

In this study, Robbe et al. investigated how CHD4/NuRD is localized to specific cardiac target genes, as neither CHD4 nor NuRD can directly bind DNA. They coupled a bioinformatics-based approach with mass spectrometry analyses to demonstrate that CHD4 interacts with the core cardiac transcription factors GATA4, NKX2-5, and TBX5 during embryonic heart development, thus providing new insights into how mutations in the CHD4 protein lead to cardiac-specific disease states.
Keywords: NuRD, CHD4, recruitment, cardiac, chromatin remodeling
Abstract
The nucleosome remodeling and deacetylase (NuRD) complex is one of the central chromatin remodeling complexes that mediates gene repression. NuRD is essential for numerous developmental events, including heart development. Clinical and genetic studies have provided direct evidence for the role of chromodomain helicase DNA-binding protein 4 (CHD4), the catalytic component of NuRD, in congenital heart disease (CHD), including atrial and ventricular septal defects. Furthermore, it has been demonstrated that CHD4 is essential for mammalian cardiomyocyte formation and function. A key unresolved question is how CHD4/NuRD is localized to specific cardiac target genes, as neither CHD4 nor NuRD can directly bind DNA. Here, we coupled a bioinformatics-based approach with mass spectrometry analyses to demonstrate that CHD4 interacts with the core cardiac transcription factors GATA4, NKX2-5, and TBX5 during embryonic heart development. Using transcriptomics and genome-wide occupancy data, we characterized the genomic landscape of GATA4, NKX2-5, and TBX5 repression and defined the direct cardiac gene targets of the GATA4–CHD4, NKX2-5–CHD4, and TBX5-CHD4 complexes. These data were used to identify putative cis-regulatory elements controlled by these complexes. We genetically interrogated two of these silencers in vivo: Acta1 and Myh11. We show that deletion of these silencers leads to inappropriate skeletal and smooth muscle gene misexpression, respectively, in the embryonic heart. These results delineate how CHD4/NuRD is localized to specific cardiac loci and explicates how mutations in the broadly expressed CHD4 protein lead to cardiac-specific disease states.
Transcriptional repression is mediated by broadly expressed multiprotein complexes that modify and remodel chromatin. Repression by these complexes is achieved by alteration of chromatin states through direct DNA sequence-specific binding of a given complex or by recruitment of the complex to defined loci via interactions with tissue-specific transcription factors. The nucleosome remodeling and deacetylase (NuRD) complex represses gene expression and is essential from flies to humans. In most cases, the histone deacetylase and ATP-dependent chromatin remodeling helicase of NuRD combine to modulate chromatin states at target genes (Wade et al. 1998, 1999; Xue et al. 1998; Zhang et al. 1998). Components of the NuRD complex differ but invariably contain either of the ATP-dependent chromodomain helicases: CHD3 or CHD4 (Watson et al. 2012; Joshi et al. 2013; Low et al. 2016; Zhang et al. 2016; Hoffmeister et al. 2017; Farnung et al. 2020). The crucial nature of CHD4 has been established through clinical studies that have shown that mutations in CHD4 lead to congenital heart disease (Zaidi et al. 2013; Homsy et al. 2015; Sifrim et al. 2016; Waldron et al. 2016; Weiss et al. 2016). In addition, genetic studies in mice have demonstrated that cardiac conditional Chd4-null mutants die at mid-gestation and that loss of CHD4-mediated repression leads to misexpression of fast skeletal and smooth muscle myofibril isoforms, cardiac sarcomere malformation, and early embryonic lethality (Gómez-Del Arco et al. 2016; Wilczewski et al. 2018).
Although CHD4 is essential for heart development, and its disease relevance has been shown, one of the central unanswered questions regarding the complex's regulatory mechanism is how CHD4/NuRD is recruited to specific cardiac gene loci, given that neither CHD4 nor NuRD binds DNA. Using a genomic approach, we identified overrepresented DNA motifs recognized by several core cardiac transcription factors at CHD4-bound genomic regions. Using parallel reaction monitoring mass spectrometry (PRM nLC-MS/MS) (Picotti et al. 2013; Federspiel et al. 2019), we confirmed that CHD4 interacts in vivo in mid-gestation hearts with GATA4, NKX2-5, and TBX5. Further analysis revealed that CHD4 and GATA4, NKX2-5, and TBX5 converge on regulatory elements genome-wide associated with transcriptional repression. These findings imply an important dual regulatory role for these critical cardiac transcription factors.
We used our data to map putative cardiac cis-regulatory elements (CREs) regulated through GATA4–CHD4, NKX2-5–CHD4, and TBX5–CHD4 complexes. Deletion of a putative CRE at the skeletal muscle Acta1 gene that contains an NKX2-5 binding site led to inappropriate Acta1 expression in fetal hearts. Similarly, deletion of a CHD4 CRE within the smooth muscle Myh11 gene containing a GATA4 binding site led to Myh11 misexpression in fetal hearts. Collectively, our results demonstrate that CHD4/NuRD is recruited to defined loci through a core set of cardiac transcription factors to repress inappropriate gene expression in developing hearts.
Results
CHD4 interacts with the core cardiac transcription factors GATA4, NKX2-5, and TBX5
One of the central issues regarding the mechanism and function of the CHD4/NuRD complex involves the recruitment of CHD4/NuRD to specific loci, given that neither CHD4 nor NuRD binds DNA directly (Fig. 1A). To address these issues, we performed motif discovery of CHD4 ChIP-seq data sets obtained from E10.5 hearts (GEO: GSE109012) (Wilczewski et al. 2018). Our analyses revealed a striking abundance of significantly overrepresented cardiac transcription factor consensus motifs in CHD4-bound regions (Fig. 1B). These sequences included binding sites for the core cardiac transcription factors GATA4, NKX2-5, and TBX5, each of which is required for cardiac development and, when mutated, causes human heart disease (Durocher et al. 1997; McCulley and Black 2012; Baban et al. 2014; Luna-Zurita et al. 2016; Akerberg et al. 2019; Jumppanen et al. 2019). Moreover, mice homozygous null for Tbx5, Gata4, and Nkx2-5 display heart defects at E10.5, the same developmental stage that requires Chd4 (Bruneau et al. 2001; Stennard et al. 2003; Watt et al. 2004; Mori et al. 2006; Maitra et al. 2009; Terada et al. 2011; Zhou et al. 2015; Gómez-Del Arco et al. 2016; Wilczewski et al. 2018).
Figure 1.

CHD4 interacts with core cardiac TFs: GATA4, NKX2-5, and TBX5. (A) Schematic of the nucleosome remodeling and deacetylase (NuRD) complex localizing to target loci through interaction with a tissue-specific cofactor. (B) Predicted co-occupancy of CHD4 and GATA4, NKX2-5, and TBX5 through significant overrepresentation of known binding motifs of each factor in CHD4-bound genomic regions. Q-values = 0.0000 generated from FDR-corrected P-value based on the cumulative hypergeometric distribution. (C–F) CHD4 associates with GATA4, NKX2-5, and TBX5 through proximity ligation assay. Each image is shown as a maximum intensity z-stack projection using a 63× oil magnification objective. The presence of PLA signal (white dots) denotes the close physical proximity of target proteins and represents a physical interaction. CHD4 and turboGFP alone (C) results in a lower frequency and intensity of PLA signal than CHD4–GATA4 (D), CHD4–NKX2-5 (E), or CHD4–TBX5 (F). (G) Flag/Myc-CHD4, turboGFP (tGFP)-NKX2-5, and tGFP-GATA4 were cotransfected into HEK293 cells; immunoprecipitation (IP) was performed with anti-Flag M2 beads to pull down the CHD4 interactome; and Western blot was performed. Anti-tGFP antibody was probed to detect NKX2-5 and GATA4 proteins, and anti-Myc antibody was probed to detect CHD4 protein. (H) CHD4 complexes affinity-purified from wild-type E10.5 mouse embryonic hearts show a significant enrichment for peptides belonging to GATA4, TBX5, or NKX2-5 when compared with purified GFP complexes, as determined by PRM-MS quantification and Student's t-test. Data are shown as mean ± SEM of triplicates. n = 3, with each replicate of >28 pooled embryonic hearts per IP. (****) P-value < 0.0001.
We confirmed CHD4 colocalization with GATA4, NKX2-5, and TBX5 by proximity ligation assays (PLAs). CHD4 was expressed with either GATA4, NKX2-5, or TBX5, or as a negative control (turboGFP) (Fig. 1C–F). In support of our genomic analyses, we found that CHD4 interacts with GATA4, NKX2-5, and TBX5 and, in all three cases, the interaction occurs predominantly in the nucleus. The interactions of CHD4 with these three transcription factors were validated by coimmunoprecipitation (co-IP) in transfected HEK293 cells (Fig. 1G; Waldron et al. 2016).
