

ABSTRACT
Cells of metazoans respond to internal and external stressors by activating stress response pathways that aim for re-establishing cellular homoeostasis or, if this cannot be achieved, triggering programmed cell death. Problems during translation, arising from defective mRNAs, tRNAs, ribosomes or protein misfolding, can activate stress response pathways as well as mRNA surveillance and ribosome quality control programs. Recently, ribosome collisions have emerged as a central signal for translational stress and shown to elicit different stress responses. Here, we review our current knowledge about the intricate mutual connections between ribosome collisions, stress response pathways and mRNA surveillance. A central factor connecting the sensing of collided ribosomes with degradation of the nascent polypeptides, dissociation of the stalled ribosomes and degradation of the mRNA by no-go or non-stop decay is the E3-ligase ZNF598. We tested whether ZNF598 also plays a role in nonsense-mediated mRNA decay (NMD) but found that it is dispensable for this translation termination-associated mRNA surveillance pathway, which in combination with other recent data argues against stable ribosome stalling at termination codons being the NMD-triggering signal.
KEYWORDS: Stress response, UPR, ISR, ribotoxic stress, translation, ribosome, ribosome collisions, quality control, mRNA surveillance, ZNF598, NGD, NSD, NMD, RQC
1. Introduction
Cells have developed sophisticated regulatory mechanisms to maintain homoeostasis under different physiological conditions and to protect themselves from adverse conditions imposed by a huge variety of extrinsic factors, including pathogens, toxic compounds, hypoxia, osmotic imbalance, nutrient starvation, UV-irradiation and various kinds of mechanical injuries. In general, stress response pathways facilitate cells to adapt to the respective stress condition appropriately and to restore cellular homoeostasis by altering gene expression, both at the transcriptional and post-transcriptional level. While stress response pathways constitute an effective protection for cells to cope with acute stresses, chronic stress conditions that perturb cellular homoeostasis over long times often lead to growth arrest and ultimately programmed cell death. Moreover, chronic activation of cellular stress responses is associated with pathogenic conditions, like neurodegeneration [1] or malignancies [2, 3]. Vice versa, infections often trigger stress responses [4, 5]. Additionally, deficiencies in cellular stress pathways are associated with several diseases [6–8], further documenting the close connection between cellular stress pathways and disease.
Many cellular stresses damage macromolecules, substantially increase damage of DNAs and in a consequence DNA repair pathways have been evolved to sustain the genomic integrity [8]. In addition, cellular stresses also damage RNAs by modifying, mutating or cleaving it, which consequently leads to the production of prematurely truncated, misfolded and/or non-functional proteins that are potentially deleterious for cells [9]. Three mRNA surveillance pathways termed nonsense-mediated mRNA decay (NMD), no-go decay (NGD) and non-stop decay (NSD) play important roles in resolving problems during translation that can arise from faulty mRNAs or defective ribosomes [9,10]. While in these three surveillance pathways the rapid degradation of the involved mRNA is well documented, the fate of the involved ribosomes and nascent polypeptides is less clear. However, various RNA damages frequently lead to ribosome collisions, which were found as a key regulator of specific stress response pathways. For example, a low dose of the translational elongation inhibitors anisomycin or emetine increases the frequency of ribosome collisions, which triggers cellular recovery through integrated stress response (ISR), whereas high doses of anisomycin, emetine, or UV irradiation incite persistent ribosome stalling, leading to apoptosis through the activation of MAPKKK cascades as a consequence of 28S rRNA damage [9,11,12].
In the past few years, ribosome collisions have been documented to be sensed by a still relatively ill-defined pathway termed ribosome-associated quality control (RQC), and mechanistic links between RQC and NGD/NSD have begun to emerge. RQC is crucial to resolve no-go or non-stop situations in the cell by leading to the rescue of the stalled ribosomes and degradation of the nascent polypeptides [13]. Recent work showed that the E3 ubiquitin ligase ZNF598 (Hel2 in yeast) plays a crucial role in NSD/NGD-associated RQC. ZNF598 ubiquitinates the 40S ribosomal subunit of collided di-ribosomes on prematurely polyadenylated mRNAs [14,15]. Furthermore, genome-wide CRISPRi screens identified the additional RQC factors GIGYF2 and 4EHP, which repress the translational initiation of faulty mRNAs and promote RQC independently of the ZNF598 axis [16]. Moreover, quantitative proteomics analysis of polysome fractions led to the identification of EDF1 as a ribosome collision sensor that is involved in the GIGYF2-4EHP axis of RQC but not in ZNF598 dependent RQC [17,18].
While a molecular link between RQC and NGD/NSD is established, it has been an open question whether RQC might also be linked to NMD, for example through ZNF598. Herein, we will first review the current literature on translation reprogramming related to different stress response pathways and then focus on the role of ribosome collisions as a sensor for activating specific cellular stress response pathways in the context of RNA quality control. Finally, we will discuss the evidence for a possible link between RQC and NMD and present our experimental data showing that ZNF598 is dispensable for NMD.
2. Stress response pathways connected with translation reprogramming
Cellular stress responses entail a series of molecular events by altering gene expression for activating appropriate pathways to clear the impairments and restore the physiological balance. Among the adjustments in gene expression, specific reprogramming of translation is crucial for the activation of stress response pathways. Interestingly, numerous stress responses impact on translation factors, such as eIF4F and eIF2, that regulate mRNA translation at initiation or elongation steps. Suppression of global protein synthesis is generally paralleled by the selective translation of mRNAs encoding specific proteins that are crucial for cellular restoration and survival [19,20]. Recent genome-wide ribosome profiling studies have demonstrated proteome complexity, alternative translation initiation and ribosome pausing during elongation, which are all manifestations of stress-induced translational reprogramming [21–23].
In the following sections, we will describe three major stress response pathways that are associated with translational reprogramming: (i) the integrated stress response, which inhibits initiation of global translation, (ii) the unfolded protein response (UPR) and (iii) the ribotoxic stress response (RSR), which activates p38/JNK signalling cascade.
2.1. Integrated Stress Response (ISR)
ISR is a conserved signalling network that restores cellular homoeostasis via translational reprogramming in response to diverse stress stimuli [24]. The hallmark of ISR is the phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α), which can be achieved by one of four different eIF2α kinase family members depending on the type of stress signal (Fig. 1). GCN2 [general control nonderepressible 2] is the kinase activated upon amino acid deficiency, proteasomal inhibition or UV-radiation, whereas haem deprivation, oxidative stress or heat shock leads to HRI [haem-regulated inhibitor] kinase-mediated eIF2α phosphorylation. Endoplasmic reticulum (ER) stress and hypoxia are sensed by PERK [double-stranded RNA-activated protein kinase (PKR)–like ER kinase] and other pathological conditions including viral infections activate PKR [protein kinase R] [25]. These four kinases all phosphorylate Ser51 of eIF2α thereby causing a global arrest of translation initiation [26,27].
Figure 1.
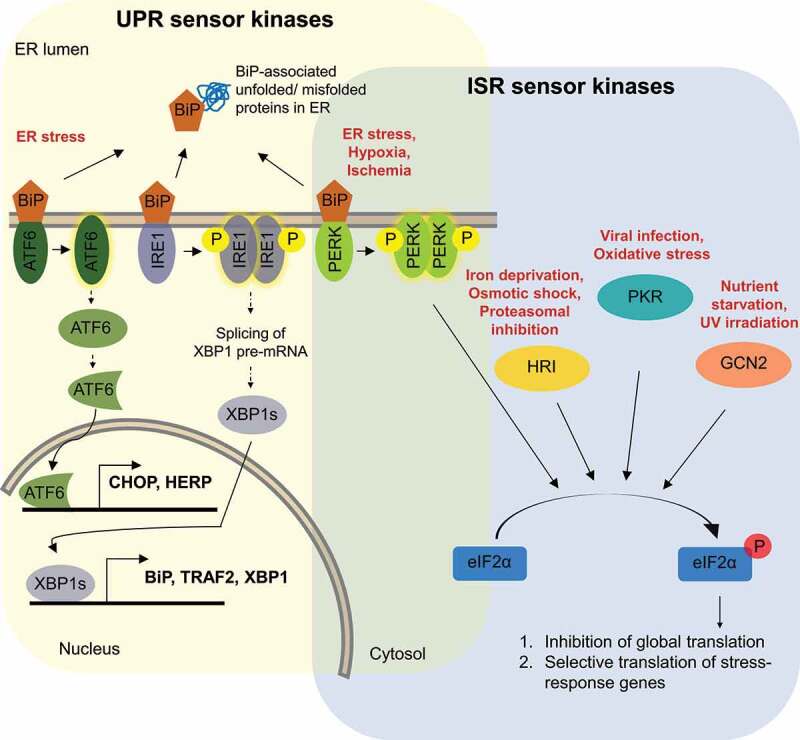
Schematic illustration of integrated stress response (ISR) and unfolded protein response (UPR) signalling cascades.
