

Abstract
Prostate cancer is characterized by a high degree of heterogeneity, which poses a major challenge to precision therapy and drug development. In this review, we discuss how nongenetic factors contribute to heterogeneity of prostate cancer. We also discuss tumor heterogeneity and phenotypic switching related to anticancer therapies. Lastly, we summarize the challenges targeting the tumor environments, and emphasize that continued exploration of tumor heterogeneity is needed in order to offer a personalized therapy for advanced prostate cancer patients.
Subject terms: Urological cancer, Prostate cancer
Introduction
Tumor heterogeneity has been a major contributor to lethal outcomes, drug resistance, and therapeutic failures, and presents a key challenge to precision medicine goals1,2. In light of the association between tumor heterogeneity and poor prognostic results, a measure of heterogeneity itself might be useful as a prognostic marker3. Although heterogeneity of prostate cancer is conventionally attributed to genetic diversity4–6, current evidence reveals that in addition to genetic factors, the tumor heterogeneity could be derived from nongenetic variabilities7.
The stroma serves as a main barrier preventing carcinogenesis in benign tissue; however, the presence of cancer cells initiates crucial changes, converting the environment into one that supports tumor growth. These changes include fibroblast recruitment, immunocytes migration, matrix remodeling, development of tumor-specific vasculature, and aberrant epigenetic landscape, and each of these changes might promote heterogeneity of tumor microenvironment (TME). Local diversity of selective pressures in TME, such as hypoxia, acidity, and growth factors, also actively shape tumor morphology. Conceivably, the distinct environmental landscape in the tumor plays a significant role in tumor heterogeneity.
TME is dynamic with spatial and temporal changes in composition in response to environmental pressures and anticancer therapies, and the continued crosstalk between tumor cells and the surrounding microenvironment is fundamental to tumor initiation, phenotypic changes, cancer progression, and therapeutic resistance. In this review, we summarize the challenges targeting the tumor environments, and emphasize that understanding nongenetic mechanisms might open novel diagnostic and therapeutic approaches with the potential to improve the efficacy of current prostate cancer treatments.
Clonal heterogeneity
Prostate cancer is a multifocal disease (Fig. 1) and each focus might have a different phenotype (intertumor heterogeneity)8. As the individual tumor volume increases over time, multiple foci might merge into a larger mass exhibiting greater tumor heterogeneity (intratumor heterogeneity). It is controversial whether those separate foci reflect a monoclonal origin9–12 or a polyclonal origin13. The studies supporting the monoclonal origin argue that both genetic and epigenetic events occur in a single ancestral cell that does not possess all the necessary mutations to transform into a cancer cell. As those cells are exposed to additional events and divide, DNA replication errors lead to daughter cells that are genetically different from each other. Hence, although the cells are heterogeneous, all derive from the same ancestor14. For instance, analysis of the whole exome sequencing and transcriptome profiles from Gleason 3 and neighboring Gleason 4 tumor foci revealed that the adjacent tumors emerged early from a common precursor and subsequently undergo independent evolution10. In a separate study, analysis of genomewide single nucleotide polymorphism and copy number variations from metastasized prostate cancers demonstrated that most cancers were of monoclonal origin and have identical copy number changes11.
Fig. 1. Intratumor and intertumor heterogeneity of prostate cancer.

Whole-mount cross-section of a radical prostatectomy specimen has two separated tumor foci. One focus is in the right anterior of the prostate (Gleason score 4 + 3 = 7, Group 3) whereas another focus is in the left posterior of the prostate (Gleason score 5 + 4 = 9, Group 5). Scale bars, 4 mm (left) and 100 µm (right). Methods: Radical prostatectomy specimens were serially sectioned into 3 mm slices and completely embedded. The case was reviewed by a single urologic pathologist (R.G.) in 2021. The following features were monitored: Gleason Score and Grade Group according to the International Society of Urological Pathology (ISUP) 2014 guidelines. The percentages of Gleason pattern 3, 4, and 5 were estimated, including presence of tertiary Gleason patterns.
In support of the polyclonal tumor origin, the chromosomal analysis and genomic DNA sequencing studies on multifocal prostate cancers have revealed that different tumor foci have independent clonal expansions5,13,15. In another study, analysis of the whole exome sequencing of 23 distinct tumor foci from 5 prostate cancer cases demonstrated that the multifocal tumors were highly heterogeneous for single nucleotide variants, copy number aberrations, and genomic rearrangements16. Similarly, a large cohort study of 89 tumor foci from 41 different patients revealed that samples from different tumor foci in the same prostatectomy specimen rarely had any shared point mutations and the same DNA copy number changes8.
Moreover, analysis of 17 tumor cells from localized lesions with different Gleason scores from 2 prostatectomy cases revealed that in patient number 1, every cell had the same TP53 mutation, which is consistent with the monoclonal model. In the patient number 2, only one cell subpopulation contained the TP53 mutation, while other cells carried different mutations, supporting a polyclonal model17. Overall, these studies indicated that the origin of prostate cancers may have a monoclonal or polyclonal origin that varies from case to case. Further research may shed more light on the generality or predominance of any of these theories.
Tumor microenvironment heterogeneity
It is well-accepted that tumorigenesis is not only dependent on genetic alterations or epigenetic modifications in cancer cells, but is also regulated by the TME18. The TME is composed of fibroblasts, pericytes, immunocytes, and endotheliocytes; each able to crosstalk with cancer cells in dynamic ways (Fig. 2). Usually, the orchestrated impact of microenvironmental components on cancer cells is characterized by the different region, and the tumorigenesis is modulated by the regional heterogeneity in the hypoxia, acidity, and cytokines in a tumor environment19,20. Moreover, the cancer-associated fibroblasts (CAFs) are among the most abundant constituents in the TME, contributing to the malignant phenotype at all levels21–25. CAFs comprise heterogeneous clusters exerting distinct functions, such as tumor growth, angiogenic process and stromal remodeling, drug resistance, and tumor metastasis26. The CAF clusters have either tumor suppressive effects27,28 or tumor promoting effects29,30. This heterogeneity might result from numerous causes, such as dynamic interaction between tumor cells and stromal cells, extracellular matrix, and cytokines and growth factors secreted into the TME31.
