

ABSTRACT
Acute coronary syndrome (ACS) is the most severe clinical manifestation of coronary heart disease.
We performed an epigenome-wide analysis of circulating CD4+ and CD8+ T cells isolated from ACS patients and healthy subjects (HS), enrolled in the DIANA clinical trial, by reduced-representation bisulphite sequencing (RRBS). In CD4+ T cells, we identified 61 differentially methylated regions (DMRs) associated with 57 annotated genes (53% hyper- and 47% hypo-methylated) by comparing ACS patients vs HS. In CD8+ T cells, we identified 613 DMRs associated with 569 annotated genes (28% hyper- and 72% hypo-methylated) in ACS patients as compared to HS. In CD4+ vs CD8+ T cells of ACS patients we identified 175 statistically significant DMRs associated with 157 annotated genes (41% hyper- and 59% hypo-methylated). From pathway analyses, we selected six differentially methylated hub genes (NFATC1, TCF7, PDGFA, PRKCB, PRKCZ, ABCA1) and assessed their expression levels by q-RT-PCR. We found an up-regulation of selected genes in ACS patients vs HS (P < 0.001). ABCA1, TCF7, PDGFA, and PRKCZ gene expression was positively associated with CK-MB serum concentrations (r = 0.75, P = 0.03; r = 0.760, P = 0.029; r = 0.72, P = 0.044; r = 0.74, P = 0.035, respectively).
This pilot study is the first single-base resolution map of DNA methylome by RRBS in CD4+ and CD8+ T cells and provides specific methylation signatures to clarify the role of aberrant methylation in ACS pathogenesis, thus supporting future research for novel epigenetic-sensitive biomarkers in the prevention and early diagnosis of this pathology.
KEYWORDS: Acute coronary syndrome, epigenetics, DNA methylation, T lymphocytes
Introduction
Acute coronary syndrome (ACS) is the most severe clinical manifestation of coronary heart disease (CHD), often leading to invalidating or fatal consequences [1,2].
Innate and adaptive immunity play an important role in the onset and progression of atherosclerosis [3,4]. Indeed, innate immunity mediates the early phases of the atherogenic process with the activation of monocytes/macrophages in the vessel wall, followed by a cascade of adaptive responses modulated by T and B lymphocytes [3,4]. The adaptive immune system has multiple proinflammatory functions, thus contributing to plaque rupture and subsequent ACS onset. In the atherosclerotic plaques both CD4+ and CD8+ T cells are present and secrete a large number of interleukins, thus exacerbating the inflammatory process [5,6]. However, in the atherosclerosis pathogenesis CD4+ T cells and subsets have a better documented role than their CD8+ T cell counterpart [7–10].
Several studies reported quantitative and functional dysregulation of CD4+ T lymphocytes in blood samples of ACS patients in correlation with incidence and severity [5,6,8,10–12]. In addition, increased levels of CD8+ T cells in the blood of CHD patients have been reported [13,14] as well as an altered maturation status, with impaired interleukin-2 production and programmed cell death-1 upregulation, in CD8+ T lymphocytes of ACS patients [15]. Moreover, in advanced atherosclerotic lesions, CD8+ T cells were abundant and outnumber as compared to CD4+ T cells [16–18]. The recent OPTICO-ACS study showed that the microenvironment of intact fibrous cap (IFC) of ACS lesions was particularly enriched of CD4+ T and CD8+ T lymphocytes with a higher amount of CD8+ T cells and related molecular effectors in thrombi of intact fibrous cap ACS culprit sites. In addition, in vitro assays of endothelial cells subjected to culture in perturbed laminar flow conditions showed an increased adhesion capacity of CD8+ T cells [19].
Genome-wide association studies identified several genetic risk loci responsible for CHD and ACS susceptibility; however, genomic variants account for only a limited percentage of the risk [20–22]. Indeed, a large part of the genetic risk remains still unexplained, thus suggesting additional mechanisms underlying CHD onset and progression [20–24]. Polymorphisms fail to account for gene–environment interactions, explaining the dynamics of epigenetic modifications in cardiovascular diseases [25]. Furthermore, other evidence showed that the reasons of the phenomenon ‘missing heritability’ could be ascribed to genome-wide and site-specific epigenetic modifications [26,27]. Epigenetic mechanisms, mainly DNA methylation, are involved in several complex and multifactorial diseases [25,28–32]. Previous studies demonstrated the association of aberrant global or gene-specific DNA methylation with cardiovascular risk factors, atherosclerosis [33,34], CHD and ACS [25,35–40]. Furthermore, several evidence have shown implication of DNA methylation variations in peripheral blood mononuclear cells (PBMNCs) as well as in different lymphocyte subpopulations both in CHD and in ACS patients [7,39–45].
To date, the diagnosis of ACS is performed through the evaluation of electrocardiogram (ECG) and serum biomarkers such as creatine kinase MB isoform (CK-MB), cardiac troponin I (cTnI) [46]. However, the sensitivity of ECGs is generally <50% and the determination of such markers has some diagnostic limitations [46]. Therefore, the identification of more sensitive and accurate biomarkers is of pivotal importance for the early ACS diagnosis.
Recently, high-throughput methylation technologies have rapidly developed, leading to the possibility to in depth-explore epigenetic changes and find novel epigenetic-sensitive tags [47]. In the current study, we evaluated genome-wide DNA methylation alterations in CD4+ and CD8+ T cells of ACS patients by reduced-representation bisulphite sequencing (RRBS), to identify differentially methylated regions (DMRs) and characterize differentially methylated genes (DMGs) as novel putative biomarkers for the early diagnosis of ACS. The flow chart of our pilot study is depicted in Figure 1.
Figure1.
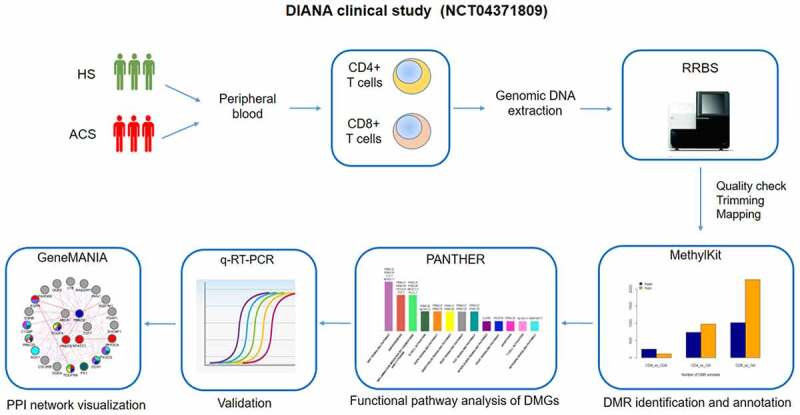
DIANA clinical study pipeline. DIANA clinical study (NCT04371809) aimed to investigate specific DNA methylation changes in circulating CD4+ and CD8+ T cells in patients with ACS as compared to HS. Genomic DNA, extracted from the two cellular subsets, was analysed through RRBS. Then, significant DMRs were identified and annotated. Functional pathway analysis was used to identify the most prevalent pathways subtending ACS. Gene expression validation of the DMGs most prevalent in disease pathways was performed by qRT-PCR. Molecular data were correlated with clinical and biochemical parameters. A biological network was built with the aim to visualize protein–protein, co-expression and functional interactions.
ACS: acute coronary syndromes; HS: healthy subjects; RRBS: reduced-representation bisulphite sequencing; DMG: differentially methylated gene; DMR: differentially methylated region; q-RT-PCR: quantitative real-time polymerase chain reaction; PPI: protein–protein interaction.
Materials and methods
Study population
The study was approved by the Institutional Ethics Committees of the University of Campania ‘L. Vanvitelli’ (prot. n. 683 of 10/10/2018) and ‘A. Cardarelli’ Hospital (prot. n. 1353 of 12/28/2018) and performed in accordance with the Helsinki Declaration. A subgroup of eight consecutive ACS patients and nine healthy subjects (HS) were selected from our ongoing DIANA clinical trial (https://www.clinicaltrials.gov/; identifier: NCT04371809). Patients with ACS including acute non-ST-elevation myocardial infarction (NSTEMI), and acute ST-elevation myocardial infarction (STEMI), were recruited at ‘A. Cardarelli’ Hospital (Naples, Italy) from March to December 2019. Diagnosis of ACS was performed on the basis of symptoms, electrocardiography findings, cardiac biomarkers, risk factors, and/or other clinical examinations [46].
Patients with congenital heart disease, cardiomyopathy, autoimmune disorders, acute infections, chronic obstructive pulmonary disease, kidney or liver diseases, hyperthyroidism, and cancers were excluded from the study to avoid confounding effects due to other clinical conditions.
