

Abstract
Pulmonary endothelial barrier dysfunction is a hallmark of clinical pulmonary edema and contributes to the development of acute lung injury (ALI). Here we reported that ruscogenin (RUS), an effective steroidal sapogenin of Radix Ophiopogon japonicus, attenuated lipopolysaccharides (LPS)-induced pulmonary endothelial barrier disruption through mediating non-muscle myosin heavy chain IIA (NMMHC IIA)‒Toll-like receptor 4 (TLR4) interactions. By in vivo and in vitro experiments, we observed that RUS administration significantly ameliorated LPS-triggered pulmonary endothelial barrier dysfunction and ALI. Moreover, we identified that RUS directly targeted NMMHC IIA on its N-terminal and head domain by serial affinity chromatography, molecular docking, biolayer interferometry, and microscale thermophoresis analyses. Downregulation of endothelial NMMHC IIA expression in vivo and in vitro abolished the protective effect of RUS. It was also observed that NMMHC IIA was dissociated from TLR4 and then activating TLR4 downstream Src/vascular endothelial cadherin (VE-cadherin) signaling in pulmonary vascular endothelial cells after LPS treatment, which could be restored by RUS. Collectively, these findings provide pharmacological evidence showing that RUS attenuates LPS-induced pulmonary endothelial barrier dysfunction by inhibiting TLR4/Src/VE-cadherin pathway through targeting NMMHC IIA and mediating NMMHC IIA‒TLR4 interactions.
KEY WORDS: Ruscogenin, Acute lung injury, Lipopolysaccharide, Endothelial barrier, Non-muscle myosin heavy chain IIA, TLR4, VE-cadherin, Interaction
Graphical abstract
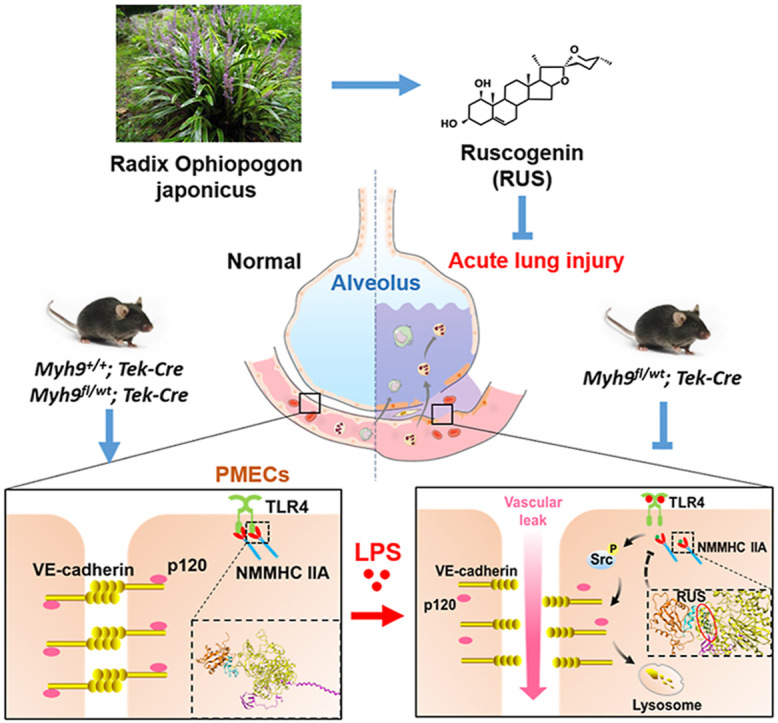
Ruscogenin impedes lipopolysaccharides (LPS)-induced non-muscle myosin heavy chain ⅡA (NMMHC IIA) dissociation from Toll-like receptor 4 (TLR4) via targeting NMMHC IIA, inhibiting activation of TLR4 downstream Src/vascular endothelial cadherin (VE-cadherin) signaling and promoting pulmonary endothelial barrier integrity.
1. Introduction
Acute lung injury (ALI) or its most advanced form, acute respiratory distress syndrome (ARDS), is a severe inflammatory pulmonary process triggered by a variety of pulmonary (pneumonia, pulmonary contusion) or nonpulmonary (sepsis, trauma) insults, characterized by diffuse alveolar damage and increased pulmonary vascular permeability1. Disruption of pulmonary endothelial barrier homeostasis resulting in extravascular accumulation of protein-rich fluids and lung edema is a key pathophysiological mechanism in ALI and ARDS2,3. Though considerable progress has been made in clarifying the mechanisms responsible for pathogenesis and resolution of ALI, the majority of therapeutic strategies to improve outcomes have been unsuccessful so far4, 5, 6, highlighting the necessity for further research and development of novel effective treatments.
Non-muscle myosin II is a motor protein essential for maintaining cellular homeostasis and participates in multiple cellular functions including cell migration, adhesion, endomitosis, and signal transduction7, 8, 9, 10. Non-muscle myosin heavy chain IIA (NMMHC IIA) encoded by Myh9 is the most widely distributed isoform of non-muscle myosin II. Previous studies have revealed that NMMHC IIA plays a significant role in regulating endothelial functions such as barrier integrity, von Willebrand factor secretion, and thrombus formation10, 11, 12, 13. However, the role of NMMHC IIA and its pharmacological intervention in lipopolysaccharides (LPS)-induced ALI and pulmonary endothelial barrier dysfunction are still limited.
Ruscogenin (RUS), a major bioactive steroidal sapogenin derived from the traditional Chinese herb Radix Ophiopogon japonicus which has been used to treat acute and chronic inflammatory and cardiovascular diseases for many years, has been found to exert prominent protective effect on ALI, and its possible mechanism could be attributed to the inhibition of tissue factor and inducible nitric oxide (NO) synthase expression and nuclear factor (NF)-κB p-p65 activation14,15. Our previous research also uncovered that RUS protected against LPS-induced lung injury and endothelial cell apoptosis via regulating Toll-like receptor 4 (TLR4) signaling, suggesting that TLR4 might be essential for the anti-inflammatory and anti-apoptotic effects of RUS16. However, whether RUS directly acts on TLR4 or TLR4 complex to regulate downstream signaling needs to be further investigated. In this study, we captured NMMHC IIA as a target protein of RUS, and with the evidence that NMMHC IIA interacts with TLR4 and regulates tight junction in brain endothelial cells by mediating downstream transduction pathway12, we speculate that NMMHC IIA‒TLR4 complex may be a potential target of RUS to mediate LPS-stimulated pulmonary endothelial barrier dysfunction.
In the present study, we explored the effects and underlying mechanisms of RUS on LPS-induced pulmonary endothelial barrier dysfunction during ALI. The results demonstrated that RUS directly bound to the N-terminal and head domain of NMMHC IIA and mediated the NMMHC IIA‒TLR4 interactions to suppress the activation of Src/vascular endothelial cadherin (VE-cadherin) signaling, protecting against LPS-induced pulmonary endothelial barrier disruption, which indicated RUS might be a promising therapeutic candidate for the treatment of ALI.
