

Abstract
Purpose:
Combinations of immune-checkpoint inhibitors (ICI) with other cancer therapies have been approved for advanced cancers in multiple indications, and numerous trials are under way to test new combinations. However, the mechanisms that account for the superiority of approved ICI combinations relative to their constituent monotherapies remain unknown.
Experimental Design:
We analyzed 13 phase III clinical trials testing combinations of ICIs with each other or other drugs in patients with advanced melanoma and lung, breast, gastric, kidney, and head and neck cancers. The clinical activity of the individual constituent therapies, measured in the same or a closely matched trial cohort, was used to compute progression-free survival (PFS) curves expected under a model of independent drug action. To identify additive or synergistic efficacy, PFS expected under this null model was compared with observed PFS by Cox regression.
Results:
PFS elicited by approved combination therapies with ICIs could be accurately predicted from monotherapy data using the independent drug action model (Pearson r = 0.98, P < 5 × 10−9, N = 4,173 patients, 8 types of cancer). We found no evidence of drug additivity or synergy except in one trial in which such interactions might have extended median PFS by 9 days.
Conclusions:
Combining ICIs with other cancer therapies affords predictable and clinically meaningful benefit by providing patients with multiple chances of response to a single agent. Conversely, there exists no evidence in phase III trials that other therapies interact with and enhance the activity of ICIs. These findings can inform the design and testing of new ICI combination therapies while emphasizing the importance of developing better predictors (biomarkers) of ICI response.
Translational Relevance.
Use of immune-checkpoint inhibitors (ICI) targeting PD-1, PD-L1, and CTLA-4 has dramatically improved survival for several previously difficult-to-treat solid cancers. Activation of immune surveillance is potentially relevant to any type of cancer, and thousands of ICI clinical trials are therefore under way, many pairing ICIs with chemotherapies, targeted drugs, and immunotherapies themselves as a means to enhance ICI activity or increase tumor immunogenicity. The superiority of some combinations relative to monotherapy in clinical trials has been widely interpreted as evidence of synergistic drug interaction; many preclinical studies are under way to understand such synergy. However, our finding that all 13 FDA-approved ICI combinations for which a test is possible conform closely to a null model of drug independence means that there is no evidence that ICIs are synergistic or even additive with each other or other drugs in humans; instead, combinations provide patients with multiple opportunities for a meaningful response to monotherapy. This makes it possible to predict the likely activity of a new combination from survival data for the constituent monotherapies, an observation of immediate utility in the design of new trials. It also raises questions about concurrent versus sequential therapy or the need for drug combinations in all patients. More generally, our findings suggest that the focus of translational research into ICIs needs to shift from studying drug interaction in atypically sensitive animal models to understanding why response is so variable in humans and how it can be predicted at the level of individual patients.
Introduction
The introduction of immunotherapy is one of the most important recent developments in oncology. Immune-checkpoint inhibitors (ICI) directed against PD-1, PD-L1, and CTLA-4 have improved survival for many but not all types of cancer (1). To increase rates and durability of responses to ICIs, thousands of combination clinical trials are under way (2). A key motivation for these trials is the hypothesis that chemotherapies, targeted drugs, and immunotherapies themselves can enhance the activity of ICIs by increasing tumor immunogenicity or through other mechanisms (3, 4). The clinical success of ICI combinations has been widely interpreted as evidence of interaction-based mechanisms, but phase III trials do not address such hypotheses. Thus, the superiority of combination ICI therapies to monotherapy in multiple indications does not resolve whether “synergy” is involved, or necessary. It is nonetheless well recognized that investigating this issue has implications for the development of new treatments and trials, as well as practical matters such as prioritization of concurrent versus sequential therapy, and whether combination therapy is necessary for all patients (5–7).
When two or more active therapies are combined, they each provide some probability of response. In general, the probability that either one of two concurrent events A and B will occur (e.g., response to therapy A or therapy B) is the sum of their individual probabilities minus the probability that both occur (P(A and/or B) = P(A) + P(B) − P(A and B) (Fig. 1). This “addition law of probability” was first applied to remission rates from monotherapy or combination therapy by the acute leukemia group B (ALGB) in 1961, and led to the conclusion that the therapeutic advantage of combining 6-mercaptopurine (6MP) and methotrexate (MTX) in the treatment of acute lymphoblastic leukemia arose from two drugs acting independently (8). Specifically, ALGB Protocol 2 observed that some patients responded to therapy 6MP but not therapy MTX, and vice versa, allowing the combination to benefit patients in both groups. Moreover, the observed combination therapy response rate was remarkably close to that predicted by the addition law of probability. Thus, it has long been known that tolerable combinations of active therapies can provide a clinically meaningful benefit to patient populations (as measured by population-averaged outcomes, such as response rate) without a requirement for pharmacologic additivity or synergy in individual patients (note that pharmacologic additivity as commonly defined using Bliss or Loewe null models is different from addition of probabilities; ref. 9).
Figure 1.