ISR is schematically illustrated in the light blue and UPR in the light yellow area, respectively. The different stresses that activate ISR and UPR are depicted in red. UPR is activated by interaction of the ER chaperone BiP with accumulating unfolded proteins, leading to autophosphorylation of IRE1 and PERK kinase and proteolytic cleavage in the Golgi of ATF6. BiP-released activated IRE1 and ATF6 lead to transcriptional activation of various stress response genes, as described in section 2.2 of the text. Activated PERK, as well as the other ISR sensors HRI, PKR and GCN2 trigger different signalling cascades that converge on phosphorylation of eIF2α, which leads to global attenuation of cap-dependent translation while concomitantly promoting translation of ISR-specific mRNAs, as described in section 2.1 of the text.
Mechanistically, eIF2· GTP and methionyl-initiator tRNA (Met-tRNAi) form a ternary complex (TC), which together with eIF3, eIF1 and eIF1A binds 40S ribosomal subunits to form 43S pre-initiation complexes. At the start codon, the 60S ribosomal subunit joins the 43S pre-initiation complex and translation can begin. Translation initiation involves GTP hydrolysis and release of Met-tRNAi to the ribosomal P site. The subsequent conversion of eIF2· GDP into eIF2· GTP is catalysed by the guanine nucleotide exchange factor (GEF) eIF2B. The ISR-triggering phosphorylation of Ser51 of the α subunit of eIF2 induces a profound structural rearrangement in eIF2α, leading to the exposure of a hydrophobic surface patch on eIF2 that displays strong affinity for an alternative binding site on eIF2B. Phosphorylated eIF2 therefore sterically interferes with proper positioning of the catalytic domain of eIF2B and becomes a potent non-competitive inhibitor of eIF2B [28,29]. Consequently, phosphorylated eIF2· GDP is not converted into eIF2· GTP anymore and since formation of the TC is a rate-limiting step in translation, global protein synthesis is reduced. However, a small number of ISR-specific mRNAs, among them the transcription factors GCN4 and ATF4, can still be efficiently translated under these conditions. ATF4 in turn promotes the transcription of the stress response genes ATF3 and CHOP (GADD153) to combat the cell stress [26,27]. The first mechanistic insight into ISR-driven selective translation stems from studies of Gcn4 in yeast [30]. GCN4 and ATF4 mRNAs both harbour inhibitory upstream open reading frames (uORFs) in their 5´-untranslated regions (5´ UTRs), which prevent initiation at their canonical AUGs [30–32]. The presence of uORFs that inhibit translation of the main ORF under normal conditions but enable it when TC becomes limiting seems to be a hallmark of ISR and has been found in several other mRNAs, including ATF5 [33], C/EBP-homologous protein (CHOP) [34], GADD34 [35] and OPHN1 in neurons [36]. However, the precise mechanism by which these mRNAs are translationally controlled remains unclear. Although it is generally assumed that ribosomes reinitiate on these mRNAs at downstream initiation codons, the possibility that the peptides encoded by the uORFs could regulate initiation at the main ORFs should also be considered [37]. In the case of mRNAs encoding the transcription regulators ATF4, ATF5, and CHOP, their translational activity/ derepression results in transcriptional changes of their target genes.
2.2. Unfolded Protein Response (UPR)
UPR is another cellular stress response pathway that is tightly coupled with transcriptional and translational reprogramming. UPR is triggered by the accumulation of unfolded (or misfolded) proteins in the ER, which can result from a wide range of stresses, including physical injuries, hypoxia, nutrient starvation, oxidative stress, or genotoxic stress [38–40]. A high burden of unfolded proteins in the ER then activates three distinct intracellular signal transduction pathways, each of which is activated by a different transmembrane ER-resident signalling component: IRE1 [inositol requiring enzyme 1], PERK, and ATF6 [activating transcription factor 6] (Fig. 1) [40]. The evolutionary most conserved of these signalling pathways is the IRE1 cascade, while the PERK and ATF6 branches have evolved later. Noteworthy, the PERK branch overlaps with the ISR [41]. Under normal physiological conditions, the ER-resident chaperone BiP/GRP78 (immunoglobulin-binding protein; also known as glucose-regulated protein 78) interacts with and inhibits these three sensors of UPR. Upon ER stress or accumulation of unfolded proteins, BiP dissociates from IRE1 and PERK, enabling them to activate downstream signalling [42].
The interaction of unfolded proteins with IRE1 triggers non-canonical cytoplasmic splicing of XBP1 (X-box binding protein 1) pre-mRNA to generate spliced mRNA, from which the transcription factor XBP1 is translated [43,44]. XBP1 activates the transcription of genes coding for different chaperones that enhance the correct folding of polypeptide chains into functional proteins and thus counteract the accumulation of unfolded proteins in the ER. Additionally, XBP1 also activates genes involved in lipid synthesis, which serves to increase the size of the ER membrane to accommodate increased protein load. Finally, XBP1 also induces genes encoding ER-associated degradation (ERAD) proteins, which move unfolded proteins back into the ER [45].
PERK activation inhibits global protein synthesis by decreasing the rate of protein translation initiation through phosphorylation of eIF2α as discussed before. Decreased translation is an important survival response in UPR, as it reduces the production of further unfolded proteins in order to enable the cell to deal with the already existing unfolded proteins. Phosphorylation of eIF2α also increases transcription of genes encoding factors important for responding to ER stress, including ATF4 and its downstream target CHOP [45].
The third branch of UPR comprises the activation of ATF6 through dissociation from BiP, that allows ATF6 to translocate to the Golgi apparatus, where it undergoes proteolytic cleavage [46]. The cleaved fragments are then translocated to the nucleus to activate transcription of several genes encoding ER-localized chaperones that are important for protein folding, including the ATF6 regulator BiP and XBP1 [40]. The ATF6-mediated activation of BiP expression creates a negative feedback loop that buffers the UPR pathway and leads to its silencing once the amount of unfolded protein is back to normal levels [47]. Collectively, these three sensors allow mammalian cells to react in a comprehensive manner to reduce abnormally high levels of unfolded proteins in the ER.
2.3. Ribotoxic Stress Response (RSR)
RSR is a conserved cellular response to damaged ribosomal RNA (23S/28S), observed in both prokaryotes and eukaryotes [11]. The conserved 3´ end of 28S rRNA, the so-called sarcin-ricin loop (SRL), is crucial for aminoacyl-tRNA binding, peptidyl transfer and ribosomal translocation. Therefore, damage at the SRL leads to translational inhibition and activation of stress kinases [11,48,49]. RSR activates the MAP kinase pathway to promote cellular recovery. Three specific protein kinases are known to recognize ribosomal damage, namely HCK [haematopoietic cell kinase], PKR [double-stranded RNA-dependent protein kinase] and ZAKα [zipper sterile alpha motif kinase]. The best characterized RSR mediator is ZAKα, a MAP3 kinase family member that is activated by various translational inhibitors (anisomycin, cycloheximide), ribotoxins (ricin, Shiga toxin type 2) and UV radiation [11,49–51].
Ribosome damage-induced ZAKα activation leads to phosphorylation of one or more MAP3 kinase cascades (Fig. 2). The MAPK family includes extracellular-receptor kinases (ERKs), p38 and c-jun N-terminal kinases (JNKs) [52]. ZAKα was first reported to activate the JNK and p38 pathways after treatment of cells with anisomycin and later it was shown that also Stx2 and ricin trigger ZAKα activation [53,54]. P38 and JNKs are well characterized stress-activated protein kinases that share structural and functional homology between yeast and mammals. They are important regulators of cell fate in response to multiple environmental stresses [52,55]. Activation of p38 induces cell-cycle arrest, whereas activation of JNK promotes apoptosis [55,56]. Upon activation, JNKs phosphorylate and activate transcription factors such as c-jun [57], ATF-2 [58,59] and Elk-1 [60,61] leading ultimately to the transcriptional activation of the immediate early genes [59,61].
Figure 2.
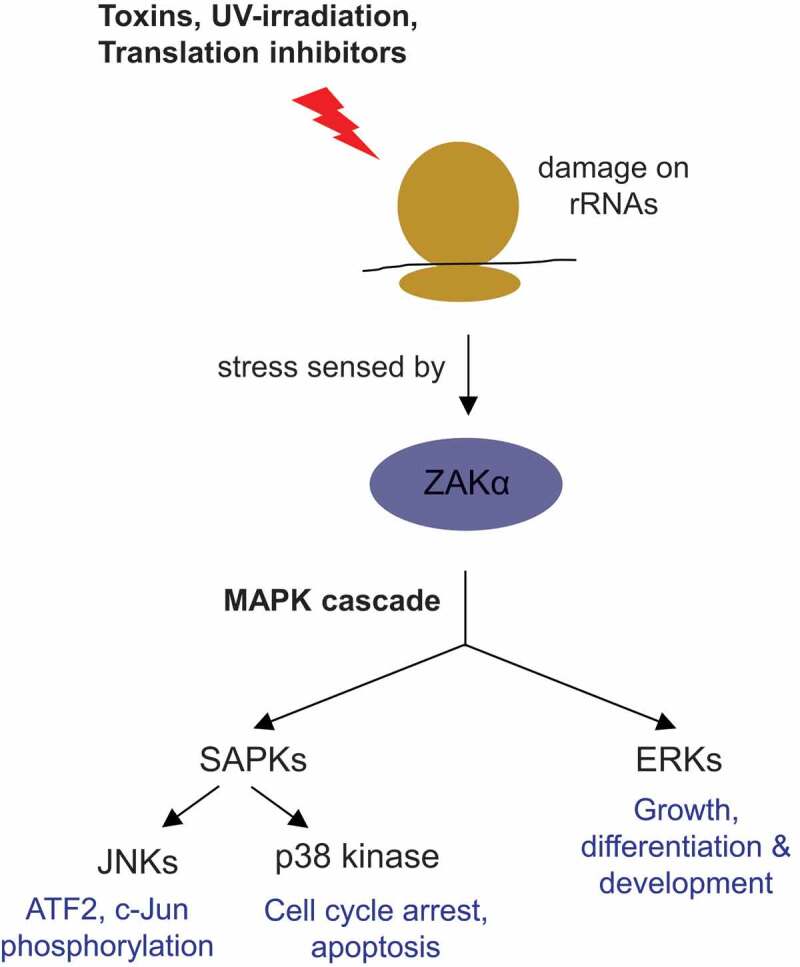
Outline of the ribotoxic stress response pathway.