2. Materials and methods
2.1. Reagents and antibodies
RUS (505836) was purchased from J&K Scientific Ltd. (Beijing, China). Dexamethasone (DEX; D1756), LPS (L2880), GenElute™ Mammalian Genomic DNA Miniprep Kits (G1N70), Duolink® In Situ PLA® Probe Anti-Rabbit PLUS (DUO92002), Duolink® In Situ PLA® Probe Anti-Mouse MINUS (DUO92004), and Duolink® In Situ Detection Reagents Red (DUO92008) were obtained from Sigma‒Aldrich (St. Louis, MO, USA). The bicinchoninic acid (BCA) protein assay kit and phenylmethanesulfonyl fluoride (PMSF) were procured from Beyotime Biotechnology (Shanghai, China). Anti-NMMHC IIA (ab75590, ab55456), anti-TLR4 (ab22048), anti-VE-cadherin (ab33168), anti-p120-catenin (ab92514), anti-Myc (ab9132), and anti-green fluorescent protein (GFP) (ab290) antibodies were purchased from Abcam (Cambridge, MA, USA). Anti-TLR4 (sc-293072) and Protein A/G PLUS-Agarose (sc-2003) were obtained from Santa Cruz Biotechnology (Texas, CA, USA). Anti-phospho-Src family (2101) and anti-Src (2123) were from Cell Signaling Technology (Boston, MA, USA). Purified rat anti-mouse CD31 (553370) was purchased from BD Biosciences (Franklin Lakes, NJ, USA). Alexa Fluor 488- and 594-conjugated secondary antibodies, Dynabeads™ Sheep Anti-Rat IgG (11035) were from Thermo Fisher Scientific (Waltham, MA, USA).
2.2. Animals
Male C57BL/6 mice (18–22 g) were provided by the Experimental Animal Center of Yangzhou University (certificate number was SCXK Jiangsu 2017-0007; Yangzhou, China). All animal experimental procedures were carried out according to the National Institutes of Health guidelines, and the protocols were approved by the Animal Ethics Committee of China Pharmaceutical University (Nanjing, China). The animals were feed at standardized temperature at 22 ± 2 °C and relative humidity at 60% ± 2%. After 3 days of adjustable feeding, the mice were randomly divided into six groups (n = 6): control group, model group, model + RUS (0.1, 0.3, and 1 mg/kg) groups, and model + DEX (1 mg/kg) group. Mice in model group were challenged with LPS (5 mg/kg) via intratracheal instillation for 6 h. RUS and DEX were administrated orally 1 h prior to LPS treatment. All mice were euthanized after LPS induction and bronchoalveolar lavage fluid (BALF) was collected for subsequent experiments.
Tek‒Cre mice and Myh9fl/fl mice were obtained from Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China). Endothelial cell (EC)-specific monoallelic deletion of NMMHC IIA (referred to as Myh9fl/wt; Tek‒Cre) mice were generated by in vitro fertilization.
2.3. Cell culture
Murine lung vascular endothelial cells (MLECs) were isolated as described previously16. MLECs were cultured in DMEM (Gibco, Grand Island, NY, USA) supplemented with 20% fetal bovine serum (FBS; Gibco), 1% MEM Non-Essential Amino Acids (Gibco), 2.5% HEPES buffer solution (1 mol/L; Gibco), endothelial cell growth supplement (0.1 mg/mL; Sigma‒Aldrich), 100 U/mL penicillin, and 100 U/mL streptomycin (Gibco). Human umbilical vascular endothelial cells (HUVECs) were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China), and cultured in RPMI 1640 supplemented with 10% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin. The cells were grown at 37 °C in a humidified incubator of 5% CO2 and 95% air.
2.4. Evans blue albumin (EBA) pulmonary transvascular flux measurement
After LPS treatment of mice via intratracheal instillation (5 mg/kg) for 4 h, EBA dye (50 mg/kg) was administered via tail vein injection. After 2 h, mice were anesthetized and intravascular EBA perfused with saline through the right ventricle for 5 min. Mouse lungs were excised, homogenized in 1 mL phosphate buffer solution (PBS), and extracted in 2 mL formamide for 18 h at 60 °C. The EBA concentration was determined based on OD620 and OD740 values.
2.5. EBA leakage analysis in MLECs
MLECs (2 × 105 cells/well) were seeded on the gelatin-coated transwell inserts of 24-well plates and cultured for 7 days. Then MLECs were pretreated with RUS for 1 h, followed by LPS stimulation for 6 h. EBA solution was added as the tracer to the transwell inserts and 4% bovine serum albumin (BSA) solution was added to the outer chamber. After 1 h, the solution in outer chamber was collected and EBA concentration was detected based on OD620 and OD740 values.
2.6. Transendothelial electrical resistance (TER) assay
MLECs (2 × 105 cells/well) seeded on the gelatin-coated transwell inserts of 24-well plates were cultured for 7 days. RUS was administrated 1 h prior to LPS exposure. After LPS treatment for 6 h, the TER value of MLECs was monitored with a Millicell-ERS voltohmmeter (Millipore, Billerica, MA, USA). The mean value was expressed in the common unit (Ω·cm2) after subtraction of the value of a blank cell-free filter.
2.7. Immunofluorescence staining
MLECs or tissue sections were fixed with 4% formaldehyde and permeabilized with 0.1% of Triton X-100. Then the samples were blocked with 5% BSA and incubated with the corresponding primary antibodies [CD31, 1:100 dilution; NMMHC IIA, 1:200 dilution; TLR4, 1:100 dilution; p-Src (Tyr416), 1:200 dilution; VE-cadherin, 1:200 dilution] overnight at 4 °C. After washed with PBS, MLECs were added with the appropriate fluorescence-conjugated secondary antibodies (Alexa Fluor 594-donkey anti-goat IgG, Alexa Fluor 488-donkey anti-goat IgG, Alexa Fluor 594-goat anti-mouse IgG, and Alexa Fluor 488-donkey anti-rabbit IgG, 1:500 dilution). Fluorescent images were visualized using a confocal laser scanning microscope (CLSM, LSM700; Zeiss, Germany).
2.8. Plasmid and siRNA transfection
Expression plasmids for GFP-tagged IQ motif, tail, N-terminal, and head domain of Myh9 were obtained from Genomeditech Co. Ltd. Myh9 siRNAs (forward, 5ʹ-GAGGCAAUGAUCACUGACUdTdT-3ʹ; reverse, 5ʹ-AGUCAGUGAUCAUUGCCUCdTdT-3ʹ) were purchased from Biomics Biotechnologies (Nantong, China). Transfection of cells with Myh9 siRNAs or plasmids was performed using ExFect Transfection Reagent (Vazyme, Nanjing, China) according to the manufacturer's instructions.
2.9. Immunofluorescence co-localization analysis
To measure the amount of co-localization between two of the stains in the images, the plugin JaCoP in Image J (National Institutes of Health, Bethesda, MD, USA) was performed in pixel matching co-localization analyses17. In brief, two different channels in one image were split and colored by Image J. Then all correlation-based co-localizations were checked, and information of microscope (CLSM, LSM700; Zeiss) and the emitted wavelength of fluorescence were filled in the JaCoP. After regulating the threshold, the co-localization was evaluated with manders’ co-localization coefficients.
2.10. Proximity ligation assays (PLA)
A PLA kit (DUO92002, DUO92004, and DUO92008; Sigma‒Aldrich) was employed to detect interactions between NMMHC IIA and TLR4 in MLECs according to the manufacturer's protocols18. Samples were incubated with primary antibodies for NMMHC IIA and TLR4. Secondary antibodies conjugated with oligonucleotides (PLA probes) were added to the reaction for subsequent ligation and rolling circle amplification. Images were observed under a CLSM (LSM700; Zeiss) and processed using ZEN imaging software.
2.11. Serial affinity chromatography
Serial affinity chromatography with some modifications was used to separate and identify the target proteins of RUS as described previously19,20. In brief, lysate from MLECs (1 mL) was shaken mildly with RUS affinity resin (30 μL) (A1, B1, and C1) for 1.5 h at 4 °C, then centrifuged at 16,000 ×g for 1 min. And the supernatants were mixed with another RUS affinity resin (30 μL) (A2, B2, and C2) at 4 °C, again for 1.5 h. Then the resulting resin was washed 3 times with 1 mL lysate buffer, resuspended in 30 μL of 2× loading buffer and heated at 95 °C for 5 min. The samples were subjected to sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and the resulting bands were stained with silver staining and then comparatively analyzed to identify specific binding proteins.
