

Summary
Studies of de novo mutation (DNM) have typically excluded some of the most repetitive and complex regions of the genome because these regions cannot be unambiguously mapped with short-read sequencing data. To better understand the genome-wide pattern of DNM, we generated long-read sequence data from an autism parent-child quad with an affected female where no pathogenic variant had been discovered in short-read Illumina sequence data. We deeply sequenced all four individuals by using three sequencing platforms (Illumina, Oxford Nanopore, and Pacific Biosciences) and three complementary technologies (Strand-seq, optical mapping, and 10X Genomics). Using long-read sequencing, we initially discovered and validated 171 DNMs across two children—a 20% increase in the number of de novo single-nucleotide variants (SNVs) and indels when compared to short-read callsets. The number of DNMs further increased by 5% when considering a more complete human reference (T2T-CHM13) because of the recovery of events in regions absent from GRCh38 (e.g., three DNMs in heterochromatic satellites). In total, we validated 195 de novo germline mutations and 23 potential post-zygotic mosaic mutations across both children; the overall true substitution rate based on this integrated callset is at least 1.41 × 10−8 substitutions per nucleotide per generation. We also identified six de novo insertions and deletions in tandem repeats, two of which represent structural variants. We demonstrate that long-read sequencing and assembly, especially when combined with a more complete reference genome, increases the number of DNMs by >25% compared to previous studies, providing a more complete catalog of DNM compared to short-read data alone.
Keywords: de novo mutation, autism, genome sequencing, long-read sequencing
Graphical abstract
Introduction
De novo mutations (DNMs) are spontaneous germline mutations that arise through a myriad of mechanisms, such as replication error, DNA damage repair, and non-allelic homologous recombination. Different mechanisms give rise to different types of mutations, the most common of which are small single-base substitutions (single-nucleotide variants [SNVs]) and insertions and deletions of a small number of bases (indels); de novo SNVs and indels have been reported at an average rate of approximately 70 DNMs per individual.1, 2, 3 Other classes of mutation, such as expansions of tandem repeats, have been estimated to be very common as well (>50 events per individual) but are currently incompletely ascertained.4 Larger mutations, such as structural variants (SVs), which affect more than 50 bp, are significantly rarer and have been observed at a rate of approximately one in every six individuals.5,6 All three classes of mutations have been implicated in autism, and it is estimated that more than 30% of all autism spectrum disorder (ASD) cases may arise as a result of DNM in a protein-coding sequence or a de novo SV.7 These estimates are based almost solely on the analysis of thousands of families via short-read whole-genome sequencing (WGS) datasets. Because long-read WGS methods have greatly increased sensitivity for SVs and large indels as well as all variant classes in repetitive loci,8,9 we expect de novo rates may have been systematically underestimated.
Mapping Illumina sequence data can successfully access approximately 84% of the genome.10 Repetitive regions, where the same 150 bp long read maps to multiple locations, are typically excluded, potentially underestimating the true mutation rate.11 In addition, Illumina sequencing is insensitive to large SVs where it is estimated that 75% of events (especially insertions) are missed in callsets generated from short-read sequencing technology.8 Previous efforts to identify de novo variation with long-read sequencing were able to call de novo SNVs and indels in five individuals, but even their highest confidence candidate set only had a true positive rate of 79% and failed to recover any de novo structural variation.6 Other studies have successfully identified de novo SVs with long-read sequencing but did not address SNVs or indels.12,13 In this study, we wished to measure the full extent of human de novo variation that exists in a family. To that end, we deeply sequenced DNA derived from blood from a family composed of two parents and their dizygotic twins with multiple long- and short-read technologies, including Pacific Biosciences (PacBio) continuous long-read (CLR) and high-fidelity (HiFi) sequencing, Oxford Nanopore Technologies (ONT) sequencing, Chromium 10X Genomic sequencing (10X), and single-cell DNA template strand sequencing (Strand-seq),14 and complemented with Bionano Genomics optical genome mapping (OGM) (Table 1). This family was selected because one of the children, the female proband, is affected with autism, and no genetic cause has been identified. This is despite extensive study by both whole-exome15 and whole-genome Illumina sequencing3 and the 2-fold increased likelihood of discovering a genetic event in females with autism.16 Here, we investigate and quantify the difference in DNM detection that can be reliably identified between short- and long-read data as well as the effect of a more complete reference genome for variant discovery. The use of multiple orthogonal sequencing technologies allows all events to be validated, producing a rigorous truth set with the potential to improve DNM detection and estimates of DNM rates.
Table 1.
Data summary
| Genomic technology | PacBio CLR | PacBio HiFi | Illumina | ONT | 10X | Strand-seq | Bionano OGM |
|---|---|---|---|---|---|---|---|
| Source | blood | blood/cellsa | blood | blood | cellsa | cellsa | cellsa |
| Platform | Sequel | Sequel 2 | Hi Seq X Ten | PromethION | Chromium | N/A | N/A |
| Metric | coverage | coverage | coverage | coverage | mean depth | number of cells | effective coverage of reference |
| Father | 55.5 | 47.1 | 37.5 | 27.5 | 73.1 | 66 | 273.5 |
| Mother | 54.5 | 43.8 | 33.5 | 29.0 | 61.4 | 63 | 256.2 |
| Proband | 74.4 | 34.0 | 41.8 | 30.8 | 64.1 | 56 | 246.2 |
| Sibling | 63.2 | 30.6 | 32.9 | 34.3 | 41.8 | 48 | 294.0 |
| Center | UW | UW | NYGC | CSHL | WU | EMBL | Radboud |
For each genomic technology—PacBio continuous long-read (CLR) and high-fidelity (HiFi) sequencing, Oxford Nanopore Technologies (ONT) sequencing, Bionano optical genome mapping (OGM)—the depth of sequencing is given for each member of the family. Coverage is based on genome size of 3.1 Gbp. UW, University of Washington; CSHL, Cold Spring Harbor Laboratory; WU, Washington University; EMBL, European Molecular Biology Laboratory.
Cells are EBV-transformed lymphoblasts.
Material and methods
Illumina sequencing and microarray data
Genome sequencing and analysis of this family was approved by the institutional review board (IRB) of the University of Washington (IRB STUDY00000383). Illumina WGS was performed on the Simons Simplex Collection (SSC) samples of a family (14455) at the New York Genome Center (NYGC) with 1 μg of DNA, an Illumina PCR-free library protocol, and sequencing on the Illumina X Ten platform. The father, mother, proband, and sibling were sequenced to an average depth of 37.47, 33.50, 41.78, and 32.90, respectively. Post-sequencing, reads were initially aligned to the reference genome (GRCh38). We applied two different single-nucleotide variant (SNV) callers—FreeBayes and GATK. We also applied a suite of structural variant (SV) callers (details in Turner et al.3) to maximize sensitivity for de novo SV mutation detection of various size ranges.3 This family was selected because we found only two variants of interest (likely gene-disrupting, missense [CADD > 30], 3′ UTR, or putative noncoding regulatory transcription factor binding site [pNCR-TFBS]) and no de novo or inherited SVs in exonic regions. The two de novo variants were a 3′ UTR event in ATP9A in the proband and a 3′ UTR in OLFM3 in the sibling (neither are candidate autism genes at this time). In addition, Illumina whole-genome sequencing (WGS), whole-exome sequencing (WES),15 and single-nucleotide polymorphism array data17 were generated as part of the SSC phase 1 study.18 No de novo variants were identified as likely pathogenic.3 This family, then, was selected for long-read sequencing for two reasons: (1) the autism case was unsolved for rare variants of large effect and (2) the unaffected and affected individuals represent fraternal twins where the female sibling was affected with autism.
Pacific Biosciences continuous long-read (CLR) sequencing
DNA from blood was sheared with a Megaruptor (Diagenode) on the 50 kbp setting. Material was prepared for PacBio sequencing with the SMRTbell Template Prep Kit 1.0 (PacBio P/N 100-259-100) (TPK1) or SMRTbell Express Template Prep Kit (P/N 101-357-000) (ExV1) following the recommended protocols. Briefly, the sample is treated for removal of single-stranded overhangs, damage repaired, end prepared, and ligated to PacBio SMRTbell adapters. TPK1 libraries have an additional step to remove imperfect SMRTbell templates, which is omitted in the ExV1 protocol. SMRTbell libraries were size selected on the BluePippin system (Sage Science) at 30 kbp or 40 kbp high pass settings. Libraries were quantified with Qubit (Thermo Fisher Scientific) and sized with FEMTO Pulse (Agilent Technologies) instruments before loading on the PacBio Sequel System with version 2.1 or 3.0 chemistries with 10 h movie acquisition times.
Pacific Biosciences high-fidelity (HiFi) sequencing
DNA from blood (all family members) or cell culture (mother, father for additional coverage) was sheared to a tight distribution with peak size of 10 or 20 kbp with gTUBEs (Covaris). We prepared SMRTbell libraries with TPK1 as described above (proband, sibling, blood; mother, father, cells), or SMRTbell Express Template Prep Kit 2.0 (P/N 100-938-900) and SMRTbell Enzyme Clean up Kit (P/N 101-746-400) (mother, father, blood), and size fractionated them on the SageELF (Sage Science) to generate tightly sized fractions. The fraction sized at 13 kbp (proband, sibling, blood), 15 kbp (mother, father, cells), or 20 kbp (mother, father, blood) was chosen for sequencing on the Sequel II system with version 1.0 (proband, sibling, blood), version 2.0EA (mother, father, cells), or version 2.0 (mother, father, blood) chemistries and 30 h movies. We processed raw subreads through the CCS workflow (PacBio SMRTLink version 7.1) to generate HiFi reads with a minimum estimated quality value (QV) of 20 (phred scaled, corresponding to an accuracy of 99%).
