azacytidine during 72 h, respectively.
2.5. DNA/RNA isolation and cDNA synthesis
Genomic DNA was extracted using Quick‐DNA Microprep Kit (ZYMO Research, Freiburg im Breisgau, Germany). Total RNA was isolated using the Direct‐zol™ RNA Miniprep Kit (ZYMO Research). DNA and RNA concentrations were measured using the NanoDrop ND‐1000 Spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA). cDNA was synthetized from 100 ng of RNA using the SuperScript II Reverse Transcriptase Kit (Invitrogen, ThermoFisher Scientific, Courtaboeuf, France).
2.6. Locus‐specific bisulfite sequencing
For methylation analyses, we used the standard bisulfite sequencing method. While this approach requires standard molecular biology apparatus, it generates reliable and consistent results in gene‐specific methylation studies. Also, compared to other global genomic bisulfite techniques, standard bisulfite sequencing method offers, in the region of interest, the ability to detect the methylation status cell by cell of all consecutive CpGs [24].
Genomic DNA was bisulfite‐converted using the EZ‐DNA Methylation Kit (ZYMO Research). The region from −650 to +150 bp relative to the transcription start site (TSS) of hTERT was amplified by PCR using GO‐Taq G2 Hot Start (Promega, Fitchburg, WI, USA). Primers were bisulfite‐specific and completely devoid of CpG sites as previously described [7, 11]. Forward and reverse primer sequences and PCR conditions are listed in Table S1. Amplicon lengths were verified, and PCR products were purified using MACHEREY‐NAGEL Extraction Kit (MACHEREY‐NAGEL, Düren, Germany). Purified amplicons were cloned into the p‐GEM‐T Easy Vector System I (Promega), and then, competent Escherichia coli (Promega) were transformed using the ligation product. Bacterial suspensions were enriched in SOC medium (New England Biolabs, Ipswich, MA, USA). Colonies were grown overnight on LB (Luria‐Bertani) Agar containing 32 μg·mL−1 Xgal, 120 μg·mL−1 IPTG, and 100 μg·mL−1 ampicillin. After white colonies' selection and checking of the DNA insertion by PCR, colonies were incubated overnight for enrichment in LB medium with 100 μg·mL−1 ampicillin at 37 °C under agitation. Plasmid DNA was isolated using Nucleospin Plasmid Kit (MACHEREY‐NAGEL). Each sample was performed in duplicate. Ten to 30 clones were extracted and sequenced. DNA sequences were analyzed using chromaspro Software (Technelysium, South Brisbane, Queensland, Australia), and bisulfite images were obtained using quma (Riken, Japan, http://quma.cdb.riken.jp) [25].
2.7. hTERT and WT1 expression analysis by quantitative real‐time PCR
cDNAs were amplified by quantitative real‐time PCR (qRT‐PCR) using Takyon™ No Rox SYBR® MasterMix dttP Blue (Eurogentec, Angers, France), and the following primer sets were used: hTERT gene, forward primer: 5′‐GCATTGGAATCAGACAGCAC‐3′ and reverse primer: 5′‐CCACGACGTAGTCCATGTTC‐3′, and housekeeping gene TBP, forward primer: 5′‐CACGAACCACGGCACTGATT‐3′ and reverse primer: 5′‐TTTTCTTGCTGCCAGTCTGGA‐3′. hTERT mRNA levels were normalized to the expression of the TBP (TATA‐box‐binding protein) gene. For Wilms' tumor 1 (WT1) gene, qRT‐PCR was performed using WT1 PrimePCR™ SYBR® Green Assay (Bio‐Rad, Des Plaines, IL, USA) according to the manufacturer's instructions. qRT‐PCR analyses were run on a Stratagene Mx3005P System (Agilent Technologies, Santa Clara, CA, USA). Each sample was performed in triplicate, and the mean value was calculated. Results were obtained using the () method [26]. Values are expressed in arbitrary units (A.U.).
2.8. Luciferase assay
Luciferase assays were performed as previously described by Gazon et al. [27]. Briefly, 293T cell line was used to set up the protocol, and then, HuT78 and MyLa cells were transfected with a plasmid DNA mixture containing 100 ng·µL−1 of pGL3‐hTERT‐378‐Luc reporter plasmid [28], 100 ng·µL−1 of pActin‐βgal, and the indicated amount of pAD/WT1‐IRES‐nAMcyan (gift from Edward McCabe, Addgene [Watertown, MA, USA] #29756). HuT78 and MyLa were electroporated using Gene Pulser XCell Electroporation Systems (Bio‐Rad). Forty‐eight hours post‐transfection, cells were washed with cold PBS and then lysed in 1× passive lysis buffer (Promega). Luciferase and β‐galactosidase assays were both performed in a Spark 10M Multiplate Reader (Tecan, Männedorf, Switzerland) with Genofax A Kit and Genofax B Kit (YELEN, Ballaison, France), and Galacto‐Star Kit (Life Technologies, Grand Island, NY, USA), respectively, as described by the manufacturer. Each experiment was performed in triplicate, and Luciferase activities were normalized for transfection efficiency based on β‐galactosidase. After WT1 overexpression, the levels of hTERT mRNA were also evaluated by qRT‐PCR.
2.9. qRT‐PCR analysis after WT1 overexpression
Total RNAs were prepared from whole cells using TRIzol (Invitrogen). Briefly, after reverse transcription (RT) using oligo‐dT 12–18 primer (Invitrogen) and SSII reverse transcriptase (Invitrogen), the abundance of transcripts was assessed by real‐time, quantitative PCR analysis using the SYBR Green PCR Master Mix (Roche Diagnostics, Meylan, France) and gene‐specific primer sets. Primer sequences for hTERT, hWT1, and hPRT‐1 are listed in Table S2. Standard curves were generated from each PCR plate for all primer pairs on the plate using a serial dilution of an appropriate experimental sample. Samples were amplified in triplicate on each plate. The conditions for the hTERT PCR were 95 °C for 30 s, 55 °C for 40 s, and 72 °C for 1 min for 45 cycles, while hWT1 and hPRT1 were amplified as previously described [29]. Data were analyzed using lightcycler® 480 Software (Roche Diagnostics). Relative mRNA levels of hTERT and hWT1 among experimental samples were determined as previously described [29].
2.10. Western blot
Western blot assay was performed according to the manufacturer's recommendations. Briefly, protein extracts from HuT78 cell line, SS PDCs 1, 2, and 3, and MCF7 cell line (positive control expressing WT1, recommended by the manufacturer) in addition to All Blue Prestained Protein ladder (Bio‐Rad) were separated by SDS/PAGE on 8–16% TGX Stain‐Free™ Protein Gels (Bio‐Rad) for approximately 45 min at 150 V in TGX buffer (Bio‐Rad). Stain‐free gels were activated by exposure to UV for 1 min. Proteins were transferred to nitrocellulose membranes (Bio‐Rad) using the Bio‐Rad Trans‐Blot Turbo Transfer System for 7 min. Total proteins on membranes were detected using the stain‐free method. Membranes were blocked with TBST with 5% BSA for 1 h. Membranes were then incubated with primary antibody (WT1 monoclonal antibody, Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1 : 500 in TBST with 5% BSA at 4 °C overnight. Excess of primary antibody was removed by washing the membranes three times in TBST for 10 min each. The secondary antibody (peroxidase‐conjugated anti‐mouse DyLight 800) diluted 1 : 5000 was incubated with the membrane in TBST with 5% BSA for 1 h. Excess of secondary antibody was removed by washing the membranes three times in TBST for 5 min each. Membranes were visualized using Bio‐Rad ChemiDoc™ Imager. Detection and quantification of bands' intensities were done using image lab Software (Bio‐Rad).