2.12. Microscale thermophoresis (MST) assay
To further confirm the specific domain of NMMHC IIA that RUS acts on, NMMHC IIA is divided into four major domains: N-terminal (aa. 29–69), head domain (aa. 83–764), IQ motif (aa. 775–835), and tail domain (aa. 842–1921) according to the analysis of Myh9 cDNA sequence. HEK293T cells were transfected with expression plasmids for GFP-tagged NMMHC IIA domains and the lysate was collected as assay buffer for MST experiments. A NanoTemper Monolith Instrument (NT.115) (NanoTemper Technologies, München, Germany) was employed for measuring thermophoresis. Samples were loaded into Monolith NT.115Pico MST standard-treated capillaries and measured using a Monolith NT.115Pico and MO.Control software (NanoTemper Technologies) at 37 °C.
2.13. Biolayer interferometry (BLI) analysis
To evaluate the binding kinetics of RUS on NMMHC IIA, the HA-tagged head domain and tail domain of recombinant human NMMHC IIA protein were purified by anti-HA affinity beads (Smart Lifesciences, China) and biotinylated using a biotinylation kit (Genemore, China) according to the manufacturer's instructions. The biotinylated protein was immobilized in the super streptavidin biosensors (Pall ForteBio, Silicon Valley, CA, USA). The binding of RUS to NMMHC IIA head and tail domains were analyzed by Octet RED96 (Pall ForteBio).
2.14. Co-immunoprecipitation (co-IP) assay
Cell lysates were incubated with anti-NMMHC IIA or an equal amount of IgG, followed by incubation with Protein A/G PLUS-Agarose (Santa Cruz Biotechnology; sc-2003). Proteins were resolved via SDS-PAGE followed by Western blot analysis.
2.15. Western blot analysis
Protein extracted from cells or lung tissues was quantified using a BCA protein assay kit. Equal amounts of protein from each group (40 μg) were separated via 10%–15% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking with 5% BSA, membranes were incubated with primary antibody overnight at 4 °C, followed by horseradish peroxidase (HRP)-conjugated secondary antibody. Bands were detected using an ECL kit and analyzed with Image Lab™ Software.
2.16. Hematoxylin and eosin (H&E) staining
Lung tissues fixed in 4% paraformaldehyde were embedded in paraffin, cut into 4 μm sections, and stained with H&E. Pathological images were examined using a digital pathological section scanner (Hamamatsu, Japan) and analyzed with NDPView2 (Hamamatsu, Japan). The lung injury score was evaluated on the basis of the following observed indicators: pulmonary hemorrhage, inflammatory infiltration and pulmonary interstitial edema. According to the severity of the disease, scores were shown as 0 (no apparent lesions), 0.5‒1 (mild), 2 (moderate), 3 (severe), and 4 (critical). The scores of each sample were accumulated and the average score of each group was calculated for statistical analysis.
2.17. Statistical analysis
All data were expressed as means ± standard deviation (SD) from at least three independent experiments and analyzed with GraphPad Prism software (version 7.0) (San Diego, CA, USA). Statistical significance was calculated with Student's t-test for comparisons between two groups or one-way analysis of variance (ANOVA) for multiple comparisons. Differences were considered significant at P values < 0.05.
3. Results
3.1. RUS improves LPS-triggered pulmonary vascular endothelial barrier disruption in vivo and in vitro
After LPS intratracheal instillation for 6 h, H&E staining of lung sections exhibited prominent alveolar collapse, congestion, and inflammatory cell infiltration, whereas RUS (0.3 and 1 mg/kg) and DEX treatment showed significant improvement in lung lesions (Fig. 1A and B). The total protein level and white blood cell count in BALF were significantly decreased in RUS intervention group compared with the ALI mouse model group (Fig. 1C and D). EBA was injected by tail vein and extent of extravasated dye was determined to evaluate pulmonary endothelial barrier function. As is shown in Fig. 1E and F, the model group exhibited obvious dye leakage after EBA injection, while RUS administration attenuated LPS-induced pulmonary vascular leakage. To evaluate whether RUS could improve the survival rate in ALI mice induced by LPS, we established an ALI model by LPS (20 mg/kg) intraperitoneal injection. The data showed that RUS significantly decreased the death rate and extended the survival time of endotoxemia mice induced by LPS (Supporting Information Fig. S1). Collectively, these results indicated that RUS could ameliorate LPS-stimulated pulmonary vascular hyperpermeability.
Figure 1.

RUS attenuates LPS-induced pulmonary endothelial barrier dysfunction. C57BL/6 mice were pre-treated with RUS (0.1, 0.3, and 1 mg/kg) for 1 h followed by LPS intratracheal instillation for 6 h. (A, B) Paraffin embedded lung tissue sections were stained with hematoxylin and eosin (H&E) and lung injury was analyzed. Scale bar = 50 μm. (C, D) After exposed to LPS for 6 h, mice were sacrificed and lungs were lavaged with PBS. The total protein content and white blood cells in bronchoalveolar lavage fluid (BALF) were analyzed. (E, F) At 4 h after LPS administration, Evans blue albumin (EBA) was given by tail intravenous injection. After 2 h, the dye was extracted from lung tissues and quantified. n = 6. (G) RUS (1 μmol/L) was administrated 1 h prior to LPS exposure in murine lung vascular endothelial cells (MLECs). After LPS treatment for 6 h, immunofluorescence of VE-cadherin and phalloidine was employed to detect the effect of RUS on LPS induced MLECs barrier disruption. The asterisks mean disruptive adherens junctions. Scale bar = 20 μm. (H, I) MLECs seeded on the gelatin-coated transwell inserts for 7 days to form a monolayer, RUS (1 μmol/L) was administrated 1 h prior to LPS challenge for 6 h, the effect of RUS on LPS-induced MLECs hyperpermeability was determined by transendothelial electrical resistance (TER) and EBA assays. n = 3. Data are presented as mean ± SD. ###P < 0.001 vs. control group; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. model group. RUS, ruscogenin; LPS, lipopolysaccharides; DEX, dexamethasone; WBC, white blood cell.
In order to assess the effect of RUS on pulmonary endothelial barrier in vitro, murine lung vascular endothelial cells (MLECs) were specifically isolated with purified rat anti-mouse CD31 monoclonal antibody-coated magnetic beads and identified by flow cytometry and immunofluorescence (Supporting Information Fig. S2). MLECs were pretreated with RUS (1 μmol/L) for 1 h, followed by LPS (5 μg/mL) incubation for 6 h. TER and EBA leakage analyses showed that LPS stimulation remarkably decreased TER value and increased EBA leakage in MLECs, and RUS treatment improved LPS-induced MLECs hyperpermeability (Fig. 1H and I). Immunostaining experiments also disclosed that RUS exerted an amelioration on LPS-stimulated disordered arrangement of the cytoskeleton and disruption of VE-cadherin structure between adjacent MLECs (Fig. 1G).
Similarly, RUS (1 mg/kg) treatment clearly relieved LPS-induced lung injury and pulmonary endothelial barrier destruction when administered at 2 and 4 h after LPS instillation (Supporting Information Fig. S3). Taken together, these results confirmed the protective effect of RUS on pulmonary endothelial barrier dysfunction and acute lung injury induced by LPS.