Oxford Nanopore Technologies sequencing
DNA from the same blood-derived DNA aliquot used for HiFi sequencing was sheared to 50 kbp with a Diagenode Megarupter following manufacturer’s recommendations. DNA was size selected via the Circulomics small read eliminator 25 kbp kit. DNA was prepared for Nanopore sequencing with the ONT 1D sequencing by ligation kit (SQK-LSK109). Briefly, 1–1.5 ug of fragmented DNA was repaired with the NEB FFPE repair kit followed by end repair and A-tailing with the NEB Ultra II end-prep kit. After an Ampure clean-up step, prepared fragments were ligated to ONT-specific adapters via the NEB blunt/TA master mix kit. The library underwent a final clean-up and was loaded onto a PromethION PRO0002 flowcell per manufacturer’s instructions. The flowcell was sequenced with standard parameters for 3 days and generated a Read N50 > 32 kbp for all samples. Base calling was performed with Guppy version 5.0.7 super accuracy model.
Strand-seq
Strand-seq libraries were prepared from four lymphoblast cell lines: SSC11453 (father), SSC11165 (mother), SSC11168 (sibling), and SSC11163 (proband). All lines were maintained in RPMI-1640 with 10% FBS, 1% Glutamax, and 1% penicillin/streptomycin. BrdU (bromodeoxyuridine; Sigma, B5002) was added to log-phase cell cultures at 40 μM or 100 μM concentrations for a period of 18 h or 24 h. Single nuclei were prepared and sorted with the BD FACSMelody cell sorter into 96-well plates for Strand-seq library production, as described (Falconer et al.;14 Sanders et al.19). The Strand-seq protocol was implemented on a Biomek FXP liquid handling robotic system, and pooled single-cell libraries were sequenced on the NextSeq500 platform (MID-mode, 75 bp paired-end protocol). After demultiplexing, Strand-seq sequencing reads were aligned to the human reference assembly GRCh38 (GCA_000001405.15_GRCh38_no_alt_analysis_set.fna) with the default parameters of BWA-MEM (version 0.7.15-r1140). Aligned BAM files were sorted by genomic position via SAMtools (version 1.7) and duplicated reads marked with sambamba (version 0.6.6). After alignment, we evaluated each single library to select only high-quality Strand-seq data for downstream analyses. Specifically, libraries with visible background reads (i.e., reads mapped to opposite direction on chromosomes that inherited template strands with the same directionality) and libraries with low (<50,000 reads) or uneven coverage were excluded, as detailed previously (Sanders et al.;19 Porubský et al.20).
Bionano Genomics
Optical genome mapping was performed as described previously (Mantere et al.21). Briefly, ultra-high molecular weight (UHMW) gDNA was isolated from frozen cell pellets, harvested from Epstein-Barr virus (EBV)-immortalized lymphocyte cell lines, following the manufacturer’s guidelines (Bionano Prep SP Frozen Cell Pellet DNA Isolation Protocol, Bionano Genomics #30268). For each sample, 750 ng of purified UHMW gDNA was labeled with DL-green fluorophores with the Direct Labeling Enzyme (DLE-1) chemistry and cleaned up with membrane adsorption (Bionano Prep Direct Label and Stain [DLS] Protocol, Bionano Genomics, #30206). Labeled gDNA samples were loaded on the Saphyr chips for linearization and imaging on the Saphyr instrument. Each flowcell was run on the maximum capacity to generate ∼1,300 Gbp of data per sample with Hg38 as the reference. The de novo assembly and SV annotation pipeline were executed with Bionano Solve software v.3.4. Fractional copy number estimates were based on the coverage-based CNV-tool and visual inspection of the events.
10X Genomics linked-read sequencing
High molecular weight (HMW) DNA was extracted from 1 million cells following a protocol outlined by 10X Genomics utilizing the salting out method. DNA was isolated with a QIAGEN MagAttract HMW DNA kit, resulting in >80 kbp DNA fragments. The HMW DNA was diluted to 1 ng/μL prior to the v2 Chromium Genome Library prep (10X Genomics). Approximately 10–15 DNA molecules were encapsulated into nanoliter droplets. DNA molecules within each droplet were tagged with a 16 nt 10X barcode and 6 nt unique molecular identifier during an isothermal incubation. The resulting barcoded fragments were converted into a sequence-ready Illumina library with an average insert size of 500 bp. We accurately determined the concentration of each 10X WGS library through qPCR (Kapa Biosystems) to produce cluster counts appropriate for the NovaSeq6000 platform (Illumina). Paired end-sequence (2×150) data were generated on a S4 300 cycle kit utilizing the XP workflow (Illumina) targeting 60× coverage (190 Gbp) providing long linked reads across the length of individual DNA molecules.
Accessible genome
The total accessible genome is based on the number of 10 kbp regions with an average mapq > 57 in both Illumina and HiFi. These calculations are based on previously described methods (Nurk et al.22), aligning CHM13 Illumina and HiFi reads to both GRCh38 and T2T-CHM13 assemblies. This results in a haploid genome size of 3.02 billion bases in T2T-CHM13 based on HiFi read alignment versus 2.63 billion bases in T2T-CHM13 based on Illumina sequence read alignment.
SNV and indel variant calling with HiFi
We aligned HiFi reads to the reference (either GRCh38 or T2T-CHM13) by using minimap2 to generate a BAM file.23 These BAM files were then used in a bifurcated pipeline to call de novo variants. For the DeepVariant portion of the pipeline, we applied DeepVariant v1.0.0 to call variants for each individual and then merged these variant files by using GLnexus.24,25 After calling, variants were filtered with GATK VariantFiltration, removing all calls with Phred-scaled quality score (QUAL) < 30.0. In addition, we used BCFtools to left-align and normalize indels. From this filtered set of variants, potential de novo variants were initially identified on the basis of genotype (father and mother genotypes were equal to 0/0 and the child’s genotype was equal to 0/1 or 1/1). For each de novo call, we used Pysam (see web resources) to count the number of reads with reference and alternate alleles in the BAM files in order to ensure the depth and allele balance were correct for each individual. Lastly, we used the following sample-level filters: father depth > 10, mother depth > 10, child depth > 10, and child genotype quality (GQ) > 20. Any remaining indels with length greater than 20 bp were also removed.
For the GATK portion of this pipeline, we applied GATK HaplotypeCaller v4.0.0 to call variants for each individual and then jointly genotyped the output by using GATK GenotypeGVCFs.26 After calling, we used GATK VariantFiltration to filter variants on the basis of three metrics: quality of depth (QD), the Phred-scaled probability that the site has no variant (QUAL), and the Z score for the Mann-Whitney rank sum test for the position of the alternate allele on reads (ReadPosRankSum). For this filtration step, variants were sorted into three groups: SNVs (QD < 2, QUAL < 30, ReadPosRankSum < −8.0), 1–2 bp indels (QD < 8, QUAL < 30, ReadPosRankSum < −20.0), and 3+ bp indels (QD < 2, QUAL < 30, ReadPosRankSum < −20.0). We then merged the three groups of variants and used BCFtools to left-align and normalize indels. From this filtered set of variants, potential de novo variants were initially identified on the basis of genotype (father and mother genotypes were equal to 0/0 and the child’s genotype was equal to 0/1 or 1/1). For each de novo call, we used Pysam to count the number of reads with reference and alternate alleles in the BAM files in order to ensure the depth and allele balance were correct for each individual. Lastly, we used the following sample-level filters: father depth > 10, mother depth > 10, child depth > 10, child allele balance > 0.25, and child GQ > 20. Any remaining indels with length greater than 20 bp were also removed.
For reads aligned to T2T-CHM13, a final filtering step was applied to remove all GATK calls within 2–100 bp of each other. This filter removed 24 calls (17 from the proband, 7 from the sibling) from the final T2T GATK callset.
SNV and indel calling with Illumina WGS
We called SNVs and indels in families by using four different callers and two different pipelines that used two (GATK and FreeBayes) or all four of the callers as previously described.3,27 Specifically, we applied GATK HaplotypeCaller v.3.5.0 FreeBayes v1.1.0, Platypus v0.8.1, and Strelka2 v2.9.2.26,28, 29, 30 In addition, multi-nucleotide mutations were called with FreeBayes and Platypus. We used post-calling BCFtools (version 1.3.1) norm to left-align and normalize indels. We partitioned the genome into the high-quality (HQ) regions, consisting of unique space as well as ancient repeats and the recent repeat (RR) regions, which consisted of repeats < 10% diverged from the consensus in RepeatMasker. Variants were only assessed in HQ portions of the genome and the RR region variants were removed from the study. Qualities of the callsets were assessed with KING for relationship checks, a variant per chromosome counter, and concordance checks for individuals with available array data.31
De novo variants were called with a custom pipeline. First, variants that were de novo based on genotype (father and mother genotypes were equal to 0/0 and the genotype in the child was 0/1 or 1/1) were retained for further assessment. Second, variants from Platypus with a filter of LowGQX or NoPassedVariantGTs were removed and Strelka2 variants had to have the filter field equal to PASS. Third, variants needed to have the support of at least two of the four callers. Fourth, variants were regenotyped with FreeBayes with default settings and needed to remain as de novo. Fifth, variants in a homopolymer A or T of length 10 or greater were removed. Sixth, we removed all variants in low-complexity regions (see web resources), recent repeats, or centromeres. Finally, we applied the following sample level filters: the father alternate allele count = 0, mother alternate allele count = 0, child allele balance > 0.25, father depth > 9, mother depth > 9, child depth > 9, and either child GQ > 20 (GATK) or sum of quality of the alternate observations (QA) > 20 (FreeBayes). For variants on the X chromosome, we separately considered variants in the pseudoautosomal regions (chrX: 10,000–2,781,479, chrX: 155,701,382–156,030,895) and the X/Y duplicatively transposed region (chrX: 89,201,803–93,120,510).