2.11. WT1 ChIP‐qPCR assay
WT1 ChIP‐qPCR assays were performed by Active Motif (Carlsbad, CA, USA). Briefly, WT1 ChIP‐qPCR assay was performed using 30 μg of chromatin obtained from cultured cells (HuT78, SS PDCs 1, 2, and 3) or primary human T lymphocytes (healthy CD4+ cells) and 8 μg of WT1 antibody sc‐192 (Santa Cruz Biotechnology). qPCRs were performed using primer pairs (Table S1) designed for the region of interest (hTERT‐323) and for two positive controls (TAL1‐2k and hTERT‐709). A negative control was also used, consisting of a primer pair that amplifies a region in a gene desert on chromosome 12 (Untr12). Data were normalized to the genomic DNA for the particular cell type.
2.12. Telomerase activity by TRAP assay
Telomerase activity was assessed in CTCL cell lines and SS PDCs (1, 2, 3, and 4) using the TRAP assay (TRAPeze Telomerase Detection Kit; S7700, Millipore, Alsace, France). Protein extracts were used to extend a synthetic telomeric DNA by PCR amplification (1 cycle of 30 °C for 30 min, followed by a telomeric PCR amplification: 95 °C for 3 min, 2 cycles of 95 °C for 20 s and 49 °C for 20 s, followed by 30 cycles of 95 °C for 20 s and 60 °C for 20 s with signal acquisition) on a Stratagene Mx3005P System (Agilent Technologies). Each sample was run in duplicate with a control DNA.
2.13. Statistical analysis
General statistical analyses were performed using the graphpad prism version 5 (San Diego, CA, USA). P values of less than 0.05 were considered statistically significant.
3. Results
3.1. CTCL cell lines and SS patients' tumor cells express hTERT
Healthy controls CD4+ and CD34+ showed hTERT expression of 0.47 and 0.95 A.U., respectively (Fig. 1). Compared to healthy controls, CTCL cell lines expressed the highest hTERT levels (ranging from 2.7 to 8.2 A.U.) (Fig. 1). In SS PDC, hTERT was expressed. While PDC 3 showed hTERT expression level similar to cell lines (6 A.U.), PDCs 1, 2, and 4 showed hTERT expression levels in the same ranges of those of healthy controls with 0.60, 0.88, and 0.50 A.U., respectively (Fig. 1). In SS patients' fresh cells, hTERT was expressed at lower levels (0.07–0.12) than healthy controls (0.47 and 0.95), except for patient 10 with 0.60 A.U. (Fig. 1). Besides, the tumor burden allowed the evaluation of hTERT expression in nontumor T cells only in half of the SS patients. No Ct values were obtained for the normal cells in these three patients tested. The hTERT expression levels reported, correlated with telomerase activity in CTCL cell lines and in SS PDC with an R 2 equal to 0.7502 (Fig. S2).
Fig. 1.
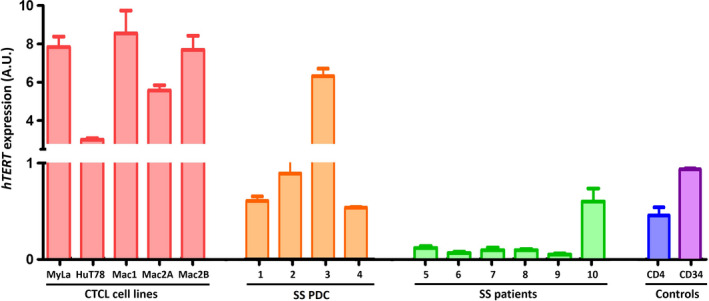
hTERT expression in cell lines and patients' cells. hTERT mRNA levels quantified by fluorescence real‐time reverse transcriptase PCR in CTCL cell lines, in SS patient‐derived cells (SS PDCs), in SS patient cells (SS patients), and in healthy CD4+ and CD34+ cells. hTERT mRNA levels were normalized to the expression of the TBP gene and expressed in arbitrary unit (A.U) ± the SEM of three independent experiments. TBP: TATA‐box‐binding protein located on 6q27. n = three independent experiments.
3.2. THOR is methylated in CTCL cell lines and SS PDC
A common hTERT promoter methylation pattern in CTCL cell lines and in SS PDC was revealed by locus‐specific bisulfite sequencing (Fig. 2). This pattern comprises a hypermethylated distal region between −650 and −150 bp from the TSS, as well as a hypomethylated proximal region between −150 and +150 bp including the TSS and the ATG start codon. The hypermethylated region in CTCL cell lines corresponds to the region recently known as THOR (Fig. 2A). Among CTCL cell lines (Fig. 2B–F), HuT78 presented the highest levels of THOR methylation with an average of 87% (Fig. 2C), followed by MyLa with 83% (Fig. 2B) and Mac1 with 64% (Fig. 2D). Mac2A (Fig. 2E) and Mac2B (Fig. 2F) showed hypermethylation levels around 49% and 45%, respectively. Regarding SS PDCs 1, 2, 3, and 4, hypermethylated THOR levels were 73%, 67%, 50%, and 53%, respectively (Fig. 2G–J). On the contrary, a very low level of methylation was observed in healthy CD4+ cells (Fig. 2K,L) and CD34+ cells (Fig. 2M,N): 11% and 7.5%, respectively. Interestingly, THOR methylation levels were significantly increased in cell lines and SS PDC compared with healthy cells (P < 0.0001). No direct correlation was observed between THOR methylation levels and hTERT mRNA levels (Fig. S3).
Fig. 2.

hTERT gene promoter methylation including THOR in CTCL cells and healthy controls. (A) hTERT gene promoter including THOR (Chr5:1 295 321–1 295 753;GRCh37/hg19) containing 52 CpG represented each by a vertical dash. (B–N) Methylation profiles of CTCL cells (red) and controls (blue): Full black dots represent methylated CpGs, whereas empty dots represent unmethylated CpGs. For CTCL cell lines: MyLa is represented in (B), HuT78 in (C), Mac1 in (D), Mac2A in (E), and Mac2B in (F). For SS PDC: PDC 1 is represented in (G), PDC 2 in (H), PDC 3 in (I), and PDC 4 in (J). Healthy CD4+ controls: Two pools are represented in (K) and (L). Normal stem/progenitor cells: Two pools of normal CD34+ cells are represented in (M) and (N). n = three independent experiments.