Fig. 2. The heterogenous tumor microenvironment.
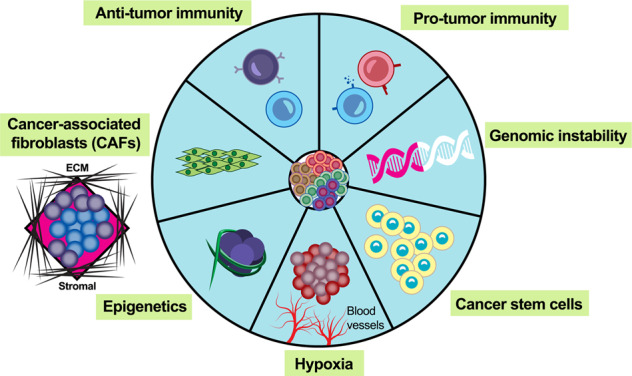
The tumor microenvironment possesses a dynamic topography within the tumor and is composed of cancer-associated fibroblasts, stromal cells, extracellular matrix, and immune cells. Hypoxic status, vasculature, and epigenetics, all may have a complex crosstalk with prostate cancer cells in dynamic ways.
Heterogeneity of CAFs is attributed, at least partially, to multiple origins. CAFs are traditionally considered to originate from the resident fibroblasts under the influence of the transforming growth factor (TGF-β) produced by stromal cells and cancer cells32 or hypoxia inducible factor (HIF)-1α pathway22. The CAF-produced TGF-β and CXCL-12 maintain the myofibroblast phenotype and promote the interaction between tumor-stroma33,34. Evidence has suggested that CAFs also derive from an endothelial-mesenchymal or epithelial-mesenchymal transition35 which is driven by TGF-β or SMAD signaling36,37. Other mature cells, including pericytes or inflammatory cells in stroma, may also transdifferentiate into CAFs through TGF-β modulated induction of mesenchymal-to-mesenchymal transition38. Another perspective is that bone marrow-derived mesenchymal stromal cells may differentiate into CAFs39. The recruitment of mesenchymal stromal cells and their activation into CAFs are facilitated by TGF-β and CXCL-12 secreted by cancer cells40,41. Lastly, cancer stem cells are found to be one of the key origins of CAFs. The transformation of cancer stem cells into stromal cells adds a new dimension that explains tumor heterogeneity42. Overall, the multiple origins contribute to the heterogeneity of CAFs.
Functional heterogeneity of cancer-associated fibroblasts
Functional diversity of prostate CAFs has been observed in some stromal specific markers. For instance, p62 plays a role in the regulation of prostate TME43. Loss of p62 in the stroma promotes prostate cancer growth and is associated with high Gleason score tumors43. CD90 is another stromal specific marker that is related to cancer progression. Stromal cells with high levels of CD90 have more tumor promoting effects than cells with low levels of CD9044. In addition, some stromal biomarkers are related to the cancer phenotypic switch. For example, CXCL-13 is expressed in tumor-associated myofibroblasts and induced by depleted androgen. The rise of CXCL-13 is related to the progress of advanced prostate cancer45. Likewise, CD105, the cell surface endoglin, is heterogeneously expressed in stromal fibroblastic cells, and CD105-positive fibroblasts promote neuroendocrine differentiation of prostate cancer46. In addition, in prostate stroma, loss of TGF-β receptor type II expression contributes to tumorigenesis, progression, and invasion44,47.
The heterogeneous population of cells in CAFs is further verified with single-cell technology23. In the human prostate, six subpopulations of CAFs have been discovered by single-cell RNA sequencing48. These CAF clusters secrete a series of different cytokines with variable immunomodulatory property. Among these clusters, one with CCL2 expression aids in the recruitment of myeloid cells to the TME and correlates with poor clinical outcomes or metastatic potential, while another cluster with CXCL-12 expression recruits mast cells, eosinophils, innate lymphoid cells, and T helper 2 cells. Those inflammatory cells contribute to tumor growth and progression through promoting tumor-associated macrophage activation48.
The heterogeneity of CAFs is also determined by the coevolution of epithelial and stromal cells and their relationship in each stage of tumor development23. CAFs obtained from prostate cancers at different stages revealed that CAF secretome evolves during prostate cancer progression49. The molecular profile of growth factors is expressed differentially at different cancer stages50. For example, the level of fibroblast growth factor 7 is high in localized prostate cancer-derived CAFs, while matrix metallopeptidase 11 and heat-shock 70 kDa protein 1 A are high in CAFs of metastatic tumors51.
Heterogeneity of CAFs has also been studied with the spatial transcriptomics, which revealed the gradients of gene expression in CAFs of multifocal prostate cancers52. In the center of tumors, the gene profile of stromal cells is related to the alteration of metabolism and oxidative stress, implying the central tumor growth relies on the energy released by the stroma52. Meanwhile, the gene profile of stromal cells in the periphery of tumor is primarily related to inflammation, a stimulator of tumor growth52–54.
In addition, in a CAF-rich tumor environment, the balance disruption between extracellular matrix proteases and proteases inhibitors may lead to more aggressive tumors, compared with a CAF-poor environment23. This matrix disruption can cause the release of growth factors/cytokines, such as fibroblast growth factor, hepatocyte growth factor, and TGF-β, which modulate cell proliferation, tumor invasion, and immune responses55,56.
Overall, the studies suggest that stromal/CAF heterogeneity in prostate TME explains the multiple functions of CAFs in the tumorigenesis. This heterogeneity over the different stages of tumor progression provides further evidence for the theory of tumor-stroma coevolution57.
Hypoxia-driven heterogeneity
In prostate cancer, vessel morphology has often been described as aberrant or disorganized58. Microvascular density can vary and higher microvascular density correlates with the metastasis, aggressive phenotype, and stage of the disease59. The disorganized vascular network results in spatial and temporal heterogeneity of tumor hypoxia60. Using oxygen electrodes to measure hypoxia at different locations of the tumor revealed that the oxygen status changes across tumor foci and even within the same tumor61. Hypoxia gene expression signature provides more reliable predictions of hypoxia-dependent changes in TME62.
