

Abstract
Cerebral ischemia reperfusion injury (IRI) induced by hemorrhagic shock and reperfusion (HSR) is the main cause of death following trauma. Previous studies indicated the neuroprotective effect of sevoflurane postconditioning (SP) in cerebral IRI. However, the mechanisms still remain elusive. Cerebral IRI models with SP were established by using HSR with C57BL/6 mice (male, 3-month-old) in vivo and by using oxygen glucose deprivation and reoxygenation (OGD/R) with HT22 cells in vitro. Postoperative cognition was evaluated by the Morris water maze, novel object recognition, and elevated plus maze tests. The role of SIRT1 was determined by using siRNA, a sensitive inhibitor (EX527), or an overexpression shRNA-GFP lentivirus. IRI caused significant disabilities of spatial learning and memory associated with enhanced cerebral infarct and neuronal apoptosis, which were effectively attenuated by SP. IRI also made a significant decrease of SIRT1 accompanied by oxidative stress, mitochondria dysfunction, and inactivated autophagy. SP or genetically overexpressing SIRT1 significantly suppressed defective autophagy, mitochondrial oxidative injury, and neuronal death caused by HSR or OGD/R. However, genetic suppression or pharmacological inhibition of SIRT1 significantly reversed the impact of SP treatment on mitochondrial DNA transcription ability and autophagy. Our results demonstrate that the loss of SIRT1 causes a sequential chain of mitochondrial dysfunction, defective autophagy, and neuronal apoptosis after IRI in the preclinical stroke models. Sevoflurane postconditioning treatment could effectively attenuate pathophysiological signatures induced by noxious stimuli, which maybe mediated by SIRT1.
1. Introduction
Approximately 1.9 million people die of hemorrhagic shock every year in the world [1]. Hemorrhagic shock reperfusion (HSR) injury occurs when restoring circulating blood volume with diluted hemoglobin concentration, which further reduced oxygen supply and aggravated tissue damage [2]. As a scenario of ischemia-reperfusion injury (IRI), HSR increases the long-term mortality, leaves serious sequelae [3, 4], such as cognitive dysfunction [5], and causes serious burden to patients' families and the society. Although early mortality is low after severe trauma, chronic critical illness is a common trajectory in survivors and is associated with poor long-term outcomes. Advancing age, shock severity, and persistent organ dysfunction are predictive variables of chronic critical illness [4]. Therefore, early identification may facilitate targeted interventions to change the trajectory of this morbid phenotype.
Silent information regulator-1 (SIRT1) is a nicotinamide adenine dinucleotide- (NAD+) dependent histone deacetylase, which can catalyze the deacetylation of acetyl lysine of histone and nonhistone substrates (such as p53 and FOXO). SIRT1 participates in the regulation of glucose and lipid metabolism, inflammation, cell aging and apoptosis, oxidative stress, and tumor formation. Numbers of studies reported that SIRT1 was downregulated in the IRI models, such as middle cerebral artery occlusion (MCAO) and reperfusion, oxygen glucose deprivation and reoxygenation (OGD/R). And it was also associated with multiple biocellular processes, including the activation of apoptosis and neuroinflammation [6], the suppression of angiogenesis [7], and mitochondrial oxidative stress and dysfunction [8]. In SIRT1−/− animals, the area of cerebral infarction was significantly increased following focal cerebral ischemia [9]. SIRT1 gene overexpression significantly alleviated hippocampal damage, including less cerebral infarction, and reduced neuronal degeneration and cognition improvement [10]. It has been regarded that SIRT1 elicits neuroprotective effect in decreasing apoptosis and neuronal damage through regulating autophagy [11]. Autophagy is a cellular homeostatic program for the turnover of cellular organelles and proteins. Emerging evidence indicates autophagy as a critical modulator for cerebral IRI [12]. However, the association between SIRT1 and autophagy in IRI is still not fully understood.
In addition to anesthesia, sevoflurane can also alleviate cerebral IRI. In the early phase after transient global cerebral IRI, the activation of antioxidant enzymes (Nrf2/HO-1) may contribute to the neuroprotection of SP [13]. Furthermore, SP alleviates neuronal death and axon demyelination in neonatal rats by regulating microglia autophagy. Therefore, SP provides long-term cognitive, learning, and memory protection [14]. In our previous study, SP improved the impairments of spatial learning and memory induced by HSR in rats [15]. SP remarkably reduced the apoptosis of hippocampal neurons through reducing the endoplasmic reticulum stress. Moreover, SP stabilized the integrity of mitochondrial structure and function via preventing the opening of the mitochondrial mPTP channel [16]. However, the role of SIRT1 in the neuroprotective impact of SP has not been clearly illustrated.
In the present study, we aimed to investigate the mechanism by which SP potentially improved the cognitive deficits following HSR in mice. The cognition and anxiety-like behaviors were evaluated following SP without or with the SIRT1 inhibitor in HSR mice. The characteristics of mitochondrial morphology and function were indicated in OGD/R with either SP or SIRT1 siRNA transfection in HT22 cells. The association between SIRT1 and autophagy in the IRI was demonstrated in both HSR and OGD/R without or with SP.
2. Materials and Methods
2.1. Animals
Male C57BL/6 mice weighing 25-30 g were provided by Laboratory Animal Center in Anhui Medical University. All experimental procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Ethics Committee for the use of experimental animals in Anhui Medical University (reference number LLSC20190765, date of approval 26/10/2019). Animals were randomly divided into specific groups depending on the different purposes. Mice were bred and housed in a temperature- and humidity-controlled room with a 12/12 h light/dark cycle. All animals had access to food and water ad libitum. One-week acclimatization period was applied before experiment employment. All behavioral tests have been performed during the light phase.
2.2. Hemorrhagic Shock and Resuscitation Mouse Model
Mice were fasted for 12 h before surgery and free to drink water. Hemorrhagic shock and resuscitation (HSR) were performed as described previously [17]. Polyethylene catheters were inserted into the right carotid artery and left jugular vein for blood extraction and infusion, respectively. Approximately 50% of the total blood volume (total blood volume [18] = weight (g) × 7%) were continuously extracted within 30 min through a two-way automatic infusion pump (Genie Touch, Kent Scientific Corporation, USA). After 1 hour, the collected blood was infused within 30 min at a constant speed. Rectal temperature was maintained at 37.0 ± 0.5°C by using a heating blanket. Finally, all catheters were removed, and the mouse was returned to its cage for recovery. The sham operation was only catheter intubation in the right carotid artery and left jugular vein without withdrawing or transfusing blood.
2.3. Intracerebroventricular Lentivirus Injection
To determine if the supplement of SIRT1 could reverse the impact of HSR, SIRT1 overexpression shRNA-GFP lentivirus (titer: 2.0 × 109 TU/ml) and control lentiviral vector (LV-CTR) (titer: 1.5 × 109 TU/ml) were purchased from GeneChem Company (Shanghai, China). After anesthesia, total 3 μl control lentiviral vector or SIRT1 overexpression shRNA-GFP lentivirus was injected in the right lateral ventricle at the rate of 0.5 μl/min. The following coordinates (relative to bregma) were used, -0.2 mm AP, -1 mm ML, and -2.5 mm DV according to the stereotaxic atlas of Paxinos and Franklin (Third edition, 2007) by using a stereotaxic apparatus (RWD Life Science Co., Ltd., China). To prevent reflux or backflow, the needle was slowly removed at 10 min after the injection. Seven days after lentivirus injection, mice were subjected to HSR.
To downregulate SIRT1, the inhibitor EX527 (i.p., 10 mg/kg) was administrated at 30 min before catheter intubation [19].
2.4. Behavioral Tests
2.4.1. Morris Water Maze Test
According to the previous report [20], the apparatus (Zhenghua Biologic, China) was consisted of a circular swimming pool (120 cm diameter and 50 cm height), a depth of 30 cm covering black platform (10 cm diameter), and a camera with a video recording system on the ceiling to capture the movement of animals in the tank. The pool was divided into four quadrants with specific conspicuous visual cues. The platform was fixed in the center of the southwest and submerged approximately 0.7 cm below the water surface.
The spatial acquisition training was started on the 3rd day after HSR and carried out for 6 successive days. For each trial, mice randomly entered into each quadrant facing the wall. If the mouse failed to find the platform within 60 sec, it was guided to the platform and allowed to stay for 10 sec. The escape latency was recorded. To evaluate memory capability, the probe test was given at 24 hours of the last training trial. After removing the platform, mice were placed in the opposite quadrant searching for 60 sec [21]. The swimming distance (cm) and the proportion of time spent in the target quadrant (%) were documented through the computer image analyzer.
2.4.2. Open Field Test
In an empty and light grey square arena (40 cm × 40 cm × 40 cm) (RWD Life Science, China), mice were placed in the center, and its behavior was recorded from 3 to 15 min [22]. The distance and the time spent in the center area were analyzed by PanLab Smart 3 software (Harvard, USA).