3.3. THOR hypermethylation is a specificity of tumor cells
In order to strengthen our findings regarding THOR methylation profiles in cultured CTCL cells, we studied the methylation status of hTERT promoter in fresh SS patient cells. For each patient, tumor cells (clonal TCRvβ or CD4‐positive cells) and normal cells used as individual controls (TCRvβ or CD4‐negative cells) were sorted and analyzed. Strikingly, THOR methylation levels were prevalently observed higher in tumor cells than in normal cells. A significant difference (P < 0.0001) was observed in patients 5 (Fig. 3A), 6 (Fig. 3B), 7 (Fig. 3C), and 9 (Fig. 3E), with an average methylation level of 46%, 35%, 42%, and 56%, respectively, in tumor cells, compared with 4%, 10%, 5%, and 13%, respectively, in normal cells. A significant difference was also found in patients 8 (Fig. 3D) and 10 (Fig. 3F) (P = 0.0455 and P = 0.0079, respectively) with lower THOR methylation levels in tumor cells (15% and 22.9%, respectively, for tumor cells; and 6.5% and 11.5%, respectively, for normal cell). Figure 3G summarizes THOR methylation levels in the aforementioned six SS patients. In all healthy cells explored (CD4+, CD34+, and SS patients' normal cells), THOR was hypomethylated with a methylation level ranging from 4 to 13% (Fig. 4). In our study, a cutoff value of 15% was used for SS patients, which is quite similar to that of 16.1% used by Lee et al. [16].
Fig. 3.
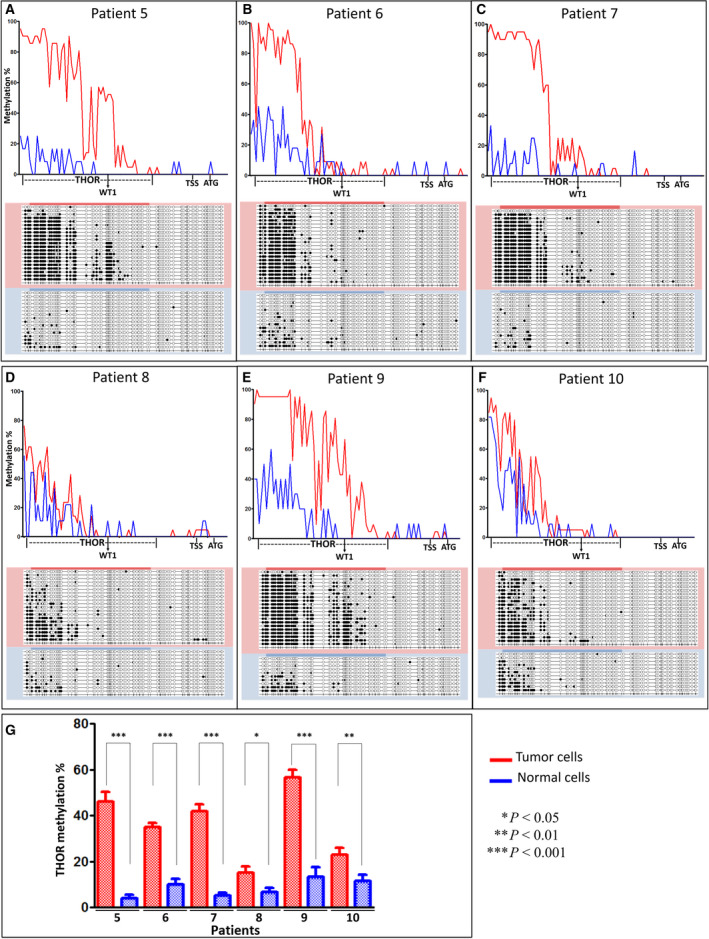
hTERT gene promoter methylation including THOR in Sézary syndrome patient cells. Graphs (A) to (F) showing the difference between methylation profiles of tumor cells (red) and normal cells (blue), in patient 5 (A), patient 6 (B), patient 7 (C), patient 8 (D), patient 9 (E), and patient 10 (F). Chart (G) showing THOR methylation levels in tumor (red) and normal (blue) cells in each of the six SS patients' cells (±SEM). Statistical significances were determined by t‐test. THOR, TERT hypermethylated oncogenic region.
Fig. 4.
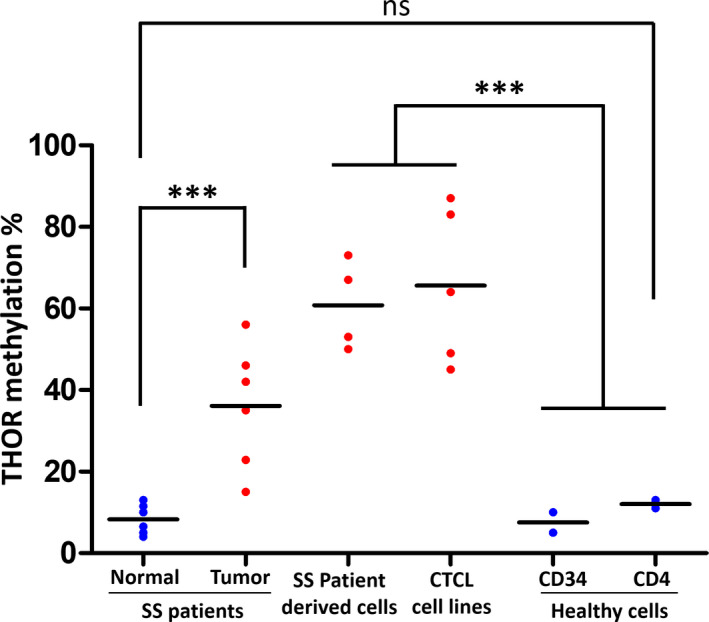
THOR methylation status. The difference in THOR methylation averages between normal cells in blue and tumor cells in red in all cells studied. This figure is a visual representation of THOR methylation in the previously mentioned CTCL cell lines, SS PDC, and SS patients. Statistical significances were determined by t‐test. n = three independent experiments.
3.4. WT1 overexpression reduces hTERT activation
Regarding the transcription factors' binding sites on THOR, while the MZF‐2 binding sites were hypermethylated in all tumor samples and the E‐box site was hypomethylated in almost all tumor samples (87%, 13/15 cell lines and patients); WT1 binding site presented different methylation levels between tumor samples (cell lines and patients). For this reason, we focused on WT1 and we assessed by qRT‐PCR the expression levels of WT1 in SS cells (Hut78 cell line and SS PDC). Interestingly, HuT78 and SS PDCs 1, 2, and 3 expressed WT1 mRNA (Fig. S4A). WT1 protein expression was verified by western blot analysis (Fig. S4B). Next, we evaluated the effect of WT1 overexpression on hTERT promoter in two aggressive MF/SS cell lines: MyLa and HuT78. We noticed in these latter that WT1 overexpression reduced significantly the hTERT activation in a dose‐dependent manner (Fig. 5A): In MyLa: P < 0.0001 with 10 and 20 μg of WT1, while in HuT78: P = 0.0051 and 0.0026 with 10 and 20 μg of WT1, respectively. Also, qRT‐PCR analyses showed a decrease in the hTERT mRNA levels after WT1 overexpression (Fig. 5B).