Ribosomal RNA damage, caused by exposure of ribosomes to toxins, translation inhibitors or UV-irradiation, is sensed by the MAP3K ZAKα. Activated ZAKα promotes the activation of two MAP kinase families, the extracellular-receptor kinases (ERKs) and the stress-activated protein kinases (SAPKs; including p38 and c-Jun N-terminal kinases (JNKs)). See section 2.3 in the text for details.
3. Ribosome collisions activate stress response pathways
It is a complex task for cells to successfully combat damages and metabolic imbalances without disturbing overall cellular homoeostasis and it is often a fine line between activation of suitable repair and survival pathways or promotion of apoptosis. Recent evidence suggests that ribosome collisions represent a key sensor for perturbation of translational homoeostasis and important regulator of stress response and cell fate [12]. Reduced translation rates and ribosome pausing for the GCN2 – phosphpo-eIF2α axis in response of amino acid starvation was measured by puromycin incorporation assays and polysome profiling [62]. Interestingly, low concentrations of translation elongation inhibitors (anisomycin or emetine) were shown to increase the frequency of ribosome collisions, which activates ISR via the ZAKα-GCN2 pathway. Searching for stress sensors of ribosome collisions, CRISPR-based loss-of-function screens in cells treated with high doses of anisomycin identified ZAKα, confirming its role in RSR as activators of MAPKKK and p38/JNK pathways upon ribosome stalling. The role of ZAKα as a sensor of collided ribosomes was further validated by induction of ribosome collisions by amino acid starvation, that activates ISR via GCN2. Above a certain overall frequency of ribosome collisions or if the stress signal is chronic, the stalled ribosomes eventually induce apoptosis via the ZAKα-MAPKK-SAPKs signalling cascade (Fig. 3) [12]. Moreover, Vind and colleagues established ZAKα as a proximal ribotoxic stress sensor, whose flexible C-terminus domain interacts to 18S rRNA helix-14 of stalled ribosome in C. elegans, as confirmed by CLIP-seq [49].
Figure 3.
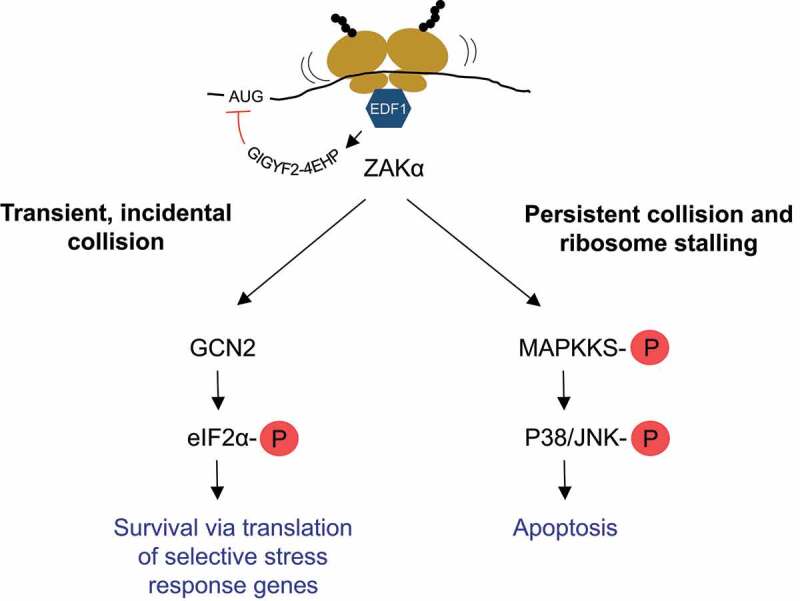
Schematic illustration of cellular responses triggered by ribosome collisions.
Binding of EDF1 to collided ribosomes promotes GIGYF2-4EHP-mediated inhibition of initiation of new ribosomes on the mRNA. The extent of collisions is sensed by ZAKα, which activates GCN2-mediated ISR by inhibiting cap-dependent translation and facilitating selective translation of stress response genes if the collisions are temporary and/or occur at low frequency. If ZAKα senses persistent collisions and stalled ribosomes, it activates MAP kinase cascades of RSR to promote apoptosis. See sections 2.3 and 3 in the text for details.
The findings reviewed above, revealed different cellular responses elicited by ribosomal collisions, but the underlying molecular mechanisms that initially sense ribosome collisions and lead to activation of the respective stress pathways have only begun to emerge. Several recent studies demonstrated a role of the E3 ubiquitin ligase ZNF598 (Hel2 in yeast) in recognition of collided ribosomes [63,64]. Translation of poly(A) sequences leads to ribosome stalling by the poly-Lys congesting the ribosomal exit tunnel, which activates RQC in a ZNF598-dependent manner [14]. ZNF598 was shown to bind specifically to collided di-ribosome structure that arises when a trailing ribosome encounters a slower leading ribosome [64]. A cryo-EM study further found that in yeast, Hel2 was predominately bound to rotated ribosomes with hybrid-state tRNAs, suggesting that this ribosome state could be the signal for aberrant translation [15]. Binding of ZNF598 to collided ribosomes then leads to the site-specific ubiquitination of the 40S ribosomal proteins RPS10 and RPS20 [14,65]. In addition to ZNF598, the 40S ribosomal subunit-associated protein RACK1 (Asc1 in yeast) regulates ubiquitylation of RPS2 and RPS3 upstream of ribosomal rescue [65]. These observations indicate that ZNF598 catalyzes the site-specific mono- or oligo-ubiquitylation of small subunit proteins in stalled ribosomes, but rather than triggering proteolysis, this ubiquitination is thought to induce the recruitment of factors required for dissociating the stalled ribosomes into the 40S and the peptidyl-tRNA associated 60S subunits. Following splitting of the ribosomal subunits, the nascent polypeptide is degraded by RQC (see section 4). Besides ZNF598-mediated ubiquitination of collided ribosomes, ribosome collisions were further shown to trigger the endonucleolytic cleavage of the mRNA [66].
Recently, quantitative proteomics analyses of collided ribosomes identified EDF1 as another sensor of ribosome collisions and the structure of EDF1 with collided ribosomes was solved by cryo-EM [18]. Additionally, EDF1 and GIGYF2 were found to be enriched on collided ribosomes in wildtype and ZNF598 knockout cells, indicating that EDF1-mediated GIGYF2-4EHP recruitment to collided ribosomes occurs independent of ZNF598 [17,18]. Earlier studies showed that GIGYF2 inhibits translation initiation by recruiting the alternative cap-binding protein 4EHP (also known as EIF4E2), which competes with eIF4E for binding the cap of the mRNA [67]. Thus, ribosome collisions activate a two-pronged response consisting of a ZNF598-driven activation of RQC to remove the stalled ribosomes and of a EDF1-GIGYF2-4EHP mediated inhibition of new ribosomes initiating translation on the affected mRNA. In addition, the stress sensor kinase ZAKα activates appropriate stress response pathways based on transient or prolonged ribosomal collisions, while for incidental collisions, the involvement of ZNF598 mediated ubiquitination and triggering of the RQC pathway has not yet been confirmed, most likely because it is challenging to capture such transient collisions (Fig. 3).
4. mRNA surveillance pathways and its links to RQC
During translation, different surveillance systems sense ribosome stalling during elongation or aberrant termination and elicit the degradation of the defective mRNAs: 1) Nonsense-mediated mRNA decay (NMD) is best known for detecting and degrading mRNAs with CDS-interrupting premature termination codons (PTCs), thereby preventing the production of C-terminally truncated proteins, although it is now well established that this pathway also regulates the stability of a significant fraction of ‘normal’ mRNAs that code for full-length functional proteins [68–70], 2) No-go decay (NGD) recognizes and degrades transcripts on which ribosomes fail to proceed within the CDS due to the presence of inhibitory stem-loop structures or in stretches of rare codons [10,63,71] and 3) Non-Stop decay (NSD) identifies and degrades transcripts lacking stop codons, on which ribosomes proceed all the way to the 3´ end of the RNA where they get stuck [72,73]. The activating signal for RNA degradation in both NSD and NGD are stalled ribosomes, which in NSD results in the recruitment of the exosomes to the RNA 3´ end through the Ski2-Ski3-Ski8 complex, while the endonuclease Cue2 initiates degradation of the mRNA in NGD [66,74,75].