2.4.3. Novel Object Recognition Test
The novel object recognition test consists of three phases with 24-hour interval: habituation, familiarization, and test phase. In our experience, the habituation phase is equal to the open field test. During the familiarization phase, a single mouse was placed in the open field arena containing two identical objects (A+A) for 10 min. During the test phase, the mouse was returned to the arena with two different objects: one is identical to the old and the other one is novel (A+B) [23]. The total exploration time and the exploration time of each object were recorded by PanLab Smart 3 software.
2.4.4. Elevated plus Maze Test
To evaluate the changes of anxiety-related behaviors in mice, elevated plus maze (EPM) test was conducted on the 14th day after HSR. The apparatus consisted of plastic open and closed arms (5 cm width and 35 cm length). The closed arm was about 15 cm height, and the height of the maze from the ground was about 40-55 cm. The animals moved freely in the elevated cross maze (RWD Life Science, China) for 10 min. The moving distance and residence time in the arms were recorded and analyzed by PanLab Smart 3 software.
2.5. Infarct Volume Assessment
After EPM test, the animals were sacrificed, and the entire brain was fast frozen at -20°C for 20 min. Then, 1 mm thick coronal sections were continuously segmented and incubated in 2% 2,3,5-tribenzene tetrachloride (TTC) for 30 min at 37°C. Brain slices were further fixed in 4% polyformaldehyde for 24 hours and then digitally scanned to a computer for analysis using ImageJ software (National Institutes of Health, USA): Infarction volume% = (infarction area × 1 mm)/(whole brain area × 1 mm) × 100%.
2.6. Oxygen and Glucose Deprivation/Reoxygenation Protocol
The mouse hippocampal neuronal cell line (HT22) (Jennio Biotech Co., Ltd., Guangzhou, China) was cultured in Dulbecco's modified Eagle's medium (DMEM) (HyClone, USA) with 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere containing 5% CO2. When reaching 70-80% confluency, cells were subjected to OGD/R. Briefly, oxygen and glucose deprivation (OGD) was carried out in an anaerobic chamber (Stem Cell, USA) containing 95% N2 and 5% CO2. Cells were incubated with glucose-free DMEM and FBS for 4 hours at 37°C. After replacing the medium to the ordinary DMEM containing 10% FBS, cells were further cultured in the CO2 incubator for 4 hours.
To genetically inhibit the expression of SIRT1, siRNA transfection was introduced 24 hours before OGD/R without or with sevoflurane postconditioning. Nonsense sequence for the SIRT1 negative control (SIRT1-NC) and siRNA against SIRT1 (si-SIRT1) were purchased from GenePharma (Shanghai, China). According to the manufacturers' instructions, HT22 cells were seeded into 6-well plates and cultured to 50-60% confluence. Then transfection was performed with 20 μM siRNA using 4 μl of jetPRIME Transfection Reagent (Polyplus-Transfection®) in 200 μl jetPRIME buffer. After 10 min, 2 ml fresh medium was added for further incubation. No-load siRNA plasmid was used as a control. The efficiency of SIRT1 intervention was confirmed by RT-PCR.
2.7. Sevoflurane Postconditioning In Vivo and In Vitro
At the onset of blood refusion, mouse was stabilized in a gas-tight anesthesia chamber with sevoflurane inhalation for 1 hour. Sevoflurane (Sevofrane®, AbbVie, Japan) was delivered by the carrier gases (5% CO2 and 95% O2, 3.5 l/min total gas flow) through the sevoflurane volatilizer (Vapor 2000, Germany). The concentration was maintained at 2.4% and measured by an anesthetic monitor (Mindray Bene View T8, Shenzhen, China). In the sham operation or HSR, mice were only exposed to the mixed gas (95% O2 and 5% CO2) for 1 hour.
For HT22 cells, at the beginning of reoxygenation with ordinary DMEM containing 10% FBS, cells were incubated in the gas-tight anesthesia chamber (Modular Incubator Chamber, Billups Rothenberg, USA) with 5% CO2 and 95% air. The percentage of sevoflurane in the chamber was maintained at 2 for 1 hour.
2.8. Evaluation of Cell Viability and Apoptosis
MTT (Beyotime Biotechnology, China) and CCK-8 (7-Seabiotech, China) assays were used to determine the viability of HT22 cells according to the manufacturer's instructions. Optical density was calculated with the absorbance measuring by Varioskan LUX microplate reader (Thermo Fisher Scientific, USA). The results were expressed as the ratio to the control values.
To detect apoptosis, HT22 cells were immunolabeled with cleaved caspase 3 (Cell Signaling Technology, USA) and identified with TdT-mediated dUTP nick-end labeling (TUNEL) (In Situ Cell Death Detection Kit TMR red, Roche, USA) according to the manufacturer's instructions. After fixation with 4% paraformaldehyde, HT22 cells were permeabilized with 0.1% Triton X-100 and then blocked with 10% normal goat serum for 2 hours at room temperature. HT22 cells were further incubated with primary antibody against cleaved caspase 3 overnight at 4°C and subsequent Alexa Fluor 488 secondary antibody (Invitrogen, USA) for 2 hours at room temperature. Finally, HT22 cells were exposed to TUNEL reagent for 1 hour at 37°C. The nucleus was recognized by DAPI (Sigma-Aldrich, USA), and the apoptotic cells were examined under a laser scanning confocal fluorescent microscope (Carl Zeiss LSM 880, Germany).
2.9. Assessment of Oxidative Stress and Mitochondrial Transmembrane Potential
Oxidative stress in the HT22 cells was measured using MDA and SOD assays (Nanjing Jiancheng, China) according to the manufacturer's recommendations. ATP content was also evaluated (Nanjing Jiancheng, China) according to the manufacturer's recommendations. MitoSOX Red (Invitrogen, USA) was used to measure the accumulation of mitochondrial ROS. JC-1 Mitochondrial Membrane Potential Dye (Invitrogen, USA) was used to evaluate mitochondrial transmembrane potential in HT22 cells. Briefly, HT22 cells were treated with MitoSOX Red or JC-1 solution at 37°C for 10-20 min and then detected by the fluorescent microscope (Axio Vert. A1, ZEISS, Germany). According the manufacturer's instructions, the average fluorescent intensities at 488 nm and 590 nm were determined by ImageJ software (National Institutes of Health, USA) and then analyzed as the red/green ratio.
2.10. Quantitative Real-Time Polymerase Chain Reaction
According to the manufacturer's protocol, total RNA from HT22 cells was extracted using TRIzol reagent (TAKARA, Japan). After reverse transcription with HiScript® III RT SuperMix (Vazyme, China), PCR was performed with StepOnePlus™ Real-Time PCR system (Applied Biosystems, USA) with the AceQ®qPCR SYBR Green Master Mix Kit (Vazyme, China). NADH dehydrogenase subunits 1 (ND1) and cytochrome c oxidase subunit 3 (COX3) were used to reflect the transcript level of mtDNA. Primers of SIRT1, ND1, and COX3 were purchased from Genepharma (Shanghai, China), and sequences were as follows: SIRT1 (F: 5′-GCAGGTTGCAGGAATCCAAA-3′, R: 5′-GGCAAGATGCTGTTGCAAAG-3′), ND1 (F: 5′-GGATCCGAGCATCTTATCCA-3′, R: 5′-GGTGGTACTCCCTCTGTAAA-3′), and COX3 (F: 5′- CGTGAAGGAAACTACCCAGG-3′, R: 5′-CGCTCAGAAGAATCCTGCAA-3′). The relative levels of SIRT1, ND1, and COX3 were normalized to internal control GAPDH with 2-ΔΔCt method. The relative mRNA level of SIRT1 was shown in the SFig. 3.
2.11. SDS-PAGE and Western Blot Analysis
Total proteins were extracted and subjected to 10% or 15% polyacrylamide gels electrophoresis. Nonspecific binding sites were blocked with 5% nonfat dry milk for 1 hour at room temperature. Then, membranes were incubated with specific primary antibodies overnight at 4°C, including SIRT1 (Cell Signaling Technology, USA), Bax (CST, USA), Bcl-2 (CST, USA), Pink1 (CST, USA), Parkin (CST, USA), LC3B (CST, USA), mtTFA (NOVUS, USA), and GAPDH (Sigma-Aldrich, USA). After the incubation with horseradish peroxidase-conjugated secondary antibodies (ZSGB-BIO, Beijing, China) for 2 hours at room temperature, the immunoreactive band signal intensity was subsequently visualized by chemiluminescence (SuperSignal™ West Femto Maximum Sensitivity Substrate) (Thermo Fisher Scientific, USA). All immunoblots were normalized for gel loading with GAPDH (Sigma-Aldrich, USA) antibody. The intensities of chemiluminescent bands were measured by ImageJ software (National Institutes of Health, USA).
