

Summary
One mechanism by which genetic factors influence complex traits and diseases is altering gene expression. Direct measurement of gene expression in relevant tissues is rarely tenable; however, genetically regulated gene expression (GReX) can be estimated using prediction models derived from large multi-omic datasets. These approaches have led to the discovery of many gene-trait associations, but whether models derived from predominantly European ancestry (EA) reference panels can map novel associations in ancestrally diverse populations remains unclear. We applied PrediXcan to impute GReX in 51,520 ancestrally diverse Population Architecture using Genomics and Epidemiology (PAGE) participants (35% African American, 45% Hispanic/Latino, 10% Asian, and 7% Hawaiian) across 25 key cardiometabolic traits and relevant tissues to identify 102 novel associations. We then compared associations in PAGE to those in a random subset of 50,000 White British participants from UK Biobank (UKBB50k) for height and body mass index (BMI). We identified 517 associations across 47 tissues in PAGE but not UKBB50k, demonstrating the importance of diverse samples in identifying trait-associated GReX. We observed that variants used in PrediXcan models were either more or less differentiated across continental-level populations than matched-control variants depending on the specific population reflecting sampling bias. Additionally, variants from identified genes specific to either PAGE or UKBB50k analyses were more ancestrally differentiated than those in genes detected in both analyses, underlining the value of population-specific discoveries. This suggests that while EA-derived transcriptome imputation models can identify new associations in non-EA populations, models derived from closely matched reference panels may yield further insights. Our findings call for more diversity in reference datasets of tissue-specific gene expression.
Keywords: PrediXcan, TWAS, ancestrally diverse, gene expression, cardiometabolic traits, PAGE
Graphical abstract
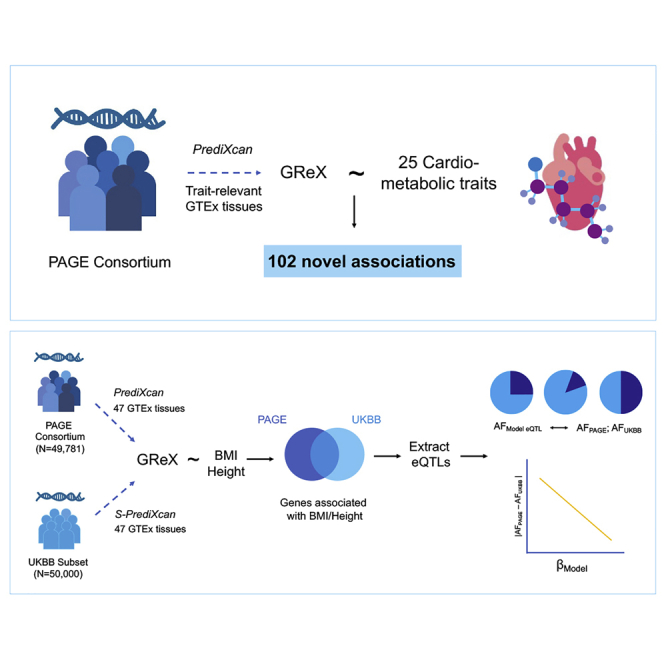
Introduction
Gene expression is tightly regulated to maintain normal biological functions. Gene expression profiles reflect a combination of cell type, developmental stage, environmental cues, and/or genetic variation, with dysregulation possibly leading to disease. Therefore, probing gene expression may yield insights into the biological mechanisms underlying a host of complex traits. However, our ability to assess gene expression is hampered by a lack of access to tissues of interest (for example, brain tissue cannot be easily obtained from healthy living people) and the high cost of assaying gene expression compared to SNP arrays. Further, the dynamic nature of gene expression means that causality may be difficult to infer from disease association studies, in which, depending on study design, differential gene expression between cases and controls may occur as a consequence of treatments or behaviors stemming from the disease being studied. Taken together, these factors mean that gene expression will likely not be directly interrogated in the appropriate tissue and cell types at the necessary scale to yield insights into disease etiology. Transcriptomic imputation (TI) offers a method to infer the genetically regulated component of tissue-specific gene expression from readily available genome-wide SNP data.
TI uses well-curated, publicly available expression quantitative trait locus (eQTL) reference panels, such as from the Genotype-Tissue Expression (GTEx) project,1 to construct tissue-specific predictions of genetically regulated gene expression (GReX). GReX can then be imputed for large, deeply phenotyped studies with genome-wide genotype data, thereby enabling association testing of broad phenotype domains. In comparison to genome-wide association studies (GWASs), TI studies offer increased biological interpretability by revealing phenotype-associated changes in expression of a particular gene in a particular tissue; by contrast, GWASs yield large regions of association, with great uncertainty about how a variant or haplotype may impact biology. TI studies may also yield insights into tissues that are traditionally difficult or impossible to access, allowing investigators to more comprehensively investigate the transcriptome. In addition, TI studies increase power to detect genetic effects by combining effects of multiple eQTLs in a region to reveal a set of candidate genes.
While TI offers great potential for insights into tissue-specific disease mechanisms, available reference panels and prediction models are overwhelmingly composed of European ancestry (EA) individuals, with application of these models largely confined to EA GWAS cohorts. GWASs have demonstrated that the practice of focusing on EA individuals hinders both analytic and translational application of findings.2 Consequently, results may not generalize to other populations, and important associations and opportunities to understand disease architecture may be missed.3, 4, 5 The few ancestrally diverse expression datasets available are limited in tissue sampling but have shown differences in gene expression by ancestral group.6 As we begin to develop tools to annotate and translate genetic associations into more biologically interpretable findings, it is important that we account for the genetic diversity present in all human populations. Here, we seek to demonstrate the feasibility of TI approaches for non-EA populations. We combine data from our large multiethnic (non-EA) GWAS7 with state-of-the art TI approaches to identify novel associated genes across 25 cardiometabolic traits (Table 1) and to demonstrate the applicability of TI approaches across populations. Our previous GWAS efforts in the Population Architecture using Genomics and Epidemiology (PAGE) study—which include genotypes from 51,520 African American, Hispanic/Latino, Asian, Native American, and Hawaiian participants—have yielded novel associations and insights into disease biology, as well as confirming > 1,400 existing GWAS associations.7 Using the PAGE summary statistics, Geoffroy et al. used S-PrediXcan in whole blood from three ancestries.5 In this paper, we seek to further these findings by employing the PrediXcan TI method.8 Although ancestry imbalance in eQTL reference data is as severe as in GWAS (for example, ∼85% of GTEx samples, 95% of eQTLGen samples, and 100% of PsychEncode samples included in eQTL analyses are EA), previous work implies that eQTLs may be more conserved across ancestries than matched, non-eQTL variants9,10 and that EA-derived PrediXcan models may have similar accuracy across populations.11 Although eQTLs are selected from EA-derived models, we determine that, for some populations, they are less ancestrally differentiated than matched SNPs on a continental level, demonstrating the need for ancestry-aware representativeness for the utility and applicability of this powerful approach for diverse populations.