3.2. RUS directly binds to NMMHC IIA N-terminal and head domains
Using the RUS affinity medium, we captured specific binding proteins for RUS from HUVEC lysates by serial affinity chromatography. The protein as indicated by SDS-PAGE obviously decreased following serial affinity chromatography (Fig. 2A). Electrospray ionisation tandem mass spectrometry (ESI‒MS) and Western blot analysis of the ∼210 kD protein identified it as NMMHC IIA (Supporting Information Fig. S4), suggesting that NMMHC IIA might be a specific binding protein of RUS. Molecular docking study was carried out to explore the interaction of RUS with NMMHC IIA and the docking prediction results revealed that RUS bound to the head domain of NMMHC IIA (Fig. 2B) with the residues such as Pro521, Pro522, Gly523, Ala526, Leu527, Glu530, Glu531, Gln550, Glu549, and Lys545 (Fig. 2C). In addition, MST analysis was performed to determine the interaction between RUS and the full length NMMHC IIA in vitro, and the equilibrium dissociation constant (Kd) value was 32.01 μmol/L, which was close to the Kd value of NMMHC II inhibitor blebbistatin (BLE) (Fig. 2D).
Figure 2.

Non-muscle myosin heavy chain IIA (NMMHC IIA) is identified as a target protein of RUS. (A) Serial affinity chromatography was used to capture the specific binding proteins of RUS and NMMHC IIA was identified as a RUS targeting protein. A comparison of proteins bound to the first resins (A1, B1, and C1) and second resins (A2, B2, and C2) showed that only the protein marked by arrow decreased significantly in amount, indicating that it was a specific binding protein. M is for the protein marker. (B) Molecular docking analysis revealed that RUS binds to the head domain of NMMHC IIA. (C) Residues that form the binding pocket of RUS are indicated. (D) Microscale thermophoresis (MST) assay was performed to detect the binding kinetics of RUS and blebbistatin (BLE) on full length NMMHC IIA.
To further investigate the specific domain of NMMHC IIA that RUS acts on, we mapped the regions in NMMHC IIA and NMMHC IIA was divided into four domains: N-terminal (aa. 29‒69), head (aa. 83‒764), IQ motif (aa. 775‒835), and tail domains (aa. 842‒1921). MST assay showed that RUS bound to the N-terminal and head domain of NMMHC IIA (Fig. 3A‒D). To further verify the interaction domain and find out whether RUS directly targets NMMHC IIA, we purified HA-tagged head domain and tail domain of NMMHC IIA protein. BLI analysis was used to detect the affinity of RUS and NMMHC IIA domains in vitro. The results suggested that RUS directly interacted with NMMHC IIA head domain in a positive dose-dependent manner and the Kd value was 88.85 μmol/L (Fig. 3E). These results indicated that NMMHC IIA was the target protein of RUS and RUS mainly bound to its head domain to mediate the biological function of NMMHC IIA.
Figure 3.

RUS directly binds to N-terminal and head domain of NMMHC IIA. (A‒D) NMMHC ⅡA is divided into four major domains: N-terminal (aa. 29‒69), head domain (aa. 83‒764), IQ motif (aa. 775‒835), and tail domain (aa. 842‒1921) according to the analysis of Myh9 cDNA sequence. HEK293T cells were transfected with expression plasmids for GFP-tagged NMMHC ⅡA domains and the lysate was collected as assay buffer for MST assays. A and B showed that RUS bound with N-terminal and head domain of NMMHC IIA in a dose-dependent manner, C and D showed that there was no binding between RUS and IQ motif and tail domain. (E) Biolayer interferometry (BLI) analysis showed that there was a directly interaction between RUS and NMMHC IIA head domain.
There are two functional binding sites in NMMHC IIA head domain, ATP-binding sites (BLE binding sites) and actin binding sites. To figure out whether RUS interacts with the same binding sites of NMMHC IIA head domain compared with BLE or actin, we constructed ATP-binding sites deletion and actin-binding sites deletion mutants. And MST results revealed that there were still combinations after ATP-binding sites and actin-binding sites deletion in NMMHC IIA head domain (Supporting Information Fig. S5), indicating that RUS interacted with the different binding sites of NMMHC IIA head domain compared with BLE and actin.
3.3. Endotoxin challenge results in elevated expression of NMMHC IIA and degradation of VE-cadherin in pulmonary endothelial cells
Then we set out to determine the role of NMMHC IIA in an LPS-induced ALI mouse model (Fig. 4). After LPS stimulation, remarkable elevation of NMMHC IIA expression in pulmonary endothelium was observed from 6 h by immunostaining (Fig. 4A and E). And TLR4 and downstream phosphorylated Src levels were enhanced after LPS treatment (Fig. 4B, C, F, and G). We further detected the contents of VE-cadherin, which was the most important adherens junctional component for maintaining endothelial barrier integrity. And the results revealed down-regulation of VE-cadherin levels following LPS treatment (Fig. 4D and H). These findings were similar to the results we obtained in MLECs (Supporting Information Fig. S6), suggesting that LPS exposure disrupts pulmonary endothelial adherens junctions through activation of TLR4/Src/VE-cadherin signaling and NMMHC IIA may play a pivotal role in this process.
Figure 4.

LPS stimulation induces an elevated expression of NMMHC IIA and downregulation of VE-cadherin in pulmonary endothelium. (A‒D) NMMHC IIA, TLR4, p-Src (Tyr416), and VE-cadherin expression in pulmonary endothelium after LPS stimulation was assessed based on immunofluorescence with anti-CD31 (red), anti-NMMHC IIA (green), anti-TLR4 (green), anti-p-Src (green), and anti-VE-cadherin (green), and visualized using confocal microscopy. Nuclei were counterstained with DAPI (blue). Scale bar = 10 μm. (E‒H) Fluorescence intensity of NMMHC IIA, TLR4, p-Src, and VE-cadherin in vessels was quantified by “Colocalization Analysis” using Image J. All data are expressed as mean ± SD, n = 3. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. 0 h group.
3.4. NMMHC IIA monoallelic deletion in endothelium attenuates LPS-induced pulmonary vascular leakage
To address the possibility that NMMHC IIA is a key factor in regulation of the vascular endothelial barrier in vivo, mice with endothelial-specific NMMHC IIA deficiency were generated. Since complete NMMHC IIA-knockout mice fail to develop a normal visceral endoderm and death occurred by embryonic day (E) 7.521, we used EC-specific NMMHC IIA monoallelic deletion (Myh9fl/wt; Tek‒Cre) mice (the construction and genotyping identification were seen in Supporting Information Fig. S7A and B) for subsequent experiments. Protein levels of NMMHC IIA in EC were examined by immunostaining (Fig. 5A), and expression of NMMHC IIA was further confirmed to decrease ∼50% via Western blot in MLECs (Fig. 5B). H&E staining of lung sections revealed no obvious abnormalities in Myh9fl/wt; Tek‒Cre mice. Mild lung injury was observed in this mouse model, compared with Myh9+/+; Tek‒Cre mice which exhibited prominent alveolar collapse, congestion, and thickening of alveolar septal walls following LPS stimulation (Fig. 5C). Myeloperoxidase (MPO) is an enzyme present mainly in neutrophils and to a lesser degree in monocytes. MPO has been demonstrated to be a local mediator of tissue damage and the marker of activation of neutrophils in various inflammatory diseases22. LPS challenge led to a marked increase in lung MPO levels in Myh9+/+; Tek‒Cre mice, which was significantly ameliorated in Myh9fl/wt; Tek‒Cre mice (Fig. 5D). EBA was injected into the tail vein and extent of extravasated dye was determined to evaluate pulmonary barrier function. Notably, Myh9fl/wt; Tek‒Cre mice exhibited improvement in LPS-induced pulmonary vasculature hyperpermeability (Fig. 5E), which was consistent with the immunostaining of albumin in lung tissues (Fig. 5F). Western blot results showed that VE-cadherin and p120-catenin levels in Myh9fl/wt; Tek‒Cre mice under endotoxin insult were significantly restored (Fig. 5G and H). These findings support that inhibition of endothelial NMMHC IIA ameliorates LPS-induced disruption of the pulmonary vascular endothelial barrier in mice.