Besides degradation of the involved mRNA, all translation-dependent mRNA surveillance systems also have evolved mechanisms to degrade the newly synthesized polypeptides, that in the case of NGD and NSD remain physically attached to the stalled ribosomes. In NGD and NSD, degradation of the nascent polypeptide and removal of the stalled ribosomes is carried out by a dedicated surveillance pathway termed ribosome-associated protein quality control (RQC) [13]. RQC involves recognition of the stalled ribosomes, dissociation of the 40S and 60S ribosomal subunits from the mRNA (i.e. ribosome rescue) and proteolysis of the nascent polypeptide chain that remains attached to the 60S subunits in the absence of proper hydrolysis from the tRNA. RQC is tightly linked with the NSD and NGD pathways: In yeast, stalled ribosomes are sensed by Hbs1 (HBS1L or GTPBP2 in mammals) and Dom34 (PELO in mammals) and Dom34 then recruits Rli1, an ATP-binding cassette protein subfamily E member 1 (ABCE1 in mammals), which separates the 40S and 60S subunit-associated with peptidyl-tRNA. The released mRNA is degraded rapidly, while Rqc2 recognizes and associates with peptidyl-tRNA–60S complex, which ultimately recruits and stabilizes the binding of the E3 ligase Ltn1 (listerin). Ltn1 catalyzes the ubiquitination of the nascent polypeptides, which leads to the recruitment of AAA ATPase Cdc48 (VCP in mammals) and its co-factors. Finally, Cdc48 extracts nascent polypeptides from the 60S ribosomal subunit and the released polypeptides will be degraded by the proteasome [13]. Noteworthy, the previously described translation repressors GIGYF2 and 4EHP were identified as additional RQC factors in a genome-wide CRISPRi screen [16].
The links between the NGD and NSD surveillance pathways and RQC are partly resolved, but whether PELO acts upstream or downstream of ZNF598 is not yet known. A recent study demonstrated that NGD and RQC both respond to ribosome collision via ZNF598-mediated ubiquitination of RPS10 [63]. Together with ZNF598-mediated proteasomal degradation of the already synthesized aberrant polypeptides, the aforementioned EDF1-GIGYF2-4EHP-mediated inhibition of translation initiation on the affected transcripts provides a cellular quality control that effectively suppresses potential detrimental effects of such aberrant polypeptides.
Given the similarities between the three translation-dependent mRNA surveillance pathways NMD, NGD and NSD with respect to their function in resolving problems during mRNA translation, the documented involvement of collided ribosomes and RQC in NGD and NSD, and the models proposing ribosome stalling during translation termination as a hallmark of NMD, it is conceivable that ribosome collisions and RQC might also be mechanistically linked to NMD. However, before delving into any details of the postulated connection between ribosome collision and NMD, we first summarize our current understanding of the molecular mechanism and the regulation of NMD.
5. NMD working models
It was first recognized in yeast and mammalian cells that mRNAs with premature termination codons (PTCs) are selectively degraded [76,77], an observation for which later the term “nonsense-mediated mRNA decay (NMD) was coined [78]. Initial screens in yeast and C. elegans revealed the first proteins required for NMD, the homologs of which were later identified in other species and additional NMD factors have been uncovered more recently by genome-wide RNAi screens in C. elegans and human cells [79]. Genome-wide transcriptome-profiling analysis revealed that NMD regulates the half-lives of 3–10% of all mRNAs in various eukaryotes [80–84], implying a role of NMD beyond quality control in general posttranscriptional gene regulation. A central factor in the NMD pathway is UPF1, an ATP-dependent RNA helicase of the SF1 superfamily, which undergoes cycles of phosphorylation and dephosphorylation and nucleates the formation of a protein complex called SURF (composed of SMG1, UPF1 and the release factors eRF1 and eRF3) at aberrantly terminating ribosomes that ultimately induces the degradation of the RNA [85–88]. Besides PTCs, additional mRNA features like upstream open reading frames (uORFs) and a long 3′ untranslated regions (UTR) have been reported to trigger NMD. They have in common that they spatially separate the termination codon (TC) from the poly(A)-binding protein (PABP), indicating that the microenvironment in which ribosomes terminate translation ultimately determines whether an mRNA is targeted by NMD [89–94].
There are two major models offering possible explanations for how NMD substrates are recognized in metazoans, but the exact mechanism is not yet fully understood. We refer to the currently prevailing model as the ‘Exon junction complex (EJC) enhanced NMD model’, in which the presence of an EJC in the 3´ UTR (i.e. downstream of the TC) plays a crucial role in activating NMD [95]. EJCs are assembled ~20–24 nucleotides upstream of spliced exon–exon boundaries during the course of intron splicing in the nucleus, and later in the cytoplasm stripped off the coding sequence by elongating ribosomes [96–99]. Therefore, EJCs located >30 nucleotides downstream of the TC on a given mRNA are thought to remain bound during translation and function as an NMD activating signal [100,101]. NMD activation occurs through UPF1 phosphorylation by the protein kinase SMG1, which is stimulated by an interaction with EJC-associated UPF2 and UPF3 positioned downstream of the TC on the mRNA [102,103]. This EJC-SURF interaction is then thought to lead to the formation of the decay-inducing (DECID) complex [87]. SMG1 phosphorylates UPF1 at several serine/threonine–glutamine (S/TQ) motifs in the N- and C-terminal regions [88,102,104–106]. Hyperphosphorylated UPF1 then binds the endonuclease SMG6 [107,108] and the heterodimer SMG5-SMG7 [109], thereby initiating the degradation of the mRNA by an endonucleolytic cleavage near the TC and by recruiting the CCR4-NOT deadenylase, respectively. In human cells, SMG6-mediated endonucleolytic cleavage appears to be the major route to elicit mRNA decay by the NMD pathway, whereas the SMG5-SMG7-mediated recruitment of CCR4-NOT might to act as a backup system [80,110].
Despite of a wealth of biochemical data that led to the above-described model, the mechanistic details of NMD still remain incompletely understood and there is evidence that NMD can also ensue in human cells in the absence of EJCs in the 3´ UTR [111]. What is the precise determinant for activating NMD in the absence of 3´ UTR located EJCs remains an important unsolved question, but the distance between the termination codon and the poly(A) tail seems to play an important role [91,111]. In fact, this ‘Termination codon to poly(A) tail distance NMD model’ could represent the evolutionary conserved mode of NMD, since it has the potential to explain NMD also in S. cerevisiae, which has no EJCs, and in C. elegans and D. melanogaster, in which EJCs exist but appear to have no function in NMD [112,113]. Based on the biochemical data documenting interactions between eRF3 and poly(A) binding protein C1 (PABPC1) as well as between eRF3 and UPF1, it has was hypothesized that NMD activation might depend on a competition between PABPC1 and UPF1 for binding to eRF3 at the terminating ribosome [92,94,114]. According to this hypothesis, long unstructured 3´ UTRs may prevent PABPC1 from efficiently interacting with eRF3 and allow UPF1 to bind eRF3 instead [101]. Interestingly in the light of clear examples of 3´UTR length-dependent NMD [111,115], high throughput transcriptome analyses aiming at identifying NMD targeted transcripts in human cells observed no general correlation between the 3´ UTR length of a transcript and the likelihood of being targeted by NMD [116,117]. However, 3´ UTR length per se is probably not an NMD eliciting signal because most 3´ UTRs are highly structured and therefore the physical distance between the TC and the poly(A) tail might not correlate with 3´ UTR length. Moreover, special binding motifs for RNA-binding proteins in the 3´ UTR of otherwise NMD sensitive mRNAs were found to efficiently protect these transcripts from NMD [118–120].
If the UPF1 versus PABPC1 competition for eRF3 interaction indeed was the determinant for whether NMD is triggered during a given translation termination event, one might expect differences in the kinetics of termination when ribosomes terminate translation on TCs that trigger NMD compared to termination at regular TCs. Indeed, there is evidence from in vitro translation approaches in yeast and rabbit reticulocyte lysates for stalling of ribosomes at NMD eliciting TCs, suggesting that NMD activation might be triggered by slow translation termination [89,121]. In contrast, toeprinting assays performed after in vitro translation in lysates of human cells detected similar ribosome occupancy at the termination codons of NMD-sensitive and NMD-insensitive mRNAs and the presence or absence of a poly(A) tail neither did affect ribosome occupancy at the TC in these assays [122]. Furthermore, ribosome profiling did also not reveal a difference in ribosome density at the TC of endogenous NMD-sensitive and NMD-insensitive mRNAs in vivo [122]. While these observations speak against stable ribosome stalling at TCs being a hallmark of NMD in human cells, they do not rule out the possibility that subtle kinetic differences leading to ribosome collisions at the TC could be involved in NMD activation.