Table 1.
Summary of traits and tissue-specific findings
| Trait | N (total or cases/controls) | Mean (SD) or % cases | Tissues | Novel genes | Significant genes |
|---|---|---|---|---|---|
| Inflammatory traits | |||||
| C-reactive protein (mg/L) | 28,520 | 4.1 (4.8) | EBV-transformed lymphocytes, liver, spleen, thyroid, whole blood |
4 | 25 |
| White blood cell count (109 cells/L) | 28,518 | 6.2 (1.9) | 1 | 180 | |
| MCHC (g/dL) | 19,803 | 32.9 (1.2) | 0 | 9 | |
| Platelets (103/μL) | 29,328 | 245 (64) | 2 | 28 | |
| Lipid traits | |||||
| HDL cholesterol (mg/dL)a | 33,063 | 51 (15) | adipose (subcutaneous and visceral omentum), liver, whole blood |
0 | 21 |
| LDL cholesterol (mg/dL)a | 32,221 | 138 (41) | 3 | 24 | |
| Triglycerides (mg/dL)a | 33,096 | 138 (92) | 2 | 28 | |
| Total cholesterol (mg/dL)a | 33,185 | 215 (46) | 1 | 27 | |
| Lifestyle traits | |||||
| Cigarettes/dayb | 15,862 | 12.5 (9.1) | braind, liver, whole blood |
6 | 7 |
| Coffee (cups/day) | 35,902 | 0.89 (1.13) | 3 | 3 | |
| Glycemic traits | |||||
| HbA1c (mmol/mol)c | 11,177 | 36.8 (4.5) | adipose (subcutaneous and visceral omentum), liver, skeletal muscle, pancreas, whole blood |
5 | 12 |
| Fasting insulin (pmol/L)c | 21,551 | 10.2 (8) | 5 | 5 | |
| Fasting glucose (mmol/L)c | 23,911 | 5.05 (0.63) | 2 | 8 | |
| Type 2 diabetes (cases/controls) | 14,042/31,683 | 30.70% | 3 | 18 | |
| Electrocardiogram traits | |||||
| QT interval (ms) | 17,348 | 410 (31) | adrenal gland, aorta, coronary artery, tibial artery, heart atrial appendage, heart left ventricle, skeletal muscle, whole blood |
6 | 27 |
| QRS interval (ms) | 17,046 | 89 (10) | 4 | 20 | |
| PR interval (ms) | 17,422 | 159 (22) | 17 | 24 | |
| Blood pressure traits | |||||
| Systolic blood pressure (mm Hg)a | 35,433 | 132 (22) | adrenal gland, aorta, coronary artery, tibial artery, heart atrial appendage, heart left ventricle, whole blood |
0 | 7 |
| Diastolic blood pressure (mm Hg)a | 35,433 | 81 (14) | 3 | 7 | |
| Hypertension (cases/controls) | 27,123/ 22,018 | 55.20% | 1 | 3 | |
| Anthropometric traits | |||||
| WaistWHRc, females | 24,838 | 0.86 (0.08) | adipose (subcutaneous and visceral omentum), braind, skeletal muscle, thyroid, whole blood |
2 | 4 |
| WHRc , males | 9,066 | 0.95 (0.7) | 5 | 5 | |
| Height (cm) | 49,781 | 164 (10) | 7 | 234 | |
| BMI (kg/m2) | 49,335 | 29 (6) | 12 | 29 | |
| Kidney traits | |||||
| eGFR (mL/min)e | 27,900 | 91 (22) | adrenal gland, aorta, coronary artery, tibial artery, heart atrial appendage, heart left ventricle, whole blood | 7 | 10 |
For each group of traits, tissues were selected based on biological knowledge. The number of total significant genes across all tissues and the number of novel associated genes are listed. MCHC, mean corpuscular hemoglobin concentration; WHR, waist to hip ratio.
Adjusted for medication use.
Non-smokers excluded from analysis.
Analyses including brain refer to all of the following tissues: amygdala, anterior cingulate cortex, caudate basal ganglia, cerebellar hemisphere, cerebellum, cortex, frontal cortex, hippocampus, hypothalamus, nucleus accumbens basal ganglia, putamen basal ganglia, spinal cord cervical c-1, substantia nigra.
Adjusted for BMI.
Estimated glomerular filtration rate (eGFR) was calculated using the CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) formula from Levey et al.12
Subjects and methods
Demographics of the PAGE sample
The PAGE study includes 51,520 participants genotyped at the Center for Inherited Disease Research (CIDR) using the Multi-Ethnic Genotyping Array (MEGA), consortium version.7 These participants come from four ancestrally diverse studies (Table S1): BioMe Biobank, the Hispanic Community Health Study/Study of Latinos (HCHS/SOL), the Multi Ethnic Cohort (MEC), and the Women’s Health Initiative (WHI), representing 35% African American, 9% Asian, 44% Hispanic/Latino, 1% Native American, 8% Native Hawaiian, and 2% other ancestries. All participants in these studies provided written informed consent to participate in genetic research, and study approvals were obtained from the institutional review boards at all participating institutions in accordance with the principals of the Declaration of Helsinki.
Imputation of gene expression in PAGE genotype samples
We imputed GReX in 51,520 individuals from the PAGE consortium, using 47 GTEx v7-derived tissue-specific prediction models (listed in Table S2). GReX was calculated as a weighted sum of SNP dosages, following standard PrediXcan methodology. GReX prediction model creation is described in detail elsewhere; notably, these models are based on the EA subset of GTEx participants.8,13 In total, our analysis included 25,861 unique genes across 47 tissues. The number of genes included in each tissue model and the number of GTEx samples used to generate the model are shown in Table S2.
Selection of trait-relevant tissues
For each of the 25 traits, tissues were selected based on etiological relevance (Table 1). For example, heart tissues were selected as relevant for electrocardiogram (EKG) phenotypes and adipose, brain, skeletal muscle, and thyroid were selected for anthropometric phenotypes. Additionally, we included whole blood for every trait because it is present throughout the body.
Association testing of gene dosages with 25 traits
We tested for association between tissue-specific GReX from trait-relevant tissues and each of our 25 outcomes—including anthropometric, inflammation, kidney function, EKG phenotypes, hypertension, lipids, glucose control, and caffeine and cigarette use (detailed in Table 1)—using generalized estimating equation (GEE) models to account for relatedness as implemented in SUGEN.14 Models were adjusted for age, sex, study center, ten principal components (PCs), and self-reported race/ethnicity. Trait harmonization, exclusion criteria, and covariates were detailed previously and are outlined in Table S3.7 An association was defined as novel if there were no known GWAS or TI (i.e., PrediXcan or TWAS) associations for that trait within one megabase (MB) of the gene as of August 5, 2019. By this definition, any gene within 1 MB of loci with significant associations in our previous work with this dataset were considered known.7
Correcting for multiple testing
Our analyses included a large number of genes, tissues, and traits. To avoid introducing spurious results, we applied multiple testing corrections. Given the high degree of eQTL sharing among tissues and of correlation in gene expression both within and across tissues, these tests cannot be considered independent, and therefore a Bonferroni correction would be overly conservative. We applied an experiment-wide false discovery rate (FDR) (all trait-tissue-gene sets) to ascertain significance using the Benjamini-Hochberg method.15 We considered tissue-specific GReX associated with a trait with an FDR < 0.05 as significant. This was equivalent to a p value of 5.02 × 10−5.
