

Abstract
Lysosome dysfunction is a shared feature of rare lysosomal storage diseases and common age-related neurodegenerative diseases. Microglia, the brain-resident macrophages, are particularly vulnerable to lysosome dysfunction because of the phagocytic stress of clearing dying neurons, myelin, and debris. CD22 is a negative regulator of microglial homeostasis in the aging mouse brain, and soluble CD22 (sCD22) is increased in the cerebrospinal fluid of patients with Niemann-Pick type C disease (NPC). However, the role of CD22 in the human brain remains unknown. In contrast to previous findings in mice, here, we show that CD22 is expressed by oligodendrocytes in the human brain and binds to sialic acid–dependent ligands on microglia. Using unbiased genetic and proteomic screens, we identify insulin-like growth factor 2 receptor (IGF2R) as the binding partner of sCD22 on human myeloid cells. Targeted truncation of IGF2R revealed that sCD22 docks near critical mannose 6-phosphate–binding domains, where it disrupts lysosomal protein trafficking. Interfering with the sCD22-IGF2R interaction using CD22 blocking antibodies ameliorated lysosome dysfunction in human NPC1 mutant induced pluripotent stem cell–derived microglia-like cells without harming oligodendrocytes in vitro. These findings reinforce the differences between mouse and human microglia and provide a candidate microglia-directed immunotherapeutic to treat NPC.
INTRODUCTION
Lysosomes are specialized organelles (1) that degrade and recycle macromolecules, connecting catabolic and anabolic metabolism. Although all tissues require lysosomes to maintain homeostasis, the metabolically demanding brain is especially reliant on lysosome function as evident by the neurological phenotypes of many lysosomal storage diseases (LSDs) (2). Neuropathological changes in rare inherited LSDs resemble those in common age-related neurodegenerative diseases. Both entities involve accumulation of lysosomal cargo due to increased substrate production, impaired substrate degradation, or reduced clearance of end products (3). For example, autosomal recessive mutations in the glucocerebrosidase gene, which encodes a lysosomal enzyme necessary for glycosphingolipid degradation, cause Gaucher’s disease, whereas heterozygous carriers are predisposed to Parkinson’s disease (4). Even when the underlying genetic etiology is different, LSDs and age-related neurodegenerative diseases display unexpected molecular overlap. Both Niemann-Pick type C disease (NPC) and Alzheimer’s disease involve protein aggregation, neuroinflammation, and disrupted lipid metabolism (5, 6, 7, 8, 9).
Patients with NPC, caused by autosomal recessive mutations in NPC1 (95% of cases) or NPC2, typically present in early childhood with motor deficits and gradual cognitive impairment. These clinical signs are associated with neurodegeneration, especially in the cerebellum, and demyelination (10). Progressive neurological decline leads to premature death in the second or third decade (11). There are currently no Food and Drug Administration (FDA)–approved therapies for NPC, necessitating new treatment approaches.
Whereas Purkinje cell loss is the classic histological finding of NPC, microglial activation has been shown to precede neurodegeneration in mouse models of disease (12–16), and inflamed microglia are found near neurons destined for death in NPC patient brains (17). These studies suggest that the neuron-extrinsic effects of dysfunctional microglia are likely key contributors to disease pathogenesis and important considerations for therapeutic development.
We previously found that CD22 is up-regulated on microglia in the aging mouse brain where it inhibits phagocytosis of protein aggregates and myelin debris (18). CD22 blockade restored microglial homeostasis and improved cognitive function in aged mice. In addition, soluble CD22 (sCD22) was found to be increased in the cerebrospinal fluid (CSF) of patients with NPC (12). Motivated by these findings, we set out to understand the role of CD22 in the human brain and to determine the translational potential of blocking CD22 to treat NPC.
RESULTS
Oligodendrocyte-derived sCD22 binds sialic acid ligands on microglia
To determine the source of sCD22 in NPC patient CSF, we mined a publicly available human single-nucleus RNA sequencing (snRNA-seq) atlas (Brain Atlas) (19). We found that CD22 is almost exclusively expressed by oligodendrocytes in the human brain (Fig. 1, A and B). This contrasts our previous findings that CD22 is expressed by microglia in the mouse brain (18), reinforcing divergence of microglial phenotypes between species. CD22 is detected in both microglia (20) and oligodendrocytes (21) from nonhuman primates (fig. S1, A and B), indicative of an evolutionary trajectory. To confirm CD22 expression in human tissue, we performed multiplexed RNA in situ hybridization (RNAscope) to probe for CD22, MOG, encoding myelin oligodendrocyte glycoprotein, and AIF1, encoding the microglia marker ionized calcium-binding adapter molecule 1 (IBA1). We detected CD22 transcripts exclusively in MOG+ oligodendrocytes and not AIF1+ microglia (Fig. 1C), aligning with the Brain Atlas. Next, we performed quantitative flow cytometry on fresh primary human cortical tissue to interrogate the native cell surface proteome of live brain cells (Fig. 1D). Whereas CD22 protein was barely detectable on the surface of microtubule-associated protein 2–positive (MAP2+) neurons, CD45+ microglia, and O4+MBP− oligodendrocyte precursor cells (OPCs), O4+MBP+ oligodendrocytes displayed high numbers of cell surface CD22 molecules similar to B cells (Fig. 1, E and F, and fig. S1C). In the absence of peripheral immune cell invasion (12), these data suggest that oligodendrocytes are the main source of sCD22 in human CSF.
Fig. 1. Oligodendrocyte-derived sCD22 binds sialic acid ligands on microglia.
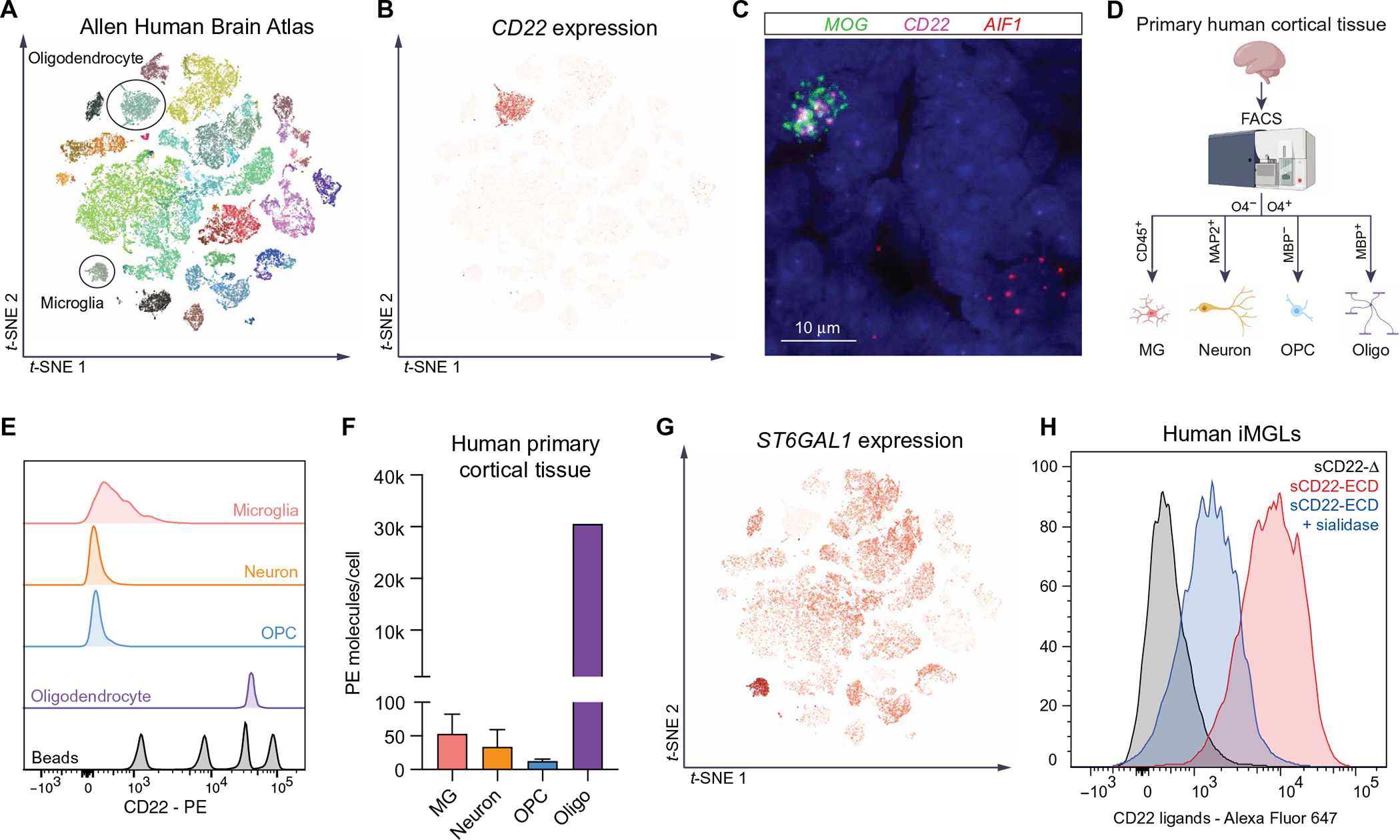
(A) t-distributed stochastic neighbor embedding (t-SNE) visualization of snRNA-seq data (Smart-seq) from multiple cortical areas of human brain colored by cell type. Data from Allen Brain Atlas (19). (B) t-SNE visualization of snRNA-seq data from multiple cortical areas of human brain colored by CD22 expression. (C) Representative image of human brain tissue probed for MOG (green), CD22 (magenta), and AIF1 (red) transcripts by multiplexed fluorescent RNAscope. Clustered puncta within 4′,6-diamidino-2-phenylindole–positive nuclei suggest true signal. (D) Schematic of FACS analysis of various cell types from fresh human primary cortical tissue. (E) Flow cytometry analysis of surface CD22 protein expression in CD45+ microglia (pink), MAP2+ neurons (orange), O4+MBP− OPCs (blue) and O4+MBP+ oligodendrocytes (purple) from fresh human primary cortical tissue (PCW 22). PE quantification beads are shown in gray. (F) Quantification of CD22-PE molecules bound to the surface of various human brain cell types calculated using PE bead standards (n = 2 biological replicates; PCWs 20 to 22; O4+MBP+ cells only detected at PCW 22). (G) t-SNE visualization of snRNA-seq data from multiple cortical areas of human brain colored by ST6GAL1 expression. (H) Flow cytometry analysis of human iMGLs stained with fluorophore-conjugated CD22 lacking its sialic acid–binding domain (sCD22-Δ, gray) or the full-length CD22 ECD (sCD22-ECD, red). In one condition, cells were pretreated with sialidase before sCD22-ECD staining (blue).
sCD22 has previously been used as a biomarker for sepsis and B cell malignancies (22, 23) and has been proposed as a biomarker of neurodegeneration in NPC (12). However, given our understanding of membrane-bound CD22 signaling and function in mice (18), we asked whether sCD22 might bind ligands in the human central nervous system (CNS) to modulate brain function. We queried the Brain Atlas for the expression of ST6GAL1, a glycosyltransferase that attaches sialic acid to growing glycan chains with an α2,6-linkage, the preferred glycan ligand for CD22 (24, 25). We found that ST6GAL1 is specifically enriched in microglia in the human CNS (Fig. 1G), suggesting that ligands on microglia might be the target of sCD22 (fig. S1D). Using fluorophore-conjugated recombinant CD22 as a staining reagent, we found that human induced pluripotent stem cell (iPSC)–derived microglia-like cells (iMGLs) and fresh autopsy-derived primary microglia express high amounts of sCD22 ligands that are sensitive to sialidase treatment (Fig. 1H and fig. S1E). In contrast, sCD22 did not bind to the surface of freshly isolated neurons or fibroblasts from patients with NPC, which minimally express ST6GAL1 (fig. S1, F and G). Together, these data support a model in which oligodendrocyte-derived sCD22 specifically binds sialic acid–dependent ligands on microglia in the human CNS (fig. S1, H and I) and raise the hypothesis that sCD22 modulates microglial function.