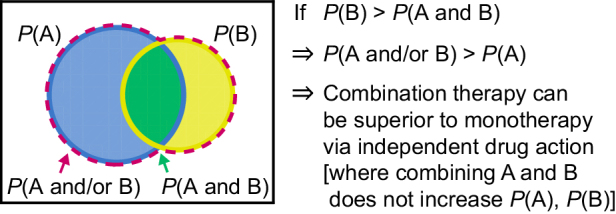
The addition law of probability and its implication for combination therapy. This Venn diagram represents a cohort of patients with cancer and their probabilities of being responsive to therapy A and/or therapy B. The mathematical formulation of this concept, P(combination) = P(A) + (1 – P(A))×P(B), was given by Frei and colleagues in 1961 (8), and recent work has adapted the concept to PFS and to account for correlations in probabilities.
Recently, we and others have shown that the concept of independent drug action can also be applied to analyzing progression-free survival (PFS) in contemporary oncology trials (10–12). Independent drug action is the null hypothesis that therapy A does not change the activity of therapy B and vice versa; “activity” can be quantified in multiple ways, and here we consider probability of PFS. The probabilities of response to two agents may be correlated, due to partial drug cross-resistance or prognostic factors affecting responses to both drugs. The greater the correlation coefficient ρ, the greater the fraction of patients are either responsive or resistant to both drugs. Accounting for correlated response probabilities can be addressed using a computational algorithm we described previously (10), but a more elegant analytical solution was recently derived by Chen and colleagues (12). Specifically, let P(A, t) and P(B, t) be the PFS probabilities of therapies A and B at time t, and ρ be the correlation between these probabilities. If therapies A and B act independently when combined [meaning that they exhibit no pharmacologic interaction and P(A, t) is not affected by the presence of B or P(B, t) by the presence of A], then the expected PFS for the combination is
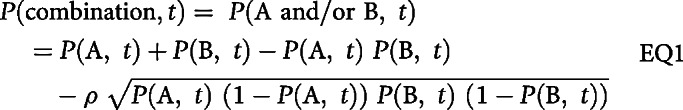 |
When ρ = 0, this equation is identical to the one used by Frei and his ALGB colleagues in the 1960s (8, 13). Evidence for pharmacologic additivity or synergy is obtained when the null model in EQ1 can be rejected. Specifically, P(combination) must surpass P(A and/or B) as calculated from single-agent activities at a specific confidence level using Cox regression; conversely, if P(combination) = P(A and/or B), then the null model cannot be rejected and additivity or synergy is not demonstrated.
An inability to reject a null hypothesis of drug independence is not incompatible with a complex process of time-varying drug response and resistance—albeit one hidden from view; nonadditive effects on the molecular states of cells are common when therapies are combined. However, if efficacy as measured in a clinical trial is insufficient to reject the null hypothesis of drug independence, then the quantitative implication is that the PFS time for each patient depends on whichever constituent of the combination was most effective for that individual, and has not been significantly altered by the presence of less effective agents or underlying molecular mechanisms. It has long been established that null models used to assess drug interaction should be mechanism-agnostic in both cell lines and trials, or else statements about synergy would vary depending on changing ideas about how drugs work rather than empirical data on efficacy (14). In the discussion of this paper, we consider the implications of the apparent discrepancy between preclinical studies on ICIs and phase III trial results.
We previously tested the model of independent drug action using data from multiple phase III clinical trials, and hundreds of patient-derived tumor xenografts. We found that many approved combination therapies produce a PFS benefit that is fully accounted for by independent action. Some combinations exhibit additivity or synergy by this definition, such as bevacizumab plus chemotherapy for metastatic ovarian or colorectal cancers (10, 15, 16). These findings in solid tumors are similar to those reported by ALGB from multiple trials in leukemia in the 1960s (8, 13). Benefit from independent action arises when drugs are active as monotherapies, patients vary substantially in their sensitivities to individual drugs, and drugs in the combination are not cross-resistant (weakly correlated; ref. 10), criteria that are met by most contemporary combination therapies. Our previous analysis primarily involved combinations of targeted therapies and chemotherapies (10) as phase III data were available at the time for only one ICI combination: ipilimumab (a CTLA-4 mAb) plus nivolumab (a PD-1 mAb) in metastatic melanoma. We found that PFS as well as changes in tumor volume were precisely consistent with independent action. Additional evidence for independent action in PD-1 inhibitor combinations was recently reported by Schmidt and colleagues (17) based on analysis of objective response rates. Here, we analyze PFS data from all clinical trials of ICI combination therapies as of April 2020 for which data on single-agent and combination arms are available and look for evidence of additive or synergistic drug interaction.
Materials and Methods
Data sources
We sought published data for all combination therapies with ICIs that were FDA approved as of April 2020. We required Kaplan–Meier plots of PFS for the combination therapy and for the constituents of the combination in matching patient cohorts. In some trials the control arm was itself a combination, which we treat as one entity (e.g., etoposide plus platinum in IMpower133; ref. 18); we did not assess drug interactions within control arms because the necessary data are rarely available. Hereafter “single agent” in most cases refers to a single drug and in other cases a control arm combination (this distinction is clear in Figs. 2 and 3). Some trials were lacking at least one monotherapy arm, in which case we searched publications and conference proceedings for a clinical trial of that therapy meeting the following criteria: (i) patients had the same type and stage of disease, (ii) patients were treated at the same or near-identical dose, and (iii) treatments involved the same line of therapy. For all combinations analyzed in this paper, treatment was first line, but for 5 of 26 monotherapy arms, data were available only in previously treated patients (second or third line). The potential impact of this difference is addressed explicitly in the results. With regard to this methodology, we note that in general, historical controls are not appropriate for trials intended to be practice-changing (19) but are acceptable for trial simulations, including those used in adaptive trials (20). In many cases, historical controls represent the only way to evaluate drug interaction for a new combination because contemporaneous data on monotherapies is not collected. Complete treatment details and patient characteristics for the trials analyzed are presented in Supplementary Tables S1 and S2.