2.12. Deacetylase Assay
Nuclear proteins from the cells were firstly extracted using a nuclear and cytoplasmic protein extraction kit (Beyotime, Shanghai, China). Then, the NAD+-dependent deacetylase activity was evaluated by using a SIRT1/Sir2 deacetylase fluorometric assay kit Ver.2 (CycLex, Medical & Biological Laboratories Co., Ltd., Japan). According to the manufacturers' instructions and the preliminary experiments, the SIRT1 activity was expressed as the rate of reaction for the first 30 min, when there was a good linear correlation between the fluorescence and the reaction time.
2.13. Transmission Electron Microscopy
After specific treatments, HT22 cells were collected by centrifugation and then fixed overnight with 2.5% glutaraldehyde at 4°C. After rinse in PBS (0.01 M, PH 7.2) for 6 hours at 4°C, samples were further fixed in 1% osmium tetroxide for 1 hour at 4°C. Following dehydration with gradient alcohol, samples were incubated in propylene oxide and epoxy resin for 2 hours at room temperature and then embedded in epoxy resin for further ultramicrosectioning (70 nm in thickness, Leica UC-7 microtome). The area of mitochondria (10000x) was analyzed by using ImageJ software. The number of mitochondria autophagy bodies (10000×) was observed in at least 10 visual fields (including intact neuronal bodies) by a JEM1400 electron microscope (JEOL, Japan).
2.14. Immunofluorescence Staining
After the behavioral observation, the mouse was deeply anesthetized, and the brain was removed and postfixed in 4% paraformaldehyde for 72 hours. Following the dehydration by using graded series of saccharose and embedding with OCT, coronal 16 μm thick sections (from -1.46 mm to -2.46 mm posterior to bregma) were made. Nonspecific binding was blocked by 10% normal goat serum. Sections were incubated with primary antibodies against SIRT1 (CST, USA), LC3B (CST, USA), TOM20 (CST, USA), 8-OHdG (Santa Cruz Biotechnology, USA), and VDAC (CST, USA) overnight at 4°C. Alexa Fluor 568 or 488 secondary antibodies (Invitrogen, USA) were further incubated for 2 hours at room temperature. The nucleus was identified with DAPI. Z-stack images were acquired by using a laser scanning confocal fluorescent microscope (20x, 40x oil, and 63x oil immersion objectives) (Carl Zeiss LSM 880, Germany) equipped with ZEN light software at 1,024 × 1,024 resolution. All quantitative analyses were performed from at least three independent experiments. Data were analyzed with ImageJ software.
2.15. Statistical Analysis
The data of the Morris water maze were analyzed by two-way analysis of variance (ANOVA) followed by the Tukey post hoc test for repeated measures, using the statistic software GraphPad Prism 8.0 (Graph Pad Software Inc., USA). Other data, such as elevated plus maze test, normalized band intensities in western blot, and quantification of immunoreactivity, were analyzed by one-way ANOVA followed by the Tukey post hoc test. Normality of the data and homogeneity of group variances were assessed using the D'Agostino-Pearson omnibus normality test, Shapiro-Wilk normality test, and Kolmogorov-Smirnov test, respectively. Statistical significance was determined if p < 0.05. Data are represented as the mean ± SD in the context.
3. Results
3.1. Sevoflurane Postconditioning Improved Cognitive Deficits and Anxiety Induced by Hemorrhagic Shock and Resuscitation
We firstly established and tested the animal model of ischemia-reperfusion injury (IRI) in mice. On postoperative day 14, hemorrhagic shock and resuscitation (HSR) caused severe cerebral infarct. The introduction of sevoflurane postconditioning (SP) at the beginning of reperfusion significantly reduced cerebral infarct size. However, when pretreated with EX527 before SP, the infarct size was significantly stronger than SPm group (F = 33.82, Figures 1(a) and 1(b)).
Figure 1.
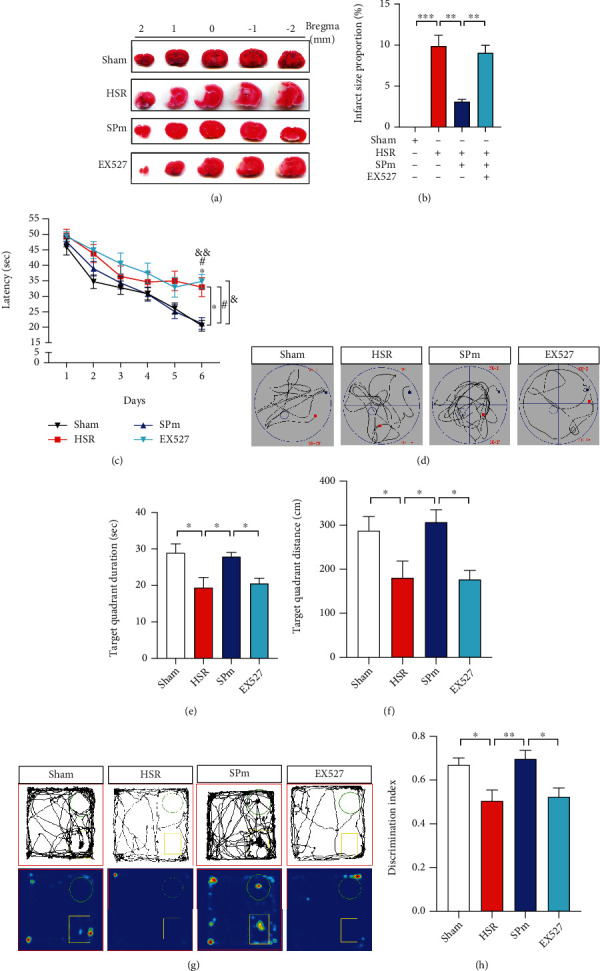
Sevoflurane postconditioning improved cognitive impairments induced by hemorrhagic shock and resuscitation (HSR). (a, b) Representative cerebral TTC staining photographs and the calculation of infarct size were made after HSR without or with sevoflurane postconditioning (SPm). EX527 (SIRT1 inhibitor) (i.p., 10 mg/kg) was administrated at 30 min before catheter intubation; then, mice were exposed to HSR with SPm. Infarct size of coronal brain sections was determined by TTC staining on postoperative day 14. HSR caused a significant cerebral infarction (white area). (c) A learning curve was made in the training session for spatial acquisition in the Morris water maze (MWM) test from postoperative day 3 to 8. (d–f) In the MWM test, the performance in the probe test included swimming route, duration, and distance in the target quadrant in the probe test. (g, h) Representative exploration route for two different objects, heat map, and discrimination index were recorded in the novel object recognition (NOR) test. The discrimination index was calculated by the ratio of time exploring the new object versus two different objects. All values are represented as mean ± SEM. n = 12 per group; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, the HSR group vs. sham group; #p < 0.05, the SPm group vs. HSR group; and &p < 0.05, the EX527 group vs. SPm group.
To evaluate the impact of HSR on the spatial learning and memory, the Morris water maze (MWM) test was performed from postoperative day 3 to 9. Especially on 6th training day, mice in the HSR group acquired longer escape latency than mice in the sham group. After SPm treatment, the escape latency was remarkably shortened. Nevertheless, mice with EX527 pretreatment needed longer time to find the platform (ptime < 0.0001, pgroup = 0.0104, and ptime∗group = 0.0189; Figure 1(c)). In the probe test for spatial memory determination, comparing with the sham group, mice after HSR swam less time (19.4 ± 9.6 sec vs. 29.0 ± 8.4 sec) and shorter distance (180.8 ± 131.7 cm vs. 287.4 ± 111.0 cm) in the target quadrant. SP significantly improved animal's spatial memory, which was indicated by swimming longer and farther. However, this improvement was effectively reversed by a single preadministration of EX527 (Figures 1(d)–1(f)).
In the novel object recognition (NOR) test, the discrimination index (DI) of mice after HSR (0.51 ± 0.17) suggested that mice spent equal time on exploring two different objects. Higher DI following SPm implied that it strengthened animals' discrimination ability to two different objects (0.70 ± 0.14). However, the addition of EX527 to SPm made a significant DI decline (F = 5.825, Figures 1(g) and 1(h)).
To investigate the impact of SP on anxiety-like behaviors, open field test (OFT) and elevated plus maze (EPM) test were initiated in HSR mice without or with SP. In the OFT, mice following HSR were less active, evidenced by shorter moving distance and less residence time in the center area. In the SPm group, there were more spontaneous activities in the center area compared with the HSR group. When mice received EX527, they performed significantly inactive in the center area (Figures 2(a)–2(c)). In the EPM test, mice in the HSR group performed anxiety-like behaviors, such as fewer entries and less residence time in the open arms. The addition of SPm significantly relieved anxiety, which indicated by higher entry frequency (Figures 2(d) and 2(e)) and longer duration (Figure 2(f)) in the open arms. However, significant less open arm entries and duration were observed in the mice with EX527 pretreatment (F = 4.008, F = 4.624).
Figure 2.