Principal-component analysis
Kinship coefficients were estimated using PC-Relate, as implemented in the R package GENESIS,16,17 and used to select unrelated individuals for accurate estimation of the PCs using SNPRelate,18 implemented in R. As was done previously,7 the relevant PCs were selected using scree plots to assess the spread of genetic ancestry within self-identified racial/ethnic clusters. A parallel coordinate plot for the first ten PCs was generated in which each PAGE individual was represented by a set of line segments connecting their PC values. The variance explained diminished with each subsequent PC, and we estimated that the top ten PCs provided sufficient information to explain the majority of genetic variation in the PAGE study population.7
eQTL by genetic ancestry
We investigated the impact of ancestral differentiation on the generalizability of effect sizes, and therefore predicted gene expression, across populations. First, we used 1:1 comparison group propensity score matching between eQTLs and matched non-eQTL sites, implemented in the “MatchIt” R package.19 A total of 23,901 polymorphic eQTLs with minor allele frequency (MAF) > 0.5% were subset from the PAGE genotype data (133 variants excluded for low frequency [>0.5%], with four being monomorphic within PAGE). For each eQTL, a matched “control” SNP was selected using a nearest-neighbor estimation based on GC percentage, distance to transcription start site (TSS), and combined allele frequency within PAGE to address possible biases. After matching, there were no differences in distance to TSS, GC percentage, or combined allele frequency (p > 0.05) between eQTL and matched controls.
After matched controls were selected, we assessed the differentiation of SNPs between continental-level genetic ancestries in PAGE by estimating the association between variant genotypes and the PCs estimated from the principal-component analysis detailed above. This univariate regression of genotypes on PCs was conducted in SUGEN. Statistically significant differentiation was determined if the resulting association had a p value < 1.07E−5 to account for multiple comparisons. Analyses were restricted to the first five PCs to ensure interpretability at a continental level for genetic ancestry. We contrasted the proportion of variants that exhibited statistically significant differentiation within the eQTLs versus the matched controls for each of these five PCs separately through logistic regression in R. We also estimated the association between the strength of differentiation (the beta from the SUGEN regression model of PCs on genotypes) with eQTL or matched-control status. Regression models contrasting eQTLs and matched controls were adjusted for GC percentage, distance to TSS, and combined allele frequency in a pooled PAGE sample to account for possible residual confounding. A sensitivity analysis was also conducted restricting variants to MAF >5% (21,682 eQTL variants and 21,682 matched-control variants). While there was no significant difference between groups in the proportion of statistically significant associations, the strength of effects as determined by the beta from the PC x genotype analysis exhibited consistent results.
Allele frequencies comparing the eQTLs and their matched controls were estimated within the 1000 Genomes Project Phase 320 data at the level of “superpopulations” (Africa, Europe, and East Asia) after removing first-degree-related individuals. The reference allele for all superpopulations was set to the minor allele (MAF <50%) in European populations to directly compare allele frequencies assuming a European discovery population.
Comparing GReX associations between PAGE and UK Biobank
To provide context and interpretability of our results, it was important to compare our PAGE PrediXcan association results to those from an equivalently sized homogeneous European population. We thus selected a random sample of 50,000 individuals of self-reported White British EA from the UK Biobank (UKBB50k). The full biobank includes ∼500,000 individuals of largely British EA origin, with self-defined information on nearly 4,000 traits. For these 50,000 UKBB50k participants, we performed GWAS of height and BMI.7 Briefly, to compare these data to our PrediXcan associations, we used S-PrediXcan13 to convert the UKBB50k GWAS summary statistics to GReX summary statistics for all 47 GTEx v7 tissues. S-PrediXcan is exactly analogous to PrediXcan,13 when linkage disequilibrium (LD) is correctly specified and may be applied directly to summary statistics rather than raw genotypes. In Europeans or populations closely matched to the PrediXcan reference panel used, we expect >98% concordance between directly computed PrediXcan association statistics and S-PrediXcan association statistics.11,13 These analyses were repeated for the full 451,305 EA individuals in UK Biobank (UKBB450k), as determined by k-means clustering. We created sex-specific residuals after regressing out age, center, genotype chip, and age-squared for BMI. Then the sex-specific residuals were inverse normalized. PCs 1–15 were adjusted for running genome-wide association analyses in Bolt-LMM.21 Results were cleaned by removing variants with info scores < 0.4, MAF < 0.001, or effective N < 30.
Allele frequency differences contributing to differences in UK Biobank and PAGE results
We compared the subset of genes that were associated with height and BMI in PAGE, UKBB50k, or both. We then extracted all eQTLs for those genes in the relevant tissues. To avoid redundancies of eQTLs between tissues, analyses were performed in parallel across tissues and in whole blood alone. Ancestry-specific allele frequencies were estimated for these sites in PAGE (as determined by self-identified race/ethnicity), and EA frequencies (as determined by superpopulation designations from 1000 Genomes) were estimated using 1000 Genomes Project Phase 3 data.20 For each gene identified in either or both of the PAGE and UKBB50k analyses, we compared allele frequencies of all SNPs in each gene prediction model. Additionally, we estimated the association between the magnitude of eQTL weights and the absolute value of allele frequency differences between populations to determine whether eQTLs with more population differentiation have different effect weights than variants with similar frequencies across populations using Pearson’s correlation.
Results
Identification of gene expression-trait associations
Across the 25 outcomes encompassing anthropometric, inflammation, kidney function, EKG phenotypes, hypertension, lipids, glucose control, and caffeine and cigarette use (detailed in Table 1), we observed a total of 1,113 tissue-specific GReX associated with a trait at FDR < 0.05 (Table 1; Tables S4 and Table S5). We observed a total of 102 novel gene-trait associations, each occurring across 1–6 trait-relevant tissues (Table S4). We identified novel genes even for traits that have previously been the focus of large-scale meta-analyses, including 41% of gene-tissue associations for BMI. In Box 1, we further explore novel findings with biologically compelling candidate genes.
Box 1. Biological relevance of select associations.