SNV and indel validation
Previously, we performed random Sanger validation of both the four-caller and two-caller DNM callset and combined this data with published validations to look at a total of 3,233 sites in a conditional inference analysis.27 We estimated our validation rate in this dataset at 99.5% and our false negative rate at 3.5%.
In this study, SNVs and indels were validated by examining the site across three different sequencing platforms—ONT, HiFi, and Illumina—aligned to the T2T-CHM13 reference (Figure S1). We used Pysam to calculate the number of reads with the reference and the alternate allele from the BAMs of reads aligned to the reference and used it to make the DNM call for both the parents and the child with the mutation. We filtered ONT reads to exclude any reads with base call QV < 10 at the site of the de novo variant—any site with more than one ONT read with the alternate allele in a parent was deemed inherited and any site with fewer than one ONT read with the alternate allele in the child was deemed a false positive. Sites were further examined across the other two sequencing platforms—in regions of read depth within two standard deviations of average, any site with one or more HiFi reads, or any site with several Illumina reads with the alternate allele in a parent was deemed inherited.
SNV and indel phasing
SNVs and indels were phased by applying WhatsHap v1.0 to aligned reads generated by PacBio HiFi, ONT, and Illumina sequencing.32 SNVs were phased in 40 kbp windows around a DNM of interest. We then used a Python script to select nearby “informative” SNVs of unambiguous parental inheritance (for example with genotype 0/0 mother, 0/1 father, and 0|1 in the child) and omit all SNVs that could not be phased or assigned to a parent. We then used these informative SNVs to determine a maternal or paternal haplotype around the DNM of interest by using an average haplotype score weighted inversely proportional to the distance from the DNM (nearby sites were weighted more highly in cases of disagreement between haplotype inheritance). This method was able to phase 194/195 (99.5%) germline DNMs in our dataset as well as 23/26 (88.5%) potential mosaic (likely postzygotic) DNM sites.
Assembly-driven detection of de novo structural variation
Assemblies for each member of this family were generated via hifiasm v0.12,33 which leverages PacBio HiFi reads generated from blood to produce haplotype-resolved assemblies. In the case of the children, we used Illumina short-read data to assign these haplotypes to a parent of origin. We used phased assembly variant (PAV) caller9 to detect SVs, indels, and SNVs in these assemblies. Detection of variants is driven by assembly to reference alignments with minimap2 v2.17 (CIGAR) and analysis of the CIGAR string. SVs were then supported by an additional run of PAV with long read aligner (lra),34 PBSV,9 subseq,9 and DeepVariant.24 Variants with detection from two or more callers (PAV [minimap2] + support caller) were then carried through to the final callset. Using this procedure, we identified 222 candidate de novo SVs, consisting of 57 deletions and 165 insertions. After these candidates were identified, we computationally analyzed alignment of parental data by using subseq over the SV region in order to determine read-based support for these events, which might have been missed in the assemblies.
STR/VNTR characterization
We applied three methods to focus specifically on de novo short tandem repeats (STRs)/variable number tandem repeats (VNTRs)—two of which have been previously described (Dolzhenko et al.,35 Sulovari et al.36). We also developed a custom k-mer-based pipeline that defines a library of uniquely mappable 30 bp k-mers (i.e., 30-mers) from the set of 21,000 phased tandem repeats of the human genome defined in Sulovari et al.36 We used seqtk (see web resources) to create the reverse complement of each haplotype-resolved STR and VNTR sequence in our library of tandem repeat sequences followed by generating all possible 30-mers by using jellyfish.37 All 30-mers were aligned with mrsFAST v3.3.8, and each was deemed uniquely mappable if and only if it mapped unambiguously to at most one specific genomic location of the unmasked GRCh38.p12 reference after allowing a hamming distance of two (i.e., command-line option -e 2).38 The vast majority of ∼21,000 polymorphic tandem repeats had at least one uniquely mappable k-mer associated with them. Next, we count each of the uniquely mappable k-mers in the Illumina short-read BAM files of each sample by using VariantBam39 to pull down reads matching the k-mer sequence and Kanalyze40 for counting the number of specific k-mers (and their reverse complements with --reverse=canonical option) across the short reads, irrespective of read mapping information and including both mapped and unmapped reads in our search space. Importantly, the information between each k-mer sequence and the GRCh38 coordinates of the STR/VNTR contigs that they originated from were stored throughout the process. After normalizing the k-mer counts by sequencing depth and GC bias coefficient (as described by Sudmant et al.41), the adjusted k-mer counts become a proxy for repeat length. The sites that appeared to have a significantly higher number of k-mer counts in either child relative to both parents were subsequently investigated as putative de novo sites. We used the CLR reads and long-range phasing information from 10X to carry out a targeted phased assembly for each locus with a putative de novo STR/VNTR.36 The loci where the targeted assembly results supported the existence of putative DNM underwent PCR validation.
Cell-line artifacts
In the process of identifying de novo SVs, we found evidence for several cell-line artifacts. We discovered these events in early exploratory phases of the project by using merged callsets from PacBio CLR (Phased-SV, SMRT-SV, PacBio structural variant calling tools [PBSV]), 10X (LongRanger v2.2.2), and Bionano (assembly-based calls from Solve). Although SV callsets were not generated from Strand-seq, we used it to find orthogonal support for variant calls. We applied subseq9 to find support for variants in aligned CLR reads for all family members to validate both the original variant call and inheritance status. Briefly, subseq expands a window around each variant, finds all reads in the region, and determines the length of the read spanning the region, which will be longer than the reference for insertions and shorter for deletions. This allows us to sensitively identify support for SVs down to two or more reads. We then selected de novo SVs with concordance from more than one technology and manually inspected them for supporting evidence across callsets, subseq, and Strand-seq. We considered genomic location, such as VNTRs and high-identity segmental duplications, which may have led to false variant calls, and we looked for clusters of SVs commonly associated with poor mapping, false calls, and poor reproducibility. Most de novo SVs could be explained by a missing parental allele or poor SV quality. However, we found several SVs that were strongly supported by using sequence data originating from cell-line-derived sources (10X, Strand-seq, Bionano) but clearly lacked support in blood-derived sources, such as CLR (by SV discovery and subseq) and Illumina whole-genome shotgun sequence detection (WSSD).
Detection of meiotic recombination breakpoints
In order to detect meiotic recombination, we realigned demultiplexed Strand-seq reads to the human reference assembly CHM13v1.0 by using default parameters of BWA-MEM (version 0.7.17-r1188). Aligned BAM files were sorted by genomic position with SAMtools (version 1.10) and duplicated reads marked with sambamba (version 1.0). Next, we phased Strand-seq reads by using StrandPhaseR with default parameters used for Illumina paired-end reads.42 We proceeded with integrative phasing by merging long-range Strand-seq haplotypes with local PacBio phasing, embedded in each long-read, with WhatsHap (versions 0.18).42 Having chromosome lengths and dense haplotypes for all family members, we set to detect all recombination breakpoints as positions where a child’s haplotype switches from matching H1 to H2 of a given parent or vice versa. In order to detect these positions, we first established what homolog in a child was inherited from either parent by calculating the level of agreement between child’s alleles and homozygous variants in each parent. Next, we compared each child’s homolog to both homologs of the corresponding parent and encoded them as 0 or 1 if they match H1 or H2, respectively. We applied a circular binary segmentation algorithm on such binary vectors by using R function “fastseg” implemented in R package fastseg (version 1.36.0) with parameter “minSeg” set to 50 and 1,000 for high-sensitivity and high-specificity breakpoint detection, respectively. Detected regions with segmentation mean ≤ 0.25 have been assigned H1 while regions with segmentation mean ≥ 0.75 have been assigned H2. Regions with segmentation mean in between these values were deemed ambiguous and were excluded. In addition, we filtered out regions shorter than 500 kbp and merged consecutive regions assigned the same haplotype.
Results
Detection of de novo variation with PacBio HiFi sequencing
In order to identify de novo SNVs and small indels (<20 bp), we initially applied three orthogonal sequencing technologies to blood-derived DNA obtained from each member of a simplex autism family (two parents and a dizygotic twin pair) where the daughter had been diagnosed with autism while the son was unaffected (Figure 1). We used Illumina and HiFi sequencing for variant discovery, while we used ONT for strictly validation purposes because of its higher error rate (Figure S1). Illumina WGS short-read data were generated and aligned to the reference GRCh38, and variants were called with both GATK HaplotypeCaller26 and FreeBayes28 via previously described best practices (Turner et al.3). De novo variants identified by both callers were included in the two-caller callset, containing 171 total DNM calls across the proband and sibling (true positive rate = 78.4%). To generate a more sensitive Illumina callset, we also included the variant callers Strelka230 and Platypus29 as described previously (Wilfert et al.27). De novo variants identified by at least three of the four Illumina callers were included in the four-caller callset, containing 144 total DNM calls across the proband and sibling (true positive rate = 91.7%). Between the two- and four-caller callsets, we identified 180 candidate DNMs. These DNMs were validated by examining the sites in both HiFi and ONT sequencing data—any sites where the variant was absent in the parents and present in the child in the orthogonal data were designated as true positive events. After validation, the Illumina callsets identified 62 total de novo SNVs and indels in the proband and 80 in the sibling (true positive rate = 78.9%), setting a lower bound for the number of DNMs present in the children.