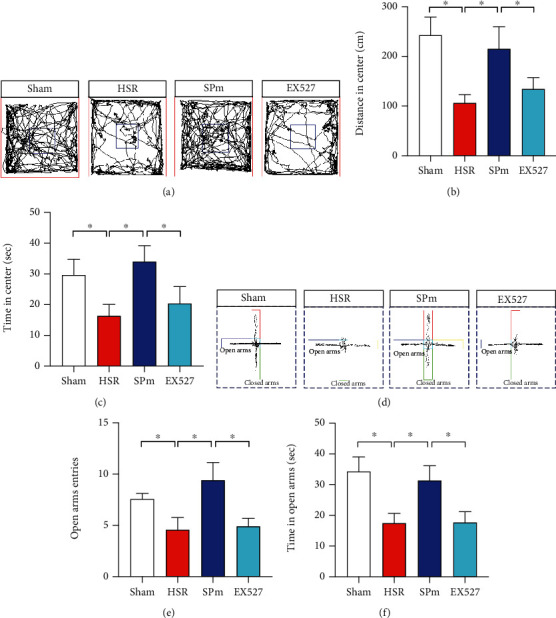
Sevoflurane postconditioning improved anxiety-like behaviors caused by HSR. (a–c) In the open field test (OFT), representative moving tracks in the arena were traced and descripted during their spontaneous activities. Particularly, moving distance and duration in the center area were further calculated as the signatures of anxiety. (d–f) In the elevated plus maze (EPM) test, representative moving tracks in both open and closed arms were traced and descripted within 10 min. To determine the anxious reactions, the entry frequency and the duration in the open arms were calculated. All values are represented as mean ± SEM. n = 12 per group, ∗p < 0.05.
3.2. Sevoflurane Postconditioning Reversed SIRT1 Loss and Cell Apoptosis Caused by IRI In Vivo and In Vitro
By using western blot analysis, consistent loss of SIRT1 was observed in IRI models in vivo and in vitro, including HSR, oxygen glucose deprivation, and reoxygenation (OGD/R) (0.59 ± 0.09-fold of Ctrl in mice; 0.46 ± 0.07-fold of Ctrl in HT22 cell, Figures 3(a) and 3(b)). Meanwhile, significant apoptosis was also observed with the increase in Bax expression and decrease in BCL2 expression (Figures 3(a) and 3(b)). After SP, changes of SIRT1, Bax, and BCL2 induced by HSR or ODG/R were significantly reversed. In addition, the deposition and distribution of SIRT1, as well as the deacetylation activity of SIRT1, were varied following OGD/R without or with SP (SFig. 1, SFig. 2). Either EX527 or si-SIRT1 significantly reduced SIRT1 expression accompanying with distinct more apoptosis (Figures 3(a) and 3(b), SFig. 3). When overexpressing SIRT1 by using lentivirus transfection before HSR, SIRT1 loss and apoptosis were significantly eliminated (Figure 3(c)).
Figure 3.
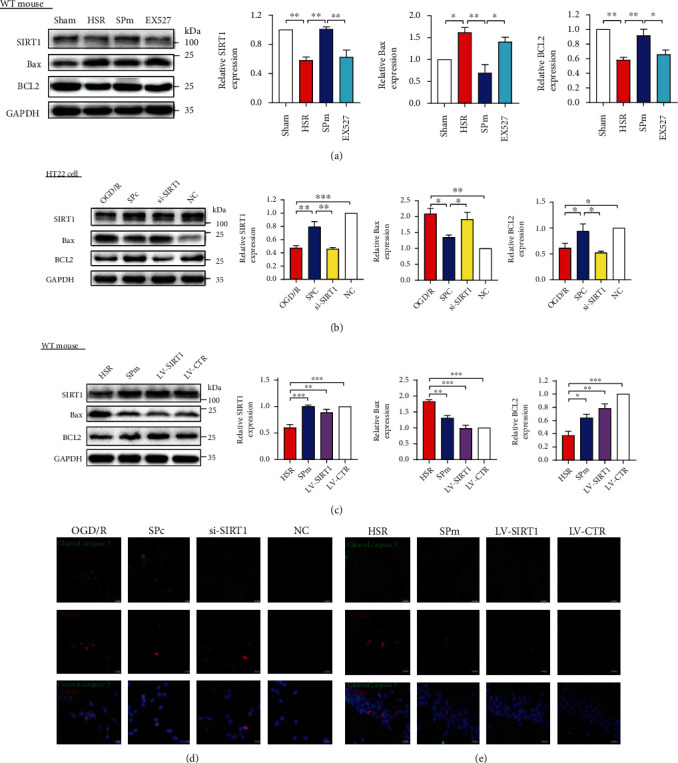
Sevoflurane postconditioning attenuated SIRT1 loss and cell apoptosis induced by ischemia-reperfusion injury in both in vivo and in vitro models. (a, b) By using western blotting analysis, protein expression levels of SIRT1, proapoptotic Bax, and antiapoptotic BCL2 were demonstrated following HSR or oxygen glucose deprivation and reoxygenation (OGD/R) with or without sevoflurane postconditioning (SP). After pretreatment with SIRT1 selective inhibitor EX527 or si-SIRT1 before SP, the levels of SIRT1 and apoptosis-related proteins were also measured. Nonsense sequence for SIRT1 was the negative control (NC). (c) After overexpressing SIRT1 through lentivirus transfection, protein levels of SIRT1, Bax, and BCL2 were evaluated in mice following HSR without SP. Control lentiviral vector (LV-CTR) was cerebrally injected without or with HSR or SPm. (d, e) Apoptotic cells induced by HSR or OGD/R were presented by double labelling with cleaved caspase 3 and TUNEL. All values are represented as mean ± SEM. n = 4‐6 per group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. Scale bar = 20 μm.
Consistent with the distinct apoptosis, cell viability was remarkably dropped after OGD/R (0.49 ± 0.03-fold of Ctrl, SFig. 4). In addition to promoting cell survival, SP prevented apoptosis and DNA damage, which was evidenced with the decreased immunoreactivities of cleaved caspase 3, TUNEL, and 8-OHdG (Figures 3(d) and 3(e), SFig. 5 & 6). However, these decreases were significantly reversed after SIRT 1 gene silencing by using si-SIRT1 transfection in HT22 cells. When HSR mice were preinjected with SIRT1 overexpression lentivirus, less distributions of cleaved caspase 3+ and TUNEL+ immuno-signals were observed in the hippocampal CA1 region.
3.3. Sevoflurane Postconditioning Improved Oxidative Stress and Mitochondrial Dysfunction Caused by IRI In Vitro
By using CCK8 assay, more survived cells were observed after SPc treatment than OGD/R (Figure 4(a)). In the HT22 cells, OGD/R made a remarkable oxidative stress, signed by the decrease of SOD production, increase of MDA accumulation (Figure 4(b)), and increase of MitoSOX immunoreactivity (Figure 4(c)). In addition, OGD/R dramatically reduced ATP production and disrupted mitochondrial membrane potentiation (MMP) determined by JC-1 assay (Figures 4(d) and 4(e)). SPc significantly alleviated oxidative stress with the evidence including more SOD yields, less MDA release, and less MitoSOX toxicity. SPc also promoted ATP production and improved mitochondrial damage through enhancing MMP. Otherwise, the effects of SPc on oxidative stress and MMP were completely reversed by genetic silence of SIRT1 with si-SIRT1.
Figure 4.