Genetic and proteomic screens elucidate CD22-IGF2R interaction
To identify sCD22-binding partners on microglia, we performed a genome-wide CRISPR-Cas9 knockout (KO) screen using the human myeloid cell line, U937 (Fig. 2A). U937 cells have previously been used to model human microglia (26) and express CD22 ligands (fig. S2A). Cas9-expressing U937 cells were infected with a library of single-guide RNAs (sgRNAs) targeting all protein-coding genes, with 10 distinct sgRNAs per gene and ~10,000 negative control sgRNAs. We stained this pool of stable single-KO cells with fluorophore-conjugated recombinant CD22 and sorted cells with high (top 5%) and low (bottom 5%) CD22 ligand expression by fluorescence-activated cell sorting (FACS) (fig. S2A). Next, we sequenced genomic DNA from each population, compared sgRNA distribution between populations, and estimated the effect size and P value for each gene KO using casTLE (27). This screen identified 66 hits with a false discovery rate of less than 10%, including ST6GAL1, a known genetic modifier of CD22 glycan ligand synthesis (Fig. 2B and table S1) (28).
Fig. 2. Genetic and proteomic screens elucidate CD22-IGF2R interaction.
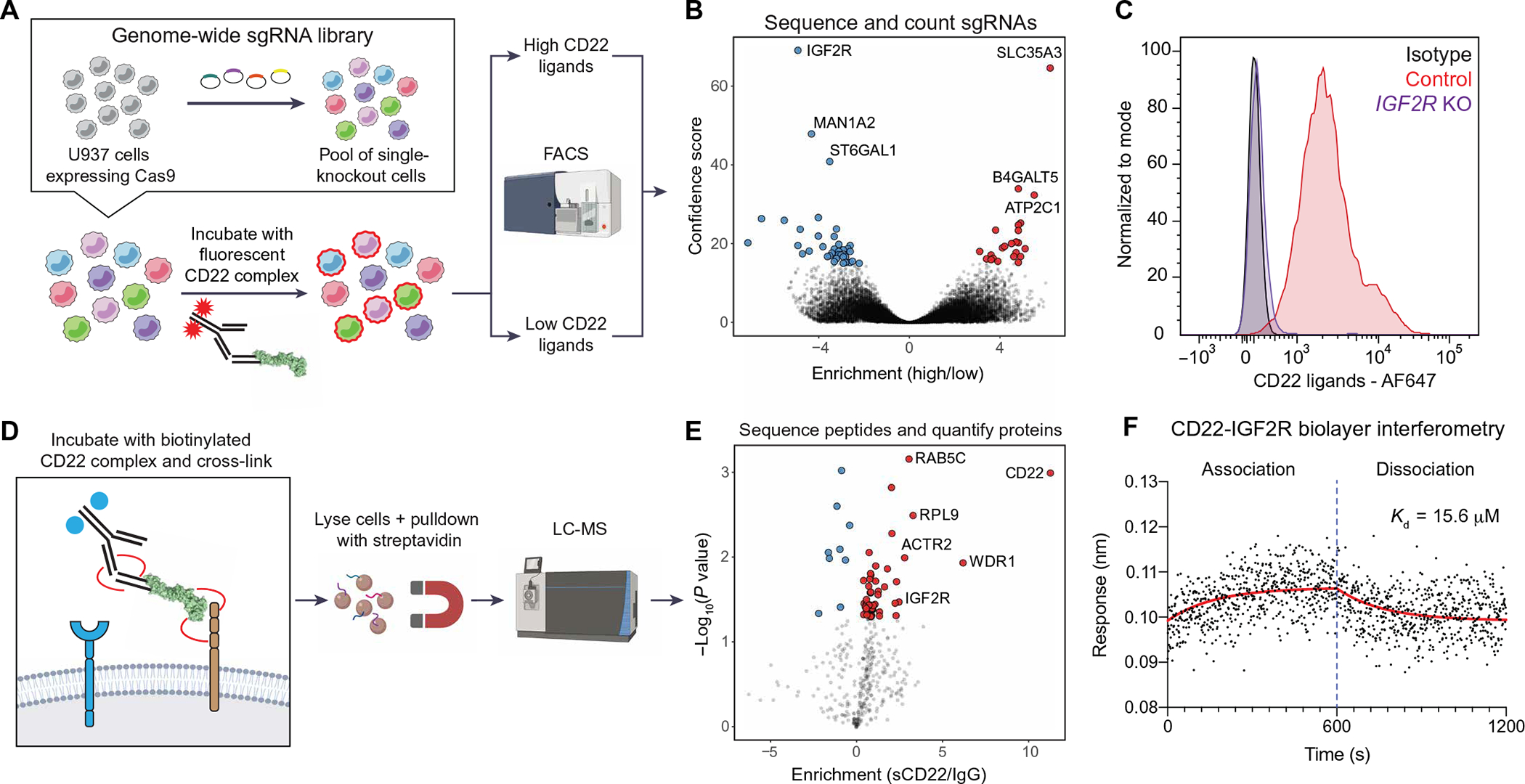
(A) Schematic of CRISPR-Cas9 screen for genetic modifiers of sCD22 binding. (B) Volcano plot of hits from CRISPR-Cas9 screen, highlighting KOs that inhibit CD22 binding (blue) and promote CD22 binding (red). (C) Flow cytometry analysis of CD22 ligand expression on U937 cells infected with a safe-targeting sgRNA (control, red) or an IGF2R-targeting sgRNA (purple). Isotype control–stained WT cells are shown in gray. AF647, Alexa Fluor 647. (D) Schematic of affinity purification LC-MS screen for direct binding partners of sCD22. (E) Volcano plot of hits from affinity purification LC-MS screen, highlighting proteins enriched in the CD22-bound fraction (red). (F) Kinetics of the CD22-IGF2R interaction determined by biolayer interferometry. Red line shows nonlinear fit of association-dissociation curve. Kd, dissociation constant.
The top hit from the KO screen that inhibited CD22 binding was insulin-like growth factor 2 receptor (IGF2R), also known as the cation-independent mannose 6-phosphate (M6P) receptor. As evident in its two names, IGF2R binds diverse ligands including the growth factor IGF2 and the lysosome-targeting glycan modification M6P. IGF2R is broadly expressed on all cell types in the human CNS, including microglia (fig. S2, B and C). Given the absence of sCD22 binding to neurons or fibroblasts (fig. S1, E and F), coexpression of IGF2R (broadly expressed) and ST6GAL1 (enriched in microglia) may be required for cell surface display of the full protein-glycan conjugate that binds sCD22. Knocking out IGF2R (fig. S2D) almost completely ablated sCD22 binding to U937 cells (Fig. 2C), consistent with the results from the genome-wide CRISPR-Cas9 KO screen. This could be due to a physical interaction between IGF2R and sCD22 or due to an upstream role of IGF2R in displaying the true sCD22-binding partner on the cell surface.
To distinguish between these two possibilities, we performed affinity purification followed by liquid chromatography–mass spectrometry (LC-MS) for direct binding partners of sCD22 (Fig. 2D). We incubated U937 cells with biotinylated recombinant CD22 and treated with a membrane-impermeable cross-linker. This fixation step is necessary to stabilize low-affinity Siglec-glycan interactions missed by traditional coimmunoprecipitation techniques (29). Next, sCD22-bound protein complexes were enriched from cell lysates with streptavidin beads (fig. S2E), stringently washed, and digested with trypsin for label-free quantification LC-MS analysis. We found several proteins enriched in the sCD22-bound fraction, including IGF2R (Fig. 2E and table S2). Other enriched proteins included CD22 (affinity reagent) and some potential nonspecific binders not known to localize to the cell surface. However, IGF2R was the only overlapping hit in orthogonal genetic and proteomic screens (Fig. 2, B and E). Using electrophoretic mobility shift assays (EMSAs) (fig. S2F) and quantitative biolayer interferometry (Fig. 2F), we confirmed a biophysical interaction between CD22 and IGF2R with micromolar affinity, comparable to that of CD22 for its native glycan ligand (30). Together, orthogonal genetic and proteomic screens identify IGF2R as a binding partner of sCD22 on human myeloid cells.
CD22 impairs M6P-dependent lysosomal trafficking
IGF2R is a large 140-kb gene with 48 exons coding for an approximately 300-kDa transmembrane protein containing multiple functional domains. To determine where sCD22 binds IGF2R and which function it might modulate, we used two truncated forms of IGF2R as decoy proteins (Fig. 3A). Decoy 1 contains both known M6P-binding sites, and decoy 2 contains the known IGF2-binding site (Fig. 3A). We preincubated fluorophore-conjugated sCD22 with these decoy proteins and assessed binding to native surface IGF2R on U937s by flow cytometry. Whereas decoy 1 (containing M6P sites) inhibited sCD22 binding, decoy 2 (containing the IGF2 site) had no effect (Fig. 3A). These data localize sCD22 binding to the N terminus of IGF2R, where it might interfere with M6P trafficking.
Fig. 3. CD22 impairs M6P-dependent lysosomal trafficking.
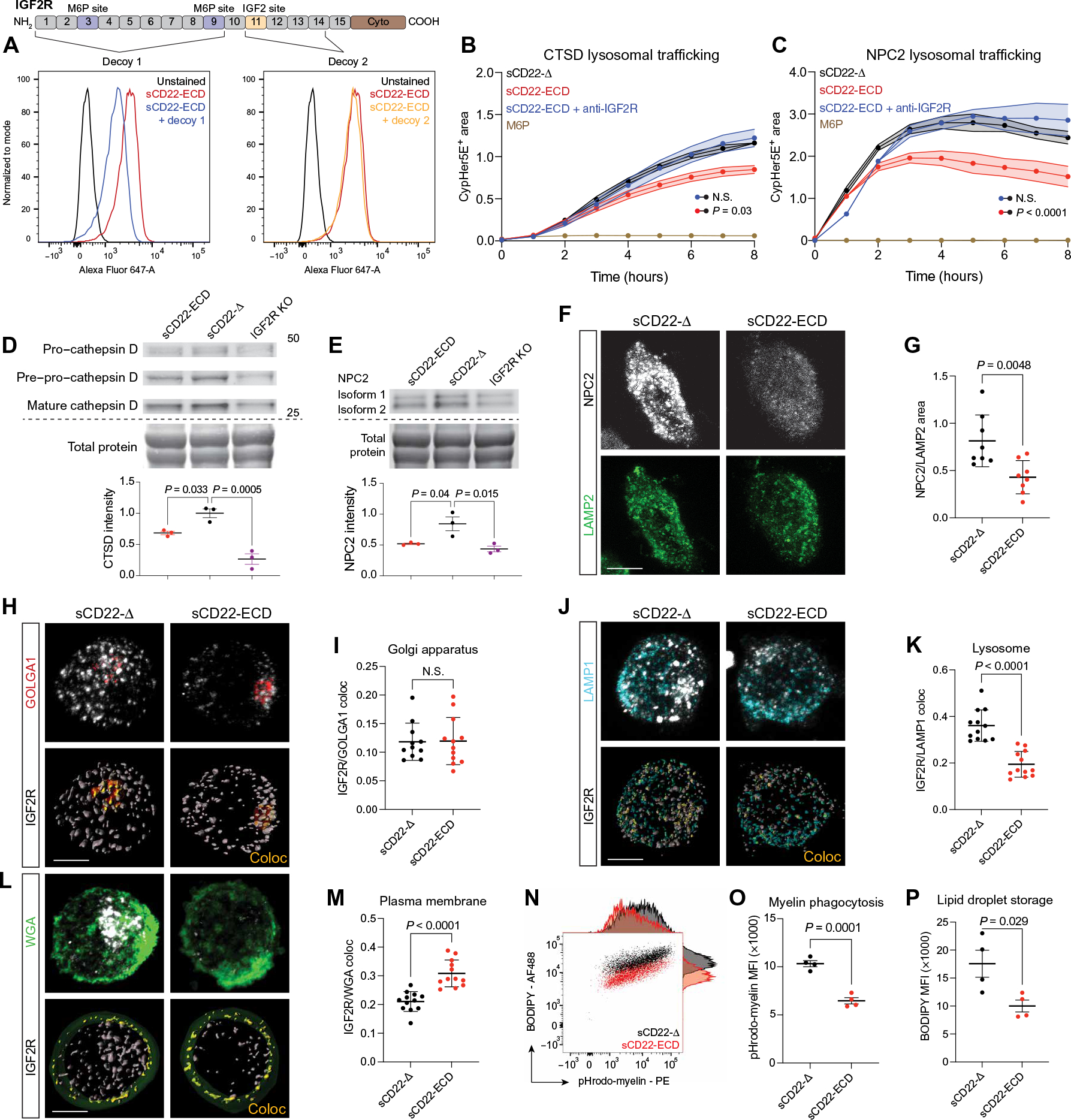
(A) Flow cytometry analysis of U937 cells stained with sCD22-ECD alone (red) or sCD22 precomplexed with a decoy peptide comprising the M6P-binding sites (blue) or the IGF2 site (orange) on IGF2R. (B) Time-lapse fluorescence microscopy analysis of cathepsin D trafficking to lysosomes in U937 cells treated with sCD22-Δ (black), sCD22-ECD (red), sCD22-ECD, and anti-IGF2R (blue) or saturating amounts of M6P (brown) (n = 3, ANOVA, means ± SEM). N.S., not significant. (C) Time-lapse fluorescence microscopy analysis of NPC2 trafficking to lysosomes in U937 cells treated with sCD22-Δ (black), sCD22-ECD (red), sCD22-ECD and anti-IGF2R (blue), or saturating amounts of M6P (brown) (n = 2, ANOVA, means ± SEM). (D) Western blot analysis of CTSD proteoform expression in WT and IGF2R KO U937 cells treated with sCD22-ECD or sCD22-Δ for 24 hours. Equal loading was confirmed across lanes by total protein stain (n = 3, one-way ANOVA, means ± SEM). (E) Western blot analysis of NPC2 expression in WT and IGF2R KO U937 cells treated with sCD22-ECD or sCD22-Δ for 24 hours. Equal loading was confirmed across lanes by total protein stain (n = 3, one-way ANOVA, means ± SEM). (F) Representative images of NPC2 (gray) and LAMP2 (green) expression in U937 cells treated with sCD22-Δ or sCD22-ECD. Scale bar, 5 μm. (G) Proportion of NPC2+ area to LAMP2+ area in U937 cells treated with sCD22-Δ or full-length sCD22-ECD (n = 8, t test, means ± SD). (H) Representative images of IGF2R (gray) colocalization (Coloc) (yellow) with the Golgi marker GOLGA1 (red) in U937 cells treated with sCD22-Δ or sCD22-ECD. Scale bar, 5 μm. (I) Proportion of IGF2R localized to the Golgi in U937 cells treated with sCD22-Δ or sCD22-ECD (n = 3 biological replicates, three to four cells quantified per replicate, t test, means ± SD). (J) Representative images of IGF2R (gray) colocalization (yellow) with the lysosomal marker LAMP1 (cyan) in U937 cells treated with sCD22-Δ or sCD22-ECD. Scale bar, 5 μm. (K) Proportion of IGF2R localized to the lysosome in U937 cells treated with sCD22-Δ or sCD22-ECD (n = 3 biological replicates, two to three cells quantified per replicate, t test, means ± SD). (L) Representative images of IGF2R (gray) colocalization (yellow) with wheat germ agglutinin (WGA) cell surface staining (green) in U937 cells treated with sCD22-Δ or sCD22-ECD. Scale bar, 5 μm. (M) Proportion of IGF2R localized to the cell surface in U937 cells treated with sCD22-Δ or sCD22-ECD (n = 3 biological replicates, three to four cells quantified per replicate, t test, means ± SD). (N) Flow cytometry analysis of iMGLs treated with sCD22-Δ or sCD22-ECD, incubated with pHrodo-myelin for 24 hours, and stained with BODIPY, with corresponding histograms. (O) Quantification of phagocytosis by pHrodo-myelin mean fluorescence intensity (MFI) in iMGLs treated with sCD22-Δ or sCD22-ECD (n = 4, t test, means ± SEM). (P) Quantification of lipid droplet storage by BODIPY MFI in iMGLs treated with sCD22-Δ or sCD22-ECD (n = 4, t test, means ± SEM).