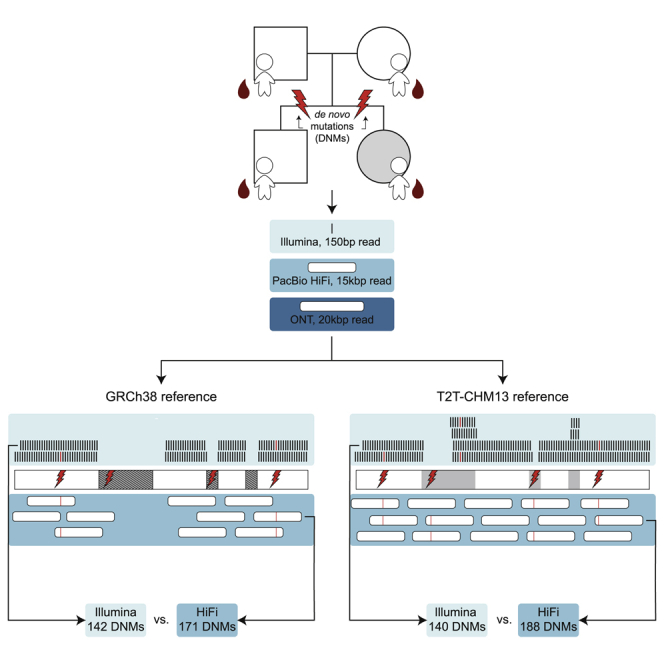
Figure 1.

De novo SNV calling and validation method
The pedigree of the quad, which consists of two parents, aged 35.53 and 35.20 (father and mother) at the time of their children’s birth, and their dizygotic twins. The proband, a female, is affected with autism, and her sibling is not. In this simplex case of autism in a female, there is an increased likelihood of finding a de novo mutation (DNM) to explain the cause of her autism. DNA derived from blood was sequenced with three different platforms: Illumina, PacBio HiFi, and ONT. Illumina and HiFi data were used for de novo discovery, and variants were validated by examination of the sites across all three sequencing technologies. True positive de novo events are exclusive to the child, whereas misclassified inherited events can be seen in at least one parent.
The limited number of validated de novo variants in the Illumina callsets was due in part to the exclusion of variants in repetitive sequence, such as repeats with greater than 90% identity (recent repeats), including low-complexity regions (LCRs)43 and centromeres, effectively restricting the callable genome to 78.6%. To identify variation missed by Illumina, we used HiFi sequencing, which generates long reads (median 15 kbp) that can unambiguously align to 88.1% of the genome. We aligned HiFi reads to GRCh38 and used two variant callers to naively identify de novo SNVs and indels. The first caller, GATK,26 identified 211 DNM calls across the proband and sibling (true positive rate 80.6%). The second caller, DeepVariant,24,25 identified 194 DNM calls across the proband and sibling (true positive rate 87.1%). Between both callsets, 217 candidate DNMs were identified and validated by examining the sites in both Illumina and ONT sequencing data. After validation, the HiFi callsets recovered 80 de novo SNVs and indels in the probands and 91 in the sibling—a 20.4% increase in the number of DNMs identified by Illumina (Figures 2A–2C).
Figure 2.

Comparison of DNM recall across short- and long-read callsets
(A) Number of DNM calls in reads aligned to GRCh38, based on the Illumina four-caller (blue), Illumina two-caller (purple), HiFi DeepVariant (yellow), and HiFi GATK (red) pipelines.
(B) Upset plot showing the concordance of de novo SNV calls across GRCh38 callsets, with the proportion of true positive (green), false positive (orange), and inherited (gray) events shown below each category. Validation status was assigned by examining the variants in ONT and HiFi or Illumina sequences, as described in the material and methods.
(C) Upset plot showing the concordance of de novo indel calls across GRCh38 callsets.
(D–F) The same analysis repeated on reads aligned to T2T-CHM13.
There were 75 DNM calls in HiFi not identified by the Illumina callers, 37 of which had support in ONT and retrospective analysis of the underlying Illumina sequence. As expected, most true DNMs exclusive to HiFi (23/37) were located in regions excluded by the Illumina pipelines; 82.6% of such calls were removed from Illumina callsets on the basis of this mask. However, removing variants in masked regions is a crucial part of the Illumina calling pipelines, as it eliminates more than 300 false positive DNM calls across both samples. Conversely, 38 false DNM calls were made only by HiFi callers, nearly three quarters of which (n = 27) were inherited variants incorrectly classified as de novo because of sequence coverage issues in one of the two parents. More than half of the inherited events (16/27) were located in clusters of less than 1 kbp. In most cases, inherited miscalls were the result of failure to sequence one of the parents’ haplotypes to sufficient coverage and could be resolved by sequencing to higher depth. For example, the mean paternal and maternal read depth in false HiFi calls is significantly lower than in validated de novo events (p = 1.01 × 10−6 and p = 7.75 × 10−8, Welch two-sample t test) (Figure S2). Thus, more permissive discovery or subsequent genotyping of the parents may further reduce the number of false calls. There were also eight DNM calls identified in Illumina missing from the HiFi callsets—all but two of these calls were identified by at least one HiFi caller but were excluded because they were either not sequenced to sufficient coverage or failed other quality filters (e.g., allele balance). In total, 179 true de novo SNVs and indels were discovered in GRCh38-aligned reads (82 in the proband and 97 in the sibling).
Detection of de novo variation with a more complete reference genome
The reference genome GRCh38 is incomplete, missing regions such as centromeres and some highly identical segmental duplications. The newly assembled T2T-CHM13 genome22 contains more than 240 Mbp of additional sequence. In order to discover de novo variation in these regions, we aligned the same Illumina and HiFi reads to the T2T-CHM13 assembly and used the same de novo calling pipelines to identify variation. Across the Illumina two- and four-caller callsets, we identified 184 DNM calls in the proband and sibling (true positive rate = 76.1%). Predictably, HiFi variant callers outperformed Illumina callers, as the HiFi GATK and DeepVariant callsets collectively identified 228 DNM calls in the proband and sibling (true positive rate = 80.3%). In total, of the 269 DNM calls made by the Illumina and HiFi callers using the T2T-aligned reads, 188 de novo SNVs and small indels had support in ONT and Illumina or HiFi sequence (Figures 2D–2F)—a 5% increase in the number of DNMs identified when compared to GRCh38 aligned reads, and only seven sites were missing as seen exclusively by callers on GRCh38 aligned reads (Figure 3). Both HiFi callers performed better on T2T-aligned reads, not only identifying more de novo sites but also generating callsets with true positive rates greater than those of the corresponding GRCh38 callsets (Figure 4A). By applying additional filters, we can improve the true positive rate by further reducing the number of false calls made in T2T-aligned data by 63% at the expense of only three true DNM calls. First, by applying a parental genotype quality filter (GQ > 25 for both parents) to the HiFi GATK callset, we can eliminate a total of 12 incorrect calls and one true call. Next, by applying a mapping quality filter (mapq > 59) to the HiFi DeepVariant callset, we can further remove seven incorrect calls and one true call. The biggest source of error, however, comes from the Illumina two-caller callset. There was a total of 33 calls identified by only the two-caller callset, 32 of which were false. By completely excluding the two-caller set from the analysis, we remove 32 incorrect calls and one true call. There is no overlap in the sites affected by these three methods—resulting in 54 sites removed from the total callset—a loss of 51/81 incorrect calls and 3/188 real DNMs. In the remaining T2T callset, there are only seven false positives and 23 inherited calls.
Figure 3.

De novo variant calling by technology and genomic region
Regions of the genome with mapping quality < 57 are highlighted in grey. HiFi mapping quality (mapq) is at the top of each chromosome and Illumina is on the bottom, along with all regions removed from Illumina callsets, including low-complexity regions, centromeres, and recent repeats highlighted in orange. The total accessible genome for GRCh38-aligned HiFi reads is 2.88 × 109 bp and for Illumina reads is 2.36 × 109 bp. The total accessible genome for T2T-CHM13-aligned HiFi reads is 3.07 × 109 bp and for Illumina reads is 2.31 × 109 bp. DNMs identified by each of the HiFi and Illumina callers are plotted in their respective locations. The chromosome 4 popout shows four true DNM calls that were made by HiFi callers but not Illumina callers and includes their annotations. The chromosome 14 popout shows three true DNM calls made by T2T callers but not GRCh38 callers.
Figure 4.

Human genome reference comparisons
(A) Concordance between T2T-CHM13 and GRCh38 callsets for each caller used. The overlap between callsets is on the left, and the proportion of true positives, false positives, and inherited events is on the right.
(B and C) Functional annotation of DNM calls across T2T and GRCh38 callsets. The three categories in (B) (repetitive regions) are separate annotations from those in (C); for example, a site that is in both a segmental duplication and a regulatory region would be included in both counts.
Most true de novo events were supported in both T2T- and GRCh38-aligned sequence, with the exception of two events that could not be lifted from GRCh38 to T2T coordinates and six events that had support in ONT and HiFi or Illumina reads aligned to GRCh38 but not when the same reads were aligned to T2T. Excluding those events, across both T2T and GRCh38 callsets, there were 88 de novo SNVs and small indels in the proband and 107 in the sibling with support in ONT and either Illumina or HiFi sequence. Eight percent of DNM calls were identified exclusively in T2T-aligned reads—three were found in centromeres, four in segmental duplications, and 12 in LCRs or recent repeats (Figure 4B). A total of four de novo SNVs were initially called in centromeres and were manually inspected in IGV (Figure S3), three of which were strongly supported in HiFi and ONT data; the fourth call was supported as de novo but the level of SNV heterozygosity for that portion of the alpha-satellite was much higher than anticipated, so we do not report it as a true de novo event.