Clinical and biochemical characteristics of ACS patients are reported in Supplementary Table 1.
HS were enrolled among the blood donors at the U.O.C. Divisione di Immunologia Clinica, Immunoematologia, Medicina Trasfusionale e Immunologia dei Trapianti, Dipartimento di Medicina Interna e Specialistica, AOU of University of Campania ‘L. Vanvitelli’ (Naples, Italy). HS had a normal physical examination and no acute or chronic diseases (including hyperlipidaemia, hypertension and diabetes mellitus, or any clinically evident signs of atherosclerosis) and had no drug administration during the four weeks prior to study enrolment. All study participants signed the written informed consent.
Sample collection and lymphocyte separation
Blood samples were collected from ACS patients within 2 h of the onset of clinical manifestations and before the use of medications, heparin, and contrast agents for coronary interventions. A total of 25 mL of peripheral venous blood, from patients and HS, was collected in EDTA tubes and processed within 1 h from the withdrawal. PBMNCs were isolated by Ficoll gradient using Histopaque®-1077 (Sigma-Aldrich) according to manufacturer’s instructions. CD4+ and CD8+ T lymphocytes were purified from PBMNCs using EasySep™ Human CD4 + T Cell Isolation Kit and EasySep™ and Human CD8 + T Cell Isolation Kit (STEMCELL Technologies), starting from 5x107/mL of PBMNCs, respectively [48].
DNA extraction
Genomic DNA was extracted from purified CD4+ and CD8+ T cells of each study participant immediately after cell isolation using DNeasy Blood & Tissue Kit (Qiagen) according to manufacturer’s protocol. DNA concentration and purity were determined by using NanoDrop spectrophotometer ND-2000 (Thermo Scientific) through the evaluation of the absorbance ratio A260/A280. DNA integrity was checked on 1% agarose gel.
RRB sequencing (RRBS)
Sequencing was performed at the Genomix4Life S.r.l. (Salerno, Italy). Briefly, 2 μg of genomic DNA were used for each library preparation. Each DNA sample was digested by MspI restriction enzyme. The digested products were purified with the GeneJet PCR Purification Kit (Thermo Fisher Scientific) and libraries were prepared by TruSeq Library Prep Kit (Illumina). Fragments were bisulphite converted using the EZ DNA Methylation-Gold Kit (Zymo Research). The converted DNA was amplified using PfuTurbo Cx Hotstart DNA Polymerase (Agilent Technologies, USA). The amplified fragments were purified by AMPure XP Beads and further quantified by the Agilent 4200 TapeStation (Agilent Technologies). Each DNA library was analysed by paired-end sequencing reads (2 × 75 cycles) on Illumina NextSeq 500 sequencing system.
Bioinformatic analysis and data visualization
Sequence post processing and alignment
RRBS fastq files were first assessed for quality using FastQC (v011.8, Babraham Bioinformatics) and then trimmed to remove both the adapter sequences and low-quality reads by using TrimGalore (v0.6.3, Babraham Bioinformatics) with the default settings. The setting designed for trimming RRBS data (–rrbs) in Trim Galore was also used in order to trim the first two bases from the 3ʹ end of the sequence, artificially added during RRBS library preparation. Reads were then mapped to the in silico bisulphite converted human reference genome (GRCh38) by using Bismark v.0.22.1 [49] and aligned by using Bowtie2 (v2.3.5.1) with non_directional parameter [50]. Only uniquely mapped reads were used to make cytosine methylation calls using Bismark methylation extractor and only methylation within CpG context was considered for further analysis. Raw datasets have been deposited to the NCBI Sequence Read Archive (SRA) database under accession number PRJNA704253.
Preliminary analysis on differentially methylated CpG (dmCpGs) sites
We performed preliminary analyses of differentially CpG (dmCpG) sites on a subset of two HS and six ACS patients for the following comparisons: CD4+ in ACS patients vs HS; CD8+ in ACS patients vs HS; CD4+ vs CD8+ T cells in ACS. We identified CpG using the coverage files from Bismark output. R package methylKit (v1.10.0) [51] was used to calculate CpG, methylation level and different methylation for each site with default parameters. In CD4+ and CD8+ T cells of HS and ACS patients, dmCpG sites were annotated using the HOMER tool (v4.11.10–24-2019) [52] with respect to gene regions (promoters, noncoding regions introns, exons, distal intergenic regions, 5ʹ and 3ʹ untranslated regions (5ʹ UTR and 3ʹ UTR). Then, annotation of CpG islands (CGIs) was obtained from the UCSC website (http://genome.ucsc.edu/hg38).
Identification and annotations of DMRs and methylation trend
After removing any potential batch effects, DMRs were identified for both T cell types with the R package methylKit (v1.10.0) [51] by comparing the differences of methylation in CpG sites of the same genomic regions in HS vs ACS patients. Particularly, we carried out the following comparisons: CD4+ T cells in ACS patients vs HS; CD8+ T cells in ACS patients vs HS; CD4+ vs CD8+ T cells in ACS. To prevent PCR bias and increase the power of the statistical test, CpG sites covering less than 10 reads or more than the 99.9th percentile of coverage distribution in each sample were filtered out. Coverage values between samples were normalized as by default and read coverage per base was calculated (Supplementary Figures 1–4). We have applied a 500 bp sliding window approach to detect DMRs, defined as those regions with more than 20% methylation differences (|ΔM|) and q-value <0.05, after applying logistic regression by using the SLIM method to correct the p-value for multiple hypothesis testing.
The R package ChIPseeker (v1.20.0) [53] was used to annotate DMRs with respect to gene regions (promoters, coding sequences, introns, distal intergenic regions, 5ʹ untranslated regions (5ʹUTR) and 3ʹ UTR) in CD4+ and CD8+ T cells from HS and ACS patients. Then, annotation of CGIs was obtained from the UCSC website (http://genome.ucsc.edu/hg38).
PANTHER functional analyses
Gene ontology and pathway analyses of DMR-related genes, located in the promoter regions, were performed by PANTHER web server (Version 15.0; http://www.pantherdb.org) [54] for the following comparisons: CD8+ T cells in ACS patients vs HS; CD4+ vs CD8+ T cells in ACS patients. We carried out gene ontology on overall annotated promoter-transcription start site (TSS) DMRs.
Expression profiling public dataset
We retrieved the expression profiling of ACS patients compared with control groups from Silbiger et al. [55] whose raw data are available at GEO (http://www.ncbi.nlm.nih.gov/geo) (GSE29532). We considered only differentially expressed genes (DEGs) with statistical significance at threshold FDR ≤0.05.
Protein–protein interaction network analysis
Since network medicine is emerging as a useful clinical approach in cardiovascular disease phenotyping [28,56,57], we built an interaction network by GeneMANIA cytoscape plugin [58] by using the most prevalent DMGs and the highest differential methylation values emerged by PANTHER analysis. Physical and co-expression interactions were selected and enrichment analysis (FDR ≤ 0.05) based on Gene Ontology (GO) Biological Process was performed.
RNA extraction and quantitative real-time PCR assay
Total RNA was extracted from PBMNCs of ACS patients and HS using TRIzol solution (Thermo Fischer Scientific), according to the manufacturer’s instructions. RNA quantity and quality were determined using a NanoDropND-1000 spectrophotometer (Thermo Fischer Scientific). RNA (500 ng) was reverse transcribed with SuperScript® III First-Strand Synthesis System for RT-PCR (Thermo Fischer Scientific) according to the manufacturer’s protocol in 20 μL reaction. The relative expression levels of mRNA were measured by CFX96 Touch Real-Time PCR Detection System (BioRad Laboratories, Ltd) using iQ™ SYBR® Green Supermix (BioRad Laboratories, Ltd) and 300 nM each primer pair. Primers were designed by Primer 3 software (http://bioinfo.ut.ee/primer3-0.4.0/) and synthesized by Life Technologies. The specificity of each oligonucleotide pair was checked with the BLAST program. Target gene expression levels were normalized using RPS18 as housekeeping gene [59]. Melt curve analysis was performed to verify a single product species. Each sample was analysed in triplicate and relative expression was assessed using the 2−ΔΔCt calculation method. Oligonucleotide sequences are reported in Supplementary Table 2.
Statistical analysis
All statistical analysis were performed using R (version 3.6.2) (www.cran.r-project.org). The Shapiro–Wilk normality test was used to assess the normality of data. The F-test was applied to evaluate the homogeneity of variance. Continuous variables were expressed as mean ± standard deviation. Unpaired Student’s t-test was used for comparison between the two groups. Categorical variables were expressed as percentage. Correlation analysis (Pearson Product–Moment Correlation Coefficient) was used to evaluate possible associations between both DNA methylation and gene expression levels with biochemical clinical parameters. A P value <0.05 was considered statistically significant.