Expression of WDFY2 (WD repeat and FYVE domain containing 2) was associated with BMI in visceral adipose tissue. WDFY2 positively regulates adipocyte differentiation by facilitating the phosphorylation and thus inactivation of the anti-adipogenic transcription factor Foxo1.22 WDFY2 also plays a role in endosomal control of AKT2 signaling, through interaction with insulin receptors (INSRs) in hepatocytes,23 and is required for insulin-stimulated AKT2 phosphorylation and glucose uptake and insulin-stimulated phosphorylation of AKT2 substrates.22 SCN11A (sodium voltage-gated channel alpha subunit 11) was associated with serum triglyceride levels in subcutaneous and visceral adipose tissue. SCN11A mediates BDNF (brain-derived neurotrophic factor) expression in the brain,24 which has been associated with obesity and food intake.25,26
Several TI signals were associated with EKG traits in relevant tissues. TMEM87B expression in adrenal gland tissue was associated with PR interval duration. TMEM87B and its close homolog, TMEM87A, encode transmembrane proteins likely involved in the complex regulation of the retrograde transport of membrane proteins from endosomes to the trans-Golgi network (TGN),27 which is centrally important across multiple cellular processes, including developmental signaling as well as transport of cytoskeletal ions and glucose.28 TMEM87B deficiency has been associated with congenital heart defects,29,30 accounting for some of the adverse cardiac phenotypes observed in recurrent 2q13 deletion,30 including restrictive cardiomyopathy and atrial septal defect.29
Expression of PCCB was associated with QT interval duration in heart atrial appendage, whole blood, and tibial artery tissue. PCCB encodes a subunit of propionyl-CoA carboxylase (PCC), an enzyme involved in fatty acid metabolism.31 PCC deficiency was previously associated with long QT syndrome,32, 33, 34, 35 and these results build upon a growing body of evidence detailing the importance of long-chain fatty acid metabolism in cardiomyocyte homeostasis, with possible implications for lipid-altering treatments and arrhythmogenesis. Of note, the beneficial effect of statins on life-threatening ventricular arrhythmias36 may be mediated by their increase of high-density lipoprotein (HDL);37,38 in contrast, the statin probucol, notable for its HDL-lowering effects, was withdrawn from the U.S. market in 1995 due to its prolongation of QT interval.39
EYA4 expression in artery aorta tissues was associated with QT interval duration. EYA4 encodes a member of the eyes absent protein family with an important role in organogenesis, likely through its function as a histone phosphatase. EYA4 mutations have been linked with ventricular size in zebrafish models, as well as withs mild abnormalities in cardiac morphology associated with late-onset congestive heart failure40 and arrhythmia41 in humans,40,42,43 highlighting the potential involvement of EYA4 in normal cardiac function as well as development, possibly through the implication of the p27/casein kinase-2α/histone deacetylase 2 transcriptional cascade.42
Consistency of TI findings in a European panel
To compare our results in diverse PAGE participants to those expected from an equivalent sample size of EA participants, we selected two traits (height and BMI) well represented in both PAGE and the UKBB50k and compared Z scores between the two analyses (height, Figure S1A; BMI, Figure S1B). We observed a highly significant (p < 1.43 × 10−6) correlation of Z scores in each tissue between the PAGE and UKBB50k studies overall, with r2BMI ≈ 0.15 and r2height ≈ 0.3 (Table S6). These tissue-specific correlations were strengthened when using the UKBB450k results.
Within any given tissue there was substantial overlap in associated genes identified in PAGE and UKBB50k (Table S7, Figure 1). Restricting to whole blood, we identified 93 genes significantly associated (pUKBB_height < 5.02 × 10−6) with height in UKBB50k GReX and found significantly higher replication of these genes in PAGE GReX than might be expected by chance (10 of 93 genes are significant [pPAGE_height < 5.02 × 10−6]: binomial p = 1.1 × 10−30; 43/93 are nominally significant [pPAGE_height < 0.05]: binomial p = 5.9 × 10−31). We see a similar enrichment of significant BMI associations across studies (2 of 19 genes are significant [binomial p = 4.6 × 10−7]; 10 of 19 are nominally significant [p = 5.9 × 10−9]).
Figure 1.

Overlap of tissue-specific associations between PAGE and UKBB
All genes significantly associated (p < 5.02 × 10−5) with BMI (A) or height (B) in PAGE or UKBB50k are displayed in the stacked bar chart. Associations significant only in PAGE are shown in red. Associations significant only in UKBB50k are blue. Associations significant in PAGE and UKBB50k are in bright purple. Associations seen in PAGE and UKBB450k are shown in dark purple.
Conversely, we identified 27 genes significantly associated in whole blood with height in PAGE, ten of which were statistically significant in UKBB50k with ten more nominally significant in UKBB50k. Four of these gene associations that are only nominally significant in UKBB50k appear to be driven primarily by strong associations within the Hispanic ancestry-specific analysis that would be underpowered in the EA-specific UKBB50k analysis (Table S8). For BMI, which is notably less polygenic than height, we identified four statistically significant genes associated in PAGE, only two of which were also significant in UKBB50k. For one of these genes, HPS5, the association was driven by Hispanic ancestry (Table S9).
eQTL differentiation reflects sampling bias
In light of these findings, we were interested in the applicability of eQTLs underlying the EA-derived models in a diverse ancestry population. To explore this, we assessed levels of population differentiation both between groups within PAGE and in contrast with a well-characterized reference dataset, the 1000 Genomes Project, specifically comparing levels of differentiation among eQTLs to those among matched controls. A total of 23,901 eQTLs and their matched-control variants (total of 47,801 variants; see material and methods) were included in this analysis with a MAF of 0.5% (see material and methods). Each variant was assessed for association with genetic ancestry at a continental level as represented by the first five PCs estimated in a principal-component analysis. Variants were determined to have statistically significant association with genetic ancestry if the association between genotype and a specific PC had a p value below 1.05 × 10−7 to account for multiple comparisons. For PC1, which represents African versus non-African ancestry, control variants were significantly more differentiated than eQTLs in terms of proportion of sites that had a significant p value for SNP x PC (t test p = 9.6 × 10−10) (Table S10). However, later PCs representing other genetic ancestries showed more differentiation for eQTLs than their matched controls.
This is at least partially explained when examining the allele frequencies for eQTLs versus their controls across continental-level populations from the 1000 Genomes Project. In European populations, the mean MAF was higher in eQTLs (22.57%) than in matched-control variants (18.66%) (Table S11). Maintaining the “minor” allele from European populations as the index allele, the average allele frequencies were higher for both eQTLs and their matched-control variants in both African (27.88% and 26.10%, respectively) and East Asian populations (25.27% and 21.83%, respectively). However, the averages do not give us a complete picture. When examining the distribution of allele frequency differences between African and European populations, as well as East Asian and European populations, more than half of the eQTL variants have a higher frequency in European versus East Asian populations (52.17%). This is not seen in the matched-control variants, which are more likely to be at a higher frequency in East Asian than in European populations, nor for either eQTLs or matched controls comparing African to European populations.