Hypoxia has been studied extensively in the diversity of the tumor phenotype. Hypoxic cells are subjected to selective pressure, with the hostile growth conditions, the most aggressive cells survive and drive tumor growth63. Moreover, hypoxia-HIF signaling can lead to mesenchymal-epithelial transition64 and promote mesenchymal reprogramming and neuroendocrine transdifferentiation in prostate cancer cells31. Low oxygen pressure and poor nutrient supply may result in histone hypermethylation, which induces the tumor epigenetic heterogeneity65 and reduces the expression of repressor element 1-silencing transcription (REST), an important epigenetic regulator. The REST in turn leads to neuroendocrine transdifferentiation66. The adaptation under hypoxia of cancer cells allows for the survival and proliferation of cancer stem cells67, and the emergence of therapy-resistant phenotypes68. The cancer stem cells in the TME mediate tumor initiation, metastasis, and therapy resistance69. In prostate cancer, four core pluripotency transcription factors, OCT4, SOX2, NANOG, and KLF4, are colocalized and required to maintain pluripotency and self-renewal of embryonic stem cells70. Hypoxia can induce the tumor phenotypic diversity by regulating the four factors71. Other studies revealed that hypoxia can also induce the electron leakage and the generation of reactive oxygen species, which subsequently cause DNA damage and further develop mutator phenotypes, such as reduced DNA repair, increased mutation rate, genomic instability, and increased metastatic capacity72,73.
Taken together, studies on tumor hypoxia heterogeneity suggest that a single tissue biopsy may not provide an accurate global hypoxic status and that biopsies of multiple regions/different time points may be needed to assess spatial and temporal heterogeneity and correctly stratify patients in this context.
Immunity-driven heterogeneity
The immune system collectively functions to recognize and defend against harmful substances. Both innate and adaptive immune systems have protumor and antitumor effects. Immune cell recruitment and localization in the TME vary greatly across lesions, thus creating spatial heterogeneity. In prostate cancer, immune cells infiltrating the tumor have different functions and compositions in different stages. It has been found that infiltration of CD4 + T cells is involved in prostate cancer progression and metastasis74 and neutrophils are associated with poor prognosis75, while invariant natural killer (NK) T cells delay prostate cancer progression by crosstalk with tumor-associated macrophages76. Also, mast cells induce more serious resistance to chemotherapy and radiotherapy in prostate cancer metastasis patients77, and the infiltration of CD8 + T cells in invasive margins is related to poor clinical outcomes78. Hence, the frequency and location of immune cells in prostate tissue might be associated with the subclonal heterogeneity of prostate cancer.
The TME of prostate cancer has been described as an immunosuppressive state. This immunosuppressive state can be characterized by poor cytolytic activity of NK cells, which may be contributed through increased TGF-β secretion by prostate tissue, and reduced antitumor immunity from activated T regulatory cells79.
This tumor immune complexity and heterogeneity is regulated by a series of factors, such as the secretion of CAFs, the extent and permeability of vasculature, and the tumor cells themselves80. Meanwhile, tumor-associated macrophages are well known for their heterogeneity and plasticity, which generally polarize toward two extremes, M1(antitumorigenesis) phenotype and M2 (protumorigenesis) phenotype81, and this heterogeneous macrophage activation is often expressed simultaneously in the TME of prostate cancer82–84. Typically, M1 macrophage activation is associated with the generation of reactive oxygen species, increased expression of IL-12, and reduced expression of IL-10. In contrast, the M2 phenotypic macrophage acquires the ability to execute protumorigenic and proangiogenic functions through the expression of IL-4, IL-13, and IL-10 cytokines85.
Growth differentiation factor 15 (GDF‐15), also known as nonsteroidal anti-inflammatory drug-inducible gene (NAG)-1 or macrophage inhibitory cytokine (MIC)-1, is a member of the TGF-β superfamily proteins. GDF-15 is induced by cellular stress conditions and responds to microenvironment stimulators86,87. GDF-15 is dysregulated not only in epithelial cancer cells, but also in the tumor stroma88, and highly expressed GDF-15 is associated with worse clinical outcomes in prostate cancer89,90. GDF-15 also exhibits the heterogenous functions of either tumor-suppressing or tumor-promoting effects91,92. In a landscape of TME, GDF-15 acts on both nonimmune cells and immune cells. For instance, GDF-15 acts on epithelial cancer cells and promotes epithelial-mesenchymal transition through the MAPK/PI3K pathway93. GDF-15 also promotes metastasis by activating the SMAD2 and SMAD3 signaling. In addition, with the condition of cellular stress, GDF-15 and other cytokines produced by fibroblasts, such as granulocyte-macrophage colony stimulating factor, interleukin-12 (IL-12), or chemokine c-x-c motif 12 (CXCL-12), act on nearby cells to promote tumor cell proliferation and tumor growth94. On the other hand, GDF-15 also acts on immune cells and promotes cancer progression by immune escape. For example, tumor-derived GDF-15 suppresses the proapoptotic activity through inhibiting TGF-β-activated kinase (TAK1) signaling to nuclear factor-κB, thereby evading macrophage-mediated immune surveillance and stimulating early cancer development86. GDF-15 preferentially inhibits M1 macrophage, thereby promoting protumorigenic activities92. GDF-15 also alters NK cell function by inhibiting TNF-alpha and interferon-gamma production and facilitates the escape of tumor cells from the immune system95. Furthermore, GDF-15 promotes the expression of PD-L1 through SMAD2 and SMAD3 signaling, thereby suppressing the activation of T cells through a programmed cell death-1 (PD-1)/PD-L1 interaction96. In general, the function of GDF-15 is complex in the TME and uncovering the pathways of GDF-15 in the tumorigenic process would lead to the development of novel therapeutics.
In addition, tumor hypoxia is essential in regulating phenotypic variations of tumor-associated macrophages. Factors released by tumor-associated macrophages play a role in processes such as tumor development, cancer immunosuppression, and angiogenesis97–99. Since tumor-associated macrophages are dependent on microenvironmental signals like hypoxia and cytokine availability, it is reasonable to suggest that phenotypic changes occur in a spatiotemporal manner85. There are even more signaling pathways that participate in macrophage function and activation, such as tumor necrosis factor, nuclear factor-κB or Toll-like receptor, which are regulated differentially in different tumor foci and further contribute to the phenotypic diversity in the tumor82.