Results
Preliminary analysis of dmCpG sites
From RRBS data, we performed a preliminary analysis to assess the feasibility to carry on an epigenome-wide analysis in CD4+ and CD8+ T cells of ACS patients, in order to identify dmCpGs annotated to genes potentially involved in ACS.
We considered six ACS patients and two HS and first quantified the amount of dmCpG sites for CD4+ and CD8+ T cells by comparing cases and controls as well as CD4+ vs CD8+ T cells in ACS condition (Supplementary Figure 5).
We found that about 70% of dmCpG sites were protein coding genes for all the comparisons. These preliminary results led us to hypothesize that specific gene regions may be more suitable to DNA methylation and may have potential regulatory roles in ACS, pointing out the importance of better characterizing DMRs.
Genomic map of differential methylation
To gain more insight into ACS-related DMRs, we carried out the genome-wide DNA methylation profiles of circulating CD4+ T and CD8+ T cells in ACS patients compared to HS, by enlarging the cohort of analysis and applying stringent bioinformatic criteria for quality improvement as described in Materials and Methods. This allows us for the DMR extraction with higher coverage and confidence level. DMR analysis was performed on eight ACS patients and seven HS.
The statistics of the libraries for each sample is reported in Supplementary Table 3. On average, across the 15 samples investigated for CD4+ T cells, 20,390,274 reads were obtained, 20,193,671 reads were retained after quality filtering, 12,050,919 reads were uniquely mapped to the reference genome with 59% mapping efficiency and the proportion of C methylated in a CpG context was 38%.
On average, across the 15 samples investigated for CD8+ T cells, 19,904,902 reads were obtained, 19,701,617 reads were retained after quality filtering, 11,690,197 reads were uniquely mapped to the reference genome with 59% mapping efficiency and the proportion of C methylated in a CpG context was 37%.
Since DNA methylation changes occurring in promoter regions, are mainly involved in the gene expression regulation [60], we focused on the inspection of DMRs located only in gene promoters.
Analysis of DMRs in CD4+ T cells of ACS patients vs HS
In CD4+ T cells of ACS patients vs HS we identified 61 DMRs (|ΔM| > 20 and q-value <0.05) associated with 57 annotated genes of which 53% (n = 32) were hyper- and 47% (n = 29) hypo-methylated in ACS patients vs HS (Supplementary Figure 6a). Among the identified DMRs, 35% (n = 21) were located within annotated gene promoters, defined as ±3 kb to the transcription start site (TSS) (Supplementary Table 4, Supplementary Figure 6b).
Analysis of DMRs in CD8+ T cells of ACS patients vs HS
In CD8+ T cells of ACS patients vs HS we retrieved 613 DMRs (|ΔM| >20 and q-value < 0.05) associated with 569 annotated genes of which 28% (n = 173) were hyper- and 72% (n = 440) hypo-methylated in ACS patients as compared to HS (Supplementary Figure 6c). Among the DMRs, located within annotated gene promoters, 18% (n = 111) were at a distance ≤1 kb from TSS, whereas 11% (n = 71) at a distance ranging between 1 and 3 kb with respect to TSS (Supplementary Table 5; Supplementary Figure 6d).
Analysis of DMRs in CD4+ vs CD8+ T cells of ACS patients
In CD4+ vs CD8+ T cells of ACS patients we identified 175 DMRs (|ΔM| >20 and q-value <0.05) associated to 157 annotated genes of which 41% (n = 72) were hyper- and 59% (n = 103) were hypo-methylated (Supplementary Figure 6e). Among the identified DMRs, located within annotated gene promoters, 29% (n = 51) were located within annotated gene promoters at a distance ≤1 kb from TSS and 14% (n = 25) were located at a distance ranging between 1 and 3 kb respect to TSS (Supplementary Table 6; Supplementary figure 6f).
Pathway functional analysis in ACS patients
Functional analysis was performed by using PANTHER web server on overall DMGs and both hypo- and hyper-methylated DMGs, respectively. We evaluated the functional characteristics and putative pathways involved in atherosclerosis and ACS pathogenesis. In particular, we selected pathways related to inflammation, angiogenesis, B and T cell activation, blood coagulation, endothelin, vascular endothelial growth factor (VEGF), epidermal growth factor receptor (EGFR) and fibroblast growth factor (FGF), integrin, interleukin and Wnt signalling pathways. GO molecular function results of DMGs in CD8+ T cells of ACS patients as compared to HS showed that binding protein and catalytic activity classes were the most prominent, followed by molecular function regulator, molecular transducer activity, transporter activity, structural molecule activity, transcription regulator activity and transporter activity processes (Figure 2).
Figure 2.
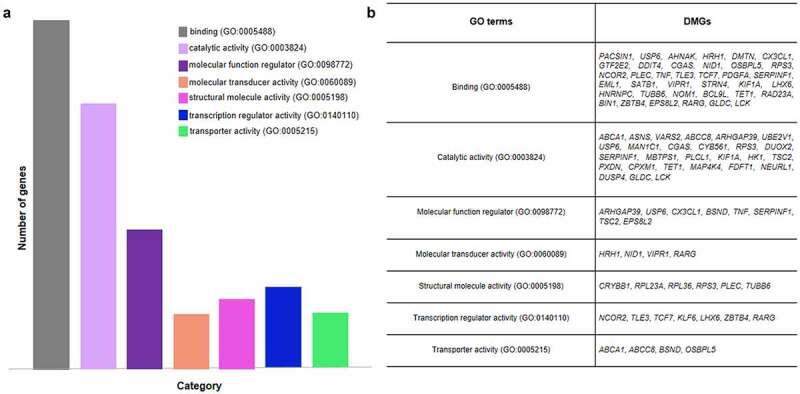
GO molecular function analysis of DMGs in CD8+ T cells of ACS patients vs HS. (a) Bar graph depicts the relative abundance of DMGs for each category. (b) The table shows the specific DMGs for each GO term. Binding, catalytic activity and transcription regulator activity are the most prevalent function of genes in the analysis.
The inspection of pathways of interest provided that in CD8+ T cells of ACS patients the highest number of DMGs were included in angiogenesis, adhesion molecule signalling and Wnt signalling pathways, thus suggesting a potential regulatory role in aberrant activation of CD8+ T cells in ACS (Figure 3). Specifically, a total of 18 genes were found, of which 8 were hypo-methylated (DOK2, PDGFA, TNF, CX3CL1, COL27A1, ACTN4, COL4A3, and CSF2RB) and 10 hyper-methylated (TCF7, MAP4K4, CELSR3, PCDHA8, FDFT1, PLCL1, DUSP4, CD247, TLE3, and LCK). Different genes such as TCF7, CELSR3, PCDHA8, and PDGFA were present in multiple pathways (Supplementary Figure 7).
Figure 3.
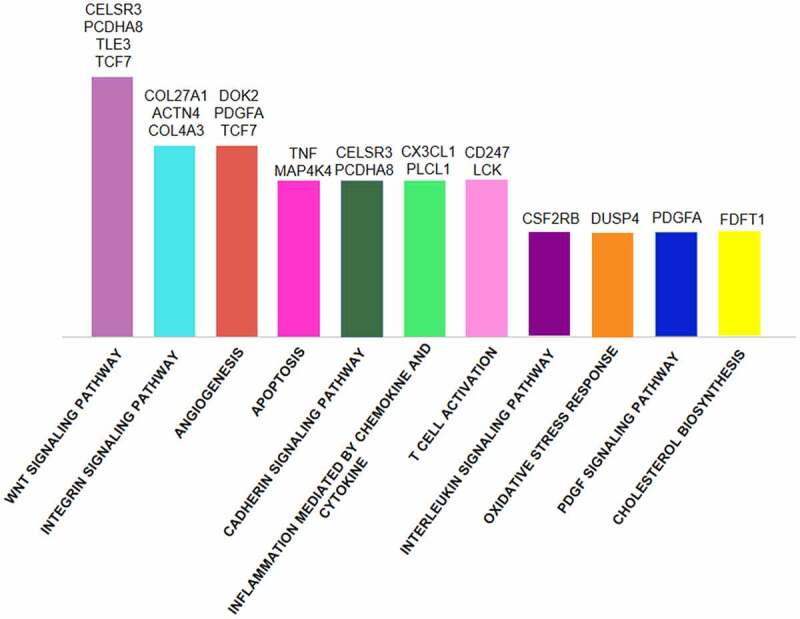
GO functional pathway analysis of overall DMGs in CD8+ T cells of ACS patients as compared to HS. The Wnt, integrin, and cadherin signalling pathways, angiogenesis, inflammation, T cell activation showed the major number of DMGs. In addition, several genes are shared by different pathways.