IGF2R typically binds M6P-modified proteins in the trans-Golgi network (TGN) and releases them in the late endosome (31). However, a fraction of M6P-modified proteins are secreted into the extracellular space and recaptured by IGF2R on the cell surface (32, 33). This minor secretion-recapture pathway has been exploited by enzyme replacement therapy (ERT) to deliver M6P-modified recombinant proteins to deficient cells (34) and more recently leveraged for generalized degradation of cell surface or extracellular proteins (35).
We asked whether sCD22 might interfere with this recapture pathway by blocking sites on the N-terminal, M6P-binding domain of IGF2R. We conjugated a pH-sensitive dye to cathepsin D (CTSD), a lysosomal protease frequently mislocalized in NPC tissues (36), increased in NPC patient serum (37), and shown to have neurotoxic effects (38–40). By flow cytometry, we detected pH-dependent fluorescent signal in wild-type (WT) but not IGF2R-KO U937 cells incubated with CTSD (fig. S3A), indicating IGF2R-dependent trafficking to the lysosome. Moreover, pretreating WT cells with saturating amounts of free M6P blocked CTSD trafficking (brown line, Fig. 3B). Compared to truncated sCD22 lacking its sialic acid–binding domain (control, sCD22-Δ, black line, Fig. 3B and fig. S3B), the full-length sCD22 extracellular domain (sCD22-ECD) impaired trafficking of CTSD to the lysosome (red line, Fig. 3B). However, cotreatment with sCD22-ECD and an IGF2R antibody that blocks CD22 (fig. S3B) but not M6P binding abrogated this effect (blue line, Fig. 3B). We found similar results with NPC2 (Fig. 3C), the M6P-modified soluble counterpart to NPC1 that assists in exporting cholesterol from the lysosome (41–43). sCD22-ECD treatment had no effect on gene expression (fig. S3D), lysosomal proteolysis (fig. S3E), transferrin endocytosis (fig. S3F), or autophagy (fig. S3G), suggestive of a specific mechanism of action in which sCD22 impairs IGF2R-dependent recapture of lysosomal proteins from the extracellular space (fig. S3H).
Next, we assessed the effect of sCD22 on endogenous lysosomal protein trafficking. Compared to binding-incompetent sCD22-Δ, sCD22-ECD treatment depleted both immature and mature proteoforms of endogenous CTSD in WT U937 cells, albeit to a lesser extent than IGF2R KO cells lacking critical M6P trafficking machinery (Fig. 3D). Similarly, sCD22-ECD depleted total NPC2 protein (Fig. 3E) and reduced the proportion of endogenous NPC2 per lysosome (Fig. 3, F and G). Mislocalized NPC2 in the CSF has been previously observed in LSDs (44) and is associated with lysosomal cholesterol accumulation (45).
IGF2R molecules are distributed throughout the TGN, endolysosomal system, and plasma membrane (46, 47) and are frequently recycled after delivering cargo to lysosomes (48, 49). Given the deleterious effects of sCD22 on both exogenous and endogenous lysosomal protein trafficking, we asked whether sCD22 might disturb the distribution of IGF2R among organelles. By immunofluorescence, sCD22-ECD had no effect on IGF2R colocalization with GOLGA1, a Golgi apparatus marker, TGN46, a TGN marker, or EEA1, an early endosome marker, relative to sCD22-Δ (Fig. 3, H and I, and fig. S3, I to L). However, sCD22-ECD treatment depleted IGF2R from Rab7+ late endosomes (fig. S3, M and N) and lysosome-associated membrane protein 1 (LAMP1+) lysosomes (Fig. 3, J and K) and increased its plasma membrane localization (Fig. 3, L and M) without a detectable change in total protein quantity (fig. S3O). These data suggest that sCD22 either promotes IGF2R trafficking from the TGN to the cell surface, bypassing the endolysosomal system, or sequesters IGF2R at the cell surface, thereby blocking exogenous lysosomal protein trafficking and reducing receptors available for TGN-to-lysosome trafficking (fig. S3P).
Properly localized lysosomal proteins are essential for the downstream processing of phagocytic cargo. To evaluate the effect of sCD22 on this process, we treated iMGLs with sCD22-ECD or sCD22-Δ and incubated the cells with pH-sensitive fluorescently labeled myelin, a phagocytic substrate abundant in the demyelinating NPC brain (50). By flow cytometry, we found that sCD22-ECD reduced myelin uptake compared to sCD22-Δ (Fig. 3, N and O). Furthermore, we observed a concomitant decrease in BODIPY signal, a marker of neutral lipid droplets (Fig. 3, N and P). Gating for only nonphagocytic cells, sCD22-ECD–treated iMGLs contained fewer lipid droplets at baseline (fig. S3Q), recapitulating a known lipid processing defect previously observed in Npc1−/− microglia (15).
Generation of a CD22 antibody that ameliorates lysosome dysfunction
Having established that sCD22 impairs IGF2R-dependent lysosomal trafficking, we asked whether blocking this interaction might rescue lysosome dysfunction in NPC. Several monoclonal antibodies (mAbs) targeting CD22 are in clinical development or FDA-approved treatments for B cell malignancies (51). However, none of these mAbs are known to block the sialic acid–binding domain critical for sCD22-IGF2R interaction (52). We screened a nearly comprehensive panel of 32 commercially available CD22 mAbs, and none exhibited any substantial effect on sCD22 binding to IGF2R (table S3). Therefore, we set out to generate blocking mAbs optimized for targeting sCD22 in the brain.
We immunized three cohorts of mice with the human CD22 ECD, confirmed high serum titers against a CD22-expressing cell line (fig. S4A) and specificity against a CD22-null cell line (fig. S4B), and isolated B cells for hybridoma fusion (Fig. 4A). Next, we selected 50 clones for intermediate to high CD22 binding, capturing a diversity of antibody phenotypes. Last, we screened a subset of 38 clones for their ability to block sCD22 binding to the surface of U937 cells. These screens yielded three promising mAbs with strong and specific CD22 binding and potent blockade of sCD22 (Fig. 4B). These clones had no effect on sCD22 binding to IGF2R KO cells (fig. S4C), indicating specific disruption of the CD22-IGF2R interaction.
Fig. 4. Generation of a CD22 antibody that ameliorates lysosome dysfunction.