In addition to DNM calling in repeats, variant calling sensitivity was also increased in functionally important regions, and T2T-aligned reads were able to identify nearly all calls made in GRCh38 (Figure 4C). In total, we identified three missense mutations (in XPO1 in the proband and USP49 and SEMA6B in the sibling), seven regulatory variants, and one 3′ UTR variant. Combining all the data, we identify 195 DNMs in the proband (n = 88) and sibling (n = 107) where the DNM status has been validated by ONT and the variant is absent in parental data, 185 of which had support in HiFi, Illumina, and ONT. Taking advantage of the available ONT and HiFi long-read data (material and methods), we successfully phased 99.5% (194/195) of the variants by considering informative single-nucleotide polymorphisms extending 20 kbp on either site of the variant position. Predictably, 72.2% (140/194) of the phased events originated in the male germline. In addition to these 195 events, another 14 sites (nine SNVs and five indels) had support in HiFi and Illumina data but not in ONT. Of those sites, five appeared to be false negatives in ONT (the affected child had no ONT reads with the alternate allele) despite having support in Illumina and HiFi data in both T2T- and GRCh38-aligned reads—these sites were not included in our final de novo callset. Another nine sites appeared inherited in ONT (one or both parents had more than one ONT read with the alternate allele).
In order to provide an estimate of false negatives and to maximize sensitivity, we measured the total number of de novo SNVs and small indels in the children by examining all of the candidate de novo calls made by DeepVariant and GATK on T2T-aligned HiFi reads. In this analysis, we removed the allele balance filter requiring the alternate allele to be present in at least 30% of the child’s reads (while retaining other filters for minimum quality and read depth) and examined all of the remaining calls in ONT and Illumina. This set, which would include mosaic DNMs, contained 209 DNMs with support across all three sequencing platforms. It included all of the 195 DNMs except 12, which were identified by only Illumina callers. If we add these 12 to the total, we identify 221 DNMs across the proband and sibling, predicting a false negative rate of 11.7% in our callset of de novo SNVs and small indels. Of the 26 variants missed in our de novo calling analysis (Table 2), all but three are allele balance (AB) < 0.35 in the affected child across all three sequencing platforms. Because of this consistently low allele balance, we suspect that these 23 variants may, in fact, represent potentially mosaic sites in the children (Figure S4). None of these 23 sites were reported in previous de novo studies of this family.3,27 This results in a mosaic mutation rate of 2.39 × 10−9 mutations per nucleotide per individual, most likely underestimating the true mosaic mutation rate because we are selecting only the highest frequency variants.44 Of the 23 potential mosaic variants, 12 were assigned to maternal haplotypes and eight were assigned to paternal haplotypes, resulting in a paternal:maternal ratio of 0.66:1, significantly different from the 2.59:1 ratio observed in the de novo germline variants (p = 0.0067, two-sample Z test). Although the mosaic sample is small, this observation is consistent with the expectation that there is no parent-of-origin bias for post-zygotic mutations.
Table 2.
Potential mosaic mutations
| Child | ID | HiFi | HiFi AB | ONT | ONT AB | Illumina | Illumina AB | Parental haplotype |
|---|---|---|---|---|---|---|---|---|
| 14455.p1 | chr2_94618830_C_A | 3/48 | 0.06 | 2/14 | 0.14 | 1/66 | 0.02 | paternal hap1 |
| 14455.p1 | chr4_1518905_G_C | 5/16 | 0.31 | 2/13 | 0.15 | 1/25 | 0.04 | maternal hap1 |
| 14455.p1 | chr5_124981762_T_C | 12/54 | 0.22 | 2/16 | 0.13 | 6/64 | 0.09 | maternal hap1 |
| 14455.p1 | chr9_42454095_C_T | 4/33 | 0.12 | 3/9 | 0.33 | 1/37 | 0.03 | maternal hap2 |
| 14455.p1 | chr17_81586404_G_C | 3/24 | 0.13 | 3/22 | 0.14 | 8/43 | 0.19 | maternal hap1 |
| 14455.p1 | chr18_15654268_G_T | 8/40 | 0.20 | 2/21 | 0.10 | 1/62 | 0.02 | paternal hap2 |
| 14455.p1 | chrX_114777954_G_A | 3/31 | 0.10 | 3/26 | 0.12 | 1/40 | 0.03 | maternal hap1 |
| 14455.s1 | chr1_2104522_A_G | 2/20 | 0.10 | 1/26 | 0.04 | 0/23 | 0.00 | paternal hap2 |
| 14455.s1 | chr2_91095600_G_T | 3/30 | 0.10 | 2/14 | 0.14 | 2/126 | 0.02 | maternal hap1 |
| 14455.s1 | chr3_96412796_A_C | 2/33 | 0.06 | 1/40 | 0.03 | 0/34 | 0.00 | maternal hap1 |
| 14455.s1 | chr6_62484584_G_A | 6/34 | 0.18 | 3/24 | 0.13 | 1/39 | 0.03 | maternal hap2 |
| 14455.s1 | chr6_70745885_CAT_C | 4/22 | 0.18 | 2/31 | 0.06 | 1/7 | 0.14 | unknown |
| 14455.s1 | chr6_127695258_G_A | 5/35 | 0.14 | 1/35 | 0.03 | 2/32 | 0.06 | paternal hap2 |
| 14455.s1 | chr6_163529274_T_C | 4/28 | 0.14 | 2/28 | 0.07 | 2/15 | 0.13 | maternal hap1 |
| 14455.s1 | chr7_2981075_TATATAG_T | 6/30 | 0.20 | 1/39 | 0.03 | 1/36 | 0.03 | maternal hap1 |
| 14455.s1 | chr7_58404995_T_A | 4/30 | 0.13 | 1/29 | 0.03 | 1/50 | 0.02 | paternal hap2 |
| 14455.s1 | chr7_58405316_C_T | 4/31 | 0.13 | 1/27 | 0.04 | 1/56 | 0.02 | paternal hap2 |
| 14455.s1 | chr7_156505941_C_A | 2/30 | 0.07 | 1/29 | 0.03 | 3/22 | 0.14 | unknown |
| 14455.s1 | chr11_50917416_C_A | 3/28 | 0.11 | 5/17 | 0.29 | 1/28 | 0.04 | paternal hap1 |
| 14455.s1 | chr14_3924466_C_CATTCCATTCCATTCT | 1/23 | 0.04 | 2/3 | 0.67 | 1/54 | 0.02 | unknown |
| 14455.s1 | chr14_10160460_G_Ta | 3/29 | 0.10 | 1/15 | 0.07 | 1/13 | 0.08 | maternal hap2 |
| 14455.s1 | chr19_5409128_C_T | 6/33 | 0.18 | 1/26 | 0.04 | 8/55 | 0.15 | paternal hap1 |
| 14455.s1 | chr22_20777172_G_Ta | 8/42 | 0.19 | 1/27 | 0.04 | 1/62 | 0.02 | maternal hap2 |
DNMs identified after removing the allele balance filter for HiFi long-read data aligned to T2T-CHM13. The number of reads with the alternate allele, total number of reads, and allele balance (AB) ratio for PacBio HiFi, ONT, and Illumina.
All variants were identified by GATK with the exception of the two variants identified by DeepVariant, denoted with the superscript.
Detection of de novo STR and VNTR events
Identification of candidate de novo expansions of STRs and VNTRs is particularly challenging with standard calling pipelines. We applied three orthogonal approaches to detect de novo events in the WGS data (Figure S5). The first approach leveraged the targeted phased assembly from Sulovari et al.;36 sequence resolved and phased STR and VNTR sites with larger repeats were examined for de novo variation in the proband and sibling, resulting in ten candidate events. The second approach used ExpansionHunter Denovo35 for identification of repeat expansions that were present in the children but not their parents, identifying three candidate events. The third approach used a custom pipeline for comparison of the number of uniquely mappable 30-mers in the parents and their children (after controlling for GC-adjusted read depth with the same genomic control regions as Sudmant et al.41), selecting sites for subsequent analysis with a higher number of k-mers in the child relative to its parents. Using these three approaches, we identified a total 15 candidate de novo STR and VNTR events, none of which was initially observed by more than one approach (Table S1). All 15 candidate events had support when validated with phased assembly generated by CLR reads haplotagged by the integrated 10X Chromium- and Strand-seq-phased variant data. The events were further validated with Sanger sequencing: six failed to sequence, five were shown to be inherited variants, but four represented true de novo events. Of the true positive events, one was a VNTR expansion in the proband (Figure 5), one was an STR deletion in the proband, one was an STR expansion in the sibling, and one appeared to be an STR expansion in both the proband and the sibling (Figure S6). The VNTR was identified by ExpansionHunter Denovo (true positive rate = 50%), one STR was identified by the 30-mer approach (true positive rate = 33%), and the remaining two STRs were identified by the approach from Sulovari et al.36 (true positive rate = 20%). All three approaches had low true positive rates and identified far fewer de novo STR and VNTR events than expected on the basis of previous reports.4
Figure 5.

De novo VNTRs
(A) The quad structure annotated with the number of repeats seen in the VNTR, as determined by PacBio CLR, 10X, and Strand-seq data in addition to Sanger sequencing, and represented on a gel.
(B) Haplotypes for every individual in the quad based on HiFi sequencing clearly show an extra copy of the motif (in red) in the proband.
(C) The 52 bp insertion in the proband compared to the parental haplotypes.
(D) The 73 bp insertion in the proband.