Apart from controlling the phenotypic diversity of tumor-associated macrophages, hypoxia also promotes immune escape by upregulating immune checkpoint proteins in tumor foci100. Accumulated studies confirm that PD-L1 binds to the PD-1 on the T cells to inhibit the immune response through induction of apoptosis and anergy in the T cells101. Hypoxia potently induces HIF-1α-dependent PD-L1 expression on tumor cells102, suggesting that PD-L1 expression can be upregulated in hypoxic tumor cells and promote immune escape from cytotoxic T cells. Restoration of T cell infiltration through inhibition of the hypoxia signaling pathway allows prostate cancer to become more susceptible to immunotherapy103. These studies suggest that coblockade of PD-L1 and HIF-1α signaling might represent a promising approach to reinforce the activity of cytotoxic T cells.
An important feature of prostate cancer is the low tumor mutation burden and thus diminished neoantigen expression compared with many other cancers104. Only 10–30% of unselected patients respond to PD-1/PD-L1 blockade105. However, a recent study investigated 1551 prostate tumors and found that 32 (3.1%) prostate cancer specimens were associated with MSI-H molecular phenotype and high tumor mutation burden. More than one-half of the patients who underwent anti-PD-1/PD-L1 therapy had a significant reduction in prostate-specific antigen levels or radiographic responses. In the multifocal sequencing analysis of six patients with MSI-H prostate cancer, the study found that two patients had an acquired MSI-H phenotype in tumors collected further down in the disease progression. Therefore, the findings provide some evidence that MSI-H molecular phenotype can be somatically acquired106. It has been shown that in colorectal cancer, MSI-H tumors exhibit greater immune cell infiltration and upregulated immune-related gene expressions, and are more immunogenic than MSI-L tumors107. Therefore, it is possible that in prostate cancer, the immune microenvironment can have dynamic changes over time and clinical states and may be altered with therapy108. For example, following androgen deprivation therapy, there is an increase in tumor-infiltrating lymphocytes in the prostate bed109 and increased PD-L1 expression levels of enzalutamide-treated prostate cancer cells110. Preclinical studies have also reported that the Fas expression and antigen presentation can be induced by docetaxel in the TME of prostate cancer111.
Taken together, the immune environment of prostate cancer is complex, heterogeneous, and dynamic during disease evolution. It can be influenced by tumor cells, oxidative stress, TME, and anticancer therapies. Exploration of immunity-mediated tumor heterogeneity may help develop precision medicine for prostate cancer.
Therapy-induced heterogeneity
Most advanced metastatic cancers remain incurable, even in those cases that the approaches of therapy are available to eliminate the majority of tumor cells. Pre-existing tumor heterogeneity increases the odds of at least some tumor cells surviving therapy-induced elimination, while ongoing diversification during treatment enables cancer cells to adapt to therapy-imposed selective pressures and facilitates development of new therapy-resistant phenotypes (Fig. 3). Clinical studies have shown that emergence of the polyploid giant cancer cells (PGCCs) phenotype could be a survival strategy for the prostate cancer population and correlates with poor response to docetaxel chemotherapy in the context of castration-resistant prostate cancer112. A large number of PGCCs can be induced at higher levels of docetaxel concentration and PGCCs exhibit increased expression of the mesenchymal marker, ZEB1, suggesting that these cells could be a possible inducer of an epithelial-mesenchymal transition and mediators of resistance in response to chemotherapeutic stress112,113.
Fig. 3. Therapy-mediated tumor heterogeneity.

Primary tumors are composed of different subclones. Some subclones are sensitive to selective pressures, including chemotherapy, androgen deprivation therapy, or immunotherapy, while some subclones are resistant to anticancer therapies. Outgrowth of resistant subclones, phenotype switch, or emergence of a new tumor phenotype are strongly related for resistance to anticancer therapies. As a result, the tumor heterogeneity plays a fundamental role in cancer progression to metastasis.
Metastatic castration-resistant prostate cancer (mCRPC) is a heterogeneous disease entity with widely varying outcomes114,115. Small cell carcinoma of the prostate is a rare entity and represents one of the most aggressive subsets of mCRPC116–119. Analysis of the whole genome and transcriptome of several mCRPC tumor biopsies from the same individual demonstrated that there is a spatial and temporal intrapatient heterogeneity of the metastatic tumors harboring adenocarcinoma, neuroendocrine carcinoma, or mixed expression phenotypes120. In the subset of mCRPC tumors, there is a phenotypic switch from an androgen receptor signaling enhanced mCRPC to an androgen receptor signaling reduced small cell carcinoma after androgen deprivation therapy. Owing to few genomic aberrations in neuroendocrine prostate cancer, it has been suggested that the neuroendocrine differentiation could be largely driven by epigenetic dysregulation or signals from the TME31. Hence, epigenetic therapy to block neuroendocrine differentiation or reverse the lineage switch and restore sensitivity to androgen deprivation therapy is a promising avenue to improve anticancer therapies, and dual targeting of adenocarcinoma and neuroendocrine carcinoma phenotypes may be needed. In a more recent study, five distinct mCRPC phenotypes were identified after androgen deprivation therapy based on the expression of well-characterized androgen receptor or neuroendocrine genes, including androgen receptor-high tumors, androgen receptor-low tumors, amphicrine tumors composed of cells coexpressing androgen receptor and neuroendocrine genes, double-negative tumors, and tumors with small cell or neuroendocrine gene expression without androgen receptor activity. It was also found that a subtype of mCRPC exhibits features of squamous cell carcinoma121. Accumulated evidence also demonstrated that therapy-mediated neuroendocrine differentiation can be induced not only by androgen deprivation therapy and chemotherapy, but also by radiotherapy122. Altogether, these studies provide further evidence that tumor heterogeneity can be induced by anticancer therapies and potentially guide future therapeutic studies and clinical trial design121.