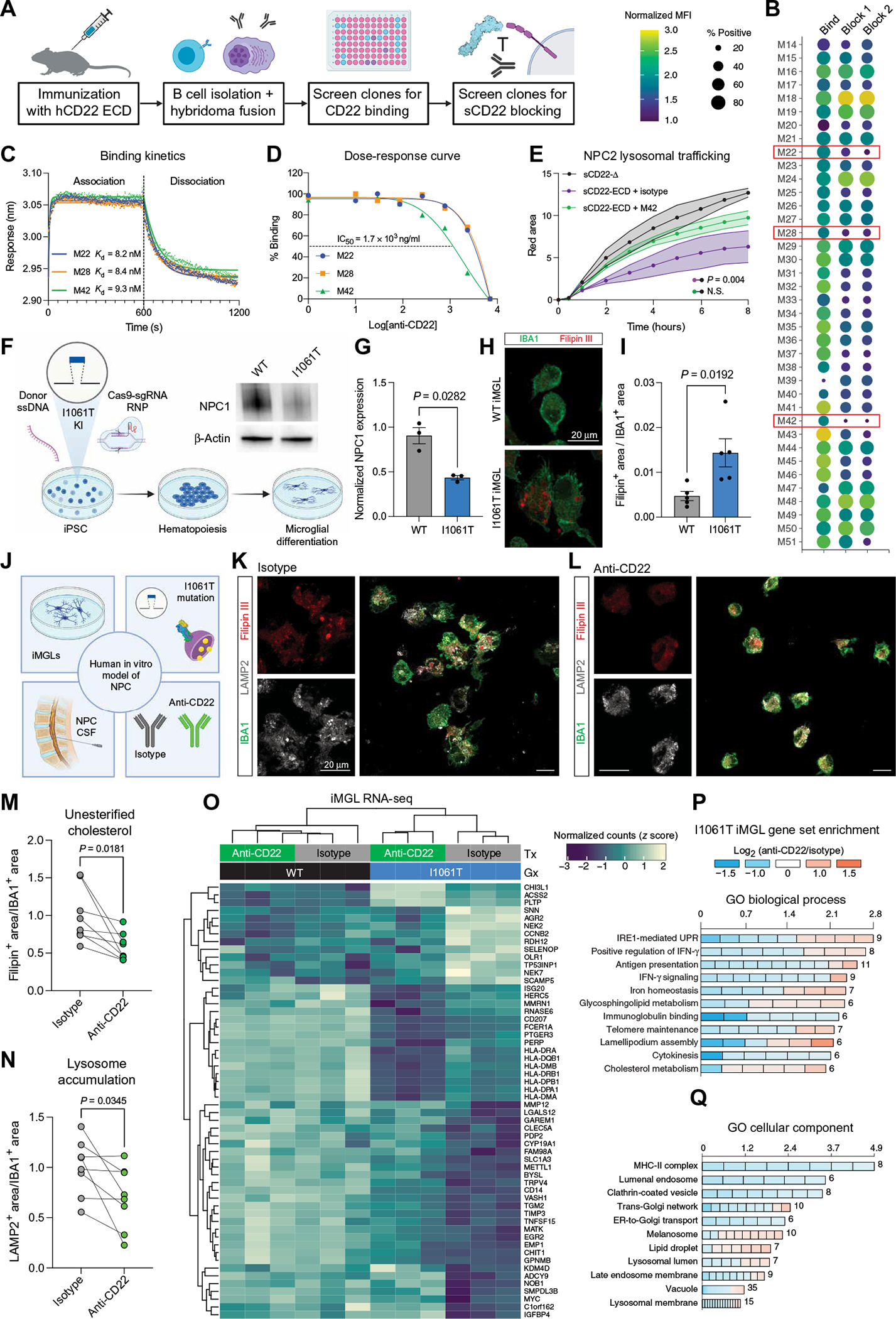
(A) Schematic of mAb generation and screening pipeline. (B) Screening results of 38 mAb clones for binding to CD22 (first column) and blocking of sCD22 to IGF2R on cell surface (second and third columns are two independent experiments). Three clones with adequate binding and potent blocking are highlighted (M22, M28, and M42). (C) Association-dissociation curves of antibody candidates binding to CD22 determined by biolayer interferometry. (D) Dose-response curves of CD22-IGF2R blockade by antibody candidates determined by flow cytometry. IC50, median inhibitory concentration. (E) Time-lapse fluorescence microscopy analysis of NPC2 trafficking to lysosomes in U937 cells treated with sCD22-Δ (gray), sCD22-ECD and an isotype control antibody (purple), or sCD22-ECD and clone M42 (green) (n = 2, ANOVA, means ± SEM). (F) Schematic of pipeline to generate isogenic WT and I1061T mutant iMGLs from iPSCs edited by CRISPR-Cas9–directed homologous recombination. After introduction of donor single-stranded DNA (ssDNA) by electroporation, a homozygous T3182C nucleotide substitution was confirmed by Sanger sequencing. NPC1 reduction was confirmed by Western blot. Mutant and isogenic control iPSCs were subsequently directed toward a hematopoietic lineage and differentiated into microglia-like cells. (G) Western blot quantification of NPC1 expression normalized to a loading control (β-actin) in WT and I1061T mutant iPSCs (n = 3, t test, means ± SEM). (H) Representative images of WT and I1061T mutant iMGLs stained for Filipin III (red, unesterified cholesterol) and IBA1 (green, microglia marker). Scale bar, 20 μm. (I) Quantification of Filipin-positive area normalized to total IBA1-positive area in WT (gray) and I1061T mutant (blue) iMGLs (n = 5 biological replicates, t test, means ± SEM). (J) Schematic of human in vitro model of microglia in NPC. Three components (iPSC-derived microglia, I1061T patient mutation, and NPC patient CSF) were combined to test the proof-of-principal in vitro efficacy of anti-CD22 in NPC. (K) Representative images of I1061T mutant iMGLs treated with NPC CSF and an isotype control antibody stained for Filipin III (red, unesterified cholesterol), LAMP2 (gray, lysosome marker), and IBA1 (green, microglia marker). Scale bars, 20 μm. (L) Representative images of I1061T mutant iMGLs treated with NPC CSF and anti-CD22 stained for Filipin III (red, unesterified cholesterol), LAMP2 (gray, lysosome marker), and IBA1 (green, microglia marker). Scale bars, 20 μm. (M) Quantification of Filipin-positive area normalized to total IBA1-positive area in isotype (gray)– and anti-CD22 (green)–treated iMGLs (n = 7 biological replicates, paired t test, means ± SEM; lines connect wells treated with the same patient’s CSF). (N) Quantification of LAMP2-positive area normalized to total IBA1-positive area in isotype (gray)– and anti-CD22 (green)–treated iMGLs (n = 7 biological replicates, paired t test, means ± SEM; lines connect wells treated with the same patient’s CSF). (O) Heatmap of normalized counts (z score) for differentially expressed genes in WT and I1061T mutant iMGLs treated with NPC CSF and isotype or anti-CD22. (P) Gene Ontology (GO) biological process enrichment analysis of differentially expressed genes between anti-CD22− and isotype–treated I1061T iMGLs. Up- or down-regulation is represented on the color scale, and the number of genes differentially expressed is indicated for each term. IRE1, inositol-requiring enzyme 1; IFN-γ, interferon-γ; UPR, unfolded protein response. (Q) GO cellular component enrichment analysis of differentially expressed genes between anti-CD22− and isotype–treated I1061T iMGLs. Up- or down-regulation is represented on the color scale, and the number of genes differentially expressed is indicated for each term. MHC-II, major histocompatibility complex class II; ER, endoplasmic reticulum.
To characterize the biophysical properties of these three candidate mAbs, we performed biolayer interferometry at various concentrations (fig. S4D) to obtain a global kinetic fit and found that each clone bound CD22 with nanomolar affinity (Fig. 4C). We selected clone M42 as our lead candidate for its potent blockade of the CD22-IGF2R interaction (Fig. 4D) and rescue of NPC2 lysosomal trafficking in U937 cells (Fig. 4E).
With this tool in hand, we sought to evaluate the therapeutic potential of sCD22 blockade in NPC. Although Npc1−/− and Npc1(I1061T) mice (53) are valuable models that recapitulate many features of the human disease, they are not suitable for evaluating CD22 function given divergent expression patterns and signaling mechanisms in mice versus humans (fig. S1, H and I). Therefore, we used iMGLs that transcriptionally and functionally resemble human microglia (54–56). Relevant for our study, iMGLs express IGF2R (fig. S4E) and ST6GAL1 (fig. S4F) and bind sCD22 in a sialic acid–dependent manner (Fig. 1H). To model the genetic defects of NPC, we introduced the most common NPC patient mutation, NPC1 I1061T, into iPSCs by CRISPR-Cas9–directed homologous recombination (Fig. 4F). Mutant iPSCs displayed ~50% reduction in NPC1 protein expression (Fig. 4G), in line with prior studies showing that I1061T encodes a misfolded protein that is rapidly degraded (57). Upon differentiation to iMGLs, the mutant line formed numerous large LAMP2+ lysosomes (fig. S5A) and massively accumulated unesterified cholesterol (Fig. 4, H and I), recapitulating the cellular hallmark of NPC. Last, to model the microglial environment and provide a physiological source of sCD22, we treated iMGLs with CSF from patients with NPC. Combining these three elements (iMGLs, I1061T mutation, and NPC patient CSF), we arrived at an in vitro model of microglia in NPC relevant for human studies (Fig. 4J).
Compared to an isotype control antibody, anti-CD22 (clone M42) reduced the burden of unesterified cholesterol deposits in mutant iMGLs (Fig. 4, K to M). This restoration of lysosomal cholesterol homeostasis was accompanied by decreased LAMP2+ lysosome accumulation (Fig. 4N), reverting I1061T iMGLs back toward the phenotype of their isogenic WT counterparts (fig. S5A). We found similar alleviation of cholesterol accumulation using WT iMGLs treated with U18666A, a specific inhibitor of NPC1 (fig. S5, B to E) (58). However, anti-CD22 treatment in U18666A cells increased LAMP2 expression (fig. S5F), indicative of distinct biological responses between pharmacologically inhibited and genetically mutated NPC1.
Microglia display profound transcriptional changes in states of impaired lysosomal function such as aging (18, 59), neurodegeneration (60–62), and LSDs (12, 63). We performed RNA-seq on WT and I1061T mutant iMGLs treated with NPC CSF and anti-CD22 or an isotype control antibody. Compared to its minimal effect in WT iMGLs, CD22 blockade broadly altered the transcriptome in I1061T mutant cells (Fig. 4O and tables S4 and S5). Specifically, anti-CD22 treatment down-regulated unfolded protein and stress response genes and up-regulated cholesterol and sphingolipid metabolism genes, partially shifting I1061T mutant iMGLs to a WT transcriptional state (Fig. 4, O and P). Anti-CD22 treatment led to the down-regulation of class II human leukocyte antigen (HLA) presentation genes beyond the low expression seen in isotype-treated I1061T iMGLs (Fig. 4O). This transcriptional change could reflect decreased antigen presentation capacity or negative feedback upon restoration of class II HLA trafficking in unburdened lysosomes. Enrichment analysis revealed that CD22 blockade decreased expression of genes associated with endolysosomal transport and increased expression of genes associated with lipid droplet localization (Fig. 4Q). Together, these findings characterize the kinetics, function, and proof-of-principle efficacy of a microglia-directed immunotherapeutic for NPC.
CD22 blockade does not disrupt oligodendrocyte maturation in vitro
Although blockade of sCD22 may be a viable therapeutic strategy to restore microglial function in patients with NPC, the potential on-target toxicity of blocking membrane-bound CD22 on oligodendrocytes may limit its clinical safety. To assess this risk, we isolated and cultured O4+ OPCs from primary human cortical tissue at postconception weeks (PCWs) 20 to 22 for in vitro differentiation to myelinating oligodendrocytes (Fig. 5A) using a previously published protocol (64). After 12 days in culture, at a time point when the cells expressed both myelin basic protein (MBP) and CD22 (Fig. 5B), we treated the cells with anti-CD22, an isotype control antibody, or an anti-MOG antibody known to cause demyelination in vitro (65). In contrast to anti-MOG, treatment with anti-CD22 did not affect oligodendrocyte proliferation or maturation as assessed by time-lapse microscopy (Fig. 5, C and D) and MBP immunofluorescence (Fig. 5, E and F). These results mitigate on-target toxicity concerns of using intrathecal CD22 antibodies to treat NPC.
Fig. 5. CD22 blockade does not disrupt oligodendrocyte maturation in vitro.