Molecular function analysis of DMGs in CD4+ vs CD8+ T cells of ACS patients showed that binding protein and catalytic activity classes were the most representative (Figure 4). Furthermore, the investigation of putative pathways involved in atherosclerosis and ACS highlighted 13 pathways of whom Wnt signalling, inflammation and angiogenesis were the most significant (Figure 5). In particular, analysis provided eight DMGs [3 hypo-methylated (PRKCB, PRKCZ, and PDGFA) and five hyper-methylated (TCF7, NFATC1, PLCL1, RAPGEF1, and IL2RB)]. PRKCZ, PRKCB, TCF7, NFATC1, and PDGFA were shared genes by different functional pathways (Supplementary Figure 8).
Figure 4.
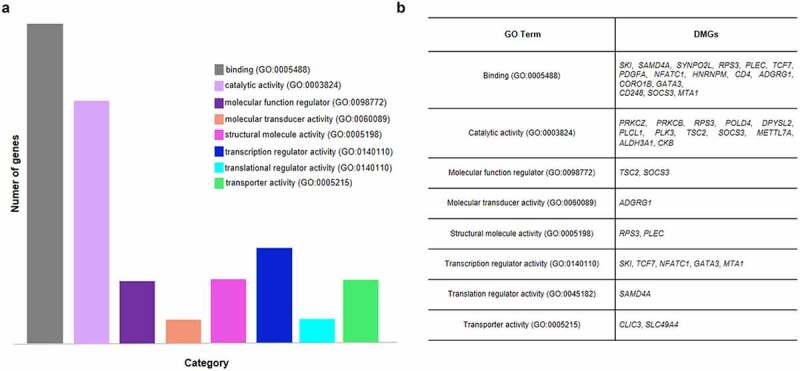
GO molecular function analysis of DMGs in CD4+ vs CD8+ T cells of ACS patients. (a) Bar graph depicts the relative abundance of DMGs for each category. (b) The table shows the specific DMGs for each GO term. Binding and catalytic activity classes are the most represented.
Figure 5.
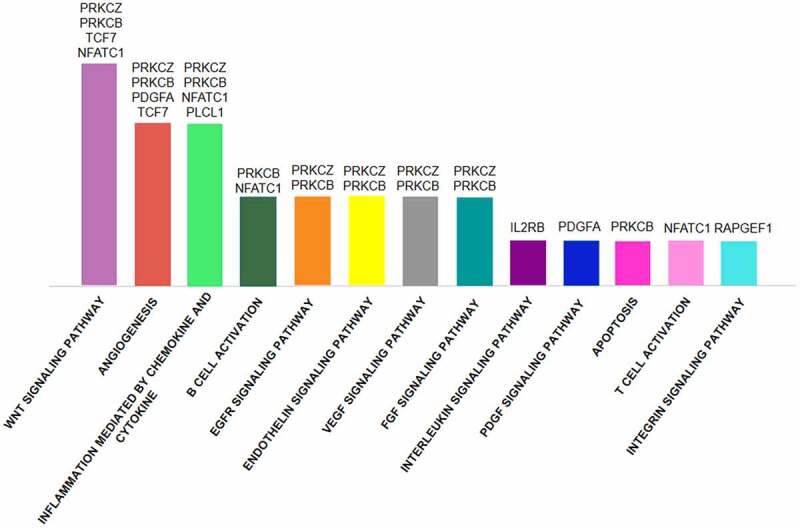
GO enrichment pathway analysis of overall DMGs in CD4+ vs CD8+ T cells of ACS patients. The Wnt signalling pathway, angiogenesis, inflammation, B cell activation, EGFR, VEGF, endothelin and FGF pathways are the most enriched and several genes are common to different pathways.
Integration of methylation and gene expression data
We integrated our methylome analysis with transcriptome data obtained from the public expression dataset GSE29532 [55]. Differentially expressed genes (DEGs) by microarray experiments in ACS patients as compared to control group were used to investigate potential aberrantly methylated regions in CD4+ and CD8+ T cells of our study population. Total identified DEGs were globally 547 unique genes.
In CD8+ T cells, the overlapping between DEGs and DMGs resulted in 13 statistically significant genes (FDR <0.05) (Supplementary Table 7). None of the DEGs at threshold of FDR <0.05 significance, overlapped with CD4+ T cells of ACS patients. Among these significant DEGs in CD8+ T cells of ACS patients, five genes (ABCA1, CDK5R1, HRH1, GFI1, and CD9) had the corresponding DMRs within the promoter region. All the genes showed a positive correlation between methylation and expression. In particular, ABCA1 and CDK5R1 were hyper-methylated/up-regulated whereas HRH1, GFI1, and CD9 were hypo-methylated/down-regulated. Although these results are outside the concept that DNA methylation is commonly associated to gene expression reduction, positive associations of DNA methylation and gene expression have also been found in hyper-methylated bivalent promoters, thus showing increased gene expression levels [61].
Gene expression profiles in PBMNCs of ACS patients
For validation experiments, we selected the top five most recurrent DMGs in ACS-associated pathways endowed with the highest differential methylation values. Specifically, we considered the following genes: NFATC1, TCF7, PDGFA, PRKCB, and PRKCZ and determined their respective mRNA levels by q-RT-PCR in PBMNCs, as more accessible biological samples, of ACS patients and HS. Interestingly, NFATC1 and TCF7 genes showed five and two hyper-methylated DMRs in ACS patients, respectively. Furthermore, PDGFA showed the highest value of differential hypo-methylation(Supplementary Figure 8).(Figure 6a-b).
Figure 6.
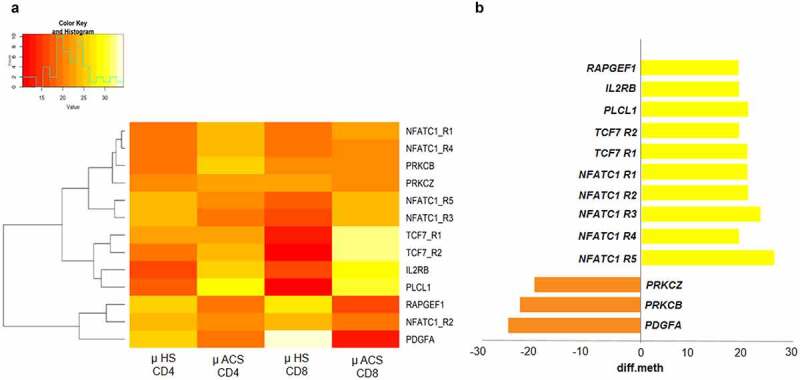
Differential of methylation in significant DMGs of ACS patients. (a) Clustering heatmap showing the mean methylation levels of the DMR associated genes in CD4+ vs CD8+ T cells in ACS patients. The colour indicates DNA methylation levels where red represents hypomethylation and yellow represents hypermethylation. (b) RRBS results show five hypermethylated DMRs in NFATC1 gene while in TCF7 gene 2 hypermethylated. In addition, PDGFA shows the highest hypomethylation value.
In addition, the analysis of external DEG dataset by Silbiger et al. [55] provided a differential expression of ABCA1 gene in PBMNCs of ACS patients vs HS (Supplementary Table 7) while our RRBS data highlighted a significant DMR located in the promoter of ABCA1 in CD8+ T cells of ACS patients from our study population. Thus, we also investigated gene expression levels of ABCA1 in our study population, since this gene was demonstrated to be altered in CHD, as our previous study reported [41].
Relative expression data showed a significant up-regulation of NFATC1 (P = 0.0002), TCF7 (P = 0.0003), PDGFA (P = 0.008), PRKCB (P = 0.0018), PRKCZ (P = 0.004), and ABCA1 (P = 0.003) of mRNA levels in patients with ACS as compared to HS (Figure 7).
Figure 7.