Figure 2.

PFS for combination therapies as observed in clinical trials and as predicted from independent activity of the therapies comprising the combination (part 1). PFS observed for each combination therapy (blue) was compared with that expected from the PFS distributions of the constituents of the combination (green and magenta) under the null hypothesis of independent drug action (black line and gray range, which reflects uncertainty in response correlation (ρ = 0.3 ± 0.2). Data from patients treated at second line or later are indicated by 2L and 2L+; all other data are from patients previously untreated for metastatic or advanced cancer. CPS, PD-L1 combined proportion score; TPS, PD-L1 tumor proportion score. Combination therapy data from (A) CheckMate 067 (31), (B) KEYNOTE-189 (32), (C) KEYNOTE-407 (33), (D) IMpassion130 (36), (E) KEYNOTE-426 (35), (F) JAVELIN Renal 101 (34), (G) IMpower133 (18) and CASPIAN (21), (H) IMpower130 (38), (I) KEYNOTE-048 (37), (J) KEYNOTE-062 (22), (K) IMpower150 (ref. 39; note the difference between expected and observed PFS is significant for left panel and not for right panel; see Supplementary Fig. S1), (L) NCT00324155 (23), and (M) KEYNOTE-022 (24, 40). Data sources, patient characteristics, and limitations are described in Supplementary Tables S1 and S2.
Figure 3.

Trials combining ICIs with other cancer therapies are consistent with, or inferior to, independent drug action. Observed PFS has a strong linear correlation with PFS expected under the null hypothesis of independent drug action, both at (A) a landmark of 12 months (R2 = 0.96; Pearson r = 0.98, P < 10−8) and (B) over all measured times (R2 = 0.98; Pearson r = 0.99), except for pembrolizumab plus dabrafenib and trametinib for BRAFV600 melanoma (24), which involved substantial dose reductions and interruptions that violated model requirements (green points).
We identified suitable combination and control monotherapy data for 11 of the 14 ICI combinations approved by the FDA (Supplementary Table S3, which also describes data limitations precluding conclusions for the three remaining combinations, including advanced endometrial cancer; Supplementary Fig. S3). Two trials in small cell lung cancer of etoposide plus platinum with or without PD-L1 blockade were merged for analysis (18, 21). We also included three ICI combination therapies lacking FDA approval where complete combination and monotherapy data were available for analysis: (i) pembrolizumab plus chemotherapy for advanced gastric and gastroesophageal junction cancers (22), (ii) ipilimumab plus dacarbazine for metastatic melanoma (23), and (iii) pembrolizumab plus dabrafenib and trametinib for metastatic BRAF-mutant melanoma (24). The latter trial did not meet our criteria for similar dosing across control and experimental arms but was included because a comparison with the predictions of independent action illustrated the negative consequences of dose reductions and interruptions.
Data extraction
Published Kaplan–Meier functions of PFS were imported into Adobe Illustrator to produce one image per treatment arm. If rasterized, figures were digitally traced in Adobe Photoshop. Kaplan–Meier curves were digitized in Wolfram Mathematica 12 using published algorithms (10). For hazard ratio calculations, individual participant data were imputed from Kaplan–Meier functions and at-risk tables using published methods (25). Supplementary code provided software and source data.
Simulation of independent action
The expected PFS distribution, under the null hypothesis of independence, was computed by two equivalent methods, being a published algorithm (10) and a published equation [ref. 12; EQ1; Supplementary code shows their consistency]. Censoring is already accounted for in the “input” data, which is Kaplan–Meier plots of PFS probability versus time. Analysis of PFS is not affected by cross-over or post-protocol therapy. As previously reported (10), our use of partial positive correlation in simulations (ρ = 0.3 ± 0.2) was empirically supported by a large database of drug responses in patient-derived tumor xenografts, and is consistent with clinical observations from sequential monotherapy trials and from comparing response rates at first and second line (8, 26–30). Partial correlations in drug response could in principle arise both from tumor-intrinsic properties, highly related mechanisms of drug action, or patient status such as age and comorbidities (i.e., prognostic factors). A conclusion that a combination involves additivity or synergy is supported if PFS is significantly superior to PFS expected from independent action (by the proportional hazards model). No current method, including those described here, can make the further distinction between additivity or synergy as pharmacologically defined based on clinical trial data.
The analysis presented here is applicable to PFS in advanced cancers. As presently formulated, this method is not applicable to adjuvant therapy or indolent tumors, scenarios in which “survival” can be strongly influenced by factors other than systemic therapy, including curative surgery or an intrinsic lack of tumor proliferation. Similar concerns apply to overall survival, as postprogression survival could be affected by subsequent lines of therapy and other factors (Supplementary Note and Supplementary Fig. S5).
Data availability
Complete source data (digitized PFS distributions) are provided in the Supplementary Code.