Taken together, these trends reflect the original sampling bias, as the eQTLs were originally identified in EA participants and therefore required a MAF within that specific population high enough to be detected given a fixed sample size. Conditioning on these allele frequencies, variants tend to be found at higher frequencies in African populations, therefore constraining population differentiation compared to matched controls. These constraints have the opposite effect for non-African ancestries compared to European ancestries due to underlying population genetics and associated allele frequencies. These trends suggest that the transferability of weights trained on eQTLs discovered in one population may depend on how the underlying genetic architecture differs between continental-level population groups with respect to the training dataset.
Genetic differentiation of eQTLs
To better understand the role of genetic differentiation for model eQTLs in application, we compared the allele frequencies of eQTLs for genes significant for height or BMI (p < 5.02 × 10−5) found in just the PAGE analysis, in just the UKBB50k analysis, or in both. For this analysis, PAGE allele frequencies were estimated pooled across the entire study, as well as stratified by self-identified race/ethnicity. These frequencies were further compared to allele frequencies in 1000 Genomes Project Phase 3 European population data. We observed that height and BMI showed different trends in the frequencies of alleles contributing to increased expression (positive eQTL weight). For BMI, the highest frequencies for both European population reference data and PAGE combined samples were found in genes identified only in PAGE (Table 2). For height, the highest frequencies for both of these datasets were found in genes identified in both PAGE and UKBB50k. This may reflect different genetic architectures by ancestry, which would contribute to the lower number of genes identified for BMI relative to height. These trends were amplified when looking only at whole blood instead of all trait-relevant tissues combined to minimize the redundancy of eQTLs across tissues (Table 3).
Table 2.
Mean frequencies of eQTL variants predicted to increase gene expression identified in PAGE, UKBB50k, or both for BMI and height in 1000 Genomes European and PAGE populations for all tissues
|
BMI (N = 207) |
Height (N = 1,112) |
||||||
|---|---|---|---|---|---|---|---|
| PAGE | Both | UKBB50k | PAGE | Both | UKBB50k | ||
| # Genes | 54 | 6 | 156 | 372 | 133 | 769 | |
| # SNPs | 2,119 | 286 | 13,303 | 21,431 | 11,202 | 83,019 | |
| 1000 Genomes European | 51.47% | 51.05% | 47.80% | 48.96% | 50.10% | 51.25% | |
| PAGE | Combined | 51.98% | 50.54% | 49.28% | 48.95% | 49.66% | 51.63% |
| African American | 52.06% | 47.82% | 49.24% | 49.03% | 49.60% | 51.48% | |
| Hispanic/Latino | 51.74% | 51.83% | 48.86% | 48.65% | 49.57% | 51.50% | |
| Asian American | 53.37% | 52.80% | 50.88% | 49.75% | 50.19% | 52.52% | |
| Native Hawaiian | 51.10% | 52.35% | 49.86% | 49.37% | 49.80% | 51.91% | |
| Native American | 51.61% | 51.14% | 48.26% | 48.76% | 49.67% | 51.38% | |
Mean allele frequencies of weighted variants oriented to a positive weight, i.e., frequency of allele predicted to increase expression of gene for all tissues (N = 48). Some SNPs may be found in multiple genes, and therefore observations are not independent.
Table 3.
Mean frequencies of eQTL variants predicted to increase gene expression identified in PAGE, UKBB50k, or both for BMI and height in 1000 Genomes European and PAGE populations in whole blood
|
BMI (N = 21) |
Height (N = 110) |
||||||
|---|---|---|---|---|---|---|---|
| PAGE | Both | UKBB50k | PAGE | Both | UKBB50k | ||
| # Genes | 2 | 2 | 17 | 17 | 10 | 83 | |
| # SNPs | 30 | 62 | 354 | 525 | 215 | 2,474 | |
| 1000 Genomes European | 60.85% | 45.81% | 48.33% | 42.47% | 53.02% | 50.59% | |
| PAGE | Combined | 58.18% | 53.88% | 50.03% | 44.99% | 51.19% | 51.56% |
| African American | 55.94% | 65.90% | 49.25% | 48.61% | 53.68% | 51.46% | |
| Hispanic/Latino | 58.84% | 45.99% | 50.48% | 42.73% | 51.93% | 51.38% | |
| Asian American | 59.37% | 49.79% | 51.57% | 44.06% | 41.53% | 52.70% | |
| Native Hawaiian | 62.24% | 52.80% | 49.05% | 43.74% | 47.62% | 51.67% | |
| Native American | 59.95% | 46.46% | 49.64% | 42.41% | 51.96% | 50.92% | |
Mean allele frequencies of weighted variants oriented to a positive weight, i.e., frequency of allele predicted to increase expression of gene in whole blood. Some SNPs may be found in multiple genes, and therefore observations are not independent.
Using only height in whole blood, we also compared the correlation between absolute allele frequency differences between 1000 Genomes European populations or the combined PAGE sample with the magnitude of the eQTL weight. Genes that were found to be significant in both analyses (UKBB50k and PAGE) had no relationship between increased weights and a significant difference in SNP frequencies (r = 0.066, p = 0.34). However, genes found in just the UKBB50k or PAGE analyses showed negative correlation at r = −0.075 (p = 1.88 × 10−4) and −0.111 (p = 0.011), respectively. This indicates that as the weights become stronger for these analyses, the allele frequencies between 1000 Genomes European populations and PAGE are less diverged. This relationship may explain partly why these genes were found in only one analysis versus both and confirm our findings that eQTLs can be less differentiated than the set of matched-control variants (p < 0.001) depending on the continent-level ancestry.
When looking at individual PCs, eQTLs for both traits in whole blood were significantly differentiated across the first five PCs (SNP x PC p value < 10−5) (Table S12). However, the degree of differentiation was PC specific and therefore population specific as well as trait specific. For example, when comparing African to non-African ancestry (PC1) among height-associated eQTLs, the highest proportion that were significantly differentiated were among PAGE-specific findings (94.72% with p < 10−5), followed by shared eQTLs between PAGE and UKBB50k (94.18%), and lastly UKBB50k-specific findings (93.86%). This is reflective of better statistical power for these genes due to the shared African ancestry in a large proportion of PAGE samples. However, when looking at PC2, which captures East Asian versus non-East Asian ancestry, there were slightly lower levels of differentiation, potentially reflecting the relatively smaller sample size of Asian participants compared to Hispanic/Latino and African American participants. For these variants, approximately 88.29% were differentiated in the shared loci, with comparable levels of differentiation among cohort-specific findings (88.29% in PAGE, 88.35% in UKBB50k). These trends are consistent when looking across all tissues (Table S13). Taken together, these results indicate the importance of diverse genetic ancestry for the identification of novel genes underlying traits, even when using models derived from EA data, but careful consideration must be taken with respect to how the sample populations’ genetic ancestry compares to the that of the training data.