Approximately 20% of individuals with advanced prostate cancer have somatic or germline mutations in DNA damage repair regulatory genes104. In prostate cancer patients with DNA repair mutations, poly-ADP-ribose polymerase (PARP) inhibitors have demonstrated promising effectiveness. Two PARP inhibitors, olaparib and rucaparib, have recently received FDA approval to treat patients with mCRPC harboring BRCA mutations123. However, the acquired PARP inhibitor resistance phenotype is an emerging clinical problem. It is crucial to understand resistance mechanisms, which will help formulate subsequent treatment strategies. Extensive preclinical studies have identified several resistance mechanisms for PARP inhibitors124. The best-documented mechanism of resistance in prostate cancer is acquired BRCA2 reversion mutations, where previously BRCA2-deficient tumor cells can achieve BRCA2 proficiency due to constant selective pressure of PARP inhibition. Analysis of circulating cell-free DNA revealed that multiclonal heterogeneity of BRCA2 reversion mutations plays a key role in resistance to PARP inhibitors125. Other inherent or acquired mechanisms to PARP inhibitor resistance include overexpression of drug-efflux transporter genes and multidrug-resistant genes and loss of the TP53-binding protein 1 and shielding factors that reactivate homologous recombination or alter intracellular drug concentrations126. Analysis of acquired PARP inhibitor-resistant tumor cells showed that some clones present with multiple mechanisms of resistance at the same time127. Moreover, PARP inhibitors may also contribute to heterogeneity of the TME or immune environment128. Hence, resistance to PARP inhibitors is an immediate result of tumor cell heterogeneity and ongoing diversification during therapy, which induce the emergence of a new tumor phenotype. The optimal way of targeting the heterogeneous PARP inhibitor-resistant clones in combinations or sequentially, should be addressed in the future.
Immunotherapies targeted T cells using engineered T lymphocytes expressing tumor-directed chimeric antigen receptor (CAR) have been designed to treat patients with various malignancies, such as neuroblastoma, B-cell lymphoma, colorectal cancer, and hepatocellular carcinoma129–132. However, clinical studies that investigated treating prostate cancer using this technology are still disappointing. For example, the Muc1 expression has been correlated with poor prognosis and an increased risk of disease recurrence133. A study found that the anti-Muc1 CAR T cells can selectively kill prostate cancer cells which expressed Muc1. However, due to heterogeneous and fluctuated expression of the Muc1 antigen on prostate cancer cells, the immunotherapy could also induce the outgrowth of target-antigen loss variant or emergence of a new tumor phenotype, thereby leading to immune escape134. Expression of prostate-specific membrane antigen (PSMA) and prostate stem cell antigen (PSCA) correlates with clinical stage, invasion, and metastasis in prostate cancer135. Both PSMA-targeting and PSCA-targeting CAR T cells have been constructed and tested in the preclinical models. Similar to anti-Muc1 CAR T cells, intratumoral application of anti-PSMA or anti-PSCA CAR T cells kills prostate cancer cells initially, but relapse was observed owing to immune escape135.
Taken together, under therapeutic selective pressure, resistance to anticancer therapies can emerge as a result of outgrowth of preexisting resistant subclones or from the evolution of a new tumor phenotype. Cotargeting the drug-sensitive subclones together with the various drug-resistant subclones and newly emerging drug-tolerant tumor cells has been explored as a strategy to induce the most durable responses.
Challenges of stroma-targeting strategies
Most conventional anticancer therapies and immunotherapy specifically target prostate cancer cells, but the TME can promote the resistance of cancer cells to these therapies. Therefore, targeting TME is an attractive strategy for the treatment of prostate cancer.
Based on the current understanding of tumor heterogeneity, drugs targeting the TME components are under development136. However, there are still significant challenges to implement strategies targeting stroma in clinical practice.
Since the complexities of tumor stroma at different stages of prostate cancer development are largely unknown, the first issue is determining the composition of the stroma and accurately modeling tumor stroma complexity and heterogeneity in a preclinical setting137. Modeling various nontumor cell types is particularly difficult because current preclinical approaches rely primarily on the implantation of tumor cells in foreign locations, where the stromal representation may differ, or in immunocompromised hosts that lack critical immune effector cells. As a result, the inclusion of different preclinical systems and the development of advanced genetic models are necessary to unravel the components of the TME further and preclinically evaluate the efficacy of combination treatments that target multiple tumor components.
In addition, developing reliable diagnostic markers that target the tumor stroma is also challenging due to the lack of clear understanding of the determinants of responsiveness. For example, despite several attempts to find prognostic biomarkers for castration-resistant prostate cancer, there are currently no confirmed predictive biomarkers available to guide therapeutic decision-making138. Tumor heterogeneity may be the reason that biomarkers for treatments targeting the tumor stroma, in general, have remained elusive.
Furthermore, the tumor’s complexity results from ongoing interaction between tumor cells and the environment throughout the course of disease progression, which adds to the challenge of spatiotemporal heterogeneity. Since cancer patients are often treated with multiple lines of therapy, serial biopsies from various locations are necessary to predict treatment responsiveness and make therapeutic decisions.
Conclusions
Heterogeneity of the TME plays a key role in prostate cancer progression. The currently available therapies for prostate cancers, including conventional therapies and immunotherapy, could lead to therapy-induced tumor heterogeneity. Further characterization of genetic and nongenetic heterogeneity will aid in the development of more effective, personalized, and targeting-specific therapies for advanced prostate cancer patients.
Author contributions
Drs. R.G. and L.C. both conceived, wrote, and edited this paper. All the authors critically read, edited, and approved the final paper.
Data availability
Any display item and related data are available upon request.
Competing interests
The authors declare no competing interests.