Results
We analyzed 13 recently reported phase III trials of drug combinations involving ICIs in melanoma, squamous and nonsquamous non–small cell lung cancers (NSCLC), small cell lung cancer, renal cell cancer, triple-negative breast cancer, gastric and gastroesophageal junction cancer, and head and neck squamous cell carcinoma, and one phase II trial in BRAF-mutant melanoma (18, 21–24, 31–40). We also updated our published analysis of ipilimumab plus nivolumab (10) in metastatic melanoma with longer follow-up data (42 months; ref. 31).
Eleven trials exhibited PFS distributions statistically indistinguishable from the prediction of the independence model (Fig. 2A–J). In the case of the IMpower150 trial in nonsquamous NSCLC (39), observed PFS exceeded the prediction of independence, and in the KEYNOTE-022 trial in BRAF-mutant melanoma (24), which had extensive dose reductions and interruptions, it was worse. For the KEYNOTE-048 trial in head and neck squamous cell carcinoma (37) and KEYNOTE-062 trial in gastric and gastroesophageal junction cancers (22), published data made it possible to analyze cohorts with or without enrichment for PD-L1 expression; both cohorts conformed to the predictions of drug independence (Fig. 2I and J). Concordance or deviation from independence was quantified by computing a hazard ratio for the comparison of observed combination therapy PFS and expected combination therapy PFS. Once again, all combination therapy trials but IMpower150 were consistent with, or inferior to, the null hypothesis of independent action, with no statistically significant difference in PFS from expectation (Supplementary Fig. S1). Moreover, observed PFS and expected PFS under the assumption of independence exhibited a strong correlation at a “landmark” of 12 months after randomization (Fig. 3A; Pearson correlation r = 0.98, P < 10−8, n = 13 trials; with time-series data, a P value can be calculated only at a single time). When observed and expected PFS were compared at all observed times, Pearson correlation was 0.99, and the mean absolute error for the independence model was less than 3% (Fig. 3B). Thus, in nearly all trials, P(combination, t) was indistinguishable from P(A and/or B, t) as computed from monotherapy data, and the null model could not be rejected. Consequently, there is little to no evidence that combining therapies A and B has increased P(A, t) or P(B, t). These findings constitute strong evidence that the dominant mode of benefit provided by approved ICI combinations is explainable by the null model of independent drug action.
A possible case of additive or synergistic drug interaction in nonsquamous NSCLC
IMpower150 evaluated first-line treatment of nonsquamous NSCLC by bevacizumab, carboplatin, and paclitaxel, with or without the PD-L1 inhibitor atezolizumab (39). In this trial, published in 2018, PFS in the ICI combination arm surpassed the expectation of independence with a hazard ratio of 0.84 (P = 0.01, n = 356; median PFS surpassed expectation by 9 days; Fig. 2K and Supplementary Fig. S1). IMpower150 did not evaluate atezolizumab alone and our initial assessment therefore used data from the 2019 OAK trial on atezolizumab monotherapy in nonsquamous NSCLC without PD-L1 preselection (41). However, this trial enrolled patients receiving atezolizumab as second-line or third-line therapy. The BIRCH trial, also published in 2018, tested atezolizumab in NSCLC of all histologies (72% nonsquamous, all tumors ≥5% PD-L1 positive; ref. 26) and found that atezolizumab was more active as first-line than as second-line or third-line therapy, consistent with the general phenomenon that previously treated tumors are less responsive to therapy. This suggests that our initial test of independence underestimated atezolizumab's single-agent activity at first line. To address this, we used clinically observed differences in atezolizumab activity by line of therapy (26) to construct a synthetic arm—as recently discussed for NSCLC (42)—of atezolizumab for first-line treatment of nonsquamous NSCLC (Fig. 2K; Supplementary Fig. S2). Under these conditions, we found that IMpower150 closely matched the expectation of independence (hazard ratio 1.04, P = 0.46, n = 356; Supplementary Fig. S1). Two competing hypotheses are therefore possible to explain the results of IMpower150: (i) atezolizumab is slightly less active at second line than first line, and independent action explains the benefit of adding atezolizumab to combination chemotherapy, and (ii) atezolizumab is equally active across first and subsequent lines of therapy, and a conclusion of drug additivity or synergy is supported. The measured differences in atezolizumab activity by line of therapy in the BIRCH trial (26) provide direct evidence for hypothesis (i). We conclude that PFS in IMpower150 is most likely consistent with the expectation of independent drug action. A more exact test of independence in cases such as this is unlikely to be possible because it would require the ethically questionable step of withholding first-line chemotherapy.
Inferiority to independent action in a combination therapy for melanoma
The KEYNOTE-022 trial tested PD-1 inhibition (pembrolizumab) plus BRAF and MEK inhibition (dabrafenib plus trametinib) for BRAF-mutant melanoma, and nearly all patients receiving this “triple combination” required dose reduction, interruption, or discontinuation due to treatment-related adverse events (24). Dose reductions or interruptions can compromise the efficacy of individual agents [lowering P(A, t) and P(B, t)], which is expected to produce combination activity inferior-to-independent action as calculated assuming no dosage compromise. This may explain why PFS observed with the dabrafenib plus trametinib plus pembrolizumab combination was substantially worse than independence (hazard ratio = 1.63, P = 0.005, n = 60). Notably, patients commenced combination therapy at full dose, and initial responses in terms of volume change were approximately as good as independence would predict (Supplementary Fig. S4; method from ref. 10). Dose interruptions were increasingly required over time due to toxicity, which is consistent with inferior efficacy observed in durability of PFS. However, the benefits expected of this combination therapy by independent action could in principle be achieved by optimally assigning patients to either PD-1 inhibition only, or BRAF plus MEK inhibition only, making development of a predictive biomarker(s) for responsiveness to PD-1 inhibition a priority in BRAF-mutant melanoma.