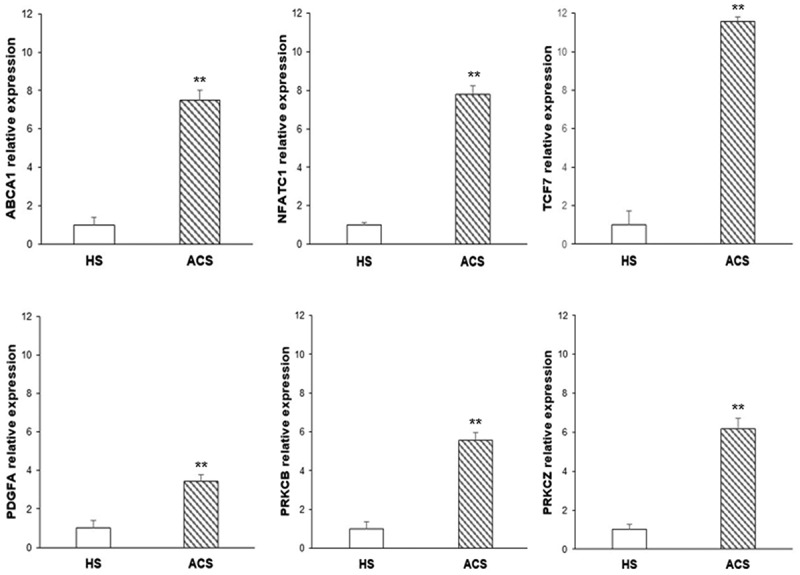
Gene expression profile evaluation. Bar graphs illustrate the relative expression of ABCA1, NFATC1, TCF7, PDGFA, PRKCB, and PRKCZ genes in PBMNCs of ACS patients as compared to HS (**P < 0.01).
Correlation analysis between DNA methylation, gene expression and clinical data
We investigated whether promoter methylation in CD4+ and CD8+ T cells and gene expression levels in PBMNCs of the six validated DMGs were individually associated with clinical parameters in ACS patients. We found meaningful cell-specific correlation signatures. In CD4+ T cells of ACS patients, concerning correlation DNA methylation/mRNA expression, we found a negative correlation between PDGFA methylation and its gene expression (r = −0.72; P = 0.044) and a positive correlation between NFATC1 methylation (r = 0.80; P = 0.0186) and its mRNA levels in PBMNCs. Moreover, we observed a positive correlation between the methylation of TCF7 and NFATC1 gene expression (r = 0.78; P = 0.022). In addition, we observed significant positive correlation between HDL cholesterol levels and PRKCZ promoter methylation (r = 0.82; P = 0.044). PRKCB promoter methylation was negatively correlated with glycaemia (r = −0.80; P = 0.015) and positively correlated with systolic blood pressure (SBP) (r = 0.84; P = 0.009). PDGFA methylation was negatively correlated with CK-MB concentrations (r = −0.79; P = 0.018) and with the gene expression of PRKCZ (r = −0.73; P = 0.039) and ABCA1 (r = −0.84; P = 0.008) (Figure 8a).
Figure 8.
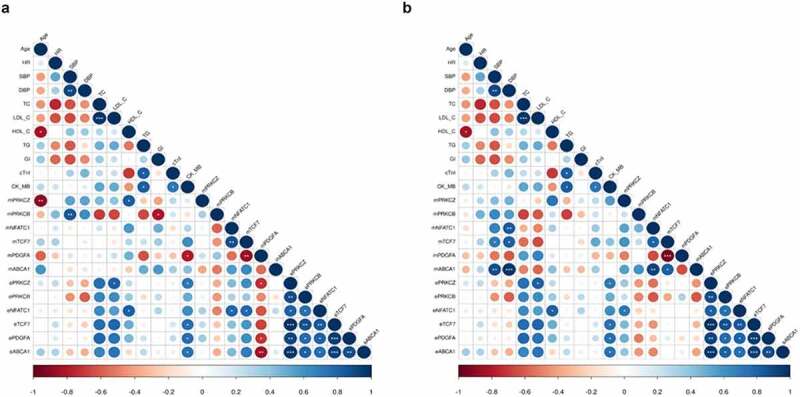
Correlation plots. Pearson’s correlation between (a) CD4+ T and (b) CD8+ T cell promoter methylation and total PBMNCs gene expression levels of the selected DMGs with clinical and biochemical parameters of ACS patients. The circle size is scaled by the correlation coefficient. Blue and red colours designate positive and negative correlations, respectively (*P < 0.05; **P < 0.01; ***P < 0.001).
ABCA1: ATP Binding Cassette Subfamily A Member 1; CK_MB: creatine kinase MB isoenzyme; cTnI: cardiac troponin I; DBP: diastolic blood pressure; Gl: glycaemia; HDL_C: high-density lipoprotein cholesterol; HR: heart rate; LDL_C: low-density lipoprotein cholesterol; NFATC1: nuclear factor of activated T cells 1; PRKCB: protein kinase C beta; PRKCZ: protein kinase C zeta; TCF7: Transcription Factor 7; SBP: systolic blood pressure; TC: total cholesterol; TG: triglycerides.
In CD8+ T cells of ACS patients promoter methylation of TCF7 and ABCA1 were positively correlated with SBP (r = 0.71, P = 0.05; r = 0.88, P = 0.003, respectively) and diastolic blood pressure (DBP) (r = 0.82, P = 0.013; r = 0.97, P < 0.001, respectively), while NFACT1 methylation was only correlated to DBP (r = 0.72; P = 0.004). Additionally, ABCA1 methylation levels showed positive correlation links with TCF7 (r = 0.82; P = 0.012) and NFATC1 (r = 0.76; P = 0.027) methylation. Finally, methylation of PRKCZ induced a potential significant transcription of NFATC1 (r = 0.72; P = 0.043) (Figure 8b).
On the other hand, statistical data showed common correlations in both T cell subsets concerning TCF7 methylation and PDGFA methylation (r = −0.87, P = 0.005 in CD4+ T cells; r = −0.94, P = 0.0004 in CD8+ T cells) and NFATC1 methylation (r = 0.78, P = 0.021 in CD4+ T cells; r = 0.72,P = 0.042 in CD8+ T cells) (Figures 8a and 8b).
Correlation analysis between gene expression in PBMNCs of ACS patients and clinical data provided positive correlations between LDL cholesterol levels and PRKCZ gene expression (r = 0.82; P = 0.046) and HDL cholesterol level and NFACT1 gene expression (r = 0.81; P = 0.048). In addition, TCF7, PDGFA, PRKCZ, and ABCA1 mRNA levels were positively correlated to CK-MB concentrations (r = 0.75, P = 0.03; r = 0.760, P = 0.029; r = 0.72, P = 0.044; r = 0.74, P = 0.035, respectively) (Figures 8a and 8b).
Protein–protein interaction network analysis
We query GeneMANIA tool [58] to predict protein–protein and co-expression interactions, and functions of NFATC1, TCF7, PRKCB, PRKCZ, PDGFA, and ABCA1 genes. Data output provided a network with 26 nodes and 138 edges. The top pathways related to the network were associated with immune response regulating cell surface receptor signalling pathway, inflammatory response, regulation of lipid metabolic process, regulation of cell adhesion, blood coagulation, protein kinase signalling, regulation of cell activation and leukocyte migration (Figure 9).
Figure 9.
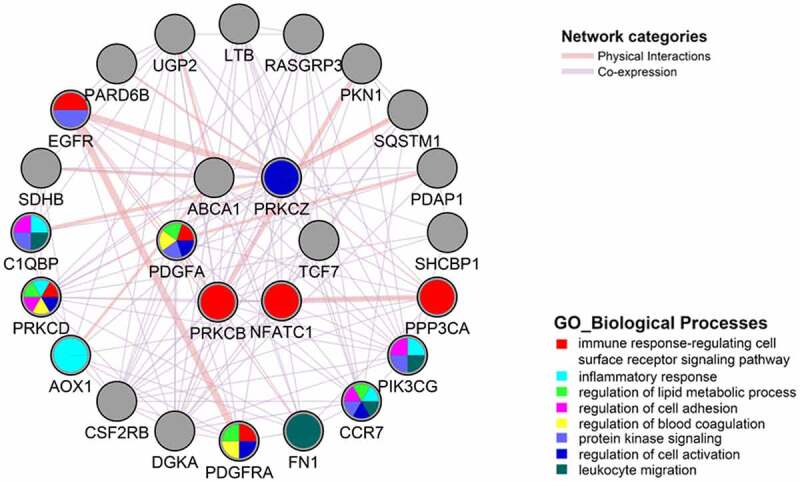
Gene interaction network constructed with GeneMANIA for the six selected DMGs. GeneMANIA PPI and co-expression network of NFATC1, TCF7, ABCA1, PRKCB, PRKCZ, PDGFA, and ABCA1 genes predicts 26 nodes and a total of 138 links representing interactions mainly to immune response regulating cell surface receptor signalling pathway, inflammatory response, regulation of lipid metabolic process, regulation of cell adhesion, blood coagulation, protein kinase signalling, regulation of cell activation and leukocyte migration The colour of the line that connects the genes depicts the interaction type (see legend) and the edge thickness depicts the interaction strength.