Ethics approval
All tumor samples were obtained from surgical resection and fixed in formalin and embedded in paraffin. The study protocol was approved by the Ethical Committee of Indiana University and performed in accordance with relevant regulations and guidelines.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.McGranahan N, et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015;7:283ra254. doi: 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 3.Brastianos, H. C. et al. Determining the Impact of Spatial Heterogeneity on Genomic Prognostic Biomarkers for Localized Prostate Cancer. Eur. Urol. Oncol. (2020). (in press). [DOI] [PubMed]
- 4.Haffner MC, et al. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 2021;18:79–92. doi: 10.1038/s41585-020-00400-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andreoiu M, Cheng L. Multifocal prostate cancer: biologic, prognostic, and therapeutic implications. Hum. Pathol. 2010;41:781–793. doi: 10.1016/j.humpath.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Cheng L, et al. Evidence of independent origin of multiple tumors from patients with prostate cancer. J. Natl Cancer Inst. 1998;90:233–237. doi: 10.1093/jnci/90.3.233. [DOI] [PubMed] [Google Scholar]
- 7.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat. Rev. Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 8.Løvf M, et al. Multifocal Primary Prostate Cancer Exhibits High Degree of Genomic Heterogeneity. Eur. Urol. 2019;75:498–505. doi: 10.1016/j.eururo.2018.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sowalsky AG, et al. Gleason Score 7 Prostate Cancers Emerge through Branched Evolution of Clonal Gleason Pattern 3 and 4. Clin. Cancer Res. 2017;23:3823–3833. doi: 10.1158/1078-0432.CCR-16-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 2009;15:559–565. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mateo J, et al. Accelerating precision medicine in metastatic prostate cancer. Nat. Cancer. 2020;1:1041–1053. doi: 10.1038/s43018-020-00141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindberg J, et al. Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. Eur. Urol. 2013;63:347–353. doi: 10.1016/j.eururo.2012.03.050. [DOI] [PubMed] [Google Scholar]
- 14.Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat. Rev. Cancer. 2018;18:19–32. doi: 10.1038/nrc.2017.102. [DOI] [PubMed] [Google Scholar]
- 15.Cooper CS, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat. Genet. 2015;47:367–372. doi: 10.1038/ng.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boutros PC, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015;47:736–745. doi: 10.1038/ng.3315. [DOI] [PubMed] [Google Scholar]
- 17.Su F, et al. Spatial Intratumor Genomic Heterogeneity within Localized Prostate Cancer Revealed by Single-nucleus Sequencing. Eur. Urol. 2018;74:551–559. doi: 10.1016/j.eururo.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Anderson NM, Simon MC. The tumor microenvironment. Curr. Biol. 2020;30:R921–r925. doi: 10.1016/j.cub.2020.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer. 2012;12:487–493. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 21.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 22.Costa A, Scholer-Dahirel A, Mechta-Grigoriou F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014;25:23–32. doi: 10.1016/j.semcancer.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Sahai E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer. 2020;20:174–186. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 25.Lin Z, Xiang X, Lu D, Xu X. Targeting tumor microenvironment as a treatment strategy for hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2020;9:794–796. doi: 10.21037/hbsn.2020.03.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mhaidly R, Mechta-Grigoriou F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin. Immunol. 2020;48:101417. doi: 10.1016/j.smim.2020.101417. [DOI] [PubMed] [Google Scholar]
- 27.Özdemir BC, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhim AD, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014;211:1503–1523. doi: 10.1084/jem.20140692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raz Y, et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018;215:3075–3093. doi: 10.1084/jem.20180818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018;15:271–286. doi: 10.1038/nrurol.2018.22. [DOI] [PubMed] [Google Scholar]
- 32.Bellomo C, Caja L, Moustakas A. Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer. 2016;115:761–769. doi: 10.1038/bjc.2016.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kojima Y, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl Acad. Sci. USA. 2010;107:20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbazán J, Matic Vignjevic D. Cancer associated fibroblasts: is the force the path to the dark side? Curr. Opin. Cell Biol. 2019;56:71–79. doi: 10.1016/j.ceb.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 35.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat. Rev. Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 36.Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 37.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009;119:1417–1419. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat. Rev. Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 39.Quante M, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Direkze NC, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64:8492–8495. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 41.Mishra PJ, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–4339. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bayik D, Lathia JD. Cancer stem cell-immune cell crosstalk in tumour progression. Nat. Rev. Cancer. 2021;21:526–536. doi: 10.1038/s41568-021-00366-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valencia T, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26:121–135. doi: 10.1016/j.ccr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiskowski MA, et al. Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 2011;71:3459–3470. doi: 10.1158/0008-5472.CAN-10-2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ammirante M, Shalapour S, Kang Y, Jamieson CA, Karin M. Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc. Natl Acad. Sci. USA. 2014;111:14776–14781. doi: 10.1073/pnas.1416498111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kato M, et al. Heterogeneous cancer-associated fibroblast population potentiates neuroendocrine differentiation and castrate resistance in a CD105-dependent manner. Oncogene. 2019;38:716–730. doi: 10.1038/s41388-018-0461-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franco OE, et al. Altered TGF-β signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011;71:1272–1281. doi: 10.1158/0008-5472.CAN-10-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vickman RE, et al. Heterogeneity of human prostate carcinoma-associated fibroblasts implicates a role for subpopulations in myeloid cell recruitment. Prostate. 2020;80:173–185. doi: 10.1002/pros.23929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frankenstein Z, et al. Stromal reactivity differentially drives tumour cell evolution and prostate cancer progression. Nat. Ecol. Evol. 2020;4:870–884. doi: 10.1038/s41559-020-1157-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 51.Eiro N, et al. Stromal factors involved in human prostate cancer development, progression and castration resistance. J. Cancer Res. Clin. Oncol. 