Fig. 5.
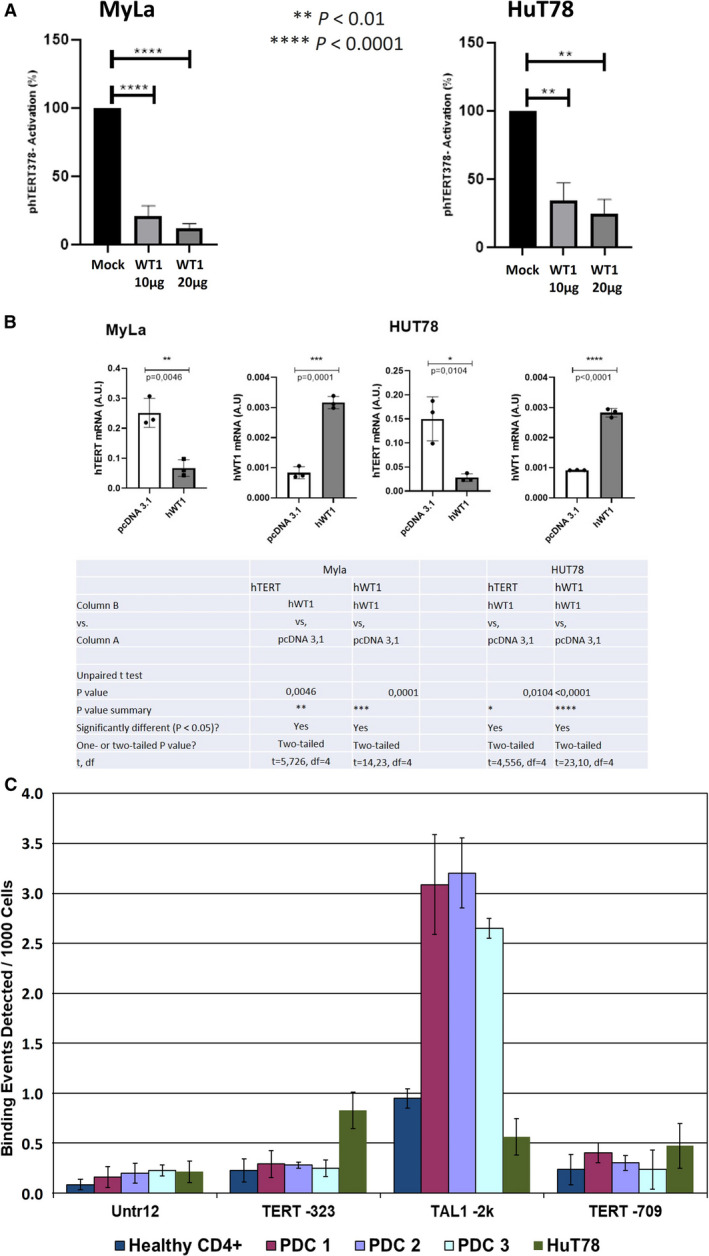
Effect of the transcription factor WT1 on hTERT promoter in CTCL. Graph (A) presents the results of luciferase assay showing the effect of empty vector (mock), 10 and 20 µg of WT1 on hTERT promoter activation in HuT78 and MyLa cell lines. Graph and table (B) show the significant decrease in hTERT mRNA expression after the overexpression of WT1 in MyLa and HuT78. Graph (C) shows the results of ChIP‐qPCR using a WT1 antibody targeting the TERT‐323 region (region of interest) in SS PDCs 1, 2, and 3, HuT78, a SS cell line, and healthy CD4+ (control), along with a negative control (Untr12) region, and two positive control regions (TAL1‐2k and TERT‐709). The results for the TAL1‐2k region confirm the efficacy of the used WT1 primer. Statistical significances were determined by t‐test. n = three independent experiments. SS PDCs, Sézary syndrome patient‐derived cells; df, degrees of freedom.
3.5. WT1 binding on hTERT promoter
The obtained results pertaining to the WT1 overexpression and its impact on hTERT expression urged us to evaluate the physical interaction between WT1 and hTERT promoter. To do so, we used a ChIP‐qPCR approach. Since this assay requires important cells' amounts, we performed it in SS PDC and in a cell line. The SS PDCs 1, 2, and 3 were selected because they derive from epigenetic therapy‐free patients. HuT78 was selected since it was the only SS cell line available in this study. A low value of WT1 binding to hTERT promoter region (−323 bp from TSS) was obtained in SS cells (Fig. 5C). This result cannot be due to technical deficiency as no binding events were detected with the negative control primers (Untr12) and significant signals were observed with the positive control primer pair TAL1 (−2k). Regarding normal CD4+ cells, we expected to detect WT1 binding on hTERT promoter; disappointingly, we observed a faint WT1 binding (−323 bp from TSS). In addition, healthy cells (normal CD4+), HuT78, and PDCs (1, 2, and 3) did not present WT1 binding signals with the additional positive control hTERT‐709.
3.6. THOR hypermethylation is insensitive to HDACi
Since two HDACi (romidepsin and vorinostat) are approved for CTCL treatment, without clear molecular investigations, we studied the effect of these two drugs focusing on hTERT expression in SS cells. hTERT expression decreased significantly (P < 0.001) in SS PDCs 1, 2, and 3 after romidepsin and vorinostat treatments (Fig. 6A). Surprisingly, in patient 4, hTERT expression was not altered the same way as in the other patients. In fact, hTERT expression increased slightly with romidepsin treatment and remained unchanged with vorinostat (Fig. 6A). Methylation levels of hTERT promoter in SS PDCs 1, 2, and 3 showed weak changes between nontreated cells (NTC) and cells treated either with vorinostat or with romidepsin (Fig. 6B). Indeed, after HDACi treatments methylation levels and profiles remained quite the same throughout the entire promoter. In patient 4, a slight decrease in THOR methylation status was observed only after romidepsin treatment in comparison with NTC, with a statistical significance of P = 0.00063. Overall, following romidepsin or vorinostat treatments, we noticed the absence of particular methylation or demethylation changes at any CpG site (Fig. S5).
Fig. 6.