6. ZNF598 is not involved in NMD
While it is well documented that ribosome collisions trigger RQC and NGD to degrade the nascent peptides and problematic mRNA, respectively, little is known about the fate of truncated polypeptides produced from PTC-containing mRNAs that are targeted for degradation by NMD. Evidence for chaperon-mediated proteasomal degradation or formation of aggresomes that are eventually cleared by autophagy has been reported for prematurely truncated polypeptides [123,124], but whether ribosome collision-directed RQC might be involved remained untested. A recent pre-print provides evidence for a proteasome-dependent degradation of polypeptides produced from NMD-sensitive transcripts involving the E3 ubiquitin ligase CNOT4, a member of the CCR4-NOT deadenylation complex, while canonical RQC factors were not identified in this screen [125]. To experimentally address whether ribosome collisions and ribosome collision-activated RQC might play a role in NMD, we investigated if ZNF598 is required for NMD. To test this hypothesis, we measured the levels of several endogenous NMD-sensitive transcripts in HEK293 Flp-In T-Rex cells in which ZNF598 was knocked-out by CRISPR-Cas9-mediated genome editing (a kind gift of Dr Aitor Garzia) [14] and compared it with wildtype (WT) HEK293 Flp-In T-Rex cells. The clonal ZNF598 knockout (KO) cell line used in our experiments had a 11 nucleotides deletion in exon 3 of the ZNF598 gene on both alleles, which created a frameshift and a PTC in the following exon compared to the corresponding sequence in the WT cells and the reference sequence from the Ensemble Genome Browser [ENSG00000167962] (Fig. 4A).
Figure 4.
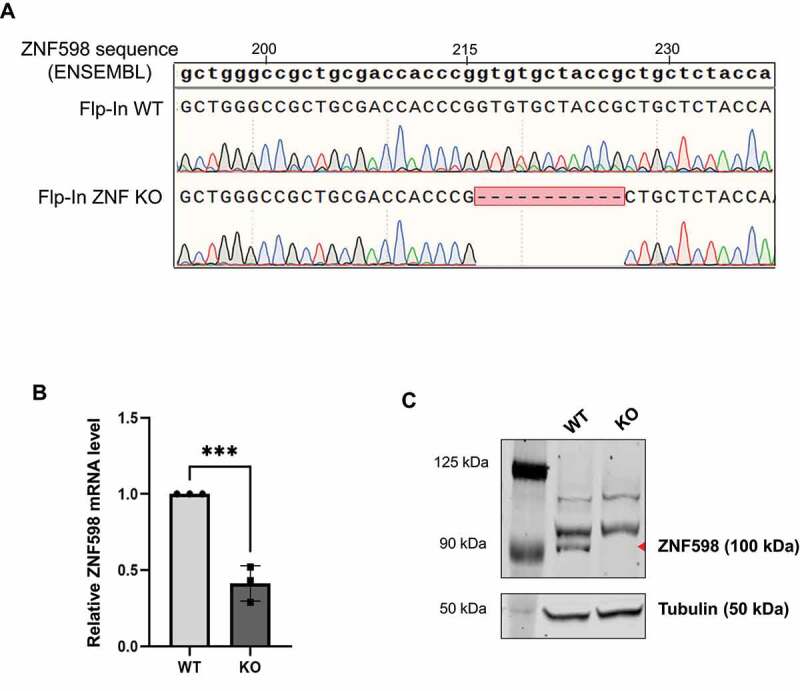
Characterization of ZNF598 KO cells.
(A) Alignment of sequences of genomic DNA isolated, amplified and sequenced from WT and ZNF598 KO cells with ZNF598 reference sequence from ENSEMBL. (B) Relative ZNF598 mRNA levels in WT and ZNF598KO cells were measured by qRT-PCR and normalized to β-actin mRNA. Mean values and SEM of three independent experiments are presented. Statistical significance was determined using a 2-way ANOVA (GraphPad Prism v9), where *** represents p ≤ 0.001. (C) Western blot analysis of ZNF598 expression in WT and KO cells. The position of ZNF598 is indicated by a red triangle, the two higher molecular weight bands are unspecific bands produced by the anti-ZNF598 antibody. The size marker is shown in the left lane and the sizes of the marker bands are indicated. The membrane was subsequently re-probed with anti-tubulin antibody, which serves as loading control (lower panel).
The PTC in exon 4 of ZNF598 in the ZNF598 KO cells is expected to render the resulting mRNA a target for the NMD pathway. Indeed, compared to the ZNF598 mRNA level measured in the WT cells, the KO cells expressed 2.5-fold less ZNF598 mRNA (Fig. 4B), consistent with this nonsense mRNA being destabilized by NMD. More importantly for our purpose, western blotting confirmed the complete absence of ZNF598 protein in the KO cells (Fig. 4C), confirming that these cells are true functional ZNF598 knockouts.
To check if knocking out ZNF598 affects NMD, we next measured the RNA levels of six different endogenous NMD-sensitive and four NMD-insensitive transcripts were measured by RT-qPCR in WT and ZNF598 KO Flp-In T-Rex HEK293 cells. Besides HNRNPL, TRA2B and BAG1, of which we measured the protein coding, NMD-insensitive mRNA isoform as well as an alternatively spliced, PTC-containing and hence NMD-sensitive mRNA isoform, we also determined the levels of GAS5, RP9P and SMG5 mRNAs, which were previously found to be targeted by NMD [80]. All measurements were normalized to the NMD-insensitive actin β mRNA and the values are shown relative to untreated WT cells, which were arbitrarily set as 1 (Fig. 5). We observed no significant differences in the RNA levels of the NMD-sensitive transcripts between untreated or DMSO treated WT and KO cells, demonstrating that ZNF598 inactivation has no influence on NMD. As a positive control for NMD inactivation, cells were treated with a SMG1 inhibitor [126], which strongly increased the levels of all six NMD-sensitive transcripts while leaving the NMD-insensitive transcripts unaffected (Fig. 5). Our conclusion that ZNF598 is not involved in NMD was corroborated by a recent study showing that ZNF598 KO did not affect the expression level of a fluorescent NMD reporter based on flow cytometry measurements [127]. That ZNF598 is dispensable for NMD, in conjunction with data indicating that EDF1, another RQC factor, is also not involved in NMD [127], argues indirectly against an involvement of ribosome collisions in NMD, which is consistent with our recently published observations indicating that NMD ensues independently of stable ribosome stalling at the TC [122].
Figure 5.
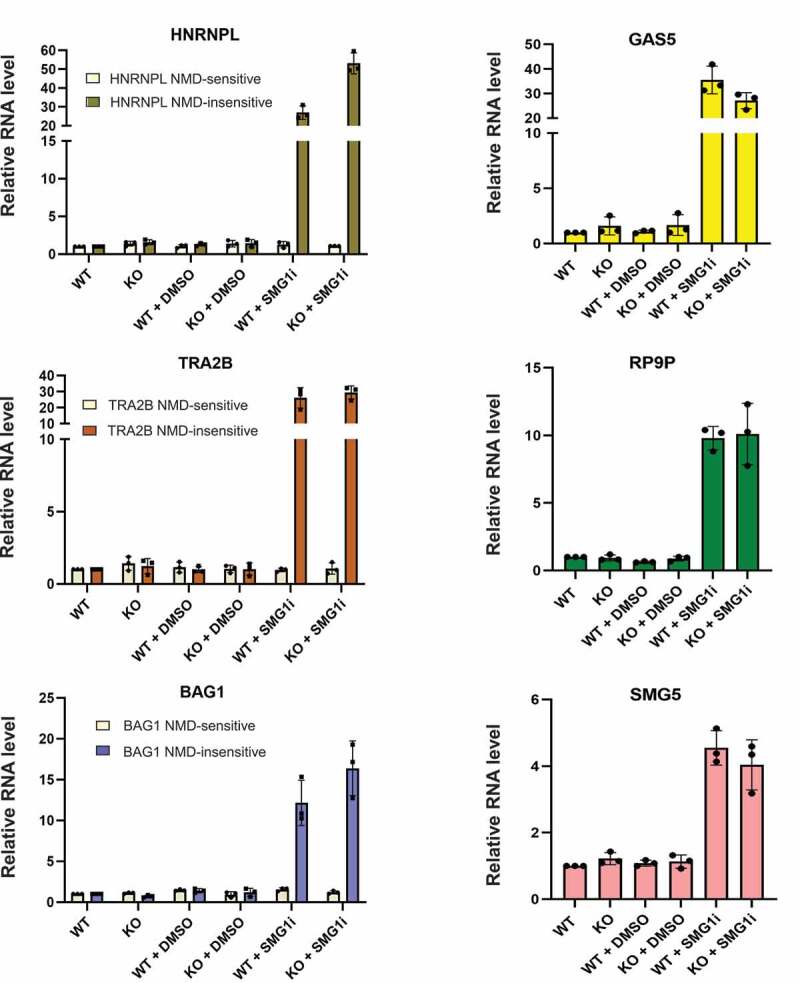
RT-qPCR analysis of known NMD substrates.
RT-qPCR analysis showing relative mRNA levels of six NMD-sensitive (HNRNPL, TRA2B and BAG1 NMD-sensitive splice isoforms and GAS5, RP9P, SMG5) and three NMD-insensitive transcripts (HNRNPL, TRA2B and BAG1 protein-coding splice isoforms) in ZNF598 WT and KO cells, normalized to β-actin mRNA. Cells were either untreated (WT, KO) or treated with DMSO or 0.6 μM SMG1 inhibitor for 24 hours. Data was analysed as in Fig. 4B, mean values and SEM of three independent experiments are shown.