In an effort to increase yield, we applied the same assembly-driven methodology used for detection of structural variation to discover indels greater than or equal to 20 bp and less than 50 bp. Variants of this size disproportionately (86% of deletions and 64% of insertions) reside in short tandem repeats in GRCh38 coordinates. We started with a set of 12,284 deletions and 13,226 insertions in the proband in addition to 12,284 and 13,124 for deletions and insertions in the unaffected sibling. We then filtered this set down to 179 deletions and 276 insertions in the proband and 167 deletion and 219 insertion events in the unaffected sibling but not in the parents. Automatic inspection of raw parental long-read alignment validation yields seven potential de novo deletions and 14 potential de novo insertions in the proband. For the unaffected sibling this estimate is three and 15 for deletions and insertions, respectively. Manual inspection of the raw reads overlapping these calls yielded two confident de novo indels in the proband and one in the unaffected sibling, which were not seen in previous analyses (Figure S7, Table S2). However, one of the indels in the proband, originally identified as a 24 bp insertion with respect to the reference, was revealed to be an expansion of an 8 bp paternal allele, yielding a total de novo insertion length of 16 bp (Figure S7B).
De novo structural variation detection
We also applied both assembly- and read-based approaches to discover SVs (events > 50 bp in length). We initially generated haplotype-resolved assemblies by using a combination of PacBio HiFi and Illumina short-read data with hifiasm v0.12 and applied the PAV caller to create a set of 9,982 deletions and 16,815 insertions in the proband.9,33 Similarly, for the sibling we started with a set of 9,999 deletions and 16,879 insertions. We used PBSV,9 subseq,9 and DeepVariant24 to provide further support in addition to a secondary analysis of PAV with lra.34 We selected all variants detected by one or more of these additional callers for our SV callset for a total of 222 candidate de novo SVs consisting of 57 deletions and 165 insertions in the proband and 74 deletions and 121 insertions in the sibling. We initially validated these events by examining read-based support via subseq to analyze parental read data, resulting in 48 potential DNMs that were visually validated by examining both PacBio HiFi and ONT alignments over the regions in IGV. Of these events, 28 were clearly inherited, eight appeared to be false positives, and the remaining 12 were absent in parental data but present in at least one read in both technologies for the proband (seven) or sibling (five). These 12 candidates were finally validated by examining the haplotype-resolved assemblies for the parents and child, inspecting realigned contigs of the 6 kbp surrounding the site. Of the 12 candidate SV events, only three appeared to be true de novo events, with two in the proband and one in the sibling (Figure S8).
In an effort to minimize the extent of manual curation, we automated this process and developed a novel pipeline that implements some of the approaches made during manual inspection (Figure S9). By using a combination of subseq, callable regions from parental PAV calls, and multiple sequence alignment of familial haplotypes, we were able to validate the same two de novo events (both insertions) in the proband in an automated fashion, but we were not able to increase sensitivity. The single de novo candidate in the unaffected sibling did not validate with the automated pipeline, as multiple haplotypes were discovered overlapping this variant. Accordingly, we reclassified this variant as a low-confidence potential de novo event. This automated pipeline, named dnSVal, is available on GitHub (see web resources). The two true de novo SVs that occurred in the proband were VNTR expansions but had not been identified with our STR/VNTR-specific approaches. Both de novo events in the proband map to genic regions (CPT1C intron and TEC exon), but neither have been functionally implicated in autism.
Because discovery of de novo SVs is still challenging, we finally considered the potential of applying both Strand-seq and Bionano Genomics as standalone technologies to increase discovery sensitivity. For Strand-seq, we used the procedure described in Ebert et al.9 to detect and phase 127 nonredundant inverted sites (median size: 38.9 kbp, min: 2.3 kbp, max: 4.3 Mbp). Because Strand-seq can unambiguously split short sequencing reads by haplotype, it makes possible the detection and phasing of large heterozygous deletions; we identified 62 redundant heterozygous deletions with respect to GRCh38 with a median size of 55.8 kbp (min: 10.1 kbp, max: 550 kbp). Considering parental genotypes, we initially identified two and four candidate inversion de novo events in the proband and sibling, respectively. However, after manual inspection of these automated inversion calls, these were determined to likely represent false positives, as they fall into regions where short Strand-seq reads map with lower confidence, such as centromeres and segmental duplications. In addition to the inversions, we detected two potentially large heterozygous deletions by using Strand-seq as an orthogonal method. However, because of lack of support in phased HiFi reads, we were unable to validate these events.
Similarly, we used a Bionano coverage-depth-based algorithm to discover three de novo SV candidates in the proband (two deletions, one insertion) and five (three deletions, two insertions) in the unaffected sibling (Table S3).21 With the exception of the deletions in the unaffected sibling, these calls are seen with relatively high frequency in the population according to Bionano controls. None of these events intersect with our read-based or assembly-driven callsets nor do they contain any support in a manual inspection of the reads underlying this region. Given that none of the Bionano calls are supported by other data, we failed to identify any true de novo events when using Bionano as a sole discovery tool. As both the Strand-seq and Bionano Genomics data were derived from cell lines (as opposed to primary material), we consider the possibility that these invalidated events may also represent additional cell-line artifacts.
Meiotic breakpoints and DNMs
Since recombination has been shown to be mutagenic in the human population,45, 46, 47 we reassessed our validated set of DNMs with respect to meiotic crossover positions in the parental haplotype. Leveraging the inherent phasing data present in Strand-seq along with the long-read PacBio sequencing data allows one to define crossover breakpoints at a fine-scale level of resolution without the need for grandparental sequence data.20,42 We defined 135 total crossover events in the proband (Figure 6A) and sibling (Figure S10) at a fine-scale of resolution (median: 12.6 kbp) (Figure 6B, Table S4). Among the children, maternal and paternal crossover events were equally distributed (69 maternal and 66 paternal) with no particular bias toward certain genomic regions. We then projected all DNMs and measured the distance of a DNM to the nearest crossover event in the maternal or paternal lineage (Figure 6C) as well as assigning the DNM to grandparental haplotypes (Figure 6D). We performed a simulation based on the observed distance distribution and found no enrichment between DNM and inferred positions of meiotic recombination events (Figure S11). This study provides a near-complete picture of the occurrence of DNMs with respect to parental homolog and meiotic recombination.
Figure 6.

Meiotic recombination and DNM
(A) A genome-wide overview of detected meiotic recombination breakpoints for the proband. Inherited segments of maternal homologs (H1-light red, H2-dark red) appear on the left side of each chromosome while inherited segments of paternal homologs (H1-light blue, H2-dark blue) appear on the right side of each chromosome. Recombination breakpoints are visible as changes from H1 to H2 segments and vice versa. Detected DNMs that could have been assigned to a single parental homolog (n = 89) are shown as empty boxes over maternal (left) and paternal (right) homologs. This individual is a female, meaning that paternal chromosome X does not recombine (striped blue box).
(B) Size distribution of detected meiotic recombination breakpoints for both the proband (n = 76) and sibling (n = 59). Median value is shown as an orange dot for both distributions.
(C) Total number of DNMs assigned to paternal (dark blue) and maternal (dark red) homologs, separately for proband (14455.p1) and sibling (14455.s1).
(D) Total number of DNMs that occurred on paternal homologs H1 (light blue) or H2 (dark blue). The same results are shown for maternal homologs H1 (light red) and H2 (dark red). Counts are reported separately for proband (14455.p1) and sibling (14455.s1). We could not determine inherited parental segments for one DNM in proband and for seven DNMs in sibling.
Discussion
We identified 195 de novo SNVs and indels in the quad: 88 in the proband and 107 in the sibling—a 35% increase from the 65 and 79 DNMs identified from our previous analysis of this family,27 which was optimized for specificity and similar to the original estimates of Kong et al.1 This contrasts with a second analysis we performed on the same family by using a two-caller Illumina approach, which was geared toward increased sensitivity and reported 131 and 138 DNMs for the proband and sibling, respectively.3 A comparison with both ONT and HiFi data for the same family, however, shows that this two-caller method demonstrates only a 78% true positive rate. Thus, while the overall numbers appear similar, the long-read data are discovering a new subset of DNMs traditionally excluded by or filtered from the short-read data. Notably, this study also widens the gap in the number of DNMs present in the proband and sibling: the total 19 DNM differential would place this quad in the 91st percentile among twins in Wilfert et al.27 and the 60th percentile in Turner et al.3 (Figure S12).
Of the 195 true DNMs that we identified, exactly seven lie on the X chromosome. Calling on the X chromosome presented a unique challenge, as we failed to identify a single DNM on the female proband’s X chromosomes. Despite there being many potential sites on the proband’s X, most failed to reach the minimum allele balance threshold, and those that did fail to meet our read depth requirements, resulting in only four calls in our final callsets, none of which were true de novo events. The male sibling, on the other hand, showed the opposite trend, with 20 DNM calls in our final callset, seven of which were validated on the basis of our criteria. The high number of calls in the sibling were the result of HiFi GATK, which alone contributed 11 sites on the X chromosome. It is likely that the increased sensitivity on the male X chromosome is an artifact of the lower average read depth—with lower read depth, one or two sequencing errors would pass the allele balance threshold, allowing a variant call to be made, as we did not set a higher threshold for AB on the male X chromosome.