Discussion
The advent of large-scale GWASs has yielded substantial insights into the genetic architecture of complex traits. Although these studies provide lists of genome-wide significant loci, these associations do not directly translate to biological mechanisms. Here, we combine GWAS summary statistics with large, well-curated eQTL reference panels through TI approaches (PrediXcan) to identify gene-level associations with directional and tissue specificity.8,44,45 In this study, we demonstrated that eQTL and TI methods are appropriate for non-EA analyses and identified novel trait-GReX associations. Our analysis leverages data from the PAGE study, including 25 clinical and behavioral traits in 51,520 non-EA individuals. To our knowledge, this is the largest trans-ancestry TI analysis performed to date.
Here, we apply TI approaches to calculate GReX in individual-level data. Our analysis of eQTL conservation across ancestries, coupled with previous evidence, suggests that these approaches may accurately be applied to non-EA populations when researchers have access to individual-level genotype data.11 Access to these individual genotypes allows researchers to estimate LD directly, rather than relying on an existing, external LD matrix.13 Specification of an appropriate ancestry-matched LD matrix is vital for summary-statistic analyses; applying publicly available LD matrices derived from European samples may lead to spurious results and further diminish the power TI in understudied populations, as we and others have previously demonstrated.5,13
We identified 1,113 gene-tissue associations, of which 102 are novel. Based on our analyses leveraging UK Biobank data, we show that many of these gene-tissue associations would not have been identified in an EA GWAS of similar size; indeed, even an analysis including all European UK Biobank (UKBB450k) samples would not identify the majority of our novel genes (Table S7; Figure 1). Our findings provide novel insights into disease architecture (Figure S2) as well as disease biology (Box 1). By comparing our findings in ∼50,000 ancestral diverse PAGE participants to 50,000 EA UKBB participants, we demonstrated that a substantial proportion of signals found in PAGE (65%) are not significantly associated in the UKBB50k dataset (Table S7). This may in part be due to the strong association of eQTLs with African ancestry (Table S12), which is only present within the PAGE sample, along with the greater phenotypic variation for these traits across ancestrally diverse samples. The larger number of associations seen in 50,000 EA samples highlights the power of applying expression models derived from the same ancestry. The vast majority of eQTL reference panels are derived from EA samples; as such, TI approaches may have reduced accuracy or applicability in non-EA samples. However, previous work has shown that eQTLs may be less differentiated by ancestry than matched, non-eQTL variants.9,10 Although we and others have demonstrated that TI prediction models have cross-ancestry applicability,11 it is likely that access to eQTL reference panels derived from matched populations will substantially improve prediction accuracy46, 47, 48, 49, 50, 51 and that release of non-EA-derived LD matrices will radically improve the applicability of existing TI prediction models. Moreover, there is an ethical imperative to improve diversity in functional genomics and transcriptomics studies; opportunities to participate in groundbreaking research approaches should be equally available regardless of race or ethnicity, and the scientific insights obtained should be applicable and accessible to all.
To date, only small non-EA eQTL reference panels are available in a few tissues, with cell-type proportions varying by individual sample and with insufficient power to derive powerful prediction models. This highlights the need to invest in ascertaining gene expression data in multi-tissue, ancestrally diverse datasets. However, we urge that as larger samples become available, ancestry-specific prediction models be derived and their relative accuracy assessed. In addition, we emphasize the need to ensure cross-ancestry applicability of methodological and translational advances; indeed, demonstration of cross-ancestry applicability should become standard for developers of new analytical tools and software designed to tackle complex trait genetics. Increasing sample sizes and assembling new cohorts for GWAS is vital to increasing diversity; however, this is not sufficient. To achieve equity in precision medicine and to ensure that biological insights into disease do not continue to exclude entire populations, deliberate efforts must be made to also diversify translational and methodological approaches. This includes expanding the ancestral diversity of functional genomic resources (for example, tissue-specific gene expression and eQTL reference data) so that new insights available from functional genomics do not compound racial and ethnic inequity in genetic research.
Acknowledgments
The Population Architecture using Genomics and Epidemiology (PAGE) program is funded by the National Human Genome Research Institute (NHGRI) with co-funding from the National Institute on Minority Health and Health Disparities (NIMHD), supported by U01HG007416 (CALiCo), U01HG007417 (ISMMS), U01HG007397 (MEC), U01HG007376 (WHI), and U01HG007419 (Coordinating Center). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The PAGE consortium thanks the staff and participants of all PAGE studies for their important contributions. The complete list of PAGE members can be found in the web resources.
Data management, integration, and dissemination; genotype imputation; ancestry deconvolution; population genetics; analysis pipelines; and study coordination were provided by the PAGE Coordinating Center (under NHGRI grants U01HG007419 and U01HG004801).
Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the NIH to The Johns Hopkins University, contract number HHSN268201200008I. Genotype data quality control and quality assurance services were provided by the Genetic Analysis Center in the Biostatistics Department of the University of Washington, through support provided by the CIDR contract.
H.M.H. received support from NHLBI training grants T32 HL007055 and T32 HL129982, American Diabetes Association Grant #1-19-PDF-045, and R01HL142825. C.J.H. received support from NHLBI training grants T32 HL129982 and T32 HL007824. C.R.G. received support from R56HG010297 and R01HG010297. A.E.J. was supported by K99 HL130580. C.K. received support from 75N92021D00001, 75N92021D00002, 75N92021D00003, 75N92021D00004, 75N92021D00005, S10OD028685, and U01HG007376. K.E.N. is supported by R01HL142302, R01HL151152, R01DK122503, R01HD057194, R01HG010297, and R01HL143885. K.L.Y. received support from R01HL149683 and R21HL140419. L.M.H. received support from R01MH118278.
Declaration of interests
E.E.K. has received speaker honoraria from Illumina, 23andMe, and Regeneron Pharmaceuticals and serves as a scientific board member for Galateo Bio.
Published: March 8, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.02.013.
Contributor Information
Heather M. Highland, Email: heather.highland@unc.edu.
Laura M. Huckins, Email: laura.huckins@mssm.edu.
Data and code availability
Summary statistics for PrediXcan and S-PrediXcan analyses are deposited in Zenodo: https://doi.org/10.5281/zenodo.5980229. All PAGE GWAS summary statistics are available through the GWAS Catalog.