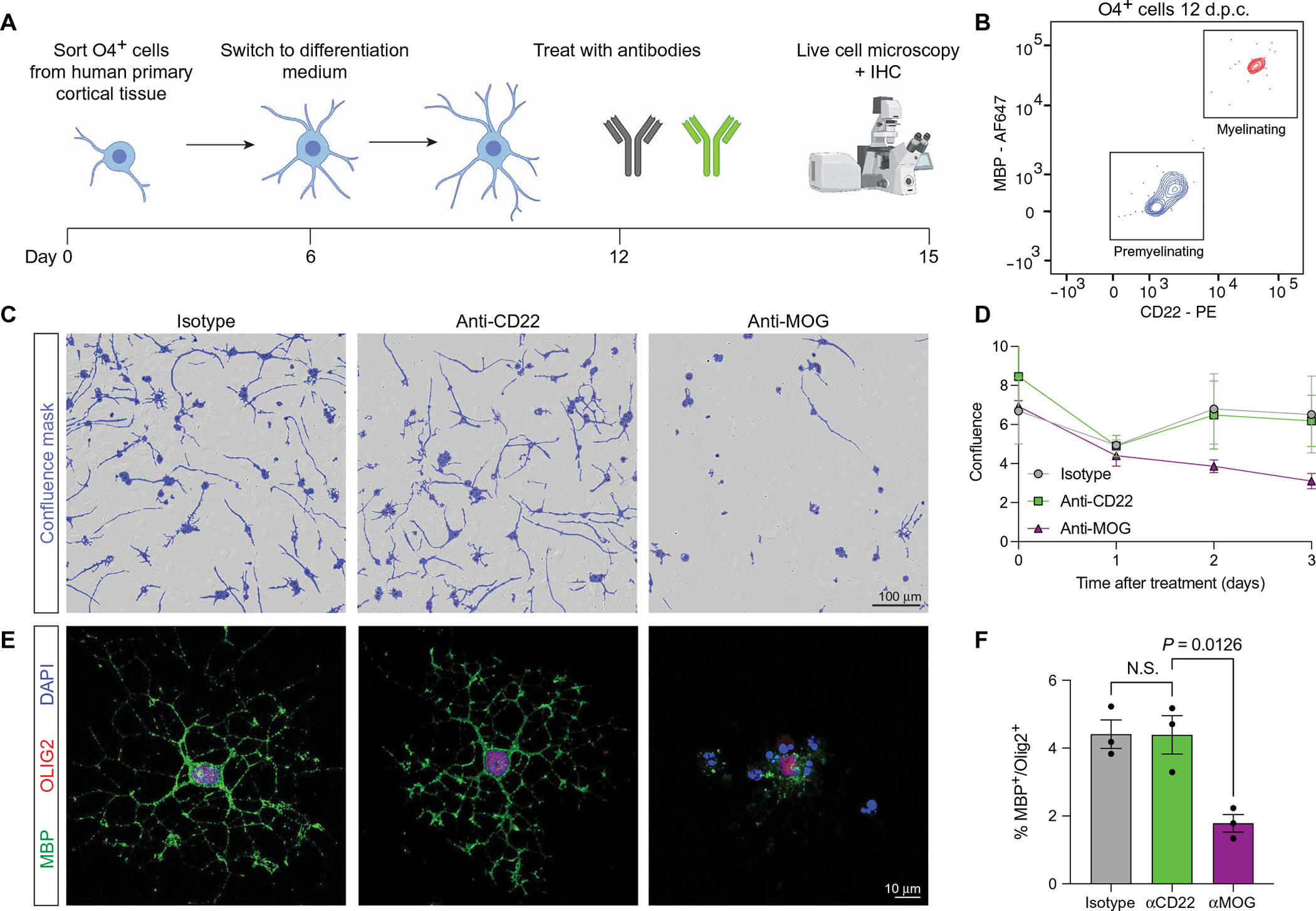
(A) Schematic of human primary cortical oligodendrocyte isolation and differentiation protocol [adapted from (64)]. IHC, immunohistochemistry. (B) Representative flow cytometry of O4+ cells after 12 days in culture (d.p.c.) at the onset of antibody treatment, showing a mixed population of premyelinating O4+MBP−CD22− cells (blue) and a subpopulation of myelinating O4+MBP+CD22+ oligodendrocytes (red). (C) Representative bright-field images of isotype-, anti-CD22−, and anti-MOG–treated oligodendrocytes on day 15 after isolation (day 3 after treatment). Confluence mask overlaid in purple. Scale bar, 100 μm. (D) Quantification of confluence in isotype (gray)–, anti-CD22 (green)–, and anti-MOG (purple)–treated cells assessed by time-lapse microscopy over 3 days (n = 3 from two separate primary tissue samples, means ± SEM). (E) Representative immunofluorescence images of isotype-, anti-CD22−, and anti-MOG–treated oligodendrocytes on day 15 after isolation (day 3 after treatment), stained for MBP (green), and OLIG2 (red). Scale bar, 10 μm. DAPI, 4′,6-diamidino-2-phenylindole. (F) Quantification of MBP+ cells among OLIG2+ nuclei in isotype (gray)–, anti-CD22 (green)–, and anti-MOG (purple)–treated cells (n = 3 from two separate primary tissue samples, one-way ANOVA, means ± SEM).
DISCUSSION
Here, we uncover a source, target, and function of sCD22 in the human brain. Specifically, we find that sCD22 is an oligodendrocyte-derived protein that binds IGF2R on microglia and impairs lysosome function. We apply these findings to NPC, where sCD22 is elevated in the CSF, and show that a CD22-blocking mAb ameliorates lysosome dysfunction in NPC1-mutant iMGLs without harming oligodendrocytes in vitro.
These findings highlight important differences between mice and humans with specific implications for translational microglia research. A cross-species RNA-seq study showed unexpected divergence of primate and rodent microglia, with notable differences in the expression of phagocytosis and complement genes (20). Moreover, the overlap of mouse and human microglial responses to neurodegeneration remains controversial (66–68). Although CD22 is highly conserved at the sequence level, we find a soluble form expressed in oligodendrocytes in humans but a membrane-bound form in microglia in mice. CD22 is expressed in both cell types in nonhuman primates, indicative of an evolutionary shift. These differences illuminate some limitations of mouse models to study microglia in human disease and reinforce the utility of humanized models for target discovery and drug development.
How does CD22 blockade restore cholesterol efflux in NPC1-defective cells? NPC1 is thought to be the dominant exporter of cholesterol from the lysosome, but parallel pathways exist through LIMP-2/SCARB2 (69), LAMP-1 (70), LAMP-2 (71), and the compensatory effect of NPC2 overexpression (72). CD22 blockade may augment the activity or lysosomal delivery of one of these alternate proteins, providing an escape pathway for cholesterol clearance in NPC1-mutant cells.
The present study focuses on the soluble form of CD22, but the function of membrane-bound CD22 on oligodendrocytes remains unknown. One clue to its function comes from the specific expression of CD22 on myelinating, but not premyelinating, oligodendrocytes. In mice, deletion of SHP-1, the downstream signaling partner of CD22, impairs oligodendrocyte differentiation and myelination (73). In humans, CD22 may facilitate SHP-1–dependent myelination. Accordingly, CD22 up-regulation in NPC may be a compensatory mechanism to overcome hypomyelination (50, 74) and oligodendrocyte maturation defects (75) caused by NPC1 mutations. How do oligodendrocytes release CD22 into the CSF? It is possible that oligodendrocytes shed sCD22 via proteolytic cleavage, secrete sCD22 as a transmembrane-null isoform (76), or package sCD22 into exosomes along with cholesterol (77, 78). Why CD22 expression was co-opted by oligodendrocytes in humans remains unclear but may be related to the protracted period of myelination in the human CNS compared to other mammals (79), requiring additional regulatory machinery. Our CD22 mAb does not alter oligodendrocyte proliferation or maturation in vitro, mitigating safety concerns without obviating the need for future in vivo toxicity studies.
We acknowledge several limitations of the present study. First, we were unable to assess in vivo efficacy of anti-CD22 in an NPC model given evolutionary divergence of CD22 expression between mice and humans. One possibility to address this limitation is the feline model of NPC, which might more closely mirror human CD22 expression patterns (80). Second, we rely on pharmacological manipulations of CD22, which may have off-target effects. A rapidly expanding genetic toolbox may facilitate more precise therapeutic targeting moving forward (81). Last, although we roughly map an interaction between the sialic acid–binding domain of CD22 and the M6P-binding domain of IGF2R, a high-resolution structural characterization of this interaction is lacking.
Among investigational drugs for NPC (82–85), the cholesterol solubilizer 2-hydroxypropyl-β-cyclodextrin (HPβCD) slowed disease progression in a phase 1/2 clinical trial (86). HPβCD slightly reduces sCD22 concentration in NPC CSF (12), which may indirectly contribute to its efficacy. Direct blockade of sCD22 may halt both microglial activation and the neurodegeneration that accompanies it. Gene therapy to restore WT NPC1 expression in neurons holds clinical promise (87), but viral transduction of microglia remains a challenging problem (88). Because sCD22 impairs exogenous lysosomal protein trafficking via IGF2R, CD22 blockade might serve as an adjuvant for ERT in other LSDs. Beyond LSDs, the CD22-IGF2R interaction may link lysosome dysfunction to lipid accumulation and microglial activation to oligodendrocyte demyelination in more common neurodegenerative diseases. Future investigations of the breadth and depth to which sCD22 impairs lysosome function in the human brain will inform the therapeutic potential of CD22-blocking mAbs.
MATERIALS AND METHODS
Study design
The aims of this study were (i) to characterize the expression, function, and mechanism of CD22 in the human brain; (ii) to develop CD22-targeting therapeutics; and (iii) to test these therapeutics in an in vitro iPSC-derived model of NPC microglia. Sample sizes, when not limited by human sample availability, were chosen on the basis of similar studies in the literature. All experiments included appropriate negative controls and, when available, validated positive controls. No data were excluded, all outliers are shown as individual data points, and end points were predetermined before initiation of experiments. None of the experiments in this study required randomization, and all immunohistochemical quantifications were performed by blinded observers. Experiments were performed with technical and biological replicates (exact number specified in the figure legends) and, when possible, validated with orthogonal techniques.
Patient samples
NPC patient CSF samples were collected at the Rush University Medical Center pursuant to institutional review board approval.
RNAscope in situ hybridization
RNA in situ hybridization was performed on fresh frozen brain tissue using the multiplex fluorescent v2 assay (Advanced Cell Diagnostics) according to the manufacturer’s protocol. Probes for CD22, MOG, and AIF1 were commercially available from the manufacturer.
Preparation of fluorescent proteins
Recombinant His-tagged human CD22 (R&D Systems, 10191-SL) was incubated with Alexa Fluor 647 (AF647) N-hydroxysuccinimide (NHS) in phosphate-buffered saline (PBS) with 0.1 M sodium bicarbonate for 1 hour at room temperature at a dye:protein ratio of 4. Excess dye was removed and buffer was exchanged using a 40K molecular weight cut-off (MWCO) Zeba spin desalting column. NPC2 or cathepsin D were tagged with CypHer5E NHS using an equivalent protocol except buffer exchanged with a 7K MWCO Zeba spin desalting column.
Quantitative flow cytometry analysis of human primary cortical brain cells
Deidentified tissue samples were obtained at Stanford University School of Medicine under a protocol approved by the Research Compliance Office at Stanford University. Brain tissue samples at PCWs 20 to 22 were delivered on ice and processed within 3 hours of the procedure. Tissue was processed according to a previously published protocol (89) to obtain a single-cell suspension. Cells were stained with the following antibodies on ice for 30 min: anti-CD45 (1:25, clone HI30, BioLegend), anti-O4 (1:20, R&D Systems), anti-IGF2R (1:10, BD Biosciences), anti-CD22 (1:20, clone HIB22, BioLegend), anti-MBP (1:100, clone SMI 99, BioLegend), anti-MAP2 (1:20, SMI 52, BioLegend), and human Fc block (1:20, BD Biosciences). After staining, cells were washed, and relevant populations were analyzed on a BD LSRFortessa. Quantibrite–phycoerythrin (PE) (BD Biosciences) beads were resuspended in FACS buffer and analyzed on a BD LSRFortessa. Using the same laser powers and detector voltages, cortical cells and Ramos cells were analyzed for CD22 expression. A standard curve was constructed to correlate PE intensity with number of PE molecules per Quantibrite bead. Last, molecule numbers on human primary cortical brain cell types were calculated by interpolation.
Flow cytometry analysis of sCD22 binding
Cells were prepared on ice at a concentration of 1 million cells/ml and incubated with directly conjugated CD22–AF647 or precomplexed CD22-Fc anti-human immunoglobulin G (IgG) AF647 at a concentration of 10 μg/ml for 30 min on ice. As a negative control for the direct staining reagent, cells were incubated with a sialic acid binding–incompetent truncated version of CD22 (sCD22-Δ; Sino Biological, 11958-H08H1) conjugated to AF647. As a negative control for the precomplexed Fc fusion staining reagent, cells were incubated with anti-human IgG secondary alone. After incubation, cells were washed with PBS and analyzed on a BD LSRFortessa.
Myelin phagocytosis and lipid droplet storage by iMGLs
WT iMGLs were plated at 50% confluence on Matrigel-coated 96-well plates and treated with pHrodo-myelin (555) (10 μg/ml) and sCD22-Δ or sCD22-ECD (10 μg/ml). After incubation of 24 hours at 37 min, cells were stained with BODIPY 493/503 (Thermo Fisher Scientific, 1:1000 from a stock solution of 1 mg/ml in DMSO) for 20 min at room temperature protected from light, washed with Dulbecco’s PBS, and analyzed on a BD Fortessa X20.