Limitations in the analysis
This report is not a meta-analysis, and it is emphatically not intended to affect current clinical practice. Instead, the work aims to illuminate the mechanistic basis for the clinical efficacy of ICI combination therapies and inform future clinical trials. Retrospective analysis as performed here has inherent methodological limitations. First, the benefit conferred by drug combinations exhibiting independence varies with the correlation in response to individual agents; we have used a range that accounts for partial cross-resistance (10) based on data obtained on many drugs in patient-derived xenografts (PDX) models (albeit not ICIs), and clinical evidence suggesting low cross-resistance between ICIs and other therapies. Specifically, among patients who have progressed on non-ICI first-line therapy, second-line ICI therapy is slightly less effective when compared with first-line ICI (26–29), whereas strong cross-resistance would imply a large reduction in efficacy. Using a range of correlation values to account for uncertainty in the correlation parameter ρ produced a range of expected PFS, with mean width ±2% (Fig. 2, gray range).
Second, although aggregate demographics were similar across matched monotherapy and combination therapy trial arms (Supplementary Table S2), the impact of patient demographics on our analysis is unknown because demographically linked individual response data are generally not published and cannot be imputed from published data. We encourage trial sponsors to investigate demographic factors themselves, as recently described by investigators from Merck (12).
A third limitation is that some of the trials we studied did not include an ICI monotherapy arm, requiring the use of data from another trial testing the same ICI in a comparable patient cohort (Supplementary Tables S1 and S2). In these cases, we matched the line of therapy and dosing to the greatest extent possible. Crucially, we permitted no imperfections that could bias against the discovery of additivity or synergy (such as monotherapy data at higher doses or in healthier cohorts), and only tolerated biases in an opposite direction, which could produce false evidence of synergy. For the KEYNOTE-407 trial in squamous NSCLC (Fig. 2C; ref. 33), monotherapy data are not available for PD-1 antibody pembrolizumab, but data in this disease are available for PD-1 antibody nivolumab, from CheckMate-017 (43). A meta-analysis of 1,887 patients with NSCLC observed no significant difference in PFS or overall survival between pembrolizumab and nivolumab (44). Nivolumab is therefore justified as a noninferior comparator for pembrolizumab. In the renal cell carcinoma trials KEYNOTE-426 (35) and JAVELIN Renal 101 (Fig. 2E and F; ref. 34), the PDGFR/VEGFR/c-Kit receptor tyrosine kinase inhibitor axitinib was used in the combination arm and the PDGFR/VEGFR/c-Kit receptor tyrosine kinase inhibitor sunitinib was used in the monotherapy arm. The assumption that axitinib is not inferior to sunitinib is supported by real-world data (45). In five cases, ICI monotherapy data were obtained from previously treated (rather than untreated) patients (arms labeled “2L”). This biases us to overestimate the likelihood of additivity or synergy, because cancer treatments are generally less active in second line than in first line. Finally, some of the trials analyzed have fewer than 2 years of follow-up, and in these cases, it may be difficult to observe drug additivity or synergy in the subset of patients expected to exhibit long-lasting responses (11). We judge this concern to be most applicable to renal cell carcinoma and PD-L1–positive NSCLC: follow-up analysis will be required as additional trial data become available. However, 2 to 3 years of follow-up was available for most of the trials analyzed, and in these cases the longer follow-up was not able to reject the independent action model.
Careful consideration of these potential limitations supports the conclusion that drug independence is almost always the more conservative model, not only from the perspective of parsimony (observed monotherapy activity explains observed combination activity; no additional hypotheses need be invoked), but also when real limitations in available data are considered. Specifically, limitations in monotherapy data “tilt the scales” in favor of synergy, and yet it is still found lacking. If we consider only trials with ideally matched data sets (line of therapy, dosage, and compounds), the correlation between data and the model of independence remained r = 0.97 overall and P = 3×10−4 for PFS at 12 months. Thus, our findings are reproducible and significant under multiple scenarios of data inclusion and exclusion.
Discussion
Based on retrospective analysis of 13 combination immunotherapy phase III trials in eight types of cancer (all of the combination ICI trials published up to April 2020 for which an analysis of independence is possible), we conclude that there is no clinical evidence of synergistic or additive interaction among ICIs or ICIs and other drugs. Thus, there is no evidence in human trial data that priming immune responses with chemotherapies or other drugs increases the activity of ICIs, as has been observed in mouse models. This must not be confused with criticism of the clinical trial results, because the conclusion that published PFS benefits are as expected is a finding that affirms the results of these trials. This includes many combinations that have established new first-line standards of care. The key conclusion from this study is instead that independent drug action is sufficient to confer the PFS benefits required for a practice-changing therapy. The fact that this benefit can be predicted from monotherapy data is of immediate use in design of future trials, as suggested by a series of recent papers from the Merck oncology group (12, 17, 46). It remains to be seen to what extent our findings in advanced solid cancers will generalize to more varied neoplasms such as hematologic malignancies, in which drug additivity does appear to be critical (47), or to adjuvant therapies for early-stage cancers. Analysis of adjuvant therapy trials will require modification of our approach.