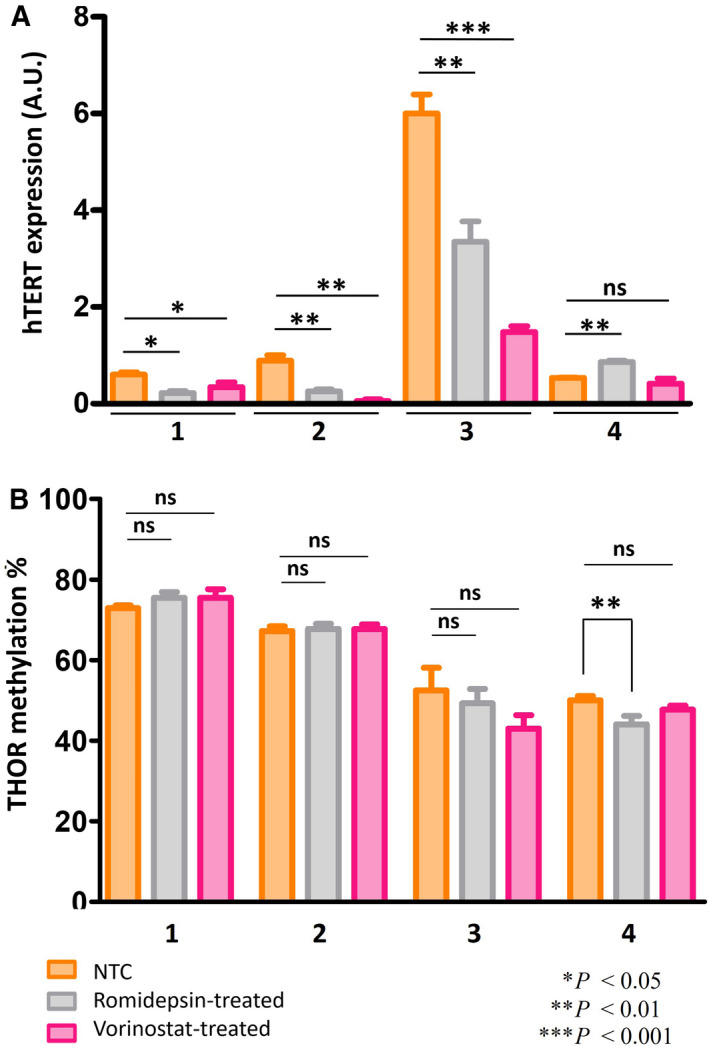
HDACi treatments in Sézary syndrome patient‐derived cells. Graph (A) shows hTERT expression in NTC, romidepsin, and vorinostat‐treated cells. Graph (B) shows THOR methylation % in NTC, romidepsin, and vorinostat‐treated cells (±SEM). Statistical significances were determined by t‐test. n = three independent experiments. HDACi, histone deacetylase inhibitors; NTC, nontreated cells.
3.7. THOR hypermethylation is insensitive to 5‐azacytidine
Since HDACi did not exert any effect on THOR methylation in SS PDC, we analyzed the effect of the demethylating agent 5‐azacytidine on hTRET expression and promoter methylation in HuT78 cell line and SS PDCs 2 and 3. While hTERT expression decreased significantly after 5‐azacytidine treatment in SS PDCs 2 and 3 (P < 0.01) and in HuT78 (P < 0.001) (Fig. 7A), the methylation status of hTERT promoter remained unchanged throughout the entire promoter, showing the same methylation levels and profiles: a highly methylated distal region and a poorly methylated proximal region (Fig. 7B).
Fig. 7.
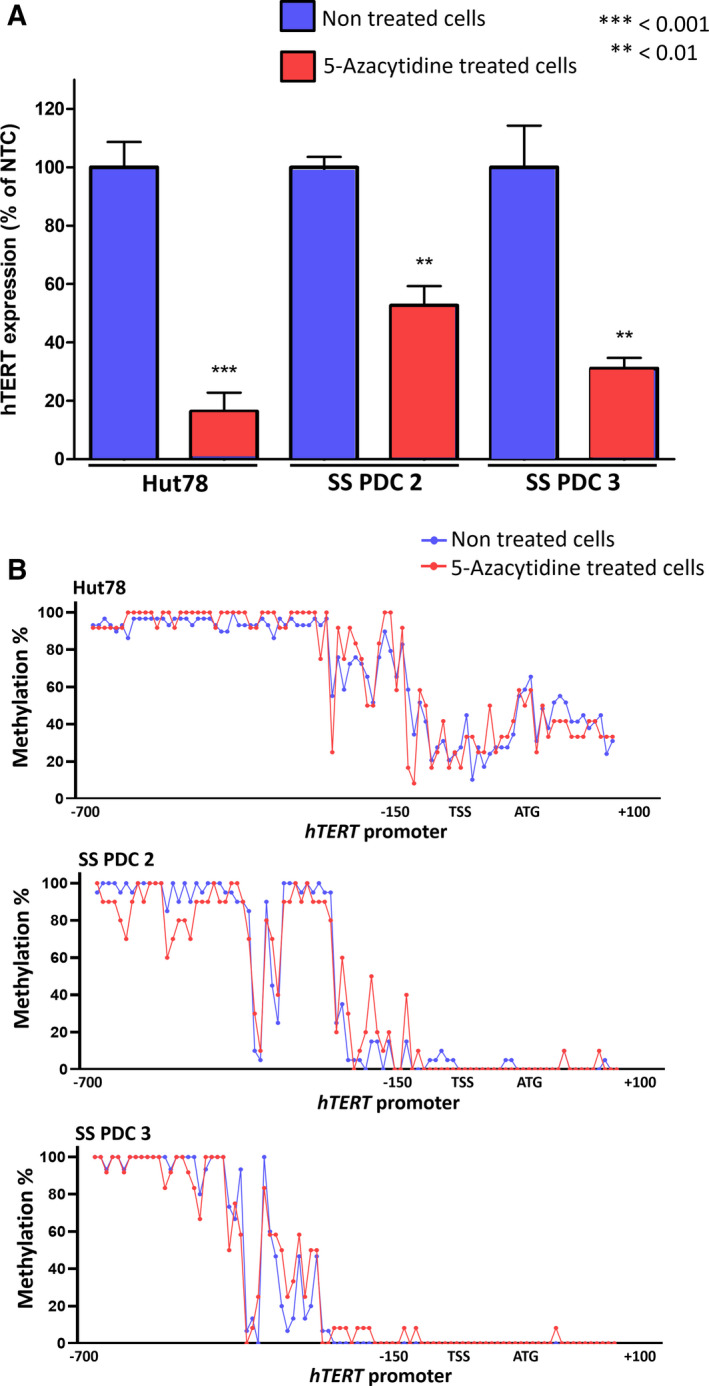
5‐azacytidine treatment in Sézary cells. Graph (A) shows hTERT expression in NTC and 5‐azacytidine‐treated cells (±SEM). Graph (B) shows THOR methylation % in NTC and 5‐azacytidine‐treated cells. Statistical significances were determined by t‐test. n = three independent experiments. ATG, start codon; NTC, nontreated cells; SS PDCs, Sézary syndrome patient‐derived cells; TSS, transcription start site.
4. Discussion
Several genetic alterations can result in aberrant hTERT expression such as hTERT gene amplifications, rearrangements, and promoter mutations [30, 31, 32, 33, 34, 35]. However, in some solid tumors, and in hematological disorders such as non‐Hodgkin lymphomas including CTCL, these known genetic alterations are rare [36, 37]. Additionally, the mechanism responsible for hTERT reactivation in these tumors remains unclear. Therefore, several epigenetic mechanisms possibly implicated in hTERT promoter regulation were investigated during the last years.