7. Conclusion and open questions
As discussed herein, cells response to many kinds of stresses mainly by reducing the overall energy consumption through inhibition of global translation, while ensuring continued protein synthesis of stress response genes. ISR hereby appears to be the central stress response pathway that shares kinase sensors of other more specialized stress response pathways. There is a fine balance between the activation of cell survival programs and the induction of apoptosis under conditions of repeated, chronic stress that prevents cells from restoring normal growth. The overall frequency of ribosome collisions in a cell is sensed and appears to be an important determinant for activation of specific stress response pathways that ultimately decide between promotion of cell survival or activation of apoptosis. Additionally, mRNA surveillance pathways were also reported to modulate the expression of ISR and UPR specific genes [128–130]. Several studies reported that aberrant transcripts generated in yeast during oxidative stress are cleared by NSD, whereas NGD influences the translation of stress response genes to sustain ER homoeostasis [128,129]. Moreover, NMD is involved in regulating UPR by degrading mRNAs encoding UPR components to prevent UPR activation by physiological, innocuous levels of unfolded proteins and in terminating the UPR response after alleviation of ER stress [130,131]. Vice versa, UPR inhibits NMD through eIF2α phosphorylation. Still much more research is needed to fully elucidate the intricate links between stress response pathways, translation regulation, mRNA surveillance and RQC at the molecular level. Providing one small piece to that puzzle, we showed here using ZNF598 KO cells that the ribosome collision sensor ZNF598 is not involved in NMD. This finding together with another recent report showing that the knockout of EDF1, another RQC factor involved in ribosome collision signalling, also does not affect NMD provides evidence against the idea of ribosome collisions occurring at PTCs and is consistent with recent findings indicating that that NMD is not triggered by stable ribosome stalling at PTCs [122]. Thus, the longstanding question about the mechanistic differences between ‘normal’ translation termination and translation termination at TCs that activate NMD still remains to be solved.
8. Material and methods
Characterization of ZNF598KO cells by sequencing of genomic DNA and immuno blotting
Flp-In T-Rex WT and ZNF598 KO cells (a kind gift of Dr. Aitor Garzia) [14] were cultured in DMEM supplemented with 10% foetal calf serum (FCS) and antibiotics. Cells were grown in 5% CO2 at 37°C. Genomic DNA was isolated using a spin column-based kit according to manufacturer’s protocol (ZYMO RESEARCH). The edited region of the ZNF598 gene was PCR amplified from the genomic DNA using CloneAmp HiFi premix and primer pair Sde57/Sde59 (see Table 1; Microsynth), which binds to exon 3 and the downstream adjacent intron. PCR products were purified from the agarose gel and sequenced using primer Sde59.
Table 1.
Primer sequences
| (A) Primers used to amplify genomic DNA of WT and ZNF598KO cells | ||
|
Primer name |
Sequence |
|
| Sde 57 (Fwd) | 5’- GCTGTGCTGCGGAGACCTG −3’ | |
| Sde 59 (Rev) |
5’- CAGGCCTCTCTCGGAAGCCG −3’ |
|
| (B) Transcript isoform-specific primers used for qRT-PCR | ||
|
Transcript |
Primer name and sequence |
|
| ZNF598 | Fwd (Sde34): 5’- AACCTCGACAAATGGTCCTG −3’ Rev (Sde35): 5’- GTCTTCGTCCTTGAGCTTCG −3’ |
|
| Hnrnpl_NMD-insensitive | Fwd (LC50): 5’- CAATCTCAGTGGACAAGGTG −3’ Rev (LC51): 5’- CTCCATATTCTGCGGGGTGA −3’ |
|
| Hnrnpl_NMD-sensitive | Fwd (LC47): 5’- GGTCGCAGTGTATGTTTGATG −3’ Rev (LC52): 5’- GGCGTTTGTTGGGGTTGCT −3’ |
|
| TRA2B_NMD-insensitive | Fwd (LC54): 5’- GAGGTTGGCAGCTTCGATT −3’ Rev (LC42): 5’- AAGCAGAACGGGATTCCC −3’ |
|
| TRA2B_NMD-sensitive | Fwd (LC43): 5’- TGGAATCAGAAAGCACTACGC −3’ Rev (LC55): 5’- GAATCTTCCTTGGAGCGAGA −3’ |
|
| BAG1_NMD-insensitive | Fwd (EK141): 5’- ACTCATATTTAAGGGAAAATCTCTG −3’ Rev (EK38): 5’- TTGGGCAGAAAACCCTGCTG −3’ |
|
| BAG1_NMD-sensitive | Fwd (EK139): 5’- CATATTTAAGGTTCTTCAACAGATA −3’ Rev (EK140): 5’- TGTTTCCATTTCCTTCAGAGA −3’ |
|
| GAS5 | Fwd (Schwi292): 5’- GCACCTTATGGACAGTTG −3’ Rev (Schwi293): 5’- GGAGCAGAACCATTAAGC −3’ |
|
| RP9P | Fwd (OM368): 5’- CAAGCGCCTGGAGTCCTTAA −3’ Rev (OM369): 5’- AGGAGGTTTTTCATAACTCGTGATCT −3’ |
|
| SMG5 | Fwd (SRE237): 5’- CCAGTGGCCGCTTCATTGTC −3’ Rev (SRE238): 5’- TGCCTCCAGGTACCGAATCC- 3’ |
|
| Actin β | Fwd (SRE61): 5’- TCCATCATGAAGTGTGACGT −3’ Rev (SRE62): 5’- TACTCCTGCTTGCTGATCCAC −3’ |
|
The knockout of ZNF598 was verified by western blotting. Briefly, 2 × 105 cell equivalents were separated on a precasted 8% Tris-Glycine SDS mini gel (Invitrogen), electroblotted to a nitrocellulose membrane and detection was performed using anti-ZNF598 antibody (GeneTex GTX119245, 1:1000 dilution). Anti-tyrosine tubulin antibody (Sigma T9028, 1: 5000 dilution) was used as loading control.
Quantitative RT-PCR
Flp-In T-Rex WT and ZNF598 KO cells were treated with either DMSO or 0.6 μM SMG1 inhibitor for 24 hours [126]. Total RNA of treated or untreated cells were extracted using TRI-reagent followed by isopropanol precipitation. cDNA was generated by reverse transcription using AffinityScript Multiple Temperature Reverse Transcriptase (Agilent). qPCR was performed using Brilliant III UltraFast SYBR Green qPCR Master Mix (Agilent) with the specific primer pairs indicated in Table 1.
Acknowledgments
We thank our lab members for stimulating discussions.
Funding Statement
S.D. is supported by a grant of the Novartis Foundation for Biomedical Research. The research in the lab of O.M. is further supported by the National Center of Competence in Research (NCCR) on RNA & Disease funded by the Swiss National Science Foundation (SNSF; 51NF40-182880), by the SNSF grant 310030-204161, and by the Kanton Bern.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Hoozemans JJ, Scheper W.. Endoplasmic reticulum: the unfolded protein response is tangled in neurodegeneration. Int J Biochem Cell Biol. 2012;44(8):1295–1298. Epub 20120504. DOI: 10.1016/j.biocel.2012.04.023. [DOI] [PubMed] [Google Scholar]
- [2].Giampietri C, Petrungaro S, Conti S, et al. Cancer Microenvironment and Endoplasmic Reticulum Stress Response. Mediators Inflamm. 2015;2015:417281. Epub 20150927. DOI: 10.1155/2015/417281. Epub 20150927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hasmim M, Messai Y, Ziani L, et al. Critical Role of Tumor Microenvironment in Shaping NK Cell Functions: Implication of Hypoxic Stress. Front Immunol. 2015;6:482. Epub 20150923. DOI: 10.3389/fimmu.2015.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chan SW. The unfolded protein response in virus infections. Front Microbiol. 2014;5:518. Epub 20140930. DOI: 10.3389/fmicb.2014.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Duwi Fanata WI, Lee SY, Lee KO. The unfolded protein response in plants: a fundamental adaptive cellular response to internal and external stresses. J Proteomics. 2013;93:356–368. Epub 20130425. DOI: 10.1016/j.jprot.2013.04.023. [DOI] [PubMed] [Google Scholar]
- [6].Chen F, Evans A, Pham J, et al. Cellular stress responses: a balancing act. Mol Cell. 2010;40:175. [DOI] [PubMed] [Google Scholar]
- [7].Fulda S, Gorman AM, Hori O, et al. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Galluzzi L, Yamazaki T, Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol. 2018;19:731–745. [DOI] [PubMed] [Google Scholar]
- [9].Yan LL, Zaher HS. How do cells cope with RNA damage and its consequences? J Biol Chem. 2019;294:15158–15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Morris C, Cluet D, Ricci EP. Ribosome dynamics and mRNA turnover, a complex relationship under constant cellular scrutiny. Wiley Interdiscip Rev RNA. 2021;12:e1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Iordanov MS, Pribnow D, Magun JL, et al. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol. 1997;17:3373–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu CC, Peterson A, Zinshteyn B, et al. Ribosome collisions trigger general stress responses to regulate fate. Cell. 2020;182:404–416 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Joazeiro CAP. Mechanisms and functions of ribosome-associated protein quality control. Nat Rev Mol Cell Biol. 2019;20:368–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Garzia A, Jafarnejad SM, Meyer C, et al. The E3 ubiquitin ligase and RNA-binding protein ZNF598 orchestrates ribosome quality control of premature polyadenylated mRNAs. Nat Commun. 2017;8:16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Matsuo Y, Ikeuchi K, Saeki Y, et al. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat Commun. 2017;8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hickey KL, Dickson K, Cogan JZ, et al. GIGYF2 and 4EHP inhibit translation initiation of defective messenger rnas to assist ribosome-associated quality control. Mol Cell. 2020;79:950–962 e956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Juszkiewicz S, Slodkowicz G, Lin Z, et al. 2020. Ribosome collisions trigger cis-acting feedback inhibition of translation initiation. eLife. 9; DOI: 10.7554/eLife.60038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sinha NK, Ordureau A, Best K, et al. EDF1 coordinates cellular responses to ribosome collisions. eLife. 2020;9. DOI: 10.7554/eLife.58828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Han NC, Kelly P, Ibba M. Translational quality control and reprogramming during stress adaptation. Exp Cell Res. 2020;394:112161. [DOI] [PubMed] [Google Scholar]