To elucidate whether CHD4 interacts with either GATA4, NKX2-5, or TBX5 in heart tissue in vivo, we performed parallel reaction monitoring MS (PRM LC-MS/MS) (Peterson et al. 2012; Justice et al. 2021; Shi et al. 2021) on immuno-affinity-purified cardiac E10.5 CHD4 interactomes isolated in the presence of Pierce universal nuclease (Conlon et al. 2012; Greco et al. 2012; Charpentier et al. 2013; Kaltenbrun et al. 2013; Waldron et al. 2016). In contrast to conventional MS/MS approaches, PRM-MS/MS is a hypothesis-driven approach using a preselected peptide to target and quantify a defined protein in a complex mixture (Supplemental Fig. S1).
Consistent with previous findings (Waldron et al. 2016), PRM LC-MS/MS analysis of E10.5 CHD4 endogenous interactomes revealed CHD4 in complex with TBX5. Our results further divulged that CHD4 is in complex with GATA4 and NKX2-5 in E10.5 hearts (Fig. 1H). To our knowledge, this is the first report of an endogenous interaction between CHD4 and NKX2-5 during heart development. Together, our data establish that CHD4 complexes in vivo with GATA4, NKX2-5, and TBX5 during a time point when CHD4 is essential for vertebrate heart development.
GATA4, NKX2-5, and TBX5 are cobound with CHD4
To determine whether GATA4, NKX2-5, and TBX5 binding sites were prevalent in CHD4 ChIP-seq peaks, we overlapped GATA4, NKX2-5, and TBX5 ChIP-seq data sets (Akerberg et al. 2019) with CHD4 ChIP-seq peaks. We observe GATA4, NKX2-5, and/or TBX5 ChIP-seq signal at CHD4-bound regions, suggesting coordinate binding between CHD4 and these factors (Fig. 2A).
Figure 2.

CHD4 co-occupies directly repressed loci with GATA4, NKX2-5, and TBX5. (A) Heat map visualizing the density of ChIP-seq signal of each transcription factor (GATA4, NKX2-5, and TBX5) across genomic regions occupied by CHD4 in developing hearts. (B) Overlap of genes bound by CHD4 and up-regulated in Chd4-null hearts. Of the 920 genes up-regulated in hearts devoid of CHD4, 475 (52%) are also bound by CHD4 at E10.5. These 475 genes are termed CHD4-repressed genes and are associated with 1082 CHD4 peaks. (C) Biological processes overrepresented in regions co-occupied and repressed by CHD4–GATA4, CHD4–NKX2-5, and CHD4–TBX5. The heat map is presented as the −log Bonferroni-corrected P-value. Gray boxes represent biological processes with a nonsignificant P-value for that group. (D) Upset plot displaying the intersection of ChIP-seq peaks shared between CHD4 and GATA4, NKX2-5, and TBX5, respectively. Each column represents a different possible overlap of the data, with the totals defined with the horizontal colored bars. CHD4 peaks that did not overlap with any transcription factor are not depicted in this plot. (E) Stacked bar plot visualizing the distribution of peaks to their associated gene feature, annotated using ChIPseeker. Columns represent transcription factor peaks associated with CHD4-repressed genes or other regions. Statistical significance was calculated using a t-test, comparing each column with GNT.
Our previous data have shown that CHD4 interacts with TBX5 to repress noncardiac gene programs (Waldron et al. 2016); thus, we hypothesized that the interactions with GATA4 and NKX2-5 have a similar consequence. Therefore, we focused on sites within genes that were also bound by CHD4 and that demonstrated increased RNA levels following CHD4 knockout, supportive of a CHD4-repressive activity (n = 1082 peaks) (Fig. 2B). Gene ontology analysis of peaks cobound by CHD4 and GATA4, NKX2-5, or TBX5 revealed potential biological differences between the CHD4 interaction with each of these transcription factors. Specifically, CHD4/GATA4- and CHD4/NKX2-5-cobound peaks were linked to genes associated with regulation of axonogenesis, muscle structure development, or cardiac muscle development. In contrast, CHD4/TBX5-cobound peaks did not demonstrate this association (Fig. 2C).
As there are well-characterized interactions between TBX5, GATA4, and NKX2-5 (Durocher et al. 1997; Jumppanen et al. 2019), we hypothesized that there are genomic loci at which CHD4 binds more than one transcription factor (Fig. 2A). Of the CHD4-bound peaks in repressed genes, more than half (n = 397) were cobound by TBX5, GATA4, and/or NKX2-5 (Fig. 2D). Furthermore, we found that 155 peaks at CHD4-repressed genes were bound by all three transcription factors in addition to CHD4 (Fig. 2D). GATA4 was found most often to be associated with CHD4-repressed regions and was also the most likely to cobind with CHD4 individually (Fig. 2D).
We next determined the genomic localization of the CHD4/transcription factor binding to determine a potential effect on gene expression. We annotated binding peaks with the nearest target gene feature (Fig. 2E). These analyses show that GATA4–CHD4, NKX2-5–CHD4, and TBX5–CHD4 are significantly enriched at intergenic, intronic, and promoter regions, suggesting that they regulate gene expression, as these sites are associated with CHD4-mediated transcriptional repression (Wilczewski et al. 2018). Motif analyses of GATA4–CHD4, NKX2-5–CHD4, and TBX5–CHD4 show an enrichment of the TEAD and CTCF binding motif (Supplemental Fig. S2A) versus motif analyses of the binding of GATA4, NKX2-5, and TBX5 alone; i.e., in the absence of CHD4 (Supplemental Fig. S2B). These findings suggest that TEAD and CTCF may act as cofactors at repressed versus activated GATA4–CHD4, NKX2-5–CHD4, and TBX5–CHD4 targets (Luna-Zurita et al. 2016).
We next sought to determine whether the number of bound transcription factors or their composition affected the magnitude of CHD4 gene repression. To this end, we examined changes in expression in CHD4/GATA-bound, CHD4/NKX2-5-bound, and/or CHD4/TBX5-bound genes (Supplemental Fig. S2C). We conclude that the magnitude of CHD4 repression is not dependent on the identity of the recruiting factor or the number of occupying factors. Thus, CHD4 interaction with GATA4, NKX2-5, and TBX5 is critical for the localization of the complex to target loci, but the composition of the interaction does not affect the extent of transcriptional repression.
NKX2-5 recruits CHD4 to repress expression of skeletal actin in embryonic hearts
Strikingly, we found that GATA4 was localized to 71% (513 of 719) of all CHD4-repressed loci (Fig. 2D). We therefore addressed whether binding of NKX2-5 or TBX5 alone and at a single site is sufficient to recruit and repress CHD4 target genes. We thus analyzed our data sets to identify putative CREs in cardiac tissue, focusing on (1) target genes that contained a single binding site for either NKX2-5 or TBX5, (2) binding sites that are conserved between mice and humans, (3) binding sites associated with a single CHD4 peak, and (4) binding sites associated with enrichment of H3K27me3 (Fig. 3; Supplemental Table S1; The ENCODE Project Consortium 2012). Among those genes meeting these four criteria was Acta1, the gene that encodes a skeletal actin isoform and is an established CHD4/NuRD target (Wilczewski et al. 2018). In normal developing hearts, cardiac actin Actc1 is the predominant actin isoform (Mayer et al. 1984), whereas Acta1, the skeletal isoform, is expressed at low to undetectable levels. Examination of the Acta1 locus identified a single putative NKX2-5 DNA-binding motif, and, consistent with our gene feature analyses, the NKX2-5 binding site is located in the Acta1 3′ UTR (Fig. 4A–C). Thus, the 3′ UTR of Acta1 was postulated to contain a cis-regulatory element (CRE) bound by NKX2-5 and CHD4 in E10.5 hearts.
Figure 3.
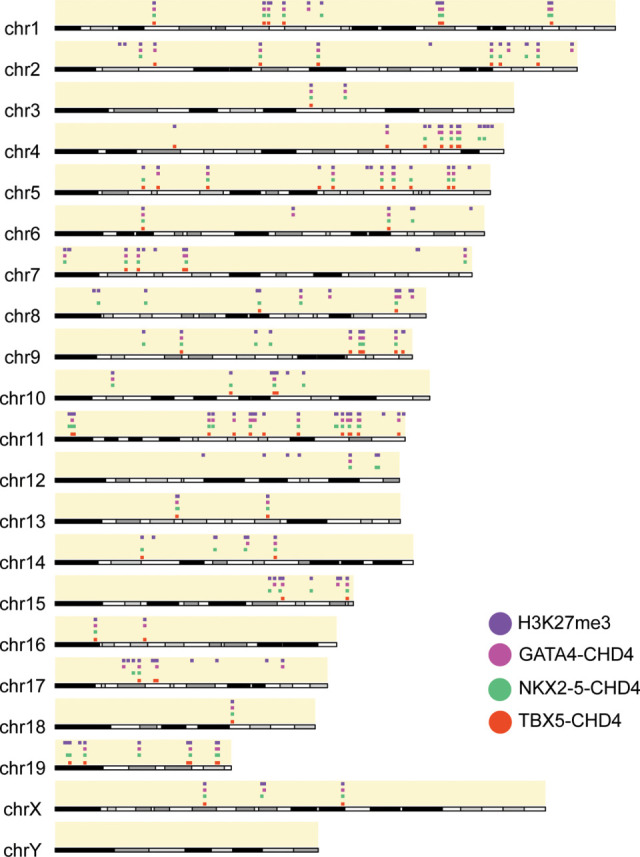
GATA4, NKX2-5, and TBX5 are associated with H3K27me3 at CHD4-repressed genes. Sites of CHD4 and transcription factor association at CHD4-repressed genes marked with H3K27me3 histone modification at a chromosome level.