Web resources
MatchIt R package, https://cran.r-project.org/package=MatchIt
PAGE members (complete list), http://www.pagestudy.org
PrediXcan models, https://predictdb.org/post/2017/11/29/gtex-v7-expression-models/
Supplemental information
References
- 1.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Popejoy A.B., Ritter D.I., Crooks K., Currey E., Fullerton S.M., Hindorff L.A., Koenig B., Ramos E.M., Sorokin E.P., Wand H., et al. Clinical Genome Resource (ClinGen) Ancestry and Diversity Working Group (ADWG) The clinical imperative for inclusivity: Race, ethnicity, and ancestry (REA) in genomics. Hum. Mutat. 2018;39:1713–1720. doi: 10.1002/humu.23644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mikhaylova A.V., Thornton T.A. Accuracy of gene expression prediction from genotype data with predixcan varies across and within continental populations. Front. Genet. 2019;10:261. doi: 10.3389/fgene.2019.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fryett J.J., Morris A.P., Cordell H.J. Investigation of prediction accuracy and the impact of sample size, ancestry, and tissue in transcriptome-wide association studies. Genet. Epidemiol. 2020;44:425–441. doi: 10.1002/gepi.22290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geoffroy E., Gregga I., Wheeler H.E. Population-Matched Transcriptome Prediction Increases TWAS Discovery and Replication Rate. iScience. 2020;23:101850. doi: 10.1016/j.isci.2020.101850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mogil L.S., Andaleon A., Badalamenti A., Dickinson S.P., Guo X., Rotter J.I., Johnson W.C., Im H.K., Liu Y., Wheeler H.E. Genetic architecture of gene expression traits across diverse populations. PLoS Genet. 2018;14:e1007586. doi: 10.1371/journal.pgen.1007586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wojcik G.L., Graff M., Nishimura K.K., Tao R., Haessler J., Gignoux C.R., Highland H.M., Patel Y.M., Sorokin E.P., Avery C.L., et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–518. doi: 10.1038/s41586-019-1310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gamazon E.R., Wheeler H.E., Shah K.P., Mozaffari S.V., Aquino-Michaels K., Carroll R.J., Eyler A.E., Denny J.C., Nicolae D.L., Cox N.J., Im H.K., GTEx Consortium A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amariuta T., Ishigaki K., Sugishita H., Ohta T., Koido M., Dey K.K., Matsuda K., Murakami Y., Price A.L., Kawakami E., et al. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat. Genet. 2020;52:1346–1354. doi: 10.1038/s41588-020-00740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang Y., Pividori M., Manichaikul A., Palmer A.A., Cox N.J., Wheeler H.E., Im H.K. Polygenic transcriptome risk scores (PTRS) can improve portability of polygenic risk scores across ancestries. Genome Biol. 2022;23:23. doi: 10.1186/s13059-021-02591-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huckins L.M., Dobbyn A., Ruderfer D.M., Hoffman G., Wang W., Pardiñas A.F., Rajagopal V.M., Als T.D.T., T Nguyen H., Girdhar K., et al. CommonMind Consortium. Schizophrenia Working Group of the Psychiatric Genomics Consortium. iPSYCH-GEMS Schizophrenia Working Group Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nat. Genet. 2019;51:659–674. doi: 10.1038/s41588-019-0364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levey A.S., Stevens L.A., Schmid C.H., Zhang Y.L., Castro A.F., Feldman H.I., Kusek J.W., Eggers P., Van Lente F., Greene T., et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbeira A.N., Dickinson S.P., Bonazzola R., Zheng J., Wheeler H.E., Torres J.M., Torstenson E.S., Shah K.P., Garcia T., Edwards T.L., et al. GTEx Consortium Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018;9:1825. doi: 10.1038/s41467-018-03621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin D.-Y., Tao R., Kalsbeek W.D., Zeng D., Gonzalez F., 2nd, Fernández-Rhodes L., Graff M., Koch G.G., North K.E., Heiss G. Genetic association analysis under complex survey sampling: the Hispanic Community Health Study/Study of Latinos. Am. J. Hum. Genet. 2014;95:675–688. doi: 10.1016/j.ajhg.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J.R. Stat. Soc. Ser. B Methodol. 1995;57:289–300. [Google Scholar]
- 16.Gogarten S.M., Sofer T., Chen H., Yu C., Brody J.A., Thornton T.A., Rice K.M., Conomos M.P. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics. 2019;35:5346–5348. doi: 10.1093/bioinformatics/btz567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aschard H., Vilhjálmsson B.J., Greliche N., Morange P.-E., Trégouët D.-A., Kraft P. Maximizing the power of principal-component analysis of correlated phenotypes in genome-wide association studies. Am. J. Hum. Genet. 2014;94:662–676. doi: 10.1016/j.ajhg.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng X., Levine D., Shen J., Gogarten S.M., Laurie C., Weir B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho D.E., Imai K., King G., Stuart E.A. Matching as Nonparametric Preprocessing for Reducing Model Dependence in Parametric Causal Inference. Polit. Anal. 2006;15:199–236. [Google Scholar]
- 20.Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R., 1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loh P.-R., Kichaev G., Gazal S., Schoech A.P., Price A.L. Mixed model association for biobank-scale data sets. Nat. Genet. 2018;50:906–908. doi: 10.1038/s41588-018-0144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fritzius T., Moelling K. Akt- and Foxo1-interacting WD-repeat-FYVE protein promotes adipogenesis. EMBO J. 2008;27:1399–1410. doi: 10.1038/emboj.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L., Li X., Zhang N., Yang X., Hou T., Fu W., Yuan F., Wang L., Wen H., Tian Y., et al. WDFY2 potentiates hepatic insulin sensitivity and controls endosomal localization of the insulin receptor and IRS1/2. Diabetes. 2020;69:1887–1902. doi: 10.2337/db19-0699. [DOI] [PubMed] [Google Scholar]
- 24.Blum R., Kafitz K.W., Konnerth A. Neurotrophin-evoked depolarization requires the sodium channel Na(V)1.9. Nature. 2002;419:687–693. doi: 10.1038/nature01085. [DOI] [PubMed] [Google Scholar]
- 25.Motamedi S., Karimi I., Jafari F. The interrelationship of metabolic syndrome and neurodegenerative diseases with focus on brain-derived neurotrophic factor (BDNF): Kill two birds with one stone. Metab. Brain Dis. 2017;32:651–665. doi: 10.1007/s11011-017-9997-0. [DOI] [PubMed] [Google Scholar]
- 26.Lebrun B., Bariohay B., Moyse E., Jean A. Brain-derived neurotrophic factor (BDNF) and food intake regulation: a minireview. Auton. Neurosci. 2006;126-127:30–38. doi: 10.1016/j.autneu.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 27.Hirata T., Fujita M., Nakamura S., Gotoh K., Motooka D., Murakami Y., Maeda Y., Kinoshita T. Post-Golgi anterograde transport requires GARP-dependent endosome-to-TGN retrograde transport. Mol. Biol. Cell. 2015;26:3071–3084. doi: 10.1091/mbc.E14-11-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chia P.Z.C., Gleeson P.A. The regulation of endosome-to-Golgi retrograde transport by tethers and scaffolds. Traffic. 2011;12:939–947. doi: 10.1111/j.1600-0854.2011.01185.x. [DOI] [PubMed] [Google Scholar]