Since hTERT promoter DNA is dense in CpG islands (Fig. 2A), numerous studies explored the role of hTERT promoter methylation in hTERT expression [11, 15, 16, 17]. A recent review of the literature mentioned that hTERT promoter does not behave in a simplistic manner [24]. Throughout hTERT promoter, the DNA methylation landscape was reportedly not uniform [11, 16, 17]. Two differentially methylated regions were identified: a region containing 52 CpGs located upstream the core promoter and a smaller region around the transcription start site (TSS). In recent investigations led to a variety of cancer cells expressing hTERT, the distal promoter region of hTERT was recurrently observed hypermethylated [16, 17]. Therefore, hTERT distal promoter DNA hypermethylation was associated with hTERT re‐expression in cancer cells and proposed as a potential biomarker [7, 12, 15, 16]. This hypermethylated region within hTERT promoter was recently named TERT hypermethylated oncological region (THOR) [16].
Our previous investigations showed that CTCL cells are hTERT‐expressing tumors, while no genetic alterations could explain this expression [19]. In this study, we reported that hTERT distal promoter was hypermethylated in all CTCL cells investigated, while the region around the TSS is unmethylated. These observations are in agreement with previous observations in different pathologies [11, 16, 17]. In our study, the comparison between the methylation patterns of hTERT promoter in CTCL tumor cells and healthy cells revealed that THOR is unmethylated in healthy CD4+ cells and in stem/progenitor cells. This observation underlined that DNA methylation alone is not sufficient to explain hTERT expression and that additional epigenetic mechanisms might be implicated [12]. Furthermore, as mentioned by Smith et al., aberrant DNA methylation was reported in carcinogenesis across a broad range of cancer types [38, 39]. Therefore, hTERT promoter hypermethylation in hTERT‐expressing cells represents a characteristic of tumor cells. Thus, nontumor cells expressing hTERT might use an alternative epigenetic mechanism to regulate hTERT expression. Our current observations in addition to previously published data allowed us to propose that hTERT promoter methylation and regulation can be related to the physiological or pathological cell status.
Classically, several binding sites for transcription factors (TFs) either activators (ETS, c‐MYC, SP1, and NFkB) or repressors (WT1 and MZF‐2) are reported within hTERT promoter. Among the repressor TFs, we recently questioned the existence of the human MZF‐2 binding sites theoretically located within THOR [40]. In acute promyelocytic leukemia, Azouz et al. showed that the methylation of the distal domain of hTERT promoter (including THOR) is associated with a decrease in WT1 binding to hTERT promoter and sustained hTERT expression [11]. As WT1 binding to hTERT promoter was reported to be methylation‐sensitive [11], we investigated the role of this binding in SS (advanced‐stage CTCL). First, we verified that WT1 mRNA and protein are expressed in CTCL. Then, we observed that WT1 overexpression reduced hTERT expression. Strengthened by this observation, we investigated the physical interaction between WT1 and THOR in SS cells. Our data suggested that hTERT modulation expression in CTCL may occur independently of WT1 binding to the THOR region. However, we are aware that low expressed or low binding levels of some TFs constitute a challenge to be identified. Hence, further investigations are required in order to confirm whether, in CTCL, the binding of this downregulating TF to THOR is methylation‐sensitive and whether other binding TFs might be present in this region.
As reported by Garsuault et al., DNA methylation is functionally linked to other epigenetic pathways, including post‐translational histone modifications. This link is mediated by a group of proteins with methyl DNA‐binding activity that localize to methylated DNA and recruit other protein complexes such as histone deacetylases (HDAC) and histone methyltransferases [41, 42]. Since the exact mechanism behind the effectiveness of HDACi treatments in SS patients remains unknown [43], we investigated first the methylation status of hTERT promoter after using two HDACi treatments approved in MF/SS patients. Interestingly, after in vitro HDACi treatments, hTERT expression levels decreased in all SS PDC, while methylation patterns of hTERT promoter including THOR remained unchanged, except for one PDC: patient 4, the only patient who had previously received romidepsin. This observation may suggest a possible drug resistance mechanism. In the other patients, THOR remained hypermethylated and hTERT proximal promoter encompassing TSS and ATG remained hypomethylated. In a previous study using vorinostat in non‐small cell lung cancer, Li et al. observed a repression of the telomerase expression and a reduction in hTERT methylation levels near the TSS (around −200 to +160 bp), but THOR was not investigated [44]. In our study, the TSS region was already hypomethylated and rationally cannot be more demethylated. Altogether, these data suggest that HDACi reduced hTERT expression only in patients who did not receive previous epigenetic therapies. Besides, we proved that other epigenetic drugs such as 5‐azacytidine, a demethylating agent, did not exert a demethylation on hTERT promoter including THOR in SS, while it reduced hTERT expression. In other pathologies, it has been reported that hTERT expression decreased after treatments with demethylating agents. This decrease could be accompanied by a promoter demethylation, suggesting that hTERT promoter demethylation could be cell type and pathology‐dependent [45, 46]. It has been also reported that demethylating drugs can exert different effects on different genes. In some genes, this effect is ‘direct’ with a decrease in the methylation levels accompanied by a corrected gene expression, whereas in other genes named ‘refractory’, DNA methylation and gene expression remain unchanged under demethylating drug pressure [47, 48, 49, 50]. Nevertheless, another group of genes respond to the demethylating drugs through some changes in gene expression, while their promoter methylation levels remain unaltered. This mechanism, termed ‘indirect’, can happen through the demethylation of a TF controlling the specific gene expression, or the demethylation of regulatory elements such as enhancers, or by a secondary response to DNA damage or reparation mechanisms, or also through histone modifications. Altogether, our results suggest that hTERT promoter methylation in CTCL is resistant to the direct effect of epigenetic drugs, indicating that these drugs can alter hTERT expression in an indirect way.
5. Conclusions
Since the mechanism behind hTERT expression in CTCL remains unknown, we undertook the first epigenetic study of hTERT promoter in this pathology. Overall, our findings strongly suggest that THOR hypermethylation is a hallmark of neoplastic CTCL cells associated with hTERT activation. Additionally, we propose that THOR hypermethylation might be used as a biomarker of cancer cells in SS patients. By adding CTCL to the list of tumors analyzed for THOR methylation, our findings represent a significant step forward toward a better understanding of the mechanisms involved in telomerase activation and its regulation by epigenetic therapies in this pathology. Moreover, our data provide a starting point for further investigations to assess the relationships between THOR methylation status, hTERT expression, and TF binding with THOR in order to fully understand the sophisticated molecular mechanism of hTERT activation in CTCL. The advent of new gene‐specific targeting tools [20] will help to establish causality between hTERT promoter DNA methylation and hTERT expression, paving the way to a better understanding of the clinical response to epigenetic drugs in advanced‐stage CTCL patients.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
AC performed DNA/RNA/protein extractions, bisulfite sequencing, tumor cell isolation, western blot, and statistical analyses, analyzed and interpreted the data, wrote the manuscript, and prepared the figures; AC, JR, JF, and YI performed qPCR analyses; JF performed TRAP assay; AC, SP, MP‐C, and FC performed flow cytometry analyses, cell cultures, and HDACi/DNMTi treatments; JMP performed luciferase assay and WT1 overexpression; MBB and APL recruited SS patients and followed them up; ES‐B, J‐MP, JPM, MB‐B, ElC, CF, and RT provided helpful advices and assisted in editing the manuscript; EdC conceived and designed the study, and revised and edited the final version of this manuscript. All authors read and approved the final version of the manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/1878‐0261.12946.