- [20].Liu B, Qian SB. Translational reprogramming in cellular stress response. Wiley Interdiscip Rev RNA. 2014;5:301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Duncan CDS, Mata J. Effects of cycloheximide on the interpretation of ribosome profiling experiments in Schizosaccharomyces pombe. Sci Rep. 2017;7:10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Moro SG, Hermans C, Ruiz-Orera J, et al. Impact of uORFs in mediating regulation of translation in stress conditions. BMC Mol Cell Biol. 2021;22(29). DOI: 10.1186/s12860-021-00363-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rubio A, Ghosh S, Mulleder M, et al. Ribosome profiling reveals ribosome stalling on tryptophan codons and ribosome queuing upon oxidative stress in fission yeast. Nucleic Acids Res. 2021;49:383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pakos-Zebrucka K, Koryga I, Mnich K, et al. The integrated stress response. EMBO Rep. 2016;17:1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Costa-Mattioli M, Walter P. 2020. The integrated stress response: from mechanism to disease. Science. 368; DOI: 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. [DOI] [PubMed] [Google Scholar]
- [27].Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bogorad AM, Lin KY, Marintchev A. Novel mechanisms of eIF2B action and regulation by eIF2alpha phosphorylation. Nucleic Acids Res. 2017;45:11962–11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kenner LR, Anand AA, Nguyen HC, et al. eIF2B-catalyzed nucleotide exchange and phosphoregulation by the integrated stress response. Science. 2019;364:491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–450. [DOI] [PubMed] [Google Scholar]
- [31].Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou D, Palam LR, Jiang L, et al. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem. 2008;283:7064–7073. [DOI] [PubMed] [Google Scholar]
- [34].Palam LR, Baird TD, Wek RC. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem. 2011;286:10939–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee YY, Cevallos RC, Jan E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J Biol Chem. 2009;284:6661–6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Di Prisco GV, Huang W, Buffington SA, et al. Translational control of mGluR-dependent long-term depression and object-place learning by eIF2alpha. Nat Neurosci. 2014;17:1073–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Morris DR, Geballe AP. Upstream open reading frames as regulators of mRNA translation. Mol Cell Biol. 2000;20:8635–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jaud M, Philippe C, Di Bella D, et al. Translational regulations in response to endoplasmic reticulum stress in cancers. Cells. 2020;9:540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. [DOI] [PubMed] [Google Scholar]
- [41].Bhattarai KR, Riaz TA, Kim HR, et al. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp Mol Med. 2021;53:151–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kopp MC, Larburu N, Durairaj V, et al. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat Struct Mol Biol. 2019;26:1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. [DOI] [PubMed] [Google Scholar]
- [44].Yoshida H, Matsui T, Yamamoto A, et al. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. [DOI] [PubMed] [Google Scholar]
- [45].Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. [DOI] [PubMed] [Google Scholar]
- [46].Shen J, Chen X, Hendershot L, et al. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. [DOI] [PubMed] [Google Scholar]
- [47].Merksamer PI, Papa FR. The UPR and cell fate at a glance. J Cell Sci. 2010;123:1003–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Garreau de Loubresse N, Prokhorova I, Holtkamp W, et al. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. [DOI] [PubMed] [Google Scholar]
- [49].Vind AC, Genzor AV, Bekker-Jensen S. Ribosomal stress-surveillance: three pathways is a magic number. Nucleic Acids Res. 2020;48:10648–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Iordanov MS, Pribnow D, Magun JL, et al. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J Biol Chem. 1998;273:15794–15803. [DOI] [PubMed] [Google Scholar]
- [51].Tesh VL. Activation of cell stress response pathways by Shiga toxins. Cell Microbiol. 2012;14:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. [DOI] [PubMed] [Google Scholar]
- [53].Jandhyala DM, Ahluwalia A, Obrig T, et al. ZAK: a MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell Microbiol. 2008;10:1468–1477. [DOI] [PubMed] [Google Scholar]
- [54].Wang X, Mader MM, Toth JE, et al. Complete inhibition of anisomycin and UV radiation but not cytokine induced JNK and p38 activation by an aryl-substituted dihydropyrrolopyrazole quinoline and mixed lineage kinase 7 small interfering RNA. J Biol Chem. 2005;280:19298–19305. [DOI] [PubMed] [Google Scholar]
- [55].Duch A, de Nadal E, Posas F. The p38 and Hog1 SAPKs control cell cycle progression in response to environmental stresses. FEBS Lett. 2012;586:2925–2931. [DOI] [PubMed] [Google Scholar]
- [56].Darling NJ, Cook SJ. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta. 2014;1843:2150–2163. [DOI] [PubMed] [Google Scholar]
- [57].Derijard B, Hibi M, Wu IH, et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. [DOI] [PubMed] [Google Scholar]
- [58].Gupta S, Campbell D, Derijard B, et al. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. [DOI] [PubMed] [Google Scholar]
- [59].van Dam H, Wilhelm D, Herr I, et al. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995;14:1798–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Whitmarsh AJ, Shore P, Sharrocks AD, et al. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. [DOI] [PubMed] [Google Scholar]
- [61].Zinck R, Cahill MA, Kracht M, et al. Protein synthesis inhibitors reveal differential regulation of mitogen-activated protein kinase and stress-activated protein kinase pathways that converge on Elk-1. Mol Cell Biol. 1995;15:4930–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Darnell AM, Subramaniam AR, O’Shea EK. Translational control through differential ribosome pausing during amino acid limitation in mammalian cells. Mol Cell. 2018;71:229–243 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ikeuchi K, Tesina P, Matsuo Y, et al. Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 2019;38. DOI: 10.15252/embj.2018100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Juszkiewicz S, Chandrasekaran V, Lin Z, et al. ZNF598 Is a quality Control Sensor of Collided Ribosomes. Mol Cell. 2018;72:469–481 e467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sundaramoorthy E, Leonard M, Mak R, et al. ZNF598 and RACK1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol Cell. 2017;65:751–760 e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Shoemaker CJ, Green R. Translation drives mRNA quality control. Nat Struct Mol Biol. 2012;19:594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Morita M, Ler LW, Fabian MR, et al. A novel 4EHP-GIGYF2 translational repressor complex is essential for mammalian development. Mol Cell Biol. 2012;32:3585–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Karousis ED, Muhlemann O. Nonsense-Mediated mRNA decay begins where translation ends. Cold Spring Harb Perspect Biol. 2019;11:a032862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kishor A, Fritz SE, Hogg JR. Nonsense-mediated mRNA decay: the challenge of telling right from wrong in a complex transcriptome. Wiley Interdiscip Rev RNA. 2019;10:e1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kurosaki T, Popp MW, Maquat LE. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol. 2019;20:406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Meydan S, Guydosh NR. A cellular handbook for collided ribosomes: surveillance pathways and collision types. Curr Genet. 2021;67:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Frischmeyer PA, van Hoof A, O’Donnell K, et al. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. [DOI] [PubMed] [Google Scholar]
- [73].van Hoof A, Frischmeyer PA, Dietz HC, et al. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–2264. [DOI] [PubMed] [Google Scholar]
- [74].D’Orazio KN, Wu CC, Sinha N, et al. 2019. The endonuclease Cue2 cleaves mRNAs at stalled ribosomes during no go decay. eLife. 8; DOI: 10.7554/eLife.49117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Graille M, Seraphin B. Surveillance pathways rescuing eukaryotic ribosomes lost in translation. Nat Rev Mol Cell Biol. 2012;13:727–735. [DOI] [PubMed] [Google Scholar]
- [76].Losson R, Lacroute F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc Natl Acad Sci U S A. 1979;76:5134–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Maquat LE, Kinniburgh AJ, Rachmilewitz EA, et al. Unstable beta-globin mRNA in mRNA-deficient beta o thalassemia. Cell. 1981;27:543–553. 0092-8674(81)90396-2 [pii]. [DOI] [PubMed] [Google Scholar]
- [78].Peltz SW, Brown AH, Jacobson A. mRNA destabilization triggered by premature translational termination depends on at least three cis-acting sequence elements and one trans-acting factor. Genes Dev. 1993;7:1737–1754. [DOI] [PubMed] [Google Scholar]
- [79].Hug N, Longman D, Caceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016;44:1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Colombo M, Karousis ED, Bourquin J, et al. Transcriptome-wide identification of NMD-targeted human ms reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA. 2017;23:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].He F, Li X, Spatrick P, et al. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5’ to 3’ mRNA decay pathways in yeast. Mol Cell. 2003;12:1439–1452. [DOI] [PubMed] [Google Scholar]