2017;143:351–359. doi: 10.1007/s00432-016-2284-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berglund E, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018;9:2419. doi: 10.1038/s41467-018-04724-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Marzo AM, et al. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer. 2007;7:256–269. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Josson S, Matsuoka Y, Chung LW, Zhau HE, Wang R. Tumor-stroma co-evolution in prostate cancer progression and metastasis. Semin. Cell Dev. Biol. 2010;21:26–32. doi: 10.1016/j.semcdb.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zahalka AH, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science. 2017;358:321–326. doi: 10.1126/science.aah5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kahn BM, et al. The vascular landscape of human cancer. J. Clin. Investig. 2021;131:e136655. doi: 10.1172/JCI136655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qiu GZ, et al. Reprogramming of the tumor in the hypoxic niche: the emerging concept and associated therapeutic strategies. Trends Pharm. Sci. 2017;38:669–686. doi: 10.1016/j.tips.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 61.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449–6465. [PubMed] [Google Scholar]
- 62.Singleton DC, Macann A, Wilson WR. Therapeutic targeting of the hypoxic tumour microenvironment. Nat. Rev. Clin. Oncol. 2021;18:751–772. doi: 10.1038/s41571-021-00539-4. [DOI] [PubMed] [Google Scholar]
- 63.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 64.Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin. Cancer Res. 2010;16:5928–5935. doi: 10.1158/1078-0432.CCR-10-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Assenov Y, Brocks D, Gerhäuser C. Intratumor heterogeneity in epigenetic patterns. Semin Cancer Biol. 2018;51:12–21. doi: 10.1016/j.semcancer.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 66.Stone L. Prostate cancer: A novel mechanism of neuroendocrine transdifferentiation. Nat. Rev. Urol. 2018;15:262–263. doi: 10.1038/nrurol.2018.42. [DOI] [PubMed] [Google Scholar]
- 67.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jing X, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer. 2019;18:157. doi: 10.1186/s12943-019-1089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prager BC, Xie Q, Bao S, Rich JN. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell. 2019;24:41–53. doi: 10.1016/j.stem.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mu P, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qian J, Rankin EB. Hypoxia-Induced Phenotypes that Mediate Tumor Heterogeneity. Adv. Exp. Med. Biol. 2019;1136:43–55. doi: 10.1007/978-3-030-12734-3_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer. 2008;8:180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 73.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu Q, et al. Factors involved in cancer metastasis: a better understanding to “seed and soil” hypothesis. Mol. Cancer. 2017;16:176. doi: 10.1186/s12943-017-0742-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu Z, et al. The Landscape of Immune Cells Infiltrating in Prostate Cancer. Front. Oncol. 2020;10:517637. doi: 10.3389/fonc.2020.517637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cortesi F, et al. Bimodal CD40/Fas-Dependent Crosstalk between iNKT Cells and Tumor-Associated Macrophages Impairs Prostate Cancer Progression. Cell Rep. 2018;22:3006–3020. doi: 10.1016/j.celrep.2018.02.058. [DOI] [PubMed] [Google Scholar]
- 77.Xie H, Li C, Dang Q, Chang LS, Li L. Infiltrating mast cells increase prostate cancer chemotherapy and radiotherapy resistances via modulation of p38/p53/p21 and ATM signals. Oncotarget. 2016;7:1341–1353. doi: 10.18632/oncotarget.6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ntala C, et al. Analysis of Prostate Cancer Tumor Microenvironment Identifies Reduced Stromal CD4 Effector T-cell Infiltration in Tumors with Pelvic Nodal Metastasis. Eur. Urol. Open Sci. 2021;29:19–29. doi: 10.1016/j.euros.2021.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sfanos KS, et al. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin. Cancer Res. 2008;14:3254–3261. doi: 10.1158/1078-0432.CCR-07-5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miranda A, et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl Acad. Sci. USA. 2019;116:9020–9029. doi: 10.1073/pnas.1818210116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gordon S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 82.Chen S, et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat. Cell Biol. 2021;23:87–98. doi: 10.1038/s41556-020-00613-6. [DOI] [PubMed] [Google Scholar]
- 83.Azizi E, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174:1293–1308.e1236. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Müller S, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18:234. doi: 10.1186/s13059-017-1362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016;126:3672–3679. doi: 10.1172/JCI84427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ratnam NM, et al. NF-κB regulates GDF-15 to suppress macrophage surveillance during early tumor development. J. Clin. Investig. 2017;127:3796–3809. doi: 10.1172/JCI91561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rybicki BA, et al. Growth and differentiation factor 15 and NF-κB expression in benign prostatic biopsies and risk of subsequent prostate cancer detection. Cancer Med. 2021;10:3013–3025. doi: 10.1002/cam4.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Felli E, Muttillo EM, Felli E. Interpatient heterogeneity in hepatic microvascular blood flow during vascular inflow occlusion (Pringle manoeuvre) Hepatobiliary Surg. Nutr. 2021;10:413–415. doi: 10.21037/hbsn-21-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown DA, et al. Macrophage inhibitory cytokine 1: a new prognostic marker in prostate cancer. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2009;15:6658–6664. doi: 10.1158/1078-0432.CCR-08-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vaňhara P, Hampl A, Kozubík A, Souček K. Growth/differentiation factor-15: prostate cancer suppressor or promoter? Prostate Cancer Prostatic Dis. 2012;15:320–328. doi: 10.1038/pcan.2012.6. [DOI] [PubMed] [Google Scholar]
- 91.Wang X, Baek SJ, Eling TE. The diverse roles of nonsteroidal anti-inflammatory drug activated gene (NAG-1/GDF15) in cancer. Biochem. Pharmacol. 2013;85:597–606. doi: 10.1016/j.bcp.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lambert JR, et al. Reduced expression of GDF-15 is associated with atrophic inflammatory lesions of the prostate. Prostate. 2015;75:255–265. doi: 10.1002/pros.22911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Peake BF, Eze SM, Yang L, Castellino RC, Nahta R. Growth differentiation factor 15 mediates epithelial mesenchymal transition and invasion of breast cancers through IGF-1R-FoxM1 signaling. Oncotarget. 2017;8:94393–94406. doi: 10.18632/oncotarget.21765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bruzzese F, et al. Local and systemic protumorigenic effects of cancer-associated fibroblast-derived GDF15. Cancer Res. 2014;74:3408–3417. doi: 10.1158/0008-5472.CAN-13-2259. [DOI] [PubMed] [Google Scholar]
- 95.Roth P, et al. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin. Cancer Res. 2010;16:3851–3859. doi: 10.1158/1078-0432.CCR-10-0705. [DOI] [PubMed] [Google Scholar]