In the past, we relied on both the two- and four-caller pipelines to identify de novo variation3,27 from autism WGS datasets in an effort to balance both specificity and sensitivity. However, this study revealed that both pipelines suffer from limitations that can make it more difficult to identify potential disease-causing variation. Across GRCh38 and T2T-CHM13, the four-caller pipeline identified 141 true DNMs with a 91.6% true positive rate, which is the highest of all callsets, but lower than our previous validations had estimated. The four-caller pipeline underestimated the number of DNMs in this family by at least 30%. The two-caller pipeline, on the other hand, suffers from both a low true positive rate (72.1%) and, when restricted to just true positive sites, underestimates the number of DNMs in this family by at least 25%. The high false positive rate in the two-caller pipeline makes it a poor choice for studies aimed to identify disease-causing mutations. Going forward, the Illumina four-caller pipeline could be optimized by removing the mask used to exclude variants in repetitive sequence and developing a new filtering schema that does not remove variants on the basis of region alone. By removing this mask, the size of the four-caller callset could be increased by at least 20% and would provide a better snapshot of the true de novo variation present in a genome.
The increase in the total number of true mutations relative to previous studies indicates that the DNM rate for SNVs and indels is most likely higher than current estimates suggest. In addition, we were able to document DNMs in centromeres and segmental duplications, two regions that are just beginning to become accessible with long-read WGS platforms. If we set the total accessible genome of our study to be the total number of 10 kbp regions with high mapping quality by HiFi (mapq ≥ 57) in T2T-CHM13, the total genome size is 3.07 billion bp. On the basis of this, we can estimate the de novo substitution rate from this one family to be approximately 1.41 × 10−8 per nucleotide per generation, which is on the high end of projected mutation rates.2,48, 49, 50 While the rate is higher than most previous genome-wide estimates, a larger number of samples will be needed to determine whether the mutation rate is particularly elevated in regions of the genome accessible only by long reads. It should be stressed, however, that the methods we used to identify de novo SNVs with long reads were still stringent, requiring an allele balance of between 0.3 and 0.7 and confirmation across two sequencing platforms. Different sequencing platform biases even among the long-read technologies will tend to underestimate variant calling for specific regions and classes of variation. Based on our false negative analysis, which revealed that there are 23 true de novo (most likely post-zygotic) SNVs that eluded our calling pipelines, the mutation rate in this family is most likely closer to 1.59 × 10−8 per nucleotide per generation.
In order to replicate these results, more families need to be sequenced with orthogonal long-read sequencing approaches. Despite the additional cost, long-read sequencing enabled us to increase our DNM discovery by ∼35%, granted us access to new regions of the genome, and allowed us to search for and verify larger mutations as well. While this study focused on DNMs, we also performed a preliminary analysis of inherited rare variants (<0.1%) by re-filtering the GATK callsets to search for variants that were present in a child and exactly one of the parents and confirmed by ONT (Figure S13). Although comparison of a larger number of long-read genomes will be required to determine true allele frequencies, this analysis suggests a comparable increase (26.4% in GRCh38) of inherited rare variants throughout the genome (Figure S14) as result of greater access to more complex and repeat rich regions of the genome. It should be noted that the number of sequencing platforms used in this study is not necessary to extensively catalog the DNM load in a trio—a combination of Illumina, ONT, and HiFi would be sufficient, and most potential mutations could be validated with much more affordable Sanger sequencing. DNM calling can be further optimized by aligning both short and long reads to the more complete T2T-CHM13 genome, which will be invaluable for estimating the mutation rate in repetitive regions of the genome.
In addition to our large de novo SNV and indel callset, we discovered three de novo STR expansions and one VNTR expansion (<50 bp) (Tables S5 and S6). This is fewer STR events than we would expect on the basis of previous projections that predict greater than 50 STR expansions per transmission.4 We also identified two small de novo SVs, insertions of 52 and 73 bp, but did not observe any larger germline SVs. Despite their importance in neurodevelopmental disease, this more comprehensive DNM analysis did not reveal any new candidate mutations to better explain the proband’s autism status. It could be the case that the underlying etiology is inherited or polygenic as the upper bound for DNM underlying autism has been estimated to account for ∼30% of cases.7 Alternatively, if the causal variant is de novo, this may mean that despite our many orthogonal methods to identify DNMs in the proband, sensitivity is not yet optimized. This is especially the case for de novo structural variation where methods for de novo SV calling among VNTRs/STRs remains a challenge despite the dramatic advances in SV detection with long reads.6,8 Another possibility, albeit less likely, is that we missed a causative mutation in a region that we are still not able to sequence and assemble, such as in the highest identity repeats. Even when we align HiFi reads to the T2T-CHM13 genome, there is still approximately 200 Mbp of sequence with coverage more than 2 standard deviations above or below the mean and with mapq less than 57; until we can accurately assign sequencing reads to these regions of the genome, we will not be able to fully catalog all classes of variation in a genome.
An important aspect of this work was that all DNM candidates were obtained from primary tissue, in this case peripheral lymphocytes from blood. It is worthwhile noting, however, that apparent de novo SVs were initially identified with other technologies including Strand-seq, 10X Genomics, and Bionano Genomics where lymphoblastoid cell culture instead of primary blood were used to obtain larger amounts of DNA or actively dividing cells for the assay. The most striking was a 147 kbp deletion event in the sibling removing all but the first exon of SETD2 (chr3-47014726-DEL-147102)—a gene previously associated with autism. This deletion was discovered from Bionano SV calls and confirmed by 10X data, but in both cases, calls originated from DNA prepared from cell-line material. We found no evidence for the variant in any sequence data derived from blood DNA, including read-depth changes in Illumina and PacBio (Figure S15A). Similarly, a second event, a 158 kbp deletion in the sibling (chr7-142644005-DEL-158289), deleted several small genes (PRSS1 and PRSS2) and had support from all three cell-line-derived datasets (Bionano, 10X, and Strand-seq), but no datasets derived from blood DNA (Figure S15B). While undetectable low levels of somatic mosaicism in the blood DNA may underlie this, it is more likely that such potentially impactful deletion events occurred in cell culture after only a few passages. This emphasizes the importance of discovery and validation of DNM variants from primary source material.
Acknowledgments
We thank Tonia Brown for assistance in editing this manuscript, Tom Maniatis and the New York Genome Center for generating Illumina WGS data, and Tychele Turner and Amy Wilfert for selection of family for study and generating Illumina callsets as published previously. This work was supported, in part, by grants from the National Institutes of Health (R01 MH101221 to E.E.E.; UM1 HG008901 to M.C.Z.; 5R50CA24389 to S.G.) and the Simons Foundation (SFARI 810018EE to E.E.E.). Y.M. is supported by funds from Sidra Medicine and Qatar National Research Fund (NPRP10-1219-160035). We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, E. Wijsman). We acknowledge funding support from the CSHL/Northwell Health Affiliation for purchase of the ONT PromethION sequencer used in this study. ONT sequencing was done in the Next Generation Sequencing Shared Resource that is part of the (NIH) Cancer Center Support grant P30-372 CA045508. W.R.M. is the Davis Family Professor of Human Genetics, and E.E.E. is an investigator of the Howard Hughes Medical Institute.
Declaration of interests
E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc.
Published: March 14, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.02.014.
Data and code availability
The accession number for all underlying raw sequence (FASTQ) files for the ONT and HiFi PacBio datasets, Strand-seq data, VCFs, and Bionano Genomics reported in this paper is SFARI Base: DS149300.
