

ABSTRACT
ZO-2 is a peripheral tight junction (TJ) protein whose silencing in renal epithelia induces cell hypertrophy. Here, we found that in ZO-2 KD MDCK cells, in compensatory renal hypertrophy triggered in rats by a unilateral nephrectomy and in liver steatosis of obese Zucker (OZ) rats, ZO-2 silencing is accompanied by the diminished activity of LATS, a kinase of the Hippo pathway, and the nuclear concentration of YAP, the final effector of this signaling route. ZO-2 appears to function as a scaffold for the Hippo pathway as it associates to LATS1. ZO-2 silencing in hypertrophic tissue is due to a diminished abundance of ZO-2 mRNA, and the Sp1 transcription factor is critical for ZO-2 transcription in renal cells. Treatment of OZ rats with metformin, an activator of AMPK that blocks JNK activity, augments ZO-2 and claudin-1 expression in the liver, reduces the paracellular permeability of hepatocytes, and serum bile acid content. Our results suggest that ZO-2 silencing is a common feature of hypertrophy, and that ZO-2 is a positive regulator of the Hippo pathway that regulates cell size. Moreover, our observations highlight the importance of AMPK, JNK, and ZO-2 as therapeutic targets for blood-bile barrier dysfunction.
KEYWORDS: ZO-2, hypertrophy, liver steatosis, Hippo pathway, Sp1, metformin
1. Introduction
Tight junctions (TJs) are cell-cell adhesion structures present at the uppermost portion of epithelial cells. TJ is a gate that regulates the passage of ions and molecules through the paracellular pathway and a fence that blocks the free diffusion within the membrane of lipids and proteins from the apical to the basolateral membrane and vice versa.1 Zonula occludens-2 (ZO-2) is a cytoplasmic TJ protein, and together with ZO-1, it forms a platform for the polymerization of the integral TJ proteins claudins into TJ strands.2 Besides this critical function at TJs, ZO-2 participates in other cellular processes, including cell size regulation (for review, see [3]).
ZO-2 silencing in renal epithelial MDCK cells induces hypertrophy by two mechanisms: prolonging the time spent in the G1 phase of the cell cycle due to an increase in the level of cyclin D1 and through an augmented rate of protein synthesis in response to the nuclear accumulation of Yes-associated protein (YAP), the final target of the Hippo pathway, and the subsequent activation of mTOR signaling. In addition, we observed that in renal compensatory hypertrophy (RCH) generated in rats after a unilateral nephrectomy (UNX), the expression of ZO-2 diminished while YAP concentrated at the nucleus.4 Now, we explore in kidney cells and tissue the mechanisms and factors that regulate ZO-2 silencing and transcription and the interaction of ZO-2 with kinases in the Hippo pathway, which regulates cell size.
To test if ZO-2 silencing is a common feature observed in organ hypertrophy, we now explore ZO-2 expression in hepatocytes from obese rats that display hypertrophy due to an excessive accumulation of lipids in the cytoplasm, also known as liver steatosis or nonalcoholic fatty liver disease. For this purpose, we employed Zucker diabetic fatty (fa/fa) rats whose genetic alteration lies on the substitution of a codon in the extracellular domain of the receptor for leptin,5 the hormone predominantly made by adipose cells that inhibits hunger (for review see [6]). Obesity in Zucker rats is inherited as a Mendelian recessive trait, and affected animals are hyperlipidemic, hypercholesterolemic, and hyperinsulinemic.7 Obese Zucker (OZ) rats are hypertriglyceridemic and insulin-resistant at seven weeks of age and since then display liver steatosis. At week 14, rats are type 2 diabetic and become diabetic with insulin deficiency at week 21.8
Fatty liver disease can remain limited to steatosis or progress toward nonalcoholic steatohepatitis, fibrosis, cirrhosis, and liver cancer.9 However, even simple steatosis poses a severe health risk as these livers show a decreased regenerative capacity10 and are unable to tolerate a variety of challenges that healthy livers can cope with, like injury by toxins11 and post-ischemic reperfusion.12,13 In addition, patients and animals with liver steatosis have elevated plasma bile acid levels,14,15 which have been attributed to a disruption in bile acid metabolism16 and hepatocellular bile acid retention17 caused by reduced canalicular bile acid secretion due to a decreased expression of the multidrug-resistant associated protein 2 (MRP2)18 and to an increase in basolateral bile acid excretion transporters OSTB and MRP1/4/5.16 In addition to these factors, we hypothesize that bile acid leakage through the paracellular space into the liver parenchyma and plasma might also be triggered by blood-bile barrier (BBiB) dysfunction due to ZO-2 silencing in the hypertrophic cells of fatty livers. In this respect, it is noteworthy that missense mutations in ZO-2 gene TJP2 that occur in the first PDZ domain of the molecule, together with mutations in BAAT, the gene for a bile acid-conjugating enzyme, induce familial hypercholanemia characterized by elevated serum bile acid concentrations, itching and fat malabsorption.19 Likewise, homozygous protein-truncating mutations in TJP2 that lead to ZO-2 absence in canalicular liver membranes and cholangiocytes caused a failure of claudin-1 localization and provoked progressive familial intrahepatic cholestasis,20,21 a severe pediatric liver disease that can develop into hepatocellular carcinoma.22,23 Here, we explored if, in the hypertrophic liver of OZ rats, the expression of ZO-2 diminishes and changes the expression or localization of kinases and targets of the Hippo pathway and the paracellular permeability of the liver.
Our results show that in the three models of hypertrophy studied: MDCK ZO-2 KD cells, RCH in rats, and the steatotic liver of OZ rats, ZO-2 silencing is accompanied by a decrease expression of active LATS and the nuclear concentration of YAP. Moreover, ZO-2 is involved in the signaling of the Hippo pathway due to its interaction with LATS1. ZO-2 silencing in hypertrophic kidneys and liver is due to a decrease in ZO-2 mRNA, and the activity of transcription factor Sp1 is critical for ZO-2 transcription. In OZ rats, activation of AMP-activated protein kinase (AMPK) with metformin restores ZO-2 and claudin-1 expression in the liver, reduces paracellular permeability of canaliculi, and total bile acid concentration in serum. This process involves the inhibition of JNK signaling, which triggers junction disassembly.24,25
2. Material and methods
2.1. Cell culture
Parental (control) and ZO-2 KD MDCK II cells were kindly provided by Alan Fanning (University of North Carolina, Chapel Hill, NC) and cultured as previously described.26 These cells stably express a mixture of three different shRNAs against ZO-2 in the pSuper vector. Parental cells instead only express the empty vector. The stable clonal ZO-2 KD MDCK cell line here employed (IC5) was obtained based on zeocin resistance.
HEK-293 T epithelial cells derived from the human embryonic kidney (ATCC, Cat. CRL-3216, Manassas, VA) were grown in a high glucose DMEM medium (Cat. 11965–118, Thermo Fisher Scientific, Waltham, MA) supplemented with 5% FBS and penicillin-streptomycin 10,000 (U/μg/ml) (Cat. A-01 In Vitro, Mexico).
2.2. Immunofluorescence
Livers from Zucker rats were excised, washed thrice with cold PBS, and cut into small pieces immersed for 2 min in 2-methyl butane, previously cooled in liquid nitrogen, and transferred to liquid nitrogen for 10 min. The tissue was then sectioned in a Leica MC1510 cryostat (Wetzlar, Germany), and 7 μm sections were placed on top of 0.5% gelatin-coated slides and left to dry at room temperature for 30 min. Sections were then fixed with 4% paraformaldehyde for 10 min and washed thrice with cold PBS. Then, liver samples were subjected to different treatments according to the antibody used. For the samples where we employed a rabbit polyclonal anti-claudin-1 (Cat. 51–9000, dilution 1:100; Invitrogen, Carlsbad, CA) or a mouse monoclonal anti-claudin-2 (Cat. Sc 293233, dilution 1:100; Santa Cruz Biotechnology, Santa Cruz, CA), the sections were fixed with 100% ethanol at −20°C for 10 min, washed thrice with cold PBS, treated with acetone at −20°C for 3 min, washed with cold PBS for 30 min and permeabilized with 0.25% Triton X-100 in PBS during 10 min. Instead, the liver sections where the rabbit antibodies against ZO-1 (Cat. 61–7300, dilution 1:300; Invitrogen, Carlsbad, CA), ZO-2 (Cat. 71–1400, dilution 1:100; Invitrogen, Carlsbad, CA), Sp1 (Cat. GTX110593, dilution 1:100; Genetex, Irvine, CA) or YAP (dilution 1:500, generously provided by Marius Sudol, Mechanobiology Institute, National University of Singapore, Singapore) were used, were first immersed in acetone at −20°C for 3 min, then washed thrice with cold PBS and permeabilized with 0.5% Triton X-100 in PBS during 15 min.
For the immunofluorescence of MDCK cells, we employed a standard procedure previously describe27 and used polyclonal rabbit antibodies against pLATS S909 (Cat. 9157, dilution 1:100; Cell Signaling, Danvers, MA) and Sp1 (Cat. GTX110593, dilution 1:200; Genetex, Irvine, CA). As secondary antibodies, we employed a donkey against rabbit IgG coupled to Alexa-Fluor 594 (Cat. A21207, dilution 1:750; Invitrogen, Carlsbad, CA). Nuclei were stained with DAPI. Images were examined in a confocal microscope (Leica TC5 SP8, Wetzlar, Germany). Quantification of Sp1 immunofluorescence intensity at the nucleus was done using ImageJ. Nuclei images stained with DAPI were used to define the region to be subsequently quantitated for Sp1 immunofluorescence.
2.3. Immunoprecipitation
Immunoprecipitation of ZO-2 was done using 1 μl of antibody against ZO-2 (Cat. 71–1400, Invitrogen, Carlsbad, CA) or LATS1 (Cat. 3477, Cell Signaling, Danvers, MA) /300 μg of protein in the lysate of parental MDCK cells, and following a protocol previously described.28 The radioimmunoprecipitation assay buffer employed contained: 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40 vol/vol, and the protease inhibitor cocktail Complete™ (Cat. 11697498001, Roche Diagnostics, Mannheim, Germany).
2.4. Pull-down assays
HEK293T cells were transfected with amino (398–962 nt), 3PSG (1595–3019 nt), or AP (3029–3923 nt) segments of cZO-2, inserted in the pcDNA4/HisMax vector as was previously reported.29 After 24 h, cells were lysed, and the extracts were subjected to affinity chromatography with Complete His-Tag Purification Columns (Cat. COHISC-RO, Sigma Aldrich, St. Louis, MO), following the manufacturer’s instructions. The purified fractions were run in an SDS-PAGE and blotted with rabbit polyclonal antibodies against 6x His tag (Cat. GTX115045, dilution 1:5000; Genetex, Irvine, CA), anti the amino segment of ZO-2 (Cat. 71–1400, dilution 1:1000; Invitrogen, Carlsbad, CA), anti the c-terminal region of ZO-2 (Cat. 00238, dilution 1:1000; BiCell Scientific, Maryland Heights, MO) and against LATS1 (Cat. 3477, dilution 1:1000; Cell Signaling, Danvers, MA).
2.5. RNA extraction and quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
An equivalent of 50 mg of kidney tissue from sham-operated and UNX rats, and of liver tissue of LZ and OZ rats was collected in TRI – Reagent (Cat. T9424; Sigma-Aldrich, St. Louis, MO). According to manufacturer instructions, total RNA was isolated using the Direct-zol MiniPrep kit (Cat. R2050; Zymo Research, Irvine, CA). Quantitative real-time reverse transcription-PCR (qRT-PCR) was performed using a one-step method with 20 ng of total RNA using the KAPA SYBR FAST One-Step qRT-PCR system (Kapa Biosystems) in a reaction volume of 10 μL. Triplicate samples were subjected to qPCR using the Step One Plus Real-Time PCR system (Cat. 4376600; Applied Biosystems, Carlsbad, CA). The PCR conditions were: after an initial cycle of 5 min at 42°C, 1 cycle of 5 min at 92°C and 40 cycles of amplification (30 s at 92°C and 30 s at 60°C) and a melt curve (15 s at 95°C; 1 min at 60°C; 15 s at 95°C). Primers used for amplification are shown in Table 1.
Table 1.