Supporting information
Fig. S1. Evaluation of tumor cells proportion before and after cell sorting.
Fig. S2. Correlation between hTERT expression level and telomerase activity.
Fig. S3. Absence of correlation between THOR methylation status and hTERT expression level.
Fig. S4. WT1 mRNA and protein expression.
Fig. S5. hTERT promoter methylation profiles after HDACi treatments.
Table S1. hTERT Bisulfite PCR conditions and primer sequences.
Table S2. Primer sequences for hTERT, hWT1 and hPRT‐1.
Table S3. Primer sequences used for WT1 ChIP‐qPCR.
Acknowledgements
We thank all the patients and healthy donors for their participation in this study, as well as Bordeaux University Hospital and the Etablissement Français du Sang (EFS) for organizing blood withdrawals and collecting samples. We also thank Dr Philippe Brunet De La Grange from the EFS for his help. Additionally, we thank Atika Zouine and Vincent Pitard from TBM core platform at Bordeaux University for technical assistance with flow cytometry (CNRS UMS3247–INSERM US005). Thanks to Dr Alexandra Kuzyk for proofreading this article. This work was supported by grants from the National Institute of Health and Medical Research (INSERM), the French Society of Dermatology (SFD), and the Ligue Régionale contre le Cancer. AC was supported by grants from Hubert Curien Partnership (PHC‐CEDRE) and ERASMUS+. JR was supported by grants from Hubert Curien Partnership (PHC‐PESSOA, PAUILF) and ERASMUS+. E.S‐B is supported by the Ligue Nationale contre le Cancer and the National Center for Scientific Research (CNRS).
Data accessibility
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. Willemze R, Cerroni L, Kempf W, Berti E, Facchetti F, Swerdlow SH & Jaffe ES (2019) The 2018 update of the WHO‐EORTC classification for primary cutaneous lymphomas. Blood 133, 1703–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, Ralfkiaer E, Chimenti S, Diaz‐Perez JL, Duncan LM et al. (2005) WHO‐EORTC classification for cutaneous lymphomas. Blood 105, 3768–3785. [DOI] [PubMed] [Google Scholar]
- 3. Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M & ESMO Guidelines Committee (2018) Primary cutaneous lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol 29 (Suppl 4), iv30–iv40. [DOI] [PubMed] [Google Scholar]
- 4. Hristov AC, Tejasvi T & Wilcox RA (2019) Mycosis fungoides and Sézary syndrome: 2019 update on diagnosis, risk‐stratification, and management. Am J Hematol 94, 1027–1041. [DOI] [PubMed] [Google Scholar]
- 5. Finkel T, Serrano M & Blasco MA (2007) The common biology of cancer and ageing. Nature 448, 767–774. [DOI] [PubMed] [Google Scholar]
- 6. Shay JW & Wright WE (2019) Telomeres and telomerase: three decades of progress. Nat Rev Genet 20, 299–309. [DOI] [PubMed] [Google Scholar]
- 7. Zinn RL, Pruitt K, Eguchi S, Baylin SB & Herman JG (2007) hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res 67, 194–201. [DOI] [PubMed] [Google Scholar]
- 8. Wang S, Hu C & Zhu J (2010) Distinct and temporal roles of nucleosomal remodeling and histone deacetylation in the repression of the hTERT gene. Mol Biol Cell 21, 821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hrdličková R, Nehyba J, Bargmann W & Bose HR (2014) Multiple tumor suppressor microRNAs regulate telomerase and TCF7, an important transcriptional regulator of the Wnt pathway. PLoS One 9, e86990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhatia S, Kaul D & Varma N (2010) Potential tumor suppressive function of miR‐196b in B‐cell lineage acute lymphoblastic leukemia. Mol Cell Biochem 340, 97–106. [DOI] [PubMed] [Google Scholar]
- 11. Azouz A, Wu Y‐L, Hillion J, Tarkanyi I, Karniguian A, Aradi J, Lanotte M, Chen G‐Q, Chehna M & Ségal‐Bendirdjian E (2010) Epigenetic plasticity of hTERT gene promoter determines retinoid capacity to repress telomerase in maturation‐resistant acute promyelocytic leukemia cells. Leukemia 24, 613–622. [DOI] [PubMed] [Google Scholar]
- 12. Garsuault D, Bouyer C, Nguyen E, Kandhari R, Prochazkova‐Carlotti M, Chevret E, Forgez P & Ségal‐Bendirdjian E (2020) Complex context relationships between DNA methylation and accessibility, histone marks, and hTERT gene expression in acute promyelocytic leukemia cells: perspectives for all‐trans retinoic acid in cancer therapy. Mol Oncol 14, 1310–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cong YS, Wen J & Bacchetti S (1999) The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum Mol Genet 8, 137–142. [DOI] [PubMed] [Google Scholar]
- 14. Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT & Benhattar J (2002) Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int J Cancer 101, 335–341. [DOI] [PubMed] [Google Scholar]
- 15. Castelo‐Branco P, Leão R, Lipman T, Campbell B, Lee D, Price A, Zhang C, Heidari A, Stephens D, Boerno S et al. (2016) A cancer specific hypermethylation signature of the TERT promoter predicts biochemical relapse in prostate cancer: a retrospective cohort study. Oncotarget 7, 57726–57736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee DD, Leão R, Komosa M, Gallo M, Zhang CH, Lipman T, Remke M, Heidari A, Nunes NM, Apolónio JD et al. (2019) DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest 129, 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rowland TJ, Bonham AJ & Cech TR (2020) Allele‐specific proximal promoter hypomethylation of the telomerase reverse transcriptase gene (TERT) associates with TERT expression in multiple cancers. Mol Oncol 14, 2358–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kyo S, Takakura M, Fujiwara T & Inoue M (2008) Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci 99, 1528–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chevret E, Andrique L, Prochazkova‐Carlotti M, Ferrer J, Cappellen D, Laharanne E, Idrissi Y, Boettiger A, Sahraoui W, Ruiz F et al. (2014) Telomerase functions beyond telomere maintenance in primary cutaneous T‐cell lymphoma. Blood 123, 1850–1859. [DOI] [PubMed] [Google Scholar]
- 20. Urbano A, Smith J, Weeks RJ & Chatterjee A (2019) Gene‐specific targeting of DNA methylation in the mammalian genome. Cancers 11, 1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poglio S, Prochazkova‐Carlotti M, Cherrier F, Gros A, Laharanne E, Pham‐Ledard A, Beylot‐Barry M & Merlio J‐P (2020) Xenograft and cell culture models of Sézary syndrome reveal cell of origin diversity and subclonal heterogeneity. Leukemia, doi: 10.1038/s41375-020-01068-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kelly‐Sell MJ, Kim YH, Straus S, Benoit B, Harrison C, Sutherland K, Armstrong R, Weng W‐K, Showe LC, Wysocka M et al. (2012) The histone deacetylase inhibitor, romidepsin, suppresses cellular immune functions of cutaneous T‐cell lymphoma patients. Am J Hematol 87, 354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fantin VR, Loboda A, Paweletz CP, Hendrickson RC, Pierce JW, Roth JA, Li L, Gooden F, Korenchuk S, Hou XS et al. (2008) Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T‐cell lymphoma. Cancer Res 68, 3785–3794. [DOI] [PubMed] [Google Scholar]