Figure 5.

Endothelium-specific NMMHC IIA monoallelic deletion attenuates LPS-induced lung vascular hyperpermeability. Myh9fl/wt; Tek‒Cre and Myh9+/+; Tek‒Cre mice were challenged with LPS (5 mg/kg) via intratracheal instillation for 6 h. (A) Endothelial-specific monoallelic deletion of NMMHC IIA was analyzed by immunofluorescent co-staining of NMMHC IIA (green) and the endothelial cell marker CD31 (red), nuclei were stained with DAPI (blue). Arrows indicate the co-localization of NMMHC IIA and CD31 (n = 3). Scale bar = 20 μm. (B) NMMHC IIA expression levels in MLECs isolated from lung tissues were detected by Western blot (n = 3). (C) Paraffin-embedded lung tissue sections were stained with H&E and lung injury was analyzed (n = 5‒8). Scale bar = 50 μm. (D) Lung myeloperoxidase (MPO) levels were determined using specific kits after LPS treatment (n = 5‒8). (E) At 4 h after LPS administration, mice were i.v. injected with EBA. After 2 h, the dye was extracted from lung tissues and quantified (n = 6). (F) Immunofluorescence for extravascular albumin (green) in lung frozen sections, nuclei were stained with DAPI (blue) (n = 4). Scale bar = 50 μm. (G, H) Western blot analyses of VE-cadherin and p120-catenin expression in lung tissues (n = 3). All data are expressed as mean ± SD. ∗P < 0.05, ∗∗∗P < 0.001.
3.5. NMMHC IIA mediates the protective effect of RUS on pulmonary endothelial barrier function
Myh9fl/wt; Tek‒Cre mice were employed to validate whether RUS regulates endothelial barrier integrity through NMMHC IIA. The total protein content, white blood cell count and percentage of monocytes in BALF were detected to evaluate the pulmonary endothelial barrier function. We observed that NMMHC IIA deficiency in endothelium significantly inhibited LPS-triggered elevation of total protein content, white blood cell count, and percentage of monocytes in BALF, but RUS-mediated improvement was abrogated in Myh9fl/wt; Tek‒Cre mice after LPS insult (Supporting Information Fig. S8A‒C). MPO levels in Myh9fl/wt; Tek‒Cre mice was lower than that in Myh9+/+; Tek‒Cre mice, but there is no significant difference. And there was also no difference after RUS administration (Fig. S8D). To further confirm the moderating effect of RUS on NMMHC IIA in vitro, NMMHC IIA was knocked down with a siRNA-specific target Myh9 (Supporting Information Fig. S9). TER and EBA leakage analyses showed that NMMHC IIA knockdown counteracted the protective effect of RUS on endothelial barrier function (Fig. 6A and B). And Western blot results also suggested that NMMHC IIA downregulation abolished the regulation of RUS on LPS-induced activation of TLR4 pathway (Fig. 6C‒F). These data support a critical role of NMMHC IIA in the modulating effect of RUS on LPS-induced pulmonary endothelial barrier dysfunction.
Figure 6.

NMMHC IIA knockdown abolishes the protective effect of RUS on LPS-induced endothelial barrier destruction through activation of TLR4 signaling. (A, B) NMMHC IIA was knockdown via small interfering RNA in HUVECs. TER and EBA leakage assays were used to determine the effect of RUS on LPS-induced hyperpermeability. (C‒F) Western blot analyses of TLR4, p-Src (Y416), VE-cadherin, and p120-catenin expression in HUVECs. Data are expressed as mean ± SD, n = 3; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
3.6. LPS-induced NMMHC IIA dissociation from TLR4
It was reported that NMMHC IIA interacts with TLR4 and regulates tight junction in brain endothelial cells by mediating downstream transduction pathway12. In this study, the duolink PLA18 was used to examine the association between NMMHC IIA and TLR4 after LPS stimulation. The results showed that NMMHC IIA dissociated from TLR4 gradually in pulmonary endothelium upon LPS stimulation for 0–24 h (Fig. 7A and B). And in MLECs we also observed a rapid dissociation of NMMHC IIA from TLR4 induced by LPS in 5 min (Fig. 7C and D), supporting the involvement of NMMHC IIA in TLR4 signaling. To further explore the mechanisms by which NMMHC IIA interactions with TLR4 regulating downstream signaling, we mapped the regions in NMMHC IIA that mediated TLR4 activity. In molecular docking analyses, the head domain of NMMHC IIA exhibited highest probability of binding to TLR4 (Fig. 8A and B). HEK293T cells were co-transfected with expression plasmids for GFP-tagged NMMHC IIA domains and Myc-tagged TLR4. Interactions of TLR4 with each individual domain of NMMHC IIA were assessed using co-IP, which showed strong binding of the N-terminal and head domains, but not the other domains, to TLR4 (Fig. 8C). BLI analysis was used to detect the affinity of TLR4 and NMMHC IIA domains in vitro. The results indicated that TLR4 directly interacted with NMMHC IIA head domain, and the Kd value was 35 μmol/L (Fig. 8D). To identify the specific interacting sites in N-terminal and head domains of NMMHC IIA, we constructed two mutants of the N-terminal and five mutants of the head domain regions according to earlier data on the most frequent mutations responsible for ∼70% Myh9-related disorders23. Notably, binding to TLR4 was abolished in Myh9 N-terminal W33C, P35A, A44P, and F41I mutants and those of the Myh9 head domain (Q372R, K373N, R702H, R705H, Q706E, and R718W), clearly indicating involvement of these residues in mediating NMMHC IIA‒TLR4 interactions (Fig. 8E and F).
Figure 7.

NMMHC IIA is dissociated from TLR4 after LPS treatment. (A) Visualization of interactions between NMMHC IIA and TLR4 in pulmonary endothelium induced by LPS using the proximity ligation assay (PLA) (red), CD31 (green), and DAPI (blue). The interactions of NMMHC IIA and TLR4 in pulmonary endothelium decreased after LPS treatment (arrows). Scale bar = 50 μm. (B) Fluorescence intensity of PLA signals in vessels was quantified using Image J. (C) Confocal microscopy of MLECs challenged with LPS for 0, 5, 10, 20, 40, and 60 min, followed by PLA reaction for NMMHC IIA‒TLR4 interactions (red signal) and DAPI (blue) staining. Scale bars = 20 μm. (D) PLA signals were quantified using Image J. Data are expressed as mean ± SD, n = 3. ∗∗P < 0.01, ∗∗∗P < 0.001.
Figure 8.

NMMHC IIA binds to TLR4 through its N-terminal and head domains. (A) Binding mode analysis of NMMHC IIA and BB loop of TLR4 TIR domain via molecular docking (blue represents NMMHC IIA and red represents the binding probability of BB loop and NMMHC IIA). (B) Homology modeling of the complex of TLR4 TIR domain and NMMHC IIA. (C) Co-IP analysis was used to determine interactions between different domains of NMMHC IIA and TLR4. (D) BLI analysis was employed to detect the direct interaction between NMMHC IIA head domain and TLR4. (E, F) HEK293T cells were co-transfected with plasmids encoding Myc-tagged TLR4 and GFP-tagged Myh9 N-terminal or head domain mutants. Cell lysates were precipitated with anti-GFP antibody and immunoprecipitates detected via Western blot using anti-Myc and anti-GFP antibodies.