Discussion
Our study aims to provide a DNA methylome signature in circulating CD4+ and CD8+ T lymphocytes from patients with ACS. This is the first study that investigates DNA methylation in ACS by RRBS approach. Moreover, we also identify ABCA1, NFATC1, TCF7, PRKCB, PRKCZ, and PDGFA genes as important sensitive-epigenetic tags in ACS.
Here, we first hypothesized cell-specific methylation alterations in CD4+ and CD8+ T cells of patients with ACS and then, for a possible clinical relapse, validated the results obtained in PBMNCs, such as leveraging a more readily obtainable biological source. Notable, mRNA levels of these genes were significantly up-regulated in ACS patients vs HS. Moreover, a further analysis showed both CD4+ and CD8+ T cell-specific correlation trends with clinical variables. Indeed, we found that PRKCZ gene expression correlated with LDL cholesterol, while NFACT1 gene expression with HDL cholesterol. In addition, TCF7, PDGFA, PRKCZ, and ABCA1 transcripts were positively correlated to circulating CK-MB concentrations.
Several evidence reported the accumulation of cytotoxic CD8+ T cells in advanced plaque as well as functional alterations of these cells in ACS patients [16–19]. Furthermore, aberrant gene-specific methylation in this T cell subset was strongly associated with cardiovascular risk factors and ACS onset [42].
In our study, top ACS-related methylation alterations in CD8+ T cells indicated a prevalence in functions like cellular adhesion, infiltration, and cell activation, suggesting that CD8+ T cells might migrate into severe atherosclerotic lesions and activate pro-inflammatory signalling pathway. In addition, our results provide that functions of CD8+ T cells may be regulated through DNA methylation. A previous study by Li et al. analysed the genome-wide aberrant DNA methylation patterns associated with ACS in blood cells by microarray [42]. Our results are in accordance with previous findings that reported a novel and reproducible association of blood methylation at 47 CpG sites in ACS patients with predominant contributions of CD8+ T cells [42]. In particular, we found in CD8+ T lymphocytes of ACS patients vs HS a match between three significant CpG sites and three DMRs, two annotated in intronic regions of PRDM16 and ZFYVE28 genes and one into the 5ʹUTR of ANKRD11 gene (Supplementary Table 5).
ATP-binding cassette transporter 1 (ABCA1) is a transporter essential for the reverse cholesterol efflux from peripheral cells and foam cells [62]. Literature data reported an association between high DNA methylation of ABCA1 promoter [63–65] and CHD risk and alterations of this transporter at mRNA level [66–69]. Indeed, ABCA1 was demonstrated to be involved in the onset of atherosclerotic plaques and compositions through an up-regulation of its transcript [68], as well as the significance of this biomarker in plaque rupture [69]. Furthermore, our recent study reported that ABCA1 mRNA was up-regulated in peripheral blood of CHD patients with non-calcified plaque [41]. RRBS data highlighted a significant hypermethylated DMR in ABCA1 promoter in CD8+ T cells of ACS patients; furthermore, gene expression analysis on PBMNCs showed a significant up-regulation of ABCA1 transcript in ACS patients, thus confirming the involvement of this protein also for high-risk plaque detection.
Nuclear factor of activated T cells 1 (NFATC1) and transcription factor 7 (TCF7) are transcription factors regulating the activation, proliferation, differentiation of T lymphocytes, and inducible expression of cytokine genes [70,71]. NFATC1 is the most expressed isoform in vascular smooth muscle cells (VSMCs) [72]. NFATC1 is involved in inflammation, vascular calcification, VSMC differentiation and proliferation [73,74]. Several studies reported that expression of NFATC1 and the activation of its downstream signalling pathways might play a critical role in phenotype transformation of VSMCs and atherosclerotic plaque formation [72,75]. A recent study showed that NFATC1 is involved in IL-33 gene expression, a cytokine highly expressed in atherosclerotic plaques and involved in the switch towards the inflammatory phenotype of endothelium and during vascular wall remodelling [76]. In addition, the expression of NFATC1 also significantly contributes to vulnerable plaque formation [77]. TCF7 gene encodes for the TCF1 protein TCF1 and activates transcription through the Wnt/beta-catenin signalling pathway. In the endothelium, the activation of βcat/TCF signalling was found both in early phases of atherogenesis and during lesion advancement, by enhancing inflammation and the transcription of critical genes involved in atherogenic endothelial phenotype [78]. In addition, β-catenin/TCF signalling was reported to be an important pathway for vascular remodelling through VSMC growth and survival stimulation [79]. Given these evidence, our results suggest a mirroring of the molecular pathological processes in circulating cells and in vascular tissue in ACS.
Although originally purified from platelets, platelet-derived growth factor (PDGF) expression can be induced in normal artery wall and in inflammatory cells infiltrating the artery in response to proatherogenetic stimuli. PDGF is a potent inducer of VSMC switch from the contractile phenotype to a proliferative and secretory phenotype [73]. In particular, in endothelial cells hyperhomocysteinemia upregulates PDGF-A mRNA levels through the demethylation of its promoter, thus leading to VSMC activation [80]. In addition, PDGF A is involved in the thickening of the plaque fibrous cap and in the detaching of thrombi from the fat-rich core matrix [81]. Indeed, high expression of mRNA levels of PDGF-A were found in occlusive atherosclerotic vessels [81]. Furthermore, findings provided the involvement of PDGF-A in ACS with an increase of both PBMNC derived secretion and serum concentrations in patients, especially in local coronary circulation [82,83]. In agreement with literature, we report a hypo-methylation of PDGFA in CD8+ T cells of ACS patients, as well as an over-expression of its mRNA in PBMNCs of such patients in correlation with CK-MB concentrations, thus suggesting a further mirroring of molecular processes underlying vascular remodelling in ACS.
Protein kinase C (PRKC) family isoforms are involved in several signal transduction pathways, and associated with multiple aspects of atherosclerosis onset and progression [74]. In particular, evidence showed that PRKC isoforms such as PRKCβ and PRKCζ regulate ECs, VSMCs, and immune system cell functions [84]. Furthermore, PRKCζ isoform is relevant for regulating CD8+ asymmetric T cell division and subsequent cell fate [85]. Our data confirm and highlight a potential specific role of CD8+ T cell dysregulation in the development of ACS.
However, our study is characterized by limitations and strengths. We selected RRBS technique for DNA methylation profiling for its ability to provide a quantitative assay of methylation of more than 1 million CpG sites at single-nucleotide resolution even when DNA sample concentration is relatively limited [86]. Indeed, RRBS can cover a large part of CpG sites across the genome including promoters, CpG shores, enhancers, exons, 5ʹand 3ʹ untranslated regions and repetitive elements as compared to array-based strategies, which utilize targeting defined regions [86,87]. In addition, RRBS is superior to detect CpGs in the relative islands and promoter regions [86]. Another strength of our investigation is the utilization of PBMNCs for the validation of DMGs since this cell population, as compared to CD4+ and CD8+ T subpopulations, is more easily obtainable following a whole-blood sampling. Finally, the population has a small sample size; thus, our findings should be validated in a larger cohort to obtain more robust associations. In addition, further studies are needed to evaluate other epigenetic modifiers since additional post-transcriptional events may influence mRNA expression levels. Future perspectives are oriented to investigate both upstream and downstream molecular interactors within the network to discover other potential candidate genes and pathways that could be differentially regulated in ACS.
Supplementary Material
Funding Statement
This work was supported by progetto Progetto di Rilevante Interesse Nazionale 2017 (2017F8ZB89), funded by Italian Ministry of Research (Napoli C.); Ricerca Corrente 2019 from ‘Italian Ministry of Health’ (Napoli C.); progetto Giovani Ricercatori (GR-2016-02364785) from ‘Italian Ministry of Health’ (Grimaldi V.);Research Competitive Grant ‘VALERE: Vanvitelli Project 2020’ funded by Italian Ministry of Research (Schiano C.); Italian Association for Cancer Research (grant number IG-23068) (Weisz A.); Regione Campania (grant GENOMAeSALUTE, POR Campania FESR 2014/2020, azione 1.5; CUP: B41C17000080007) (Weisz A.). Infante T. is a PhD student of Translational Medicine supported by Educational Grant from the University of Campania ‘Luigi Vanvitelli,’ Naples, Italy. The abstract of this article was accepted as e-poster presentation at ESC Congress 2021.
Supplementary material
Supplemental data for this article can be accessed here.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the author(s).