Primers used for amplification
| Sample | Gene | Primer Forward | Primer Reverse | Ta (°C) |
|---|---|---|---|---|
| Kidney tissue | TJP2 | 5ʹ – CTTTCAACTACTCCAAGTCAAACC – 3’ | 5ʹ – GCTATTTCAATCCTCGCATTCTG – 3’ | 62 |
| Kidney tissue | GAPDH | 5ʹ – GACCCCTTCATTGACCTCAAC – 3’ | 5ʹ – GTGGCAGTGATGGCATGGAC – 3’ | 66 |
| MDCK cells | TJP2 | 5ʹ – CTTTCAACTACTCCAAGTCAAACC – 3’ | 5ʹ – GCTATTTCAATCCTCGCATTCTG – 3’ | 62 |
| MDCK cells | PRP0 | 5ʹ – TACAACCCTGAAGTGCTTGAC – 3’ | 5ʹ – GCAGATGGATCAGCCAAGAAG- 3’ | 64 |
| Liver tissue | GAPDH | 5ʹ – GACCCCTTCATTGACCTCAAC – 3’ | 5ʹ – GTGGCAGTGATGGCATGGAC – 3’ | 66 |
| Liver tissue | ACTIN | 5ʹ – GCTCGTCGTCGACAACGGCT – 3’ | 5ʹ – CAAACATGATCTGGGTCATCTTCTC – 3’ | 66 |
| Liver tissue | TJP2 | 5ʹ – CTCTATACACGTCTGCTCGG- 3’ | 5ʹ – GCTGGTGGAAATGATGTTGG – 3’ | 64 |
The relative abundance of ZO-2 mRNA was expressed as UNX vs. sham compared to gapdh or prp0 (large ribosomal proteinP0) mRNA from MDCK cells and was calculated using the 2− ΔΔct method.
2.6. Western blot
Western blots were done as previously reported,30 with the loading buffer as the sole variation. For the Western blots of lysates from MDCK cells, we employed a commercial sample buffer (Cat. NP0008; Invitrogen, Carlsbad, CA); for Western blots of kidney nuclear fractions from rats that were sham-operated or had a UNX, and for the Western blots of livers from Zucker lean and obese rats, we employed Laemmli buffer (150 mM Tris-HCl, pH 6.8; 300 mM DTT, 6% SDS, 0.3% bromophenol blue, 30% glycerol and 20% 2-mercaptoethanol). The following primary rabbit polyclonal antibodies were used: anti-ZO-2 (Cat. 71–1400, dilution 1:1000; Invitrogen, Carlsbad, CA), anti claudin-1 (Cat. 51–9000, dilution 1:1000; Invitrogen, Carlsbad, CA) anti Sp1 (Cat. GTX110593, dilution 1:1000; Genetex, Irvine, CA), anti-LC3-I/LC3-II (Cat. 2775, dilution 1:1000; Cell Signaling, Danvers, MA), anti pLATS S909 (Cat. 9157, dilution 1:1000; Cell Signaling, Danvers, MA), anti LATS1 (Cat. 3477, dilution 1:1000; Cell Signaling, Danvers, MA), anti JNK (Cat. 9252, dilution 1:500; Cell Signaling, Danvers, MA), and anti pJNK (Cat. ab131499, dilution 1:500; Abcam, Cambridge, MA). We also employed a rabbit monoclonal antibody against LATS2 (Cat. 5888, dilution 1:500; Cell Signaling, Danvers, MA) and mouse monoclonals anti claudin-2 (Cat. Sc 293233, dilution 1:2000; Santa Cruz Biotechnology, Santa Cruz, CA), lamin B1 (Cat. 33–2,000, dilution 1:2,000; Invitrogen, Carlsbad, CA) and tubulin (Cat. T7816, dilution 1:20,000; Sigma Aldrich, San Louis, MO). As secondary antibodies, we employed a goat IgG against rabbit (Cat. 9162; Sigma Aldrich, San Louis, MO) or mouse (Cat. 62–6520; Invitrogen, Carlsbad, CA) coupled to horseradish peroxidase that was followed by Immobilon chemiluminescence detection (Cat. WVKLS 0500; Darmstadt, Germany).
In Figure 1Ab for the Western blot detection of SAV in a ZO-2 immunoprecipitate, we employed the TidyBlot Western blot Detection Reagent:HRP (Cat. STAR209, BioRad, Hercules, CA), which specifically binds to native (non-reduced) antibodies, in contrast to conventional secondary antibodies. This reagent allowed us to detect SAV that weights 45 kDa without interference from denatured IgG heavy chains (50 kDa) from primary antibodies released from beads during immunoprecipitation experiments.
Figure 1.
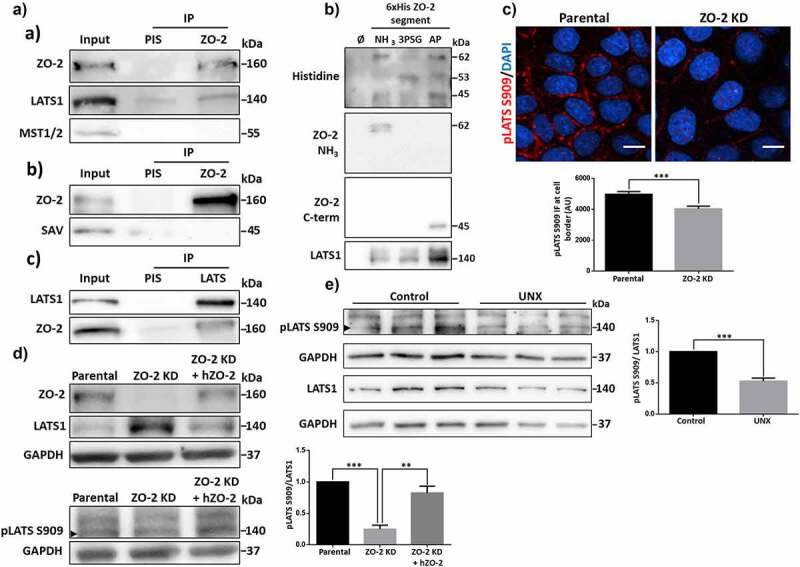
ZO-2 interacts with the kinase LATS1, and its presence favors LATS activation and concentration at the cell border. A) In parental MDCK cells, ZO-2 was immunoprecipitated, and a subsequent immunoblot was done with antibodies against ZO-2, LATS1, MST1/2, or SAV (a and b). A LATS1 immunoprecipitate was also blotted with antibodies against LATS1 and ZO-2 (c). As a secondary antibody for detecting SAV in the ZO-2 immunoprecipitate, we employed the TidyBlot Western blot Detection Reagent:HRP, which binds to non-reduced antibodies, in order to avoid interference from denatured IgG heavy chains (50 kDa). B) Western blot analysis of pull-downs of amino, 3PSG, and AP segments of cZO-2. First panel, anti histidine antibody recognizes bands of 62, 53, and 45 kDa that respectively correspond to the expected molecular weight of the amino, 3PSG, and AP segments of cZO-2. Second panel, the antibody against the amino segment of ZO-2 recognizes the 62 kDa band present in the pull-down of the ZO-2 amino segment. Third panel, the antibody against the c-terminal segment of ZO-2 recognizes the 45 kDa band present in the pull-down of the AP segment of ZO-2. Fourth panel, anti LATS1 antibody gives a strong signal in the pull-down of the AP segment of ZO-2. C) Immunofluorescence detection of active LATS1 (pLATS S909) in parental and ZO-2 KD MDCK cells. Nuclei were stained with DAPI. Bar, 10 μm. Upper panel, representative images; lower panel, quantitation of immunofluorescence for pLATS S909 at the cell borders. Results are mean ± SEM from at least three independent experiments. Statistical analysis was done with Mann Whitney test. ***p < .001. D) Western blot of LATS1 (upper panel) and pLATS S909 (lower panel, arrowhead), in parental and ZO-2 KD MDCK transfected or not with an hZO-2 construct not susceptible to the shRNAs against ZO-2. GAPDH was employed as a loading control. Left, representative images; right quantitative analysis. Results are mean ± SEM from at least three independent experiments. Parental cells data normalized to 1. Statistical analysis was done with One-way ANOVA followed by Tukey’s multiple comparisons post-hoc test, **p < .01, ***p < .001. E) Western blot of LATS1 and pLATS S909 (arrowhead) in lysates derived from kidneys from 11-week-old rats that at 8 weeks of age were subjected to UNX or sham operation (control) and tested 3 weeks later. GAPDH was employed as a loading control. Left, representative images; right quantitative analysis. Results are mean ± SEM from at least three independent experiments. Control kidney data normalized to 1. Statistical analysis was done with Student’s t-test, ***p < .001.
2.7. Uninephrectomy (UNX)
8 week–old Wistar rats with bodyweight between 200–300 g were anesthetized with pentobarbital (30 mg/kg). Then, an incision was done in the abdominal wall under sterile conditions to expose the right kidney. The renal pedicle was isolated, and the right kidney was removed. After surgery, the rats were returned to their cage and given food and water ad libitum. Three weeks after surgery, the remaining kidney undergoing renal compensatory hypertrophy was removed, and the animal was killed with intraperitoneal pentobarbital sodium (100 mg/kg). The kidneys were frozen in liquid nitrogen. Half of each kidney was later processed for Western blot experiments, and the rest was used for immunofluorescence assays.
2.8. Isolation of nuclear fractions from kidney tissue
A previously described protocol was employed with minor modifications.31 Briefly, fresh kidneys were cut into small rectangles with a sharp knife. Slices were incubated for 20 min in buffer A [3 mM MgCl2, 0.32 M sucrose, and 10 mM PBS containing Complete inhibitor cocktail (Cat. 04693132001; Roche Applied Science, Madison, WI)] at 4°C. Slices were then homogenized with 25 strokes in a Dounce immersed in ice. Then, homogenates were filtered through a Falcon® 100 µm Cell Strainer (Cat. 352360; Falcon, Corning, NY). The obtained cell suspension was centrifuged at 1100 x g for 20 min at 4°C, and the cellular pellet was resuspended in 10 mL of buffer B (20 mM Tris-HCl pH 7.6, 4 mM KH2PO4, 3 mM MgCl2, and 1.16 M sucrose) at 4°C. The cell suspension was transferred to a new tube over 1.5 mL of a 2 M sucrose cushion (20 mM Tris-HCl pH 7.6, 4 mM KH2PO4, 3 mM MgCl2, and 2 M sucrose) and centrifuged at 159,784 x g for 100 min at 4°C. The supernatant and sucrose cushion was carefully removed, and the pellet containing the purified nuclei was resuspended in RIPA buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) and sonicated for 30 s. The nuclear extracts were kept frozen at −70°C until used.
2.9. miRNA RT-qPCR analysis
Small RNA from frozen kidneys was extracted using the miRVana™ miRNA isolation kit (Cat. AM1560; Life Technologies Corporation, Carlsbad, CA) following the manufacturer’s instructions. Small RNA integrity was confirmed by denaturing polyacrylamide gel electrophoresis as previously described.32 Steady-state miRNA levels were quantified by the stem-loop RT-qPCR method33 using Maxima SYBR™ Green/ROX qPCR Master Mix (Cat. K0223; Thermo Fisher Scientific Inc. Waltham, MA) in a Rotor Gene™ 3000 thermocycler (Corbett Research Pty Ltd., Australia). Specific miR-23a and miR-23b primers were designed according to34 based on the respective template sequences obtained from the miRBASE database.35 miR-124 primers were used as previously described,36 and U6 rRNA was used as an endogenous reference.37 Primer sequences are listed in Supplementary Table 1. PCR amplicons were sequenced using the BigDye Terminator v3.1 Sequencing Kit (Cat. 4337455; Thermo Fisher Scientific Inc., Waltham, MA) in an Applied Biosystems Genetic Analyzer 310 automatic sequencer.