3.7. RUS inhibits LPS-induced activation of TLR4/Src/VE-cadherin signaling by modulating NMMHC IIA‒TLR4 interactions
To determine the regulation effect of RUS on NMMHC IIA‒TLR4 interactions, PLA assay was used in vivo (Fig. 9). We observed that RUS significantly impeded LPS-stimulated NMMHC IIA dissociation from TLR4 in pulmonary endothelium (Fig. 9A and D). We also performed immunofluorescence co-localization, co-IP, and PLA assays in MLECs. The results indicated that RUS effectively reversed LPS-stimulated disruptive interactions between NMMHC IIA and TLR4 in MLECs (Fig. 9B, C, E, and F). And Western blot results suggested the inhibition of RUS on NMMHC IIA and LPS-induced activation of TLR4/Src/VE-cadherin signal pathway (Fig. 9G‒J). These data implied that RUS suppressed TLR4 signaling activation induced by LPS through stabilizing NMMHC IIA‒TLR4 interactions.
Figure 9.

RUS suppresses LPS-induced NMMHC IIA dissociation from TLR4 and activation of TLR4/Src/VE-cadherin pathway. (A) Visualization of interactions between NMMHC IIA and TLR4 in pulmonary endothelium detected by PLA (red), CD31 (green) and DAPI (blue). Scale bar = 10 μm. (B) Immunofluorescence of NMMHC IIA (green) and TLR4 (red) in MLECs. Scale bar = 20 μm. (C) Visualization of interactions between NMMHC IIA and TLR4 in MLECs induced by LPS using the PLA (red) and nucleus was stained with DAPI (blue). Scale bar = 20 μm. (D) Fluorescence intensity of PLA signals in vessels in (A) was quantified using Image J. (E) PLA signals in MLECs in (C) were quantified using Image J. (F) NMMHC IIA was immunoprecipitated from whole cell lysates and immunoblotted for TLR4. (G‒J) Western blot analyses of NMMHC IIA, TLR4, p-Src (Y416), and VE-cadherin expression in MLECs. All data are expressed as mean ± SD, n = 3; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
4. Discussion
Increased pulmonary vascular permeability which leads to flooding of the alveoli with proteinaceous fluid and lung edema is the hallmark of ALI/ARDS3. Decreased lung compliance caused by lung oedema and elevation of pulmonary dead space resulted from impaired gas exchange are independent predictors of mortality in ALI/ARDS24. In the present study, we showed that the natural product RUS exerted significant protective effect of ALI through improving pulmonary endothelial hyperpermeability. By serial affinity chromatography, biolayer interferometry, microscale thermophoresis assays, and endothelial specific knockdown, we identified that NMMHC IIA was a direct binding target of RUS and the mechanism by which RUS attenuated LPS-induced endothelial barrier disruption was mainly attributed to the modulation of NMMHC IIA‒TLR4 interactions by targeting NMMHC IIA N-terminal and head domain.
TLR4 is well known for recognizing LPS and contributes to inflammation by initiating cellular release of cytokines and chemokines25,26. TLR4 signaling plays a key role in LPS-induced neutrophil accumulation and activation, which leads to increased microvascular permeability in lung injury27,28. Furthermore, TLR4 signaling is not only involved in the development of ALI characterized by increased pulmonary vascular permeability or viral and bacterial infectious diseases29,30, but also associated with some other diseases such as neurodegenerative disease, rheumatic disease and endocrine disease31, 32, 33. It is of great importance to further explore the regulating mechanism of TLR4 signaling. Structurally, TLR4 contains an extracellular ligand-binding domain (ECD), single transmembrane α-helix domain, and intracellular Toll/IL-1 receptor domain (TIR) responsible for downstream signaling34. Despite identification of the downstream pathway mediators of TLR4 signaling, the mechanisms underlying initiation of signaling induced by ligand binding-mediated dimerization remain further investigation. Our previous work has indicated that RUS played a protective role against LPS-induced endothelial cell apoptosis via regulating TLR4 signaling16, but whether RUS directly binds to TLR4 or TLR4 complex to regulate downstream signaling is not fully elucidated. In this study, we found that the target protein of RUS, NMMHC IIA, acted as a TLR4 inhibitory partner through interactions with TLR4 via its N-terminal and head domains under physiological conditions (Fig. 8).
NMMHC IIA is a motor protein with contractile property, which exhibits a very asymmetric shape with two major domains: the globular head at the N-terminal which includes actin-binding sites and the C-terminal α-helical rod domain (tail domain) which accounts for the formation of bipolar filaments35, 36, 37. NMMHC IIA participates in a range of cellular functions, such as migration, adhesion, phagocytosis, cytokinesis, and signal transduction, some of which depend on regulation of multiple protein‒protein interactions. For instance, Arf-like protein is required for cell migration through interactions with NMMHC IIA38, and association between the chemokine receptor, CXCR4, NMMHC IIA accounts for cellular chemotaxis39. A previous study by our group demonstrated that NMMHC IIA bound to TNF receptor 2 and mediated signal internalization in TNF-α-stimulated endothelial cells to regulate tissue factor expression and venous thrombosis10. Previous researches have shown that the regions of NMMHC IIA interacted with other proteins mainly locate in the tail domain, which may be involved in the function of cell migration and intracellular transport40, 41, 42. There are relatively less reports about the head functional domain of NMMHC IIA in protein‒protein interactions. In our present study, we found that RUS acted as a NMMHC IIA inhibitor by directly binding to its N-terminal and head domain (Fig. 3). Interestingly, there was also a directly interaction between TLR4 and NMMHC IIA N-terminal and head domain. Upon LPS binding to ECD of TLR4 to induce dimerization, interactions between NMMHC IIA and TLR4 are disrupted, thus initiating downstream signaling. And RUS could suppress this process by stabilizing NMMHC IIA‒TLR4 interactions (Fig. 9). Although studies have shown that the head domain of NMMHC IIA involved in the protein‒protein interactions plays a role in controlling cell shape or cell division35,43, we found that the head domain of NMMHC IIA might also be connected with the regulation of intracellular signal transduction. These findings enrich our knowledge of the regulatory mechanisms underlying TLR4 signaling and provide evidence on the potentially significant role of NMMHC IIA in pathological processes.
As is well-known, adherens junctions maintain tight association of neighboring pulmonary endothelial cells via VE-cadherin-mediated interactions. The cytoplasmic tail domain of VE-cadherin interacts with numerous intracellular partners, including β-catenin, plakoglobin (γ-catenin), and p120-catenin, which contribute to adherens junctions strength44. β-Catenin and plakoglobin are reported to form structural bridges between VE-cadherin and the actin cytoskeleton while p120-catenin was initially identified as a Src kinase substrate for cadherin clustering45, 46, 47. Moreover, VE-cadherin levels are regulated by p120-catenin, which interacts with the juxtamembrane domain and inhibits clathrin-mediated endocytosis and lysosomal degradation of VE-cadherin48. Src is activated in the settings of ALI, and stimulation of Src kinase activity is critical in the induction of endothelial hyperpermeability49,50. Consistent with earlier results, we observed an increase in Src phosphorylation at Tyr416 following LPS challenge, which could be attenuated by RUS administration through the inhibition of NMMHC IIA and dissociation from TLR4. Meanwhile RUS treatment prevented LPS-induced disruption of VE-cadherin at intercellular junctions and decreased protein expression of VE-cadherin, potentially due to blockade of Src phosphorylation and stabilization of interactions between p120-catenin and VE-cadherin.
The integrity of pulmonary vascular endothelial barrier is critical for maintaining lung homeostasis. Loss of barrier integrity is a pathophysiological hallmark of clinically relevant lung injury which occurs most commonly from sepsis, burn injury, and bacterial or viral infections. Based on our present results, we propose that the target protein of RUS, NMMHC IIA, may act as a key mediator in LPS-induced ALI through regulation of pulmonary endothelial barrier by modulating TLR4 signaling. These findings not merely provide broad insights into lung injury characterized by pulmonary vascular hyperpermeability, but also point to new therapeutic strategies for serious diseases associated with TLR4 signaling.