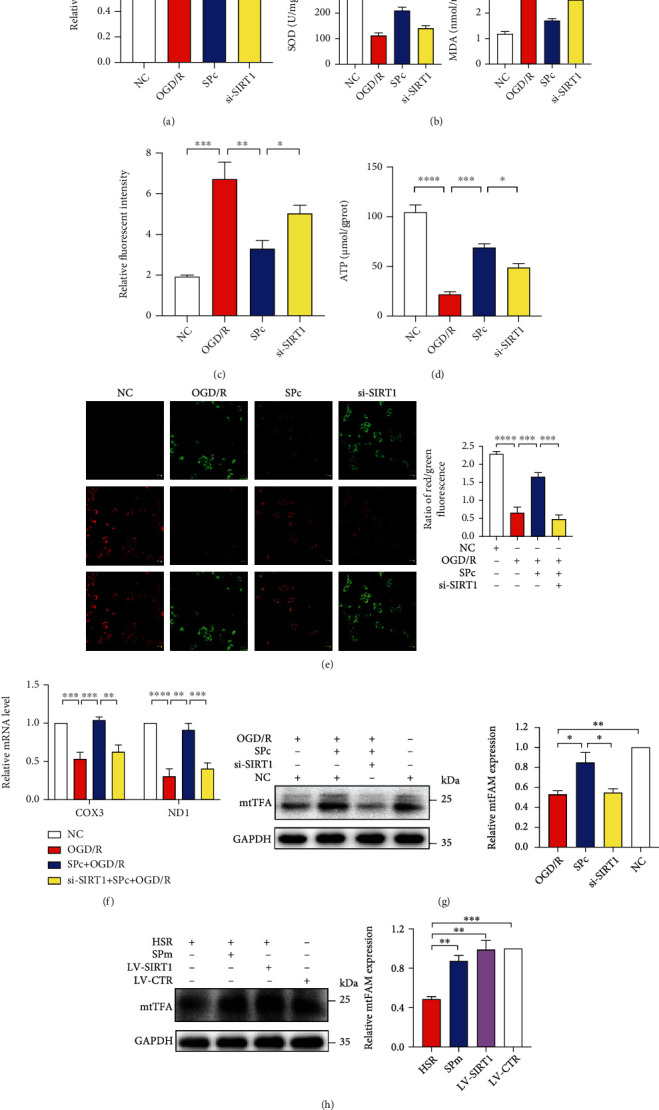
Sevoflurane postconditioning alleviated oxidative stress and mitochondrial dysfunction caused by OGD/R. (a) During OGD/R, sevoflurane postconditioning (SPc) increased cell viability in the CCK8 assays. Transfection of siRNA against SIRT1 (si-SIRT1) was introduced 24 hours before OGD/R with SPc. Nonsense sequence for SIRT1 was the negative control (NC). (b–d) SPc relieved oxidative stress with more SOD, less burdens of MDA and MitoSOX, and more ATP production. (e) Representative photographs and the quantification of mitochondria membrane potentiation (MMP) was determined by JC-1 assay in HT22 cells following OGD/R with SP treatment. Scale bar = 20 μm. (f) Relative mitochondria DNA copy numbers of COX3 and ND1 were quantified by RT-qPCR after SPc with si-SIRT1 treatments. (g, h) Relative mitochondrial transcription factor A (mtTFA) protein expressions in HT22 cells and hippocampi following IRI with SIRT1 down- and upregulation. All values are represented as mean ± SEM. n = 4‐6 per group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
To further investigate the transcript level of mtDNA during IRI without or with SPc, mitochondrial copy number including NADH dehydrogenase subunits 1 (ND1) and cytochrome c oxidase subunit 3 (COX3) was determined. OGD/R strikingly suppressed mitochondrial DNA transcript ability, which was boosted by SPc (Figure 4(f)). Additionally, both OGD/R and HSR decreased the expression of mitochondria transcription factor A (mtTFA). After SP, the increases of mtTFA were noted in both HT22 cells and mouse hippocampi. However, there was a prominent drop of mtTFA after SIRT1 gene silencing. The overexpression of SIRT1 by lentivirus transfection made a comparable level of mtTFA to SPm (Figures 4(g) and 4(h)).
3.4. Sevoflurane Postconditioning Reversed Autophagy Caused by IRI In Vitro
Under TEM observation, OGD/R made mitochondria swelling and hypertrophy, as well as mitochondrial ridge disruption (Figure 5(a)). Meanwhile, less mitophagy events were noted (Figure 5(b)). The effect of SPc or si-SIRT1 on autophagy was further determined by immunofluorescent staining of LC3B (Figures 5(c)–5(e)). Furthermore, the levels of autophagy markers, such as LC3B, PINK1, Parkin, and P62, were significantly reduced following OGD/R in HT22 cells (Figure 5(f)). SPc successfully prevented mitochondria against swelling, ridge disruption, and vacuolization. SPc also improved the suppression of LC3B, PINK1, and Parkin induced by OGD/R. Otherwise, all these changes were reversed after the introduction of si-SIRT1 to SPc.
Figure 5.
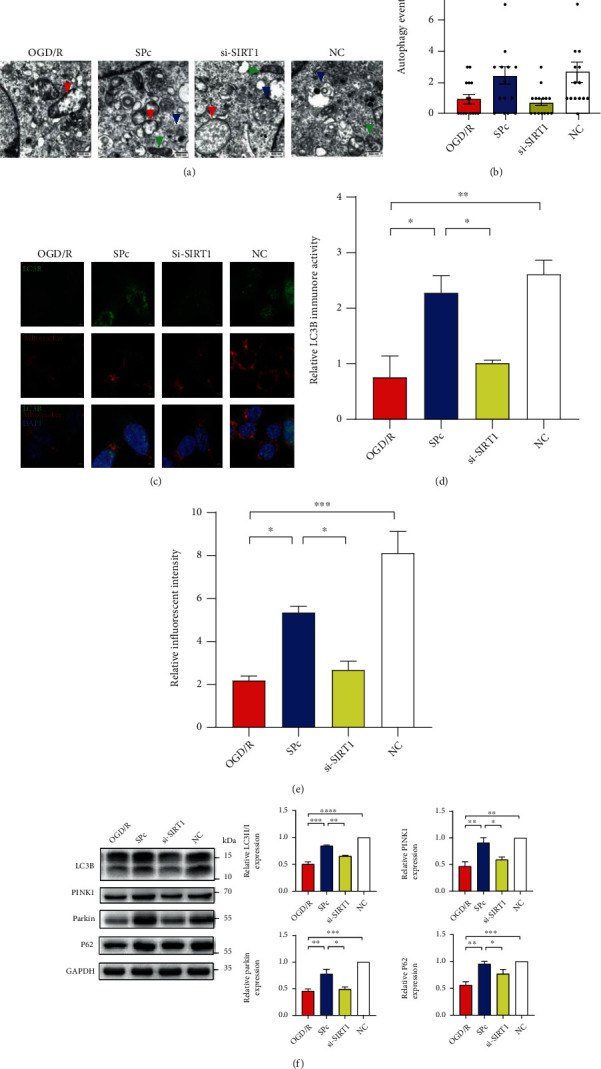
SIRT1 silence reversed the effect of sevoflurane postconditioning on autophagy during OGD/R. (a) Mitochondrial morphological changes under TEM following OGD/R and SPc. Green arrow, healthy mitochondria; red arrow, injured mitochondria; blue arrow, mitophagy. Scale bar = 500 nm. Sevoflurane postconditioning was performed at the beginning of reoxygenation in HT22 cells (SPc). During OGD/R, transfection of siRNA against SIRT1 (si-SIRT1) was introduced 24 hours before OGD/R with SPc. Nonsense sequence for SIRT1 was the negative control (NC). (b) By using electron microscopy images, autophagy-like events were quantified in HT22 cells after OGD/R and SPc, n = 4 neurons from 3 mice per each group. (c–e) The deposition and distribution of LC3B in HT22 cells were quantified after OGD/R and SPc. Scale bar = 5 μm. Mitochondria were labelled with MitoTracker. The relative fluorescent intensity of MitoTracker was quantified. (f) Relative levels of autophagy-related proteins were determined in HT22 cells after OGD/R with SPc. All values are represented as mean ± SEM. n = 4‐6 per group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
3.5. Sevoflurane Postconditioning Manipulated Autophagy through SIRT1 Intervention with Pharmacological Inhibition or Genetic Overexpression In Vivo
Firstly, the impact of SP on autophagy in IRI was consistently demonstrated in the animal model of HSR (Figures 6(a) and 6(b)). After the systemic administration of EX527, there were noted decreases of LC3B, PINK1, and Parkin (Figure 6(a)). The effect of SPm without or with SIRT1 intervention on autophagy was also presented by the varied distributions of LC3B in the hippocampal CA1 region (Figure 6(c)).
Figure 6.