Figure 4.

NKX2-5 recruits CHD4 to repress expression of skeletal actin in embryonic hearts. (A) NKX2-5-mediated recruitment of CHD4 to target repressor downstream from the TTS in the Acta1 locus. The putative NKX2-5 motif was identified through motif analysis. (B) Visualization by IGV browser of ChIP-seq signal across the Acta1 locus. The repressor region is highlighted with a yellow rectangle. (C) Sequence conservation of the NKX2-5 motif in the CRISPR-deleted region between humans and mice. The red box denotes predicted NKX2-5 binding motif and shows strong conservation with the human sequence. (D) Survival ratio of each phenotype at E10.5 and P0 (postnatal day 0). Eight litters for E10.5 and seven litters for P0 were examined. (E) Relative expression of Acta1 in E10.5 embryonic hearts when the repressor region was excised from one or both alleles. (F) Histological analysis of E10.5 WT, Acta1Δ3′/+, and Acta1Δ3′/Δ3′ hearts. Boxed areas are enlarged at the right. Acta1Δ3′/+ and Acta1Δ3′/Δ3′ hearts exhibit a thinner compact myocardium (c.m.) layer compared with WT controls (red line segments), and the Acta1Δ3′/Δ3′ hearts also display fewer myocardial trabecula (tr.) compared with WT and Acta1Δ3′/+ hearts (blue arrows). (G,H) Quantification of compact myocardium thickness in the right (G) and left (H) ventricles. Data are presented as mean ± SEM; significance was assessed using an unpaired two-tailed Student's t-test. (*) P-value < 0.05, (**) P-value < 0.01, (***) P-value < 0.001. n = 5 nonpooled hearts per genotype for qRT-PCR. Data were normalized to expression of Pgk1. n = 6 per genotype for histological analyses. Scale bars, 40 μm.
To genetically interrogate whether Acta1 is repressed in vivo in cardiac tissue by NKX2-5–CHD4, we used CRISPR/Cas9 technologies to generate mice containing a deletion of the putative CRE containing an endogenous NKX2-5 binding site in the Acta1 3′ UTR (Acta1Δ3′) (Fig. 4A–D; Supplemental Fig. S3A). These studies reveal that the loss of a single copy of the CRE containing the NKX2-5 binding site in the 3′ UTR leads to Acta1 misexpression in E10.5 hearts in a dominant manner (Fig. 4E), with mRNA levels of Acta1 showing a significant increase relative to wild-type littermate controls (Fig. 4E). Thus, a single copy of an NKX2-5 binding site is sufficient to recruit CHD4 and repress expression of a skeletal actin isoform in developing mammalian hearts.
Repression of Acta1 is essential for normal cardiac development
Mice heterozygous for Acta1Δ3′ (Acta1Δ3′/+) were found to be viable and fertile. Although homozygous Acta1Δ3′ (Acta1Δ3′/Δ3′) hearts showed only a modest increase in Acta1 over heterozygous Acta1Δ3′ hearts (Fig. 4E), we recovered no homozygous Acta1Δ3′ (Acta1Δ3′/Δ3′) mice postnatally (Fig. 4D). Histological analyses of Acta1Δ3′/+ and Acta1Δ3′/Δ3′ E10.5 hearts revealed a decrease in thickness of the outside wall of the left and right ventricles (Fig. 4F–H). By E12.5, Acta1Δ3′/+ and Acta1Δ3′/Δ3′ hearts had undergone a dramatic remodeling of the right ventricle, with the outer wall of Acta1Δ3′/+ indistinguishable from wild-type littermates, while Acta1Δ3′/Δ3′ hearts (in opposition to E10.5) showed an increase in thickness (Supplemental Fig. S3B–D). As with E10.5, the left ventricle in Acta1Δ3′/+ and Acta1Δ3′/Δ3′ hearts remained thinner versus controls. Phenotypes similar to those found in Acta1Δ3′/+ and Acta1Δ3′/Δ3′ E12.5 hearts were also observed at E16.5 (Supplemental Fig. S3E). Together, these data demonstrate that repression of Acta1 is essential for normal cardiac development.
To determine the transcriptional consequence of misexpressing Acta1 in embryonic hearts, we performed transcriptional profiling (RNA-seq) on E10.5 heart tissue derived from Acta1Δ3′/Δ3′ and wild-type littermates. Analyses revealed a total of 73 genes dysregulated in Acta1Δ3′/Δ3′ hearts (Fig. 5A); two genes (including Acta1) were up-regulated (Fig. 5A,B), and an additional 71 genes were down-regulated (Fig. 5A,C–F). Changes in expression for a subset of the down-regulated genes—Snta1, Taf10, Gata6, and Ctf1—were verified by qRT-PCR (Fig. 5C–F). Gene ontology (GO) analyses of the dysregulated genes in Acta1-expressing hearts (Fig. 5G) revealed that the genes most overrepresented are those involved in metabolic processes. These data suggest that inappropriate expression of the skeletal muscle gene Acta1 disrupts essential metabolic processes, leading to a malformed heart and, ultimately, embryonic death.
Figure 5.

Transcriptional profiling (RNA-seq) of E10.5 heart tissue derived from Acta1Δ3′/Δ3′ and wild-type littermates. (A) Volcano plot of identified genes. Differential genes (adjusted P < 0.05) are labeled in blue, and nonchanged genes are labeled in red. Genes with log2 (fold change) < 0.5 are down-regulated in Acta1Δ3′/Δ3′, and genes with log2 (fold change) > 0.5 are up-regulated in Acta1Δ3′/Δ3′. Genes of interests are labeled. (B) Normalized counts of Acta1 from the RNA-seq. (C–F) Relative expression of Snta1 (C), Taf10 (D), Gata6 (E), and Ctf1 (F) in E10.5 WT and Acta1Δ3′/Δ3′ embryonic hearts. Data are presented as mean ± SEM, and significance was assessed using an unpaired two-tailed Student's t-test. (*) P-value < 0.05, (**) P-value < 0.01, (***) P-value < 0.001. n = 5 nonpooled hearts per genotype. (G) Gene ontology (GO) term analyses for differentially down-regulated genes in Acta1Δ3′/Δ3′ hearts.
Deletion of a GATA4-CHD4 site in Myh11 leads to its misexpression in developing mouse hearts
To test the function of GATA4 in CHD4-mediated gene repression, we identified potential CREs containing GATA4 binding sites. Our screen revealed a single GATA4 binding site in the third intron of Myh11, the gene encoding the smooth muscle myosin heavy chain (SMMHC) protein (Fig. 6A,B), an established target of CHD4 (Wilczewski et al. 2018). The canonical GATA4 DNA-binding motif in Myh11 intron 3 was shown to be conserved between mice and humans; to be co-occupied by CHD4 and GATA4, NKX2-5, and TBX5 during fetal heart development; and to be enriched for H3K27me3 (Fig. 6B,C). Consistently, we found Myh11 expression to be increased significantly in GATA4 mutant hearts versus littermate controls (Supplemental Fig. S4). Interestingly, even though we observed strong binding for NKX2-5 and TBX5, we failed to identify a putative binding sequence for either NKX2-5 or TBX5 in the third intron of Myh11 in mice or humans. Thus, it may be that GATA4 recruits NKX2-5 and TBX5 as well as CHD4 to this region.
Figure 6.