- 29.Yu H.-C., Coughlin C.R., Geiger E.A., Salvador B.J., Elias E.R., Cavanaugh J.L., Chatfield K.C., Miyamoto S.D., Shaikh T.H. Discovery of a potentially deleterious variant in TMEM87B in a patient with a hemizygous 2q13 microdeletion suggests a recessive condition characterized by congenital heart disease and restrictive cardiomyopathy. Cold Spring Harb. Mol. Case Stud. 2016;2:a000844. doi: 10.1101/mcs.a000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell M.W., Raeker M.O., Geisler S.B., Thomas P.E., Simmons T.A., Bernat J.A., Thorsson T., Innis J.W. Functional analysis of candidate genes in 2q13 deletion syndrome implicates FBLN7 and TMEM87B deficiency in congenital heart defects and FBLN7 in craniofacial malformations. Hum. Mol. Genet. 2014;23:4272–4284. doi: 10.1093/hmg/ddu144. [DOI] [PubMed] [Google Scholar]
- 31.Wongkittichote P., Ah Mew N., Chapman K.A. Propionyl-CoA carboxylase - A review. Mol. Genet. Metab. 2017;122:145–152. doi: 10.1016/j.ymgme.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duras E., İrdem A., Özkaya O. Long QT syndrome diagnosed in two sisters with propionic acidemia: a case report. J. Pediatr. Endocrinol. Metab. 2017;30:1133–1136. doi: 10.1515/jpem-2016-0469. [DOI] [PubMed] [Google Scholar]
- 33.Kovacevic A., Garbade S.F., Hoffmann G.F., Gorenflo M., Kölker S., Staufner C. Cardiac phenotype in propionic acidemia - Results of an observational monocentric study. Mol. Genet. Metab. 2020;130:41–48. doi: 10.1016/j.ymgme.2020.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Bodi I., Grünert S.C., Becker N., Stoelzle-Feix S., Spiekerkoetter U., Zehender M., Bugger H., Bode C., Odening K.E. Mechanisms of acquired long QT syndrome in patients with propionic academia. Heart Rhythm. 2016;13:1335–1345. doi: 10.1016/j.hrthm.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 35.Baumgartner D., Scholl-Bürgi S., Sass J.O., Sperl W., Schweigmann U., Stein J.-I., Karall D. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J. Pediatr. 2007;150:192–197, 197.e1. doi: 10.1016/j.jpeds.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 36.Kostapanos M.S., Liberopoulos E.N., Goudevenos J.A., Mikhailidis D.P., Elisaf M.S. Do statins have an antiarrhythmic activity? Cardiovasc. Res. 2007;75:10–20. doi: 10.1016/j.cardiores.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 37.Den Ruijter H.M., Franssen R., Verkerk A.O., van Wijk D.F., Vaessen S.F., Holleboom A.G., Levels J.H., Opthof T., Sungnoon R., Stroes E.S., et al. Reconstituted high-density lipoprotein shortens cardiac repolarization. J. Am. Coll. Cardiol. 2011;58:40–44. doi: 10.1016/j.jacc.2010.11.072. [DOI] [PubMed] [Google Scholar]
- 38.Kirchhof P., Fabritz L. High-density lipoprotein shortens the ventricular action potential. A novel explanation for how statins prevent sudden arrhythmic death? J. Am. Coll. Cardiol. 2011;58:45–47. doi: 10.1016/j.jacc.2010.12.048. [DOI] [PubMed] [Google Scholar]
- 39.Yamashita S., Matsuzawa Y. Where are we with probucol: a new life for an old drug? Atherosclerosis. 2009;207:16–23. doi: 10.1016/j.atherosclerosis.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 40.Schönberger J., Wang L., Shin J.T., Kim S.D., Depreux F.F.S., Zhu H., Zon L., Pizard A., Kim J.B., Macrae C.A., et al. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat. Genet. 2005;37:418–422. doi: 10.1038/ng1527. [DOI] [PubMed] [Google Scholar]
- 41.Roselli C., Chaffin M.D., Weng L.-C., Aeschbacher S., Ahlberg G., Albert C.M., Almgren P., Alonso A., Anderson C.D., Aragam K.G., et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 2018;50:1225–1233. doi: 10.1038/s41588-018-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abe S., Takeda H., Nishio S.-Y., Usami S.-I. Sensorineural hearing loss and mild cardiac phenotype caused by an EYA4 mutation. Hum. Genome Var. 2018;5:23. doi: 10.1038/s41439-018-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schönberger J., Levy H., Grünig E., Sangwatanaroj S., Fatkin D., MacRae C., Stäcker H., Halpin C., Eavey R., Philbin E.F., et al. Dilated cardiomyopathy and sensorineural hearing loss: a heritable syndrome that maps to 6q23-24. Circulation. 2000;101:1812–1818. doi: 10.1161/01.cir.101.15.1812. [DOI] [PubMed] [Google Scholar]
- 44.GTEx Consortium Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gay N.R., Gloudemans M., Antonio M.L., Abell N.S., Balliu B., Park Y., Martin A.R., Musharoff S., Rao A.S., Aguet F., et al. GTEx Consortium Impact of admixture and ancestry on eQTL analysis and GWAS colocalization in GTEx. Genome Biol. 2020;21:233. doi: 10.1186/s13059-020-02113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong Y., Perera M.A., Gamazon E.R. On using local ancestry to characterize the genetic architecture of human traits: genetic regulation of gene expression in multiethnic or admixed populations. Am. J. Hum. Genet. 2019;104:1097–1115. doi: 10.1016/j.ajhg.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keys K.L., Mak A.C.Y., White M.J., Eckalbar W.L., Dahl A.W., Mefford J., Mikhaylova A.V., Contreras M.G., Elhawary J.R., Eng C., et al. On the cross-population generalizability of gene expression prediction models. PLoS Genet. 2020;16:e1008927. doi: 10.1371/journal.pgen.1008927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shang L., Smith J.A., Zhao W., Kho M., Turner S.T., Mosley T.H., Kardia S.L.R., Zhou X. Genetic Architecture of Gene Expression in European and African Americans: An eQTL Mapping Study in GENOA. Am. J. Hum. Genet. 2020;106:496–512. doi: 10.1016/j.ajhg.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petty L.E., Highland H.M., Gamazon E.R., Hu H., Karhade M., Chen H.-H., de Vries P.S., Grove M.L., Aguilar D., Bell G.I., et al. Functionally oriented analysis of cardiometabolic traits in a trans-ethnic sample. Hum. Mol. Genet. 2019;28:1212–1224. doi: 10.1093/hmg/ddy435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Natri H.M., Hudjashov G., Jacobs G., Kusuma P., Saag L., Darusallam C.C., Metspalu M., Sudoyo H., Cox M.P., Gallego Romero I., Banovich N.E. Genetic architecture of gene regulation in Indonesian populations identifies QTLs associated with global and local ancestries. Am. J. Hum. Genet. 2022;109:50–65. doi: 10.1016/j.ajhg.2021.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics for PrediXcan and S-PrediXcan analyses are deposited in Zenodo: https://doi.org/10.5281/zenodo.5980229. All PAGE GWAS summary statistics are available through the GWAS Catalog.