- [82].Mendell JT, Sharifi NA, Meyers JL, et al. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–1078. [DOI] [PubMed] [Google Scholar]
- [83].Rehwinkel J, Letunic I, Raes J, et al. Nonsense-mediated m decay factors act in concert to regulate common m targets. RNA. 2005;11:1530–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Yepiskoposyan H, Aeschimann F, Nilsson D, et al. Autoregulation of the nonsense-mediated m decay pathway in human cells. RNA. 2011;17:2108–2118. rna.030247.111 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Denning G, Jamieson L, Maquat LE, et al. Cloning of a novel phosphatidylinositol kinase-related kinase: characterization of the human SMG-1 RNA surveillance protein. J Biol Chem. 2001;276:22709–22714. [DOI] [PubMed] [Google Scholar]
- [86].Grimson A, O’Connor S, Newman CL, et al. SMG-1 is a phosphatidylinositol kinase-related protein kinase required for nonsense-mediated mRNA Decay in Caenorhabditis elegans. Mol Cell Biol. 2004;24:7483–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kashima I, Yamashita A, Izumi N, et al. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006;20:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Yamashita A, Ohnishi T, Kashima I, et al. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 2001;15:2215–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Amrani N, Ganesan R, Kervestin S, et al. A faux 3′-UTR promotes aberrant termination and triggers nonsense- mediated mRNA decay. Nature. 2004;432:112–118. [DOI] [PubMed] [Google Scholar]
- [90].Behm-Ansmant I, Gatfield D, Rehwinkel J, et al. A conserved role for cytoplasmic poly(A)-binding protein 1 (PABPC1) in nonsense-mediated mRNA decay. EMBO J. 2007;26:1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Eberle AB, Stalder L, Mathys H, et al. Posttranscriptional gene regulation by spatial rearrangement of the 3’ untranslated region. PLoS Biol. 2008;6:e92. 07-PLBI-RA-3782 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Ivanov PV, Gehring NH, Kunz JB, et al. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 2008;27:736–747. emboj200817 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Silva AL, Ribeiro P, Inacio A, et al. Proximity of the poly(A)-binding protein to a premature termination codon inhibits mammalian nonsense-mediated m decay. RNA. 2008;14:563–576. rna.815108 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Singh G, Rebbapragada I, Lykke-Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 2008;6:e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99. [DOI] [PubMed] [Google Scholar]
- [96].Gehring NH, Lamprinaki S, Kulozik AE, et al. Disassembly of exon junction complexes by PYM. Cell. 2009;137:536–548. S0092-8674(09)00256-6 [pii]. [DOI] [PubMed] [Google Scholar]
- [97].Le Hir H. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 2000;19:6860–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sauliere J, Murigneux V, Wang Z, et al. CLIP-seq of eIF4AIII reveals transcriptome-wide mapping of the human exon junction complex. Nat Struct Mol Biol. 2012;19:1124–1131. nsmb.2420 [pii]. [DOI] [PubMed] [Google Scholar]
- [99].Singh G, Kucukural A, Cenik C, et al. The ular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012;151:750–764. S0092-8674(12)01220-2 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Rebbapragada I, Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr Opin Cell Biol. 2009;21:394–402. S0955-0674(09)00061-1 [pii]. [DOI] [PubMed] [Google Scholar]
- [101].Stalder L, Muhlemann O. The meaning of nonsense. Trends Cell Biol. 2008;18:315–321. S0962-8924(08)00139-6 [pii]. [DOI] [PubMed] [Google Scholar]
- [102].Chakrabarti S, Jayachandran U, Bonneau F, et al. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol Cell. 2011;41:693–703. [DOI] [PubMed] [Google Scholar]
- [103].Melero R, Buchwald G, Castano R, et al. The cryo-EM structure of the UPF-EJC complex shows UPF1 poised toward the RNA 3’ end. Nat Struct Mol Biol. 2012;19:498–505. S491-492. nsmb.2287 [pii]. [DOI] [PubMed] [Google Scholar]
- [104].Fiorini F, Bonneau F, Le Hir H. Biochemical characterization of the RNA helicase UPF1 involved in nonsense-mediated mRNA decay. Methods Enzymol. 2012;511:255–274. B978-0-12-396546-2.00012-7 [pii]. [DOI] [PubMed] [Google Scholar]
- [105].Okada-Katsuhata Y, Yamashita A, Kutsuzawa K, et al. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012;40:1251–1266. gkr791 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Yamashita A, Izumi N, Kashima I, et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 2009;23:1091–1105. 23/9/1091 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Eberle AB, Lykke-Andersen S, Muhlemann O, et al. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol. 2009;16:49–55. nsmb.1530[pii]. [DOI] [PubMed] [Google Scholar]
- [108].Huntzinger E, Kashima I, Fauser M, et al. SMG6 is the catalytic endonuclease that cleaves ms containing nonsense codons in metazoan. RNA. 2008;14:2609–2617. rna.1386208 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Jonas S, Weichenrieder O, Izaurralde E. An unusual arrangement of two 14-3-3-like domains in the SMG5-SMG7 heterodimer is required for efficient nonsense-mediated mRNA decay. Genes Dev. 2013;27:211–225. 27/2/211 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Lykke-Andersen S, Chen Y, Ardal BR, et al. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev. 2014;28:2498–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Buhler M, Steiner S, Mohn F, et al. EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3’ UTR length. Nat Struct Mol Biol. 2006;13:462–464. [DOI] [PubMed] [Google Scholar]
- [112].Gatfield D. Nonsense-mediated mRNA decay in Drosophila: at the intersection of the yeast and mammalian pathways. EMBO J. 2003;22:3960–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Longman D, Plasterk RH, Johnstone IL, et al. Mechanistic insights and identification of two novel factors in the C. elegans NMD pathway. Genes Dev. 2007;21:1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Muhlemann O, Jensen TH. mRNP quality control goes regulatory. Trends Genet. 2012;28:70–77. S0168-9525(11)00185-5 [pii]. [DOI] [PubMed] [Google Scholar]
- [115].Hogg JR, Goff SP. Upf1 senses 3ʹUTR length to potentiate mRNA decay. Cell. 2010;143:379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Karousis ED, Gypas F, Zavolan M, et al. Nanopore sequencing reveals endogenous NMD-targeted isoforms in human cells. Genome Biol. 2021;22:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Tani H, Imamachi N, Salam KA, et al. Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability. RNA Biol. 2012;9:1370–1379. 22360 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Fritz SE, Ranganathan S, Wang CD, et al. The RNA-binding protein PTBP1 promotes ATPase-dependent dissociation of the RNA helicase UPF1 to protect transcripts from nonsense-mediated mRNA decay. J Biol Chem. 2020;295:11613–11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Ge Z, Quek BL, Beemon KL, et al. Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. eLife. 2016;5:e11155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kishor A, Ge Z, Hogg JR. hnRNP L-dependent protection of normal mRNAs from NMD subverts quality control in B cell lymphoma. EMBO J. 2018;38. DOI: 10.15252/embj.201899128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Peixeiro I, Inacio A, Barbosa C, et al. Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG-proximal nonsense mutations. Nucleic Acids Res. 2012;40:1160–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Karousis ED, Gurzeler LA, Annibaldis G, et al. Human NMD ensues independently of stable ribosome stalling. Nat Commun. 2020;11:4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kuroha K, Ando K, Nakagawa R, et al. The Upf factor complex interacts with aberrant products derived from mRNAs containing a premature termination codon and facilitates their proteasomal degradation. J Biol Chem. 2013;288:28630–28640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Park Y, Park J, Hwang HJ, et al. Nonsense-mediated mRNA decay factor UPF1 promotes aggresome formation. Nat Commun. 2020;11:3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Inglis AJ, Galvez Merchan G, Pal A, et al. Coupled protein quality control during nonsense mediated mRNA decay. bioRxiv. 2022. DOI: 10.1101/2021.12.22.473893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Gopalsamy A, Bennett EM, Shi M, et al. Identification of pyrimidine derivatives as hSMG-1 inhibitors. Bioorg Med Chem Lett. 2012;22:6636–6641. [DOI] [PubMed] [Google Scholar]
- [127].Zinshteyn B, Sinha NK, Enam SU, et al. Translational repression of NMD targets by GIGYF2 and EIF4E2. PLoS Genet. 2021;17:e1009813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Guydosh NR, Kimmig P, Walter P, et al. 2017. Regulated Ire1-dependent mRNA decay requires no-go mRNA degradation to maintain endoplasmic reticulum homeostasis in S. pombe. eLife. 6; DOI: 10.7554/eLife.29216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Jamar NH, Kritsiligkou P, Grant CM. The non-stop decay mRNA surveillance pathway is required for oxidative stress tolerance. Nucleic Acids Res. 2017;45:6881–6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Nasif S, Contu L, Muhlemann O. Beyond quality control: the role of nonsense-mediated mRNA decay (NMD) in regulating gene expression. Semin Cell Dev Biol. 2018;75:78–87. [DOI] [PubMed] [Google Scholar]
- [131].Goetz AE, Wilkinson M. Stress and the nonsense-mediated RNA decay pathway. Cell Mol Life Sci. 2017;74:3509–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]