2.10. LC3-II flux assay
OZ and LZ rats subjected to a 24 h starvation period were euthanized by cervical dislocation. Then the livers were removed and placed at 4°C in PBS pH 7.4. Slices of 200 μm from the whole liver were obtained using a vibratome series 1000 sectioning System Microtome (Cat. 054026; Technical Products International, Saint Louis, MO) and collected in PBS pH 7.4 Slices were then incubated in 12 well plates with 0.5 ml of DMEM and 10% newborn calf serum. In half of the wells, NH4Cl was added at a final concentration of 10 mM from a 2 M stock solution in water. The plates containing the liver slices were then incubated for 2 h at 37°C in a CO2 incubator with occasional swirling. The tissue and medium suspension from each well were placed in 1.5 ml tubes and centrifuged at 1,000 x g for 5 min at 4°C. The supernatant was discarded, and the tissue was washed with ice-cold PBS. After another centrifugation at 1,000 x g for 5 min at 4°C, the supernatant was discarded and the tissue bathed in RIPA buffer (25 mM Tris- HCl, pH 7.6, 150 mM NaCl, 5 mM EDTA, 0.1% Triton X-100, 1% sodium deoxycholate and 0.1% SDS) containing the protease inhibitor cocktail Complete (Cat. 11697498001; Roche Diagnostics, Manheim, Germany) was homogenized with 30 strokes in a dounce immersed in ice. After centrifugation at 15,142 x g for 15 min, the samples supernatants were recovered, the protein quantitated, run in a 15% acrylamide SDS-PAGE, and processed by immunoblot with antibodies against LC3-I and LC3-II. Actin was used as a loading control.
2.11. Zucker diabetic fatty rats
12 week-old male, obese or lean Zucker Diabetic Fatty (ZDF) rats were obtained from the Claude Bernard animal facility of the University of Puebla. All protocols involving animals were approved by the Institutional Animal Care and Use Committee and followed the guidelines published for animal care and use of laboratory animals by the National Institutes of Health. Rats were maintained in a 12:12 dark-light cycle at 22°C and had free access to LabDiet 5008 and water.
Three days before the animals were sacrificed, rats were challenged with an intraperitoneal bolus of glucose. For this purpose, early in the morning, blood glucose was tested by tail puncture using the reactive strip Contour TS system (Bayer). This measure was considered a spontaneous glucose test. Then, animals were deprived of lab chow but had free access to tap water. Fasting blood glucose was measured six hours later before the intraperitoneal glucose administration (2 mg/Kg). Afterward, blood glucose levels were evaluated every 30 minutes for two hours. Obese ZDF rats with blood glucose levels over 200 mg/dL were excluded because they were considered diabetic. Therefore, only obese non-diabetic and lean ZDF rats (OZ and LZ) were used in these experiments.
Rats were sacrificed by cervical dislocation after induction of anesthesia with sevoflurane (3–5%). An anterior thoracotomy granted access to organs. The heart was excised and perfused in a Langendorft system with a 37°C tyrode solution to remove blood. Immediately after, the liver was removed and perfused through the portal vein until complete exsanguination. Then the liver was processed for Western blot, electron microscopy, and immunofluorescence. Blood samples were obtained directly from the heart just before organ removal. 400 µl of blood were placed in an Eppendorf tube and centrifuged to 3500 RPM. The supernatant was recovered and froze to −20°C.
2.12. Metformin treatment of obese ZDF rats
Twelve male obese ZDF rats aging 6 weeks were caged individually and organized into two groups. All animals had free access to LabDiet 5008 and water. Consumption of water was measured and averaged during the first seven days of acclimation. Once rats aged 7 weeks, in one group of 6 rats, the drinking water was daily supplemented with 300 mg/kg/day of metformin (Cat. D150959; Sigma-Aldrich, St. Louis, MO). This metformin dose was chosen because it had been previously employed to treat nonalcoholic hepatic steatosis in ZDF rats.8 The control group had no changes in the original housing conditions. Bodyweight was controlled two times per week through all the protocols (Supplementary Fig. 3A). When rats reached 12 weeks of age, a glucose tolerance test was performed, and three days after, the animals were sacrificed, and the livers were weighed (Supplementary Fig. 3B) and processed for Western blot, electron microscopy, or immunofluorescence.
2.13. Total bile acids assay
The quantitative determination of total bile acids in serum from LZ and OZ rats treated or not with metformin was done in vitro with the Diazyme Total Bile Acid Assay Kit (Cat. DZ042A; Diazyme, Poway, CA), following the manufacturer’s instructions. Blood samples were obtained directly from the heart of the rats just before organ removal.
2.14. Liver lanthanum permeability determined by electron microscopy
To determine the paracellular permeability of the liver to lanthanum, we followed a protocol previously described.38 Briefly, livers were first flushed with a physiological solution to clear the tissue from the blood. Then we employed a perfusion/fixation solution of 2.5% glutaraldehyde/4% lanthanum nitrate in 0.1 mol/L cacodylate buffer, pH 7.8. The lanthanum nitrate solution was prepared by dissolving lanthanum nitrate in water (8%, wt/vol) at 65°C and diluting 1:1 with 0.2 mol/L cacodylate buffer pH 7.8. After perfusion, livers were excised, cut into fragments, and incubated in the perfusion/fixation solution. Then, tissue samples were rinsed for 30 min in 0.1 mol/L cacodylates buffer/4% nitrate lanthanum and postfixed in 1% osmium tetroxide, containing 4% nitrate lanthanum, for 1 h at 4°C. Samples were then dehydrated in graded ethanol and propylene oxide, embedded in Polybed epoxy resins, and polymerized at 60°C for 24 h. Thin sections (60 nm) were stained at room temperature for 20 min with uranyl acetate and subsequently for 2 min with lead citrate before examination using a Jeol JEM-1011 transmission electron microscope.
The permeability of the TJ present in the hepatocytes of ZDF rats was evaluated by the penetration of lanthanum, an electron-dense heavy metal tracer, across the junction, as previously reported.38 Normal TJs prevent lanthanum diffusion into the canaliculi; hence if bile canaliculi contained lanthanum particles, TJs were considered leaky.
3. Results
3.1. ZO-2 interacts with LATS1, and the absence of ZO-2 reduces LATS phosphorylation and concentration at the cell borders
Previously, we demonstrated that ZO-2 silencing in MDCK cells reduced the phosphorylation of YAP, the downstream target of the Hippo pathway.4 This observation prompted us to explore if ZO-2 is associated with proteins of the Hippo pathway. Upon immunoprecipitation of ZO-2, we detected that neither the kinase MST1/2 (mammalian Ste2-like) nor its associated scaffold protein Salvador (SAV) coimmunoprecipitate with ZO-2 (Figure 1Aa,b). Instead, the kinase LATS1 (large tumor suppressor 1) is present in a ZO-2 immunoprecipitate (Figure 1Aa), and correspondingly ZO-2 is detected in a LATS1 immunoprecipitate (Figure 1Ac).
To further confirm ZO-2 interaction with LATS1, we made a pull-down assay in the human kidney cell line HEK293T. We choose these cells because they display a better transfection efficiency than MDCK cells. We transfected HEK293T cells with the amino (coding PDZ domains 1, 2, and 3), 3PSG (coding the PDZ3, SH3, and GuK domains), or AP (coding the acidic and proline-rich regions) constructs of cZO-2, introduced in the pcDNA4/HisMax vector. The corresponding proteins were purified from extracts of HEK293T cell using Ni affinity columns, run in SDS-PAGE, and blotted with antibodies against histidine and the amino or carboxyl segments of ZO-2 (Figure 1B). The antibody against histidine reveals bands of 62, 53, and 45 kDa that respectively correspond to the amino, 3PSG, and AP segments of cZO-2. The identity of the amino and carboxyl ZO-2 pulled-down segments was further confirmed with the respective anti-ZO-2 antibodies that recognize these sections of the protein, as indicated by the manufacturers, while there is no commercial ZO-2 antibody that can recognize the 3PSG segment. Blotting against LATS1 gives a robust signal in the pull-down of the AP segment of ZO-2, thus indicating that LATS1 strongly associates with this ZO-2 segment (Figure 1B). Taken together, the immunoprecipitation and pull-down assays indicate that ZO-2 interacts with LATS1, a kinase of the Hippo pathway involved in cell size regulation.
Next, we explored if the lack of ZO-2 affected LATS1 localization in MDCK cells. By immunofluorescence, we observed that in parental cells, phosphorylated (S909) and hence active LATS1/2 is present at the cell borders, while in ZO-2 KD cells, its expression is reduced (Figure 1C). Unfortunately, we could not analyze the total LATS1 distribution since the antibodies against it do not work for immunofluorescence (data not shown).
By Western blot, we observe that in the absence of ZO-2, the amount of phosphorylated LATS1/2/total LATS1 diminishes, and this effect is reversed by the transfection of MDCK KD cells with an hZO-2 construct not susceptible to the shRNAs against ZO-2, thus indicating that ZO-2 promotes the localization of active LATS1/2 at the cell border (Figure 1D). We did not analyze LATS2 because available commercial antibodies do not detect LATS2 in MDCK cells (Suppl. Fig. 1).
Previously, we demonstrated in rats that after a UNX, ZO-2 expression diminished, while the amount of nuclear YAP increased in the remaining kidney.4 Therefore, we now analyzed if the content of phosphorylated LATS1/2 changed after a UNX, finding that 3 weeks later, the remaining rat kidney displayed a lower amount of phosphorylated LATS (Figure 1E).
These results suggest that ZO-2 interaction with LATS1 favors its phosphorylation in kidney epithelial cells both in vivo and in culture.
3.2. ZO-2 expression is regulated at the transcriptional level by Sp1
Since ZO-2 content diminishes in the remaining kidney after a UNX,4 we now explored by quantitative RT-PCR if this effect was due to a decrease in ZO-2 mRNA. Figure 2A shows that three weeks after the UNX, the amount of ZO-2 mRNA in the remaining kidneys decreases by 36% compared to the kidneys from sham-operated animals of the same age (control).
Figure 2.
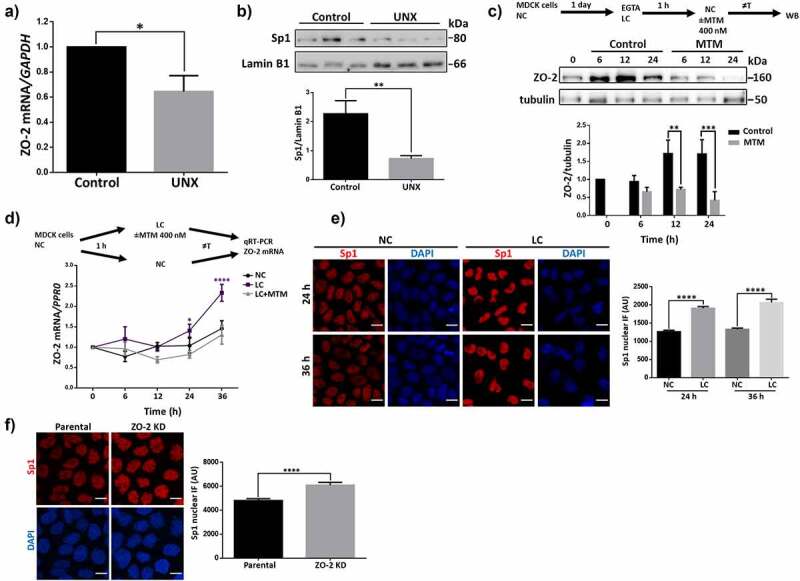
Sp1 transcription factor regulates the expression of ZO-2 protein and mRNA. A) qRT-PCR amplification of ZO-2 from the kidney of sham-operated (control) rats or that had undergone a UNX 3 weeks ago. Control data normalized to 1. Statistical analysis was done with Student’s t-test, *p < .05. Results are mean ± SEM from at least three independent experiments. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified as a control. B) Western blot of Sp1 present in nuclear fractions derived from the kidney of sham-operated (control) rats or with a UNX done three weeks before. Lamin B1 was employed as a loading control. Upper panel, representative images of three independent experiments; lower panel, densitometric analysis. Results are mean ± SEM from at least three independent experiments. Statistical analysis done with Student’s t-test, **p = .0074. C) The increase in ZO-2 protein observed after MDCK monolayers are switched from LC media containing 2 mM EGTA to NC media, to induce TJ proteins synthesis and assembly, was not present in monolayers incubated with 400 nM MTM. Upper panel, a scheme of the experimental procedure and representative Western blot of three independent experiments; lower panel, densitometric analysis. Results are mean ± SEM. Control value at time 0 normalized to 1. Statistical analysis was done with Two-way ANOVA followed by Fisher’s LSD test, **p < .01, ***p < .001. D) In monolayers transferred to LC, the amount of ZO-2 mRNA increases through a process mediated by MTM. Upper panel, the scheme of the experimental procedure. Lower panel, amount of ZO-2 mRNA was determined by RT-qPCR. PRP0 mRNA was used as control. Data were normalized against NC values at time 0. Results are presented as mean ± SEM from at least three independent experiments. Values at time 0 normalized to 1. Statistical analysis was done with Two-way ANOVA followed by Tukey’s multiple comparisons post-hoc test, *p < .05, ****p < .0001. E) Immunofluorescence detection of Sp1 in the nuclei of parental MDCK cells after 24 h of incubation in NC or LC condition. Nuclei were stained with DAPI. Bar, 10 μm. Left panel, representative images; right panel, quantitation of immunofluorescence for Sp1 at the nuclei. The nuclei of 190 cells per condition derived from three independent experiments were quantitated. Statistical analysis was done with Kruskal-Wallis test followed by Dunn’s multiple comparisons test, ****p < .0001. F) Immunofluorescence detection of Sp1 in the nuclei of parental and ZO-2 KD MDCK cells. Nuclei were stained with DAPI. Bar, 10 μm. Left panel, representative images; right panel, quantitation of immunofluorescence for Sp1 at the nuclei. The nuclei of 100 cells per condition derived from three independent experiments were quantitated. Statistical analysis was done with Mann Whitney test, ****p < .0001.