5. Conclusions
Our data indicate that the natural steroidal sapogenin RUS may be a potential inhibitor of NMMHC IIA. With the anti-inflammatory and anti-thrombotic effects that have been reported15,51, RUS protected against LPS-induced pulmonary endothelial barrier dysfunction by targeting NMMHC IIA and modulating NMMHC IIA‒TLR4 interactions (Fig. 10). Our findings demonstrate that NMMHC IIA may be the potential target for the protection of endothelial barrier destruction and for RUS to exert efficacy against LPS-induced ALI.
Figure 10.

The graphic illustration of the mechanism of RUS ameliorating LPS-induced pulmonary endothelial barrier dysfunction. NMMHC IIA binds to TLR4 through its N-terminal and head domains as an inactive complex under physiological conditions. Upon LPS binding to TLR4, NMMHC IIA is dissociated from TLR4, initiating downstream signaling and leading to endothelial barrier dysfunction by disruption of VE-cadherin junctions. RUS effectively prevents LPS-induced pulmonary endothelial barrier disruption by targeting NMMHC IIA and modulating NMMHC IIA‒TLR4 interactions. PMECs, pulmonary vascular endothelial cells; p120, p120-catenin.
Acknowledgments
The present study was supported by the National Natural Science Foundation of China (No. 81773971, China), and Double First-Class University Project (No. CPU2018GF07, China).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.09.017.
Contributor Information
Yuanyuan Zhang, Email: yuanyuanzhang@cpu.edu.cn.
Junping Kou, Email: junpingkou@cpu.edu.cn.
Author contributions
Yunhao Wu and Junping Kou conceived this project and design the experiments. Yunhao Wu and Xiu Yu performed most experiments and interpreted the data. Shuaishuai Gong, Yuwei Wang and Yalin Huang performed part of in vitro experiments. Siyu Jiang, Yuanli Xia, Jiahui Tang, and Fang Li performed part of in vivo experiments. Yuanyuan Zhang and Boyang Yu conceived this project. Yunhao Wu wrote the manuscript. Junping Kou and Yuanyuan Zhang revised the manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Thompson B.T., Chambers R.C., Liu K.D. Acute respiratory distress syndrome. N Engl J Med. 2017;377:562–572. doi: 10.1056/NEJMra1608077. [DOI] [PubMed] [Google Scholar]
- 2.Ranieri V.M., Rubenfeld G.D., Thompson B.T., Ferguson N.D., Caldwell E., Fan E., et al. Acute respiratory distress syndrome: the Berlin Definition. J Am Med Assoc. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 3.Millar F.R., Summers C., Griffiths M.J., Toshner M.R., Proudfoot A.G. The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax. 2016;71:462–473. doi: 10.1136/thoraxjnl-2015-207461. [DOI] [PubMed] [Google Scholar]
- 4.Dong W., He B., Qian H., Liu Q., Wang D., Li J., et al. RAB26-dependent autophagy protects adherens junctional integrity in acute lung injury. Autophagy. 2018;14:1677–1692. doi: 10.1080/15548627.2018.1476811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gong H., Rehman J., Tang H., Wary K., Mittal M., Chaturvedi P., et al. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest. 2015;125:652–664. doi: 10.1172/JCI77701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slutsky A.S., Ranieri V.M. Ventilator-induced lung injury. N Engl J Med. 2013;369:2126–2136. doi: 10.1056/NEJMra1208707. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z., Van Rossen E., Timmermans J.P., Geerts A., van Grunsven L.A., Reynaert H. Distinct roles for non-muscle myosin II isoforms in mouse hepatic stellate cells. J Hepatol. 2011;54:132–141. doi: 10.1016/j.jhep.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 8.Vicente-Manzanares M., Ma X., Adelstein R.S., Horwitz A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badirou I., Pan J., Legrand C., Wang A., Lordier L., Boukour S., et al. Carboxyl-terminal-dependent recruitment of nonmuscle myosin II to megakaryocyte contractile ring during polyploidization. Blood. 2014;124:2564–2568. doi: 10.1182/blood-2014-06-584995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhai K., Tang Y., Zhang Y., Li F., Wang Y., Cao Z., et al. NMMHC IIA inhibition impedes tissue factor expression and venous thrombosis via Akt/GSK3β‒NF-κB signalling pathways in the endothelium. Thromb Haemost. 2015;114:173–185. doi: 10.1160/TH14-10-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stefani C., Gonzalez-Rodriguez D., Senju Y., Doye A., Efimova N., Janel S., et al. Ezrin enhances line tension along transcellular tunnel edges via NMIIa driven actomyosin cable formation. Nat Commun. 2017;8:15839. doi: 10.1038/ncomms15839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lv Y., Liu W., Ruan Z., Xu Z., Fu L. Myosin IIA regulated tight junction in oxygen glucose-deprived brain endothelial cells via activation of TLR4/PI3K/Akt/JNK1/2/14-3-3ε/NF-κB/MMP9 signal transduction pathway. Cell Mol Neurobiol. 2019;39:301–319. doi: 10.1007/s10571-019-00654-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li P., Wei G., Cao Y., Deng Q., Han X., Huang X., et al. Myosin IIa is critical for cAMP-mediated endothelial secretion of von Willebrand factor. Blood. 2018;131:686–698. doi: 10.1182/blood-2017-08-802140. [DOI] [PubMed] [Google Scholar]
- 14.Kou J., Sun Y., Lin Y., Cheng Z., Zheng W., Yu B., et al. Anti-inflammatory activities of aqueous extract from Radix Ophiopogon japonicus and its two constituents. Biol Pharm Bull. 2005;28:1234–1238. doi: 10.1248/bpb.28.1234. [DOI] [PubMed] [Google Scholar]
- 15.Sun Q., Chen L., Gao M., Jiang W., Shao F., Li J., et al. Ruscogenin inhibits lipopolysaccharide-induced acute lung injury in mice: involvement of tissue factor, inducible NO synthase and nuclear factor (NF)-κB. Int Immunopharmacol. 2012;12:88–93. doi: 10.1016/j.intimp.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 16.Wu Y., Wang Y., Gong S., Tang J., Zhang J., Li F., et al. Ruscogenin alleviates LPS-induced pulmonary endothelial cell apoptosis by suppressing TLR4 signaling. Biomed Pharmacother. 2020;125:109868. doi: 10.1016/j.biopha.2020.109868. [DOI] [PubMed] [Google Scholar]
- 17.Bolte S., Cordelières F.P. A guided tour into subcellular co-localization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 18.Gullberg M., Andersson A.C. Visualization and quantification of protein‒protein interactions in cells and tissues. Nat Methods. 2010;7:641–647. [Google Scholar]
- 19.Yamamoto K., Yamazaki A., Takeuchi M., Tanaka A. A versatile method of identifying specific binding proteins on affinity resins. Anal Biochem. 2006;352:15–23. doi: 10.1016/j.ab.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 20.Ju J., Qi Z., Cai X., Cao P., Huang Y., Wang S., et al. The apoptotic effects of toosendanin are partially mediated by activation of deoxycytidine kinase in HL-60 cells. PLoS One. 2012;7:e52536. doi: 10.1371/journal.pone.0052536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conti M.A., Even-Ram S., Liu C., Yamada K.M., Adelstein R.S. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J Biol Chem. 2004;279:41263–41266. doi: 10.1074/jbc.C400352200. [DOI] [PubMed] [Google Scholar]