GATA4 is required for MYH11 expression in embryonic hearts. (A) Schematic demonstrating GATA4-mediated recruitment of CHD4 to the target repressor in the third intron of Myh11. A putative GATA4 binding motif was identified through motif analysis using HOMER. (B) Visualization by IGV browser of ChIP-seq signal of GATA4, NKX2-5, TBX5, CHD4, and H3K27me3 across the Myh11 locus. The repressor region is highlighted with a yellow rectangle. (C) Sequence conservation of the GATA4 motif in the CRISPR-deleted region between humans and mice. The red box denotes the predicted GATA4 binding motif and shows strong conservation with the human sequence. (D) Relative expression of Myh11 in E10.5 embryonic hearts when the repressor region was excised in one or both alleles. (E) Immunohistochemistry (IHC) staining on E10.5 WT, Myh11Δi3/+, and Myh11Δi3/Δi3 hearts. Tropomyosin (TMY) stained the cardiomyocytes, MYH11 was stained in red, and DAPI stained the nuclei. (F) Histological analysis of E10.5 WT, Myh11Δi3/+, and Myh11Δi3/Δi3 hearts. Boxed areas are enlarged at the right. Myh11Δi3/+ and Myh11Δi3/Δi3 hearts exhibit a thinner compact myocardium (c.m.) layer in the right ventricles compared with WT controls (red line segments), and the Myh11Δi3/Δi3 hearts also display more myocardial trabecula (tr.) compared with WT and Myh11Δi3/+ hearts (blue arrows). (G,H) Quantification of compact myocardium thickness in the right (G) and left (H) ventricles. Data are presented as mean ± SEM, and significance was assessed with unpaired two-tailed Student's t-test. (*) P-value < 0.05, (**) P-value < 0.01. n = 3 nonpooled hearts per genotype for qRT-PCR. Data were normalized to expression of Pgk1. n = 6 per genotype for the histological analyses and IHC staining. Scale bars, 40 μm.
To determine the role of the potential cooperativity of all three cofactors in CHD4-mediated transcriptional repression, we used CRISPR/Cas9 technologies to delete the GATA4 binding region in mice (Myh11Δi3) (Fig. 6A–C; Supplemental Fig. S5A,B; Hashimoto et al. 2016). Strikingly, we found that deletion of the Myh11 silencer was sufficient to cause a dramatic up-regulation of Myh11 in a dominant manner (Fig. 6D). Myh11 expression increased ∼70-fold in E10.5 heart tissue derived from heterozygous Myh11Δi3/+ mice, whereas in mice homozygous for Myh11Δi3/Δi3, the deletion increased Myh11 expression by >200-fold. We further confirmed MYH11 misexpression in Myh11Δi3/+ and Myh11Δi3/Δi3 E10.5 hearts by immunohistochemistry (Fig. 6E). Collectively, these data define a novel CRE of Myh11 that is regulated by the GATA4–CHD4 complex in association with NKX2-5 and TBX5, and we further show that Myh11Δi3 CRE is essential to repress Myh11 in developing hearts.
Cardiac misexpression of MYH11 leads to global changes in cardiac gene expression
To determine the consequences of misexpressing the smooth muscle gene Myh11 in developing mammalian hearts, we conducted histological analyses on Myh11Δi3/+ and Myh11Δi3/Δi3 E10.5 hearts. Analyses reveal that Myh11Δi3/+ and Myh11Δi3/Δi3 hearts have increased thickness of the outer wall of the right ventricle, while the left ventricle remained undistinguishable from wild-type littermates (Fig. 6F–H). Transcriptional profiling of Myh11Δi3/Δi3 and wild-type littermate E10.5 hearts showed that morphological defects in Myh11Δi3/Δi3 hearts are associated with dysregulation of 3653 genes, 48% (1763 of 3653) of which were down-regulated in Myh11Δi3/Δi3, and 52% (1890 of 3653), including Myh11, of which were up-regulated (Fig. 7A–F). Of note, we found that deletion of the Myh11Δi3/Δi3 CRE, an element contained in intron 3 of the Myh11 gene, led to misregulation of three genes flanking the Myh11 locus (Ercc4, Pam, and Abcc1) (Supplemental Fig. S6), thus suggesting that Myh11Δi3/Δi3 leads to broad changes in the chromatin landscape.
Figure 7.

Transcriptional profiling (RNA-seq) of E10.5 heart tissue derived from Myh11Δi3/Δi3 and wild-type littermates. (A) Volcano plot of identified genes. Differential genes (adjusted P < 0.05) are labeled in blue, and nonchanged genes are labeled in red. Genes with log2 (fold change) < 0.5 are down-regulated in Myh11Δi3/Δi3 hearts, and genes with log2 (fold change) > 0.5 are up-regulated in Myh11Δi3/Δi3 hearts. Genes of interests are labeled. (B) Normalized counts of Myh11 from the RNA-seq. (C–F) Relative expression of Tnni3 (C), Tnnt1 (D), Hand1 (E), and Tbx5 (F) in E10.5 WT and Myh11Δi3/Δi3 embryonic hearts. Data are presented as mean ± SEM, and significance was assessed using an unpaired two-tailed Student's t-test. (*) P-value < 0.05, (**) P-value < 0.01, (***) P-value < 0.001. n = 3 nonpooled hearts per genotype. (G) Circular plot of representative differentially expressed genes in Myh11Δi3/Δi3 hearts, simultaneously presenting a detailed view of the relationships between expression changes (left semicircle perimeter) and enriched biological processes (right semicircle perimeter).
From our transcriptional profiling, we found that genes down-regulated in Myh11Δi3/Δi3 are predominantly associated with muscle assembly and contraction, while up-regulated genes were associated with heart morphogenesis and angiogenesis (Fig. 7G). Surprisingly, the anatomical and transcriptional changes associated with Myh11Δi3/Δi3 were compensated for at later stages of cardiac development, and we found that in adult Myh11Δi3/+ and Myh11Δi3/Δi3 hearts, Myh11 levels are equivalent to that of wildtype littermate controls (Supplemental Fig. S5C). Consistently, we recovered Myh11Δi3/+ and Myh11Δi3/Δi3 live-born mice at the expected Mendelian ratios (Supplemental Fig. S5B). In sum, these data demonstrate that repression of Myh11 in E10.5 hearts is required for early cardiac muscle assembly, but the hearts undergo compensatory changes, shutting down Myh11 misexpression and forming fully functional hearts by birth.
Discussion
Although CHD4 and the NuRD complex are well established to function in repressing transcription, how CHD4/NuRD is recruited to specific loci remained unknown. Here, we demonstrate that the core cardiac transcription factors GATA4, NKX2-5, and TBX5 interact with and recruit CHD4 to defined genomic locations associated with H3K27me3. Our findings reveal that NKX2-5, a cardiac transcription factor that has long been understood to function as a transcriptional activator (McCulley and Black 2012), can also function as a cardiac transcriptional repressor. These findings are congruent with recent studies showing that TBX5 and GATA4 can interact with CHD4 in human induced pluripotent stem cells (iPSC) differentiated into cardiomyocytes (Gonzalez-Teran et al. 2022). Our work further shows that NKX2-5 and GATA4 function to repress genes incompatible with heart development and function. Because mutations in GATA4, NKX2-5, and TBX5 cause a range of congenital heart diseases (Basson et al. 1997; Li et al. 1997; Furtado et al. 2017; Steimle and Moskowitz 2017; Zhang et al. 2017; Behiry et al. 2019), our findings imply that the respective patient phenotypes are due not only to loss of cardiac gene expression but also to misexpression of noncardiac genes in the developing hearts.
Our data support the hypothesis that mutations in regulatory regions essential for CHD4-mediated repression act in a dominant manner. This may provide one mechanism for the prevalence of CHD in humans with one mutated copy of NKX2-5, GATA4, or TBX5. Our findings that CHD4 complexes with temporally and spatially regulated cardiac transcription factors further explicates how mutations in the broadly expressed CHD4 protein lead to cardiac-specific disease states.
The finding that GATA4, NKX2-5, and TBX5 can recruit CHD4 to the majority of CHD4-repressed target genes in the heart is broadly consistent with studies in B cells demonstrating interaction of CHD4 with lineage-specific transcription factors (Yoshida et al. 2019). Identifying the region of CHD4 that interacts with tissue-specific transcription factors is essential in understanding the molecular basis of cardiac disease associated with mutations in CHD4. However, analyses of GATA4, NKX2-5, TBX5, or the B cell transcription factors that interact with CHD4 failed to uncover any shared sequence homology, conserved sequences, or common motifs, even at low stringency, thus raising the question of how this diverse set of transcription factors interacts with CHD4. Moreover, our analyses of the CHD4 interactome failed to identify any proteins that may function as adapter proteins. Based on these observations, we favor a model in which these sets of transcription factors recognize a secondary structure within CHD4, possibly in or adjacent to the PHD and/or CD domains (Mansfield et al. 2011; Watson et al. 2012; Low et al. 2016; Zhang et al. 2016; Farnung et al. 2020).