Transcription of ZO-2 mRNA is driven in humans by alternative promoters PA and PC that give rise to two ZO-2 isoforms: ZO-2A and ZO-2 C.39 PA in the hTJP2 gene has 27 Sp1 and 6 Zeste sites.39 Hence, we next analyzed the expression of the Sp1 transcription factor in renal tubular cells from sham-operated and UNX rats. We performed a cell fractionation assay of kidney tissue and analyzed the amount of Sp1 present in nuclear fractions, finding a decreased Sp1 content in the nucleus of kidney tissue after a UNX compared to sham-operated rats (control) (Figure 2B).
To test the importance of Sp1 for ZO-2 transcription, we next induced TJ formation in MDCK cells in the presence of the gene-selective Sp1 inhibitor, mithramycin (MTM).40 First, we transferred monolayers cultured for 24 h in normal calcium (NC) media (1.8 mM Ca2+), to low calcium media (LC, 1–5 μM Ca2+) containing 2 mM EGTA for 1 h, to induce TJ disassembly and endocytosis, as previously reported.41 Then, the monolayers were transferred back to NC media to induce TJ formation in the presence or absence of 400 nM MTM. Figure 2C shows that the increase in ZO-2 observed after the monolayers were transferred from LC to NC was not present in monolayers incubated with MTM. Next, we analyzed by qRT-PCR the amount of TJP2 mRNA, observing that in cells cultured in the LC condition, the amount of mRNA reaches significantly higher values than in cells maintained in the NC condition, resembling what we had previously observed for TJP1 mRNA42 (Figure 2D). Moreover, we observed that MTM treatment inhibited the increase in ZO-2 mRNA observed in the LC condition.
To further confirm these results, we analyzed by immunofluorescence the expression of Sp1 in MDCK monolayers after 24 or 36 h of incubation in NC or LC conditions. Figure 2E shows that the expression of Sp1 is higher in the LC than in the NC condition at both time points, in agreement with the increased amount of ZO-2 mRNA found in the LC condition.
Next, we analyzed by immunofluorescence Sp1 expression in MDCK ZO-2 KD cells and observed an increased expression compared to parental cells (figure 2F). These results suggest that the presence of the shRNAs against ZO-2 that trigger the cleavage of the ZO-2 mRNA or represses its translation in ZO-2 KD cells induces a mechanism that augments Sp1 expression.
Altogether, our results strongly suggest that Sp1 regulates the transcription of ZO-2.
3.3. ZO-2 silencing in the kidney after a unilateral nephrectomy is not regulated by miRNAs −23a, −23b, −124 or −137
Next, we analyzed if ZO-2 silencing after a UNX could also be the result of miRNA regulation. miRNAs are endogenous small non-coding RNAs of 20–25 nucleotides associated with argonaute proteins target sites at the 3ʹ untranslated region (3ʹ-UTR) of mRNAs, promoting translational repression and degradation of targeted mRNAs. miRNAs regulate the expression of genes involved in embryonic development and diverse cellular functions, and their deregulation is involved in the initiation and progress of many diseases, including cancer (for review, see [43]).
An in silico analysis done with TargetScan (www.targetscan.org) revealed that the 3ʹ-UTR of ZO-2 mRNA has conserved target sites for miR-23a and b, and miR-124 in rat, mouse, dog, and human, while the target site for miR-137 is not conserved in the dog (data not shown). miRs −23a and −23b that only differ by 1 nucleotide (g19) outside of the seed region, respectively inhibit and increase endothelial permeability;44 miR-137 blocks the expression of ZO-2 and increases the permeability of brain microvascular cells,45 and miR-124 functions as a tumor suppressor and exhibits a polymorphism associated to the risk and clinicopathological characteristics of colorectal cancer.46
By RT-qPCR in rat kidneys, we searched for the presence of miR-23a, miR-23b, miR-137, and miR-124, finding no expression of miR-137 (data not shown), and no difference in the level of expression of miRs −23a, −23b, and −124 between animals with a UNX or sham-operated (Figure 3)
Figure 3.
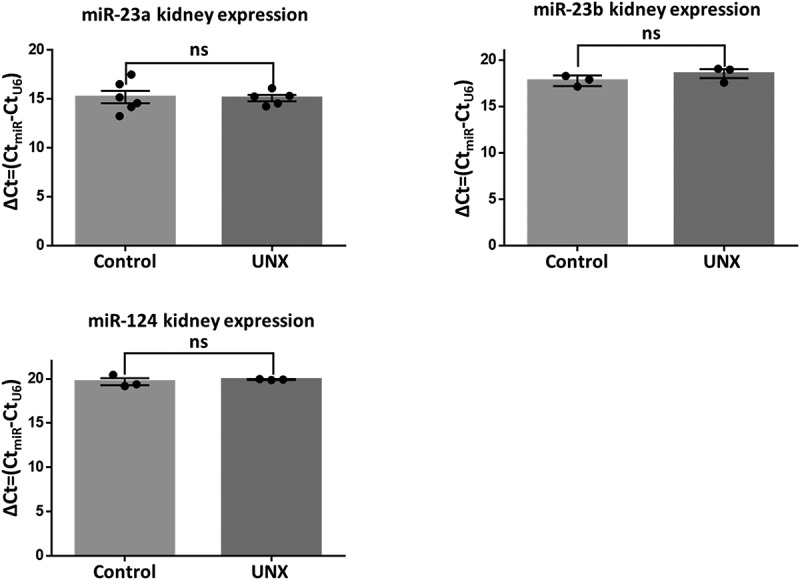
miRs −23a, −23b, and −124 that target the 3ʹ UTR of rat TJP2 mRNA are expressed at the same level in rats with a UNX or sham-operated. RT-qPCR analysis of miRs −23a, −23b, and −124 done in the kidneys of rats 3 weeks after a UNX or a sham operation. Data derived from three animals per experimental condition. Results are mean ± SEM. Statistical analysis was done with Student’s t-test. ns, non-significant.
Our results indicate that in the kidney, ZO-2 silencing after a UNX is due to transcriptional inhibition regulated by a decreased expression of Sp1 and not by translational repression with miRs −23a, −23b, −124, or −137.
3.4. In the steatotic liver of OZ rats, the expression of ZO-2 in hepatocytes is silenced while YAP concentrates at the nuclei
To further study if ZO-2 silencing is a common feature of organ hypertrophy, we next studied the steatotic liver of 12 week-old OZ rats. As a control, we employed lean Zucker (LZ) rats of the same age. Figure 4Aa shows in LZ rats that ZO-2 gives a pair of dots staining pattern that limits the apical membrane of adjacent hepatocytes forming the lumen of bile canaliculi and of “railroad tracks” in cholangiocytes that delineate the intrahepatic small and large bile ducts.47 In the liver of OZ rats, we observed by immunofluorescence that the expression of ZO-2 diminished (Figure 4ab). Since ZO-1 and ZO-2 play a redundant role as platforms for polymerizing claudins at the plasma membrane,2 we next analyzed whether ZO-1 was affected, finding that in OZ rats’ liver, the expression of ZO-1 was higher than in LZ rats (Figures 4Ac and d).
Figure 4.
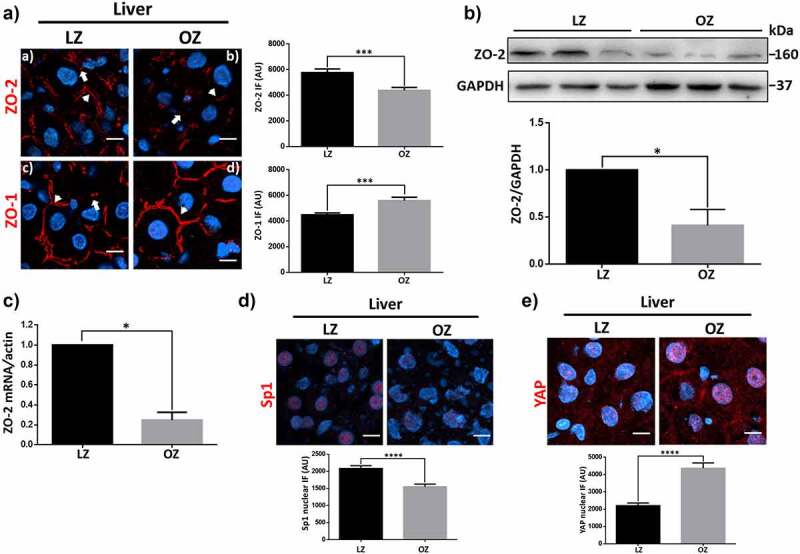
In the liver of OZ rats with steatosis, TJP2 mRNA and ZO-2 protein diminish while YAP concentrates at the nucleus of hepatocytes. A) Frozen sections from livers of lean (LZ) and obese Zucker rats (OZ) of 12 weeks of age were processed for immunofluorescence with antibodies against ZO-1 and ZO-2. Nuclei were stained with DAPI. Bar, 10 μm. Arrows indicate TJ staining as a pair of dots; arrowheads show TJ staining at parallel strands along intrahepatic bile ducts. Left panel, representative images; right panel, quantitative analysis. Statistical analysis was done with GraphPad Prism 5, Mann Whitney test, ***p < .001. B) Western blot of ZO-2 present in a liver lysate derived from LZ and OZ rats. Upper panel, representative Western blot; lower panel, quantitative analysis of three independent experiments. Results are mean ± SEM. Statistical analysis was done with the student’s t-test. *p = .03. C) qRT-PCR amplification of TJP2 mRNA from LZ and OZ rats. Data of LZ rat normalized to 1. Statistical analysis was done with GraphPad Prism 5, Student’s t-test, *p < .05. Results were obtained from three independent experiments. Data were normalized against the geometric mean of actin mRNA. D) Frozen sections from livers of LZ and OZ of 12 weeks of age were processed for immunofluorescence with antibodies against Sp1. Nuclei were stained with DAPI. Bar, 10 μm. Upper panel, representative images; lower panel, quantitation of immunofluorescence for nuclear Sp1. The nuclei of 200 cells per condition derived from three independent experiments were quantitated. Statistical analysis was done with Mann Whitney test, ****p < .0001. E) Frozen sections from livers of LZ and OZ of 12 weeks of age were processed for immunofluorescence with antibodies against YAP. Nuclei were stained with DAPI. Bar, 10 μm. Upper panel, representative images; lower panel, quantitation of immunofluorescence for nuclear YAP. The nuclei of 100 cells per condition derived from three independent experiments were quantitated. Statistical analysis was done with Mann Whitney test, ****p < .0001.
The diminished expression of ZO-2 in the steatotic liver was further confirmed with a Western blot against ZO-2 done in liver lysates derived from LZ and OZ rats (Figure 4B). ZO-2 silencing is apparently due to transcriptional regulation, as the level of ZO-2 mRNA in the liver diminishes in OZ compared to LZ rats (Figure 4C). In addition, we observed a decreased expression of Sp1 in the nuclei of OZ rats compared to LZ rats (Figure 4D).
In rats, we had previously shown that in the remaining kidney after a UNX, ZO-2 silencing correlated with an increasing amount of nuclear YAP.4 Therefore, we now analyzed by immunofluorescence the expression of nuclear YAP in the liver of OZ rats, observing an increased expression compared to the liver of LZ animals (Figure 4E). Hence, these results indicate that ZO-2 silencing coupled to YAP concentration at the nucleus are common features present in hypertrophic organs.