Single-agent ICIs have been remarkable in achieving long-lasting “treatment-free survival” for subsets of patients (31, 48). Consequently, synergistic interaction is unnecessary for ICIs to elevate long-term survival when added to established therapies, provided they have single-agent activity in the disease of interest. This explains why ICIs have partnered most effectively with different other drugs in different diseases: it presently appears that the best “partner” therapy to ICIs are whichever non-ICI regimen is itself most effective in a given indication, subject to tolerability. The corollary to this is that the success of a given ICI combination (e.g., PD-1 plus axitinib in renal cell carcinoma) cannot be expected to transfer to a different disease unless each constituent is individually active in that disease.
Widespread evidence of drug independence in human clinical trials would appear to conflict with data from mouse studies, many of which report that ICIs synergize with a wide range of agents including established and investigational cancer therapies, immune modulators, repurposed drugs, and diet (49–53). Why might this be true? The most obvious difference between preclinical analysis of ICIs and clinical trials is that the former studies are performed primarily in one of a small number of syngeneic and genetically engineered mouse models, each of which is homogeneous genetically (or nearly so). In contrast, patient populations are genetically heterogeneous and exhibit high variability in drug response. Interpatient heterogeneity strongly impacts the efficacy of combination therapy in human populations (8, 10, 13), but this phenomenon is largely inaccessible to mouse models except in the case of panels representing (at least approximately) the diversity of patient populations. This has become possible in the case of PDXs and targeted anticancer therapies (54). However, animals used for traditional PDX modeling are immunocompromised and cannot be used to evaluate ICIs. Thus, even if drug interactions identified in unusually sensitive preclinical models do occur in humans, we speculate that interpatient heterogeneity is sufficient that too few patients receive benefit from both drugs for additivity or synergy to be evident at the population level (10). A corollary is that understanding the molecular basis of patient-to-patient heterogeneity should make it possible to identify patient subgroups with higher monotherapy response rates and more opportunity for favorable drug interaction.
Calculating the benefits expected from drug independence provides a sound strategy for the design of ICI combinations; prediction is desirable as the number of possible combinations grows (12, 46). If the predicted benefits of independence are insufficient for a new combination to exceed standard of care, then based on available data, the combination is likely to fail. These observations are of immediate use in helping trialists and drug developers prioritize regimens entering trials. As monotherapy data become available for stratified subgroups (e.g., PD-L1 status, prior treatment, and patient demographics), it may also be possible to predict the activity of combination therapies in specific patient subsets. Of course, users of the theory presented here must also use appropriate criteria in selecting monotherapy data with regard to patient cohorts, line of therapy, etc. Adverse effects remain the primary unknown, and better methods for predicting, preventing, or mitigating toxicity are therefore a priority. If a combination is poorly tolerated, as may be apparent in a phase I trial (55), then it is ground to anticipate “inferior-to-independent” efficacy in subsequent trials. This likely explains the negative result for triple BRAF–MEK–PD1 inhibitor combination in the KEYNOTE-022 trial (24). Future research could test whether the independent action model can predict adverse responses from combinations following collection or publication of demographically linked data on adverse events to monotherapies and their correlation.
The analysis presented addresses two clinical questions left unresolved by trial reports themselves (5–7). First, for diseases where an ICI combination therapy is approved, is combination therapy optimal for all patients? Our findings imply that the benefits of ICIs in approved combination therapies apply to a subset of patients no larger than those responsive to single-agent ICIs. Use of ICI combination therapy is therefore justified as a “bet hedging” strategy because sufficiently accurate biomarkers of ICI response are lacking. In the future, development of more accurate biomarkers would support two types of clinical trials. For indications in which unstratified populations do not exhibit a significant benefit from an ICI combination (e.g., pembrolizumab plus chemotherapy in gastric cancer; ref. 22), ICI combinations might still be tested in subgroups with the highest responsiveness to single-agent ICIs, as identified by appropriate biomarkers. This is a circumstance in which benefit from independent action is simply more likely. For indications in which ICI combinations are approved, noninferiority trials could test whether ICI (and accompanying toxicities) can be spared for subgroups identified as resistant to that ICI.
Second, can sequential use of drugs in approved combinations provide treatment benefits similar to those of using the drugs simultaneously? For drugs acting by independent action, the answer is likely to be yes, but only if patients are consistently able to switch to a second line of therapy. In cancers such as NSCLC, an overriding consideration is that patients with rapidly progressing disease may be unable to receive a second-line therapy, which is a compelling reason for use of first-line combination therapy irrespective of mechanism of action (synergy or independence; ref. 7). In clinical settings in which patients are routinely able to receive a second line of therapy, independent drug action supports testing “sequential combinations” involving switching from a first agent to the second upon progression. This hypothesis was tested directly by ECOG1193 (56), which established sequential chemotherapy for metastatic breast cancer as noninferior to upfront combination chemotherapy, while imposing less toxicity on patients and so improving quality of life.