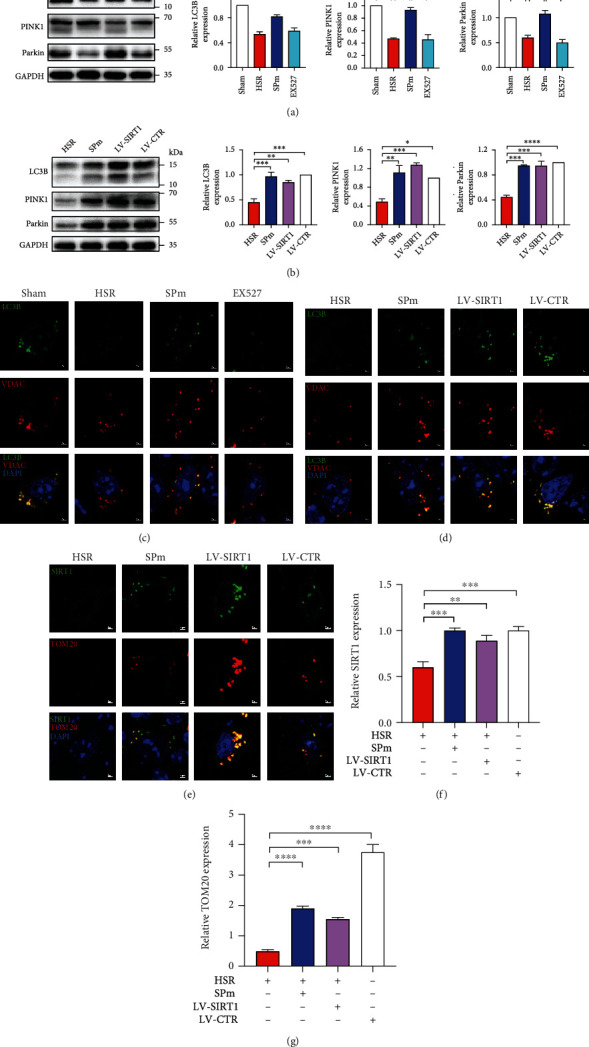
Regulation of autophagy and SIRT1 manipulation by sevoflurane postconditioning during HSR. (a, b) By using western blot analysis, relative levels of autophagy-related proteins were quantified in the hippocampi after HSR with SPm. Sevoflurane postconditioning was performed at the onset of blood refusion in mice (SPm). EX527 (SIRT1 inhibitor) (i.p., 10 mg/kg) was administrated at 30 min before catheters intubation; then, mice were exposed to HSR with SPm. After overexpressing SIRT1 through lentivirus transfection, mice were exposed to HSR without SPm. Control lentiviral vector (LV-CTR) was cerebrally injected without HSR or SPm. (c, d) The localization of LC3B was presented in the hippocampal CA1 region following SPm with pharmacological inhibition or genetic overexpression of SIRT1. (e–g) The quantification of SRIT1 and TOM20 in the hippocampus was presented following HSR without and with genetic overexpression of SIRT1. All values are represented as the mean ± SEM. n = 4‐6 per group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
To further investigate the effect of SIRT1 on autophagy, SIRT1 overexpression lentivirus was ventricularly injected one week before HSR. SPm and LV-SIRT1 comparably increased the protein expressions of LC3B, PINK1, and Parkin, as well as the distribution of LC3B in the hippocampal CA1 region (Figures 6(b) and 6(d)). Furthermore, significant deposition of SIRT1 was noted in the same region, associated with remarkable colocalization with TOM20 (Figures 6(e)–6(g)).
4. Discussion
We successfully demonstrated the neuroprotective effect of sevoflurane postconditioning (SP) in two ischemia reperfusion injury (IRI) models, hemorrhagic shock and resuscitation (HSR), and oxygen and glucose deprivation (OGD/R). SP significantly prevented the impairments of spatial learning and memory, as well as anxiety-like behaviors. It also improved the oxidative damage, mitochondrial dysfunction, and defective autophagy induced by IRI. However, the impact of SP on IRI was consistently reversed by the posttranslational modification of SIRT1 by pharmacological inhibitor or gene silencing.
The mechanisms of IRI are mainly related to oxidative stress, calcium imbalance, mitochondrial injury, excessive inflammatory response, endoplasmic reticulum stress, and programmed cell death. In the common animal model of cerebral IRI, such as middle cerebral artery occlusion (MCAO), distinct cerebral infarction and cognitive dysfunction are usually presented with apoptosis, neuroinflammation, oxidative stress, and mitochondrial dysfunction [8–11, 24]. In this study, HSR was used as another mouse model of IRI. It caused significant cerebral infarctions, spatial learning, and memory deficits, as well as object recognition disability. Meanwhile, mice after HSR also performed anxiety-like behaviors, including less active and more anxiety.
On the other hand, oxygen glucose deprivation and reoxygenation (OGD/R), as an in vitro model of IRI, suppressed cell viability with increased oxidative stress injury in HT22 cells. Mitochondria are a major target in hypoxic/ischemic injury. At the early stage of reperfusion, there is a promotion in the opening of mitochondrial membrane permeability transition pore (mPTP) [25] and increases in the releases of cytochrome c and apoptosis inducing factor. At a long period following ischemia-reperfusion, a large number of ROS interrupts the activities of respiratory chain complexes I, II, III, and IV. And the dysfunction of mitochondrial respiratory chain further increases the production and accumulation of ROS, triggering a series of cell damage [26], such as mitochondrial DNA (mtDNA) oxidative damage [27]. The mtDNA damage directly decreased the encoding of respiratory chain related proteins, which contributed to the dysfunctions of oxidative respiratory chain and ATP production. Consequently, ROS were further increased, so that oxidative damage was aggravated [28].
Disturbance of mitochondria function resulted in neuronal apoptosis and neurological dysfunction after cerebral infarction. In this study, consistent cell apoptosis was observed in HT22 cells after OGD/R and in the hippocampal CA1 region after HSR, respectively. In addition to the disrupted mitochondrial membrane potentiation and ATP generation, OGD/R also interrupted mitochondrial DNA (mtDNA) transcription. Because mtDNA is close to the respiratory chain and lacks histone protection, it is more vulnerable to the oxidative stress [29, 30]. Consistent to the findings in the aging process [27], we found that cerebral IRI made mtDNA oxidative damage, such as decreases in NADH dehydrogenase subunits 1 (ND1) and cytochrome c oxidase subunit 3 (COX3). Studies have shown that mtDNA oxidative damage can be detected 1 hour after IR, and cell death can be observed 6 hours later [31], suggesting that mtDNA oxidative damage may be one of the factors leading to early IR injury. Mitochondrial transcription factor A (mtTFA) is essential for mitochondrial biogenesis and plays a critical role in the maintenance of mitochondrial DNA replication and transcription [32]. However, mtTFA expression was consistently decreased in both HSR and OGD/R.
SIRTs are class III histone deacetylases and sorted into groups I–IV (SIRT1–3, SIRT4, SIRT5, and SIRT6/7). SIRT1 impacts multiple processes including chromatin remodeling, DNA repair, cell survival, and neurogenesis. It is essential for the maintenance of brain integrity, normal learning, memory, and synaptic plasticity in animals [33]. At 48 hours after focal ischemia, larger infarct volumes were displayed in SIRT1−/− mice than the wild-type counterparts [9]. In contrast to SIRT1, SIRT2 and SIRT3 show diverse force in IR. At the early phase of ischemic stroke, SIRT2 mediates myelin-dependent neuronal dysfunction. In both mild and severe MCAO, there were no significant differences in neither infarct volumes nor inflammatory cell count between SIRT2−/− and wild-type mice. Especially at 48 hours after reperfusion, SIRT2−/− mice showed less neurological damage in the experimental stroke models [34]. It also reported that a compensatory rise in SIRT1 rather than the loss of SIRT3 after stroke contributed to the significant neuroprotective effect against IR or stroke injury in SIRT3−/− mice [35]. By using two experimental IRI models (HSR and OGD/R), we found a significant decrease of SRIT1 associated with cell apoptosis and mitochondrial oxidative damage, which were strikingly improved after SIRT1 genetic overexpression or SP treatment.
In response to hypoxic-ischemic conditions, SIRT1 has a pivotal role in maintaining mitochondrial homeostasis by activating many downstream targets, such as regulating peroxisome proliferator-activated receptor γ coactivator α (PGC-1α) for mitochondrial biosynthesis, increasing the expression of ROS degrading enzymes [36], inhibiting p53 induced apoptosis through deacetylation [37], and regulating molecular mediators such as hypoxia-inducible factor 1α (HIF-1α), c-MYC, and AMPK [38], as well as SIRT1/2/3-Foxo3a-MnSOD/PGC-1 signaling pathways [39]. SIRT1 also plays a critical role in the turnover of defective mitochondria through mitophagy [40]. To promote neuronal survival in cerebral ischemia, mitophagy is initiated to eliminate damaged mitochondria through autophagy [12]. SIRT1 activation and autophagy induction contribute to the anti-inflammatory and neuroprotective activities of urolithin A in brain cells (BV2 microglia and APPSwe-transfected ReNcell VM human neural cells) exposing to lipopolysaccharide. Those changes are reversed in the presence of EX527 (SIRT1 inhibitor) and chloroquine (autophagy inhibitor) [41]. In our study, both SIRT1 and autophagy were suppressed by HSR and OGD/R and associated with cognitive dysfunction, mitochondrial dysfunction, and neuronal apoptosis. The overexpression of SIRT1 by shRNA-GFP lentivirus successfully improved the pathological disruptions induced by HSR.
During starvation, LC3 cycles between nucleus and cytoplasm depending on its nuclear deacetylation by SIRT1. Consistent to other cases [42, 43], we also found the dominant distribution of LC3 in the nucleus of intact hippocampal neuronal cells. However, IR suppressed the redistribution of LC3, which was partially attributed to SIRT1 loss. In addition to senescence and aging [44], translocation of SIRT1 from its original nuclear resides to cytosolic puncta was also found in IR in this study. The colocalization of SIRT1 and LC3 supports the new identify of SIRT1 as a new substrate of nuclear autophagy. SIRT1 degradation is mediated by LC3 with the involvement of nucleus-to-cytoplasm shuttling of SIRT1 and autophagosome-lysosome degradation [43, 45]. Interestingly, although SIRT1 prevents DNA damage and oxidative stress through the induction of autophagy, SIRT inhibits corticosterone-induced autophagy with subsequent enforced cell apoptosis [46]. Therefore, a further study is needed to clarify the relationship between SIRT1 and LC3 in the development of IRI.