A central question from our findings is how TBX5, GATA4, and NKX2-5 can discriminate between the up-regulation and down-regulation of target genes. We favor a model in which the interactome of TBX5, GATA4, and NKX2-5 is spatially and temporally regulated during heart development, potentially through post-translational modifications. This model is supported by findings with TBX20, an essential cardiac protein that interacts with the cardiac transcription factor CASZ1 in a temporally regulated manner (Kennedy et al. 2017). Alternatively, the choice to up-regulate versus down-regulate may reflect the composition of the chromatin remodeling complex. For example, the composition of the BAF complex is altered by changes in levels of BMP4 (Hota et al. 2022). Testing these models through the generation and characterization of cardiac phosphoproteomics will be critical to address these issues.
CHD4/NuRD is essential for numerous developmental events, such as ensuring proper timing of the switch from stem cell lineages to differentiated cell types, maintaining cell differentiation, and activating DNA damage response pathways (Larsen et al. 2010; Polo et al. 2010; Scimone et al. 2010; Hosokawa et al. 2013; O'Shaughnessy and Hendrich 2013; Sparmann et al. 2013; Chudnovsky et al. 2014; O'Shaughnessy-Kirwan et al. 2015; Gómez-Del Arco et al. 2016; Zhao et al. 2017; Ostapcuk et al. 2018; Arends et al. 2019; McKenzie et al. 2019; Yoshida et al. 2019; Hou et al. 2020; Sreenivasan et al. 2021). We propose that one of the generalized functions of CHD4/NuRD is to repress the inappropriate activation of developmental programs of a given tissue or cell type. We suggest that alterations in CHD4 recruitment by tissue-specific transcription factors lead to the wide range of CHD4-associated disease states.
Materials and methods
Mice
Chd4flox/flox mice were obtained from Dr. Katia Georgeopolos (Williams et al. 2004). Nkx2-5Cre/+ mice were obtained from Robert Schwartz (Moses et al. 2001). Chd4 conditional knockout mice and control littermates were obtained by breeding female Chd4flox/flox mice to male Chd4flox/+; Nkx2-5Cre/+ mice. CRISPR/Cas9-mediated genome editing was performed by the University of North Carolina Animal Models Core Facility. CRISPR founder mice were bred to wild-type C57BL/6J female mice for two generations. Heterozygous F2 mice were interbred to generate embryos. Research was approved by the Institutional Animal Care and Use Committee at the University of North Carolina and conformed to the Guide for the Care and Use of Laboratory Animals.
CRISPR/Cas9-mediated mouse engineering and breeding
Cas9 guide RNAs flanking the desired deletion regions were identified using Benchling software. Three guide RNAs at each end of the target sequence were selected for activity testing. Guide RNAs were cloned into a T7 promoter vector followed by in vitro transcription and spin column purification. Functional testing was performed by transfecting a mouse embryonic fibroblast cell line with guide RNA and Cas9 protein. The guide RNA target site was amplified from transfected cells and analyzed by ICE (Synthego). One guide RNA at each end of the target sequence was selected, and a donor oligonucleotide was included to facilitate homologous recombination to produce a clean deletion event between the guide RNA cut sites. C57BL/6J zygotes were electroporated with 1.2 μM Cas9 protein, 47 ng/μL each guide RNA, and 400 ng/μL donor oligonucleotide and implanted in recipient pseudopregnant females. Resulting pups were screened by PCR and sequencing for the presence of the deletion allele. Male founders with the correct deletion were mated to wild-type C57BL/6J females for germline transmission of the deletion allele. The founding Acta1Δ3′ and Myh11Δi3 males were bred to wild-type mice for two generations, and the genotypes of Acta1Δ3′ and Myh11Δi3 founding males and all F2 offspring were confirmed by sequencing and PCR/restriction digests. F2 mice were intercrossed, and hearts derived from homozygous, heterozygous, and wild-type littermates were collected and assayed for Acta1 and Myh11 misexpression, respectively.
Motif discovery
De novo motif discovery on CHD4 chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) regions (GSE109012) (Wilczewski et al. 2018) from embryonic day (E)10.5 cardiac tissue was performed using hypergeometric optimization of motif enrichment (HOMER) (Heinz et al. 2010).
Immunoaffinity purification
E10.5 wild-type CD1 hearts (minimum 28 hearts per IP, three separate biological replicates) were harvested in cold PBS, snap-frozen, and cryolysed as previously described (Kaltenbrun et al. 2013). Frozen tissue powder was resuspended in optimized lysis buffer (5 mL/g powder; 20 mM K-HEPES at pH7.4, 0.11 M KOAC, 2 mM MgCl2, 0.1% Tween 20, 1 μM ZnCl2, 1 mM CaCl2, 0.5% Triton X-100, 150 mM NaCl, protease inhibitor [Sigma], phosphatase inhibitor [Sigma]). To reduce the possibility that interacting proteins are recruited by nucleic acids, universal nucleases (Thermo Fisher 88700) were used in the lysis process. CHD4 complexes were solubilized and immunoaffinity-purified as previously described (Cristea and Chait 2011; Kaltenbrun et al. 2013) using rabbit anti-CHD4 antibody (Abcam ab72418) or negative control custom rabbit anti-GFP antibody (Cristea et al. 2005) with elution for 10 min at 95°C. The immunoisolated proteins were resolved (∼1 cm) by SDS-PAGE and visualized by Coomassie blue. Each lane was subjected to in-gel digestion with trypsin as previously reported (Waldron et al. 2016).
For in vitro coimmunoprecipitation (co-IP) assay, Flag/Myc-CHD4, TurboGFP-NKX2-5, and TurboGFP-GATA4 constructs were cotransfected into HEK293 cells, and co-IP was performed as previously reported (Waldron et al. 2016) by using anti-Flag magnetic beads (Origene TA150042) with elution buffer for 10 min at 95°C. To reduce the possibility that interacting proteins are recruited by nucleic acids, universal nucleases (Thermo Fisher 88700) were used in the lysis process. The immunoisolated proteins were subjected to Western blot, rabbit anti-Myc-HRP antibody (1:2500; Abcam ab1326) was probed to detect CHD4 protein, and anti-TurboGFP (1:1000; Origene TA150041) was probed followed by probing anti-IgG-HRP secondary antibody (1:10,000; Jackson Immunoresearch) to detect NKX2-5 and GATA4 proteins. Antibody–antigen complexes were visualized using an ECL Western blotting analysis system (Amersham).
Parallel reaction monitoring (PRM) nLC-MS/MS
Immunoisolated proteins were resolved (∼1 cm) by SDS-PAGE and visualized by Coomassie blue. Each lane was subjected to in-gel digestion with trypsin as previously reported (Waldron et al. 2016). Desalted peptides (2 µL of each) were analyzed by nano-liquid chromatography (nLC)-MS/MS using a Dionex Ultimate 3000 nRSLC system directly coupled to a Q-exactive HF orbitrap mass spectrometer (Thermo Fisher Scientific) equipped with an Easy-Spray ion source (Thermo Fisher Scientific). Peptide mixtures were evaporated in vacuo and resuspended in 1% trifluoroacetic acid/4% acetonitrile/95% water, loaded onto a 50-cm-long column with a 75-µm internal diameter (PepMap), and separated over a 60-min gradient with a mobile phase containing aqueous acetonitrile and 0.1% formic acid programmed from 1%–3% acetonitrile over 12 min, then 3%–35% acetonitrile over 60 min, and then 35%–97% acetonitrile over 1 min, followed by 10 min at 97% acetonitrile and 97%–70% over 1 min, all at a flow rate of 250 nL/min.
The PRM analysis was performed as previously reported (Justice et al. 2021; Shi et al. 2021). Briefly, the method consisted of targeted MS2 scans at a resolution of 60,000 and with an AGC value of 1 × 106, a maximum injection time of 500 msec, a 0.8 m/z isolation window, a fixed first mass of 150 m/z, and normalized collision energy of 27, which were recorded as profile data. The targeted MS2 methods were controlled with a timed inclusion list containing the target precursor m/z value, charge, and a retention time window that were determined from shotgun analysis.
RNA sequencing and qRT-PCR
RNA sequencing and analysis were performed from mouse embryonic hearts as described previously (Wilczewski et al. 2018). Briefly, RNA from individual E10.5 hearts (N = 3 or 5 per genotype) was isolated using the RNAqueous-Micro total RNA isolation kit following the manufacturer's recommended protocols (Invitrogen). Purified poly-A RNA that had undergone two rounds of oligo-dT selection was converted into cDNA and used to generate RNA-seq libraries. Libraries were sequenced (75-bp paired-end reads; Illumina HiSeq 2500) to a target depth of >30 million reads. Reads were aligned to the mm10 reference genome using STAR via the bcbio-nextgen RNA sequencing pipeline. RNA-seq analysis was performed using DESeq2 (DESeq2_1.18.1) in R (3.4.3). Genes that had a >0.5 log2 (fold change) and an adjusted P-value < 0.05 were considered statistically significant.