References
- [1].Sanchis-Gomar F, Perez-Quilis C, Leischik R, et al. Epidemiology of coronary heart disease and acute coronary syndrome. Ann Transl Med. 2016;4(13):256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].GBD Causes of death collaborators . Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the global burden of disease study 2016. Lancet. 2016;2017(390):1151–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Abdolmaleki F, Gheibi Hayat SM, Bianconi V, et al. Atherosclerosis and immunity: a perspective. Trends Cardiovasc Med. 2019;29(6):363–371. [DOI] [PubMed] [Google Scholar]
- [4].Flego D, Liuzzo G, Weyand CM, et al. Adaptive immunity dysregulation in acute coronary syndromes: from cellular and molecular basis to clinical implications. J Am Coll Cardiol. 2016;68(19):2107–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Saigusa R, Winkels H, Ley K.. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;17:387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang H, Liu Z, Shao J, et al. Immune and inflammation in acute coronary syndrome: molecular mechanisms and therapeutic implications. J Immunol Res. 2020;2020:4904217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhu L, Jia L, Liu Z, et al. Elevated methylation of FOXP3 (forkhead box P3)-TSDR (regulatory T-cell–specific demethylated region) is associated with increased risk for adverse outcomes in patients with acute coronary syndrome. Hypertension. 2019;74(3):581–589. [DOI] [PubMed] [Google Scholar]
- [8].Ruggio A, Pedicino D, Flego D, et al. Correlation between CD4+CD28null T lymphocytes, regulatory T cells and plaque rupture: an optical coherence tomography study in acute coronary syndromes. Int J Cardiol. 2019;276:289–292. [DOI] [PubMed] [Google Scholar]
- [9].Jiang L, Chen F, Hu X, et al. Decreased helios expression in regulatory T cells in acute coronary syndrome. Dis Markers. 2017;2017:7909407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hasib L, Lundberg AK, Zachrisson H, et al. Functional and homeostatic defects of regulatory T cells in patients with coronary artery disease. J Intern Med. 2016;279(1):63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Angelini G, Flego D, Vinci R, et al. Matrix metalloproteinase-9 might affect adaptive immunity in non-ST segment elevation acute coronary syndromes by increasing CD31 cleavage on CD4+ T-cells. Eur Heart J. 2018;39(13):1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Flego D, Severino A, Trotta F, et al. Increased PTPN22 expression and defective CREB activation impair regulatory T-cell differentiation in non-ST-segment elevation acute coronary syndromes. J Am Coll Cardiol. 2015;65(12):1175–1186. [DOI] [PubMed] [Google Scholar]
- [13].Bergström I, Backteman K, Lundberg A, et al. Persistent accumulation of interferon-γ-producing CD8+CD56+ T cells in blood from patients with coronary artery disease. Atherosclerosis. 2012;224(2):515–520. [DOI] [PubMed] [Google Scholar]
- [14].Hwang Y, Yu HT, Kim D-H, et al. Expansion of CD8+ T cells lacking the IL-6 receptor α chain in patients with coronary artery diseases (CAD). Atherosclerosis. 2016;249:44–51. [DOI] [PubMed] [Google Scholar]
- [15].Zidar DA, Mudd JC, Juchnowski S, et al. Altered maturation status and possible immune exhaustion of CD8 T lymphocytes in the peripheral blood of patients presenting with acute coronary syndromes. Arterioscler Thromb Vasc Biol. 2016;36(2):389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fernandez DM, Rahman AH, Fernandez NF, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25(10):1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Grivel JC, Ivanova O, Pinegina N, et al. Activation of T lymphocytes in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2011;31(12):2929–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gewaltig J, Kummer M, Koella C, et al. Requirements for CD8 T-cell migration into the human arterial wall. Hum Pathol. 2008;39(12):1756–1762. [DOI] [PubMed] [Google Scholar]
- [19].Leistner DM, Kränkel N, Meteva D, et al. Differential immunological signature at the culprit site distinguishes acute coronary syndrome with intact from acute coronary syndrome with ruptured fibrous cap: results from the prospective translational OPTICO-ACS study. Eur Heart J. 2020;41(37):3549–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Erdmann J, Kessler T, Munoz Venegas L, et al. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018;114(9):1241–1257. [DOI] [PubMed] [Google Scholar]
- [21].Clarke SL, Assimes TL.. Genome-wide association studies of coronary artery disease: recent progress and challenges ahead. Curr Atheroscler Rep. 2018;20(9):47. [DOI] [PubMed] [Google Scholar]
- [22].Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Musunuru K, Kathiresan S. Genetics of common, complex coronary artery disease. Cell. 2019;177(1):132–145. [DOI] [PubMed] [Google Scholar]
- [24].van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122(3):433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Aavik E, Babu M, Ylä-Herttuala S. DNA methylation processes in atheosclerotic plaque. Atherosclerosis. 2019;281:168–179. [DOI] [PubMed] [Google Scholar]
- [26].Nikpay M, Stewart AFR, McPherson R. Partitioning the heritability of coronary artery disease highlights the importance of immune-mediated processes and epigenetic sites associated with transcriptional activity. Cardiovasc Res. 2017;113(8):973–983. [DOI] [PubMed] [Google Scholar]
- [27].Nikpay M, Beehler K, Valsesia A, et al. Genome-wide identification of circulating-miRNA expression quantitative trait loci reveals the role of several miRNAs in the regulation of Cardiometabolic phenotypes. Cardiovasc Res. 2019;115(11):1629–1645. [DOI] [PubMed] [Google Scholar]
- [28].Mansueto G, Benincasa G, Della Mura N, et al. Epigenetic-sensitive liquid biomarkers and personalised therapy in advanced heart failure: a focus on cell-free DNA and microRNAs. J Clin Pathol. 2020;73(9):535–543. [DOI] [PubMed] [Google Scholar]
- [29].Napoli C, Benincasa G, Donatelli F, et al. Precision medicine in distinct heart failure phenotypes: focus on clinical epigenetics. Am Heart J. 2020;224:113–128. [DOI] [PubMed] [Google Scholar]
- [30].Schiano C, Benincasa G, Franzese M, et al. Epigenetic-sensitive pathways in personalized therapy of major cardiovascular diseases. Pharmacol Ther. 2020;210:107514. [DOI] [PubMed] [Google Scholar]
- [31].Infante T, Forte E, Schiano C, et al. An integrated approach to coronary heart disease diagnosis and clinical management. Am J Transl Res. 2017;9(7):3148–3166. [PMC free article] [PubMed] [Google Scholar]
- [32].Infante T, Forte E, Punzo B, et al. Correlation of circulating miR-765, miR-93-5p, and miR-433-3p to obstructive coronary heart disease evaluated by cardiac computed tomography. Am J Cardiol. 2019;124(2):176–182. [DOI] [PubMed] [Google Scholar]
- [33].Napoli C, Lerman LO, de Nigris F, et al. Rethinking primary prevention of atherosclerosis-related diseases. Circulation. 2006;114(23):2517–2527. [DOI] [PubMed] [Google Scholar]
- [34].Napoli C, Crudele V, Soricelli A, et al. Primary prevention of atherosclerosis: a clinical challenge for the reversal of epigenetic mechanisms? Circulation. 2012;125(19):2363–2373. [DOI] [PubMed] [Google Scholar]
- [35].Fernández-Sanlés A, Sayols-Baixeras S, Curcio S, et al. Age-independent cardiovascular risk, an epigenome-wide approach: the REGICOR study (REgistre gIroní del COR). Arterioscler Thromb Vasc Biol. 2018;38(3):645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Prasher D, Greenway SC, Singh RB. The impact of epigenetics on cardiovascular disease. Biochem Cell Biol. 2020;98(1):12–22. [DOI] [PubMed] [Google Scholar]
- [37].Zhong J, Agha G, Baccarelli AA. The role of DNA methylation in cardiovascular risk and disease: methodological aspects, study design, and data analysis for epidemiological studies. Circ Res. 2016;118(1):119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Duan L, Hu J, Xiong X, et al. The role of DNA methylation in coronary artery disease. Gene. 2018;646:91–97. [DOI] [PubMed] [Google Scholar]
- [39].Li D, Yan J, Yuan Y, et al. Genome-wide DNA methylome alterations in acute coronary syndrome. Int J Mol Med. 2018;41(1):220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schiano C, Benincasa G, Infante T, et al. Integrated analysis of DNA methylation profile of HLA-G gene and imaging in coronary heart disease: pilot study. PLoS One. 2020;15(8):e0236951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Infante T, Forte E, Schiano C, et al. Evidence of association of circulating epigenetic-sensitive biomarkers with suspected coronary heart disease evaluated by Cardiac Computed Tomography. PLoS One. 2019;14(1):e0210909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li J, Zhu X, Yu K, et al. Genome-wide analysis of DNA methylation and acute coronary syndrome. Circ Res. 2017;120(11):1754–1767. [DOI] [PubMed] [Google Scholar]