Even though the effect of sevoflurane on cognition were controversial, such as the deleterious impact of sevoflurane exposure in the neonatal period [47], numbers of studies reported the protective effect of SP in varied IRI models. In myocardial IRI, SP reduces the production of mitochondrial ROS, increases the content of mitochondrial ATP, and thus reduces apoptosis through activating the JAK2-STAT3 pathway [48]. For transient global cerebral ischemia in rats, SP plays an anti-inflammatory role by blocking the TLR-4/NF-κB pathway [49]. SP also attenuates OGD/R-induced autophagosome accumulation in the SHSY-5Y cell line [50]. Our previous studies demonstrated that SP prevented cerebral damage against HSR in rats [15, 16]. SP efficiently improved the spatial learning and memory ability by stabilizing the integrity of mitochondrial structure and function. Furthermore, the downregulation of endoplasmic reticulum stress also involved in the attenuated hippocampal neuronal apoptosis induced by SP through the IRE1α/caspase-12 pathway [16]. In this study, the employment of 1-hour SP dramatically prevented the cognitive and mental disorders induced by HSR in mice, as well as mitochondrial dysfunction and neuronal apoptosis attributed to OGD/R in HT22 cells. Pharmacological inhibition with EX527 or genetic suppression with siRNA effectively reversed the neuroprotective actions of SP in cognitive dysfunction, mitochondrial oxidative damage, and autophagy disturbance. Otherwise, it is notable to investigate if SP-induced neuroprotection could be overturned when autophagy was suppressed with specific inhibitors.
5. Conclusions
In this study, we showed that SP prevented the cognitive dysfunction and anxiety caused by HSR. SP improved neuronal apoptosis by attenuating mitochondrial oxidative stress and improving autophagy. SIRT1 potentially plays an essential role in the neuroprotective effect of SP against IRI. Our findings suggest that SIRT1 may be a functional target in modulating the oxidative stress and autophagy in IRI.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (NSFC 81801050), Basic and clinical cooperative research promotion program of Anhui Medical University (2019xkjT026), and Anhui Medical University promotion program of scientific research platform and base construction (2020xkjT060). We thank the technical support from the center for Scientific Research of Anhui Medical University. We thank Dr. Chanjuan Chen for revising the entire manuscript.
Contributor Information
Xianwen Hu, Email: huxianwen001@163.com.
Chunxia Huang, Email: huangchunxia@ahmu.edu.cn.
Data Availability
The data used to support the findings of this study are available from the corresponding authors upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
Authors' Contributions
J.S., X.H., and Q.L. performed the experiment. J.W., Y.Z., Y.C., M.C., and C.Q. analyzed the data. Y.Z. revised the manuscript. X.H. and C.H. designed the experiments and then drafted and revised the manuscript. All authors have read and approved the final manuscript. Jianwei Shu, Xiaotong Huang, and Qizhi Liao contributed equally to this work.
Supplementary Materials
Figure S1: sevoflurane postconditioning varied the deposition and distribution of SIRT1 following IRI. Figure S2: relative deacetylation activity of SIRT1 after sevoflurane postconditioning in HT22 cells. Figure S3: relative mRNA level of SIRT1 after siRNA transfection in HT22 cells. Figure S4: sevoflurane postconditioning promoted the cell viability following OGD/R. Figure S5: sevoflurane postconditioning attenuated the apoptosis induced by OGD/R. Figure S6: sevoflurane postconditioning attenuated DNA damage induced by OGD/R.
References
- 1.Lozano R., Naghavi M., Foreman K., et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet . 2012;380(9859):2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cannon J. W. Hemorrhagic shock. The New England journal of medicine. . 2018;378(4):370–379. doi: 10.1056/NEJMra1705649. [DOI] [PubMed] [Google Scholar]
- 3.Mitra B., Gabbe B. J., Kaukonen K. M., Olaussen A., Cooper D. J., Cameron P. A. Long-term outcomes of patients receiving a massive transfusion after trauma. Shock (Augusta, Ga) . 2014;42(4):307–312. doi: 10.1097/SHK.0000000000000219. [DOI] [PubMed] [Google Scholar]
- 4.Mira J. C., Cuschieri J., Ozrazgat-Baslanti T., et al. The epidemiology of chronic critical illness after severe traumatic injury at two level-one trauma centers. Critical Care Medicine . 2017;45(12):1989–1996. doi: 10.1097/CCM.0000000000002697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes C. G., Morandi A., Girard T. D., et al. Association between endothelial dysfunction and acute brain dysfunction during critical illness. Anesthesiology . 2013;118(3):631–639. doi: 10.1097/ALN.0b013e31827bd193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li M., Li S. C., Dou B. K., et al. Cycloastragenol upregulates SIRT1 expression, attenuates apoptosis and suppresses neuroinflammation after brain ischemia. Acta Pharmacologica Sinica . 2020;41(8):1025–1032. doi: 10.1038/s41401-020-0386-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng X. W., Shan C. S., Xu Q. Q., et al. Buyang Huanwu decoction targets SIRT1/VEGF pathway to promote angiogenesis after cerebral ischemia/reperfusion injury. Frontiers in Neuroscience . 2018;12:p. 911. doi: 10.3389/fnins.2018.00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duan J., Cui J., Zheng H., et al. Aralia taibaiensis protects against I/R-induced brain cell injury through the Akt/SIRT1/FOXO3a pathway. Oxidative Medicine and Cellular Longevity . 2019;2019:18. doi: 10.1155/2019/7609765.7609765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernandez-Jimenez M., Hurtado O., Cuartero M. I., et al. Silent information regulator 1 protects the brain against cerebral ischemic damage. Stroke; a Journal of Cerebral Circulation . 2013;44(8):2333–2337. doi: 10.1161/STROKEAHA.113.001715. [DOI] [PubMed] [Google Scholar]
- 10.Xian W., Li T., Li L., Hu L., Cao J. Maresin 1 attenuates the inflammatory response and mitochondrial damage in mice with cerebral ischemia/reperfusion in a SIRT1-dependent manner. Brain Research . 2019;1711:83–90. doi: 10.1016/j.brainres.2019.01.013. [DOI] [PubMed] [Google Scholar]
- 11.Chen W., Wang H., Zhu Z., Feng J., Chen L. Exosome-shuttled circSHOC2 from IPASs regulates neuronal autophagy and ameliorates ischemic brain injury via the miR-7670-3p/SIRT1 axis. Molecular Therapy--Nucleic Acids . 2020;22:657–672. doi: 10.1016/j.omtn.2020.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan H., Zhang X., Hu W., et al. Histamine H3 receptors aggravate cerebral ischaemic injury by histamine- independent mechanisms. Nature Communications . 2014;5(1):p. 3334. doi: 10.1038/ncomms4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H., Park Y. H., Jeon Y. T., et al. Sevoflurane post-conditioning increases nuclear factor erythroid 2-related factor and haemoxygenase-1 expression via protein kinase C pathway in a rat model of transient global cerebral ischaemia. British Journal of Anaesthesia . 2015;114(2):307–318. doi: 10.1093/bja/aeu268. [DOI] [PubMed] [Google Scholar]
- 14.Xue H., Zhang Y. H., Gao Q. S., et al. Sevoflurane post-conditioning ameliorates neuronal deficits and axon demyelination after neonatal hypoxic ischemic brain injury: role of microglia/macrophage. Cellular and Molecular Neurobiology . 2021;41(8):1801–1816. doi: 10.1007/s10571-020-00949-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu X., Zhang M., Duan X., et al. Sevoflurane postconditioning improves the spatial learning and memory impairments induced by hemorrhagic shock and resuscitation through suppressing IRE1α-caspase-12-mediated endoplasmic reticulum stress pathway. Neuroscience Letters . 2018;685:160–166. doi: 10.1016/j.neulet.2018.08.035. [DOI] [PubMed] [Google Scholar]
- 16.Hu X., Wang J., Zhang L., Zhang Q., Duan X., Zhang Y. Postconditioning with sevoflurane ameliorates spatial learning and memory deficit via attenuating endoplasmic reticulum stress induced neuron apoptosis in a rat model of hemorrhage shock and resuscitation. Brain Research . 2018;1696:49–55. doi: 10.1016/j.brainres.2018.05.047. [DOI] [PubMed] [Google Scholar]
- 17.Tang Y., Xia X. F., Zhang Y., et al. Establishment of an experimental mouse model of trauma-hemorrhagic shock. Experimental Animals . 2012;61(4):417–425. doi: 10.1538/expanim.61.417. [DOI] [PubMed] [Google Scholar]
- 18.de Waard M. C., van der Pluijm I., Zuiderveen Borgesius N., et al. Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathologica . 2010;120(4):461–475. doi: 10.1007/s00401-010-0715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H., Shen L., Lv T., et al. Salidroside attenuates dextran sulfate sodium-induced colitis in mice via SIRT1/FoxOs signaling pathway. European Journal of Pharmacology . 2019;861:p. 172591. doi: 10.1016/j.ejphar.2019.172591. [DOI] [PubMed] [Google Scholar]