3.5. In the steatotic liver of OZ rats, autolysosome activity is impaired
In the UNX model of renal hypertrophy, YAP induces an increase in cell size through mTOR activation, leading to the phosphorylation of p70S6 kinase (S6K),4 which in turn phosphorylates S6, a component of the 40S ribosomal subunit. Both S6K48–50 and S651 are critical regulators of cell size. Here, we observed that in the liver of OZ compared to LZ rats, the amount of p70S6K, and its degree of phosphorylation, did not change, while the phosphorylation of S6 diminished in OZ rats (Supplemental Fig. 2). These results indicate that the hypertrophy of the liver in OZ rats is not mediated by mTOR activation.
Hypertrophy is triggered by an imbalance between protein synthesis and degradation,4 and lysosomal alkalinization is known to induce hypertrophy by reducing protein degradation.52,53 Hence, we next analyzed lysosome functionality in OZ hepatocytes compared to LZ rats with an ex vivo LC3 flux assay. LC3-I protein is conjugated to phosphatidylethanolamine (PE) to form LC3-II. The latter is specifically targeted to the phagophore and remains associated with its expanding membrane, sealed autophagosomes, and mature autolysosomes. Upon fusion of autophagosomes with lysosomes, the luminal pool of LC3-II is degraded. Therefore, if there is an impaired lysosomal activity, LC3-II degradation is blocked, and the ratio of LC3-II/LC3-I augments (for review, see [54]). This assay was done in liver slices derived from OZ and LZ rats after a 24 h starvation period since autophagic protein degradation is enhanced during starvation.55 To cause a reduction in the autophagic flux, we used 100 mM NH4Cl, which increases the pH inside the lysosome and inhibits autophagosome-lysosome fusion. Supplementary figure 2B shows that in the liver of starved LZ rats, treatment with NH4Cl increases the ratio of LC3-II/LC3-I as expected due to the inhibition of LC3-II degradation. Instead, this increase of LC3-II/LC3-I ratio is not observed in starved OZ rats after NH4Cl treatment.
These results indicate a lower abundance of functional autolysosomes in the liver of OZ rats compared to LZ animals, suggesting the contribution of this condition to the development of liver hypertrophy.
3.6. The activation of AMPK with metformin augments in the liver of OZ rats, the expression of ZO-2 and active LATS, and diminishes the nuclear concentration of YAP
To test the importance of ZO-2 silencing in the development of leaky TJ in hepatic steatotic cells, we tried to induce the re-expression of ZO-2 at the TJs of liver cells. Our strategy for this purpose was the activation of AMPK. Previous work had shown that activation of this serine/threonine kinase promotes cell polarization56 and TJ assembly57,58 and the re-localization of ZO-2 from the cytoplasm to the borders of MDCK cells cultured in a condition of LC.59 Therefore, we treated 7 week-old OZ rats with metformin, an antidiabetic drug that activates AMPK in hepatocytes.60 We administered a daily dose of 300 mg/kg/day of metformin until the animals reached 12 weeks of age and were sacrificed. By immunofluorescence, we found that in the liver of OZ rats treated with metformin, ZO-2 expression recovers (Figure 5Aa-c), and by Western blot, we observed an increased expression of ZO-2 in OZ rats treated with metformin compared to OZ and LZ animals (Figure 5B).
Figure 5.
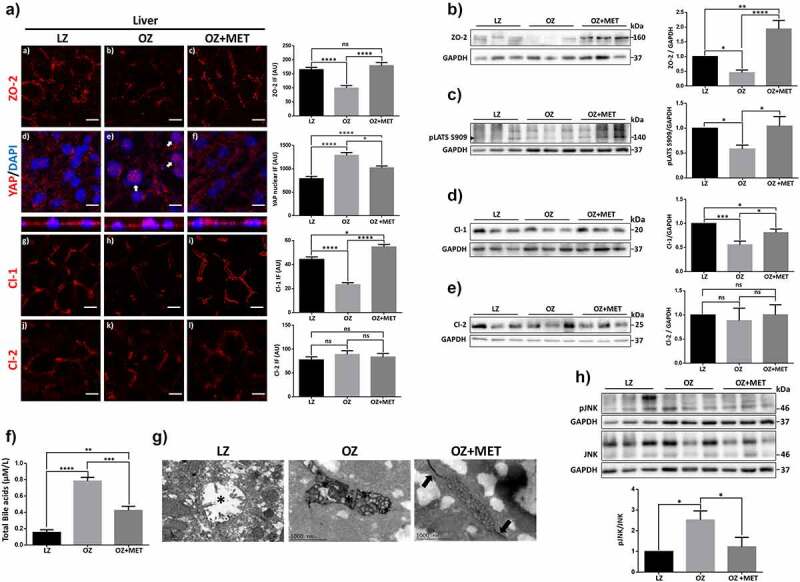
Treatment of OZ rats with metformin restores ZO-2, and claudin-1 expression in the liver reduces the permeability of bile canaliculi and total bile acid concentration in serum and diminishes JNK activation. 7-week-old OZ rats received or not a daily dose of 300 mg/kg/day of metformin (MET) until they reached 12 weeks of age and were sacrificed together with 12 week-old LZ rats. 3 rats were employed per experimental condition. Livers were excised and then processed as explained in A-F. A) Immunofluorescence with antibodies against ZO-2, YAP and claudins −1 (Cl-1) and −2 (Cl-2). Nuclei were stained with DAPI. Left panel, representative images, bar, 10 μM; right panel, quantitation of immunofluorescence. For ZO-2, Cl-1 and Cl-2, 20 bile canaliculi per condition derived from three independent experiments were quantitated. Statistical analysis was done with Kruskal-Wallis test followed by Dunn’s multiple comparisons test, *p < .05, ****p < .0001. For YAP, the nuclei of 100 cells per condition derived from three independent experiments were quantitated. Statistical analysis was done with Kruskal-Wallis test followed by Dunn’s multiple comparisons test, *p < .05, ****p < .0001. B) Western blot with antibodies against ZO-2. GAPDH was employed as a loading control. Six rats per cohort were employed and at least two blots were analyzed per each rat. Left panel, representative Western blot; right panel, quantitative analysis. Results are mean ± SEM from at least three independent experiments. LZ data normalized to 1. Statistical analysis was done with One-way ANOVA followed by Fisher’s LSD, *p < .05, **p < .001, ****p < .0001. C) Western blot with antibodies against pLATS S909 (arrowhead). GAPDH was employed as a loading control. Six rats per cohort were employed and at least two blots were analyzed per each rat. Left panel, representative Western blot; right panel, quantitative analysis. Here we quantitated pLATS S909/GAPDH and not pLATS S909/LATS1, because the antibody against LATS1 does not work in rat liver. Results are mean ± SEM from at least three independent experiments. LZ data normalized to 1. Statistical analysis was done with One-way ANOVA followed by Fisher’s LSD, *p < .05. D) Western blot with antibodies against claudin-1. GAPDH was employed as a loading control. Six rats per cohort were employed, and at least two blots were analyzed per rat. Left panel, representative Western blot; right panel, quantitative analysis. Results are mean ± SEM from at least three independent experiments. LZ data normalized to 1. Statistical analysis was done with One-way ANOVA followed by Fisher’s LSD, *p < .05, ***p < .001. E) Western blot with antibodies against claudin-2. GAPDH was employed as a loading control. Six rats per cohort were employed, and at least two blots were analyzed per rat. Left panel, representative Western blot; right panel, quantitative analysis. Results are mean ± SEM from at least three independent experiments. LZ data normalized to 1. Statistical analysis was done with One-way ANOVA followed by Fisher’s LSD. F) Quantification of total bile acids in serum of LZ and OZ rats treated or not with metformin as mentioned above. Results are presented as mean ± SEM. Statistical analysis was done with One-way ANOVA followed by Tukey’s multiple comparison post-hoc tests, **p < .01, ***p < .001, ****p < .0001. G) Representative images of the penetration of lanthanum into the canalicular liver lumen. In LZ rats, no accumulation of lanthanum is observed in the canalicular lumen (asterisk). OZ rats, canalicular lumen with intense lanthanum staining (asterisk). OZ rats treated with metformin, TJ area that limits the canalicular lumen shows the accumulation of lanthanum (arrows), together with a discrete lanthanum infiltration into the canalicular lumen (asterisk). H) Western blot with antibodies against pJNK and JNK. GAPDH was employed as a loading control. Six rats per cohort were employed and at least two blots were analyzed per rat. Left panel, representative Western blot; right panel, quantitative analysis. Results are mean ± SEM from at least three independent experiments. LZ data normalized to 1. Statistical analysis was done with One-way ANOVA followed by Fisher’s LSD, *p < .05.
Since we had observed that YAP concentrates at the nuclei of hepatocytes in OZ rats (Figure 4E), we next tested if this effect could be reversed by metformin treatment. Figure 5Ad-f shows by immunofluorescence that the activation of AMPK with metformin diminished the nuclear accumulation of YAP in OZ rats. This observation, together with the newfound interaction between ZO-2 and LATS1 (Figure 1A), prompted us to test the expression of active LATS in the steatotic liver. Figure 5C shows by Western blot a decrease in pLATS in the liver of OZ rats in comparison to LZ, which recovers after metformin treatment.
Our results indicate that ZO-2 silencing in the hypertrophic liver is coupled to LATS inactivation and YAP nuclear concentration and that this process can be reversed by AMPK activation with metformin.
3.7. Metformin treatment in OZ rats restores claudin-1 expression, and ameliorates the increased paracellular permeability of hepatocytes and the augmented content of bile acids in serum
Progressive familial intrahepatic cholestasis (PFIC), has several different genetic etiologies including in PFIC type 4, mutations in TJP2.20,22,23,61 These mutations in TJP2 generate premature termination codons that lead to the absence of ZO-2 in liver tissue and a significant reduction in claudin-1 but not claudin-2 in the liver.21 Therefore, we next examined if, in OZ rats, the reduction of ZO-2 in the liver was accompanied by changes in the expression of claudins −1 and −2. By immunofluorescence, we observed in OZ compared to LZ rats a reduction in claudin-1 expression that was reversed after OZ animals were treated with metformin (Figure 5Ag-i), while no changes were observed in claudin-2 expression (Figure 5Aj-l). By Western blot, we confirmed these observations (Figure 5D,E).
Since elevated plasma/serum bile acid levels are present in animals and patients with liver steatosis14,15 as well as in children with PFIC-4,21 we next analyzed if treatment with metformin that augments the expression of ZO-2 in the liver can reduce in OZ rats, the abundance of bile acids in serum. figure 5F shows that the amount of bile acids in serum augments in OZ rats compared to LZ animals and that this value diminishes after metformin treatment. This result suggests that in OZ rats, the escape of bile acids through the paracellular space into the liver parenchyma and serum could be a consequence of the reduced expression of ZO-2 that leads to the development of a compromised BBiB.
To assess TJ permeability in hepatocytes, we evaluated by electron microscopy the penetration of lanthanum, an electron-dense paracellular marker, across the junction. Figure 5G shows that no accumulation of lanthanum is observed in the liver in the canalicular lumen of LZ rats. Instead, in the liver of OZ rats, the canalicular lumen display intense lanthanum staining, whereas, in those treated with metformin, lanthanum accumulates around the area that limits the canaliculi, infiltrating into them, albeit with less intensity than in the liver of OZ rats.
Altogether, these results indicate that treatment of OZ rats with metformin augments the expression of ZO-2 and claudin-1 in hepatocytes and partially restores the BBiB.
3.8. Metformin reversal of BBiB dysfunction encompasses inhibition of JNK
Stress-activated c-Jun N-terminal kinase (JNK) triggers TJ disassembly triggered by extracellular calcium depletion,24 while JNK inhibition attenuates dextran sulfate sodium (DSS)-induced intestinal TJ barrier disruption.25 On the other hand, metformin via AMPKα1 activation inhibits JNK signaling and blocks, in consequence, intestinal barrier dysfunction.62 Therefore, we next analyzed whether metformin decreased the amount of active JNK in the liver of OZ rats. Figure 5H shows an increase in phosphorylated and hence active JNK in OZ rats’ liver compared to LZ animals, which diminishes upon treatment with metformin. Hence, these results suggest that the mechanism through which metformin protects against ZO-2 silencing and BBiB dysfunction in the steatotic liver involves inhibition of JNK signaling.