Is it possible to move beyond independent drug action and improve patient outcomes using drugs that enhance ICI activity? As mentioned above, we hypothesize that the key to achieving drug additivity or synergy is increasing rates of response to the constituent monotherapies via patient stratification. If we can identify patients who respond to all or the majority of drugs in a combination (57), we are likely to enrich for favorable interaction, which may not occur to a statistically detectable degree in unstratified populations. Thus, our findings emphasize the importance of achieving greater precision in cancer immunotherapy based on discovery and validation of response biomarkers.
Authors' Disclosures
A.C. Palmer reports personal fees from Merck & Co outside the submitted work. P.K. Sorger reports grants from NCI and Burroughs Wellcome Fund during the conduct of the study, as well as personal fees from Glencoe Software, Applied Biomath, RareCyte Inc., NanoString, Merck, and Montai Health outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
We thank Chris Chidley, Deborah Plana, Geoffrey Fell, Lorenzo Trippa, and Brian Alexander for help with this project and all the investigators and patients who participated in the clinical trials analyzed in this work. This work was funded by NCI grants U54-CA225088 to PKS and K08-CA222663 and R37-CA258829 to B. Izar. B. Izar is supported by the Burroughs Wellcome Fund Career Award for Medical Scientists.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked "advertisement" in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
A.C. Palmer: Formal analysis. B. Izar: Formal analysis. H. Hwangbo: Formal analysis. P.K. Sorger: Formal analysis.
References
- 1. Darvin P, Toor SM, Nair VS, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med 2018;50:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN Guidelines with the level of evidence. Cancers 2020;12:738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol 2019;30:219–35. [DOI] [PubMed] [Google Scholar]
- 4. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018;8:1069–86. [DOI] [PubMed] [Google Scholar]
- 5. Escudier B. Combination therapy as first-line treatment in metastatic renal-cell carcinoma. N Engl J Med 2019;380:1176–8. [DOI] [PubMed] [Google Scholar]
- 6. Procopio G, Sepe P, Claps M, de Braud F, Verzoni E. Should we use combination therapy for all advanced renal cell carcinoma? Lancet Oncol 2019;20:1331–2. [DOI] [PubMed] [Google Scholar]
- 7. Goldberg SB, Herbst RS. Should chemotherapy plus immune checkpoint inhibition be the standard front-line therapy for metastatic NSCLC? Cancer 2018;124:4592–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frei E, Freireich EJ, Cehan E, Pinkel D, Holland JF, Selawry O, et al. Studies of sequential and combination antimetabolite therapy in acute leukemia: 6-mercaptopurine and methotrexate. Blood 1961;18:431–54. [Google Scholar]
- 9. Gaddum JH. Pharmacology. 1st ed.Oxford: Oxford University Press; 1940. [Google Scholar]
- 10. Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 2017;171:1678–91.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doroshow JH, Simon RM. On the design of combination cancer therapy. Cell 2017;171:1476–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen C, Liu F, Ren Y, Suttner L, Sun Z, Shentu Y, et al. Independent drug action and its statistical implications for development of combination therapies. Contemp Clin Trials 2020;98:106126. [DOI] [PubMed] [Google Scholar]
- 13. Frei E, Karon M, Levin RH, Freireich EJ, Taylor RJ, Hananian J, et al. The effectiveness of combinations of antileukemic agents in inducing and maintaining remission in children with acute leukemia. Blood 1965;26:642–56. [PubMed] [Google Scholar]
- 14. Berenbaum MC. What is synergy? Pharmacol Rev 1989;41:93–141. [PubMed] [Google Scholar]
- 15. Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol 2012;30:2039–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giantonio BJ, Catalano PJ, Meropol NJ, O'Dwyer PJ, Mitchell EP, Alberts SR, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol 2007;25:1539–44. [DOI] [PubMed] [Google Scholar]
- 17. Schmidt EV, Chisamore MJ, Chaney MF, Maradeo ME, Anderson J, Baltus GA, et al. Assessment of clinical activity of PD-1 checkpoint inhibitor combination therapies reported in clinical trials. JAMA Netw Open 2020;3:e1920833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med 2018;379:2220–9. [DOI] [PubMed] [Google Scholar]
- 19. Sacks H, Chalmers TC, Smith H. Randomized versus historical controls for clinical trials. Am J Med 1982;72:233–40. [DOI] [PubMed] [Google Scholar]
- 20. Ghadessi M, Tang R, Zhou J, Liu R, Wang C, Toyoizumi K, et al. A roadmap to using historical controls in clinical trials – by Drug Information Association Adaptive Design Scientific Working Group (DIA-ADSWG). Orphanet J Rare Dis 2020;15:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet 2019;394:1929–39. [DOI] [PubMed] [Google Scholar]
- 22. Tabernero J, Van Cutsem E, Bang Y-J, Fuchs C, Wyrwicz L, Lee KW, et al. Pembrolizumab with or without chemotherapy versus chemotherapy for advanced gastric or gastroesophageal junction (G/GEJ) adenocarcinoma: The phase III KEYNOTE-062 study. J Clin Oncol 2019;37:LBA4007. [Google Scholar]
- 23. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26. [DOI] [PubMed] [Google Scholar]
- 24. Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med 2019;25:941–6. [DOI] [PubMed] [Google Scholar]