- 24. Lee DD, Komosa M, Nunes NM & Tabori U (2020) DNA methylation of the TERT promoter and its impact on human cancer. Curr Opin Genet Dev 60, 17–24. [DOI] [PubMed] [Google Scholar]
- 25. Kumaki Y, Oda M & Okano M (2008) QUMA: quantification tool for methylation analysis. Nucleic Acids Res 36 (Web Server issue), W170–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Livak KJ & Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 27. Gazon H, Belrose G, Terol M, Meniane J‐C, Mesnard J‐M, Césaire R & Peloponese J‐M (2016) Impaired expression of DICER and some microRNAs in HBZ expressing cells from acute adult T‐cell leukemia patients. Oncotarget 7, 30258–30275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takakura M, Kyo S, Inoue M, Wright WE & Shay JW (2005) Function of AP‐1 in transcription of the telomerase reverse transcriptase gene (TERT) in human and mouse cells. Mol Cell Biol 25, 8037–8043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terol M, Gazon H, Lemasson I, Duc‐Dodon M, Barbeau B, Césaire R, Mesnard J‐M & Péloponèse J‐M Jr (2017) HBZ‐mediated shift of JunD from growth suppressor to tumor promoter in leukemic cells by inhibition of ribosomal protein S25 expression. Leukemia 31, 2235–2243. [DOI] [PubMed] [Google Scholar]
- 30. Cao Y, Bryan TM & Reddel RR (2008) Increased copy number of the TERT and TERC telomerase subunit genes in cancer cells. Cancer Sci 99, 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, Krämer A, Roncaioli JL, Sand F, Heuckmann JM et al. (2015) Telomerase activation by genomic rearrangements in high‐risk neuroblastoma. Nature 526, 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, van Noesel MM, George RE, Tytgat GAM, Molenaar JJ et al. (2015) TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet 47, 1411–1414. [DOI] [PubMed] [Google Scholar]
- 33. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K et al. (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961. [DOI] [PubMed] [Google Scholar]
- 34. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L & Garraway LA (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L et al. (2013) Frequency of TERT promoter mutations in human cancers. Nat Commun 4, 2185. [DOI] [PubMed] [Google Scholar]
- 36. Ropio J, Merlio J‐P, Soares P & Chevret E (2016) Telomerase activation in hematological malignancies. Genes 7, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lam G, Xian RR, Li Y, Burns KH & Beemon KL (2016) Lack of TERT Promoter mutations in human B‐cell non‐Hodgkin lymphoma. Genes 7, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LT‐Y, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE et al. (2013) Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chatterjee A, Stockwell PA, Rodger EJ, Duncan EJ, Parry MF, Weeks RJ & Morison IM (2015) Genome‐wide DNA methylation map of human neutrophils reveals widespread inter‐individual epigenetic variation. Sci Rep 5, 17328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chebly A, Peloponese J‐M, Ségal‐Bendirdjian E, Merlio J‐P, Tomb R & Chevret E (2020) hMZF‐2, the elusive transcription factor. Front Genet 11, 581115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN & Bird A (1998) Transcriptional repression by the methyl‐CpG‐binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. [DOI] [PubMed] [Google Scholar]
- 42. Tamaru H & Selker EU (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa . Nature 414, 277–283. [DOI] [PubMed] [Google Scholar]
- 43. Mervis JS & McGee JS (2019) Epigenetic therapy and dermatologic disease: moving beyond CTCL. J Dermatol Treat 30, 68–73. [DOI] [PubMed] [Google Scholar]
- 44. Li C‐T, Hsiao Y‐M, Wu T‐C, Lin Y‐W, Yeh K‐T & Ko J‐L (2011) Vorinostat, SAHA, represses telomerase activity via epigenetic regulation of telomerase reverse transcriptase in non‐small cell lung cancer cells. J Cell Biochem 112, 3044–3053. [DOI] [PubMed] [Google Scholar]
- 45. Tao S‐F, Zhang C‐S, Guo X‐L, Xu Y, Zhang S‐S, Song J‐R, Li R, Wu M‐C & Wei L‐X (2012) Anti‐tumor effect of 5‐aza‐2’‐deoxycytidine by inhibiting telomerase activity in hepatocellular carcinoma cells. World J Gastroenterol. 18, 2334–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pettigrew KA, Armstrong RN, Colyer HAA, Zhang S‐D, Rea IM, Jones RE, Baird DM & Mills KI (2012) Differential TERT promoter methylation and response to 5‐aza‐2’‐deoxycytidine in acute myeloid leukemia cell lines: TERT expression, telomerase activity, telomere length, and cell death. Genes Chromosomes Cancer 51, 768–780. [DOI] [PubMed] [Google Scholar]
- 47. Evans IC, Barnes JL, Garner IM, Pearce DR, Maher TM, Shiwen X, Renzoni EA, Wells AU, Denton CP, Laurent GJ et al. (1979) Epigenetic regulation of cyclooxygenase‐2 by methylation of c8orf4 in pulmonary fibrosis. Clin Sci (Lond) 2016 , 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee SH, Kim J, Kim W‐H & Lee YM (2009) Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene 28, 184–194. [DOI] [PubMed] [Google Scholar]
- 49. Seelan RS, Mukhopadhyay P, Pisano MM & Greene RM (2018) Effects of 5‐Aza‐2′‐deoxycytidine (decitabine) on gene expression. Drug Metab Rev 50, 193–207. [DOI] [PubMed] [Google Scholar]
- 50. Wozniak RJ, Klimecki WT, Lau SS, Feinstein Y & Futscher BW (2007) 5‐Aza‐2’‐deoxycytidine‐mediated reductions in G9A histone methyltransferase and histone H3 K9 di‐methylation levels are linked to tumor suppressor gene reactivation. Oncogene 26, 77–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Evaluation of tumor cells proportion before and after cell sorting.
Fig. S2. Correlation between hTERT expression level and telomerase activity.
Fig. S3. Absence of correlation between THOR methylation status and hTERT expression level.
Fig. S4. WT1 mRNA and protein expression.
Fig. S5. hTERT promoter methylation profiles after HDACi treatments.
Table S1. hTERT Bisulfite PCR conditions and primer sequences.
Table S2. Primer sequences for hTERT, hWT1 and hPRT‐1.
Table S3. Primer sequences used for WT1 ChIP‐qPCR.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.