4. Discussion
Our previous observation showed that in MDCK cells, ZO-2 silencing reduced the phosphorylation of YAP, the downstream target of the Hippo pathway,4 prompting us to explore the connection between ZO-2 and this signaling pathway. We demonstrate the interaction between ZO-2 and LATS1, a kinase responsible for YAP phosphorylation by co-immunoprecipitation and pull-down assays. Moreover, we show that in the two models of hypertrophy analyzed: RCH and liver steatosis, ZO-2 silencing occurs together with a reduction in the amount of active LATS and the concentration of YAP at the nuclei (Figure 6). Altogether, these observations suggest that ZO-2 functions as a scaffold that facilitates LATS activation and promotes signaling through the Hippo pathway to regulate cell size.
Figure 6.
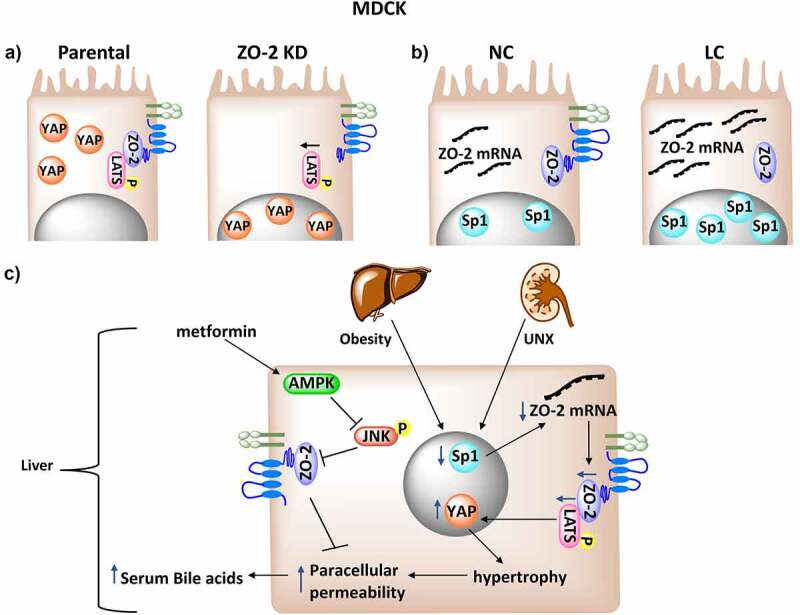
ZO-2 silencing diminishes LATS activation, favoring the nuclear concentration of YAP and the development of hypertrophy. A) ZO-2 functions as a scaffold at the TJ that facilitates LATS phosphorylation and inhibits the nuclear concentration of YAP. B) In confluent cultures of MDCK cells incubated in medium with low calcium (LC), TJs cannot be established, but the cells accumulate SP1 transcription factor and ZO-2 mRNA. C) In hypertrophic kidney and in the liver with steatosis, ZO-2 silencing is generated by a decrease in ZO-2 transcription probably associated to a diminished nuclear content of Sp1 transcription factor. ZO-2 interacts with LATS, and the reduced expression of ZO-2 blocks LATS phosphorylation and activation facilitating the nuclear concentration of YAP. In the steatotic liver this condition triggers an increase in paracellular permeability of hepatocytes and the presence of bile acids in serum. Metformin, via AMPKα1 activation inhibits JNK signaling that promotes TJ barrier dysfunction.
We found that neither the kinase MST1/2 nor its associated scaffold protein Salvador (SAV) coimmunoprecipitate with ZO-2. However, it would be important in the future to test if the interaction of LATS1 with MST1/2 or MAP4Ks, another family of kinases that phosphorylates LATS1,63 diminishes in the absence of ZO-2.
Here, we explored if ZO-2 silencing in hypertrophy is regulated at the transcriptional or translational level. We did not find evidence of translational repression by miRs −23a, −23b, −124, or −137 but observed a decrease in ZO-2 mRNA in the hypertrophic rat kidney. Transcription by alternative promoters PA and PC generates ZO-2 isoforms A and C.39 Both are expressed in normal cells, but only the latter that lacks 23 amino acids at the amino terminus is maintained in cancerous pancreatic cells,39 suggesting that isoform ZO-2A might be critical for TJ function in non-cancerous epithelial cells. Numerous Sp1 sites are present in PA,39 and Sp1 is a transcription factor ubiquitously expressed that functions as an activator of a wide variety of genes, including housekeeping, cell cycle regulators, and tissue-restricted genes (for review see [64]). Hence, we analyzed the expression of the Sp1 transcription factor in the hypertrophic kidney and MDCK cells. Our observation of a decreased amount of this transcription factor in the kidney after a UNX prompted us to employ MTM in MDCK cells to study the effect of Sp1 inhibition on ZO-2 expression. Our results revealed that MTM blocked the increase in ZO-2 mRNA when the culture was maintained in the LC condition, and the increase in ZO-2 protein was observed after the cells were transferred from LC to NC media. These observations confirm that Sp1 regulates ZO-2 expression at the transcriptional level and suggest that a decreased expression of Sp1 silences ZO-2 in the hypertrophic kidney. The increased ZO-2 mRNA observed in cells cultured in LC compared to those maintained in NC resembles a similar observation previously made with ZO-1 in MDCK cells.42 It appears that if TJs are not established due to the lack of extracellular calcium, the cells accumulate ZO-2 mRNA as if they were building a reserve in order to guarantee its future synthesis. Previously we observed a similar situation for CD1 mRNA, where the amount increased when MDCK cells were not proliferating due to their culture in a low serum condition (0.1% serum) compared to proliferating cells maintained in 10% serum.65
Hypertrophy develops due to an imbalance between the rates of protein synthesis and degradation,52,53 and in hypertrophic ZO-2 KD MDCK cells, we observed an increased mTOR pathway activity, which augmented the protein synthesis rate.4 However, in OZ rats, we found that the phosphorylation of S6 diminished, indicating that liver hypertrophy is not mediated by mTOR activation. Instead, we found in OZ rats livers a lower abundance of functional autolysosomes than in LZ rats, thus suggesting that this condition contributes to the development of liver hypertrophy. These observations are in agreement with a report indicating that in primary fetal hepatocytes derived from ZDF rats with a fa gene frequency of 0.75, the synthesis of proteins is significantly lower than in cultures with 0.0 fa gene frequency.66 In addition, they are in line with the finding that NH4Cl inhibits protein synthesis in 0.0 but not in 0.75 fa gene frequency cultures, suggesting that fetal hepatocytes bearing the fa gene have a genetic error in lysosomal function.66 This genetic mistake that decreases lysosomal degradation might be crucial for the development of hypertrophy in hepatocytes. It could also be related to the observation that treatments that restrict food intake in OZ rats reduce body weight but cannot normalize these animals’ obese body composition, which is 50% lipid versus under 20% in lean littermates.67
Another mechanism that could trigger the hypertrophy of hepatocytes in OZ rats relies on the observation that steatotic livers up-regulate the cyclin-dependent kinase inhibitor p21 due to the over-expression of the tumor suppressor p53 and a lowered phosphorylation of retinoblastoma protein. All these alterations lead to a diminished cell division11 that might facilitate a hypertrophic phenotype.
We observe that the hypertrophy generated in OZ rats liver induces ZO-2 silencing, and previously reported that hypertrophy in kidney triggered by UNX also induces ZO-2 silencing.4 Hence, we hypothesize that hypertrophy, independently of how it was generated, silences ZO-2.
In ZO-2 KD MDCK cells, we observed that transfection of ZO-2 allowed cells to recover their standard size.4 This observation prompted us to employ metformin to induce OZ rats to re-express ZO-2 in liver TJs. Metformin is a derivative of guanidine, the active ingredient of G. officinalis, also known as French lilac, which in medieval times was used to treat diabetes.68 In OZ rats with steatosis and diabetes, previous reports reveal that treatment with metformin mitigates liver steatosis and decreases plasma triacylglycerol (TAG) concentrations at 21 weeks of treatment but does not modify energy intake or expenditure, the evolution of diabetes, or the expression in the liver of genes of fatty acid metabolism.8
Metformin,60 AICAR (5-aminoimidazole-4-carboxamide riboside), and the kinase LKB1 (ortholog of PAR4 in C. elegans and D. melanogaster) activate the serine/threonine kinase AMPK through different procedures. For example, metformin that employs an adenine nucleotide-independent mechanism69 increases the phosphorylation of Thr-172 regulatory site on AMPK α-catalytic subunit in human hepatic cells.70 Instead, AICAR is a nucleoside that converts to the nucleotide ZMP, which mimics the effects of AMP on AMPK,71,72 and LKB1 phosphorylates Thr-172 in AMPK.73,74
AMPK is a cellular energy sensor activated during metabolic stresses such as hypoxia, ischemia, glucose deprivation, or exercise.75,76 Activation of AMPK with LKB1, AICAR, or metformin favors the appearance of TJ proteins. Thus, LKB1 activation induces the polarization of single intestinal epithelial cells that display a redistribution of the TJ protein ZO-1 in a dotted circle peripheral to the brush border.56 Activation of AMPK with AICAR facilitates TJ assembly under conditions of standard extracellular Ca2+ concentrations, partially protects the TJs from disassembly induced by Ca2+ depletion, and even initiates TJ assembly in the absence of Ca2+.57,58 Moreover, we specifically showed that AMPK activation with AICAR triggers the re-localization of ZO-2 from the cytoplasm to the borders of MDCK cells cultured in a condition of low calcium.59 Metformin restores junction assembly in the intestinal epithelium and protects against intestinal barrier dysfunction in experimental colitis induced by DSS and patients with ulcerative colitis.62,77 These observations agree with our results showing that treatment of OZ rats with metformin up-regulates ZO-2 and claudin-1 expression in the liver, reduces the serum bile acid content, and the paracellular permeability of hepatocyte TJs.
The mechanism through which AMPK activation by metformin enhances the expression of ZO-2 protein in the liver of steatotic OZ rats appears to involve the inhibition of JNK activation since the augmented level of phosphorylated JNK found in OZ rats compared to LZ animals decreased after metformin treatment. These results agree with observations done in intestinal Caco-2 monolayers where metformin activation of AMPKα1 suppresses DSS-induced JNK activation and augments the expression of ZO-1 and occludin.62
The formation of bile acids is one of the main functions of the liver. Bile acids are transported across the hepatocytes apical membrane into the lumen of bile canaliculi. Then, via extrahepatic bile ducts reach the intestinal tract, having a fundamental role in the solubilization of dietary lipids. Bile acids are powerful detergents, and TJs prevent their leakage to the basolateral surface and the underlying parenchyma of hepatocytes and cholangiocytes. In the liver claudins −1, −2 and −3 have been observed in hepatocyte TJs.78,79,80 Claudin-2 has been more thoroughly studied, demonstrating its importance for the paracellular water flow required for proper bile composition, as a claudin-2 deficiency in mice increased bile concentration and promoted cholesterol gallstone formation.81 In humans, the role of ZO-2 in liver TJs appears to be critical and non-redundant since homozygote20,22 and compound heterozygote23,61 mutations in TJP2 cause PFIC-4. This disease, characterized by fluctuating jaundice, persistent cholestasis, pruritus, and malabsorption, affects young children and is an end-stage liver disease that leads to cirrhosis and hepatocellular carcinoma.22,23 In PFIC-4, the mutations of TJP2 that ablate ZO-2 expression induce a severe disease only in the liver, even though ZO-2 is a protein normally expressed in all epithelial cells. These observations suggest that in the human liver, ZO-2 plays a non-redundant role at TJs that cannot be compensated by other TJ proteins like, for example, ZO-1 or ZO-3.
In mice, ZO-2 absence is lethal during embryogenesis, as ZO-2 knock-out mice die shortly after implantation, showing decreased proliferation at embryonic day 6.5 (E6.5) and increased apoptosis at E7.5.82 Thus, ZO-2 appears to be essential for the extraembryonic tissue and not for the mice embryo, as lethality was rescued when ZO-2 −/− embryonic stem cells were injected into wild-type blastocysts.83 However, the resulting chimeric mice developed a compromised blood-testis barrier in adulthood.83
In mice, the liver-specific deletion of TJP2 does not trigger a complete disruption of the BBiB but reduces claudin-1 protein expression and induces mild progressive cholestasis characterized by a diminished expression of bile acid transporter Abc11/Bsep and detoxification enzyme Cyp2b10.84 These mice are, however, more susceptible to liver injury.84 In contrast, the ZO-1/ZO-2 double mutant mice die by 6 weeks of age and display a disrupted BBiB that does not form TJs.85 These observations suggest that in mice, ZO-2 is important for the BBiB, however, its function in the liver can be compensated by ZO-1. In rats, a similar situation is expected, since the liver of OZ rats showed a decreased amount of ZO-2 accompanied by an increased expression of ZO-1, in comparison to LZ animals.