- 96.Peng H, Li Z, Fu J, Zhou R. Growth and differentiation factor 15 regulates PD-L1 expression in glioblastoma. Cancer Manag. Res. 2019;11:2653–2661. doi: 10.2147/CMAR.S192095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Casazza A, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24:695–709. doi: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 98.Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat. Rev. Cancer. 2020;20:516–531. doi: 10.1038/s41568-020-0273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Calderaro J. Dual angiogenesis and PD-1 blockade in hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2020;9:350–352. doi: 10.21037/hbsn.2019.10.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer. 2016;2:758–770. doi: 10.1016/j.trecan.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 101.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74:665–674. doi: 10.1158/0008-5472.CAN-13-0992. [DOI] [PubMed] [Google Scholar]
- 103.Jayaprakash P, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018;128:5137–5149. doi: 10.1172/JCI96268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Armenia J, et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018;50:645–651. doi: 10.1038/s41588-018-0078-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mehra N, Gerritsen W. Now the dust has settled over immune checkpoint blockade in metastatic prostate cancer. Ann. Oncol. 2018;29:1620–1622. doi: 10.1093/annonc/mdy239. [DOI] [PubMed] [Google Scholar]
- 106.Abida W, et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019;5:471–478. doi: 10.1001/jamaoncol.2018.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grasso CS, et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Disco. 2018;8:730–749. doi: 10.1158/2159-8290.CD-17-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cha HR, Lee JH, Ponnazhagan S. Revisiting immunotherapy: a focus on prostate cancer. Cancer Res. 2020;80:1615–1623. doi: 10.1158/0008-5472.CAN-19-2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thoma C. Prostate cancer: Towards effective combination of ADT and immunotherapy. Nat. Rev. Urol. 2016;13:300. doi: 10.1038/nrurol.2016.80. [DOI] [PubMed] [Google Scholar]
- 110.Calagua C, et al. Expression of PD-L1 in Hormone-naïve and Treated Prostate Cancer Patients Receiving Neoadjuvant Abiraterone Acetate plus Prednisone and Leuprolide. Clin. Cancer Res. 2017;23:6812–6822. doi: 10.1158/1078-0432.CCR-17-0807. [DOI] [PubMed] [Google Scholar]
- 111.Bilusic M, Madan RA, Gulley JL. Immunotherapy of prostate cancer: facts and hopes. Clin. Cancer Res. 2017;23:6764–6770. doi: 10.1158/1078-0432.CCR-17-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pienta KJ, Hammarlund EU, Brown JS, Amend SR, Axelrod RM. Cancer recurrence and lethality are enabled by enhanced survival and reversible cell cycle arrest of polyaneuploid cells. Proc. Natl Acad. Sci USA. 2021;18:e2020838118. doi: 10.1073/pnas.2020838118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Larsen JE, et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Investig. 2016;126:3219–3235. doi: 10.1172/JCI76725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ryan CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013;368:138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Robinson D, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aparicio AM, et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016;22:1520–1530. doi: 10.1158/1078-0432.CCR-15-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Beltran H, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kumar A, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016;22:369–378. doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Priemer DS, et al. Neuroendocrine Tumors of the Prostate: Emerging Insights from Molecular Data and Updates to the 2016 World Health Organization Classification. Endocr. Pathol. 2016;27:123–135. doi: 10.1007/s12022-016-9421-z. [DOI] [PubMed] [Google Scholar]
- 120.Aggarwal RR, et al. Whole-Genome and Transcriptional Analysis of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer Demonstrates Intraclass Heterogeneity. Mol. Cancer Res. 2019;17:1235–1240. doi: 10.1158/1541-7786.MCR-18-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Labrecque MP, et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019;129:4492–4505. doi: 10.1172/JCI128212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Owens JL, et al. Targeting Protein Arginine Methyltransferase 5 Suppresses Radiation-induced Neuroendocrine Differentiation and Sensitizes Prostate Cancer Cells to Radiation. Mol. Cancer. Ther. 2022;21:448–459. doi: 10.1158/1535-7163.MCT-21-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schmidt KT, Huitema ADR, Chau CH, Figg WD. Resistance to second-generation androgen receptor antagonists in prostate cancer. Nat. Rev. Urol. 2021;18:209–226. doi: 10.1038/s41585-021-00438-4. [DOI] [PubMed] [Google Scholar]
- 124.Francica P, Rottenberg S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018;10:101. doi: 10.1186/s13073-018-0612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Quigley D, et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Disco. 2017;7:999–1005. doi: 10.1158/2159-8290.CD-17-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Noordermeer SM, van Attikum H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019;29:820–834. doi: 10.1016/j.tcb.2019.07.008. [DOI] [PubMed] [Google Scholar]
- 127.Färkkilä A, et al. Heterogeneity and Clonal Evolution of Acquired PARP Inhibitor Resistance in TP53- and BRCA1-Deficient Cells. Cancer Res. 2021;81:2774–2787. doi: 10.1158/0008-5472.CAN-20-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gillessen S, Bristow RG. The tip of the iceberg: predicting PARP inhibitor efficacy in prostate cancer. Lancet Oncol. 2020;21:17–19. doi: 10.1016/S1470-2045(19)30780-6. [DOI] [PubMed] [Google Scholar]
- 129.Pule MA, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Savoldo B, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Eso Y, Seno H. Optimization of immunotherapy for patients with hepatobiliary cancer. Hepatobiliary Surg. Nutr. 2021;10:717–719. doi: 10.21037/hbsn-21-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Okamoto K, Uetake H. Current status of treatment for colorectal liver metastases in the United Kingdom. Hepatobiliary Surg. Nutr. 2021;10:116–118. doi: 10.21037/hbsn.2020.04.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lapointe J, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl Acad. Sci. USA. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Morello A, Sadelain M, Adusumilli PS. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Disco. 2016;6:133–146. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wolf P, Alzubi J, Gratzke C, Cathomen T. The potential of CAR T cell therapy for prostate cancer. Nat. Rev. Urol. 2021;18:556–571. doi: 10.1038/s41585-021-00488-8. [DOI] [PubMed] [Google Scholar]
- 136.Karlou M, Tzelepi V, Efstathiou E. Therapeutic targeting of the prostate cancer microenvironment. Nat. Rev. Urol. 2010;7:494–509. doi: 10.1038/nrurol.2010.134. [DOI] [PubMed] [Google Scholar]
- 137.Singh M, Ferrara N. Modeling and predicting clinical efficacy for drugs targeting the tumor milieu. Nat. Biotechnol. 2012;30:648–657. doi: 10.1038/nbt.2286. [DOI] [PubMed] [Google Scholar]
- 138.Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018;15:366–381. doi: 10.1038/s41571-018-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any display item and related data are available upon request.