Web resources
Low complexity regions, https://github.com/lh3/varcmp/blob/master/scripts/LCR-hs38.bed.gz
Seqtk, https://github.com/lh3/seqtk
Supplemental information
References
- 1.Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., Magnusson G., Gudjonsson S.A., Sigurdsson A., Jonasdottir A., Jonasdottir A., et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jónsson H., Sulem P., Kehr B., Kristmundsdottir S., Zink F., Hjartarson E., Hardarson M.T., Hjorleifsson K.E., Eggertsson H.P., Gudjonsson S.A., et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–522. doi: 10.1038/nature24018. [DOI] [PubMed] [Google Scholar]
- 3.Turner T.N., Coe B.P., Dickel D.E., Hoekzema K., Nelson B.J., Zody M.C., Kronenberg Z.N., Hormozdiari F., Raja A., Pennacchio L.A., et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell. 2017;171:710–722.e12. doi: 10.1016/j.cell.2017.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitra I., Huang B., Mousavi N., Ma N., Lamkin M., Yanicky R., Shleizer-Burko S., Lohmueller K.E., Gymrek M. Patterns of de novo tandem repeat mutations and their role in autism. Nature. 2021;589:246–250. doi: 10.1038/s41586-020-03078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belyeu J.R., Brand H., Wang H., Zhao X., Pedersen B.S., Feusier J., Gupta M., Nicholas T.J., Brown J., Baird L., et al. De novo structural mutation rates and gamete-of-origin biases revealed through genome sequencing of 2,396 families. Am. J. Hum. Genet. 2021;108:597–607. doi: 10.1016/j.ajhg.2021.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pauper M., Kucuk E., Wenger A.M., Chakraborty S., Baybayan P., Kwint M., van der Sanden B., Nelen M.R., Derks R., Brunner H.G., et al. Long-read trio sequencing of individuals with unsolved intellectual disability. Eur. J. Hum. Genet. 2021;29:637–648. doi: 10.1038/s41431-020-00770-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iossifov I., O’Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E., et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaisson M.J.P., Sanders A.D., Zhao X., Malhotra A., Porubsky D., Rausch T., Gardner E.J., Rodriguez O.L., Guo L., Collins R.L., et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019;10:1784. doi: 10.1038/s41467-018-08148-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ebert P., Audano P.A., Zhu Q., Rodriguez-Martin B., Porubsky D., Bonder M.J., Sulovari A., Ebler J., Zhou W., Serra Mari R., et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science. 2021;372:6537. doi: 10.1126/science.abf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Telenti A., Pierce L.C.T., Biggs W.H., di Iulio J., Wong E.H.M., Fabani M.M., Kirkness E.F., Moustafa A., Shah N., Xie C., et al. Deep sequencing of 10,000 human genomes. Proc. Natl. Acad. Sci. USA. 2016;113:11901–11906. doi: 10.1073/pnas.1613365113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ebbert M.T.W., Jensen T.D., Jansen-West K., Sens J.P., Reddy J.S., Ridge P.G., Kauwe J.S.K., Belzil V., Pregent L., Carrasquillo M.M., et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol. 2019;20:97. doi: 10.1186/s13059-019-1707-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merker J.D., Wenger A.M., Sneddon T., Grove M., Zappala Z., Fresard L., Waggott D., Utiramerur S., Hou Y., Smith K.S., et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med. 2018;20:159–163. doi: 10.1038/gim.2017.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reiner J., Pisani L., Qiao W., Singh R., Yang Y., Shi L., Khan W.A., Sebra R., Cohen N., Babu A., et al. Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of a Bardet-Biedl Syndrome 9 (BBS9) deletion. NPJ Genom. Med. 2018;3:3. doi: 10.1038/s41525-017-0042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falconer E., Hills M., Naumann U., Poon S.S.S., Chavez E.A., Sanders A.D., Zhao Y., Hirst M., Lansdorp P.M. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods. 2012;9:1107–1112. doi: 10.1038/nmeth.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krumm N., Turner T.N., Baker C., Vives L., Mohajeri K., Witherspoon K., Raja A., Coe B.P., Stessman H.A., He Z.-X., et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015;47:582–588. doi: 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy D., Ronemus M., Yamrom B., Lee Y.H., Leotta A., Kendall J., Marks S., Lakshmi B., Pai D., Ye K., et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 17.Sanders S.J., He X., Willsey A.J., Ercan-Sencicek A.G., Samocha K.E., Cicek A.E., Murtha M.T., Bal V.H., Bishop S.L., Dong S., et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischbach G.D., Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Sanders A.D., Falconer E., Hills M., Spierings D.C.J., Lansdorp P.M. Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat. Protoc. 2017;12:1151–1176. doi: 10.1038/nprot.2017.029. [DOI] [PubMed] [Google Scholar]
- 20.Porubský D., Sanders A.D., van Wietmarschen N., Falconer E., Hills M., Spierings D.C.J., Bevova M.R., Guryev V., Lansdorp P.M. Direct chromosome-length haplotyping by single-cell sequencing. Genome Res. 2016;26:1565–1574. doi: 10.1101/gr.209841.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mantere T., Neveling K., Pebrel-Richard C., Benoist M., van der Zande G., Kater-Baats E., Baatout I., van Beek R., Yammine T., Oorsprong M., et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am. J. Hum. Genet. 2021;108:1409–1422. doi: 10.1016/j.ajhg.2021.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nurk S., Koren S., Rhie A., Rautiainen M., Bzikadze A.V., Mikheenko A., Vollger M.R., Altemose N., Uralsky L., Gershman A., et al. The complete sequence of a human genome. Preprint at bioRxiv. 2021 doi: 10.1101/2021.05.26.445798. [DOI] [Google Scholar]
- 23.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poplin R., Chang P.-C., Alexander D., Schwartz S., Colthurst T., Ku A., Newburger D., Dijamco J., Nguyen N., Afshar P.T., et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018;36:983–987. doi: 10.1038/nbt.4235. [DOI] [PubMed] [Google Scholar]
- 25.Yun T., Li H., Chang P.-C., Lin M.F., Carroll A., McLean C.Y. Accurate, scalable cohort variant calls using DeepVariant and GLnexus. Bioinformatics. 2021;36:5582–5589. doi: 10.1093/bioinformatics/btaa1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poplin R., Ruano-Rubio V., DePristo M.A., Fennell T.J., Carneiro M.O., Van der Auwera G.A., Kling D.E., Gauthier L.D., Levy-Moonshine A., Roazen D., et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Preprint at bioRxiv. 2017 doi: 10.1101/201178. [DOI] [Google Scholar]
- 27.Wilfert A.B., Turner T.N., Murali S.C., Hsieh P., Sulovari A., Wang T., Coe B.P., Guo H., Hoekzema K., Bakken T.E., et al. Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat. Genet. 2021;53:1125–1134. doi: 10.1038/s41588-021-00899-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. Preprint at arXiv. 2012 https://arxiv.org/abs/1207.3907 arXiv:1207.3907 [q-bio.GN] [Google Scholar]
- 29.Rimmer A., Phan H., Mathieson I., Iqbal Z., Twigg S.R.F., Wilkie A.O.M., McVean G., Lunter G., WGS500 Consortium Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014;46:912–918. doi: 10.1038/ng.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim S., Scheffler K., Halpern A.L., Bekritsky M.A., Noh E., Källberg M., Chen X., Kim Y., Beyter D., Krusche P., Saunders C.T. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods. 2018;15:591–594. doi: 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 31.Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin M., Patterson M., Garg S.O., Fischer S., Pisanti N., Klau G.W., Schöenhuth A., Marschall T. WhatsHap: fast and accurate read-based phasing. Preprint at bioRxiv. 2016 doi: 10.1101/085050. [DOI] [Google Scholar]
- 33.Cheng H., Concepcion G.T., Feng X., Zhang H., Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 2021;18:170–175. doi: 10.1038/s41592-020-01056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren J., Chaisson M.J.P. lra: A long read aligner for sequences and contigs. PLoS Comput. Biol. 2021;17:e1009078. doi: 10.1371/journal.pcbi.1009078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolzhenko E., Bennett M.F., Richmond P.A., Trost B., Chen S., van Vugt J.J.F.A., Nguyen C., Narzisi G., Gainullin V.G., Gross A.M., et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 2020;21:102. doi: 10.1186/s13059-020-02017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sulovari A., Li R., Audano P.A., Porubsky D., Vollger M.R., Logsdon G.A., Warren W.C., Pollen A.A., Chaisson M.J.P., Eichler E.E., Human Genome Structural Variation Consortium Human-specific tandem repeat expansion and differential gene expression during primate evolution. Proc. Natl. Acad. Sci. USA. 2019;116:23243–23253. doi: 10.1073/pnas.1912175116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marçais G., Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hach F., Hormozdiari F., Alkan C., Hormozdiari F., Birol I., Eichler E.E., Sahinalp S.C. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wala J., Zhang C.-Z., Meyerson M., Beroukhim R. VariantBam: filtering and profiling of next-generational sequencing data using region-specific rules. Bioinformatics. 2016;32:2029–2031. doi: 10.1093/bioinformatics/btw111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Audano P., Vannberg F. KAnalyze: a fast versatile pipelined k-mer toolkit. Bioinformatics. 2014;30:2070–2072. doi: 10.1093/bioinformatics/btu152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sudmant P.H., Mallick S., Nelson B.J., Hormozdiari F., Krumm N., Huddleston J., Coe B.P., Baker C., Nordenfelt S., Bamshad M., et al. Global diversity, population stratification, and selection of human copy-number variation. Science. 2015;349:aab3761. doi: 10.1126/science.aab3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porubsky D., Garg S., Sanders A.D., Korbel J.O., Guryev V., Lansdorp P.M., Marschall T. Dense and accurate whole-chromosome haplotyping of individual genomes. Nat. Commun. 2017;8:1293. doi: 10.1038/s41467-017-01389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30:2843–2851. doi: 10.1093/bioinformatics/btu356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muyas F., Zapata L., Guigó R., Ossowski S. The rate and spectrum of mosaic mutations during embryogenesis revealed by RNA sequencing of 49 tissues. Genome Med. 2020;12:49. doi: 10.1186/s13073-020-00746-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halldorsson B.V., Palsson G., Stefansson O.A., Jonsson H., Hardarson M.T., Eggertsson H.P., Gunnarsson B., Oddsson A., Halldorsson G.H., Zink F., et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science. 2019;363:eaau1043. doi: 10.1126/science.aau1043. [DOI] [PubMed] [Google Scholar]
- 46.Arbeithuber B., Betancourt A.J., Ebner T., Tiemann-Boege I. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc. Natl. Acad. Sci. USA. 2015;112:2109–2114. doi: 10.1073/pnas.1416622112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Francioli L.C., Polak P.P., Koren A., Menelaou A., Chun S., Renkens I., van Duijn C.M., Swertz M., Wijmenga C., van Ommen G., et al. Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 2015;47:822–826. doi: 10.1038/ng.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kessler M.D., Loesch D.P., Perry J.A., Heard-Costa N.L., Taliun D., Cade B.E., Wang H., Daya M., Ziniti J., Datta S., et al. De novo mutations across 1,465 diverse genomes reveal mutational insights and reductions in the Amish founder population. Proc. Natl. Acad. Sci. USA. 2020;117:2560–2569. doi: 10.1073/pnas.1902766117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldmann J.M., Wong W.S.W., Pinelli M., Farrah T., Bodian D., Stittrich A.B., Glusman G., Vissers L.E.L.M., Hoischen A., Roach J.C., et al. Parent-of-origin-specific signatures of de novo mutations. Nat. Genet. 2016;48:935–939. doi: 10.1038/ng.3597. [DOI] [PubMed] [Google Scholar]
- 50.Conrad D.F., Keebler J.E.M., DePristo M.A., Lindsay S.J., Zhang Y., Casals F., Idaghdour Y., Hartl C.L., Torroja C., Garimella K.V., et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for all underlying raw sequence (FASTQ) files for the ONT and HiFi PacBio datasets, Strand-seq data, VCFs, and Bionano Genomics reported in this paper is SFARI Base: DS149300.