Finding that ZO-2 expression diminishes in the steatotic liver and that metformin restoration of ZO-2 expression is accompanied by a decreased serum content of bile acids strongly suggests that ZO-2 can be a therapeutic target for treating liver conditions where the BBiB is compromised. In this regard, it is noteworthy the case of intrahepatic cholestasis of pregnancy (ICP), a disease where women have pruritus, hepatic impairment, and elevated serum bile acids that can lead to spontaneous preterm delivery and stillbirth (for review see [86]). In this disease that has a complete resolution after delivery and high recurrence rates in subsequent pregnancies, the affected women have heterozygous mutations in TJP2 or one of the four genes for liver transporters: ABCB4, ABCB11, ABCC2, and ATP8B1,87 known to generate PFIC in patients with homozygous protein-truncating mutations.20 However, patients only manifest the cholestatic phenotype when pregnant, typically in the third trimester, when they might have gained the most weight. Also, the coexistence of ICP has been observed with acute fatty liver of pregnancy.88,89 Our observation showing that ZO-2 content diminishes in the hypertrophic liver speculatively suggest that in pregnant women with a steatotic liver, a decrease in ZO-2 expression could facilitate the development of intrahepatic cholestasis if the woman harbors a heterozygous mutation in the genes for TJP2 or the liver transporters ABCB4, ABCB11, ABCC2 or ATP8B1.
In summary, our results demonstrate that ZO-2 interacts with LATS1 and promotes its activation, whereas ZO-2 silencing in hypertrophic kidney and liver leads to a decrease in LATS phosphorylation and the nuclear concentration of YAP. These results portray ZO-2 as a novel regulator of the Hippo pathway and explain why ZO-2 silencing in MDCK cells leads to the previously observed increase in cell size.4 Our observations highlight the importance of AMPK, JNK, and ZO-2 as therapeutic intervention targets for the restoration of BBiB dysfunction.
Supplementary Material
Acknowledgments
This work was supported by grants to L.G.M from the Miguel Alemán Valdés Foundation 2018, SEP-Cinvestav FIDSC2018/33 and National Council of Science and Technology of Mexico (Conacyt) FORDECYT-PRONACES-140644/2020. L.G.G., H.G.G, C.H.G and S.I.R.G were recipients of doctoral fellowship from the Conacyt (340209, 282075, 407499 and 455381) and H.G.G. was also a recipient of a scholarship from Mexiquense Council of Science and Technology (Comecyt, 2018AD0009-11).
Funding Statement
This work was supported by the Consejo Nacional de Ciencia y Tecnología [FORDECYT-PRONACES-140644/2020]; Fundación Miguel Alemán, A.C. [2018]; SEP-CINVESTAV [FIDSC2018/33].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Mandel LJ, Bacallao R, Zampighi G.. Uncoupling of the molecular ‘fence’ and paracellular ‘gate’ functions in epithelial tight junctions. Nature. 1993;361(6412):1–23. doi: 10.1038/361552a0. [DOI] [PubMed] [Google Scholar]
- 2.Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M, Tsukita S, et al. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006;126(4):741–754. doi: 10.1016/j.cell.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez-Mariscal L, Gallego-Gutiérrez H, González-González L, Hernández-Guzmán C.. ZO-2 Is a master regulator of gene expression, cell proliferation, cytoarchitecture, and cell size. Int J Mol Sci. 2019;20(17):4128. doi: 10.3390/ijms20174128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dominguez-Calderon A, Ávila-Flores A, Ponce A, López-Bayghen E, Calderón-Salinas J-V, Luis Reyes J, Chávez-Munguía B, Segovia J, Angulo C, Ramírez L, et al. ZO-2 silencing induces renal hypertrophy through a cell cycle mechanism and the activation of YAP and the mTOR pathway. Mol Biol Cell. 2016;27(10):1581–1595. doi: 10.1091/mbc.E15-08-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iida M, Murakami T, Ishida K, Mizuno A, Kuwajima M, Shima K. Substitution at codon 269 (glutamine –> proline) of the leptin receptor (OB-R) cDNA is the only mutation found in the Zucker fatty (fa/fa) rat. Biochem Biophys Res Commun. 1996;224(2):597–604. doi: 10.1006/bbrc.1996.1070. [DOI] [PubMed] [Google Scholar]
- 6.Kelesidis T, Kelesidis I, Chou S, and Mantzoros CS. Narrative review: the role of leptin in human physiology: emerging clinical applications. Ann Intern Med. 2010;152(2):93–100. doi: 10.7326/0003-4819-152-2-201001190-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kava R, Greenwood MRC, Johnson PR. Zucker (fa/fa) Rat. ILAR J. 1990;32(3):4–8. doi: 10.1093/ilar.32.3.4. [DOI] [Google Scholar]
- 8.Forcheron F, Abdallah P, Basset A, Del Carmine P, Haffar G, Beylot M. Nonalcoholic hepatic steatosis in Zucker diabetic rats: spontaneous evolution and effects of metformin and fenofibrate. Obesity (Silver Spring). 2009;17(7):1381–1389. doi: 10.1038/oby.2008.661. [DOI] [PubMed] [Google Scholar]
- 9.Matteoni CA, Younossi Z, Gramlich T, Boparai N, Liu Y, Mccullough A. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity☆☆. Gastroenterology. 1999;116(6):1413–1419. doi: 10.1016/S0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 10.Yang SQ, Lin HZ, Mandal AK, Huang J, Diehl AM. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathophysiology. Hepatology. 2001;34(4 Pt 1):694–706. doi: 10.1053/jhep.2001.28054. [DOI] [PubMed] [Google Scholar]
- 11.Donthamsetty S, Bhave VS, Mitra MS, Latendresse JR, Mehendale HM. Nonalcoholic fatty liver sensitizes rats to carbon tetrachloride hepatotoxicity. Hepatology. 2007;45(2):391–403. doi: 10.1002/hep.21530. [DOI] [PubMed] [Google Scholar]
- 12.McCormack L, Dutkowski P, El-Badry AM, Clavien PA. Liver transplantation using fatty livers: always feasible? J Hepatol. 2011;54(5):1055–1062. doi: 10.1016/j.jhep.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Kloek JJ, Marechal X, Roelofsen J, Houtkooper RH, van Kuilenburg AB, Kulik W, Bezemer R, Neviere R, van Gulik TM, Heger M. Cholestasis is associated with hepatic microvascular dysfunction and aberrant energy metabolism before and during ischemia-reperfusion. Antioxid Redox Signal. 2012;17(8):1109–1123. doi: 10.1089/ars.2011.4291. [DOI] [PubMed] [Google Scholar]
- 14.Dasarathy S, Yang Y, McCullough AJ, Marczewski S, Bennett C, Kalhan SC. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol. 2011;23(5):382–388. doi: 10.1097/MEG.0b013e328345c8c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalhan SC, Guo L, Edmison J, Dasarathy S, McCullough AJ, Hanson RW, Milburn M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism. 2011;60(3):404–413. doi: 10.1016/j.metabol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka N, Matsubara T, Krausz KW, Patterson AD, Gonzalez FJ. Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology. 2012;56(1):118–129. doi: 10.1002/hep.25630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin IV, Schmitt J, Minkenberg A, Mertens JC, Stieger B, Mullhaupt B, Geier A. Bile acid retention and activation of endogenous hepatic farnesoid-X-receptor in the pathogenesis of fatty liver disease in ob/ob-mice. Biol Chem. 2010;391(12):1441–1449. doi: 10.1515/bc.2010.141. [DOI] [PubMed] [Google Scholar]
- 18.Pizarro M, Balasubramaniyan N, Solis N, Solar A, Duarte I, Miquel JF, Suchy FJ, Trauner M, Accatino L, Ananthanarayanan M, et al. Bile secretory function in the obese Zucker rat: evidence of cholestasis and altered canalicular transport function. Gut. 2004;53(12):1837–1843. doi: 10.1136/gut.2003.037689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carlton VE, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson DL, Strauss KA, Shneider BL, Lim WA, Salen G, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34(1):91–96. doi: 10.1038/ng1147. [DOI] [PubMed] [Google Scholar]
- 20.Sambrotta M, Thompson RJ. Mutations in TJP2, encoding zona occludens 2, and liver disease. Tissue Barriers. 2015;3(3):e1026537. doi: 10.1080/21688370.2015.1026537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrotta M, Strautnieks S, Papouli E, Rushton P, Clark BE, Parry DA, Logan CV, Newbury LJ, Kamath BM, Ling S, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. 2014;46(4):326–328. doi: 10.1038/ng.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vij M, Shanmugam NP, Reddy MS, Sankaranarayanan S, Rela M. Paediatric hepatocellular carcinoma in tight junction protein 2 (TJP2) deficiency. Virchows Arch. 2017;471(5):679–683. doi: 10.1007/s00428-017-2204-1. [DOI] [PubMed] [Google Scholar]
- 23.Zhou S, Hertel PM, Finegold MJ, Wang L, Kerkar N, Wang J, Wong LJC, Plon SE, Sambrotta M, Foskett P, et al. Hepatocellular carcinoma associated with tight-junction protein 2 deficiency. Hepatology. 2015;62(6):1914–1916. doi: 10.1002/hep.27872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naydenov NG, Hopkins AM, Ivanov AI. c-Jun N-terminal kinase mediates disassembly of apical junctions in model intestinal epithelia. Cell Cycle. 2009;8(13):2110–2121. doi: 10.4161/cc.8.13.8928. [DOI] [PubMed] [Google Scholar]
- 25.Samak G, Chaudhry K, Gangwar R, Narayanan D, Jaggar J, Rao R. Calcium/Ask1/MKK7/JNK2/c-Src signalling cascade mediates disruption of intestinal epithelial tight junctions by dextran sulfate sodium. Biochem J. 2015;465(3):503–515. doi: 10.1042/BJ20140450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Itallie CM, Fanning AS, Bridges A, Anderson JM. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell. 2009;20(17):3930–3940. doi: 10.1091/mbc.e09-04-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hernandez S, Chavez Munguia B, Gonzalez-Mariscal L. ZO-2 silencing in epithelial cells perturbs the gate and fence function of tight junctions and leads to an atypical monolayer architecture. Exp Cell Res. 2007;313(8):1533–1547. doi: 10.1016/j.yexcr.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 28.Raya-Sandino A, Castillo-Kauil A, Domínguez-Calderón A, Alarcón L, Flores-Benitez D, Cuellar-Perez F, López-Bayghen B, Chávez-Munguía B, Vázquez-Prado J, González-Mariscal L, et al. Zonula occludens-2 regulates Rho proteins activity and the development of epithelial cytoarchitecture and barrier function. Biochim Biophys Acta. 2017;1864(10):1714–1733. doi: 10.1016/j.bbamcr.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 29.Betanzos A, Huerta M, Lopez-Bayghen E, Azuara E, Amerena J, González-Mariscal L. The tight junction protein ZO-2 associates with Jun, Fos and C/EBP transcription factors in epithelial cells. Exp Cell Res. 2004;292(1):51–66. doi: 10.1016/j.yexcr.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Quiros M, Alarcón L, Ponce A, Giannakouros T, González-Mariscal L. The intracellular fate of zonula occludens 2 is regulated by the phosphorylation of SR repeats and the phosphorylation/O-GlcNAcylation of S257. Mol Biol Cell. 2013;24(16):2528–2543. doi: 10.1091/mbc.e13-04-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aleman V, Osorio B, Chavez O, Rendon A, Mornet D, Martinez D. Subcellular localization of Dp71 dystrophin isoforms in cultured hippocampal neurons and forebrain astrocytes. Histochem Cell Biol. 2001;115(3):243–254. doi: 10.1007/s004180000221. [DOI] [PubMed] [Google Scholar]
- 32.Petrov A, Tsa A, Puglisi JD. Analysis of RNA by analytical polyacrylamide gel electrophoresis. Methods Enzymol. 2013;530:301–313. [DOI] [PubMed] [Google Scholar]
- 33.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33(20):e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kramer MF. Stem-loop RT-qPCR for miRNAs. Curr Protoc Mol Biol. 2011. Chapter 15;Unit 15 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36(Database issue):D154–8. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng T, Shao F, Wu Q, Zhang X, Xu D, Qian K, Xie Y, Wang S, Xu N, Wang Y, et al. miR-124 downregulation leads to breast cancer progression via LncRNA-MALAT1 regulation and CDK4/E2F1 signal activation. Oncotarget. 2016;7(13):16205–16216. doi: 10.18632/oncotarget.7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzalez-Torres A, Banuelos-Villegas EG, Martinez-Acuna N, Sulpice E, Gidrol X, Alvarez-Salas LM. MYPT1 is targeted by miR-145 inhibiting viability, migration and invasion in 2D and 3D HeLa cultures. Biochem Biophys Res Commun. 2018;507(1–4):348–354. doi: 10.1016/j.bbrc.2018.11.039. [DOI] [PubMed] [Google Scholar]