- 25. Rahman R, Ventz S, Fell G, Vanderbeek AM, Trippa L, Alexander BM. Divining responder populations from survival data. Ann Oncol 2019;30:1005–13. [DOI] [PubMed] [Google Scholar]
- 26. Peters S, Gettinger S, Johnson ML, Jänne PA, Garassino MC, Christoph D, et al. Phase II trial of atezolizumab as first-line or subsequent therapy for patients with programmed death-ligand 1-selected advanced non-small-cell lung cancer (BIRCH). J Clin Oncol 2017;35:2781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84. [DOI] [PubMed] [Google Scholar]
- 28. Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387:1540–50. [DOI] [PubMed] [Google Scholar]
- 29. Hui R, Garon EB, Goldman JW, Leighl NB, Hellmann MD, Patnaik A, et al. Pembrolizumab as first-line therapy for patients with PD-L1-positive advanced non-small cell lung cancer: a phase 1 trial. Ann Oncol 2017;28:874–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berge EM, Lu X, Maxson D, Baron AE, Gadgeel SM, Solomon BJ, et al. Clinical benefit from pemetrexed before and after crizotinib exposure and from crizotinib before and after pemetrexed exposure in patients with anaplastic lymphoma kinase-positive non-small-cell lung cancer. Clin Lung Cancer 2013;14:636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med 2018;378:2078–92. [DOI] [PubMed] [Google Scholar]
- 33. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med 2018;379:2040–51. [DOI] [PubMed] [Google Scholar]
- 34. Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019;380:1103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019;380:1116–27. [DOI] [PubMed] [Google Scholar]
- 36. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 2018;379:2108–21. [DOI] [PubMed] [Google Scholar]
- 37. Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet 2019;394:1915–28. [DOI] [PubMed] [Google Scholar]
- 38. West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2019;20:924–37. [DOI] [PubMed] [Google Scholar]
- 39. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med 2018;378:2288–301. [DOI] [PubMed] [Google Scholar]
- 40. Ferrucci PF, Di Giacomo AM, Del Vecchio M, Atkinson V, Schmidt H, Schachter J, et al. KEYNOTE-022 part 3: a randomized, double-blind, phase 2 study of pembrolizumab, dabrafenib, and trametinib in BRAF-mutant melanoma. J Immunother Cancer 2020;8:e001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 2017;389:255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dron L, Golchi S, Hsu G, Thorlund K. Minimizing control group allocation in randomized trials using dynamic borrowing of external control data—an application to second line therapy for non-small cell lung cancer. Contemp Clin Trials Commun 2019;16:100446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 2015;373:123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peng T-R, Tsai F-P, Wu T-W. Indirect comparison between pembrolizumab and nivolumab for the treatment of non-small cell lung cancer: a meta-analysis of randomized clinical trials. Int Immunopharmacol 2017;49:85–94. [DOI] [PubMed] [Google Scholar]
- 45. Konishi S, Hatakeyama S, Tanaka T, Ikehata Y, Tanaka T, Fujita N, et al. Comparison of axitinib and sunitinib as first-line therapies for metastatic renal cell carcinoma: a real-world multicenter analysis. Med Oncol 2018;36:6. [DOI] [PubMed] [Google Scholar]
- 46. Sun LZ, Wu C, Li X, Chen C, Schmidt EV. Independent action models and prediction of combination treatment effects for response rate, duration of response and tumor size change in oncology drug development. Contemporary Clinical Trials 2021;106:106434. [DOI] [PubMed] [Google Scholar]
- 47. Palmer AC, Chidley C, Sorger PK. A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. eLife 2019;8:e50036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Melero I, Berman DM, Aznar MA, Korman AJ, Pérez Gracia JL, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer 2015;15:457–72. [DOI] [PubMed] [Google Scholar]
- 50. Rios-Doria J, Durham N, Wetzel L, Rothstein R, Chesebrough J, Holoweckyj N, et al. Doxil synergizes with cancer immunotherapies to enhance antitumor responses in syngeneic mouse models. Neoplasia 2015;17:661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luchtel RA, Bhagat T, Pradhan K, Jacobs WR, Levine M, Verma A, et al. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc Natl Acad Sci U S A 2020;117:1666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Luo F, Luo M, Rong Q-X, Zhang H, Chen Z, Wang F, et al. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J Immunother Cancer 2019;7:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ferrere G, Alou MT, Liu P, Goubet A-G, Fidelle M, Kepp O, et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight 2021;6:145207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 2015;21:1318–25. [DOI] [PubMed] [Google Scholar]
- 55. Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH, Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 2019;25:936–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sledge GW, Neuberg D, Bernardo P, Ingle JN, Martino S, Rowinsky EK, et al. Phase III trial of doxorubicin, paclitaxel, and the combination of doxorubicin and paclitaxel as front-line chemotherapy for metastatic breast cancer: an intergroup trial (E1193). J Clin Oncol 2003;21:588–92. [DOI] [PubMed] [Google Scholar]
- 57. Tunger A, Sommer U, Wehner R, Kubasch AS, Grimm M-O, Bachmann MP, et al. The evolving landscape of biomarkers for anti-PD-1 or anti-PD-L1 therapy. J Clin Med 2019;8:1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Complete source data (digitized PFS distributions) are provided in the Supplementary Code.