CRISPR-Cas9 screen
The 10-sgRNA-per-gene CRISPR-Cas9 deletion library was synthesized, cloned, and infected into Cas9-expressing U937 cells as previously described (27). Briefly, ~300 million U937 cells in stably expression EF1α-Cas9-BLAST were infected with the 10 guide/gene sgRNA genome (Addgene, catalog nos. 101926 to 101934) at a multiplicity of infection < 1. Infected cells underwent puromycin selection (1.5 μg/ml) for 5 days after which point puromycin was removed and cells were resuspended in normal growth medium without puromycin. After selection, sgRNA infection was confirmed by flow cytometry, which indicated >90% of cells expressed the mCherry reporter. Sufficient sgRNA library representation was confirmed by deep sequencing after selection. Cells were cultured and maintained at 1000× coverage for 1 week. Staining reagent was prepared by precomplexing recombinant CD22-Fc fusion protein with anti-human IgG AF647 at an equimolar ratio on ice for 30 min. Cells were washed and resuspended at 10 million cells/ml and incubated with precomplex at 10 μg/ml for 30 min on ice. Cells were washed and prepared FACS on a BD FACSAria III. Cells expressing the top 5% (15 million cells) and bottom 5% (15 million cells) of CD22 ligands were sorted for downstream processing. Genomic DNA was extracted for all populations separately using a QIAGEN Blood Midi Kit. Deep sequencing of sgRNA sequences on an Illumina NextSeq was used to monitor library composition. Guide composition was analyzed and compared to the plasmid library and between conditions using casTLE (https://bitbucket.org/dmorgens/castle). Briefly, casTLE compares each set of gene-targeting guides to the negative controls, comprising nontargeting and nongenic (“safe-targeting”) sgRNAs, which have been shown to more aptly control for on-target toxicity due to endonuclease-induced DNA damage. The enrichment of individual guides was calculated as the log ratio between high and low populations, and gene-level effects were calculated from 10 guides targeting each gene. P values were then calculated by permutating the targeting guides.
Electrophoretic mobility shift assay
rCD22 was prelabeled using NHS-AF647 as described above. A binding reaction (10 μl) consisting of 100 ng of rCD22-AF647 with 0×, 1×, or 3× molar equivalents of IGF2R-N-term in 1× EMSA buffer [10 mM Hepes (pH 7.5), 20 mM KCl, 1 mM MgCl2, and 1 mM dithiothreitol] was incubated at 25°C for 60 min to allow binding. To visualize complex formation, 1 μl of 50% glycerol was added to each sample and then run in a 4% native polyacrylamide gel electrophoresis (PAGE) at 100 V. After electrophoresis, gels were directly scanned on a LI-COR Odyssey in the 700 nm channel.
Biolayer interferometry
Fortebio Octet anti-mouse Fc capture biosensors were loaded with CD22 mAb supernatant and associated into recombinant His-tagged human CD22 ECD analyte at seven different concentrations (100 nM, serially diluted 1:3). Binding constants and global fits were calculated for the association and dissociation of the analyte according to the manufacturer’s protocol. For CD22-IGF2R kinetic analysis, anti-human Fc capture biosensors were loaded with recombinant human CD22 Fc-fusion protein (R&D Systems) and associated into recombinant His-tagged human IGF2R analyte at seven different concentrations (1000 nM, serially diluted 1:3). Unloaded parallel reference sensors were used to monitor and subtract background interactions between analytes and sensor.
Western blot
Cell lysates were prepared by resuspending and incubating cell pellets on ice in RIPA Lysis Buffer with cOmplete protease inhibitor cocktail and spun at 17,000g for 10 min. The supernatant was collected, and protein concentration was measured with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). An aliquot containing 20 μg of protein from each sample was mixed with 4× loading buffer (Thermo Fisher Scientific), 2.5% beta-mercaptoethanol (MilliporeSigma), and boiled for 5 min at 95°C before being subjected to SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). The membrane was first stained for total protein with Revert (LI-COR). The membrane was destained, blocked with Intercept Blocking Buffer (LI-COR), and stained overnight at 4°C with primary antibodies at the designated concentrations: rabbit anti-NPC2 (1:1000; R&D Systems, AF8644), rabbit anti-CTSD [1:1000; Cell Signaling Technology (CST), 2284S], or rabbit anti-LC3B (1:1000; CST, 2775S). The membrane was washed, stained with IRDye-conjugated secondary antibodies (1:15,000, LI-COR), and imaged on the Odyssey CLx (LI-COR).
Exogenous lysosomal protein recapture assay
Recombinant His-tagged CTSD (R&D Systems, 1014-AS) or NPC2 (Sino Biological, 13341-H08H) was conjugated to CypHer5E NHS (GE Life Sciences) as described above. U937 cells were pretreated with sCD22-ECD (10 μg/ml), sCD22-Δ (10 μg/ml), sCD22-ECD and anti-IGF2R (10 μg/ml; Abcam, ab124767), or 500 mM M6P for 30 min in Dulbecco’s modified Eagle’s medium (DMEM) with l-glutamine and 10% fetal bovine serum (FBS) at 37°. After pretreatment, CypHer5E-conjugated lysosomal proteins were added at 0.5 μg/ml, and fluorescence was monitored for 8 hours by time-lapse fluorescence microscopy (Incucyte S3).
Immunocytochemistry
U937 cells were grown on poly-l-lysine–coated eight-well chambered coverslips (ibidi GmbH) in DMEM with l-glutamine and 10% FBS in the presence of 10 μg/ml sCD22-ECD or sCD22-delta for 24 hours. For surface staining, live cells were stained with wheat germ agglutinin (WGA)–fluorescein (1:100, Vector Laboratories) for 30 min on ice before fixation. Cells were gently washed and fixed onto the chamber slide with 4% paraformaldehyde at room temperature for 15 min. After 1 hour of blocking and permeabilization in PBS with 0.05% Triton X-100 and 1% bovine serum albumin (BSA), cells were stained with the following combinations of primary antibodies in the same blocking medium at 4° overnight: rabbit anti-NPC2 (1:100; R&D Systems, AF8644) and mouse anti-LAMP2 (1:100; Santa Cruz Biotechnology, H4B4); mouse anti-IGF2R (1:100; Abcam, 2G11) and rabbit anti-Rab7 (1:100; Abcam, ab126712); mouse anti-IGF2R and rabbit anti–Golgin-97 (1:100; CST, D8P2K); mouse anti-IGF2R and rabbit anti-TGN46 (1:100; Abcam, 174280); mouse anti-IGF2R and rabbit anti-EEA1 (1:100; CST, C45B10); and mouse anti-IGF2R and rabbit anti-LAMP1 (1:100; CST, D2D11). Cells were then washed and stained with secondary antibodies (1:250, Thermo Fisher Scientific) for 2 hours. Filipin III (0.2 mg/ml; MilliporeSigma) staining was performed concurrently with secondary staining. Cells were mounted with VECTASHIELD Vibrance Antifade Mounting Medium (Vector Laboratories) before imaging on a confocal laser scanning microscope (Zeiss LSM 900).
Subcellular colocalization analysis
Images were analyzed using Imaris (Bitplane). Briefly, a surface was created for organelle signals (WGA, GOLGA1, RAB7, EEA1, TGN46, and LAMP1). This surface was used as a mask for the IGF2R channel. Surfaces were created for IGF2R in the masked channel and for total IGF2R. The colocalization index was taken as a ratio of the total volumes of these two surfaces.
Antibody generation
Three cohorts (Balb/c with alhydrogel adjuvant, Balb/c with Ribi adjuvant, and NZBW with alhydrogel adjuvant) of three mice each were immunized with recombinant His-tagged human CD22 ECD (R&D Systems). Serum titers against CD22-expressing and CD22-KO Ramos cells were checked on day 21. Splenocytes from mice with high serum titers were isolated on day 28 and subjected to hybridoma fusion. Supernatants from hybridoma libraries were screened against CD22-expressing cells, and libraries with high titers were clone sorted into plates. Of ~1400 clones, 38 were selected for intermediate to high CD22 binding and assayed for sCD22 blocking. Briefly, WT of IGF2R KO U937 cells were incubated with fluorescently labeled sCD22 in the presence of each anti-CD22 clone (3 μg/ml). Cells were analyzed by flow cytometry, and clones that efficiently blocked sCD22 binding to WT but not IGF2R KO cells were selected for downstream analysis. These three clones (M22, M28, and M42) were subjected to biolayer interferometry to calculate a dissociation constant and dose-dependent sCD22 blocking to calculate median inhibitory concentration. These screens yielded M42 as a candidate based on strong and specific CD22 binding and on strong and specific sCD22 blocking.
Differentiation of human microglia-like cells from iPSCs
iMGLs were differentiated according to a previously published protocol (55). Briefly, iPSCs were differentiated to hematopoietic progenitor cells using the STEMdiff Hematopoietic kit for 10 to 15 days before passage into microglia differentiation medium including DMEM/F12, 2× insulin transferrin-selenite, 2× B27, 0.5× N2, 1× glutamax, 1× nonessential amino acids, 400 μM monothioglycerol, and human insulin (5 μg/ml). Cultures were maintained in this basal medium supplemented with interleukin-34 (100 ng/ml), transforming growth factor–β1 (TGF-β1; 50 ng/ml), and macrophage colony-stimulating factor (25 ng/ml) (PeproTech) for 28 days. For the last 3 days in culture, two additional cytokines were added [CD200 (100 ng/ml, Novoprotein) and CX3CL1 (100 ng/ml, PeproTech)] to mature the microglia in a homeostatic brain-like environment.
iMGL immunocytochemistry
iMGLs were differentiated as described above. Eight-well chamber slides (Millicell) were coated with fibronectin, and iMGLs were plated at 100,000 cells per well in 200 μl of the aforementioned microglia medium without TGF-β. Cells were allowed to attach for 1 hour, and the medium was replaced with fresh medium containing the following treatments: U18666A (5 μM, Tocris), CSF from patients with NPC (10% volume), and mouse IgG1 isotype control (10 μg/ml, MOPC21, BioXCell) or mouse IgG1 anti-human CD22-blocking antibody (10 μg/ml, M42). After incubation for 24 hours at 37°, cells were gently washed with PBS, fixed with 4% paraformaldehyde for 15 min at room temperature, and gently permeabilized with 0.05% Triton X-100 in PBS with 1% BSA for 10 min at 4°. Cells were then washed to remove detergent and blocked with PBS containing 1% BSA for 1 hour at room temperature. Primary antibodies (mouse anti-LAMP2, 1:100; Santa Cruz Biotechnology, sc-18822; goat anti-IBA1, 1:200; Abcam, ab5076) were added in blocking buffer and incubated overnight at 4°. Cells were washed with PBS and incubated with blocking solution containing the following components for 1 hour at room temperature: donkey anti-mouse IgG AF647 (1:400, Thermo Fisher Scientific), donkey anti-goat IgG Alexa Fluor 488 (1:400, Thermo Fisher Scientific), Filipin III (0.2 mg/ml, Sigma-Aldrich). Slides were dried for 2 hours in the dark, coverslipped with fluoromount, dried overnight, and imaged using the Olympus FV3000RS confocal microscope.
iMGL bulk RNA-seq samples
Eight-well chamber slides (Millicell) were coated with fibronectin, and WT and I1061T mutant iMGLs were plated at 100,000 cells per well in 200 μl of the aforementioned microglia medium without TGF-β. Cells were allowed to attach for 1 hour, and the medium was replaced with fresh medium containing CSF from patients with NPC (10% volume) and mouse IgG1 isotype control (10 μg/ml, MOPC21, BioXCell) or mouse IgG1 anti-human CD22 blocking antibody (10 μg/ml, M42). After incubation for 24 hours at 37°, total RNA was isolated using an RNeasy Micro Plus kit (Qiagen). A total of 100,000 iMGLs were lysed in 350 μl of RLT buffer containing β-mercaptoethanol, and RNA was isolated per the manufacturer’s instructions. RNA integrity was assessed on a Bioanalyzer (Agilent), and high-quality samples were used for library preparation. Complementary DNA synthesis and amplification was performed using the SmartSeq v.4 Ultra-low input kit (Takara), and libraries were tagmented, adaptor-ligated, and indexed using the Nextera XT kit (Illumina). After normalization and pooling, libraries were sequenced on a NextSeq 550 (Illumina) using paired-end 150–base pair reads. Libraries were sequenced to a depth of ~20 million reads per sample. Raw sequencing files were demultiplexed with bcl2fastq, reads were trimmed with cutadapt and aligned using STAR, the count matrix was generated using SummarizedExperiment, and differential expression analysis was performed using edgeR using the TidyTranscriptomics package. Gene Ontology enrichment was performed using the TopGO package (Bioconductor) and visualized with the CellPlot package (Dieterich lab).
Statistical analysis
Statistical analyses were performed using GraphPad Prism (v9) or RStudio. Two-sided Student’s t test was used for two group comparisons, and analysis of variance (ANOVA) with Tukey multiple hypothesis correction was used for three or more group comparisons. Uncertainty is expressed as means ± 2 SDs or SE.