- 38.Lora L, Mazzon E, Martines D, Fries W, Muraca M, Martin A, d’Odorico A, Naccarato R, Citi S. Hepatocyte tight-junctional permeability is increased in rat experimental colitis. Gastroenterology. 1997;113(4):1347–1354. doi: 10.1053/gast.1997.v113.pm9322530. [DOI] [PubMed] [Google Scholar]
- 39.Chlenski A, Ketels KV, Engeriser JL, Talamonti MS, Tsao M-S, Koutnikova H, Oyasu R, Scarpelli DG. zo-2 gene alternative promoters in normal and neoplastic human pancreatic duct cells. Int J Cancer. 1999;83(3):349–358. doi: 10.1002/(SICI)1097-0215(19991029)83:3<349::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 40.Sleiman SF, Langley BC, Basso M, Berlin J, Xia L, Payappilly JB, Kharel MK, Guo H, Marsh JL, Thompson LM, et al. Mithramycin is a gene-selective Sp1 inhibitor that identifies a biological intersection between cancer and neurodegeneration. J Neurosci. 2011;31(18):6858–6870. doi: 10.1523/JNEUROSCI.0710-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez-Palomo A, Meza I, Beaty G, Cereijido M. Experimental modulation of occluding junctions in a cultured transporting epithelium. J Cell Biol. 1980;87(3 Pt 1):736–745. doi: 10.1083/jcb.87.3.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez-Mariscal L, Islas S, Contreras RG, García-Villegas MR, Betanzos A, Vega J, Diaz-Quiñónez A, Martín-Orozco N, Ortiz-Navarrete V, Cereijido M, et al. Molecular characterization of the tight junction protein ZO-1 in MDCK cells. Exp Cell Res. 1999;248(1):97–109. doi: 10.1006/excr.1999.4392. [DOI] [PubMed] [Google Scholar]
- 43.Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, Zhao Y, Lu Y, Ritchie W, Grau G, Vadas MA, Gamble JR. The poly-cistronic miR-23-27-24 complexes target endothelial cell junctions: differential functional and molecular effects of miR-23a and miR-23b. Mol Ther Nucleic Acids. 2016;5(8):e354. doi: 10.1038/mtna.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu H, Xue Y, Wang P, Liu X, Ma J, Zheng J, Li Z, Li Z, Cai H, Liu Y, et al. Knockdown of long non-coding RNA XIST increases blood-tumor barrier permeability and inhibits glioma angiogenesis by targeting miR-137. Oncogenesis. 2017;6(3):e303. doi: 10.1038/oncsis.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao XR, Wang H-P, Zhang S-L, Wang M-X, Zhu Z-S. Pri-miR-124 rs531564 polymorphism and colorectal cancer risk. Sci Rep. 2015;5(1):14818. doi: 10.1038/srep14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103(3):755–766. doi: 10.1083/jcb.103.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 Kinase: a Regulator of Cell Size. Science. 1999;285(5436):2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- 49.Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998;17(22):6649–6659. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, Sonenberg N, Kelly PA, Sotiropoulos A, Pende M, et al. Atrophy of S6K1(-/-) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol. 2005;7(3):286–294. doi: 10.1038/ncb1231. [DOI] [PubMed] [Google Scholar]
- 51.Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19(18):2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ling H, et al. Role of lysosomal cathepsin activities in cell hypertrophy induced by NH4Cl in cultured renal proximal tubule cells. J Am Soc Nephrol. 1996;7(1):73–80. doi: 10.1681/ASN.V7173. [DOI] [PubMed] [Google Scholar]
- 53.Jurkovitz CT, Vamvakas S, Gekle M, Schaefer L, Teschner M, Schaefer RM, Heidland A. Influence of ammonia and pH on protein and amino acid metabolism in LLC-PK1 cells. Kidney Int. 1992;42(3):595–601. doi: 10.1038/ki.1992.323. [DOI] [PubMed] [Google Scholar]
- 54.Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk VI, Kaushik S, Klionsky DJ. In search of an “autophagomometer”. Autophagy. 2009;5(5):585–589. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- 55.Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K, Yoshida M, et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7(7):727–736. doi: 10.4161/auto.7.7.15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baas AF, Kuipers J, van der Wel NN, Batlle E, Koerten HK, Peters PJ, Clevers HC. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell. 2004;116(3):457–466. doi: 10.1016/S0092-8674(04)00114-X. [DOI] [PubMed] [Google Scholar]
- 57.Zhang L, Li J, Young LH, Caplan MJ. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc Natl Acad Sci U S A. 2006;103(46):17272–17277. doi: 10.1073/pnas.0608531103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng B, Cantley LC. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc Natl Acad Sci U S A. 2007;104(3):819–822. doi: 10.1073/pnas.0610157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amaya E, Alarcon L, Martin-Tapia D, Cuellar-Perez F, Cano-Cortina M, Ortega-Olvera JM, Cisneros B, Rodriguez AJ, Gamba G, Gonzalez-Mariscal L. Activation of the Ca(2+) sensing receptor and the PKC/WNK4 downstream signaling cascade induces incorporation of ZO-2 to tight junctions and its separation from 14-3-3. Mol Biol Cell. 2019;15;30(18):2377-2398. doi: 10.1091/mbc.E18-09-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ge T, Zhang X, Xiao Y, Wang Y, Zhang T. Novel compound heterozygote mutations of TJP2 in a Chinese child with progressive cholestatic liver disease. BMC Med Genet. 2019;20(1):18. doi: 10.1186/s12881-019-0753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deng J, Zeng L, Lai X, Li J, Liu L, Lin Q, Chen Y. Metformin protects against intestinal barrier dysfunction via AMPKα1-dependent inhibition of JNK signalling activation. J Cell Mol Med. 2018;22(1):546–557. doi: 10.1111/jcmm.13342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, Park HW, Mo J-S, Lu W, Lu S, et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun. 2015;6(1):8357. doi: 10.1038/ncomms9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Connor L, Gilmour J, Bonifer C. The role of the ubiquitously expressed transcription factor Sp1 in tissue-specific transcriptional regulation and in disease. Yale J Biol Med. 2016;89:513–525. [PMC free article] [PubMed] [Google Scholar]
- 65.Tapia R, Huerta M, Islas S, Avila-Flores A, Lopez-Bayghen E, Weiske J, Huber O, González-Mariscal L. Zona occludens-2 inhibits cyclin D1 expression and cell proliferation and exhibits changes in localization along the cell cycle. Mol Biol Cell. 2009;20(3):1102–1117. doi: 10.1091/mbc.e08-03-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goldstein AL, Palmer JE, Johnson PR. Primary cultures of fetal hepatocytes from the genetically obese Zucker rat: protein synthesis. Vitro. 1981;17(8):651–655. doi: 10.1007/BF02628400. [DOI] [PubMed] [Google Scholar]
- 67.Cleary MP, Vasselli JR, Greenwood MR. Development of obesity in Zucker obese (fafa) rat in absence of hyperphagia. Am J Physiol. 1980;238:E284–92. [DOI] [PubMed] [Google Scholar]
- 68.Witters LA. The blooming of the French lilac. J Clin Invest. 2001;108(8):1105–1107. doi: 10.1172/JCI14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51(8):2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 70.Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H, Brecher P, Ruderman NB, Cohen RA. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279(46):47898–47905. doi: 10.1074/jbc.M408149200. [DOI] [PubMed] [Google Scholar]
- 71.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229(2):558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 72.Henin N, Vincent MF, Gruber HE, Van den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995;9(7):541–546. doi: 10.1096/fasebj.9.7.7737463. [DOI] [PubMed] [Google Scholar]
- 73.Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci U S A. 2003;100(15):8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2(4):28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546(1):113–120. doi: 10.1016/S0014-5793(03)00560-X. [DOI] [PubMed] [Google Scholar]
- 76.Coven DL, Hu X, Cong L, Bergeron R, Shulman GI, Hardie DG, Young LH. Physiological role of AMP-activated protein kinase in the heart: graded activation during exercise. Am J Physiol Endocrinol Metab. 2003;285(3):E629–36. doi: 10.1152/ajpendo.00171.2003. [DOI] [PubMed] [Google Scholar]
- 77.Chen L, Wang J, You Q, He S, Meng Q, Gao J, Wu X, Shen Y, Sun Y, Wu X, et al. Activating AMPK to restore tight junction assembly in intestinal epithelium and to attenuate experimental colitis by metformin. Front Pharmacol. 2018;9:761. doi: 10.3389/fphar.2018.00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and −2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141(7):1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rahner C, Mitic LL, Anderson JM. Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology. 2001;120(2):411–422. doi: 10.1053/gast.2001.21736. [DOI] [PubMed] [Google Scholar]
- 80.Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci U S A. 1999;96(2):511–516. doi: 10.1073/pnas.96.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matsumoto K, Imasato M, Yamazaki Y, Tanaka H, Watanabe M, Eguchi H, Nagano H, Hikita H, Tatsumi T, Takehara T, et al. Claudin 2 deficiency reduces bile flow and increases susceptibility to cholesterol gallstone disease in mice. Gastroenterology. 2014;147(5):1134–45 e10. doi: 10.1053/j.gastro.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 82.Xu J, Kausalya PJ, Phua DCY, Ali SM, Hossain Z, Hunziker W. Early embryonic lethality of mice lacking ZO-2, but Not ZO-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol Cell Biol. 2008;28(5):1669–1678. doi: 10.1128/MCB.00891-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu J, Anuar F, Mohamed Ali S, Ng MY, Phua DCY, Hunziker W. Zona occludens-2 is critical for blood-testis barrier integrity and male fertility. Mol Biol Cell. 2009;20(20):4268–4277. doi: 10.1091/mbc.e08-12-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu J, Kausalya PJ, Van Hul N, Caldez MJ, Xu S, Ong AGM, Woo WL, Mohamed Ali S, Kaldis P, Hunziker W, et al. Protective functions of ZO-2/Tjp2 expressed in hepatocytes and cholangiocytes against liver injury and cholestasis. Gastroenterology. 2021;160(6):2103–2118. doi: 10.1053/j.gastro.2021.01.027. [DOI] [PubMed] [Google Scholar]
- 85.Itoh M, Terada M, Sugimoto H. The zonula occludens protein family regulates the hepatic barrier system in the murine liver. Biochim Biophys Acta Mol Basis Dis. 2021;1867(1):165994. doi: 10.1016/j.bbadis.2020.165994. [DOI] [PubMed] [Google Scholar]
- 86.Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15(17):2049–2066. doi: 10.3748/wjg.15.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dixon PH, Sambrotta M, Chambers J, Taylor-Harris P, Syngelaki A, Nicolaides K, Knisely AS, Thompson RJ, Williamson C. An expanded role for heterozygous mutations of ABCB4, ABCB11, ATP8B1, ABCC2 and TJP2 in intrahepatic cholestasis of pregnancy. Sci Rep. 2017;7(1):11823. doi: 10.1038/s41598-017-11626-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vanjak D, Moreau R, Roche-Sicot J, Soulier A, Sicot C. Intrahepatic cholestasis of pregnancy and acute fatty liver of pregnancy. An unusual but favorable association? Gastroenterology. 1991;100(4):1123–1125. doi: 10.1016/0016-5085(91)90292-S. [DOI] [PubMed] [Google Scholar]
- 89.English N, Rao J. Acute fatty liver of pregnancy with hypoglycaemia, diabetes insipidus and pancreatitis, preceded by intrahepatic cholestasis of pregnancy. BMJ Case Rep. 2015;2015(apr15 1):bcr2015209649–bcr2015209649. doi: 10.1136/bcr-2015-209649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.