For qRT-PCR, cDNA synthesis was performed using random hexamers and SuperScript IV reverse transcriptase (Invitrogen). Quantitative PCR was performed using PowerUP SYBR Green master mix (Thermo Fisher) at standard cycling conditions on a QuantStudio 6 Flex instrument (Thermo Fisher) with the following primers: Myh11 F1 (GCTAATCCACCCCCGGAGTA), Myh11 R1 (TCGCTGAGCTGCCCTTTCT), Acta1 F1 (GTGACCACAGCTGAACGTG), Acta1 R1 (CCAGGGAGGAGGAAGAGG), Snta1 F1 (AGATGATGGCACGAGTCTCC), Snta1 R1 (GGTGGAGGTGAGCAGTAGGA), Taf10 F1 (CCCACGCATAATTCGGCTCA), Taf10 R1 (GGGGACAGAGGGAAAGACAAAT), Gata6 F1 (TCAGGGGTAGGGGCATCAG), Gata6 R1 (TTGAGACCCCAGGAATGCAC), Ctf1 F1 (CAGAGGGAGGGAAGTCTGGA), Ctf1 R1 (AGCCCAAGAACACACAGGAC), Tnni3 F1 (GGCTGATGAGAGCAGCGAT), Tnni3 R1 (GACGTCCTTCAGAGCACAGT), Tnnt1 F1 (ATGGGAGCTCATTTTGGGGG), Tnnt1 R1 (CTCCACACAGCAGGTCATGT), Hand1 F1 (GCCTACTTGATGGACGTGCT), Hand1 R1 (ACCATGGCTTTTGGGGTTGA), Tbx5 F1 (AGGAATGTTCCAGCACGGAG), Tbx5 R1 (GAGGTTACAACGGGCGATCT), Pgk1 F1 (GTCGTGATGAGGGTGGACTT), and Pgk1 R1 (AAGGACAACGGACTTGGCTC).
Proximity ligation assay (PLA)
A standardized procedure for PLA was used (Jalili et al. 2018). Briefly, HEK293 cells were seeded on circular coverslips and transfected with specific plasmids for 72 h using a 3:1 ratio of PEI to pDNA. Cells were fixed with 4% paraformaldehyde for 10 min, followed by permeabilization and blocking (10% heat-inactivated goat serum, 1% Triton X-100) for 1 h. Cells were then probed with two specific primary antibodies raised in different species—mouse anti-TurboGFP (1:250; Origene TA150041)/rabbit anti-Flag (1:500; Sigma F7425)—overnight at 4°C. The cells were incubated with PLA probes for 1 h at 37°C, with ligase for 30 min at 37°C, and with polymerase for 100 min at 37°C, based on the manufacturer's protocols. Cells were stained with DAPI and then mounted with PermaFluor mounting medium (Thermo Scientific TA030FM). Images were captured on a Zeiss LSM 700 laser scanning confocal microscope.
DNA constructs
Full-length human CHD4 tagged at the C terminus with Flag/Myc construct was obtained from Origene (RC224232). Full-length human GATA4 cDNA was amplified with 5′ primer (ATTAGCGATCGCCATGTATCAG) and 3′ primer (CGTACGCGTCGCAGTGAT). Amplicons were digested with restriction enzymes AsiSI/MluI and inserted into pCMV6-AC-TurboGFP vector (Origene PS100010). Full-length human NKX2-5 cDNA was amplified with 5′ primer (ATTAGGATCCATGTTCCCCAGCCCTG) and 3′ primer (GTCGACTCACCAGGCTCGGATACCAT). Amplicons were digested with restriction enzymes AsiSI/MluI and inserted into pCMV6-AC-TurboGFP vector (Origene PS100010).
Histological sectioning and immunohistochemistry
Embryos were fixed in 4% PFA and paraffin-embedded. Paraffin sections (10 µm) were dewaxed, rehydrated, and stained with hematoxylin and eosin (H&E) using standard protocols (Dorr et al. 2015). Histology sections were imaged on an Olympus BX61 microscope. Digital images were used for measurement using ImageJ (NIH) software, and at least three independent sections were analyzed for each genotype at E10.5 and E12.5. Immunohistochemistry staining and imaging for MYH11 of E10.5 hearts were performed as previously reported (Wilczewski et al. 2018).
NGS data sets used
The following NGS data sets were used in this study: GATA4 genome occupancy at E12.5 in developing mouse hearts (GEO: GSE52123) (He et al. 2014), NKX2-5 genome occupancy at E12.5 in developing mouse hearts (GEO: GSE124008) (Akerberg et al. 2019), TBX5 genome occupancy at E12.5 in developing mouse hearts (GEO: GSE124008) (Akerberg et al. 2019), CHD4 genome occupancy at E10.5 in developing mouse hearts (GEO: GSE109012) (Wilczewski et al. 2018), CHD4 transcriptomics at E10.5 in developing mouse hearts (GEO: GSE109012) (Wilczewski et al. 2018), and H3K27me3 histone modification at E10.5 in developing mouse hearts (GEO: GSE86693) (The ENCODE Project Consortium 2012).
Genome annotation and co-occupancy analysis
BAM and BED files were obtained from the Gene Expression Omnibus aligned to mm10. Peaks were called using MACS2 (v2.2.7.1) (Zhang et al. 2008) using default settings with a Q-value of 0.01. High-confidence peaks appearing in both replicates were retained for downstream analysis. Peak annotation was conducted with HOMER (v4.10) (Heinz et al. 2010). Peaks were annotated using the ChIPseeker (v1.24.0) R package (Yu et al. 2015). Biological process GO terms for peak regions were generated using R package ClusterProfiler (Qian et al. 2012). Gene tracks were visualized using IGV (Robinson et al. 2011). Peak overlaps were determined using BedTools, and the upset plot was generated using UpSetR v1.4.0 (Conway et al. 2017) in R. Overlaps between CHD4, GATA4, NKX2-5, TBX5, and H3K4me3 were plotted in relation to their chromosomal location using the karyoploteR (v1.14.0) R package (Gel and Serra 2017).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health/National Heart, Lung, and Blood Institute grants R01HL156424 to F.L.C, R01HD089275 to F.L.C. and I.M.C., and 2UM1HL098166 to W.T.P. We thank Reviewer #3 for requesting the analyses that led to Supplemental Figures S2, A and B, and S6.
Author contributions: Z.L.R., W.S., and F.L.C. designed most of the experiments. Z.L.R., W.S., and C.M.W. performed RNA-seq, B.N.A. performed the ChIP-seq. Z.L.R., W.S., L.K.W., C.M.W., A.J.H., and B.N.A. performed and interpreted the bioinformatic analyses. Z.L.R., C.M.W., and X.S. performed the IP/PRM-MS experiments and analyses. Z.L.R. and W.S. generated the knockout mouse lines and performed the qRT-PCR. W.S. and A.P.S. performed the histological analyses. W.T.P., I.M.C., I.J.D., and F.L.C. supervised the project. Z.L.R., W.S., L.K.W., and F.L.C. wrote the manuscript with feedback from all authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.349154.121.
Competing interest statement
The authors declare no competing interests.