- 20.Wang J., Zhu S., Lu W., et al. Varenicline improved laparotomy-induced cognitive impairment by restoring mitophagy in aged mice. European Journal of Pharmacology . 2022;916:p. 174524. doi: 10.1016/j.ejphar.2021.174524. [DOI] [PubMed] [Google Scholar]
- 21.Li D., Huang Y., Cheng B., Su J., Zhou W. X., Zhang Y. X. Streptozotocin induces mild cognitive impairment at appropriate doses in mice as determined by long-term potentiation and the Morris water maze. Journal of Alzheimer's disease : JAD. . 2016;54(1):89–98. doi: 10.3233/JAD-150979. [DOI] [PubMed] [Google Scholar]
- 22.Kraeuter A. K., Guest P. C., Sarnyai Z. The open field test for measuring locomotor activity and anxiety-like behavior. Methods in Molecular Biology . 2019;1916:99–103. doi: 10.1007/978-1-4939-8994-2_9. [DOI] [PubMed] [Google Scholar]
- 23.Wolf A., Bauer B., Abner E. L., Ashkenazy-Frolinger T., Hartz A. M. A comprehensive behavioral test battery to assess learning and memory in 129S6/Tg2576 mice. PLoS One . 2016;11(1, article e0147733) doi: 10.1371/journal.pone.0147733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z. T., Hu C. Y., Dai F. B., Tang F., Tang C. L. Mechanisms and status of research on the protective effects of traditional Chinese medicine against ischemic brain injury. Tradit Med Res. . 2022;7(1):p. 6. doi: 10.53388/TMR20211021250. [DOI] [Google Scholar]
- 25.Wang X. R., Ding R., Tao T. Q., et al. Myofibrillogenesis regulator 1 rescues Renal ischemia/reperfusion injury by recruitment of PI3K-dependent P-AKT to mitochondria. Shock (Augusta, Ga) . 2016;46(5):531–540. doi: 10.1097/SHK.0000000000000658. [DOI] [PubMed] [Google Scholar]
- 26.Qu J., Chen W., Hu R., Feng H. The injury and therapy of reactive oxygen species in intracerebral hemorrhage looking at mitochondria. Oxidative Medicine and Cellular Longevity . 2016;2016:9. doi: 10.1155/2016/2592935.2592935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinto M., Pickrell A. M., Wang X., et al. Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS-dependent but p53/p21-independent manner. Cell Death and Differentiation . 2017;24(2):288–299. doi: 10.1038/cdd.2016.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Q., Ren J., Li G., et al. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death & Disease . 2018;9(3):p. 403. doi: 10.1038/s41419-018-0436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Q., Zhou Q., Wu J., Wu X., Ren J. The role of mitochondrial DNA in the development of ischemia reperfusion injury. Shock (Augusta, Ga) . 2019;51(1):52–59. doi: 10.1097/SHK.0000000000001190. [DOI] [PubMed] [Google Scholar]
- 30.West A. P., Shadel G. S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nature Reviews. Immunology . 2017;17(6):363–375. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan X., Zhang L., Jiang Y., et al. Postconditioning ameliorates mitochondrial DNA damage and deletion after renal ischemic injury. Nephrology, Dialysis, Transplantation . 2013;28(11):2754–2765. doi: 10.1093/ndt/gft278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Busch K. B., Kowald A., Spelbrink J. N. Quality matters: how does mitochondrial network dynamics and quality control impact on mtDNA integrity? Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences . 2014;369(1646):p. 20130442. doi: 10.1098/rstb.2013.0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michan S., Li Y., Chou M. M., et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. The Journal of neuroscience : the official journal of the Society for Neuroscience. . 2010;30(29):9695–9707. doi: 10.1523/JNEUROSCI.0027-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krey L., Lühder F., Kusch K., et al. Knockout of silent information regulator 2 (SIRT2) preserves neurological function after experimental stroke in mice. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism . 2015;35(12):2080–2088. doi: 10.1038/jcbfm.2015.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verma R., Ritzel R. M., Crapser J., Friedler B. D., McCullough L. D. Evaluation of the neuroprotective effect of Sirt3 in experimental stroke. Translational Stroke Research . 2019;10(1):57–66. doi: 10.1007/s12975-017-0603-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang H., Aminian A., Hassan M., et al. Gastric bypass surgery improves the skeletal muscle ceramide/S1P ratio and upregulates the AMPK/ SIRT1/ PGC-1α pathway in Zucker diabetic fatty rats. Obesity Surgery . 2019;29(7):2158–2165. doi: 10.1007/s11695-019-03800-z. [DOI] [PubMed] [Google Scholar]
- 37.Carloni S., Riparini G., Buonocore G., Balduini W. Rapid modulation of the silent information regulator 1 by melatonin after hypoxia-ischemia in the neonatal rat brain. Journal of Pineal Research . 2017;63(3):p. e12434. doi: 10.1111/jpi.12434. [DOI] [PubMed] [Google Scholar]
- 38.Ham P. B., 3rd, Raju R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Progress in Neurobiology . 2017;157:92–116. doi: 10.1016/j.pneurobio.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie W., Zhu T., Zhou P., et al. Notoginseng leaf triterpenes ameliorates OGD/R-induced neuronal injury via SIRT1/2/3-Foxo3a-MnSOD/PGC-1α signaling pathways mediated by the NAMPT-NAD pathway. Oxidative Medicine and Cellular Longevity . 2020;2020:15. doi: 10.1155/2020/7308386.7308386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang B. L. Sirt1 and the mitochondria. Molecules and Cells . 2016;39(2):87–95. doi: 10.14348/molcells.2016.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Velagapudi R., Lepiarz I., El‐Bakoush A., et al. Induction of autophagy and activation of SIRT-1 deacetylation mechanisms mediate neuroprotection by the pomegranate metabolite urolithin A in BV2 microglia and differentiated 3D human neural progenitor cells. Molecular Nutrition & Food Research . 2019;63(10, article e1801237) doi: 10.1002/mnfr.201801237. [DOI] [PubMed] [Google Scholar]
- 42.Huang R., Xu Y., Wan W., et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Molecular Cell . 2015;57(3):456–466. doi: 10.1016/j.molcel.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 43.Tan J. X., Finkel T. Autophagy goes nuclear. Nature Cell Biology . 2020;22(10):1159–1161. doi: 10.1038/s41556-020-00587-5. [DOI] [PubMed] [Google Scholar]
- 44.Xu C., Wang L., Fozouni P., et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nature Cell Biology . 2020;22(10):1170–1179. doi: 10.1038/s41556-020-00579-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L., Xu C., Johansen T., Berger S. L., Dou Z. SIRT1 - a new mammalian substrate of nuclear autophagy. Autophagy . 2021;17(2):593–595. doi: 10.1080/15548627.2020.1860541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang Y., Botchway B. O. A., Hu Z., Fang M. Overexpression of SIRT1 inhibits corticosterone-induced autophagy. Neuroscience . 2019;411:11–22. doi: 10.1016/j.neuroscience.2019.05.035. [DOI] [PubMed] [Google Scholar]
- 47.Song S. Y., Meng X. W., Xia Z., et al. Cognitive impairment and transcriptomic profile in hippocampus of young mice after multiple neonatal exposures to sevoflurane. Aging . 2019;11(19):8386–8417. doi: 10.18632/aging.102326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu J., Yu J., Xie P., et al. Sevoflurane postconditioning protects the myocardium against ischemia/reperfusion injury via activation of the JAK2-STAT3 pathway. PeerJ . 2017;5, article e3196 doi: 10.7717/peerj.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hwang J. W., Jeon Y. T., Lim Y. J., Park H. P. Sevoflurane postconditioning-induced anti-inflammation via inhibition of the Toll-like receptor-4/nuclear factor kappa B pathway contributes to neuroprotection against transient global cerebral ischemia in rats. International Journal of Molecular Sciences . 2017;18(11):p. 2347. doi: 10.3390/ijms18112347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng A., Lu Y., Huang Q., Zuo Z. Attenuating oxygen-glucose deprivation-caused autophagosome accumulation may be involved in sevoflurane postconditioning-induced protection in human neuron-like cells. European Journal of Pharmacology . 2019;849:84–95. doi: 10.1016/j.ejphar.2019.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: sevoflurane postconditioning varied the deposition and distribution of SIRT1 following IRI. Figure S2: relative deacetylation activity of SIRT1 after sevoflurane postconditioning in HT22 cells. Figure S3: relative mRNA level of SIRT1 after siRNA transfection in HT22 cells. Figure S4: sevoflurane postconditioning promoted the cell viability following OGD/R. Figure S5: sevoflurane postconditioning attenuated the apoptosis induced by OGD/R. Figure S6: sevoflurane postconditioning attenuated DNA damage induced by OGD/R.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding authors upon request.