- 22.Aratani Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys. 2018;640:47–52. doi: 10.1016/j.abb.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Pecci A., Ma X., Savoia A., Adelstein R.S. MYH9: structure, functions and role of non-muscle myosin IIA in human disease. Gene. 2018;664:152–167. doi: 10.1016/j.gene.2018.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nuckton T.J., Alonso J.A., Kallet R.H., Daniel B.M., Pittet J.F., Eisner M.D., et al. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med. 2002;346:1281–1286. doi: 10.1056/NEJMoa012835. [DOI] [PubMed] [Google Scholar]
- 25.Ryu J.K., Kim S.J., Rah S.H., Kang J.I., Jung H.E., Lee D., et al. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4‒MD2 for efficient LPS recognition and transfer. Immunity. 2017;46:38–50. doi: 10.1016/j.immuni.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 26.Rosadini C.V., Kagan J.C. Early innate immune responses to bacterial LPS. Curr Opin Immunol. 2017;44:14–19. doi: 10.1016/j.coi.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andonegui G., Bonder C.S., Green F., Mullaly S.C., Zbytnuik L., Raharjo E., et al. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest. 2003;111:1011–1020. doi: 10.1172/JCI16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu G., Malik A.B., Minshall R.D. Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit Care Med. 2010;38:194–201. doi: 10.1097/CCM.0b013e3181bc7c17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirey K.A., Lai W., Scott A.J., Lipsky M., Mistry P., Pletneva L.M., et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. 2013;497:498–502. doi: 10.1038/nature12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takizawa H., Fritsch K., Kovtonyuk L.V., Saito Y., Yakkala C., Jacobs K., et al. Pathogen-induced TLR4‒TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell. 2017;21:225–240. doi: 10.1016/j.stem.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 31.Perez-Pardo P., Dodiya H.B., Engen P.A., Forsyth C.B., Huschens A.M., Shaikh M., et al. Role of TLR4 in the gut‒brain axis in Parkinson's disease: a translational study from men to mice. Gut. 2019;68:829–843. doi: 10.1136/gutjnl-2018-316844. [DOI] [PubMed] [Google Scholar]
- 32.Ma K., Li J., Wang X., Lin X., Du W., Yang X., et al. TLR4+CXCR4+ plasma cells drive nephritis development in systemic lupus erythematosus. Ann Rheum Dis. 2018;77:1498–1506. doi: 10.1136/annrheumdis-2018-213615. [DOI] [PubMed] [Google Scholar]
- 33.Ji Y., Sun S., Shrestha N., Darragh L.B., Shirakawa J., Xing Y., et al. Toll-like receptors TLR2 and TLR4 block the replication of pancreatic β cells in diet-induced obesity. Nat Immunol. 2019;20:677–686. doi: 10.1038/s41590-019-0396-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawai T., Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 35.Smith A.S., Pal K., Nowak R.B., Demenko A., Zaninetti C., Da Costa L., et al. MYH9-related disease mutations cause abnormal red blood cell morphology through increased myosin‒actin binding at the membrane. Am J Hematol. 2019;94:667–677. doi: 10.1002/ajh.25472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pal K., Nowak R., Billington N., Liu R., Ghosh A., Sellers J.R., et al. Megakaryocyte migration defects due to nonmuscle myosin IIA mutations underlie thrombocytopenia in MYH9-related disease. Blood. 2020;135:1887–1898. doi: 10.1182/blood.2019003064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franke J.D., Dong F., Rickoll W.L., Kelley M.J., Kiehart D.P. Rod mutations associated with MYH9-related disorders disrupt nonmuscle myosin-IIA assembly. Blood. 2005;105:161–169. doi: 10.1182/blood-2004-06-2067. [DOI] [PubMed] [Google Scholar]
- 38.Casalou C., Seixas C., Portelinha A., Pintado P., Barros M., Ramalho J.S., et al. Arl13b and the non-muscle myosin heavy chain IIA are required for circular dorsal ruffle formation and cell migration. J Cell Sci. 2014;127:2709–2722. doi: 10.1242/jcs.143446. [DOI] [PubMed] [Google Scholar]
- 39.Rey M., Vicente-Manzanares M., Viedma F., Yáñez-Mó M., Urzainqui A., Barreiro O., et al. Cutting edge: association of the motor protein nonmuscle myosin heavy chain-IIA with the C terminus of the chemokine receptor CXCR4 in T lymphocytes. J Immunol. 2002;169:5410–5414. doi: 10.4049/jimmunol.169.10.5410. [DOI] [PubMed] [Google Scholar]
- 40.Dahan I., Yearim A., Touboul Y., Ravid S. The tumor suppressor Lgl1 regulates NMII-A cellular distribution and focal adhesion morphology to optimize cell migration. Mol Biol Cell. 2012;23:591–601. doi: 10.1091/mbc.E11-01-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiss B., Duelli A., Radnai L., Kékesi K.A., Katona G., Nyitray L. Crystal structure of the S100A4‒nonmuscle myosin IIA tail fragment complex reveals an asymmetric target binding mechanism. Proc Natl Acad Sci U S A. 2012;109:6048–6053. doi: 10.1073/pnas.1114732109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petrosyan A., Ali M.F., Verma S.K., Cheng H., Cheng P.W. Non-muscle myosin IIA transports a Golgi glycosyltransferase to the endoplasmic reticulum by binding to its cytoplasmic tail. Int J Biochem Cell Biol. 2012;44:1153–1165. doi: 10.1016/j.biocel.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Obungu V.H., Lee Burns A., Agarwal S.K., Chandrasekharapa S.C., Adelstein R.S., Marx S.J. Menin, a tumor suppressor, associates with nonmuscle myosin II-A heavy chain. Oncogene. 2003;22:6347–6358. doi: 10.1038/sj.onc.1206658. [DOI] [PubMed] [Google Scholar]
- 44.Lampugnani M.G., Dejana E., Giampietro C. Vascular endothelial (VE)-cadherin, endothelial adherens junctions, and vascular disease. Cold Spring Harb Perspect Biol. 2018;10:a029322. doi: 10.1101/cshperspect.a029322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leckband D.E., de Rooij J. Cadherin adhesion and mechanotransduction. Annu Rev Cell Dev Biol. 2014;30:291–315. doi: 10.1146/annurev-cellbio-100913-013212. [DOI] [PubMed] [Google Scholar]
- 46.Su W., Kowalczyk A.P. The VE-cadherin cytoplasmic domain undergoes proteolytic processing during endocytosis. Mol Biol Cell. 2017;28:76–84. doi: 10.1091/mbc.E16-09-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reynolds A.B., Roczniak-Ferguson A. Emerging roles for p120-catenin in cell adhesion and cancer. Oncogene. 2004;23:7947–7956. doi: 10.1038/sj.onc.1208161. [DOI] [PubMed] [Google Scholar]
- 48.Nanes B.A., Grimsley-Myers C.M., Cadwell C.M., Robinson B.S., Lowery A.M., Vincent P.A., et al. p120-catenin regulates VE-cadherin endocytosis and degradation induced by the Kaposi sarcoma-associated ubiquitin ligase K5. Mol Biol Cell. 2017;28:30–40. doi: 10.1091/mbc.E16-06-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oyaizu T., Fung S.Y., Shiozaki A., Guan Z., Zhang Q., dos Santos C.C., et al. Src tyrosine kinase inhibition prevents pulmonary ischemia-reperfusion-induced acute lung injury. Intensive Care Med. 2012;38:894–905. doi: 10.1007/s00134-012-2498-z. [DOI] [PubMed] [Google Scholar]
- 50.Orsenigo F., Giampietro C., Ferrari A., Corada M., Galaup A., Sigismund S., et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. 2012;3:1208. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bi L.Q., Zhu R., Kong H., Wu S.L., Li N., Zuo X.R., et al. Ruscogenin attenuates monocrotaline-induced pulmonary hypertension in rats. Int Immunopharmacol. 2013;16:7–16. doi: 10.1016/j.intimp.2013.03.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.