Supplementary Material
Acknowledgments:
We thank the patients and their families for donating tissue samples used for this research and J. Cottrell at the NIH NeuroBioBank at the University of Maryland for collecting tissue samples. We thank the members of the Wyss-Coray, Blurton-Jones, and Bertozzi laboratories for feedback and support; S. Pfeffer, M. Abu-Remaileh, and U. Medoh for critical reading of the manuscript; T. Branon and S. Han for helpful discussions; H. Zhang and K. Dickey for laboratory management; B. Lehallier for RNA-seq analysis scripts; M. Macauley for reagents; L. Kean for helpful discussions; and B. Carter for flow cytometry technical expertise. Schematics were created with BioRender.com.
Funding:
This work was funded by the Department of Veterans Affairs (T.W.-C.), the National Institute on Aging (RF1-AG064897–02 to T.W.-C., F30AG060638 to J.V.P., and T32AG000266 to M.S.H.), the UCI-ADRC (NIA P30 AG066519 to M.B.-J.), the National Institute of General Medical Sciences (R01-GM058867 to C.R.B. and K00CA212454 to N.M.R.), the NOMIS Foundation (T.W.-C.), the Glenn Foundation for Aging Research (T.W.-C.), the NYSCF Robertson Stem Cell Investigator Award (S.P.P.), the Stanford Wu Tsai Neurosciences Big Idea Project on Human Brain Organogenesis (S.P.P.), the Zuckerberg Initiative Ben Barres Investigator Award (S.P.P.) and the Wu Tsai Neurosciences Institute (T.W.-C. and C.R.B.).
Footnotes
Competing interests: C.R.B. is a cofounder and Scientific Advisory Board member of Lycia Therapeutics, Palleon Pharmaceuticals, Enable Bioscience, InterVenn Bio, and Redwood Bioscience (a subsidiary of Catalent) and a member of the Board of Directors of Eli Lilly and Company. M.B.-J. is a cofounder of NovoGlia Inc. and coinventor of patent application WO/2018/160496, related to the differentiation of stem cells into microglia. T.W.-C., J.V.P., M.S.H., and C.R.B. are coinventors on a patent application related to the work in this paper (US16/956,339, “Compositions and methods for treating age related disesases”). T.W.-C. and J.V.P. are coinventors on a patent application related to the work in this paper (PCT/US21/32875, “Compositions and methods for treating lysosomal storage disorders”).
SUPPLEMENTARY MATERIALS
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Raw sequencing data are available on NCBI GEO (accession number GSE184243).
REFERENCES AND NOTES
- 1.de Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F, Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60, 604–617 (1955). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Platt FM, d’Azzo A, Davidson BL, Neufeld EF, Tifft CJ, Lysosomal storage diseases. Nat. Rev. Dis. Primers. 4, 27 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Ballabio A, Bonifacino JS, Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R, Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 351, 1972–1977 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Reddy JV, Ganley IG, Pfeffer SR, Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLOS ONE 1, e19 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cologna SM, Jiang X-S, Backlund PS, Cluzeau CVM, Dail MK, Yanjanin NM, Siebel S, Toth CL, Jun H-S, Wassif CA, Yergey AL, Porter FD, Quantitative proteomic analysis of Niemann-Pick disease, type C1 cerebellum identifies protein biomarkers and provides pathological insight. PLOS ONE 7, e47845 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malnar M, Hecimovic S, Mattsson N, Zetterberg H, Bidirectional links between Alzheimer’s disease and Niemann-Pick type C disease. Neurobiol. Dis. 72(Pt. A), 37–47 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Jin L-W, Shie F-S, Maezawa I, Vincent I, Bird T, Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am. J. Pathol. 164, 975–985 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torres S, García-Ruiz CM, Fernandez-Checa JC, Mitochondrial Cholesterol in Alzheimer’s disease and Niemann-Pick type C disease. Front. Neurol 10, 1168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walterfang M, Fahey M, Desmond P, Wood A, Seal ML, Steward C, Adamson C, Kokkinos C, Fietz M, Velakoulis D, White and gray matter alterations in adults with Niemann-Pick disease type C: A cross-sectional study. Neurology 75, 49–56 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Bianconi SE, Hammond DI, Farhat NY, Dang Do A, Jenkins K, Cougnoux A, Martin K, Porter FD, Evaluation of age of death in Niemann-Pick disease, type C: Utility of disease support group websites to understand natural history. Mol. Genet. Metab. 126, 466–469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cougnoux A, Drummond RA, Collar AL, Iben JR, Salman A, Westgarth H, Wassif CA, Cawley NX, Farhat NY, Ozato K, Lionakis MS, Porter FD, Microglia activation in Niemann–Pick disease, type C1 is amendable to therapeutic intervention. Hum. Mol. Genet. 27, 2076–2089 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kavetsky L, Green KK, Boyle BR, Yousufzai FAK, Padron ZM, Melli SE, Kuhnel VL, Jackson HM, Blanco RE, Howell GR, Soto I, Increased interactions and engulfment of dendrites by microglia precede Purkinje cell degeneration in a mouse model of Niemann Pick Type-C. Sci. Rep. 9, 14722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabandé-Rodríguez E, Pérez-Cañamás A, Soto-Huelin B, Mitroi DN, Sánchez-Redondo S, Martínez-Sáez E, Venero C, Peinado H, Ledesma MD, Lipid-induced lysosomal damage after demyelination corrupts microglia protective function in lysosomal storage disorders. EMBO J. 38, e99553 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colombo A, Dinkel L, Müller SA, Sebastian Monasor L, Schifferer M, Cantuti-Castelvetri L, König J, Vidatic L, Bremova-Ertl T, Lieberman AP, Hecimovic S, Simons M, Lichtenthaler SF, Strupp M, Schneider SA, Tahirovic S, Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia. Nat. Commun. 12, 1158 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baudry M, Yao Y, Simmons D, Liu J, Bi X, Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: Immunohistochemical observations of microglia and astroglia. Exp. Neurol. 184, 887–903 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Cologna SM, Cluzeau CVM, Yanjanin NM, Blank PS, Dail MK, Siebel S, Toth CL, Wassif CA, Lieberman AP, Porter FD, Human and mouse neuroinflammation markers in Niemann-Pick disease, type C1. J. Inherit. Metab. Dis. 37, 83–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, Li L, Lee DP, Morgens DW, Yang AC, Shuken SR, Gate D, Scott M, Khatri P, Luo J, Bertozzi CR, Bassik MC, Wyss-Coray T, CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568, 187–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, Yao Z, Eggermont J, Höllt T, Levi BP, Shehata SI, Aevermann B, Beller A, Bertagnolli D, Brouner K, Casper T, Cobbs C, Dalley R, Dee N, Ding S-L, Ellenbogen RG, Fong O, Garren E, Goldy J, Gwinn RP, Hirschstein D, Keene CD, Keshk M, Ko AL, Lathia K, Mahfouz A, Maltzer Z, McGraw M, Nguyen TN, Nyhus J, Ojemann JG, Oldre A, Parry S, Reynolds S, Rimorin C, Shapovalova NV, Somasundaram S, Szafer A, Thomsen ER, Tieu M, Quon G, Scheuermann RH, Yuste R, Sunkin SM, Lelieveldt B, Feng D, Ng L, Bernard A, Hawrylycz M, Phillips JW, Tasic B, Zeng H, Jones AR, Koch C, Lein ES, Conserved cell types with divergent features in human versus mouse cortex. Nature , 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, Balic A, Giladi A, Sheban F, Dutertre C-A, Pfeifle C, Peri F, Raffo-Romero A, Vizioli J, Matiasek K, Scheiwe C, Meckel S, Mätz-Rensing K, van der Meer F, Thormodsson FR, Stadelmann C, Zilkha N, Kimchi T, Ginhoux F, Ulitsky I, Erny D, Amit I, Prinz M, Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell 179, 1609–1622.e16 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Khrameeva E, Kurochkin I, Han D, Guijarro P, Kanton S, Santel M, Qian Z, Rong S, Mazin P, Sabirov M, Bulat M, Efimova O, Tkachev A, Guo S, Sherwood CC, Camp JG, Pääbo S, Treutlein B, Khaitovich P, Single-cell-resolution transcriptome map of human, chimpanzee, bonobo, and macaque brains. Genome Res. 30, 776–789 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang Y-N, Cai X, Zhou H-M, Jin W-D, Zhang M, Zhang Y, Du X-X, Chen Z-HK, Diagnostic and prognostic roles of soluble CD22 in patients with Gram-negative bacterial sepsis. Hepatobiliary Pancreat. Dis. Int. 14, 523–529 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Matsushita K, Margulies I, Onda M, Nagata S, Stetler-Stevenson M, Kreitman RJ, Soluble CD22 as a tumor marker for hairy cell leukemia. Blood 112, 2272–2277 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Powell LD, Sgroi D, Sjoberg ER, Stamenkovic I, Varki A, Natural ligands of the B cell adhesion molecule CD22 beta carry N-linked oligosaccharides with alpha-2,6-linked sialic acids that are required for recognition. J. Biol. Chem. 268, 7019–7027 (1993). [PubMed] [Google Scholar]
- 25.Ereño-Orbea J, Sicard T, Cui H, Mazhab-Jafari MT, Benlekbir S, Guarné A, Rubinstein JL, Julien J-P, Molecular basis of human CD22 function and therapeutic targeting. Nat. Commun. 8, 746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haney MS, Bohlen CJ, Morgens DW, Ousey JA, Barkal AA, Tsui CK, Ego BK, Levin R, Kamber RA, Collins H, Tucker A, Li A, Vorselen D, Labitigan L, Crane E, Boyle E, Jiang L, Chan J, Rincon E, Greenleaf WJ, Li B, Snyder MP, Weissman IL, Theriot JA, Collins SR, Barres BA, Bassik MC, Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nat. Genet. 50, 1716–1727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgens DW, Deans RM, Li A, Bassik MC, Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 34, 634–636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hennet T, Chui D, Paulson JC, Marth JD, Immune regulation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. U.S.A. 95, 4504–4509 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramya TNC, Weerapana E, Liao L, Zeng Y, Tateno H, Liao L, Yates JR 3rd, Cravatt BF, Paulson JC, In situ trans ligands of CD22 identified by glycan-protein photocross-linking-enabled proteomics. Mol. Cell. Proteomics 9, 1339–1351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powell LD, Jain RK, Matta KL, Sabesan S, Varki A, Characterization of sialyloligosaccharide binding by recombinant soluble and native cell-associated CD22. Evidence for a minimal structural recognition motif and the potential importance of multisite binding. J. Biol. Chem. 270, 7523–7532 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Kornfeld S, Structure and function of the mannose 6-phosphate/insulin like growth factor II receptors. Annu. Rev. Biochem. 61, 307–330 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Fratantoni JC, Hall CW, Neufeld EF, Hurler and Hunter syndromes: Mutual correction of the defect in cultured fibroblasts. Science 162, 570–572 (1968). [DOI] [PubMed] [Google Scholar]
- 33.Vladutiu GD, Rattazzi MC, Excretion-reuptake route of beta-hexosaminidase in normal and I-cell disease cultured fibroblasts. J. Clin. Invest. 63, 595–601 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neufeld EF, in Fabry Disease: Perspectives from 5 Years of FOS, Mehta A, Beck M, Sunder-Plassmann G, Eds. (Oxford PharmaGenesis, 2011). [PubMed] [Google Scholar]
- 35.Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, Bertozzi CR, Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amritraj A, Peake K, Kodam A, Salio C, Merighi A, Vance JE, Kar S, Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice. Am. J. Pathol. 175, 2540–2556 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cluzeau CVM, Watkins-Chow DE, Fu R, Borate B, Yanjanin N, Dail MK, Davidson CD, Walkley SU, Ory DS, Wassif CA, Pavan WJ, Porter FD, Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum. Mol. Genet. 21, 3632–3646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim S, Ock J, Kim AK, Lee HW, Cho J-Y, Kim DR, Park J-Y, Suk K, Neurotoxicity of microglial cathepsin D revealed by secretome analysis. J. Neurochem. 103, 2640–2650 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Cataldo AM, Nixon RA, Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc. Natl. Acad. Sci. U.S.A. 87, 3861–3865 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amritraj A, Wang Y, Revett TJ, Vergote D, Westaway D, Kar S, Role of cathepsin D in U18666A-induced neuronal cell death: Potential implication in Niemann-Pick type C disease pathogenesis. J. Biol. Chem. 288, 3136–3152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sleat DE, Wiseman JA, El-Banna M, Price SM, Verot L, Shen MM, Tint GS, Vanier MT, Walkley SU, Lobel P, Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. U.S.A. 101, 5886–5891 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winkler MBL, Kidmose RT, Szomek M, Thaysen K, Rawson S, Muench SP, Wüstner D, Pedersen BP, Structural insight into eukaryotic sterol transport through niemann-pick type C proteins. Cell 179, 485–497.e18 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Qian H, Wu X, Du X, Yao X, Zhao X, Lee J, Yang H, Yan N, Structural basis of Low-pH-Dependent lysosomal cholesterol egress by NPC1 and NPC2. Cell 182, 98–111.e18 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Sleat DE, Tannous A, Sohar I, Wiseman JA, Zheng H, Qian M, Zhao C, Xin W, Barone R, Sims KB, Moore DF, Lobel P, Proteomic Analysis of brain and cerebrospinal fluid from the three major forms of neuronal ceroid lipofuscinosis reveals potential biomarkers. J. Proteome Res. 16, 3787–3804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willenborg M, Schmidt CK, Braun P, Landgrebe J, von Figura K, Saftig P, Eskelinen E-L, Mannose 6-phosphate receptors, Niemann-Pick C2 protein, and lysosomal cholesterol accumulation. J. Lipid Res. 46, 2559–2569 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Griffiths G, Hoflack B, Simons K, Mellman I, Kornfeld S, The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell 52, 329–341 (1988). [DOI] [PubMed] [Google Scholar]