- [43].Agha G, Mendelson MM, Ward-Caviness CK, et al. Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation. 2019;140(8):645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yang J, Yuan X, Lv C, et al. Methylation of the FOXP3 upstream enhancer as a clinical indicator of defective regulatory T cells in patients with acute coronary syndrome. Am J Transl Res. 2016;8(12):5298–5308. [PMC free article] [PubMed] [Google Scholar]
- [45].Nguyen A, Mamarbachi M, Turcot V, et al. Lower methylation of the ANGPTL2 gene in leukocytes from post-acute coronary syndrome patients. PLoS One. 2016;11(4):e0153920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Collet JP, Thiele H, Barbato E, et al., ESC Scientific Document Group . 2020 ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J. 2021;42(14):1289–1367. [DOI] [PubMed] [Google Scholar]
- [47].Sarda S, Hannenhalli S. Next-generation sequencing and epigenomics research: a hammer in search of nails. Genomics Inform. 2014;12(1):2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Benincasa G, Franzese M, Schiano C, et al. DNA methylation profiling of CD4+/CD8+ T cells reveals pathogenic mechanisms in increasing hyperglycemia: PIRAMIDE pilot study. Ann Med Surg (Lond). 2020;60:218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Akalin A, Kormaksson M, Li S, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yu G, Wang L-G, He Q-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31(14):2382–2383. [DOI] [PubMed] [Google Scholar]
- [54].Mi H, Poudel S, Muruganujan A, et al. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44(D1):D336–D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Silbiger VN, Luchessi AD, Hirata RD, et al. Novel genes detected by transcriptional profiling from whole-blood cells in patients with early onset of acute coronary syndrome. Clin Chim Acta. 2013;421:184–190. [DOI] [PubMed] [Google Scholar]
- [56].Benincasa G, Marfella R, Della Mura N, et al. Strengths and opportunities of network medicine in cardiovascular diseases. Circ J. 2020;84(2):144–152. [DOI] [PubMed] [Google Scholar]
- [57].Infante T, Del Viscovo L, De Rimini ML, et al. Network medicine: a clinical approach for precision medicine and personalized therapy in coronary heart disease. J Atheroscler Thromb. 2020;27(4):279–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Montojo J, Zuberi K, Rodriguez H, et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26(22):2927–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rienzo M, Schiano C, Casamassimi A, et al. Identification of valid reference housekeeping genes for gene expression analysis in tumor neovascularization studies. Clin Transl Oncol. 2013;15(3):211–218. [DOI] [PubMed] [Google Scholar]
- [60].Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. [DOI] [PubMed] [Google Scholar]
- [61].Bernhart SH, Kretzmer H, Holdt LM, et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci Rep. 2016;6(1):37393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wang N, Westerterp M. ABC Transporters, Cholesterol Efflux, and Implications for Cardiovascular Diseases. Adv Exp Med Biol. 2020;1276:67–83. [DOI] [PubMed] [Google Scholar]
- [63].Ma SC, Zhang HP, Kong FQ, et al. Integration of gene expression and DNA methylation profiles provides a molecular subtype for risk assessment in atherosclerosis. Mol Med Rep. 2016;13(6):4791–4799. [DOI] [PubMed] [Google Scholar]
- [64].Guay SP, Légaré C, Houde AA, et al. Acetylsalicylic acid, aging and coronary artery disease are associated with ABCA1 DNA methylation in men. Clin Epigenetics. 2014;6(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ghaznavi H, Mahmoodi K, Soltanpour MS. A preliminary study of the association between the ABCA1 gene promoter DNA methylation and coronary artery disease risk. Mol Biol Res Commun. 2018;7(2):59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Demina EP, Miroshnikova VV, Rodygina TI, et al. ABCA1 gene expression in peripheral blood lymphocytes and macrophages in patients with atherosclerosis. Mol Biol (Mosk). 2011;45(2):289–293. [PubMed] [Google Scholar]
- [67].Maiwald S, Zwetsloot PP, Sivapalaratnam S, et al. Monocyte gene expression and coronary artery disease. Curr Opin Clin Nutr Metab Care. 2013;16(4):411–417. [DOI] [PubMed] [Google Scholar]
- [68].Liu HF, Cui KF, Wang JP, et al. Significance of ABCA1 in human carotid atherosclerotic plaques. Exp Ther Med. 2012;4(2):297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Heo SH, Lee EH, Park HH, et al. Differences between the molecular mechanisms underlying ruptured and non-ruptured carotid plaques, and the significance of ABCA1. J Stroke. 2018;20(1):80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Vaeth M, Feske S. NFAT control of immune function: new Frontiers for an abiding trooper. F1000Res. 2018;7:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Garcia-Perez L, Famili F, Cordes M, et al. Functional definition of a transcription factor hierarchy regulating T cell lineage commitment. Sci Adv. 2020;6(31):eaaw7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Goettsch C, Rauner M, Hamann C, et al. Nuclear factor of activated T cells mediates oxidised LDL-induced calcification of vascular smooth muscle cells. Diabetologia. 2011;54(10):2690–2701. [DOI] [PubMed] [Google Scholar]
- [73].Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801. [DOI] [PubMed] [Google Scholar]
- [74].Guo X, Zhou C, Sun N. The neuropeptide catestatin promotes vascular smooth muscle cell proliferation through the Ca2+-calcineurin-NFAT signaling pathway. Biochem Biophys Res Commun. 2011;407(4):807–812. [DOI] [PubMed] [Google Scholar]
- [75].Zhong W, Li B, Yang P, et al. CD137-CD137L interaction modulates neointima formation and the phenotype transformation of vascular smooth muscle cells via NFATc1 signaling. Mol Cell Biochem. 2018;439(1–2):65–74. [DOI] [PubMed] [Google Scholar]
- [76].Govatati S, Pichavaram P, Janjanam J, et al. NFATc1-E2F1-LMCD1-mediated IL-33 Expression by thrombin is required for injury-induced neointima formation. Arterioscler Thromb Vasc Biol. 2019;39(6):1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Weng J, Wang C, Zhong W, et al. Activation of CD137 signaling promotes angiogenesis in atherosclerosis via modulating endothelial Smad1/5-NFATc1 pathway. J Am Heart Assoc. 2017;6(3):e004756. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [78].Gelfand BD, Meller J, Pryor AW, et al. Hemodynamic activation of beta-catenin and T-cell-specific transcription factor signaling in vascular endothelium regulates fibronectin expression. Arterioscler Thromb Vasc Biol. 2011;31(7):1625–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Jeon KI, Jono H, Miller CL, et al. Ca2+/calmodulin-stimulated PDE1 regulates the beta-catenin/TCF signaling through PP2A B56 gamma subunit in proliferating vascular smooth muscle cells. FEBS J. 2010;277(24):5026–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhang D, Chen Y, Xie X, et al. Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet-derived growth factor in endothelial cells. J Mol Cell Cardiol. 2012;53(4):487–496. [DOI] [PubMed] [Google Scholar]
- [81].Zhang Y, Zhang W, Wang KQ, et al. Expression of platelet-derived growth factor in the vascular walls of patients with lower extremity arterial occlusive disease. Exp Ther Med. 2015;9(4):1223–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Campioni D, Zauli G, Gambetti S, et al. In vitro characterization of circulating endothelial progenitor cells isolated from patients with acute coronary syndrome. PLoS One. 2013;8(2):e56377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Pang S, Tao Z, Min X, et al. Correlation between the serum platelet-derived growth factor, angiopoietin-1, and severity of coronary heart disease. Cardiol Res Pract. 2020;2020:3602608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Fan H-C, Fernández-Hernando C, Lai J-H. Protein kinase C isoforms in atherosclerosis: pro- or anti-inflammatory? Biochem Pharmacol. 2014;88(2):139–149. [DOI] [PubMed] [Google Scholar]
- [85].Metz PJ, Lopez J, Kim SH, et al. Regulation of asymmetric division by atypical protein kinase c influences early specification of CD8(+) T lymphocyte fates. Sci Rep. 2016;6(1):19182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Walker DL, Bhagwate AV, Baheti S, et al. DNA methylation profiling: comparison of genome-wide sequencing methods and the infinium human methylation 450 bead chip. Epigenomics. 2015;7(8):1287–1302. [DOI] [PubMed] [Google Scholar]
- [87].Sun Z, Cunningham J, Slager S, et al. Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis. Epigenomics. 2015;7(5):813–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.