References
- Akerberg BN, Gu F, VanDusen NJ, Zhang X, Dong R, Li K, Zhang B, Zhou B, Sethi I, Ma Q, et al. 2019. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat Commun 10: 4907. 10.1038/s41467-019-12812-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arends T, Dege C, Bortnick A, Danhorn T, Knapp JR, Jia H, Harmacek L, Fleenor CJ, Straign D, Walton K, et al. 2019. CHD4 is essential for transcriptional repression and lineage progression in B lymphopoiesis. Proc Natl Acad Sci 116: 10927–10936. 10.1073/pnas.1821301116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baban A, Pitto L, Pulignani S, Cresci M, Mariani L, Gambacciani C, Digilio MC, Pongiglione G, Albanese S. 2014. Holt-Oram syndrome with intermediate atrioventricular canal defect, and aortic coarctation: functional characterization of a de novo TBX5 mutation. Am J Med Genet A 164A: 1419–1424. 10.1002/ajmg.a.36459 [DOI] [PubMed] [Google Scholar]
- Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, et al. 1997. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15: 30–35. 10.1038/ng0197-30 [DOI] [PubMed] [Google Scholar]
- Behiry EG, Al-Azzouny MA, Sabry D, Behairy OG, Salem NE. 2019. Association of NKX2-5, GATA4, and TBX5 polymorphisms with congenital heart disease in Egyptian children. Mol Genet Genomic Med 7: e612. 10.1002/mgg3.612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, et al. 2001. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106: 709–721. 10.1016/S0092-8674(01)00493-7 [DOI] [PubMed] [Google Scholar]
- Charpentier MS, Christine KS, Amin NM, Dorr KM, Kushner EJ, Bautch VL, Taylor JM, Conlon FL. 2013. CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev Cell 25: 132–143. 10.1016/j.devcel.2013.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudnovsky Y, Kim D, Zheng S, Whyte WA, Bansal M, Bray MA, Gopal S, Theisen MA, Bilodeau S, Thiru P, et al. 2014. ZFHX4 interacts with the NuRD core member CHD4 and regulates the glioblastoma tumor-initiating cell state. Cell Rep 6: 313–324. 10.1016/j.celrep.2013.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon FL, Miteva Y, Kaltenbrun E, Waldron L, Greco TM, Cristea IM. 2012. Immunoisolation of protein complexes from Xenopus. Methods Mol Biol 917: 369–390. 10.1007/978-1-61779-992-1_21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Lex A, Gehlenborg N. 2017. Upsetr: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33: 2938–2940. 10.1093/bioinformatics/btx364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristea IM, Chait BT. 2011. Affinity purification of protein complexes. Cold Spring Harb Protoc 2011: pdb.prot5611. 10.1101/pdb.prot5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristea IM, Williams R, Chait BT, Rout MP. 2005. Fluorescent proteins as proteomic probes. Mol Cell Proteomics 4: 1933–1941. 10.1074/mcp.M500227-MCP200 [DOI] [PubMed] [Google Scholar]
- Dorr KM, Amin NM, Kuchenbrod LM, Labiner H, Charpentier MS, Pevny LH, Wessels A, Conlon FL. 2015. Casz1 is required for cardiomyocyte G1-to-S phase progression during mammalian cardiac development. Development 142: 2037–2047. 10.1242/dev.119107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher D, Charron F, Warren R, Schwartz RJ, Nemer M. 1997. The cardiac transcription factors Nkx2-5 and GATA-4 are mutual cofactors. EMBO J 16: 5687–5696. 10.1093/emboj/16.18.5687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnung L, Ochmann M, Cramer P. 2020. Nucleosome-CHD4 chromatin remodeler structure maps human disease mutations. Elife 9: e56178. 10.7554/eLife.56178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federspiel JD, Tandon P, Wilczewski CM, Wasson L, Herring LE, Venkatesh SS, Cristea IM, Conlon FL. 2019. Conservation and divergence of protein pathways in the vertebrate heart. PLoS Biol 17: e3000437. 10.1371/journal.pbio.3000437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furtado MB, Wilmanns JC, Chandran A, Perera J, Hon O, Biben C, Willow TJ, Nim HT, Kaur G, Simonds S, et al. 2017. Point mutations in murine Nkx2-5 phenocopy human congenital heart disease and induce pathogenic Wnt signaling. JCI Insight 2: e88271. 10.1172/jci.insight.88271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gel B, Serra E. 2017. Karyoploter: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33: 3088–3090. 10.1093/bioinformatics/btx346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Del Arco P, Perdiguero E, Yunes-Leites PS, Acín-Pérez R, Zeini M, Garcia-Gomez A, Sreenivasan K, Jiménez-Alcázar M, Segalés J, López-Maderuelo D, et al. 2016. The chromatin remodeling complex Chd4/NuRD controls striated muscle identity and metabolic homeostasis. Cell Metab 23: 881–892. 10.1016/j.cmet.2016.04.008 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Teran B, Pittman M, Felix F, Thomas R, Richmond-Buccola D, Hüttenhain R, Choudhary K, Moroni E, Costa MW, Huang Y, et al. 2022. Transcription factor protein interactomes reveal genetic determinants in heart disease. Cell 185: 794–814.e30. 10.1016/j.cell.2022.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco TM, Miteva Y, Conlon FL, Cristea IM. 2012. Complementary proteomic analysis of protein complexes. Methods Mol Biol 917: 391–407. 10.1007/978-1-61779-992-1_22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Yamashita Y, Takemoto T. 2016. Electroporation of Cas9 protein/sgRNA into early pronuclear zygotes generates non-mosaic mutants in the mouse. Dev Biol 418: 1–9. 10.1016/j.ydbio.2016.07.017 [DOI] [PubMed] [Google Scholar]
- He A, Gu F, Hu Y, Ma Q, Ye LY, Akiyama JA, Visel A, Pennacchio LA, Pu WT. 2014. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat Commun 5: 4907. 10.1038/ncomms5907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeister H, Fuchs A, Erdel F, Pinz S, Gröbner-Ferreira R, Bruckmann A, Deutzmann R, Schwartz U, Maldonado R, Huber C, et al. 2017. CHD3 and CHD4 form distinct NuRD complexes with different yet overlapping functionality. Nucleic Acids Res 45: 10534–10554. 10.1093/nar/gkx711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. 2015. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350: 1262–1266. 10.1126/science.aac9396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa H, Tanaka T, Suzuki Y, Iwamura C, Ohkubo S, Endoh K, Kato M, Endo Y, Onodera A, Tumes DJ, et al. 2013. Functionally distinct Gata3/Chd4 complexes coordinately establish T helper 2 (Th2) cell identity. Proc Natl Acad Sci 110: 4691–4696. 10.1073/pnas.1220865110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hota SK, Rao KS, Blair AP, Khalilimeybodi A, Hu KM, Thomas R, So K, Kameswaran V, Xu J, Polacco BJ, et al. 2022. Brahma safeguards canalization of cardiac mesoderm differentiation. Nature 602: 129–134. 10.1038/s41586-021-04336-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou T, Cao Z, Zhang J, Tang M, Tian Y, Li Y, Lu X, Chen Y, Wang H, Wei FZ, et al. 2020. SIRT6 coordinates with CHD4 to promote chromatin relaxation and DNA repair. Nucleic Acids Res 48: 2982–3000. 10.1093/nar/gkaa006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili R, Horecka J, Swartz JR, Davis RW, Persson HHJ. 2018. Streamlined circular proximity ligation assay provides high stringency and compatibility with low-affinity antibodies. Proc Natl Acad Sci 115: E925–E933. 10.1073/pnas.1718283115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, Cristea IM. 2013. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol 9: 672. 10.1038/msb.2013.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumppanen M, Kinnunen SM, Välimäki MJ, Talman V, Auno S, Bruun T, Boije Af Gennäs G, Xhaard H, Aumüller IB, Ruskoaho H, et al. 2019. Synthesis, identification, and structure–activity relationship analysis of GATA4 and NKX2-5 protein–protein interaction modulators. J Med Chem 62: 8284–8310. 10.1021/acs.jmedchem.9b01086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice JL, Kennedy MA, Hutton JE, Liu D, Song B, Phelan B, Cristea IM. 2021. Systematic profiling of protein complex dynamics reveals DNA-PK phosphorylation of IFI16 en route to herpesvirus immunity. Sci Adv 7: eabg6680. 10.1126/sciadv.abg6680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbrun E, Greco TM, Slagle CE, Kennedy LM, Li T, Cristea IM, Conlon FL. 2013. A Gro/TLE–NuRD corepressor complex facilitates Tbx20-dependent transcriptional repression. J Proteome Res 12: 5395–5409. 10.1021/pr400818c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy L, Kaltenbrun E, Greco TM, Temple B, Herring LE, Cristea IM, Conlon FL. 2017. Formation of a TBX20-–CASZ1 protein complex is protective against dilated cardiomyopathy and critical for cardiac homeostasis. PLoS Genet 13: e1007011. 10.1371/journal.pgen.1007011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen DH, Poinsignon C, Gudjonsson T, Dinant C, Payne MR, Hari FJ, Rendtlew Danielsen JM, Menard P, Sand JC, Stucki M, et al. 2010. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol 190: 731–740. 10.1083/jcb.200912135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, et al. 1997. Holt-Oram syndrome is caused by mutations in TBX5, a member of the brachyury (T) gene family. Nat Genet 15: 21–29. 10.1038/ng0197-21 [DOI] [PubMed] [Google Scholar]