- 47.Waguri S, Dewitte F, Le Borgne R, Rouillé Y, Uchiyama Y, Dubremetz J-F, Hoflack B, Visualization of TGN to endosome trafficking through fluorescently labeled MPR and AP-1 in living cells. Mol. Biol. Cell 14, 142–155 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dahms NM, Lobel P, Kornfeld S, Mannose 6-phosphate receptors and lysosomal enzyme targeting. J. Biol. Chem. 264, 12115–12118 (1989). [PubMed] [Google Scholar]
- 49.Lin SX, Mallet WG, Huang AY, Maxfield FR, Endocytosed cation-independent mannose 6-phosphate receptor traffics via the endocytic recycling compartment en route to the trans-Golgi network and a subpopulation of late endosomes. Mol. Biol. Cell 15, 721–733 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weintraub H, Abramovici A, Sandbank U, Booth AD, Pentchev PG, Sela B, Dysmyelination in NCTR-Balb/C mouse mutant with a lysosomal storage disorder. Morphological survey. Acta Neuropathol. 74, 374–381 (1987). [DOI] [PubMed] [Google Scholar]
- 51.Samra B, Jabbour E, Ravandi F, Kantarjian H, Short NJ, Evolving therapy of adult acute lymphoblastic leukemia: State-of-the-art treatment and future directions. J. Hematol. Oncol. 13, 70 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meyer SJ, Linder AT, Brandl C, Nitschke L, B cell siglecs-news on signaling and its interplay with ligand binding. Front. Immunol. 9, 2820 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Praggastis M, Tortelli B, Zhang J, Fujiwara H, Sidhu R, Chacko A, Chen Z, Chung C, Lieberman AP, Sikora J, Davidson C, Walkley SU, Pipalia NH, Maxfield FR, Schaffer JE, Ory DS, A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 35, 8091–8106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, Caraway CA, Fote GM, Madany AM, Agrawal A, Kayed R, Gylys KH, Cahalan MD, Cummings BJ, Antel JP, Mortazavi A, Carson MJ, Poon WW, Blurton-Jones M, iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94, 278–293.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, Blurton-Jones M, Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 13, 67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hasselmann J, Coburn MA, England W, Figueroa Velez DX, Kiani Shabestari S, Tu CH, McQuade A, Kolahdouzan M, Echeverria K, Claes C, Nakayama T, Azevedo R, Coufal NG, Han CZ, Cummings BJ, Davtyan H, Glass CK, Healy LM, Gandhi SP, Spitale RC, Blurton-Jones M, Development of a chimeric model to study and manipulate human microglia in vivo. Neuron 103, 1016–1033.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gelsthorpe ME, Baumann N, Millard E, Gale SE, Langmade SJ, Schaffer JE, Ory DS, Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 283, 8229–8236 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu F, Liang Q, Abi-Mosleh L, Das A, De Brabander JK, Goldstein JL, Brown MS, Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. eLife 4, e12177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, Edinger AL, Jung S, Rossner MJ, Simons M, Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995–998 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai L-H, Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M, Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, Grün D, Kroshilina AA, Dionne D, Sarkis RA, Cosgrove GR, Helgager J, Golden JA, Pennell PB, Prinz M, Vonsattel JPG, Teich AF, Schneider JA, Bennett DA, Regev A, Elyaman W, Bradshaw EM, De Jager PL, Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 11, 6129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cougnoux A, Drummond RA, Fellmeth M, Navid F, Collar AL, Iben J, Kulkarni AB, Pickel J, Schiffmann R, Wassif CA, Cawley NX, Lionakis MS, Porter FD, Unique molecular signature in mucolipidosis type IV microglia. J. Neuroinflammation 16, 276 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Douvaras P, Fossati V, Generation and isolation of oligodendrocyte progenitor cells from human pluripotent stem cells. Nat. Protoc. 10, 1143–1154 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Peschl P, Schanda K, Zeka B, Given K, Böhm D, Ruprecht K, Saiz A, Lutterotti A, Rostásy K, Höftberger R, Berger T, Macklin W, Lassmann H, Bradl M, Bennett JL, Reindl M, Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J. Neuroinflammation 14, 208 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerrits E, Heng Y, Boddeke EWGM, Eggen BJL, Transcriptional profiling of microglia; current state of the art and future perspectives. Glia 68, 740–755 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, Fourne Y, Matthews PM, Theys T, Mancuso R, de Strooper B, Fiers M, Single-nucleus RNA-Seq is not suitable for detection of microglial activation genes in humans. Cell Rep. 32, 108189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ, Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177, 1873–1887.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heybrock S, Kanerva K, Meng Y, Ing C, Liang A, Xiong Z-J, Weng X, Ah Kim Y, Collins R, Trimble W, Pomès R, Privé GG, Annaert W, Schwake M, Heeren J, Lüllmann-Rauch R, Grinstein S, Ikonen E, Saftig P, Neculai D, Lysosomal integral membrane protein-2 (LIMP-2/SCARB2) is involved in lysosomal cholesterol export. Nat. Commun. 10, 3521 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singhal A, Szente L, Hildreth JEK, Song B, Hydroxypropyl-beta and -gamma cyclodextrins rescue cholesterol accumulation in Niemann–Pick C1 mutant cell via lysosome-associated membrane protein 1. Cell Death Dis. 9, 1019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li J, Pfeffer SR, Lysosomal membrane glycoproteins bind cholesterol and contribute to lysosomal cholesterol export. eLife 5, e21635 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Twu Y-C, Lee T-S, Lin Y-L, Hsu S-M, Wang Y-H, Liao C-Y, Wang C-K, Liang Y-C, Liao Y-J, Niemann-Pick Type C2 protein mediates hepatic stellate cells activation by regulating free cholesterol accumulation. Int. J. Mol. Sci. 17, 1122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Massa PT, Saha S, Wu C, Jarosinski KW, Expression and function of the protein tyrosine phosphatase SHP-1 in oligodendrocytes. Glia 29, 376–385 (2000). [PubMed] [Google Scholar]
- 74.Kodachi T, Matsumoto S, Mizuguchi M, Osaka H, Kanai N, Nanba E, Ohno K, Yamagata T, Severe demyelination in a patient with a late infantile form of Niemann-Pick disease type C. Neuropathology 37, 426–430 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Takikita S, Fukuda T, Mohri I, Yagi T, Suzuki K, Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J. Neuropathol. Exp. Neurol. 63, 660–673 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Ray TA, Cochran K, Kozlowski C, Wang J, Alexander G, Cady MA, Spencer WJ, Ruzycki PA, Clark BS, Laeremans A, He M-X, Wang X, Park E, Hao Y, Iannaccone A, Hu G, Fedrigo O, Skiba NP, Arshavsky VY, Kay JN, Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 11, 3328 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Strauss K, Goebel C, Runz H, Möbius W, Weiss S, Feussner I, Simons M, Schneider A, Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-Pick type C disease. J. Biol. Chem. 285, 26279–26288 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hane M, Chen DY, Varki A, Human-specific microglial Siglec-11 transcript variant has the potential to affect polysialic acid-mediated brain functions at a distance. Glycobiology 31, 231–242 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miller DJ, Duka T, Stimpson CD, Schapiro SJ, Baze WB, McArthur MJ, Fobbs AJ, Sousa AMM, Sestan N, Wildman DE, Lipovich L, Kuzawa CW, Hof PR, Sherwood CC, Prolonged myelination in human neocortical evolution. Proc. Natl. Acad. Sci. U.S.A. 109, 16480–16485 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vite CH, Ding W, Bryan C, O’Donnell P, Cullen K, Aleman D, Haskins ME, Van Winkle T, Clinical, electrophysiological, and serum biochemical measures of progressive neurological and hepatic dysfunction in feline Niemann-Pick type C disease. Pediatr. Res. 64, 544–549 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davidson CD, Gibson AL, Gu T, Baxter LL, Deverman BE, Beadle K, Incao AA, Rodriguez-Gil JL, Fujiwara H, Jiang X, Chandler RJ, Ory DS, Gradinaru V, Venditti CP, Pavan WJ, Improved systemic AAV gene therapy with a neurotrophic capsid in Niemann-Pick disease type C1 mice. Life Sci Alliance 4, e202101040 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patterson MC, Garver WS, Giugliani R, Imrie J, Jahnova H, Meaney FJ, Nadjar Y, Vanier MT, Moneuse P, Morand O, Rosenberg D, Schwierin B, Héron B, Long-term survival outcomes of patients with Niemann-Pick disease type C receiving miglustat treatment: A large retrospective observational study. J. Inherit. Metab. Dis. 43, 1060–1069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kirkegaard T, Gray J, Priestman DA, Wallom K-L, Atkins J, Olsen OD, Klein A, Drndarski S, Petersen NHT, Ingemann L, Smith DA, Morris L, Bornæs C, Jørgensen SH, Williams I, Hinsby A, Arenz C, Begley D, Jäättelä M, Platt FM, Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 8, 355ra118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alam MS, Getz M, Haldar K, Chronic administration of an HDAC inhibitor treats both neurological and systemic Niemann-Pick type C disease in a mouse model. Sci. Transl. Med. 8, 326ra23 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Kaya E, Smith DA, Smith C, Morris L, Bremova-Ertl T, Cortina-Borja M, Fineran P, Morten KJ, Poulton J, Boland B, Spencer J, Strupp M, Platt FM, Acetyl-Leucine slows disease progression in lysosomal storage disorders. bioRxiv 2020.05.20.105973 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ory DS, Ottinger EA, Farhat NY, King KA, Jiang X, Weissfeld L, Berry-Kravis E, Davidson CD, Bianconi S, Keener LA, Rao R, Soldatos A, Sidhu R, Walters KA, Xu X, Thurm A, Solomon B, Pavan WJ, Machielse BN, Kao M, Silber SA, McKew JC, Brewer CC, Vite CH, Walkley SU, Austin CP, Porter FD, Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: A non-randomised, open-label, phase 1–2 trial. Lancet 390, 1758–1768 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hughes MP, Smith DA, Morris L, Fletcher C, Colaco A, Huebecker M, Tordo J, Palomar N, Massaro G, Henckaerts E, Waddington SN, Platt FM, Rahim AA, AAV9 intracerebroventricular gene therapy improves lifespan, locomotor function and pathology in a mouse model of Niemann–Pick type C1 disease. Hum. Mol. Genet. 27, 3079–3098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maes ME, Colombo G, Schulz R, Siegert S, Targeting microglia with lentivirus and AAV: Recent advances and remaining challenges. Neurosci. Lett. 707, 134310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, Li G, Duncan JA III, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MGH, Barres BA, Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Loh KH, Stawski PS, Draycott AS, Udeshi ND, Lehrman EK, Wilton DK, Svinkina T, Deerinck TJ, Ellisman MH, Stevens B, Carr SA, Ting AY, Proteomic analysis of unbounded cellular compartments: Synaptic clefts. Cell 166, 1295–1307.e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K, Carr SA, Ting AY, Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 11, 456–475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cox J, Mann M, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 93.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J, The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Raw sequencing data are available on NCBI GEO (accession number GSE184243).