- Low JK, Webb SR, Silva AP, Saathoff H, Ryan DP, Torrado M, Brofelth M, Parker BL, Shepherd NE, Mackay JP. 2016. CHD4 is a peripheral component of the nucleosome remodeling and deacetylase complex. J Biol Chem 291: 15853–15866. 10.1074/jbc.M115.707018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MA, He D, Small EM, Mileikovsky M, et al. 2016. Complex interdependence regulates heterotypic transcription factor distribution and coordinates cardiogenesis. Cell 164: 999–1014. 10.1016/j.cell.2016.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, Garg V. 2009. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol 326: 368–377. 10.1016/j.ydbio.2008.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield RE, Musselman CA, Kwan AH, Oliver SS, Garske AL, Davrazou F, Denu JM, Kutateladze TG, Mackay JP. 2011. Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4 and methylated H3K9. J Biol Chem 286: 11779–11791. 10.1074/jbc.M110.208207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer Y, Czosnek H, Zeelon PE, Yaffe D, Nudel U. 1984. Expression of the genes coding for the skeletal muscle and cardiac actions in the heart. Nucleic Acids Res 12: 1087–1100. 10.1093/nar/12.2.1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulley DJ, Black BL. 2012. Transcription factor pathways and congenital heart disease. Curr Top Dev Biol 100: 253–277. 10.1016/B978-0-12-387786-4.00008-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie LD, LeClair JW, Miller KN, Strong AD, Chan HL, Oates EL, Ligon KL, Brennan CW, Chheda MG. 2019. CHD4 regulates the DNA damage response and RAD51 expression in glioblastoma. Sci Rep 9: 4444. 10.1038/s41598-019-40327-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori AD, Zhu Y, Vahora I, Nieman B, Koshiba-Takeuchi K, Davidson L, Pizard A, Seidman JG, Seidman CE, Chen XJ, et al. 2006. Tbx5-dependent rheostatic control of cardiac gene expression and morphogenesis. Dev Biol 297: 566–586. 10.1016/j.ydbio.2006.05.023 [DOI] [PubMed] [Google Scholar]
- Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. 2001. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 31: 176–180. 10.1002/gene.10022 [DOI] [PubMed] [Google Scholar]
- O'Shaughnessy A, Hendrich B. 2013. CHD4 in the DNA-damage response and cell cycle progression: not so NuRDy now. Biochem Soc Trans 41: 777–782. 10.1042/BST20130027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shaughnessy-Kirwan A, Signolet J, Costello I, Gharbi S, Hendrich B. 2015. Constraint of gene expression by the chromatin remodelling protein CHD4 facilitates lineage specification. Development 142: 2586–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostapcuk V, Mohn F, Carl SH, Basters A, Hess D, Iesmantavicius V, Lampersberger L, Flemr M, Pandey A, Thomä NH, et al. 2018. Activity-dependent neuroprotective protein recruits HP1 and CHD4 to control lineage-specifying genes. Nature 557: 739–743. 10.1038/s41586-018-0153-8 [DOI] [PubMed] [Google Scholar]
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. 2012. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics 11: 1475–1488. 10.1074/mcp.O112.020131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Clément-Ziza M, Lam H, Campbell DS, Schmidt A, Deutsch EW, Röst H, Sun Z, Rinner O, Reiter L, et al. 2013. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 494: 266–270. 10.1038/nature11835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. 2010. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J 29: 3130–3139. 10.1038/emboj.2010.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J-D, Srivastava D. 2012. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485: 593–598. 10.1038/nature11044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimone ML, Meisel J, Reddien PW. 2010. The Mi-2-like Smed-CHD4 gene is required for stem cell differentiation in the planarian Schmidtea mediterranea. Development 137: 1231–1241. 10.1242/dev.042051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Sheng X, Dorr KM, Hutton JE, Emerson JI, Davies HA, Andrade TD, Wasson LK, Greco TM, Hashimoto Y, et al. 2021. Cardiac proteomics reveals sex chromosome-dependent differences between males and females that arise prior to gonad formation. Dev Cell 56: 3019–3034.e7. 10.1016/j.devcel.2021.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, et al. 2016. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet 48: 1060–1065. 10.1038/ng.3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A, Xie Y, Verhoeven E, Vermeulen M, Lancini C, Gargiulo G, Hulsman D, Mann M, Knoblich JA, van Lohuizen M. 2013. The chromodomain helicase Chd4 is required for Polycomb-mediated inhibition of astroglial differentiation. EMBO J 32: 1598–1612. 10.1038/emboj.2013.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasan K, Rodríguez-delaRosa A, Kim J, Mesquita D, Segales J, Arco PG, Espejo I, Ianni A, Di Croce L, Relaix F, et al. 2021. CHD4 ensures stem cell lineage fidelity during skeletal muscle regeneration. Stem Cell Reports 16: 2089–2098. 10.1016/j.stemcr.2021.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steimle JD, Moskowitz IP. 2017. TBX5: a key regulator of heart development. Curr Top Dev Biol 122: 195–221. 10.1016/bs.ctdb.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennard FA, Costa MW, Elliott DA, Rankin S, Haast SJ, Lai D, McDonald LP, Niederreither K, Dolle P, Bruneau BG, et al. 2003. Cardiac T-box factor Tbx20 directly interacts with Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the developing heart. Dev Biol 262: 206–224. 10.1016/S0012-1606(03)00385-3 [DOI] [PubMed] [Google Scholar]
- Terada R, Warren S, Lu JT, Chien KR, Wessels A, Kasahara H. 2011. Ablation of Nkx2-5 at mid-embryonic stage results in premature lethality and cardiac malformation. Cardiovasc Res 91: 289–299. 10.1093/cvr/cvr037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade PA, Jones PL, Vermaak D, Wolffe AP. 1998. A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr Biol 8: 843–848. 10.1016/S0960-9822(98)70328-8 [DOI] [PubMed] [Google Scholar]
- Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP. 1999. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet 23: 62–66. 10.1038/12664 [DOI] [PubMed] [Google Scholar]
- Waldron L, Steimle JD, Greco TM, Gomez NC, Dorr KM, Kweon J, Temple B, Yang XH, Wilczewski CM, Davis IJ, et al. 2016. The cardiac TBX5 interactome reveals a chromatin remodeling network essential for cardiac septation. Dev Cell 36: 262–275. 10.1016/j.devcel.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson AA, Mahajan P, Mertens HD, Deery MJ, Zhang W, Pham P, Du X, Bartke T, Zhang W, Edlich C, et al. 2012. The PHD and chromo domains regulate the ATPase activity of the human chromatin remodeler CHD4. J Mol Biol 422: 3–17. 10.1016/j.jmb.2012.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Battle MA, Li J, Duncan SA. 2004. GATA4 is essential for formation of the proepicardium and regulates cardiogenesis. Proc Natl Acad Sci 101: 12573–12578. 10.1073/pnas.0400752101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss K, Terhal PA, Cohen L, Bruccoleri M, Irving M, Martinez AF, Rosenfeld JA, Machol K, Yang Y, Liu P, et al. 2016. De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am J Hum Genet 99: 934–941. 10.1016/j.ajhg.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczewski CM, Hepperla AJ, Shimbo T, Wasson L, Robbe ZL, Davis IJ, Wade PA, Conlon FL. 2018. CHD4 and the NuRD complex directly control cardiac sarcomere formation. Proc Natl Acad Sci 115: 6727–6732. 10.1073/pnas.1722219115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CJ, Naito T, Arco PG, Seavitt JR, Cashman SM, De Souza B, Qi X, Keables P, Von Andrian UH, Georgopoulos K. 2004. The chromatin remodeler Mi-2β is required for CD4 expression and T cell development. Immunity 20: 719–733. 10.1016/j.immuni.2004.05.005 [DOI] [PubMed] [Google Scholar]
- Xue Y, Wong J, Moreno GT, Young MK, Côté J, Wang W. 1998. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell 2: 851–861. 10.1016/S1097-2765(00)80299-3 [DOI] [PubMed] [Google Scholar]
- Yoshida T, Hu Y, Zhang Z, Emmanuel AO, Galani K, Muhire B, Snippert HJ, Williams CJ, Tolstorukov MY, Gounari F, et al. 2019. Chromatin restriction by the nucleosome remodeler Mi-2β and functional interplay with lineage-specific transcription regulators control B-cell differentiation. Genes Dev 33: 763–781. 10.1101/gad.321901.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, He QY. 2015. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31: 2382–2383. 10.1093/bioinformatics/btv145 [DOI] [PubMed] [Google Scholar]
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. 2013. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498: 220–223. 10.1038/nature12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, LeRoy G, Seelig H, Lane W, Reinberg D. 1998. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 95: 279–289. 10.1016/S0092-8674(00)81758-4 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Aubert A, Gomez de Segura JM, Karuppasamy M, Basu S, Murthy AS, Diamante A, Drury TA, Balmer J, Cramard J, et al. 2016. The nucleosome remodeling and deacetylase complex NuRD is built from preformed catalytically active sub-modules. J Mol Biol 428: 2931–2942. 10.1016/j.jmb.2016.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ai F, Zheng J, Peng B. 2017. Associations of GATA4 genetic mutations with the risk of congenital heart disease: a meta-analysis. Medicine 96: e6857. 10.1097/MD.0000000000006857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Han Z, Liu X, Gu J, Tang F, Wei G, Jin Y. 2017. The chromatin remodeler Chd4 maintains embryonic stem cell identity by controlling pluripotency- and differentiation-associated genes. J Biol Chem 292: 8507–8519. 10.1074/jbc.M116.770248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Liu J, Olson P, Zhang K, Wynne J, Xie L. 2015. Tbx5 and Osr1 interact to regulate posterior second heart field cell cycle progression for cardiac septation. J Mol Cell Cardiol 85: 1–12. 10.1016/j.yjmcc.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.